

Abstract
Congenital cardiovascular malformations represent the most common type of birth defect and encompass a spectrum of anomalies that range from mild to severe. The etiology of congenital heart disease (CHD) is becoming increasingly defined based upon prior epidemiologic studies that supported the importance of genetic contributors and technological advances of the human genome. These have led to the discovery of a growing number of disease-contributing genetic abnormalities in individuals affected by CHD. The ever-growing population of adult CHD survivors, which are the result of reductions in mortality from CHD during childhood, and this newfound genetic knowledge have led to important questions regarding recurrence risks, the mechanisms by which these defects occur, the potential for novel approaches for prevention and the prediction of long-term cardiovascular morbidity in adult CHD survivors. Here, we will review the current status of genetic models that accurately model human CHD as they provide an important tool to answer these questions and test novel therapeutic strategies.
Keywords: congenital heart disease, animal models, cellular models, genetics
Introduction
Congenital heart disease (CHD) affects 2–3% of newborn infants when including cases of isolated bicuspid aortic valve (BAV) (Pierpont et al. 2018). The impact of this most common type of birth defects is increasing with tremendous improvements in the clinical management of individuals affected by CHD. This improved survival has resulted in a growing population of adult CHD survivors who now outnumber children with CHD. Unfortunately, these adult survivors often suffer from co-morbid conditions and are at risk for neurodevelopmental and cognitive deficits, cardiac arrhythmias, and ventricular dysfunction which contribute to their early mortality (Russell et al. 2018). Epidemiologic studies investigating the etiology of CHD have implicated both genetic and environmental factors. With recent advancement in next generation sequencing technologies, a genetic abnormality, defined as chromosomal aneuploidy or a pathogenic chromosomal copy number or single gene variant, is able to be detected in approximately 1/3 of CHD cases (Akhirome et al. 2017). While the majority of CHD cases remain unexplained, the discoveries of these genetic contributors have led to the development of genetic models of CHD.
In vivo and in vitro model systems are important tools to understand disease pathogenesis and progression as well as to investigate its underlying mechanisms. In addition, these models can serve to test preclinical therapeutic interventions. Accordingly, the best models recapitulate the anatomy and physiology of the disease being studied in humans and are also cost-effective and easy to generate and manipulate. Another aspect of a translational genetic model is that the genetic abnormality has a strong and consistent association with the disease phenotype in humans. In the fields of cardiac developmental biology and CHD, numerous animal models from Drosophila melanogaster (fruit fly), Danio rerio (zebrafish), Xenopus laevis (frog), Gallus gallus (chick) to small (mice and rats) and large mammals (sheep, dogs and pigs) have been utilized along with in vitro cell culture models.
Each of these models have their own benefits and limitations. As the focus of this review is the development of genetic models of human CHD, we have emphasized the role of murine models which are amenable to reverse genetics, share a high degree of sequence conservation to humans and have similar stages of cardiac morphogenesis along with adult cardiac structures and physiology. In addition, the field has amassed over 25 years of experience studying cardiac development and disease in mouse models. The murine model is not without its limitations primarily related to the time involved to generate and characterize a new mouse, which is both time-consuming and expensive. In addition, the models of CHD in mice often result in perinatal lethality limiting our ability to study long-term cardiovascular sequelae from the genetic abnormality. Alternatively, zebrafish, a lower species vertebrate model, is quite popular due to its low cost of housing, easily available genetic tools, the embryos lack of dependence upon a functional cardiovascular system and their optical transparency which allows for more detailed analysis of cellular mechanisms. However, the utility of relatively primitive two-chambered heart of the zebrafish is limiting for studying more complex structural abnormalities found in human CHD (Bakkers 2011). Similarly, the fruit fly has similar benefits in regards to being high throughput and lower cost but again is limited by its primitive cardiac structure (Zhu et al. 2017b). In vitro non-cardiac cell-based models have been used to study potentially pathogenic sequence variants identified in affected individuals with CHD, but have numerous limitations in regards to the biologic relevance of these observations. More recently, patient-specific induced pluripotent stem cells (iPSCs) have begun to be investigated as a model to study human CHD (Mital 2016; Mercer and Evans 2017). Somewhat similar to the weaknesses related to zebrafish and fruit fly in regards to modeling the structural and morphogenetic aspects of CHD, these models are the most genetically similar and may allow for the investigation of the most relevant molecular mechanisms.
In this chapter, we discuss the genetic models of CHD that are linked to well-established monogenic causes of CHD with a focus on the murine model system where these CHD subtypes can morphologically be recapitulated. We have divided these models according to subtypes of CHD including cardiac septation defects, heart valve malformations and conotruncal heart defects and discussed a subset of these models in more depth to highlight the insights that can be obtained. We also discuss the development of in vivo genetic models of more complex CHD, which are unlikely to be monogenic in etiology, and look forward to the use of other model systems in lower species and in vitro modeling using iPSCs.
Murine models of cardiac septation defects
Formation of a four-chambered heart with its separate pulmonary and systemic circulations requires the proper development of atrial and ventricular septa. Defects of cardiac septation are amongst the most common forms of CHD and include atrial septal defects (ASD), ventricular septal defects (VSD) and atrioventricular septal defects (AVSD) (Figure 1). Many ASD and VSD are small and close spontaneously, while hemodynamically significant ASD and VSD and all AVSD require cardiac intervention. Genetic abnormalities associated with cardiac septation defects have been well-described and include pathogenic variants in single genes. These findings have been the basis for the generation of numerous mouse models, which have provided important insights into disease mechanisms (Table). Studies in families with autosomal dominant inherited CHD have linked pathogenic sequence variants in the cardiac transcription factors, TBX5, NKX2–5 and GATA4, with cardiac septation defects in humans (reviewed in Pierpont et al. 2018).
Figure 1. Murine models of cardiac septation defects:
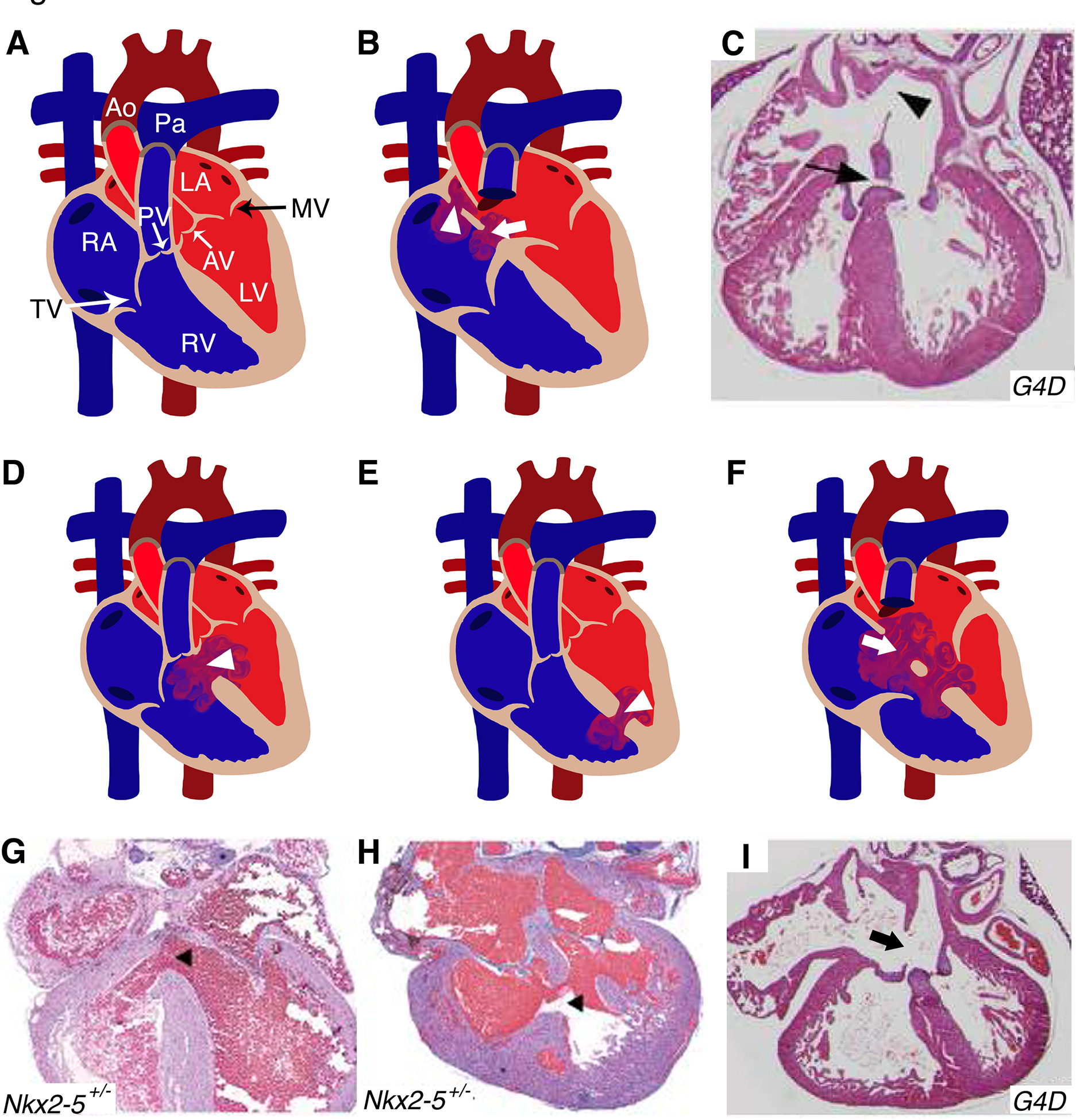
(A) Illustration depicts a cross-sectional view of a normal and mature four chambered heart. (B) Location of primum (arrowhead) and secundum (arrow) ASD in human heart diagram which has been recapitulated in Gata4Δex2/WT (G4D) mouse model (C). (D, E) Schematics display similar features of perimembranous and muscular VSDs (arrowhead) seen in human CHD patients were also detected in Nkx2–5+/− mice (G, H). (F) The diagram represents AVSD phenotype with single valve (arrow) noted in humans, resembled by the G4D murine model (I). RA: right atrium; RV: right ventricle; LA: left atrium; LV: left ventricle; MV: mitral valve; AV: aortic valve; PV: pulmonary valve; TV: tricuspid valve; Ao: Aorta; Pa: Pulmonary artery; ASD: atrial septal defect; VSD: ventricular septal defect; AVSD: atrioventricular septal defect. (C, F, I) and (G, H) have been adapted from Rajagopal et al. 2007 and Winston et al. 2010 with permission.
Table.
Murine models of congenital heart disease
| Cardiac septation defects | ||||||
|---|---|---|---|---|---|---|
| Gaie | OMIM # | human CHD phenotype | Syndrome with associated CHD | Mouse Genotype | Mouse phenotype | References |
| ACVR1(ALK2) | 102576 | AVSD | Alk2fl/−, Tie2-Cre | AVSD, VSD | (Wang et al. 2005) | |
| CITED2 | 602937 | ASD, VSD | Cited2−/− | ASD, VSD, DORV, TA | (Bamforth et al. 2001) | |
| CREBBP | 600140 | ASD, VSD, CoA, PS, BAV | Rubinstein-Taybi syndrome | CBP+/− | ASD, VSD, BAV | (Oike et al. 1999) |
| EP300 | 602700 | ASD, VSD, CoA, PS, BAV | Rubinstein-Taybi syndrome | EP300+/AS | ASD, VSD | (Shikama et al. 2003) |
| GATA4 | 600576 | ASD, PS, TOF, VSD, AVSD | GataΔex2/wt | ASD, VSD, AVSD | (Rajagopal et al. 2007) | |
| Gata4G295Ski/wt | ASD, AS, PS | (Misra et al. 2012; LaHaye et al. 2019) | ||||
| KMT2D | 602113 | AS, CoA, ASD, VSD, BAV, HLHS, TOF | Kabuki syndrome | Kmt2fl/fl, Mef2c-AHF-Cre | VSD | (Ang et al. 2016a) |
| NIPBL | 608667 | ASD, VSD, PS | Comelia de Lange syndrome | Nipbl+/− | ASD, VSD | (Kawauchi et al. 2009) |
| NKX2–5 | 600584 | ASD, atrioventricular conduction delay, TOF, VSD, HLHS | Nkx2. 5+/− | ASD, PFO, VSD, AVSD, BAV, AS | (Biben et al. 2000; Winston et al. 2010) | |
| Nkx2 S+/R52G | ASD, VSD, AVSD, Ebstein’s anomaly, atrioventricular block, tricuspid valve atresia | (Ashraf et al. 2014; Chowdhury et al. 2015) | ||||
| Nkx2.5R141C/+ | ASD, atrioventricular block, VSD | (Zakariyah et al. 2017) | ||||
| PTPN11 | 176876 | PS, AVSD, CoA, ASD, VSD, TOF, left ventricular outflow tract obstruction | Noonan syndrome | Ptpn11D6IG/+ | ASD, AVSD, DORV | (Araki et al. 2004) |
| SHOC2 | 602775 | PS, AVSD, CoA, ASD, VSD, TOF | Noonan syndrome | Sur-8Δ/fl, Tie2-Cre | VSD, DORV, TGA | (Yi et al. 2010) |
| TBX5 | 601620 | ASD, VSD | Holt-Oram syndrome | Tbx5del/+ | ASD, atrioventricular block, VSD | (Bruneau et al. 2001) |
| Tbx5flox/flox, Tie2-Cre | ASD, PFO | (Nadeau et al. 2010) | ||||
| 190685 | ASD, VSD, AVSD, TOF | Down syndrome | Tc1 | VSD, AVSD | (O’Doherty et al. 2005) | |
| Dp(10)1Yey/+,Dp(16)1Yey/+,Dp(17)1Yey/+ | VSD, AVSD | (Yu et al. 2010) | ||||
|
Dp1Tyb Dp3Tyb |
VSD, AVSD, DORV | (Lana-Elola et al. 2016) | ||||
| Cardiac valve malformations | ||||||
| Gene | OMIM # | human CHD phenotype | Syndrome with associated CHD | Mouse Genotype | Mouse phenotype | References |
| DCHS1 | 603057 | MVP | Dchs1+/− | MVP | (Durst et al. 2015) | |
| GATA5 | 611496 | BAV | Gatay−/− Gatafl/fl; Tie2-Cre | BAV, AS | (Laforest et al. 2011) | |
| GATA6 | 601656 | TA, ASD, AVSD, TOF, BAV | Gata6− Gata6wt/fl; Isll-Crs | BAV | (Gharibeh et al. 2018) | |
| MATR3 | 164015 | BAV, CoA, PDA | Matr3+/− | BAV, CoA, PDA, VSD, DORV | (Quintero-Rivera et al. 2015) | |
| NOTCH1 | 190198` | BAV, AS, HLHS, TOF, PS, CAVD | Notch1+/− | BAV, CAVD, aortic aneurysm | (Nigam and Srivastava 2009; Nus et al. 2011; Bosse et al. 2013; Koenig et al. 2017) | |
| Notch1fl/fl , Nfatc1-enCre | BAV | (MacGrogan et al. 2016; Wang et al. 2017) | ||||
| Notch1+/− mTRG2 | CAVD, AS | (Theodoris et al. 2017) | ||||
| Nos3−/−; Notch1+/− | BAV, AS, AR, CAVD, TOF | (Bosse et al. 2013; Koenig et al. 2016) | ||||
| SMAD6 | 602931 | BAV, AS, CoA | Smad−/− | cardiac valve hyperplasia | (Galvin et al. 2000) | |
| Conotruncal and aortic arch artery defects | ||||||
| Gene | OMIM # | human CHD phenotype | Syndrome with associated CHD | Mouse Genotype | Mouse phenotype | References |
| CHD7 | 608892 | TOF, DORV, VSD, ASD, TA, PS, AS, MS, TS | CHARGE syndrome | Chd7+/− | IAA, aortic arch defects | (Randall et al. 2009) |
| CRKL | 602007 | TOF, TA, IAA, VSD, aortic arch defects | 22q11 deletion syndrome | Crkol−/− | IAA, VSD, overriding aorta, DORV | (Guns et al. 2001) |
| FOXC1 | 601090 | TOF | Foxc1−/− | CoA, semilunar valve dysplasia, IAA, VSD | (Winnier et al. 1999) | |
| FOXC2 | 602402 | TOF | Foxc2−/− | IAA, VSD | (Winnier et al. 1999) | |
| FOXH1 | 603621 | TOF, VSD | Foxh1C/− | right isomerism, ASD, VSD, TGA, DORV | (Yamamoto et al. 2003) | |
| JAG1 | 601920 | TOF, PS, ASD, VSD | Allagille syndrome | Jag1fl/fl , Islet1-Cre Jaglfl/fl , Mef2c-AHF-Cre | DORV, PS, TA, ASD, VSD, aortic arch defects | (High et al. 2009) |
| TBX1 | 602054 | TOF, TA, IAA, VSD, aortic arch defects | 22q11 deletion syndrome | Df1−/+ | Aortic arch defects, VSD | (Lindsay et al. 1999) |
| Tbx1Neo2/Neo | TOF, TA, DORV, IAA, VSD, aortic arch defects | (Zhang and Baldini 2008) | ||||
| Tbx1neo/neo | TA, IAA, VSD, aortic arch defects | (Hu et al. 2004) | ||||
| Tbx1+/− | IAA, aortic arch defects | (Lindsay et al. 2001; Zhang et al. 2005) | ||||
| ZFPM2(FOG2) | 603693 | TOF, DORV | Fog2−/− | TOF, ASD, VSD | (Tevosian et al. 2000; Pizzuti et al. 2003) | |
| Other cardiac defects | ||||||
| Gene | OMIM # | human CHD phenotype | Syndrome with associated CHD | Mouse Gmotype | Mouse phenotype | References |
| ELN | 130160 | SVAS | Williams-Beuren syndrome | Eln+/− | SVAS | (Li et al. 1998) |
| FBN1 | 134797 | BAV, AR, MVP, aortic aneurysm, aortic dissection | Marfan syndrome | Fbn1C1039G/+ | MVP, aortic aneurysm | (Ng et al. 2004; Habashi et al. 2006) |
AS, aortic valve stenosis; ASD, atrial septal defect; AR, aortic valve regurgitation; AVSD, atrioventricular septal defect; BAV, bicuspid aortic valve; CAVD, calcific aortic valve disease; CHD, congenital heart diseas; CoA, coarctation of aorta; DORV, double outlet right ventricle; HLHS, hypoplastic left heart syndrome; IAA, interrupted aortic arch; MS, mitral valve stenosis; MVP, mitral valve prolapse; PDA, patent ductus arteriosus; PFO, patent foremen ovale; PS, pulmonic valve stenosis; SVAS, supravalvar aortic stenosis; TA, truncus arteriosus; TGA, transposition of great arteries; TOF, tetralogy of Fallot; TS tricuspid valve stenosis; VSD, ventricular septal defect.
TBX5
Pathogenic sequence variants in TBX5 have been linked to Holt-Oram syndrome, which is characterized by upper limb and cardiac abnormalities (Basson et al. 1997; Li et al. 1997; Basson et al. 1999). Cardiac defects are found in 75–85% of individuals with Holt-Oram syndrome including ASD (to the point of common atrium), VSD, and defects in atrioventricular conduction that can progress to complete heart block (Basson et al. 1994; Newbury-Ecob et al. 1996; Bruneau et al. 1999). The majority of heterozygous pathogenic variants result in a loss-of-function allele and this has led to the successful development of a murine model for Holt-Oram syndrome which recapitulates the majority of human disease phenotypes.
Mice heterozygous for a Tbx5 mutant allele, which deletes exon 3 that harbors the T-box domain, was generated by breeding Tbx5lox/+ from Black Swiss background to mice with a constitutively active Cre recombinase (EIIaCre from 129SvJ strain) (Bruneau et al. 2001). The resultant Tbx5del/+ progeny were viable but displayed a 40% perinatal lethality. This perinatal lethality was increased in Tbx5del/+, when the Tbx5lox/+ mice were from 129SvEv background indicating a role for genetic modifiers. The surviving adult Tbx5del/+ mice from a mixed background (Black Swiss and 129SvJ) demonstrated large ASD along with evidence of varying degrees of atrioventricular block. Interestingly, VSD were found only in Tbx5del/+ embryos or neonatal mice, likely representing the reason for observed perinatal mortality. Another publication demonstrated that complete deletion of Tbx5 from endocardial and endocardial-derived cells utilizing Tie2Cre mice resulted in secundum ASD while endocardial-specific heterozygous Tbx5 (eTbx5+/−) deletion demonstrated a 65% incidence of patent foramen ovale (PFO), suggesting that PFO is an attenuated manifestation of ASD (Nadeau et al. 2010).
NKX2–5
Disease-causing sequence variants in NKX2–5 were the first reported cause of non-syndromic CHD by studying 4 kindred with autosomal dominant disease (Schott et al. 1998). The predominant phenotype in affected family members was a secundum ASD along with atrioventricular conduction abnormalities. Since then, a range of cardiac abnormalities, including VSD, tetralogy of Fallot (TOF), pulmonary valve atresia, subvalvular aortic stenosis, and Ebstein’s anomaly have been correlated with heterozygous NKX2–5 mutations (Stallmeyer et al. 2010; Ellesoe et al. 2016). Heterozygous Nkx2–5 knockout mice displayed a spectrum of cardiac developmental phenotypes that are dependent upon the genetic background. In the FVB/N and 129/Sv backgrounds, Nkx2–5+/− mice have PFO, atrial septal aneurysms, and BAV with aortic stenosis (Biben et al. 2000). While Nkx2–5+/− mice in C57Bl/6 background demonstrate a high incidence (~40%) of secundum ASD and both muscular and membranous VSD (Figure 1, D, E, G, H) (Winston et al. 2010). Additional mice harboring specific human disease-associated variants in Nkx2–5 have also been reported and they display a spectrum of cardiac phenotypes which mimic human disease (Ashraf et al. 2014; Chowdhury et al. 2015; Zakariyah et al. 2017).
GATA4
A similar human genetics approach used for TBX5 and NKX2–5 led to the discovery that heterozygous mutations in GATA4 caused cardiac septation defects, primarily secundum ASD and membranous VSD (Garg et al. 2003). Multiple disease-causing, likely loss-of-function genetic variants in GATA4, have been reported for a range of cardiac defects including AVSD, pulmonic valve stenosis, and TOF (Okubo et al. 2004; Hirayama-Yamada et al. 2005; Sarkozy et al. 2005; Nemer et al. 2006; Schluterman et al. 2007). Mice harboring a mutant Gata4 allele (Gata4Δex2/WT), was generated in which the start codon and almost half of the coding region is deleted (Rajagopal et al. 2007). While mice heterozygous for this allele display no cardiac phenotypes in a mixed genetic background, when bred into C57Bl6 background display ~50% perinatal lethality. Analysis of Gata4Δex2/WT embryonic hearts identified CHD in 76% including AVSD; primum and secundum ASD; inlet, membranous and muscular VSD; and right ventricular hypoplasia (Figure 1 B, C, F, I). The incidence of CHD decreased to 30% when this mutant allele was backcrossed into FVB genetic background. Mice harboring a human CHD-segregating mutation (G296S) in GATA4 have also been reported and display a PFO in 80% of adult mice along with stenosis of the aortic and pulmonic valve at a lower penetrance (Misra et al. 2012; LaHaye et al. 2019).
In combination, these models of a common form of CHD have provided important insights for in vivo modeling of human CHD. First, the mouse model recapitulates structural malformations found in humans. Second, these models demonstrated that one genetic mutation could result in multiple cardiac abnormalities, a septation defect and atrioventricular block with mutations in NKX2–5 or TBX5 and a septation defect and pulmonary valve stenosis with a GATA4 mutation. Another important point was the variability of phenotypes found in these murine models with alterations in the genetic background supporting the role of modifier genes. Finally, an interesting observation that was suggested is that PFO, which is present in 10–25% of the population (Fisher et al. 1995; Homma et al. 2016), may represent a mild form of ASD and therefore may also have genetic contributors.
Murine models of cardiac valve malformations
Proper development of heart valves is important for unidirectional blood flow in humans and other species with chambered hearts. Congenital valve malformations are the most frequent type of CHD and bicuspid aortic valve (BAV) is the most common with a population prevalence of 1.3% (Verma and Siu 2014). In BAV, there is improper septation or fusion of two of the three aortic leaflets (right coronary, left coronary and non-coronary) (Figure 2) and is associated with the development of adult-onset complications including aortic valve stenosis (AVS) and regurgitation, calcific aortic valve disease (CAVD) and aortopathy. BAV can be subdivided into three morphologic categories, R-L (fusion of right and left coronary leaflets) which is most common in humans, R-NC (fusion of right and non-coronary) and L-NC (fusion of left and non-coronary). In addition, congenital stenosis of the aortic or pulmonic valve are common types of CHD. For each of these conditions, genetic etiologies in humans have been described and murine models developed (Table).
Figure 2. Mouse models for aortic valve malformations:
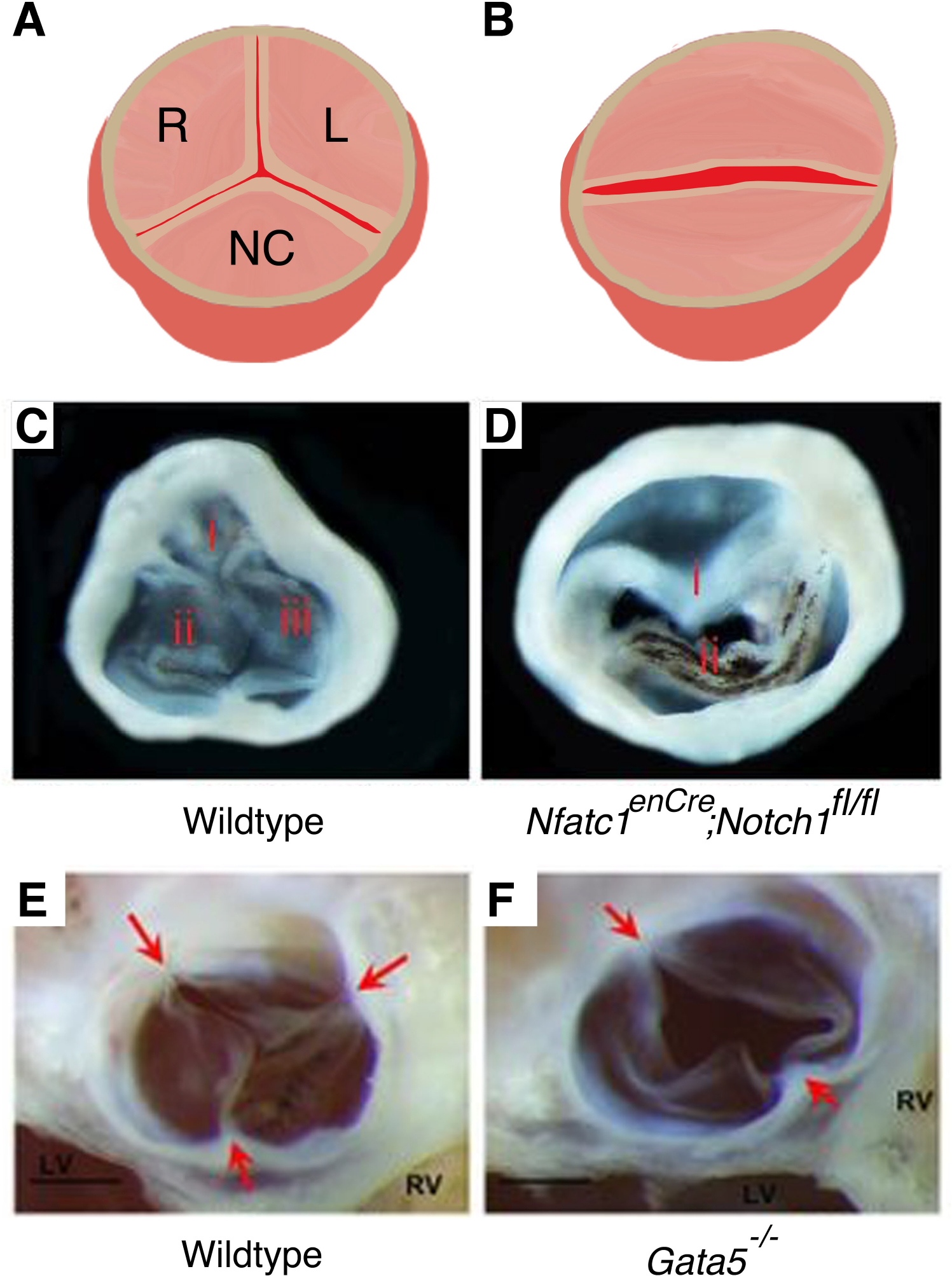
Illustrations represent a tricuspid aortic valve (A) and a bicuspid aortic valve, BAV (B). Right (R), left (L) and noncoronary (NC) leaflets are labelled. BAV were demonstrated in endothelial specific deletion of Notch1 (Nfatc1enCre; Notch1fl/fl) (D) and in Gata5−/− (F) mice compared to normal three leaflet structure found in wildtype mice (C, E). (C, D) and (E, F) have been adapted from Wang et al. 2017 and Laforest et al. 2011 with permission.
NOTCH1
By studying multi-generational families with autosomal dominant inherited cardiac disease, pathologic loss-of-function sequence variants in NOTCH1 were the first reported genetic cause of BAV and its associated aortic disease. Since then, other groups have also identified different NOTCH1 mutations that cause BAV, other forms of left-sided CHD and TOF (McBride et al. 2008; Kerstjens-Frederikse et al. 2016; Page et al. 2019). Mice heterozygous for Notch1 display minimal aortic disease with a low incidence of BAV and mild CAVD (Nigam and Srivastava 2009; Nus et al. 2011; Bosse et al. 2013). The BAV phenotype is more penetrant with complete deletion of Notch1 from valve endothelial cells (after endocardial to mesenchymal transformation) using Nfatc1enCre as these mice display a ~30% incidence of R-L BAV by one group (Figure 2C, D) but other investigators reported both R-L and R-NC BAV (MacGrogan et al. 2016; Wang et al. 2017). In addition, the analysis of Notch1+/− mice lacking telomerase activity by genetic deletion of Terc (telomerase RNA component; mTR) displayed robust CAVD with associated AVS (Theodoris et al. 2017). Similarly, the generation of Notch1+/− mice in the setting of complete deletion of endothelial nitric oxide synthase (Nos3−/−) resulted in high incidence of BAV with associated AVS and regurgitation and molecular changes consistent with CAVD (Bosse et al. 2013). Interestingly, Nos3−/−;Notch1+/− mice display significant perinatal lethality which was due to TOF-like phenotypes (Koenig et al. 2016). The role of genetic background in murine models has also been seen with Notch1+/− mice, where dilation of the aortic root is seen when backcrossed into a 129SvEv strain (Koenig et al. 2017).
GATA5 and GATA6
In addition to NOTCH1, GATA5, a member of the GATA family of transcription factors, has also been implicated in BAV where predicted damaging sequence variants in GATA5 have been identified in affected individuals by several investigators (Padang et al. 2012; Bonachea et al. 2014; Shi et al. 2014). Genetic deletion of Gata5 in mice led to a partially penetrant R-NC BAV phenotype (25%) with associated valve stenosis (Figure 2E–F) (Laforest et al. 2011). Further studies revealed that endothelial cell-specific deletion of Gata5 utilizing Tie2-Cre in a mixed genetic background (129SV/C57Bl6) recapitulated the R-NC BAV (21%) phenotype of Gata5−/− mice. Of note, BAV identified in humans with GATA5 variants had both R-NC and R-L BAV. Similar to NOTCH1, disease-causing variants in GATA5 have been reported in a spectrum of CHD (Jiang et al. 2013; Wei et al. 2013; Kassab et al. 2016).
Interestingly another member of the GATA transcription factor family has also been implicated in BAV. While mutations in GATA6 are implicated as a cause of truncus arteriosus (TA) and identified in patients with septal defects and TOF, murine models display a BAV phenotype (Kodo et al. 2009; Maitra et al. 2010). Mice haploinsufficient for Gata6 display 56% incidence of BAV in males and 27% incidence in females due to R-L fusion. Further analysis revealed that second heart field (SHF) specific deletion of Gata6 utilizing Isl1-Cre mice can recapitulate the BAV phenotype indicating SHF-specific role for Gata6 in valve development (Gharibeh et al. 2018). The conotruncal heart defects identified in humans are only found in Gata4+/−;Gata5+/− and Gata5+/−;Gata6+/− murine embryos, as they display double outlet right ventricle (DORV) and overriding aorta (Laforest and Nemer 2011).
These mouse models of BAV provided important tools to gain insight into the disease. First, generation of murine models of R-L and R-NC BAV imply that these phenotypes have distinct embryologic origins and clinical observations have suggested that they have unique long term co-morbidities (Mahadevia et al. 2014). These murine models also display the common complications of BAV, which include CAVD and aortopathy. Accordingly, the models will serve as important tools that can be used to study the mechanisms of disease progression and potentially test novel therapies. A final observation is the genotype-phenotype discrepancy in leaflet fusion phenotype found in mouse models versus humans with corresponding genetic mutations.
Murine models of conotruncal and aortic arch artery defects
Malformations of the cardiac outflow tract and aortic arch include a range of CHD from TOF, DORV, TA to interrupted aortic arch (IAA) (Figure 3). The etiology for majority of conotruncal and aortic arch defects remains unknown, however, a known genetic risk factor is 22q11 microdeletion, which accounts for approximately 12% of conotruncal heart defects (Momma 2010).
Figure 3. Murine models for conotruncal and aortic arch artery defects.
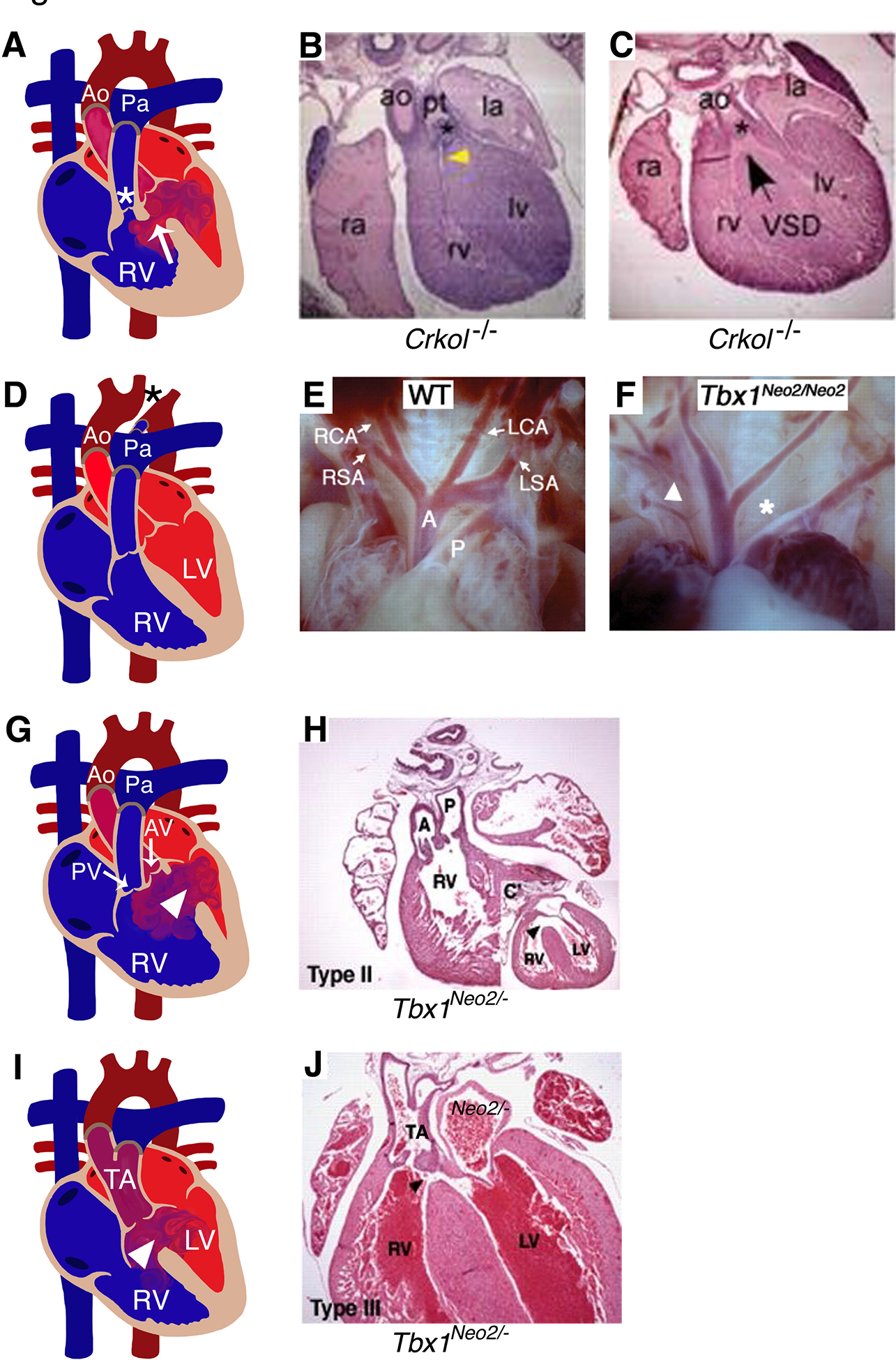
(A) Diagram illustrates TOF, characterized by right ventricular hypertrophy, VSD (arrow), stenosis of pulmonary valve and sub-valvar area (*) in human heart. Comparable TOF phenotype was observed in mouse model of Crkol−/− at embryonic day 16.5 shown in B and C. (D, F) IAA (*) detected in human patients were recapitulated in Tbx1Neo2/Neo2 mouse model at E18.5 compared to wildtype mouse (E). (G, I) Representative diagram of a diseased heart display DORV and TA with VSD (arrow head) in human (G, I). Similar phenotypes were observed in Tbx1Neo2/Neo2 mice (H, J) at E18.5. RA: right atrium; RV: right ventricle; LA: left atrium; LV: left ventricle; AV: aortic valve; Ao/A: Aorta; Pa/P: Pulmonary artery; TOF: tetralogy of Fallot; IAA: Interrupted aortic arch; DORV: double outlet right ventricle; TA: truncus arteriosus; VSD: ventricular septal defect. (E, F, H, J) have been adapted from Guris et al. 2001 and Zhang and Baldini 2008 with permission.
22q11 Deletion Syndrome/TBX1/CRKL
Approximately 75% of patients with 22q11 deletion syndrome (22q11DS), who commonly harbor a ~3Mb heterozygous deletion of chromosome 22q11.2, are born with CHD, primarily conotruncal heart defects and aortic arch anomalies (Yamagishi and Srivastava 2003). Identifying the causative gene for CHD associated with 22q11DS was an important step in the investigation of genetic and developmental basis for CHD. Although molecular genetic studies revealed that >40 genes resided in the crucial 22q11.2 locus, direct sequencing failed to detect mutations in any genes in patients with 22q11DS (Lindsay 2001). Therefore, mouse models for 22q11DS were generated by creating orthologous chromosomal deletions. By using this genetically engineered mouse (Df1), which harbored a heterozygous deletion of the orthologous 22q11 region, investigators were successful in reproducing aortic arch abnormalities and some conotruncal heart defects in Df1/+ mice that recapitulated those in human 22q11DS (Lindsay et al. 1999). Subsequent studies demonstrated that pathogenic variants in TBX1, a gene located in chromosome 22q11.2, as the cause of cardiovascular defects that occur in 22q11DS (Lindsay et al. 2001; Merscher et al. 2001).
Heterozygosity of Tbx1 caused aortic arch anomalies, whereas homozygosity of Tbx1 led to most of the phenotypes observed in 22q11DS, including not only conotruncal and aortic arch defects but also thymic hypoplasia, parathyroid hypoplasia, cleft palate and abnormal facial features that are characteristic of this syndrome (Hu et al. 2004; Zhang and Baldini 2008). Several animal models with mutant Tbx1 alleles have also been generated (Table). Homozygous Tbx1 null mouse embryos are associated with 100% neonatal lethality with cardiovascular defects including conotruncal heart defects. However, mice harboring hypomorphic alleles of Tbx1 that express varying levels of Tbx1 display a range of defects including TOF, DORV, TA and IAA (Figure 3D–J) (Hu et al. 2004; Zhang and Baldini 2008).
CRKL, a gene encoding CRK like proto-oncogene, adaptor protein also maps within the commonly deleted region for 22q11DS. Mice homozygous for a targeted null mutation at Crkol locus (Crkol−/−) showed abnormal aortic arteries and malalignment of the outflow tract including IAA, overriding aorta and DORV (Figure 3A–C) (Guris et al. 2001). While TBX1 is considered to be the major genetic determinant for several phenotypes associated with 22q11DS, CRKL may also play a role (Guris et al. 2006; Moon et al. 2006).
Moreover, novel therapeutic strategies have been proposed using Tbx1 mutant mouse models. Canonical Wnt/β-catenin has major roles in cardiac outflow tract development that may act upstream of Tbx1. By gene expression profiling, Tbx1 and β-catenin were found to have opposing effects on anterior heart field (AHF) differentiation and consistent with this, genetic rescue experiments between Tbx1 and β-catenin in the AHF found that TA observed in Tbx1 conditional null embryos was partially rescued by loss of one allele of the β-catenin gene in the Mef2c-AHF-Cre domain (Racedo et al. 2017). While these genetic approaches are important, it is difficult to translate them to the clinical setting. A recent study using a high throughput screening approach identified vitamin B12 as a molecule capable of enhancing Tbx1 gene expression (Lania et al. 2016). Using Tbx1lacZ/+ mice as a readout, vitamin B12 was shown to increase expression of Tbx1 in vivo and partially rescue the Tbx1 haploinsufficient phenotype.
FOXC1, FOXC2, FOXH1 (Forkhead transcription factors)
The forkhead transcription factors Foxc1, Foxc2 and Foxh1 have been shown to recognize and bind to cis-regulatory elements in Tbx1 promoter and directly promote Tbx1 expression (Maeda et al. 2006). Mutations in FOXC1, FOXC2 and FOXH1 were identified in patients with TOF (Roessler et al. 2008; Topf et al. 2014). Mice null for Foxc1 have coarctation of the aorta and aortic and pulmonary valve dysplasia as well as IAA type B while Foxc2-null mice have IAA type A and type B (Winnier et al. 1999). In addition, Foxc1 and Foxc2 compound mutant mice also show coarctation of the aorta, aortic and pulmonary valve dysplasia, IAA type A and B (Winnier et al. 1999). Mice deficient for Foxh1, which mediates Nodal signaling, demonstrate defects in anterior-posterior patterning and in node formation (Yamamoto et al. 2001). Conditional deletion of Foxh1 in mice manifests severe conotruncal heart defects including transposition of the great arteries (TGA) and DORV (Yamamoto et al. 2003).
These studies demonstrate the utility of mouse models to generate models of CHD that involve malformations of multiple cardiovascular structures including cardiac septa, valves, and great vessels. In addition, they show the value of genetically engineered murine models to identify the CHD-causing genes within a contiguous gene deletion syndrome or pathologic copy number variation (CNV). Lastly, they show how models can be used to define molecular mechanisms which are then used to identify novel genetic or pharmacologic therapies that may ameliorate the severity of the CHD phenotype.
Murine models of complex congenital heart defects
The development of murine models which mimic the complex forms of CHD have been more difficult to develop. Most success has been obtained with heterotaxy syndrome, which is linked to mutation of genes involved in cilia morphogenesis. A classic example of this is the genetic association with mutations in ZIC3, which encodes a gene downstream of the left-right signaling pathway, in patients with X-linked heterotaxy (D’Alessandro et al. 2013). Consistent with the human phenotype mice, null for Zic3 and those harboring a hypomorphic allele show the spectrum of left-right patterning cardiac defects including TGA, single ventricle/DORV, AVSD and abnormal systemic venous connections along with other neural tube and skeletal malformations (Purandare et al. 2002; Haaning et al. 2013). Numerous genetic murine models of left-right patterning defects have also been reported that recapitulate human disease (Pierpont et al. 2018).
Another major subtype of left-sided human CHD is hypoplastic left heart syndrome (HLHS). HLHS is a severe form of CHD that accounts for 4–8% of all CHD, and is characterized by hypoplasia of the left ventricle, aorta, and other left-sided cardiac structures including atresia or stenosis of both the aortic and mitral valves (Figure 4). In spite of significant advances in the management of HLHS, mortality and morbidity remain significant, and its etiology is not well understood. Although genetic syndromes such as Turner and Jacobsen syndrome are associated with HLHS, role of individual genes are unknown. While mouse knockouts of Hand1 and Hand2, transcription factors expressed in developing ventricles, have provided critical insights in ventricular morphogenesis, they were unable to replicate the HLHS phenotype (McFadden et al. 2005). As no major genetic driver for HLHS has yet been identified, no monogenetic animal model for HLHS exists to study its molecular basis and an oligogenic etiology has been proposed (McBride et al. 2005).
Figure 4. Models for hypoplastic left heart syndrome (HLHS).
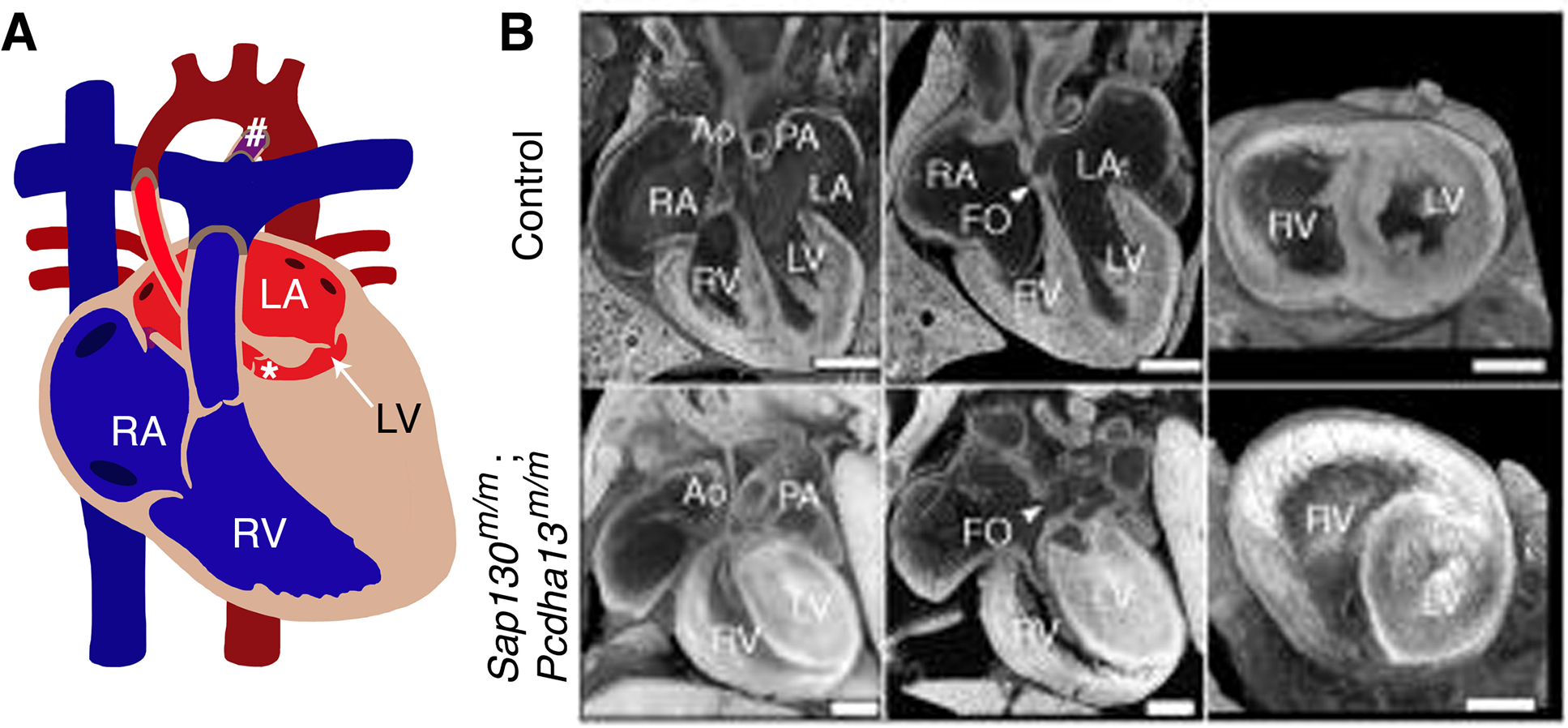
(A) Diagram represents a heart with HLHS, characterized by small/hypoplastic left ventricle (arrow) and aorta (*) and PDA (#). B represents similar features of HLHS phenotype in mouse model of Sap130m/m; Pcdha13m/m compared to littermate controls at postnatal day 0 (P0). RA: right atrium; RV: right ventricle; LA: left atrium; LV: left ventricle; Ao: Aorta; Pa: Pulmonary artery; PDA: Patent ductus arteriosus; FO: foramen ovale. (B) has been adapted from Liu et al. 2017 with permission.
A recent study described HLHS in mutant mice and the identification of genes causing HLHS (Liu et al. 2017). Mutations in Sap130, which encodes Sin3A-associated protein in the histone deacetylase complex, and Pcdha9 which encodes the cell-adhesion protein protocadherinA9, were validated by CRISPR-Cas9 genome editing in mice, revealing digenic etiology for HLHS. They also identified one individual with HLHS who harbored SAP130 and PCDHA13 mutations. 26% of Sap130m/m;Pcdha9m/m mutant mice exhibit phenotypes associated with HLHS where Sap130 mediates left ventricular hypoplasia, and Pcdha9 increases penetrance of aortic valve abnormalities, suggesting synergistic interactions between Sap130 and Pcdha9 mutations resulting in HLHS (Figure 4). Of note, a subset of the embryos displayed DORV with hypoplastic left ventricle as opposed to the strict morphologic definition of HLHS. These studies raise the question if digenic or oligogenic murine models will need to be developed to accurately recapitulate some of these more complex forms of CHD.
Other approaches for genetic modeling of congenital heart disease
Rapid advancements in next generation sequencing approaches have aided in the discovery of novel etiologies for CHD. The NHLBI-funded Pediatric Cardiac Genomics Consortium (PCGC) and an international study led by the Welcome Trust have performed whole exome sequencing of nearly 5,000 individuals with CHD (Sifrim et al. 2016; Jin 2017). These studies have uncovered numerous potential CHD-causing genes which are estimated to be ~400, which does not incorporate the potential different variants within each gene that may have differential effects. Accordingly, the sheer number of variants requires genetic models that allow for a more high throughput analysis. Along these lines, the zebrafish and the fruit fly have emerged as in vivo and iPSCs as in vitro models, and the details of each are discussed below.
Zebrafish and Fruit Fly
The zebrafish (Danio rerio) has emerged as an important vertebrate model to study cardiovascular disease. In the two-chambered zebrafish heart, deoxygenated blood flows through single atrium and ventricle, which pumps blood through the gills for oxygenation. Despite the anatomic differences between the two-chambered zebrafish and four-chambered mammalian heart, several studies have indicated similar gene regulatory networks driving cardiogenesis (Asnani and Peterson 2014). Eighty-two percent of human disease related genes (listed in OMIM) have at least one orthologue in zebrafish and these are well-suited for genetic manipulation as well as morpholino-induced gene knockdown to simulate human cardiac disease (Howe et al. 2013). In addition, zebrafish larvae are transparent, which allows easy visualization of the heart and vasculature during development (Brown et al. 2016). The zebrafish model has already been utilized to model cardiac phenotypes associated with genes implicated in non-syndromic (e.g. GATA4, GATA6, MYH6, NKX2–5, and TBX20) and syndromic CHD (e.g. PTPN11, NIPBL, CHD7, TBX1, and TBX5) (reviewed in (Grant et al. 2017)). While the exact cardiac morphologic phenotypes are not often recapitulated, these high throughput studies highlight the ability for this model to screen for potential disease genes and genetic variants.
A relatively simple organism, the fruit fly (Drosophila melanogaster), is also being utilized in genetic studies. Although it is difficult to directly compare fly developmental defects to specific CHD subtypes, this can serve as a testing platform for disease-associated genes identified from patient genomic sequencing. Functional homologs of 75% of human disease associated genes are also represented in the fly genome (Reiter et al. 2001). In a recent study, a cardiac-specific, 4XHand-Gal4 driver, was combined with fly lines carrying UAS-Gene-IR RNAi based silencing constructs to examine cardiac structure and function for fly homologs. One hundred and thirty-four candidate disease genes published by the PCGC and 52% of these homologs demonstrated their involvement in cardiac development and function in flies (Zhu et al. 2017a). In addition, the Drosophila model system may be useful to test for gene-gene interactions that are important for heart formation which may be useful in attempts to define the oligogenic nature of complex CHD (as discussed above) (Vogler and Bodmer 2015).
Induced pluripotent stem cells (iPSC)
Various studies based on transgenic mouse models have verified genes involved in cardiac development. However, complete deletion of gene is often required to generate cardiac phenotypes in mouse model, which does not reflect the pathogenic point mutation and haploinsufficiency commonly observed in human CHD (Musunuru et al. 2018). Human-induced pluripotent stem cells (hiPSC) provide a unique in vitro platform to study human cells as they can be differentiated and expanded into multiple somatic cell types. When hiPSCs are derived from patient, they not only carry the exact pathogenic mutation in the human genome but also the genetic background, which may serve as a modifier of the CHD phenotype. These properties have uniquely qualified the hiPSCs model to study human disease in a dish (Mital 2016). Additionally, introduction and/or correction of genetic variants is possible with the genome editing tools which allows researchers to generate new disease models and determine therapeutic strategies for individuals. Initial progress has been made utilizing hiPSCs for comprehensive understanding of cardiomyopathies, arrhythmias, vascular disorders, along with metabolic risk factors for ischemic heart disease (Musunuru et al. 2018).
As discussed above, mutations in GATA4 and NOTCH1 have been linked to cardiac septal defects and aortic valve disease, respectively. In both cases, patient derived iPSCs have been used to define the underlying molecular mechanism for these diseases. Generation and analysis of iPSC-derived cardiomyocytes from patients with heterozygous GATA4 G296S missense mutation demonstrated that the mutant cells displayed decreased contractility and abnormal calcium handling along with dysregulation of Shh signaling, a pathway implicated in cardiac septation that was recently shown to be downstream of Gata4 in mice (Hoffmann et al. 2009; Ang et al. 2016b; Zhou et al. 2017). They also used multiple genomic approaches to investigate the underlying mechanisms. As anticipated, they found a disruption of the cooperative regulation of a cardiac gene program mediated by Gata4 and Tbx5 but surprisingly also noted an upregulation of an endocardial/endothelial gene program, demonstrating the multitude of cellular effects from a single point mutation. A similar approach was used to investigate NOTCH1 mutations, where iPSC-derived endothelial cells from patients with NOTCH1 mutations were examined. Using an in vitro model of vascular shear stress, and these mutant cells failed to activate anti-osteogenic, anti-inflammatory, and anti-oxidant pathways that were induced in control cells (Theodoris et al. 2015). This observation was consistent with observed susceptibility towards calcification of the BAV. Supravalvular aortic and pulmonary artery stenosis can occur due to mutations in ELN (elastin) or as part of Williams-Beuren syndrome, a continguous gene deletion syndrome at chromosome 7q11.23 where ELN resides. Disease-causing mutations in ELN were modeled using hiPSC-derived smooth muscle cells (SMCs) and demonstrated less differentiated SMCs, with impaired formation of actin filaments and vascular tubes, along with increased proliferation rates (Ge et al. 2012; Kinnear et al. 2013). The generation of these cellular models for NOTCH1 and ELN mutation-mediated disease have resulted in a novel tool to screen for novel pharmacologic therapies.
Apart from monogenic diseases, hiPSC studies are also important to delineate complex CHD caused by multiple genetic and environmental factors, such as HLHS. Accordingly, investigators have generated iPSC derived-cardiomyocytes from patients with HLHS and found that these cells display a diminished yield of cardiomyocytes, a reduced beating rate, disorganized sarcomeres and sarcoplasmic reticulum along with downregulation of NOTCH signaling components, which were interestingly rescued by nitric oxide stimulation (Jiang et al. 2014; Hrstka et al. 2017; Yang et al. 2017). Together these observations show the advantages of iPSCs as an in vitro model to understand cellular events leading to morphogenetic defects along with generating an important tool to investigate new therapies. In addition, iPSC cells may serve as a promising strategy for the functional examination of the predicted disease-causing variants in the non-coding genome that are identified from whole genome sequencing approaches in the CHD population.
Concluding Remarks
With the sequencing of human genome and technological advances in genome analysis, there has been the discovery of numerous monogenic causes of non-syndromic and syndromic CHD. While cardiac defects span a spectrum of malformations involving the four-chambered heart, genetic manipulation of mice has been successful in recapitulating these phenotypes from septation defects, valve abnormalities to conotruncal heart defects. These models have served to define disease mechanisms and have also allowed for a better understanding of genotype-phenotype correlations and the importance of modifiers that result in phenotypic variability. In addition, novel gene-environment effects have been discovered where maternal exercise is able to rescue the ASD phenotype in Nkx2–5 haploinsufficient mice and Notch1 haploinsufficient embryos exposed to maternal diabetes display a highly penetrant VSD phenotype (Schulkey et al. 2015; Basu et al. 2017). Also for mice that survive to adulthood, they allow for investigation into the role of cardiac developmental genes in adult-onset cardiovascular complications.
The recent large-scale sequencing efforts in human CHD have also demonstrated its complex genetic architecture. These studies have frequently identified likely pathogenic CNV and rare and predicted pathogenic sequence variants in children with CHD. For the majority of cases, the functional significance of the proposed candidate genes and genetic variants in causing CHD is unknown. Accordingly, analysis of this large number of genetic abnormalities is not amenable using the murine model system, which has been the gold standard. Therefore, it will be important to use additional in vivo (fruit fly and zebrafish) and in vitro (iPSC) model systems to functionally characterize these new potentially pathologic variants. Ultimately as the field moves forward, it may be necessary to generate larger animal models of CHD (i.e. pig) using gene-editing techniques (Ryu et al. 2018) as this will allow for surgical correction of these defects and long term studies to better understand the mechanisms of disease found in adult CHD survivors.
Acknowledgements
The authors thank Dr. Madhumita Basu for critical review of the manuscript. V.G. is supported by funding from the Abigail Wexner Research Institute at Nationwide Children’s Hospital and grants from the National Institutes of Health (R01HL121797 and R01HL132801). J.Y. is supported by funding from the Japan Heart Foundation/Bayer Yahukin Research Grant Abroad.
References
- Akhirome E, Walton NA, Nogee JM, Jay PY. 2017. The Complex Genetic Basis of Congenital Heart Defects. Circ J 81: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang SY, Uebersohn A, Spencer CI, Huang Y, Lee JE, Ge K, Bruneau BG. 2016a. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development 143: 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI et al. 2016b. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 167: 1734–1749 e1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA et al. 2004. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10: 849–857. [DOI] [PubMed] [Google Scholar]
- Ashraf H, Pradhan L, Chang EI, Terada R, Ryan NJ, Briggs LE, Chowdhury R, Zarate MA, Sugi Y, Nam HJ et al. 2014. A mouse model of human congenital heart disease: high incidence of diverse cardiac anomalies and ventricular noncompaction produced by heterozygous Nkx2–5 homeodomain missense mutation. Circ Cardiovasc Genet 7: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnani A, Peterson RT. 2014. The zebrafish as a tool to identify novel therapies for human cardiovascular disease. Dis Model Mech 7: 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkers J 2011. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res 91: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, Farza H, Henderson DJ, Hurst HC, Bhattacharya S. 2001. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat Genet 29: 469–474. [DOI] [PubMed] [Google Scholar]
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J et al. 1997. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15: 30–35. [DOI] [PubMed] [Google Scholar]
- Basson CT, Cowley GS, Solomon SD, Weissman B, Poznanski AK, Traill TA, Seidman JG, Seidman CE. 1994. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med 330: 885–891. [DOI] [PubMed] [Google Scholar]
- Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G et al. 1999. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc Natl Acad Sci U S A 96: 2919–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu M, Zhu JY, LaHaye S, Majumdar U, Jiao K, Han Z, Garg V. 2017. Epigenetic mechanisms underlying maternal diabetes-associated risk of congenital heart disease. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Koentgen F, Robb L, Feneley M et al. 2000. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2–5. Circ Res 87: 888–895. [DOI] [PubMed] [Google Scholar]
- Bonachea EM, Chang SW, Zender G, LaHaye S, Fitzgerald-Butt S, McBride KL, Garg V. 2014. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr Res 76: 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, LaHaye S, Tao G, Lucchesi PA, Lincoln J et al. 2013. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol 60: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Samsa LA, Qian L, Liu J. 2016. Advances in the Study of Heart Development and Disease Using Zebrafish. J Cardiovasc Dev Dis 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. 1999. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol 211: 100–108. [DOI] [PubMed] [Google Scholar]
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE et al. 2001. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106: 709–721. [DOI] [PubMed] [Google Scholar]
- Chowdhury R, Ashraf H, Melanson M, Tanada Y, Nguyen M, Silberbach M, Wakimoto H, Benson DW, Anderson RH, Kasahara H. 2015. Mouse Model of Human Congenital Heart Disease: Progressive Atrioventricular Block Induced by a Heterozygous Nkx2–5 Homeodomain Missense Mutation. Circ Arrhythm Electrophysiol 8: 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro LC, Latney BC, Paluru PC, Goldmuntz E. 2013. The phenotypic spectrum of ZIC3 mutations includes isolated d-transposition of the great arteries and double outlet right ventricle. Am J Med Genet A 161A: 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, Salani M, Talkowski ME, Brand H, Perrocheau M et al. 2015. Mutations in DCHS1 cause mitral valve prolapse. Nature 525: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellesoe SG, Johansen MM, Bjerre JV, Hjortdal VE, Brunak S, Larsen LA. 2016. Familial Atrial Septal Defect and Sudden Cardiac Death: Identification of a Novel NKX2–5 Mutation and a Review of the Literature. Congenit Heart Dis 11: 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher DC, Fisher EA, Budd JH, Rosen SE, Goldman ME. 1995. The incidence of patent foramen ovale in 1,000 consecutive patients. A contrast transesophageal echocardiography study. Chest 107: 1504–1509. [DOI] [PubMed] [Google Scholar]
- Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA Jr. et al. 2000. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet 24: 171–174. [DOI] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K et al. 2003. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424: 443–447. [DOI] [PubMed] [Google Scholar]
- Ge X, Ren Y, Bartulos O, Lee MY, Yue Z, Kim KY, Li W, Amos PJ, Bozkulak EC, Iyer A et al. 2012. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 126: 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharibeh L, Komati H, Bosse Y, Boodhwani M, Heydarpour M, Fortier M, Hassanzadeh R, Ngu J, Mathieu P, Body S et al. 2018. GATA6 Regulates Aortic Valve Remodeling, and Its Haploinsufficiency Leads to Right-Left Type Bicuspid Aortic Valve. Circulation 138: 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MG, Patterson VL, Grimes DT, Burdine RD. 2017. Modeling Syndromic Congenital Heart Defects in Zebrafish. Curr Top Dev Biol 124: 1–40. [DOI] [PubMed] [Google Scholar]
- Guris DL, Duester G, Papaioannou VE, Imamoto A. 2006. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell 10: 81–92. [DOI] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. 2001. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet 27: 293–298. [DOI] [PubMed] [Google Scholar]
- Haaning AM, Quinn ME, Ware SM. 2013. Heterotaxy-spectrum heart defects in Zic3 hypomorphic mice. Pediatr Res 74: 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C et al. 2006. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312: 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. 2009. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. J Clin Invest 119: 1986–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M et al. 2005. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 135: 47–52. [DOI] [PubMed] [Google Scholar]
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. 2009. sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 136: 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma S, Messe SR, Rundek T, Sun YP, Franke J, Davidson K, Sievert H, Sacco RL, Di Tullio MR. 2016. Patent foramen ovale. Nat Rev Dis Primers 2: 15086. [DOI] [PubMed] [Google Scholar]
- Howe K Clark MD Torroja CF Torrance J Berthelot C Muffato M Collins JE Humphray S McLaren K Matthews L et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496: 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrstka SC, Li X, Nelson TJ, Wanek Program Genetics Pipeline G. 2017. NOTCH1-Dependent Nitric Oxide Signaling Deficiency in Hypoplastic Left Heart Syndrome Revealed Through Patient-Specific Phenotypes Detected in Bioengineered Cardiogenesis. Stem Cells 35: 1106–1119. [DOI] [PubMed] [Google Scholar]
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. 2004. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 131: 5491–5502. [DOI] [PubMed] [Google Scholar]
- Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ, Fang WY, Chen XZ, Zhang W, Wang XZ, Yang YQ. 2013. Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int J Cardiol 165: 570–573. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Habibollah S, Tilgner K, Collin J, Barta T, Al-Aama JY, Tesarov L, Hussain R, Trafford AW, Kirkwood G et al. 2014. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS-derived cardiac myocytes. Stem Cells Transl Med 3: 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC et al. 2017. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nature Genetics 49: 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassab K, Hariri H, Gharibeh L, Fahed AC, Zein M, El-Rassy I, Nemer M, El-Rassi I, Bitar F, Nemer G. 2016. GATA5 mutation homozygosity linked to a double outlet right ventricle phenotype in a Lebanese patient. Mol Genet Genomic Med 4: 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA et al. 2009. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 5: e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstjens-Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, Klein Wassink-Ruiter JS, van der Zwaag PA, du Marchie Sarvaas GJ, Bergman KA et al. 2016. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med 18: 914–923. [DOI] [PubMed] [Google Scholar]
- Kinnear C, Chang WY, Khattak S, Hinek A, Thompson T, de Carvalho Rodrigues D, Kennedy K, Mahmut N, Pasceri P, Stanford WL et al. 2013. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med 2: 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R, Yamagishi H. 2009. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci U S A 106: 13933–13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig SN, Bosse K, Majumdar U, Bonachea EM, Radtke F, Garg V. 2016. Endothelial Notch1 Is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. J Am Heart Assoc 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig SN, LaHaye S, Feller JD, Rowland P, Hor KN, Trask AJ, Janssen PM, Radtke F, Lilly B, Garg V. 2017. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B, Andelfinger G, Nemer M. 2011. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest 121: 2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B, Nemer M. 2011. GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev Biol 358: 368–378. [DOI] [PubMed] [Google Scholar]
- LaHaye S, Majumdar U, Yasuhara J, Koenig SN, Matos-Nieves A, Kumar R, Garg V. 2019. Developmental origins for semilunar valve stenosis identified in mice harboring congenital heart disease-associated GATA4 mutation. Dis Model Mech. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana-Elola E, Watson-Scales S, Slender A, Gibbins D, Martineau A, Douglas C, Mohun T, Fisher EM, Tybulewicz V. 2016. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lania G, Bresciani A, Bisbocci M, Francone A, Colonna V, Altamura S, Baldini A. 2016. Vitamin B12 ameliorates the phenotype of a mouse model of DiGeorge syndrome. Hum Mol Genet 25: 4369–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. 1998. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102: 1783–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T et al. 1997. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15: 21–29. [DOI] [PubMed] [Google Scholar]
- Lindsay EA. 2001. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet 2: 858–868. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A. 1999. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401: 379–383. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ et al. 2001. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410: 97–101. [DOI] [PubMed] [Google Scholar]
- Liu X, Yagi H, Saeed S, Bais AS, Gabriel GC, Chen Z, Peterson KA, Li Y, Schwartz MC, Reynolds WT et al. 2017. The complex genetics of hypoplastic left heart syndrome. Nat Genet 49: 1152–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGrogan D, D’Amato G, Travisano S, Martinez-Poveda B, Luxan G, Del Monte-Nieto G, Papoutsi T, Sbroggio M, Bou V, Gomez-Del Arco P et al. 2016. Sequential Ligand-Dependent Notch Signaling Activation Regulates Valve Primordium Formation and Morphogenesis. Circ Res 118: 1480–1497. [DOI] [PubMed] [Google Scholar]
- Maeda J, Yamagishi H, McAnally J, Yamagishi C and Srivastava D (2006) Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev. Dyn, 235, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevia R, Barker AJ, Schnell S, Entezari P, Kansal P, Fedak PW, Malaisrie SC, McCarthy P, Collins J, Carr J et al. 2014. Bicuspid aortic cusp fusion morphology alters aortic three-dimensional outflow patterns, wall shear stress, and expression of aortopathy. Circulation 129: 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Koenig SN, Srivastava D, Garg V. 2010. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res 68: 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KL, Pignatelli R, Lewin M, Ho T, Fernbach S, Menesses A, Lam W, Leal SM, Kaplan N, Schliekelman P et al. 2005. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am J Med Genet A 134A: 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, Belmont JW, Cole SE. 2008. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet 17: 2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Barbosa AC, Richardson JA, Schneider MD, Srivastava D, Olson EN. 2005. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 132: 189–201. [DOI] [PubMed] [Google Scholar]
- Mercer EJ, Evans T. 2017. Congenital heart disease in a dish: progress toward understanding patient-specific mutations. J Thorac Dis 9: E510–E513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S et al. 2001. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104: 619–629. [DOI] [PubMed] [Google Scholar]
- Misra C, Sachan N, McNally CR, Koenig SN, Nichols HA, Guggilam A, Lucchesi PA, Pu WT, Srivastava D, Garg V. 2012. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet 8: e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital S 2016. Human Pluripotent Stem Cells to Model Congenital Heart Disease. in Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology (eds. Nakanishi T, Markwald RR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H), pp. 321–327, Tokyo. [Google Scholar]
- Momma K 2010. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 105: 1617–1624. [DOI] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. 2006. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell 10: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC, American Heart Association Council on Functional G, Translational B et al. 2018. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 11: e000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau M, Georges RO, Laforest B, Yamak A, Lefebvre C, Beauregard J, Paradis P, Bruneau BG, Andelfinger G, Nemer M. 2010. An endocardial pathway involving Tbx5, Gata4, and Nos3 required for atrial septum formation. Proc Natl Acad Sci U S A 107: 19356–19361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F. 2006. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 27: 293–294. [DOI] [PubMed] [Google Scholar]
- Newbury-Ecob RA, Leanage R, Raeburn JA, Young ID. 1996. Holt-Oram syndrome: a clinical genetic study. J Med Genet 33: 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP et al. 2004. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 114: 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam V, Srivastava D. 2009. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol 47: 828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nus M, MacGrogan D, Martinez-Poveda B, Benito Y, Casanova JC, Fernandez-Aviles F, Bermejo J, de la Pompa JL. 2011. Diet-induced aortic valve disease in mice haploinsufficient for the Notch pathway effector RBPJK/CSL. Arterioscler Thromb Vasc Biol 31: 1580–1588. [DOI] [PubMed] [Google Scholar]
- O’Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D et al. 2005. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309: 2033–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, Yamamura K. 1999. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet 8: 387–396. [DOI] [PubMed] [Google Scholar]
- Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N et al. 2004. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet 41: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C. 2012. Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol 53: 277–281. [DOI] [PubMed] [Google Scholar]
- Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, Sutcliffe L, Topf A, Bourgey M, Bourque G et al. 2019. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ Res 124: 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A et al. 2018. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association. Circulation 138: e653–e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzuti A, Sarkozy A, Newton AL, Conti E, Flex E, Digilio MC, Amati F, Gianni D, Tandoi C, Marino B et al. 2003. Mutations of ZFPM2/FOG2 gene in sporadic cases of tetralogy of Fallot. Hum Mutat 22: 372–377. [DOI] [PubMed] [Google Scholar]
- Purandare SM, Ware SM, Kwan KM, Gebbia M, Bassi MT, Deng JM, Vogel H, Behringer RR, Belmont JW, Casey B. 2002. A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development 129: 2293–2302. [DOI] [PubMed] [Google Scholar]
- Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, Lee JH, Higgins AW, Anchan RM, Roberts AE, Seong IS, Fan X, Lage K et al. 2015. MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet 24: 2375–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racedo SE, Hasten E, Lin M, Devakanmalai GS, Guo T, Ozbudak EM, Cai CL, Zheng D, Morrow BE. 2017. Reduced dosage of beta-catenin provides significant rescue of cardiac outflow tract anomalies in a Tbx1 conditional null mouse model of 22q11.2 deletion syndrome. PLoS Genet 13: e1006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D et al. 2007. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol 43: 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall V, McCue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, Vitelli F, Prescott K, Shaw-Smith C, Devriendt K et al. 2009. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest 119: 3301–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. 2001. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res 11: 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Ouspenskaia MV, Karkera JD, Velez JI, Kantipong A, Lacbawan F, Bowers P, Belmont JW, Towbin JA, Goldmuntz E et al. 2008. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet 83: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MW, Chung WK, Kaltman JR, Miller TA. 2018. Advances in the Understanding of the Genetic Determinants of Congenital Heart Disease and Their Impact on Clinical Outcomes. J Am Heart Assoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Prather RS, Lee K. 2018. Use of gene-editing technology to introduce targeted modifications in pigs. J Anim Sci Biotechnol 9: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B. 2005. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet 42: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluterman MK, Krysiak AE, Kathiriya IS, Abate N, Chandalia M, Srivastava D, Garg V. 2007. Screening and biochemical analysis of GATA4 sequence variations identified in patients with congenital heart disease. Am J Med Genet A 143A: 817–823. [DOI] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. 1998. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science 281: 108–111. [DOI] [PubMed] [Google Scholar]
- Schulkey CE, Regmi SD, Magnan RA, Danzo MT, Luther H, Hutchinson AK, Panzer AA, Grady MM, Wilson DB, Jay PY. 2015. The maternal-age-associated risk of congenital heart disease is modifiable. Nature 520: 230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Wang Q et al. 2014. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med 33: 1219–1226. [DOI] [PubMed] [Google Scholar]
- Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth JF, Marino S, Wittwer J, Scheidweiler A, Eckner R. 2003. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J 22: 5175–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Hitz M-P, Wilsdon A, Breckpot J, Turki SHA, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ et al. 2016. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nature Genetics 48: 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallmeyer B, Fenge H, Nowak-Gottl U, Schulze-Bahr E. 2010. Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet 78: 533–540. [DOI] [PubMed] [Google Scholar]
- Tevosian SG, Deconinck AE, Tanaka M, Schinke M, Litovsky SH, Izumo S, Fujiwara Y, Orkin SH. 2000. FOG-2, a cofactor for GATA transcription factors, is essential for heart morphogenesis and development of coronary vessels from epicardium. Cell 101: 729–739. [DOI] [PubMed] [Google Scholar]
- Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, Srivastava D. 2015. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 160: 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoris CV, Mourkioti F, Huang Y, Ranade SS, Liu L, Blau HM, Srivastava D. 2017. Long telomeres protect against age-dependent cardiac disease caused by NOTCH1 haploinsufficiency. J Clin Invest 127: 1683–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topf A, Griffin HR, Glen E, Soemedi R, Brown DL, Hall D, Rahman TJ, Eloranta JJ, Jungst C, Stuart AG et al. 2014. Functionally significant, rare transcription factor variants in tetralogy of Fallot. PLoS One 9: e95453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Siu SC. 2014. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med 370: 1920–1929. [DOI] [PubMed] [Google Scholar]
- Vogler G, Bodmer R. 2015. Cellular Mechanisms of Drosophila Heart Morphogenesis. J Cardiovasc Dev Dis 2: 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Sridurongrit S, Dudas M, Thomas P, Nagy A, Schneider MD, Epstein JA, Kaartinen V. 2005. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol 286: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu B, Farrar E, Lui W, Lu P, Zhang D, Alfieri CM, Mao K, Chu M, Yang D et al. 2017. Notch-Tnf signalling is required for development and homeostasis of arterial valves. Eur Heart J 38: 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li RG, Xu YJ, Yang YQ. 2013. GATA5 loss-of-function mutations underlie tetralogy of fallot. Int J Med Sci 10: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnier GE, Kume T, Deng K, Rogers R, Bundy J, Raines C, Walter MA, Hogan BL, Conway SJ. 1999. Roles for the winged helix transcription factors MF1 and MFH1 in cardiovascular development revealed by nonallelic noncomplementation of null alleles. Dev Biol 213: 418–431. [DOI] [PubMed] [Google Scholar]
- Winston JB, Erlich JM, Green CA, Aluko A, Kaiser KA, Takematsu M, Barlow RS, Sureka AO, LaPage MJ, Janss LL et al. 2010. Heterogeneity of genetic modifiers ensures normal cardiac development. Circulation 121: 1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Srivastava D. 2003. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med 9: 383–389. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Meno C, Sakai Y, Shiratori H, Mochida K, Ikawa Y, Saijoh Y, Hamada H. 2001. The transcription factor FoxH1 (FAST) mediates Nodal signaling during anterior-posterior patterning and node formation in the mouse. Genes Dev 15: 1242–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Mine N, Mochida K, Sakai Y, Saijoh Y, Meno C, Hamada H. 2003. Nodal signaling induces the midline barrier by activating Nodal expression in the lateral plate. Development 130: 1795–1804. [DOI] [PubMed] [Google Scholar]
- Yang C, Xu Y, Yu M, Lee D, Alharti S, Hellen N, Ahmad Shaik N, Banaganapalli B, Sheikh Ali Mohamoud H, Elango R et al. 2017. Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum Mol Genet 26: 3031–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Chen M, Wu X, Yang X, Xu T, Zhuang Y, Han M, Xu R. 2010. Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev Dyn 239: 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N et al. 2010. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet 19: 2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakariyah AF, Rajgara RF, Veinot JP, Skerjanc IS, Burgon PG. 2017. Congenital heart defect causing mutation in Nkx2.5 displays in vivo functional deficit. J Mol Cell Cardiol 105: 89–98. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Baldini A. 2008. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum Mol Genet 17: 150–157. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A et al. 2005. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 132: 5307–5315. [DOI] [PubMed] [Google Scholar]
- Zhou L, Liu J, Xiang M, Olson P, Guzzetta A, Zhang K, Moskowitz IP, Xie L. 2017. Gata4 potentiates second heart field proliferation and Hedgehog signaling for cardiac septation. Proceedings of the National Academy of Sciences 114: E1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JY, Fu Y, Nettleton M, Richman A, Han Z. 2017a. High throughput in vivo functional validation of candidate congenital heart disease genes in Drosophila. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JY, Fu Y, Richman A, Han Z. 2017b. Validating Candidate Congenital Heart Disease Genes in Drosophila. Bio Protoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]