

Abstract
As the technical ability for genetic diagnosis continues to improve, an increasing number of diagnoses are being made in infancy or as early as the neonatal period. Many of these diagnoses are known to be associated with developmental delay and intellectual disability - features which would not be clinically detectable at the time of diagnosis. Others may be associated with intellectual impairment but the incidence and severity are yet to be fully described. These newly-diagnosed neonates and infants therefore represent an emerging group of patients at high risk for neurodevelopmental disabilities. While there are well-established developmental supports for high-risk infants – particularly preterm infants – after discharge from the neonatal intensive care unit, programs specifically for infants with genetic diagnoses are rare. And while prior research has demonstrated the positive effect of early developmental interventions on outcomes among preterm infants, the impact of such supports for infants with genetic disorders, who are largely full-term, remains to be understood. We therefore review the literature regarding existing developmental assessment and intervention approaches for children with genetic disorders, evaluating these in the context of current developmental supports post-discharge for preterm infants. Further research is needed into the role of developmental support programs for early assessment and intervention in high-risk neonates diagnosed with rare genetic disorders.
Table of Contents Summary:
Infants with rare genetic disorders represent a newly-identified population at risk for developmental disabilities. We therefore review optimal developmental supports for this population.
Introduction:
Medical conditions presenting in the newborn period have the potential to substantially impact developmental outcomes. Both preterm birth and low birth weight are known to confer a risk for developmental delay (1, 2) as do major congenital anomalies (3-5) and many genetic conditions including inborn errors of metabolism (6-11). Developmental screening and intervention programs, such as early intervention, have been shown to improve outcomes not only for preterm infants (12-14), but also for developmental delays noted in otherwise healthy newborns (15). Earlier identification and therapy appears to be important for optimal outcomes in many developmental disorders such as autism spectrum disorders or neurosensory conditions such as hearing loss (16, 17). Universal developmental surveillance is now recommended from birth to age three by the American Academy of Pediatrics (18). Many infants at risk for developmental delays are admitted to the neonatal intensive care unit (NICU), where it has been shown that these risks are not restricted to only low birth weight or preterm infants (3-5, 19). However, while preterm infants may qualify for developmental follow-up programs that facilitate improved outcomes (12-14), many infants diagnosed with genetic syndromes are born full-term and are consequently not routinely eligible for these structured infant follow-up programs.
Concurrently, efforts at genetic diagnosis in the NICU have been accelerating, with multiple studies showing a high diagnostic yield of exome sequencing (ES), genome sequencing (GS) or of large gene panels in this population (20-26). In addition, population-based analyses demonstrate that genetic disorders affect a large proportion of infants admitted to the NICU (27, 28). Common indications for a genetic evaluation leading to the use of ES/GS for diagnosis include the presence of multiple congenital anomalies, hypotonia, seizures, newborn screen results suggesting inborn errors of metabolism, and dysmorphic facial features or other findings suggestive of a genetic syndrome. Despite the increasing number of infants diagnosed with genetic disorders putting them at high risk for neurodevelopmental disabilities, the need for, and potential impact of early developmental supports on this population has not been assessed. The purpose of this review is therefore to present and evaluate a role for developmental follow-up and intervention for those diagnosed with genetic syndromes in the neonatal or infant period.
The Developmental Implications of Genetic Syndromes
Many genetic syndromes carry a substantial risk for developmental delay and intellectual disability, in addition to the medical sequelae of these disorders such as critical congenital heart disease or epilepsy that can also affect developmental and cognitive outcomes. Chromosomal disorders such as Down Syndrome, Williams-Beuren Syndrome, and 22q11 deletion syndrome, with which clinicians have considerable experience because they are detectable by older genetic diagnostic techniques such as karyotype, are linked to high rates of intellectual disability (29, 30). Indeed, about 40% of children and young adults tested at a developmental medicine clinic over a 15 year period were found to have genetic conditions, even before widespread adoption of sequencing-based tests such as gene panels or ES (31). More recently-discovered genetic disorders also often carry substantial developmental impact, evidenced by the high diagnostic yield of ES for patients with neurodevelopmental disorders (32) and the high rate of novel disease gene discovery in this population (33, 34).
As mentioned, multiple recent studies have demonstrated the high diagnostic yield of ES in infants (20, 22, 24-26). Upon our review of 102 diagnoses made in one recent, large study of exome sequencing for diagnosis in the NICU (20) 66/102 or 65% of these diagnoses are associated with a risk of developmental delay/intellectual disability (not including gross motor delay due to neuromuscular disease); many may also be at risk for neurosensory issues such as impaired hearing and vision that potentially impact development. Additionally, inborn errors of metabolism, classically diagnosed by newborn screening and follow-up biochemical testing, have been associated with developmental delays or psychiatric disorders, even those diseases that are not typically characterized by the production of neurotoxic metabolites, such as fatty acid oxidation disorders (9, 10). Should these neonates with genetic conditions survive infancy, it is likely that their service needs will be high. Although often incompletely described, particularly for extremely rare genetic disorders, the developmental prognosis of these genetic syndromes is highly important to parents, particularly those of young children who must now imagine or plan for life with a child who may never be able to live independently (35-37).
Although increasing numbers of infants are now receiving genetic diagnoses, the implications of such diagnoses are not completely understood. Further research is needed to characterize the neurodevelopmental profiles of genetic disorders (7) as there is a paucity of data on developmental outcomes, particularly regarding functional abilities such as the ability to live independently or maintain employment, even for relatively common genetic syndromes such as Down syndrome, Williams-Beuren syndrome and the 22q11 deletion syndrome (36-38). The developmental potential of children with genetic syndromes spans a broad spectrum that is influenced by many factors, from the presence of medical comorbidities, to the genotype of the individual, to the home environment and parental educational achievement. Standardized, longitudinal assessments are useful in providing insight into the developmental phenotype of these rare disorders, and from this work, important observations can be made, such as relative strengths and weaknesses and the evolution of the developmental phenotype over time (39, 40). This understanding is critical to provide effectuve therapies and improve developmental outcomes, as it has been recognized that even children with common genetic disorders such as Down Syndrome or Williams-Beuren Syndrome often have unmet needs (41).
In particular, it remains to be determined how our understanding of the phenotypic spectra of neurodevelopmental genetic disorders may be influenced by expanded access to diagnostic genetic testing, particularly in the neonatal period. Historically, such patients may not have been referred to a genetics clinic until several years of age, by which point developmental delays and intellectual disability are more apparent. Based upon facial dysmorphia or other clinical features, the geneticist would order a targeted genetic test to diagnose a specific condition. These disorders were therefore defined by these classically-presenting patients, who may represent a more severe end of the spectrum. As testing is broadened to include those with milder features, the phenotypic spectrum for many conditions has broadened, particularly in the era of “reverse phenotyping”, in which a molecular genetic diagnosis (such as a pathogenic variant in a causative gene) is made first and it is subsequently determined whether or not the patient matches the genotype (42). This occurs particularly often for neonates, in whom reverse phenotyping has become increasingly prevalent with the widespread application of ES/GS and the lack of measures for diagnosing neurodevelopmental disability at birth. Indeed, a review of individuals with the 22q11 deletion syndrome found that those diagnosed after age two were identified due to language and developmental delays, whereas those diagnosed in the neonatal period were found in the setting of cardiac defects and hypocalcemia (43). Neonates with genetic disorders thus represent a high risk population, though they may not be as closely monitored from a developmental perspective as are preterm infants. It has been previously noted that infants with normal birth weight admitted to the NICU have high rates of post-discharge resource utilization, particularly if congenital anomalies are present (44), although they may be lacking a medical home providing developmental surveillance and support. As a framework for developmental follow-up for preterm infants and certain other high-risk populations has been previously been established, we now review this approach in order to provide context for an assessment of developmental supports for neonates with genetic disorders.
Follow-up for Preterm Infants
It is well-established that prematurity, in addition to very low birth weight, carries a risk for impaired developmental outcomes, and there are evidence-based interventions to mitigate this risk (12-14). Particularly, referral to early intervention and monitoring in developmental follow-up programs have been shown to be helpful for these infants, with one large study randomizing low birth weight preterm infants to either standard pediatric follow-up or participation in a program of developmental and family support showing increased IQ scores and decreased behavioral problems in the intervention group (14). Developmental follow-up programs for preterm infants help to identify gaps in early intervention programming and to help ensure that these children receive the services that they need to reach their full potential. More rapid enrollment in early intervention and greater service intensity have also been associated with significantly better developmental outcomes (12), and a recent Cochrane review of developmental intervention programs for preterm infants demonstrates improvement in cognitive outcomes by preschool age (13). In addition, early intervention programs have been shown to have a positive impact on behavioral problems in preterm infants (45) and on parental stress (46).
Structured developmental follow-up for preterm infants is formally recognized as an important component of NICU discharge in a 2008 policy statement by the American Academy of Pediatrics (47). Infant follow-up programs typically include a multidisciplinary team of providers, with a neonatologist, developmental specialist, registered nurse, and clinic coordinator/case manager commonly present at each clinic visit and physical and occupational therapists, nutritionists, and social workers also often present (48). Typical assessments performed at the visit include growth measurements, structured developmental assessments such as the Bayley Scales of Infant and Toddler Development, Third edition (49) or others (50), evaluation of neurological status, nutritional and feeding management, and assessment by occupational/physical/speech therapists (48). These visits are usually initiated one to four months after NICU discharge and occur until infants are two to three years of age (48). Domains addressed during these visits include the infant’s physical and mental health, cognitive abilities, quality of life (51), and family support.
Developmental Support for other High-Risk Infants
In addition to infants born preterm, other populations of infants have been identified as high risk for developmental disabilities and structured neurodevelopmental follow-up has been recommended for these infants as well (47, 51). Follow-up programs in this context monitor and support development in addition to collecting data on long-term outcomes for clinical care and research purposes. Neurodevelopmental support and outcomes for infants with congenital heart disease have been previously well-reviewed in a scientific statement from the American Heart Association and formal follow-up has been a recommendation since 2012 (52). Notably, it is highlighted that for this population, just as for infants with genetic disorders (some of whom will also have congenital heart disease), early identification of delays and prompt intervention is essential to promote optimal outcomes, and in order to do this, support is needed outside of the primary care office. A framework is therefore presented for developmental surveillance and referral of high risk congenital heart disease patients for formal evaluation (52). Similarly, term infants with hypoxic-ischemic encephalopathy have also been recognized as high risk for developmental delays and disabilities, and follow-up programs for these infants have also been developed (53), and have been critical in the assessment of response to treatments such as therapeutic hypothermia (54). Programs for infants with congenital heart disease or hypoxic-ischemic encephalopathy often resemble preterm infant follow-up programs, with the addition of an appropriate subspecialist, such as a cardiologist or neurologist.
Although they represent a more heterogeneous and less systematically-studied population, infants with major congenital anomalies requiring surgery, such as congenital diaphragmatic hernia (55), esophageal atresia (56), and omphalocele (57), may also have developmental impairments. Studies of these cohorts may include infants with genetic disorders, though typically not in large enough numbers to draw real conclusions. Nevertheless, the importance of developmental follow-up programs has been emphasized for pediatric surgical patients, many of whom will have genetic syndromes (58). It is important to recognize, however, that there are many infants with Mendelian genetic disorders found in the neonatal period that do not have major congenital anomalies and would therefore not be captured by a surveillance program designed specifically for infants with surgical conditions.
Developmental Support for Infants with Genetic Syndromes
Neurodevelopmental outcomes have been recognized as highly important to the families of infants deemed high risk for any reason, genetic or otherwise (37, 50). A survey of individuals presenting to a Down syndrome specialty clinic found that behavioral and language issues were the top two concerns of parents (59). However, in our review of the literature on structured developmental assessments and/or supports for infants with genetic disorders, the few studies identified were primarily observational, retrospective, and disease-specific. They do, however, suggest that early diagnosis of genetic conditions provides a unique opportunity to improve developmental outcomes. Assessments performed for children with the 22q11 deletion syndrome found high rates of developmental delay that were not associated with the presence of congenital heart disease, and the authors advocate for early evaluation for delays and initiation of developmental therapies in this syndrome (60). A prospective study of individuals with Down syndrome, assessing neurodevelopment, adaptive functioning, and motor skills from infancy to school-age found that higher skills detected in infancy were associated with improved functioning at age 10.7 years (40). Further studies are greatly needed, particularly those that are prospective, include structured developmental assessments with standardized tools, and include infants with ultra-rare genetic disorders. Prior follow-up studies of infants with major congenital anomalies have excluded those with genetic syndromes to avoid a negative influence on developmental outcomes in their analyses (though it is possible that certain infants included in these studies had an undiagnosed genetic condition, as they were evaluated prior to the more widespread use of exome or genome sequencing for neonatal diagnosis) (4, 5). This may represent a fatalistic attitude towards infants with diagnosed genetic conditions, if the belief is that these developmental outcomes are not highly modifiable.
In fact, developmental support and intervention show great promise to improve outcomes when applied to infants with genetic syndromes, particularly those who are diagnosed early in life. Families of children with a diagnosed condition have been shown to report a more positive experience with early intervention services compared to families of children referred for non-specific developmental delay, suggesting that earlier diagnosis of genetic syndromes in infancy may also have a positive impact on this experience (61). While not required for the provision of developmental services, a genetic diagnosis found in the neonatal period (or even prenatally) allows for surveillance, early identification of risk for developmental disability, and prompt referral. Case reports of an early diagnosis of rare genetic syndromes have illustrated developmental progress over time and suggest that early developmental support is helpful (62-64). A recent study demonstrated improved outcomes in a group of adolescents with Down syndrome who had received early intervention compared to controls (65). Also informative is the transforming developmental prognosis of Down syndrome, as infants are now commonly diagnosed before birth and therefore are closely monitored from a very young age. Current data demonstrates improvement in language outcomes for this condition and suggests that this is related to improved access to therapies (36); opportunities also now exist for college and employment that did not previously (66). Down Syndrome specialty clinics play an important role in this transformed prognosis, as these clinics closely monitor developmental and behavioral issues (59). This paradigm of referral to a specialty clinic to serve as a medical home and to closely monitor developmental progress after the diagnosis of a genetic disorder has also been utilized in the 22q11 deletion syndrome (37), though is unlikely to exist for infants with ultra-rare disorders.
The framework set forth by preterm follow-up programs naturally lends itself to infants with rare genetic disorders. This approach allows for developmental surveillance and early intervention for this high risk population. Indeed, infants with genetic or metabolic disorders have been identified as a group warranting structured developmental follow-up (51), though further research into the optimal delivery of such a program is much needed. Building off of prior examples of high risk infant follow-up programs, a program targeting infants with genetic disorders would involve a multidisciplinary approach (Figure 1), including a neonatologist, clinical geneticist, developmental psychologist or other specialist as well as physical and speech therapists with expertise in the management of infants. Structured developmental assessments would allow for careful monitoring of developmental progress, and issue specific to infancy such as sleep and feeding (both commonly perturbed in infants with genetic conditions (67)) can be addressed. Developmental support programs would also serve to identify areas of unmet developmental needs and aid families in addressing these gaps in care. For example, diagnosing features of autism in young children with genetic syndromes is thought to be highly important for their management (7), particularly as earlier recognition and treatment of autism spectrum disorders results in improved outcomes (16); indeed, identification of autism spectrum disorders resulted in referral for Applied Behavioral Analysis therapy for children with Down Syndrome presenting to a Down Syndrome specialty clinic – a service that they were not receiving previously (59).
Figure 1. A developmental home for infants with rare genetic disorders.

Incorporation of this approach to infants diagnosed with genetic conditions putting them at risk of developmental disorders may help to improve developmental outcomes.
A developmental home for infants with rare genetic disorders would serve multiple other purposes in addition to developmental surveillance. One important function of a developmental home for these infants involves parental support (68). While important at all ages, parental support is particularly crucial in the first three years of life and especially for the parents of infants with genetic disorders, as they face adjustment not only to a new family structure but also to a new and likely foreign diagnosis. Parents of infants with genetic syndromes experience stress (69), the degree and experience of which may be syndrome-dependent. For example, parents of children with Fragile X and Prader-Willi syndromes were shown to have higher levels of stress correlated to a more external locus of control related to the behavioral difficulties with these conditions (69), and mothers of infants with prenatally-diagnosed variants of uncertain significance found by chromosomal microarray experience stress surrounding early development and potential (70). Involvement in a structured infant follow-up program can lead to improved guidance and support for parents, and the inclusion of a clinical geneticist in this program allows for this to occur in the context of the underlying genetic condition. Creating a specialty clinic serving as a “developmental home” for infants with genetic disorders may also help to alleviate parental stress via an increased feeling of empowerment (69, 71). Additionally, while at this time, rare disease families often network via social media and disease-specific support groups, which can be invaluable resources for parental emotional support and education, they can also be intimidating and overwhelming and are likely not sufficient (72, 73). Providing families with information to help them connect with other parents and to interpret and process the information they receive by these venues is another valuable role provided by a developmental follow-up program. Importantly, addressing parental stress and emotional health also improves outcomes for the child (74).
Currently, care for infants with genetic conditions known to cause developmental disabilities is largely supportive (11), though the optimal definition of such “supportive” care has not been developed or thoroughly assessed. While the exciting opportunity for novel therapies for genetic disorders, such as gene therapy, hematopoetic stem cell transplant, or enzyme replacement therapy should not be minimized, it is counterproductive to concurrently ignore the potential to improve supportive therapies while these “targeted” treatments – a goal of precision medicine – are sought. These therapies often take years to develop and in the meantime, precious opportunity for improved developmental intervention during early critical windows of brain development is lost. Even if a targeted treatment were available for each and every genetic disorder, comprehensive developmental follow-up programs for these patients would be essential to provide accurate natural history data and a benchmark for assessing response to therapy, as has been shown in the Neonatal Research Network (50). The “developmental home” will also contribute to increased understanding of the developmental prognosis, particularly for rare and novel genetic disorders (64), which in may lead to improving outcomes (39, 41). The increasing usage of genome-wide assessments such as ES or GS may uncover variants of unknown significance rather than a diagnosis; while this presents certain challenges to management of these infants, their inclusion in a developmental surveillance program may help elucidate the phenotypic impact of these variants. Although not the focus of this article, developmental surveillance and support also plays a role in the management of older children receiving genetic diagnoses.
Future Directions
Early diagnosis of genetic disorders known to confer a risk for developmental delay and intellectual disability presents a unique opportunity for surveillance and intervention. Applying the comprehensive approach to follow-up that currently exists for preterm or low birth weight infants and infants with major congenital anomalies or hypoxic ischemic encephalopathy (4, 5, 52) to infants diagnosed with genetic disorders potentially will improve developmental functioning and the quality of life for the children and their families. Relying solely on a primary care-based model for referral to early intervention services is unlikely to be sufficient, since a high percentage of patients needing developmental services are neither referred to early intervention nor evaluated (75, 76). A proposed model for care includes: 1) developmental screening by a psychologist or provider specially trained to evaluate early developmental outcomes and behavior, 2) assessment by a neonatologist specializing in medical and developmental follow-up of high-risk infants and 3) work-up by a clinical geneticist with knowledge of developmental risks associated with the underlying genetic diagnosis (Figure 1). The sequential assessments performed in the developmental home would lead to targeted therapies for motor, language and cognitive development. Ideally, such follow-up should be family-centered, with an emphasis on enhancing communication among community-based pediatricians, early intervention providers, and parents, and integration of care across settings (71, 77). At the same time, these programs would provide valuable information on neurodevelopmental outcomes through comprehensive, high-quality data collection in infancy and early childhood, information lacking even for more common genetic conditions (37).
While precedent for this type of program exists in preterm infant follow-up clinics and in Down syndrome or 22q11 deletion disorder specialty clinics (37, 59, 60) and infant follow-up programs may include genetic syndromes as a criterion for referral (51, 78), the utility of more widespread application of this model to include all infants with genetic disorders placing them at risk for developmental disability warrants further study. Indeed, standardization of follow-up practices for high-risk infants to allow for pooling of data and benchmarking of outcomes, in addition to measuring quality of life, social, and functional outcomes (rather than purely developmental/intellectual measurements), has been identified as research gap in this field (51). Challenges to this approach include access and cost, with underserved communities in particular facing barriers such as the cost of genetic testing or the cost of either establishment of such a developmental home or travel to be seen at a specialty center providing this service. As the cost of genetic testing continues to decrease, this particular challenge may be attenuated in the future. Multiple prior studies have attempted to demonstrate the economic benefits of neonatal genetic diagnosis by exome sequencing, though enhanced developmental support is typically not considered in these calculations (79-81). While there are substantial costs associated with providing a developmental support program (48), if the cost savings associated with follow-up care of preterm infants (by allowing earlier discharge from the NICU and preventing costly hospitalizations down the road) (48) and by preventing the immeasurable costs of not intervening (51) are also seen in developmental follow-up of term infants, this could be another argument for increased uptake and insurance coverage of such testing. These programs could be developed around existing NICU follow-up infrastructure, as has been recommended for follow-up of infants with congenital heart disease, which helps to mitigate the cost of starting a program de novo (82). While universal genetic screening of neonates is a complicated issue, the creation of a developmental home for infants who do undergo genetic testing and are diagnosed could potentially represent an additional benefit of a molecular genetic diagnosis (Figure 2) and support broader efforts towards genetic diagnosis in the neonatal period.
Figure 2. The impact of a molecular diagnosis.
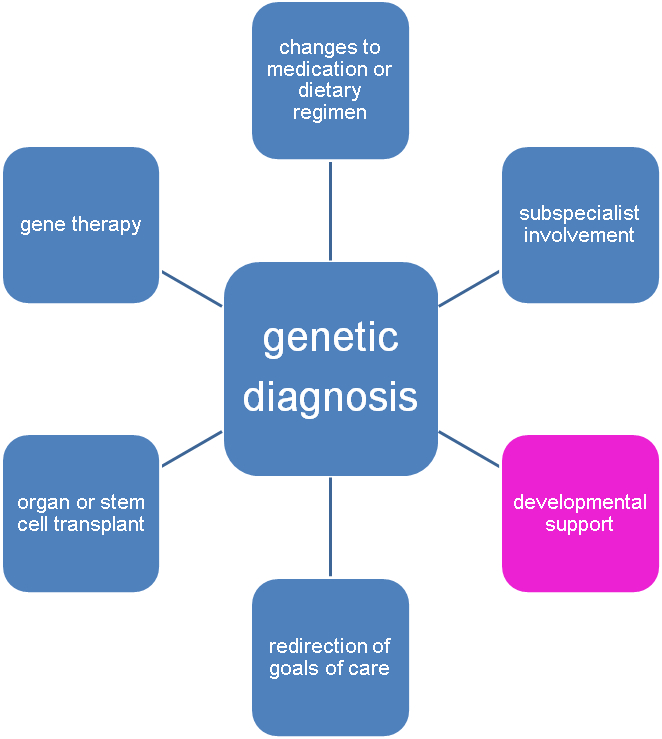
Identifying the genetic cause of an individual’s condition can lead to improved care in multiple ways, with developmental support being an important component.
Acknowledgments
Funding Source: NIH T32GM007748 [MHW]
Abbreviations:
- ES
exome sequencing
- GS
genome sequencing
- NICU
neonatal intensive care unit
Footnotes
Potential Conflicts of Interest: The authors have no conflicts of interest relevant to this article to disclose.
References
- 1.McCormick MC, Litt JS, Smith VC, Zupancic JA. Prematurity: an overview and public health implications. Annu Rev Public Health. 2011;32:367–79. [DOI] [PubMed] [Google Scholar]
- 2.Spittle AJ, Orton J. Cerebral palsy and developmental coordination disorder in children born preterm. Semin Fetal Neonatal Med. 2014;19(2):84–9. [DOI] [PubMed] [Google Scholar]
- 3.Stolwijk LJ, Lemmers PM, Harmsen M, Groenendaal F, de Vries LS, van der Zee DC, et al. Neurodevelopmental Outcomes After Neonatal Surgery for Major Noncardiac Anomalies. Pediatrics. 2016;137(2):e20151728. [DOI] [PubMed] [Google Scholar]
- 4.Gischler SJ, Mazer P, Duivenvoorden HJ, van Dijk M, Bax NM, Hazebroek FW, et al. Interdisciplinary structural follow-up of surgical newborns: a prospective evaluation. J Pediatr Surg. 2009;44(7):1382–9. [DOI] [PubMed] [Google Scholar]
- 5.Laing S, Walker K, Ungerer J, Badawi N, Spence K. Early development of children with major birth defects requiring newborn surgery. J Paediatr Child Health. 2011;47(3):140–7. [DOI] [PubMed] [Google Scholar]
- 6.Vaux KK, Jones KL, Jones MC, Schelley S, Hudgins L. Developmental outcome in Kabuki syndrome. Am J Med Genet A. 2005;132A(3):263–4. [DOI] [PubMed] [Google Scholar]
- 7.Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53(10):852–73. [DOI] [PubMed] [Google Scholar]
- 8.McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome 1999. September 23 [Updated 2013 Feb 28]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1523/ [Google Scholar]
- 9.Waisbren SE, Landau Y, Wilson J, Vockley J. Neuropsychological outcomes in fatty acid oxidation disorders: 85 cases detected by newborn screening. Dev Disabil Res Rev. 2013;17(3):260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waisbren SE, Gropman AL, Members of the UDC, Batshaw ML. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J Inherit Metab Dis. 2016;39(4):573–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Karnebeek CD, Bowden K, Berry-Kravis E. Treatment of Neurogenetic Developmental Conditions: From 2016 into the Future. Pediatr Neurol. 2016;65:1–13. [DOI] [PubMed] [Google Scholar]
- 12.Litt JS, Glymour MM, Hauser-Cram P, Hehir T, McCormick MC. Early Intervention Services Improve School-age Functional Outcome Among Neonatal Intensive Care Unit Graduates. Acad Pediatr. 2018;18(4):468–74. [DOI] [PubMed] [Google Scholar]
- 13.Spittle A, Orton J, Anderson PJ, Boyd R, Doyle LW. Early developmental intervention programmes provided post hospital discharge to prevent motor and cognitive impairment in preterm infants. Cochrane Database Syst Rev. 2015(11):CD005495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enhancing the outcomes of low-birth-weight, premature infants. A multisite, randomized trial. The Infant Health and Development Program. JAMA. 1990;263(22):3035–42. [DOI] [PubMed] [Google Scholar]
- 15.Bailey DB, Hebbeler K, Spiker D, Scarborough A, Mallik S, Nelson L. Thirty-six-month outcomes for families of children who have disabilities and participated in early intervention. Pediatrics. 2005;116(6):1346–52. [DOI] [PubMed] [Google Scholar]
- 16.Rogers SJ, Vismara L, Wagner AL, McCormick C, Young G, Ozonoff S. Autism treatment in the first year of life: a pilot study of infant start, a parent-implemented intervention for symptomatic infants. J Autism Dev Disord. 2014;44(12):2981–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshinaga-Itano C, Sedey AL, Wiggin M, Chung W. Early Hearing Detection and Vocabulary of Children With Hearing Loss. Pediatrics. 2017;140(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Children With Special Needs Project Advisory Committee, Section on Developmental Behavioral Pediatrics, Bright Futures Steering Committee and Medical Home Initiatives for Council on Children With Disabilities. Identifying infants and young children with developmental disorders in the medical home: an algorithm for developmental surveillance and screening. Pediatrics. 2006;118(1):405–20. [DOI] [PubMed] [Google Scholar]
- 19.Ramadan G, Paul N, Morton M, Peacock JL, Greenough A. Outcome of ventilated infants born at term without major congenital abnormalities. Eur J Pediatr. 2012;171(2):331–6. [DOI] [PubMed] [Google Scholar]
- 20.Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA pediatrics. 2017:e173438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Diemen CC, Kerstjens-Frederikse WS, Bergman KA, de Koning TJ, Sikkema-Raddatz B, van der Velde JK, et al. Rapid Targeted Genomics in Critically Ill Newborns. Pediatrics. 2017;140(4):2854. [DOI] [PubMed] [Google Scholar]
- 22.Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4(154):154ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3(5):377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith LD, Willig LK, Kingsmore SF. Whole-Exome Sequencing and Whole-Genome Sequencing in Critically Ill Neonates Suspected to Have Single-Gene Disorders. Cold Spring Harb Perspect Med. 2015;6(2):a023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18(11):1090–6. [DOI] [PubMed] [Google Scholar]
- 27.Wojcik MH, Schwartz TS, Yamin I, Edward HL, Genetti CA, Towne MC, et al. Genetic disorders and mortality in infancy and early childhood: delayed diagnoses and missed opportunities. Genet Med. 2018;20:1396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malam F, Hartley T, Gillespie MK, Armour CM, Bariciak E, Graham GE, et al. Benchmarking outcomes in the Neonatal Intensive Care Unit: Cytogenetic and molecular diagnostic rates in a retrospective cohort. Am J Med Genet A. 2017. [DOI] [PubMed] [Google Scholar]
- 29.Martens MA, Wilson SJ, Reutens DC. Research Review: Williams syndrome: a critical review of the cognitive, behavioral, and neuroanatomical phenotype. J Child Psychol Psychiatry. 2008;49(6):576–608. [DOI] [PubMed] [Google Scholar]
- 30.Patterson T, Rapsey CM, Glue P. Systematic review of cognitive development across childhood in Down syndrome: implications for treatment interventions. J Intellect Disabil Res. 2013;57(4):306–18. [DOI] [PubMed] [Google Scholar]
- 31.Best S, Rosser E, Bajaj M. Fifteen years of genetic testing from a London developmental clinic. Arch Dis Child. 2017;102(11):1014–8. [DOI] [PubMed] [Google Scholar]
- 32.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Science translational medicine. 2014;6(265):265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385(9975):1305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grein KA, Glidden LM. Predicting well-being longitudinally for mothers rearing offspring with intellectual and developmental disabilities. J Intellect Disabil Res. 2015;59(7):622–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Graaf G, Levine SP, Goldstein R, Skotko BG. Parents' perceptions of functional abilities in people with Down syndrome. Am J Med Genet A. 2018. [DOI] [PubMed] [Google Scholar]
- 37.Swillen A. The importance of understanding cognitive trajectories: the case of 22q11.2 deletion syndrome. Curr Opin Psychiatry. 2016;29(2):133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palikara O, Ashworth M, Van Herwegen J. Addressing the Educational Needs of Children with Williams Syndrome: A Rather Neglected Area of Research? J Autism Dev Disord. 2018;48(9):3256–9. [DOI] [PubMed] [Google Scholar]
- 39.Zwanenburg RJ, Ruiter SA, van den Heuvel ER, Flapper BC, Van Ravenswaaij-Arts CM. Developmental phenotype in Phelan-McDermid (22q13.3 deletion) syndrome: a systematic and prospective study in 34 children. J Neurodev Disord. 2016;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marchal JP, Maurice-Stam H, Houtzager BA, Rutgers van Rozenburg-Marres SL, Oostrom KJ, Grootenhuis MA, et al. Growing up with Down syndrome: Development from 6 months to 10.7 years. Res Dev Disabil. 2016;59:437–50. [DOI] [PubMed] [Google Scholar]
- 41.Van Herwegen J, Ashworth M, Palikara O. Parental views on special educational needs provision: Cross-syndrome comparisons in Williams Syndrome, Down Syndrome, and Autism Spectrum Disorders. Res Dev Disabil. 2018;80:102–11. [DOI] [PubMed] [Google Scholar]
- 42.Alkuraya FS. Natural human knockouts and the era of genotype to phenotype. Genome Med. 2015;7(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancrini C, Puliafito P, Digilio MC, Soresina A, Martino S, Rondelli R, et al. Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr. 2014;164(6):1475–80.e2. [DOI] [PubMed] [Google Scholar]
- 44.Gray JE, McCormick MC, Richardson DK, Ringer S. Normal birth weight intensive care unit survivors: outcome assessment. Pediatrics. 1996;97(6 Pt 1):832–8. [PubMed] [Google Scholar]
- 45.Nordhov SM, Rønning JA, Ulvund SE, Dahl LB, Kaaresen PI. Early intervention improves behavioral outcomes for preterm infants: randomized controlled trial. Pediatrics. 2012;129(1):e9–e16. [DOI] [PubMed] [Google Scholar]
- 46.Kaaresen PI, Rønning JA, Ulvund SE, Dahl LB. A randomized, controlled trial of the effectiveness of an early-intervention program in reducing parenting stress after preterm birth. Pediatrics. 2006;118(1):e9–19. [DOI] [PubMed] [Google Scholar]
- 47.Committee on Fetus and Newborn. Hospital discharge of the high-risk neonate. Pediatrics. 2008;122(5):1119–26. [DOI] [PubMed] [Google Scholar]
- 48.Kuppala VS, Tabangin M, Haberman B, Steichen J, Yolton K. Current state of high-risk infant follow-up care in the United States: results of a national survey of academic follow-up programs. J Perinatol. 2012;32(4):293–8. [DOI] [PubMed] [Google Scholar]
- 49.Bayley N. Bayley Scales of Infant and Toddler Development Manual . 3rd edn. San Antonio: TTPC, 2006. [Google Scholar]
- 50.Vohr BR, O'Shea M, Wright LL. Longitudinal multicenter follow-up of high-risk infants: why, who, when, and what to assess. Semin Perinatol. 2003;27(4):333–42. [DOI] [PubMed] [Google Scholar]
- 51.Doyle LW, Anderson PJ, Battin M, Bowen JR, Brown N, Callanan C, et al. Long term follow up of high risk children: who, why and how? BMC Pediatr. 2014;14:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126(9):1143–72. [DOI] [PubMed] [Google Scholar]
- 53.Robertson CM, Perlman M. Follow-up of the term infant after hypoxic-ischemic encephalopathy. Paediatr Child Health. 2006;11(5):278–82. [PMC free article] [PubMed] [Google Scholar]
- 54.Shankaran S, Laptook AR, Pappas A, McDonald SA, Das A, Tyson JE, et al. Effect of Depth and Duration of Cooling on Death or Disability at Age 18 Months Among Neonates With Hypoxic-Ischemic Encephalopathy: A Randomized Clinical Trial. JAMA. 2017;318(1):57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wynn J, Aspelund G, Zygmunt A, Stolar CJ, Mychaliska G, Butcher J, et al. Developmental outcomes of children with congenital diaphragmatic hernia: a multicenter prospective study. J Pediatr Surg. 2013;48(10):1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harmsen WJ, Aarsen FJ, van der Cammen-van Zijp MHM, van Rosmalen JM, Wijnen RMH, Tibboel D, et al. Developmental problems in patients with oesophageal atresia: a longitudinal follow-up study. Arch Dis Child Fetal Neonatal Ed. 2017;102(3):F214–F9. [DOI] [PubMed] [Google Scholar]
- 57.Danzer E, Gerdes M, D'Agostino JA, Bernbaum J, Siegle J, Hoffman C, et al. Prospective, interdisciplinary follow-up of children with prenatally diagnosed giant omphalocele: short-term neurodevelopmental outcome. J Pediatr Surg. 2010;45(4):718–23. [DOI] [PubMed] [Google Scholar]
- 58.IJsselstijn H, Gischler SJ, Wijnen RMH, Tibboel D. Assessment and significance of long-term outcomes in pediatric surgery. Semin Pediatr Surg. 2017;26(5):281–5. [DOI] [PubMed] [Google Scholar]
- 59.Skotko BG, Davidson EJ, Weintraub GS. Contributions of a specialty clinic for children and adolescents with Down syndrome. Am J Med Genet A. 2013;161A(3):430–7. [DOI] [PubMed] [Google Scholar]
- 60.Gerdes M, Solot C, Wang PP, McDonald-McGinn DM, Zackai EH. Taking advantage of early diagnosis: preschool children with the 22q11.2 deletion. Genet Med. 2001;3(1):40–4. [DOI] [PubMed] [Google Scholar]
- 61.Javalkar K, Litt JS. Reason for Referral Predicts Utilization and Perceived Impact of Early Intervention Services. J Dev Behav Pediatr. 2017;38(9):706–13. [DOI] [PubMed] [Google Scholar]
- 62.Nacinovich R, Villa N, Redaelli S, Broggi F, Bomba M, Stoppa P, et al. Interstitial 11q deletion: genomic characterization and neuropsychiatric follow up from early infancy to adolescence and literature review. BMC Res Notes. 2014;7:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arias-Llorente RP, Rodriguez-Dehli C, López-Martínez A, Riaño-Galán I. Focal Dermal Hypoplasia Due to De Novo Mutation c.1061T>C(p.Leu354Pro) in the PORCN Gene: Importance of Early Diagnosis and Multidisciplinary Follow-Up. Fetal Pediatr Pathol. 2015;34(6):375–82. [DOI] [PubMed] [Google Scholar]
- 64.Wojcik MH, Linnea K, Stoler JM, Rappaport L. Updating the neurodevelopmental profile of Alazami syndrome: Illustrating the role of developmental assessment in rare genetic disorders. Am J Med Genet A. 2019. doi: 10.1002/ajmg.a.61189. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Connolly BH, Morgan SB, Russell FF, Fulliton WL. A longitudinal study of children with Down syndrome who experienced early intervention programming. Phys Ther. 1993;73(3):170–9; discussion 9-81. [DOI] [PubMed] [Google Scholar]
- 66.Saul RA, Meredith SH. Beyond the Genetic Diagnosis: Providing Parents What They Want to Know. Pediatr Rev. 2016;37(7):269–78. [DOI] [PubMed] [Google Scholar]
- 67.Abel EA, Tonnsen BL. Sleep phenotypes in infants and toddlers with neurogenetic syndromes. Sleep Med. 2017;38:130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Purdy IB, Craig JW, Zeanah P. NICU discharge planning and beyond: recommendations for parent psychosocial support. J Perinatol. 2015;35 Suppl 1:S24–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lanfranchi S, Vianello R. Stress, locus of control, and family cohesion and adaptability in parents of children with Down, Williams, Fragile X, and Prader-Willi syndromes. Am J Intellect Dev Disabil. 2012;117(3):207–24. [DOI] [PubMed] [Google Scholar]
- 70.Werner-Lin A, Walser S, Barg FK, Bernhardt BA. "They Can't Find Anything Wrong with Him, Yet": Mothers' experiences of parenting an infant with a prenatally diagnosed copy number variant (CNV). Am J Med Genet A. 2017;173(2):444–51. [DOI] [PubMed] [Google Scholar]
- 71.Minnes P, Perry A, Weiss JA. Predictors of distress and well-being in parents of young children with developmental delays and disabilities: the importance of parent perceptions. J Intellect Disabil Res. 2015;59(6):551–60. [DOI] [PubMed] [Google Scholar]
- 72.Plumridge G, Metcalfe A, Coad J, Gill P. The role of support groups in facilitating families in coping with a genetic condition and in discussion of genetic risk information. Health Expect. 2012;15(3):255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cacioppo CN, Conway LJ, Mehta D, Krantz ID, Noon SE. Attitudes about the use of internet support groups and the impact among parents of children with Cornelia de Lange syndrome. Am J Med Genet C Semin Med Genet. 2016;172(2):229–36. [DOI] [PubMed] [Google Scholar]
- 74.McManus BM, Poehlmann J. Maternal depression and perceived social support as predictors of cognitive function trajectories during the first 3 years of life for preterm infants in Wisconsin. Child Care Health Dev. 2012;38(3):425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Conroy K, Rea C, Kovacikova GI, Sprecher E, Reisinger E, Durant H, et al. Ensuring Timely Connection to Early Intervention for Young Children With Developmental Delays. Pediatrics. 2018;142(1). [DOI] [PubMed] [Google Scholar]
- 76.King TM, Tandon SD, Macias MM, Healy JA, Duncan PM, Swigonski NL, et al. Implementing developmental screening and referrals: lessons learned from a national project. Pediatrics. 2010;125(2):350–60. [DOI] [PubMed] [Google Scholar]
- 77.Puthussery S, Chutiyami M, Tseng PC, Kilby L, Kapadia J. Effectiveness of early intervention programs for parents of preterm infants: a meta-review of systematic reviews. BMC Pediatr. 2018;18(1):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lipner HS, Huron RF. Developmental and Interprofessional Care of the Preterm Infant: Neonatal Intensive Care Unit Through High-Risk Infant Follow-up. Pediatr Clin North Am. 2018;65(1):135–41. [DOI] [PubMed] [Google Scholar]
- 79.Stark Z, Schofield D, Alam K, Wilson W, Mupfeki N, Macciocca I, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19(8):867–74. [DOI] [PubMed] [Google Scholar]
- 80.Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, et al. Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA pediatrics. 2017;171(9):855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stark Z, Schofield D, Martyn M, Rynehart L, Shrestha R, Alam K, et al. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Genet Med. 2019;21(1):173–80. [DOI] [PubMed] [Google Scholar]
- 82.Chorna O, Baldwin HS, Neumaier J, Gogliotti S, Powers D, Mouvery A, et al. Feasibility of a Team Approach to Complex Congenital Heart Defect Neurodevelopmental Follow-Up: Early Experience of a Combined Cardiology/Neonatal Intensive Care Unit Follow-Up Program. Circ Cardiovasc Qual Outcomes. 2016;9(4):432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]