

Abstract
Transcription and replication of the influenza virus RNA genome is catalysed by the viral heterotrimeric RNA-dependent RNA polymerase in the context of viral ribonucleoprotein (vRNP) complexes. Atomic resolution structures of the viral RNA synthesis machinery have offered insights into the initiation mechanisms of viral transcription and genome replication, and the interaction of the viral RNA polymerase with host RNA polymerase II, which is required for the initiation of viral transcription. Replication of the viral RNA genome by the viral RNA polymerase depends on host ANP32A, and host-specific sequence differences in ANP32A underlie the poor activity of avian influenza virus polymerases in mammalian cells. A failure to faithfully copy the viral genome segments can lead to the production of aberrant viral RNA products, such as defective interfering (DI) RNAs and mini viral RNAs (mvRNAs). Both aberrant RNA types have been implicated in innate immune responses against influenza virus infection. This chapter discusses recent insights into the structure-function relationship of the viral RNA polymerase and its role in determining host range and virulence.
Introduction
Infections with influenza viruses type A and B are responsible for annual seasonal epidemics leading to about half a million deaths worldwide. Type influenza A viruses also cause occasional pandemics which can claim millions of lives. By contrast, influenza C type infections are milder and influenza type D viruses, which infect pigs and cattle, have not been associated with human disease (Krammer et al. 2018; Taubenberger et al. 2019).
The influenza virus genome consists of eight (influenza A and B viruses) or seven (influenza C and D viruses) segments of single-stranded negative-sense viral RNA (vRNA) (Fig. 1A). Each segment consists of conserved, partially complementary 5’ and 3’ termini, segment-specific untranslated regions (UTR) and one or more open-reading frames (ORF) (Fig. 1A). The vRNA segments are assembled into viral ribonucleoprotein (vRNP) complexes, in which the 5’ and 3’ termini are bound by a viral RNA-dependent RNA polymerase (RdRp) and the rest of the RNA is bound by oligomeric viral nucleoprotein (NP), forming a double-helical rod-like structure (Fig. 1B, C) (Eisfeld et al. 2015). Internal base pairing within the segments allows the formation of secondary RNA structures (Ferhadian et al. 2018). Influenza viruses must express a full complement of their genome segments in the infected cell in order to be productively infectious. The segments can be contributed by one virion or, in the case of a defective particle, multiple virions.
Figure 1. Influenza virus RNAs and vRNP structure.
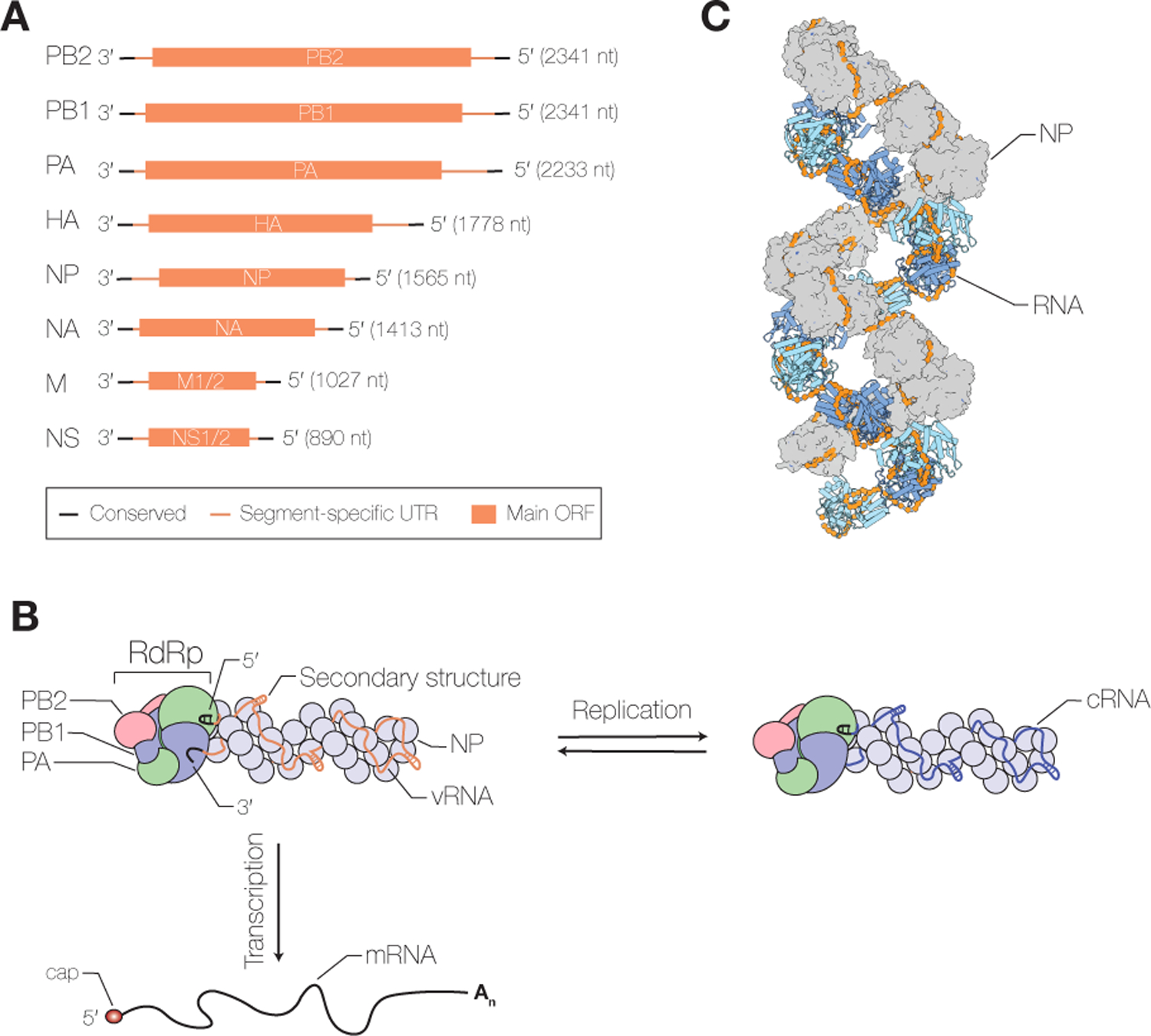
A) Influenza virus vRNAs. The influenza A virus genome consists of eight segments of single-stranded negative sense vRNA. Each segment contains conserved 5’ and 3’ termini, segment-specific untranslated regions (UTR) and at least one large open reading frame (ORF). B) Schematic of influenza virus vRNP and transcription and replication. vRNA segments are assembled into vRNPs with multiple copies of NP and a single copy of the viral RNA-dependent RNA polymerase (RdRp), composed of PB1, PB2 and PA subunits, that binds the conserved 5’ and 3’ vRNA termini. The RNA polymerase, in the context of vRNPs, transcribes vRNA into viral mRNA, which contains a 5’ cap-1 structure (cap) and a 3’ polyA tail (An), and replicates vRNA through a complementary RNA (cRNA) replicative intermediate, which is assembled with NP and polymerase into cRNP. C) Structure of the influenza virus vRNP. NP protomers are shown in surface and cartoon representation (PDB 4BBL).
After fusion of viral and endosomal membranes during influenza virus entry, vRNPs are released into the cytoplasm and transported into the nucleus where viral transcription takes place (Walker and Fodor 2019). It is in the context of nuclear vRNPs that the viral RNA polymerase transcribes the vRNA into positive-sense capped and polyadenylated mRNA for translation of viral proteins (Fig. 1B). The influenza A virus genome encodes 10 major and several auxiliary proteins. Viral protein expression is essential for the replication of the viral RNA genome by the viral RNA polymerase, which proceeds through a complementary RNA (cRNA) replicative intermediate (Fig. 1B) (Te Velthuis and Fodor 2016; Pflug et al. 2017).
Recent structural and functional studies of the influenza RNA synthesis machinery have provided novel insights into the mechanisms by which influenza viruses transcribe and replicate their RNA genome (Te Velthuis and Fodor 2016; Pflug et al. 2017; Lo et al. 2018). These studies have answered fundamental questions about influenza RNA polymerase function and contributed to our understanding of the dependence of the influenza virus on host factors for the transcription and replication of its genome. This chapter discusses how RNA polymerase structures inform us about the function of the RNA synthesis machinery and its role in defining viral virulence and host range.
Architecture of vRNPs
vRNPs are composed of one single-stranded vRNA segment, a copy of the RNA polymerase bound to the partially complementary 5’ and 3’ vRNA termini, and multiple copies of NP that are arranged in a double helical conformation (Fig. 1B, C). Currently, only low-resolution structural models of vRNPs are available that differ in their helical parameters and handedness and do not allow unambiguous mapping of the path of the RNA and NP-NP contacts within vRNPs (Arranz et al. 2012; Moeller et al. 2012). Studies aimed at obtaining high-resolution vRNP structures are hampered by the heterogeneity in vRNA length (890 to 2341 nucleotides for influenza A virus; Fig. 1A) and high plasticity of the NP-NP interactions, which leads to the formation of curves and kinks in vRNPs, and complicates computational processing of cryo-EM data (Gallagher et al. 2017).
NP is a crescent-shaped molecule that is comprised of a head and body domain and a 28 amino acid-long tail loop. NP forms oligomers when the tail loop of one NP inserts into the body domain of a neighbouring NP (Ye et al. 2006; Ng et al. 2008; Turrell et al. 2013). A basic groove separating the head and body domains represents the RNA binding surface. Phosphorylation and dephosphorylation of NP by cellular kinases and phosphatases affects NP oligomerisation and viral replication (Hutchinson et al. 2012; Chenavas et al. 2013; Mondal et al. 2015; Turrell et al. 2015; Mondal et al. 2017). The footprint of NP on vRNA is about 24 nucleotides (Ortega et al. 2000; Hutchinson et al. 2012), but it is not known how many nucleotides make direct contact with NP and how the RNA is arranged in vRNPs. Ribonuclease digestion studies show that the RNA in vRNP is susceptible to hydrolytic cleavage suggesting that vRNA is bound on the outside surface of NP. Mapping studies of NP-vRNA interactions have identified regions of low and high NP density (Lee et al. 2017; Williams et al. 2018), and chemical probing of the vRNA structure in vRNPs shows that vRNPs packaged into virions can accommodate secondary structures in their vRNAs (Dadonaite et al. 2019), demonstrating that NP does not associate with vRNA evenly. The NP helix in vRNPs thus likely provides a scaffold for the formation of secondary RNA structures. These RNA structures can facilitate intersegments RNA-RNA interactions and selective packaging of the vRNPs into virions (Dadonaite et al. 2019).
Architecture of the RNA polymerase
The influenza virus RNA polymerase is composed of the proteins polymerase basic 1 (PB1), PB2, and polymerase acidic (PA) (P3 in influenza C and D viruses) (Fig. 2A) (Te Velthuis and Fodor 2016; Pflug et al. 2017; Lo et al. 2018). X-ray crystallography and cryo-EM studies showed that the RNA polymerase consists of a core domain and several peripheral domains that are connected to the core through highly flexible linkers (Pflug et al. 2014; Reich et al. 2014; Chang et al. 2015; Hengrung et al. 2015; Thierry et al. 2016; Fan et al. 2019; Peng et al. 2019). The PB1 subunit contributes most to the core domain, and contains the fingers, palm and thumb subdomains that are characteristic of RNA polymerases, and a priming loop, a beta-hairpin structure that protrudes from the thumb domain into the polymerase active site and that is typical of RNA polymerases that can initiate RNA synthesis de novo (Fig. 2B) (Te Velthuis 2014). The PA subunit is composed of two domains. The N-terminal domain contains endonuclease activity and is connected to the polymerase core via a flexible linker. By contrast, the C-terminal domain is intimately associated with PB1 and tightly integrated into the thumb subdomain of the polymerase core. The N-terminal third of PB2 is also part of the polymerase core, but the C-terminal two thirds form multiple flexible domains, including the cap-binding, mid-link, 627 and nuclear localization signal (NLS) domains. All subunits contribute to the formation of the channels that facilitate template RNA entry and exit, NTP entry, and product RNA exit (Fig. 2B).
Figure 2. Structure of the influenza virus RNA polymerase.
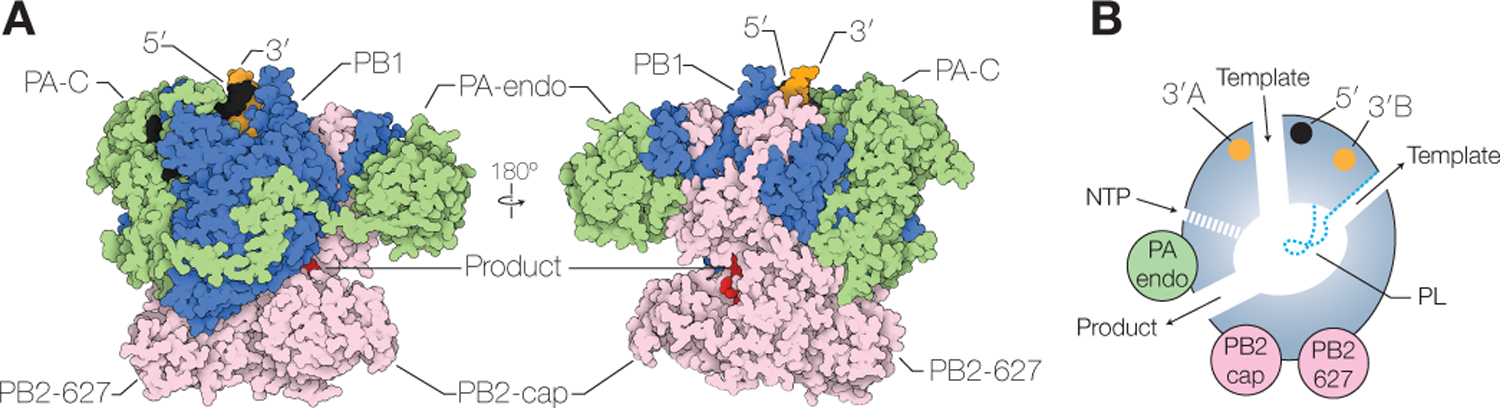
A) Surface presentation of the influenza A virus RNA polymerase (PDB 6RR7) bound to capped RNA primer and 5’ and 3’ vRNA termini. The subunits of the RNA polymerase are coloured as in Figure 1. B) Schematic model of the influenza A virus RNA polymerase showing the core chamber of the polymerase and its connection to the solvent via template and NTP entry channels, and template and product exit channels. The location of the priming loop (PL) as well as the 3’ and 5’ end binding pockets are indicated.
The polymerase contains several sites for binding viral RNA (Fig. 2). The 5’ end of vRNA and cRNA are tightly bound near the template entry channel of the polymerase by a pocket composed of amino acid residues contributed by the PB1 and PA subunits (Pflug et al. 2014; Reich et al. 2014; Robb et al. 2016; Fan et al. 2019; Peng et al. 2019). The 3’ end of the vRNA can associate with two different sites (the A-site and B-site) on the surface of the polymerase, while the 3’ end of cRNA has been shown to associate with the B-site only (Fig. 2B) (Pflug et al. 2014; Reich et al. 2014; Fan et al. 2019; Peng et al. 2019). The functional significance of these surface 3’ end binding sites remains to be determined, but they likely provide i) a pre-initiation site from which the 3’ end of vRNA enters the polymerase active site (the A-site), ii) a pre-initiation site from which the 3’ end of cRNA enters the polymerase active site (the B-site), or iii) a site to accommodate the 3’ end of the RNA template once it emerges from the polymerase active site after being copied (the B-site).
Transcription
Transcription is a primer-dependent process that involves the generation of 5’ capped and 3’ polyadenylated mRNA molecules using the negative-sense vRNA segments as templates by the viral RNA polymerase (Fig. 1B). Unlike the RNA polymerases from non-segmented negative-sense RNA viruses, the influenza virus RNA polymerase cannot synthesize and methylate cap structures. To ensure that influenza virus mRNAs contain a cap-1 structure at their 5’ terminus, the RNA polymerase needs to perform several key steps. First, the RNA polymerase associated with vRNPs, i.e. the resident polymerase, binds the C-terminal domain (CTD) of the large subunit of host RNA polymerase II (Pol II) in order to bring vRNPs in close proximity to nascent host 5’ capped RNAs (Fig. 3A) (Engelhardt et al. 2005; Martinez-Alonso et al. 2016; Lukarska et al. 2017; Serna Martin et al. 2018). Pol II CTD interacts with the viral RNA polymerase at several binding sites (Lukarska et al. 2017; Serna Martin et al. 2018), which stabilizes the PB2 cap-binding and PA endonuclease domains in a transcription-ready conformation (Serna Martin et al. 2018). Second, the PB2 cap-binding domain binds the 5’ cap of the nascent host RNA and the PA endonuclease domain cleaves the mRNA 10–14 bases downstream of the 5’ cap (Fig. 3A). Third, the 3’ end of the resulting capped RNA primer is inserted into the polymerase active site where it base-pairs with the 3’ end of the vRNA template (Fig. 3B). Initiation of viral mRNA synthesis occurs at the penultimate or third base from the 3’ end of the vRNA, depending on where the 3’ end of the primer has base-paired with the vRNA (Pflug et al. 2018; Te Velthuis and Oymans 2018; Kouba et al. 2019). A poor transition from initiation to elongation may result in early-termination at the fourth base from the 3’ end of the vRNA. The RNA polymerase can use the partially extended capped primer to restart viral transcription, which results in transcripts that contain two or more copies of the first four residues of the 3’ end of the vRNA template (Te Velthuis and Oymans 2018; De Vlugt et al. 2019). Extension of the capped primer results in the formation of a 9 base-pair RNA duplex downstream of the active site. To make space for this duplex, the priming loop, which is not required for transcription, is extruded from the polymerase (Te Velthuis et al. 2016; Kouba et al. 2019). The vRNA template and nascent mRNA are separated, with Tyr207 of the PB2 lid domain playing a key role, and template and product are directed towards their respective exit channels (Kouba et al. 2019). Finally, termination of the viral transcription product occurs by stuttering of the RNA polymerase on a sequence of uridine residues near the 5’ end of the vRNA template, leading to the addition of a polyA tail (Robertson et al. 1981; Poon et al. 1999).
Figure 3. Transcription by the influenza virus RNA polymerase.
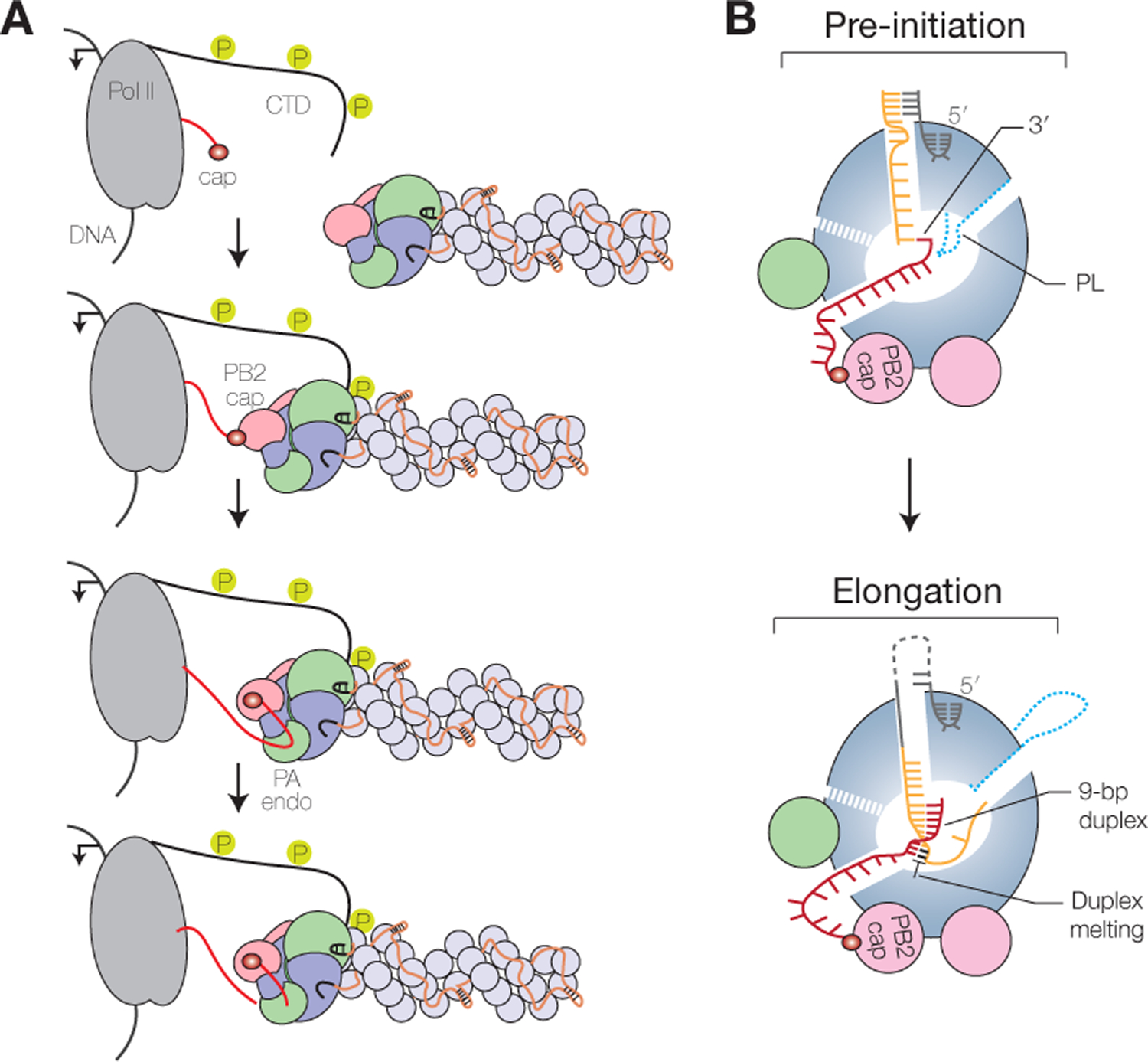
A) Association of the influenza virus RNA polymerase with the serine-5 phosphorylated C-terminal domain (CTD) of host RNA polymerase II (Pol II) and cap-snatching. B) Schematic model of the influenza virus RNA polymerase showing the binding of capped RNA primer and template prior to transcription initiation (top) and elongation and duplex unwinding during transcription (bottom). PL, priming loop.
Replication
Replication of the vRNA segments by the viral RNA polymerase is a two-step process (Fig. 4A, B). In the first step, vRNA is copied into a cRNA replicative intermediate (Fig. 4A). This step is initiated by entry of the 3’ end into the active site, presumably from the A-site (Fig. 2B) (Robb et al. 2019). Next, the viral polymerase uses de novo initiation at the 3’ terminal UC of the vRNA template to generate a pppApG dinucleotide (Fig. 4C). Initiation is dependent on the priming loop, which contributes to the stabilisation of the initiation complex (Te Velthuis et al. 2016). After initiation, the priming loop is likely extruded from the active site in order to make space for extension of the nascent cRNA, as has been observed during transcription elongation (Kouba et al. 2019), since truncation of the priming loop stimulates elongation (Te Velthuis et al. 2016). As the nascent cRNA emerges from the RNA polymerase, the cRNA assembles into a complementary ribonucleoprotein (cRNP) complex, a vRNP-like structure, in which a newly produced RNA polymerase associates with the terminal cRNA ends and oligomeric NP binds the rest of the cRNA stabilising it from degradation (Fig. 4A) (Vreede et al. 2004; York et al. 2013). Expression of RNA polymerase and NP is indeed essential for the accumulation of cRNA during infection.
Figure 4. Replication by the influenza virus RNA polymerase.
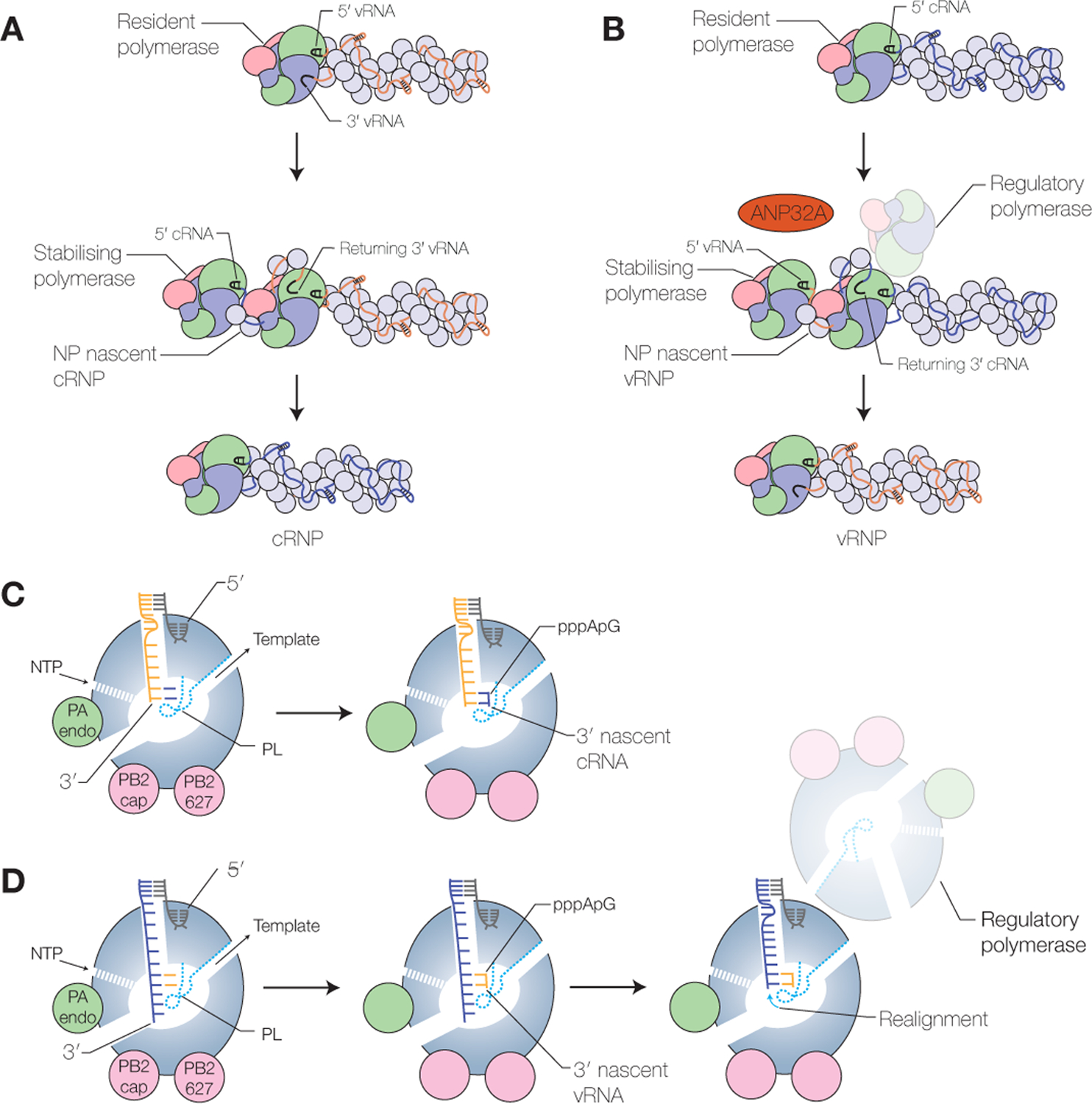
A) Synthesis of cRNA by the influenza virus RNA polymerase and co-replicative assembly of cRNP. B) Synthesis of vRNA by the influenza virus RNA polymerase and co-replicative assembly of vRNP. Template realignment is promoted by a regulatory polymerase. ANP32A associates with RNA-free RNA polymerase and may regulate dimerization of the RNA polymerase. C) Schematic model of the influenza virus RNA polymerase showing replication initiation during cRNA synthesis. D) Schematic model of the influenza virus RNA polymerase showing replication initiation and realignment during vRNA synthesis.
In the second step of replication, cRNA in the context of cRNPs acts as a template for the synthesis of vRNA (Fig. 4B). The 3’ end of the cRNA has been shown to bind at the B-site on the surface of the polymerase (Fig. 2B), which suggests that the 3’ cRNA terminus uses a different pre-initiation site before the 3’ end of the cRNA enters the active site and initiation occurs (Fan et al. 2019; Peng et al. 2019). By contrast to initiation on the vRNA template, the polymerase initiates internally at positions 4 and 5 of the cRNA template to produce a pppApG dinucleotide (Fig. 4D). The dinucleotide next acts as a primer for terminal initiation at positions 1 and 2 of cRNA after backtracking of the cRNA template (Deng et al. 2006) with the aid of the priming loop and a regulatory polymerase (Oymans and Te Velthuis 2018; Fan et al. 2019). The regulatory polymerase forms a dimer with the resident cRNP-associated polymerase which carries out replication. Several studies have shown that the influenza virus RNA polymerase can dimerize and that genome replication requires a second polymerase in addition to the polymerase that resides in the cRNP (Jorba et al. 2008; York et al. 2013; Chang et al. 2015; Fan et al. 2019; Peng et al. 2019). In the influenza A virus polymerase, PA residues 352–356 of both polymerases, and PB2 residues 71–76 of one polymerase and PA residue 347 of the second polymerase contribute to the dimer interface (Chang et al. 2015; Fan et al. 2019). Mutation of the dimer interface or its disruption with a nanobody inhibit vRNA synthesis by reducing the efficiency of cRNA template backtracking, and thereby realignment of the pppApG and terminal pppApG-primed initiation (Fan et al. 2019). Specifically, analysis of the priming loop shows that the beta-hairpin adopts different positions dependent on the oligomeric state of the polymerase. When the polymerase is in a monomeric form, the priming loop resides in a position that would promote internal initiation, but upon dimerization the priming loop changes position and facilitates backtracking of the template RNA to allow terminal pppApG-primed initiation of vRNA synthesis (Fan et al. 2019). The 5’ end of the vRNA product is most likely captured by a stabilising polymerase as soon as it emerges from the active site of the replicating cRNP-resident polymerase, initiating the assembly of the vRNA with NP into vRNP (Fig. 4B). Whether it is the same or two different polymerase heterotrimers that carry out regulation and stabilisation remains to be determined.
Host factors and adaptive mutations
Influenza virus polymerase function depends on association with host factors (Peacock et al. 2019). The transcriptional activity of the viral RNA polymerase is dependent on host Pol II for capped RNA primers that facilitate transcription initiation (Plotch et al. 1981) and structural studies have shown that the interaction of the viral polymerase with the CTD of Pol II is important for stabilising a conformation of the viral polymerase that is competent in cap-snatching and transcription initiation (Fig. 3A) (Lukarska et al. 2017; Serna Martin et al. 2018). ANP32 proteins have been identified as further host factors that are essential for viral polymerase activity. Simultaneous knock out of both ANP32A and ANP32B isoforms in human cells results in cells that are unable to support influenza virus polymerase activity in mini-genome assays and virus growth (Staller et al. 2019; Zhang et al. 2019). ANP32 proteins have been linked to viral genome replication, particularly the second step involving the synthesis of vRNA from the cRNA template (Fig. 4B) (Sugiyama et al. 2015), suggesting that ANP32 proteins are important factors for the replicase function of the polymerase. ANP32A can directly interact with the apo form of the viral polymerase (Sugiyama et al. 2015; Baker et al. 2018; Staller et al. 2019), in agreement with a hypothesis that ANP32A may help recruit a second polymerase for vRNP formation. However, the molecular details of how ANP32A protein interacts with the replicase remain unknown.
The dependence of the viral polymerase on ANP32 proteins plays a critical role in the host restriction of avian influenza virus polymerases in mammalian cells (Long et al. 2016). A key determinant of host range is PB2 amino acid 627. This residue is a glutamic acid in avian influenza virus polymerases (Subbarao et al. 1993), but an adaptive mutation to lysine allows the avian influenza polymerase to function efficiently in a mammalian cell. In addition to the E627K change, alternative adaptive mutations have been observed. Most notably, the swine origin 2009 pandemic virus retained 627E in its avian PB2 gene and used an adaptation at position 591 to increase viral replication (Mehle and Doudna 2009; Yamada et al. 2010). These adaptive changes are linked to a host-specific difference in the ANP32 proteins. Specifically, avian ANP32A contains a 33-amino acids insertion downstream of amino acid 175. This insertion, which consists of six unique amino acids representing a hydrophobic SUMO interaction motif (SIM)-like sequence and a 27 amino acid long duplication of amino acids 149 to 175, is critical for the support of PB2 627E-containing polymerase (Long et al. 2016; Domingues and Hale 2017; Baker et al. 2018). Human ANP32A and ANP32B variants as well avian ANP32A splice forms that lack this sequence are unable to support the activity of an avian-adapted influenza polymerase with PB2 627E. Currently, the molecular basis of how the E627K mutation allows the polymerase to work with the mammalian version of ANP32A proteins remains unknown.
DI RNAs, mvRNAs and svRNAs
In addition to the synthesis of full-length copies of the vRNA segments, influenza virus replication produces defective interfering (DI) RNAs, mini viral RNAs (mvRNAs) and small viral RNAs (svRNAs) (Fig. 5A). DI RNAs were first described in the context of defective interfering particles (DIP), which are virions that are able to interfere with the production of wild-type virus (Dimmock and Easton 2014). The DI RNAs have large internal deletions but usually retain at least 150–200 nucleotides from both termini, including the conserved 5’ and 3’ terminal sequences (Jennings et al. 1983). DI RNAs are encapsidated by multiple NP molecules and are bound by a single copy of the RNA polymerase, similar to full-length viral genome segments (Fig. 5A) (Coloma et al. 2009). DI RNAs retain sufficient sequence information to be packaged into virions replacing the parent full-length vRNA segment.
Figure 5. Aberrant RNA synthesis by the influenza virus RNA polymerase.
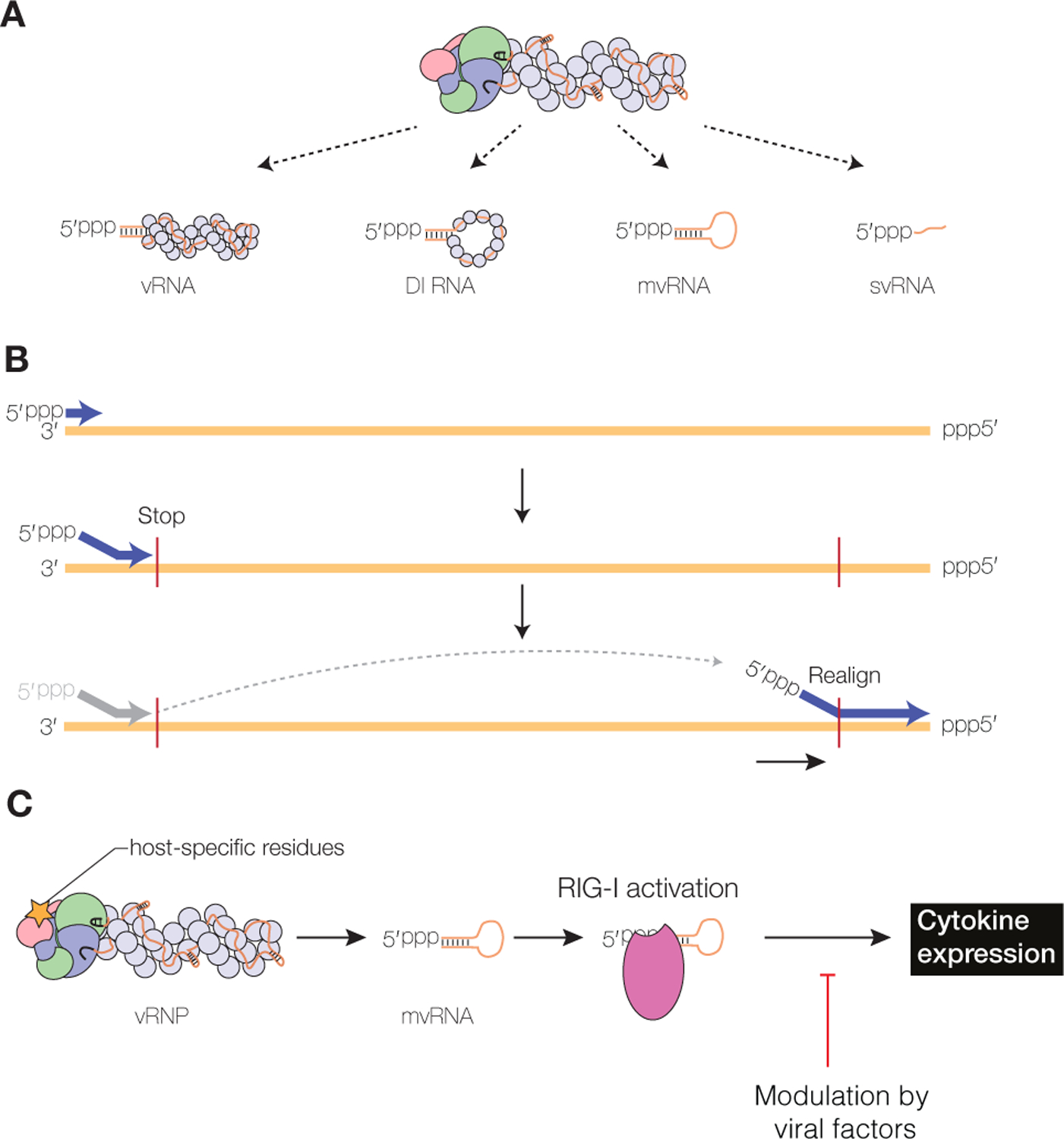
A) In addition to full-length vRNAs, the influenza virus RNA polymerase produces DI RNAs, mvRNAs and svRNAs. B) Synthesis of DI RNAs and mvRNAs uses an intramolecular copy-choice mechanism. C) Host-specific mutation in the viral RNA polymerase are linked to mvRNA levels. mvRNAs can be bound by RIG-I and induce RIG-I activation and cytokine expression.
Next generation sequencing of DI RNAs has shown that deletions can occur in any genome segment, but that some segments are more prone to give rise to DI RNAs than other segments. Specifically, DI RNAs derived from the three polymerase encoding segments are most frequently observed (Saira et al. 2013; Alnaji et al. 2019). DI RNAs are generated through a still poorly defined mechanism, in which the viral polymerase ceases product elongation at one site of the viral RNA template, only to resume elongation at a downstream site, which results in a failure to copy the interlaying stretch of the template (Fig. 5B). It has been hypothesised that an intra-molecular copy-choice mechanism could be involved, in which resumption of RNA synthesis could be dependent on base-pairing between the 3’ end of the nascent product and the RNA template. However, a recent analysis of the sequences that flank the deletion (i.e., polymerase stop and restart site) found no evidence for the enrichment of direct repeat sequences at junctions, which argues against a significant role for direct repeats in DI RNA formation (Alnaji et al. 2019). Interestingly, DI RNAs encode open reading frames and could express viral protein fragments or novel proteins that may have a function in the viral replication cycle (Akkina et al. 1984; Boergeling et al. 2015).
mvRNAs are approximately 50–125 nt long aberrant replication products with very large internal deletions, retaining the conserved 5ʹ and 3ʹ termini (Fig. 5A). In contrast to DI RNAs, mvRNAs can be replicated and transcribed in the absence of NP (Turrell et al. 2013; Te Velthuis et al. 2018). Consequently, they are not (fully) encapsidated and likely not packaged into virions. Formation of mvRNAs appears to follow an intra-molecular copy-choice mechanism that does not require perfect complementarity similar to DI formation (Fig. 5B) and that is more prevalent during the replication of some vRNA segments than others, suggesting a dependence on RNA sequence and/or structure (Te Velthuis et al. 2018). The generation of mvRNAs has been linked to the dysregulation of replication, for example by an imbalance in the polymerase-NP ratio, as well as to specific amino acids in the polymerase (Te Velthuis et al. 2018; Liu et al. 2019). Specifically, an amino acid change increasing polymerase fidelity has been shown to decrease mvRNA production, while amino acids characteristic of avian influenza A virus polymerases, increase mvRNA production in mammalian cells (Te Velthuis et al. 2018).
By contrast to DI RNAs and mvRNAs, svRNAs are 22–27 nt long RNAs that only contain the 5ʹ terminus of vRNAs (Fig. 5A). They likely represent early termination products of vRNA synthesis from a cRNA template. svRNAs have been found in cultured infected cells as well as in infected mouse lung cells, and may play a role in regulating viral replication (Perez et al. 2010; Perez et al. 2012; Koire et al. 2016). In particular, it has been shown that svRNA synthesis can influence vRNA synthesis by binding to the viral RNA polymerase (Perez et al. 2012). The viral non-structural protein 2 (NS2/NEP) may be involved in regulating svRNA expression (Perez et al. 2010; Liu et al. 2019).
Innate immune responses
The innate immune response plays a critical role in the outcome of influenza A virus infections. In particular, a dysregulated immune response underpins the high virulence of pandemic and avian influenza virus infections in humans (de Jong et al. 2006; Kash et al. 2006; Forero et al. 2015). Key cytokines involved in influenza A virus infections include type I and III interferons, such as interferon beta and lambda (Klinkhammer et al. 2018). These cytokines are typically expressed in response to the detection of viral RNA by the cellular pathogen sensor retinoic acid-inducible gene I (RIG-I) which is activated by binding short double-stranded RNA containing a 5’ di- or triphosphate. Influenza vRNA and cRNA contain 5’ triphosphate and partially complementary termini which could be bound by RIG-I (Yoneyama et al. 2004; Kato et al. 2006; Pichlmair et al. 2006). In agreement with viral RNA being important for RIG-I activation, correlation between the activity of polymerase and the expression of cytokines has been observed in a number of studies (Forero et al. 2015; Li et al. 2018; Russell et al. 2019). Specifically, the polymerases of the 1918 H1N1 pandemic virus and H5N1 avian influenza viruses have been linked to excessive cytokine production (Forero et al. 2015; Li et al. 2018).
Genomic viral RNA has been shown to be a poor inducer of RIG-I activation (Rehwinkel et al. 2010), likely because the binding of the viral RNA polymerase and NP molecules to the viral genome segments precludes RIG-I binding. svRNAs do not affect the innate immune responses during viral infections, likely because they are single-stranded and/or readily bound by the viral RNA polymerase (Perez et al. 2010). By contrast, mvRNAs and some DI RNAs are efficiently bound by RIG-I and able to induce innate immune responses (Baum et al. 2010; Te Velthuis et al. 2018). Avian and pandemic influenza viruses were found to produce high levels of mvRNAs, which correlated with the activation of antiviral genes, suggesting that production of mvRNAs by unadapted avian influenza virus polymerases in mammalian cells could be a biomarker for the “cytokine storm” phenomenon that underlies the high virulence associated with infections with these viruses (Fig. 5C). However, reduced accumulation of defective viral RNA during infection of patients with pandemic 2009 viruses has been linked to severe outcome, suggesting that low levels of defective RNAs are beneficiary through activating a protective level of antiviral innate immune responses (Vasilijevic et al. 2017). The propensity of the RNA polymerase to generate subgenomic RNAs such as mvRNAs and DI RNAs is likely to be one of the critical factors in determining the magnitude of an innate immune response generated against a particular strain of influenza virus, which helps to explain previous observations showing that the RNA polymerase contributes to influenza virus virulence.
Concluding remarks
Recent years witnessed an unprecedented progress in understanding the structure of the influenza virus RNA polymerase, providing insights into the function of the RNA synthesis machinery in transcription of viral genes, replication of the viral RNA genome, as well in determining host range and virulence. High-resolution structures solved by using cryo-EM and crystallography offer insights into the atomic details of the initiation of transcription and replication by the influenza virus RNA polymerase. Understanding the interplay between the virus and the host and in particular the dependence of viral transcription and replication on host factors provide novel insights into the adaptation of influenza A viruses to mammalian hosts. The identification of defective viral RNAs, such as mvRNAs, and their link to innate immune activation, furthers our understanding of the molecular basis of the virulence of influenza viruses. However, many long-standing questions still remain unanswered, including how the ANP32 proteins promote the replicase function of the polymerase, how defective viral RNAs such as DI RNAs and mvRNAs are generated and what the contribution of the defective RNAs is to disease severity in humans.
Acknowledgements
E. Fodor is supported by UK Medical Research Council programme grant MR/R009945/1. A. te Velthuis is supported by joint Wellcome Trust and Royal Society grant 206579/Z/17/Z, Isaac Newton Trust grant 17.37(r), and the National Institutes of Health grant R21AI147172.
References
- Akkina RK, Chambers TM, Nayak DP. 1984. Expression of defective-interfering influenza virus-specific transcripts and polypeptides in infected cells. Journal of virology 51: 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnaji FG, Holmes JR, Rendon G, Vera JC, Fields CJ, Martin BE, Brooke CB. 2019. Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses. Journal of virology 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, Martin-Benito J. 2012. The structure of native influenza virion ribonucleoproteins. Science 338: 1634–1637. [DOI] [PubMed] [Google Scholar]
- Baker SF, Ledwith MP, Mehle A. 2018. Differential Splicing of ANP32A in Birds Alters Its Ability to Stimulate RNA Synthesis by Restricted Influenza Polymerase. Cell Rep 24: 2581–2588 e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A, Sachidanandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proceedings of the National Academy of Sciences of the United States of America 107: 16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boergeling Y, Rozhdestvensky TS, Schmolke M, Resa-Infante P, Robeck T, Randau G, Wolff T, Gabriel G, Brosius J, Ludwig S. 2015. Evidence for a Novel Mechanism of Influenza Virus-Induced Type I Interferon Expression by a Defective RNA-Encoded Protein. PLoS pathogens 11: e1004924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Sun D, Liang H, Wang J, Li J, Guo L, Wang X, Guan C, Boruah BM, Yuan L et al. 2015. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 A resolution. Mol Cell 57: 925–935. [DOI] [PubMed] [Google Scholar]
- Chenavas S, Estrozi LF, Slama-Schwok A, Delmas B, Di Primo C, Baudin F, Li X, Crepin T, Ruigrok RW. 2013. Monomeric nucleoprotein of influenza A virus. PLoS pathogens 9: e1003275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coloma R, Valpuesta JM, Arranz R, Carrascosa JL, Ortin J, Martin-Benito J. 2009. The Structure of a Biologically Active Influenza Virus Ribonucleoprotein Complex. PLoS pathogens 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadonaite B, Gilberston B, Knight ML, Trifkovic S, Rockman S, Laederach A, Brown LE, Fodor E, Bauer DLV. 2019. The structure of the influenza A virus genome. Nature Microbiology. [DOI] [PMC free article] [PubMed]
- de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC et al. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12: 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlugt C, Sikora D, Rocheleau L, Pelchat M. 2019. Priming and realignment by the influenza a virus RdRp is dependent on the length of the host primers and the extent of base pairing to viral RNA. Virology 536: 91–100. [DOI] [PubMed] [Google Scholar]
- Deng T, Vreede FT, Brownlee GG. 2006. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. Journal of virology 80: 2337–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock NJ, Easton AJ. 2014. Defective interfering influenza virus RNAs: time to reevaluate their clinical potential as broad-spectrum antivirals? Journal of virology 88: 5217–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues P, Hale BG. 2017. Functional Insights into ANP32A-Dependent Influenza A Virus Polymerase Host Restriction. Cell Rep 20: 2538–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Neumann G, Kawaoka Y. 2015. At the centre: influenza A virus ribonucleoproteins. Nat Rev Microbiol 13: 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. Journal of virology 79: 5812–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Walker AP, Carrique L, Keown JR, Serna Martin I, Karia D, Sharps J, Hengrung N, Pardon E, Steyaert J et al. 2019. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 573: 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferhadian D, Contrant M, Printz-Schweigert A, Smyth RP, Paillart JC, Marquet R. 2018. Structural and Functional Motifs in Influenza Virus RNAs. Front Microbiol 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero A, Tisoncik-Go J, Watanabe T, Zhong G, Hatta M, Tchitchek N, Selinger C, Chang J, Barker K, Morrison J et al. 2015. The 1918 Influenza Virus PB2 Protein Enhances Virulence through the Disruption of Inflammatory and Wnt-Mediated Signaling in Mice. Journal of virology 90: 2240–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JR, Torian U, McCraw DM, Harris AK. 2017. Structural studies of influenza virus RNPs by electron microscopy indicate molecular contortions within NP supra-structures. J Struct Biol 197: 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengrung N, El Omari K, Serna Martin I, Vreede FT, Cusack S, Rambo RP, Vonrhein C, Bricogne G, Stuart DI, Grimes JM et al. 2015. Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature 527: 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS pathogens 8: e1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings PA, Finch JT, Winter G, Robertson JS. 1983. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 34: 619–627. [DOI] [PubMed] [Google Scholar]
- Jorba N, Area E, Ortin J. 2008. Oligomerization of the influenza virus polymerase complex in vivo. The Journal of general virology 89: 520–524. [DOI] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A et al. 2006. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443: 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441: 101–105. [DOI] [PubMed] [Google Scholar]
- Klinkhammer J, Schnepf D, Ye L, Schwaderlapp M, Gad HH, Hartmann R, Garcin D, Mahlakoiv T, Staeheli P. 2018. IFN-lambda prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koire A, Gilbert BE, Sucgang R, Zechiedrich L. 2016. Repurposed Transcriptomic Data Reveal Small Viral RNA Produced by Influenza Virus during Infection in Mice. PloS one 11: e0165729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouba T, Drncova P, Cusack S. 2019. Structural snapshots of actively transcribing influenza polymerase. Nature structural & molecular biology 26: 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG et al. 2018. Influenza. Nat Rev Dis Primers 4: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Le Sage V, Nanni AV, Snyder DJ, Cooper VS, Lakdawala SS. 2017. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic acids research 45: 8968–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Bradley KC, Long JS, Frise R, Ashcroft JW, Hartgroves LC, Shelton H, Makris S, Johansson C, Cao B et al. 2018. Internal genes of a highly pathogenic H5N1 influenza virus determine high viral replication in myeloid cells and severe outcome of infection in mice. PLoS pathogens 14: e1006821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Lu Y, Liu Q, Zhou Y. 2019. Inhibition of Ongoing Influenza A Virus Replication Reveals Different Mechanisms of RIG-I Activation. Journal of virology 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CY, Tang YS, Shaw PC. 2018. Structure and Function of Influenza Virus Ribonucleoprotein. Subcell Biochem 88: 95–128. [DOI] [PubMed] [Google Scholar]
- Long JS, Giotis ES, Moncorge O, Frise R, Mistry B, James J, Morisson M, Iqbal M, Vignal A, Skinner MA et al. 2016. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 529: 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukarska M, Fournier G, Pflug A, Resa-Infante P, Reich S, Naffakh N, Cusack S. 2017. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature 541: 117–121. [DOI] [PubMed] [Google Scholar]
- Martinez-Alonso M, Hengrung N, Fodor E. 2016. RNA-Free and Ribonucleoprotein-Associated Influenza Virus Polymerases Directly Bind the Serine-5-Phosphorylated Carboxyl-Terminal Domain of Host RNA Polymerase II. Journal of virology 90: 6014–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Doudna JA. 2009. Adaptive strategies of the influenza virus polymerase for replication in humans. Proceedings of the National Academy of Sciences of the United States of America 106: 21312–21316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. 2012. Organization of the influenza virus replication machinery. Science 338: 1631–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal A, Dawson AR, Potts GK, Freiberger EC, Baker SF, Moser LA, Bernard KA, Coon JJ, Mehle A. 2017. Influenza virus recruits host protein kinase C to control assembly and activity of its replication machinery. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal A, Potts GK, Dawson AR, Coon JJ, Mehle A. 2015. Phosphorylation at the homotypic interface regulates nucleoprotein oligomerization and assembly of the influenza virus replication machinery. PLoS pathogens 11: e1004826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A et al. 2008. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 22: 3638–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J, Martin-Benito J, Zurcher T, Valpuesta JM, Carrascosa JL, Ortin J. 2000. Ultrastructural and functional analyses of recombinant influenza virus ribonucleoproteins suggest dimerization of nucleoprotein during virus amplification. Journal of virology 74: 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oymans J, Te Velthuis AJW. 2018. A Mechanism for Priming and Realignment during Influenza A Virus Replication. Journal of virology 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock TP, Sheppard CM, Staller E, Barclay WS. 2019. Host Determinants of Influenza RNA Synthesis. Annu Rev Virol [DOI] [PubMed]
- Peng Q, Liu Y, Peng R, Wang M, Yang W, Song H, Chen Y, Liu S, Han M, Zhang X et al. 2019. Structural insight into RNA synthesis by influenza D polymerase. Nat Microbiol [DOI] [PubMed]
- Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, Garcia-Sastre A, tenOever BR. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proceedings of the National Academy of Sciences of the United States of America 107: 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JT, Zlatev I, Aggarwal S, Subramanian S, Sachidanandam R, Kim B, Manoharan M, Tenoever BR. 2012. A Small-RNA Enhancer of Viral Polymerase Activity. Journal of virology 86: 13475–13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflug A, Gaudon S, Resa-Infante P, Lethier M, Reich S, Schulze WM, Cusack S. 2018. Capped RNA primer binding to influenza polymerase and implications for the mechanism of cap-binding inhibitors. Nucleic acids research 46: 956–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflug A, Guilligay D, Reich S, Cusack S. 2014. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 516: 355–360. [DOI] [PubMed] [Google Scholar]
- Pflug A, Lukarska M, Resa-Infante P, Reich S, Cusack S. 2017. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res 234: 103–117. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314: 997–1001. [DOI] [PubMed] [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, Krug RM. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23: 847–858. [DOI] [PubMed] [Google Scholar]
- Poon LL, Pritlove DC, Fodor E, Brownlee GG. 1999. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. Journal of virology 73: 3473–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E et al. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140: 397–408. [DOI] [PubMed] [Google Scholar]
- Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crepin T, Hart D, Lunardi T, Nanao M, Ruigrok RW et al. 2014. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 516: 361–366. [DOI] [PubMed] [Google Scholar]
- Robb NC, Te Velthuis AJ, Wieneke R, Tampe R, Cordes T, Fodor E, Kapanidis AN. 2016. Single-molecule FRET reveals the pre-initiation and initiation conformations of influenza virus promoter RNA. Nucleic acids research 44: 10304–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb NC, Te Velthuis AJW, Fodor E, Kapanidis AN. 2019. Real-time analysis of single influenza virus replication complexes reveals large promoter-dependent differences in initiation dynamics. Nucleic acids research 47: 6466–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JS, Schubert M, Lazzarini RA. 1981. Polyadenylation sites for influenza virus mRNA. Journal of virology 38: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Elshina E, Kowalsky JR, Te Velthuis AJW, Bloom JD. 2019. Single-Cell Virus Sequencing of Influenza Infections That Trigger Innate Immunity. Journal of virology 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V et al. 2013. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. Journal of virology 87: 8064–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serna Martin I, Hengrung N, Renner M, Sharps J, Martinez-Alonso M, Masiulis S, Grimes JM, Fodor E. 2018. A Mechanism for the Activation of the Influenza Virus Transcriptase. Mol Cell 70: 1101–1110 e1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller E, Sheppard CM, Neasham PJ, Mistry B, Peacock TP, Goldhill DH, Long JS, Barclay WS. 2019. ANP32 Proteins Are Essential for Influenza Virus Replication in Human Cells. Journal of virology 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. Journal of virology 67: 1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama K, Kawaguchi A, Okuwaki M, Nagata K. 2015. pp32 and APRIL are host cell-derived regulators of influenza virus RNA synthesis from cRNA. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Kash JC, Morens DM. 2019. The 1918 influenza pandemic: 100 years of questions answered and unanswered. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Velthuis AJ. 2014. Common and unique features of viral RNA-dependent polymerases. Cellular and molecular life sciences : CMLS. [DOI] [PMC free article] [PubMed]
- Te Velthuis AJ, Fodor E. 2016. Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol 14: 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Velthuis AJ, Robb NC, Kapanidis AN, Fodor E. 2016. The role of the priming loop in influenza A virus RNA synthesis. Nat Microbiol 1: 16029. [DOI] [PubMed] [Google Scholar]
- Te Velthuis AJW, Long JC, Bauer DLV, Fan RLY, Yen HL, Sharps J, Siegers JY, Killip MJ, French H, Oliva-Martin MJ et al. 2018. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat Microbiol 3: 1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Velthuis AJW, Oymans J. 2018. Initiation, Elongation, and Realignment during Influenza Virus mRNA Synthesis. Journal of virology 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, Pflug A, Hengrung N, El Omari K, Baudin F et al. 2016. Influenza Polymerase Can Adopt an Alternative Configuration Involving a Radical Repacking of PB2 Domains. Mol Cell 61: 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrell L, Hutchinson EC, Vreede FT, Fodor E. 2015. Regulation of influenza A virus nucleoprotein oligomerization by phosphorylation. Journal of virology 89: 1452–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrell L, Lyall JW, Tiley LS, Fodor E, Vreede FT. 2013. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nature communications 4: 1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilijevic J, Zamarreno N, Oliveros JC, Rodriguez-Frandsen A, Gomez G, Rodriguez G, Perez-Ruiz M, Rey S, Barba I, Pozo F et al. 2017. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS pathogens 13: e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreede FT, Jung TE, Brownlee GG. 2004. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. Journal of virology 78: 9568–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AP, Fodor E. 2019. Interplay between Influenza Virus and the Host RNA Polymerase II Transcriptional Machinery. Trends Microbiol 27: 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GD, Townsend D, Wylie KM, Kim PJ, Amarasinghe GK, Kutluay SB, Boon ACM. 2018. Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nature communications 9: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Hatta M, Staker BL, Watanabe S, Imai M, Shinya K, Sakai-Tagawa Y, Ito M, Ozawa M, Watanabe T et al. 2010. Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS pathogens 6: e1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444: 1078–1082. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737. [DOI] [PubMed] [Google Scholar]
- York A, Hengrung N, Vreede FT, Huiskonen JT, Fodor E. 2013. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proceedings of the National Academy of Sciences of the United States of America 110: E4238–E4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang Z, Wang Y, Wang M, Wang X, Zhang X, Ji S, Du C, Chen H, Wang X. 2019. Fundamental Contribution and Host Range Determination of ANP32A and ANP32B in Influenza A Virus Polymerase Activity. Journal of virology 93. [DOI] [PMC free article] [PubMed] [Google Scholar]