

Acutely decompensated pulmonary and cardiovascular physiology secondary to SARS-CoV2 infection presents one of the gravest threats to human wellbeing in the past century. The most expeditious route to develop urgently needed therapeutic options for this disease is to repurpose drugs known to be safe in humans. SARS-CoV2 dysregulates renin-angiotensin-aldosterone system (RAAS) homeostasis, elevating angiotensin II (AngII) levels. Since hyperactivation of the angiotensin II type 1 receptor (AT1R) appears to be a major contributor to adverse outcomes in patients with COVID-19-related acute respiratory distress syndrome (ARDS), approaches to dampen excess AngII activity are currently in clinical trials for COVID-19, including the angiotensin receptor blocker (ARB) losartan. Sustained activation of AT1R by its endogenous ligand, the octapeptide hormone Angiotensin II (AngII), leads to heterotrimeric Gq protein signaling that culminates in potent vasoconstriction, cardiomyocyte hypertrophy, and fibrosis in the heart and lungs. However, the pleiotropic signaling downstream of the AT1R includes not only vasoconstrictive but also cardioprotective pathways. Therefore, we propose that an even more effective therapeutic option for severe acute COVID‑19 infections would be to selectively modulate rather than to categorically block AT1R signaling, a concept known as “biased signaling” (Figure 1). Notably, this strategy mimics built-in regulatory mechanisms of the RAAS system and can be achieved with a drug candidate with established safety in humans.
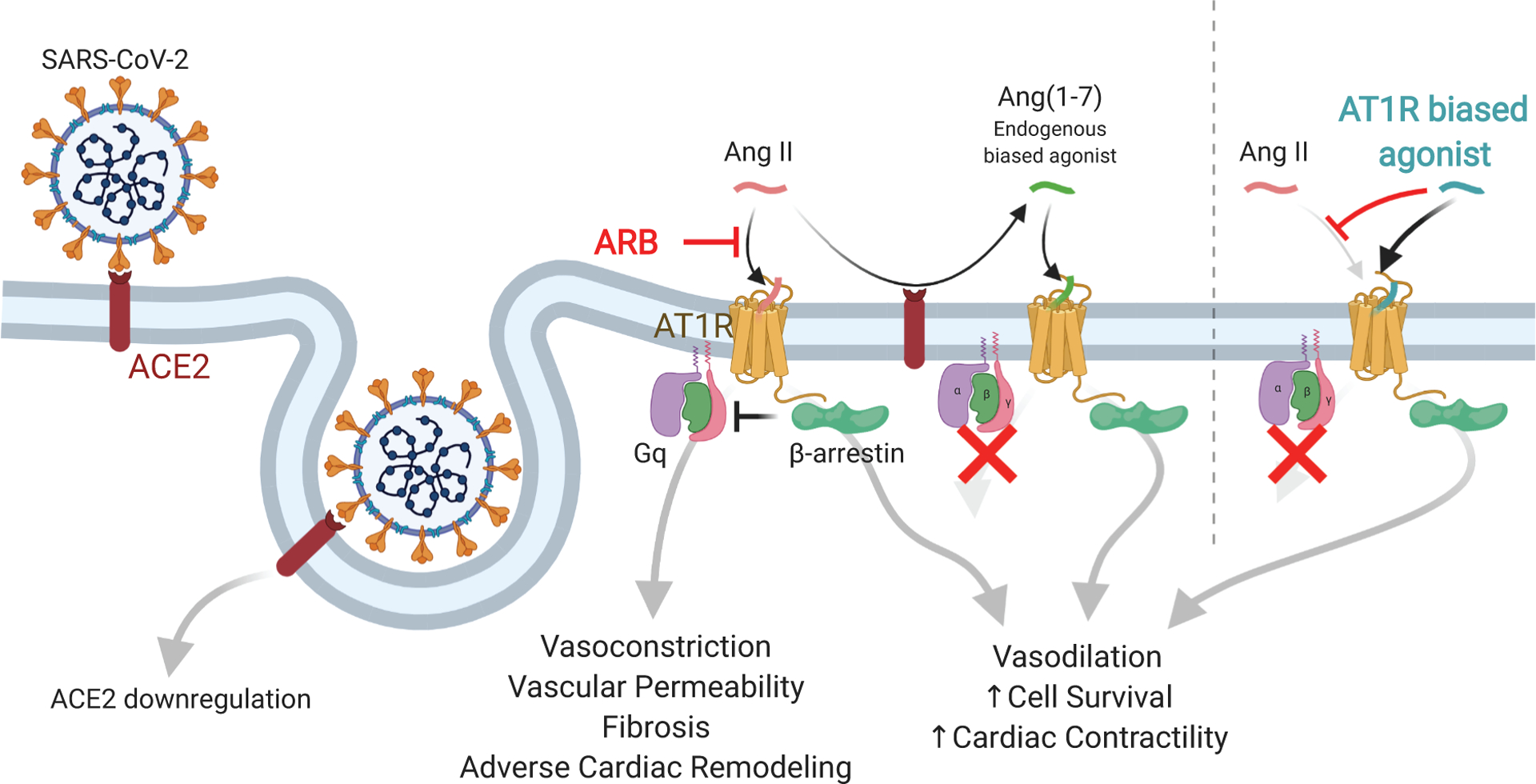
SARS-CoV2 enters cells via ACE2, leading to downregulation of ACE2 activity. Angiotensin II (AngII) signals through the Angiotensin II type 1 receptor (AT1R) via classic Gq signaling to drive vasoconstriction and adverse cellular responses in heart failure and acute lung injury. AngII action is tempered by β-arrestin. Cleavage of AngII by ACE2 (Angiotensin Converting Enzyme 2) both decreases AngII levels and leads to production of Ang(1–7), an endogenous β-arrestin biased agonist of the AT1R with potentially therapeutically beneficial effects. Compared to angiotensin receptor blockers (ARBs), synthetic biased agonists of the AT1R, which have improved potency and pharmacodynamic properties compared to Ang(1–7), may have a more favorable profile in patients with COVID-19 due to the fact that they both block AngII activity and induce β-arrestin signaling.
The traditional model for activation of G protein-coupled receptors (GPCR) involves the binding of a hormone to the receptor, which induces coupling to a G protein transducer to stimulate the production of second messengers that result in cellular and physiological responses. More recently, it has been appreciated that GPCR signaling involves not only G protein transducer activation but also the recruitment and activation of another adaptor and scaffold protein known as β-arrestin. The ability to direct a GPCR to selectively signal through a G-protein mediated pathway or a β-arrestin-mediated pathway is known as biased signaling (Figure 1). In this context, AT1R stimulates cellular signaling not only via Gq proteins but also via β-arrestins. However, in contrast to Gq signaling that can lead to vasoconstriction, increased vascular permeability, fibrosis and acute organ damage in the heart, lungs and kidneys, β-arrestin-mediated AT1R signaling has beneficial anti-inflammatory, anti-apoptotic, and vasodilatory effects1 (Figure 1). Whereas AngII activates both the Gq and β-arrestin signaling arms, certain “β-arrestin-biased” ligands of the AT1R are able to selectively activate only β-arrestin pathways.
The cellular receptor mediating viral entry of both SARS-CoV and SARS-CoV2 is Angiotensin Converting Enzyme 2 (ACE2), which plays a direct role in RAAS homeostasis by cleaving a single residue from AngII to generate Ang(1–7) (Figure 1). Binding of the virus to ACE2 leads to its internalization and downregulation of ACE2,2 which disrupts RAAS homeostasis through two related mechanisms. First, reducing the levels of active ACE2 in the plasma membrane slows AngII degradation, prolonging its potentially harmful effects. Second, reducing ACE2 availability lowers levels of Ang(1–7), which competes with AngII for receptor binding and has protective cardiopulmonary properties in its own right. Ang(1–7) promotes vasodilation, decreases inflammation and fibrosis, and, importantly, attenuated lung injury in an ARDS animal model.3 Intriguingly, Ang(1–7) acts as a β-arrestin-biased ligand for the AT1R.4
Several lines of evidence suggest that interference in RAAS homeostasis by SARS-CoV2 infection contributes to the severe ARDS and cardiomyopathies that many COVID-19 patients develop. Though not definitive, genetic studies have implicated inappropriate activation of the RAAS in the severity of ARDS. More directly, ACE2 activity is protective in preclinical models of acute lung injury and ARDS.2, 3 These observations have spurred clinical trials to test multiple approaches designed to modulate elevated levels of AngII-induced signaling for COVID-19 infections, including the ARB losartan and recombinant human ACE2 (rhACE2). However, these therapeutic approaches do not explicitly harness the beneficial β-arrestin-mediated signaling pathways promoted by Ang(1–7) itself.
We hypothesize that an AT1R ligand that blocks AngII activation of Gq signaling, but further activates protective β-arrestin signaling, could lessen the tissue damage in the lung and heart that occurs with fulminant disease accompanying COVID-19. While Ang(1–7) itself has poor pharmacokinetic properties, other AT1R ligands have been developed that exhibit the same β-arrestin-biased pharmacological profile but are more potent, efficacious, and metabolically stable (Figure 1). β-Arrestin-biased angiotensin II receptor agonists have been evaluated previously in phase IIb clinical trials for acute heart failure. Whereas this approach did not confer benefit over placebo, no safety issues were seen, and post-hoc analyses suggested a reduced serum creatinine and trends towards improved 180-day outcomes in patients with elevated baseline systolic blood pressure.5 The potential benefit of this approach in heart failure may therefore be specific to patients with elevated renin-angiotensin signaling, a population also at significantly higher risk for adverse outcomes from COVID-19.
Drug repurposing is one of the few viable strategies to tackle the surge of acutely decompensating patients with COVID-19. It is now well established that inappropriate AngII signaling worsens outcomes in ARDS pathophysiology, especially in the context of SARS-CoV infections. Whereas the potential of ARBs to mitigate aberrant AT1R signaling in COVID-19 has received considerable attention, we propose that the cellular signaling mechanisms of this receptor could be harnessed even more optimally by modulating it with biased ligands. Such precise modulation of AT1R signaling may also provide therapeutic options for future respiratory pandemics by acting on a central host signaling pathway contributing to ARDS pathophysiology, and should be explored expeditiously for its therapeutic potential for COVID-19.
Sources of Funding
Funding was provided by the Searle Scholars Program (AM) and NIH (Grants HL056687 and HL075443 to HAR, HL16037 to RJL). RJL is an investigator with the Howard Hughes Medical Institute.
Footnotes
Disclosures
TRV027 is a β-arrestin-biased angiotensin II receptor agonist developed by Trevena, Inc. HAR is a scientific co-founder of and owner of stock in Trevena. RJL is a scientific co-founder of and owner of options in Trevena. AM and LMW report no conflicts.
References
- 1.Kim KS, Abraham D, Williams B, Violin JD, Mao L, Rockman HA. beta-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am J Physiol Heart Circ Physiol. 2012; 303:H1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imai Y, Kuba K, Penninger JM. The renin-angiotensin system in acute respiratory distress syndrome. Drug Discov Today Dis Mech. 2006; 3:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wosten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, van Goor H, Kamilic J, Florquin S, Bos AP. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1–7) or an angiotensin II receptor antagonist. J Pathol. 2011; 225:618–627. [DOI] [PubMed] [Google Scholar]
- 4.Teixeira LB, Parreiras ESLT, Bruder-Nascimento T, Duarte DA, Simoes SC, Costa RM, Rodriguez DY, Ferreira PAB, Silva CAA, Abrao EP, et al. Ang-(1–7) is an endogenous beta-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci Rep. 2017; 7:11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotter G, Davison BA, Butler J, Collins SP, Ezekowitz JA, Felker GM, Filippatos G, Levy PD, Metra M, Ponikowski P, et al. Relationship between baseline systolic blood pressure and long-term outcomes in acute heart failure patients treated with TRV027: an exploratory subgroup analysis of BLAST-AHF. Clin Res Cardiol. 2018; 107:170–181. [DOI] [PubMed] [Google Scholar]