

Abstract
PURPOSE
Anaplastic thyroid carcinoma is an aggressive malignancy that is almost always fatal and lacks effective systemic treatment options for patients with BRAF-wild type disease. As part of a phase I/II study in patients with advanced/metastatic solid tumors, patients with anaplastic thyroid carcinoma were treated with spartalizumab, a humanized monoclonal antibody against the programmed death-1 (PD-1) receptor.
METHODS
We enrolled patients with locally advanced and/or metastatic anaplastic thyroid carcinoma in a phase II cohort of the study. Patients received 400 mg spartalizumab intravenously, once every 4 weeks. The overall response rate was determined according to RECIST v1.1.
RESULTS
Forty-two patients were enrolled. Adverse events were consistent with those previously observed with PD-1 blockade. Most common treatment-related adverse events were diarrhea (12%), pruritus (12%), fatigue (7%), and pyrexia (7%). The overall response rate was 19%, including three patients with a complete response and five with a partial response. Most patients had baseline tumor biopsies positive for PD-L1 expression (n = 28/40 evaluable), and response rates were higher in PD-L1–positive (8/28; 29%) versus PD-L1–negative (0/12; 0%) patients. The highest rate of response was observed in the subset of patients with PD-L1 ≥ 50% (6/17; 35%). Responses were seen in both BRAF-nonmutant and BRAF-mutant patients and were durable, with a 1-year survival of 52.1% in the PD-L1–positive population.
CONCLUSION
To our knowledge, this is the first clinical trial to show responsiveness of anaplastic thyroid carcinoma to PD-1 blockade.
INTRODUCTION
Undifferentiated (anaplastic) thyroid carcinoma (ATC) is a rare, aggressive form of thyroid cancer with nearly 100% disease-specific mortality and no standard therapeutic approach.1,2 Existing treatment options include surgery, radiation, and chemotherapy, used in combination when possible.1 The benefits of aggressive multimodality therapy are typically short lived and survival benefit is unclear, with a median overall survival (OS) of only 5 months.1–4 Mutations affecting BRAF are found in approximately 27%–45%5–7 of ATC, and combination therapy with the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib was recently approved by the US Food and Drug Administration for this subset of patients.8 This is the only systemic therapy in the modern era to have shown meaningful activity in ATC, albeit in a subset of patients. A critical unmet need remains for effective therapy for patients with BRAF wild-type disease.
Drugs targeting the interaction between the programmed death-1 (PD-1) receptor and its ligands, programmed death-ligand 1/2 (PD-L1/L2), have shown clinical activity in a broad range of tumor types, particularly in cancers demonstrating PD-L1 expression.9–11 Approximately 22%–29% of ATC tumor samples have been reported to express PD-L1,12,13 suggesting targeting the PD-1/PD-L1 axis may have promise. Indeed, a positive response to combined BRAF and PD-1–directed therapy was reported for one patient with BRAF-mutant ATC,14 and a small retrospective single-institution cohort reported on responses in a subset of patients with ATC with prior disease progression on kinase inhibitors with the addition of pembrolizumab.15
Spartalizumab (PDR001) is a humanized immunoglobulin 4 monoclonal antibody that binds PD-1 with subnanomolar activity and blocks interaction with PD-L1 and PD-L2.16 This first-in-human phase I/II study was designed to investigate the safety and efficacy of spartalizumab in patients with advanced or metastatic solid tumors. Here, we describe the results from the phase II part of the study in patients with ATC.
METHODS
Clinical Study Design and Oversight
This was a phase I/II, international, multicenter, open-label study of spartalizumab in patients with advanced solid tumors (ClinicalTrails.gov identifier: NCT02404441). The data cutoff date was February 1, 2019.
This Novartis-sponsored study was performed in compliance with Good Clinical Practice guidelines and in accordance with the Declaration of Helsinki. The protocol and all amendments were reviewed and approved by an independent ethics committee and/or institutional review board at each participating site. Written informed consent was obtained from all patients before the study start.
Study Objectives
The primary objective of the phase I part of the study was to determine the recommended phase II dose/maximum tolerated dose for spartalizumab; these findings have been reported.16 The primary objective for the phase II part was to estimate the antitumor activity of spartalizumab (overall response rate [ORR] according to Response Evaluation Criteria in Solid Tumors [RECIST] v1.1). Secondary and exploratory objectives included characterizing the safety and tolerability of spartalizumab, characterizing the pharmacokinetic profile of spartalizumab, further evaluating the antitumor activity of spartalizumab (ORR according to immune-related response criteria [irRC], progression-free survival [PFS] and OS, duration of response, and disease control rate), and assessing the pharmacodynamic effect and potential predictors of efficacy.
Patients
Patients eligible for the phase II part of the study had locally advanced/metastatic solid tumors; this report describes patients with ATC. Central pathology review was performed to confirm the diagnosis but was not required for the start of study treatment. Adult patients with at least one measurable lesion as determined by RECIST v1.1 and a tumor amenable to biopsy were enrolled. All patients had an Eastern Cooperative Oncology Group (ECOG) performance status of ≤ 1 and provided consent to tumor biopsy at baseline and during treatment. Amendments were made to the study protocol during recruitment: patients with locally advanced disease were considered for chemoradiation and/or surgery before entering the study if those therapies were in the best interest of the patient, disease progression on prior treatment was not required, and patients with short-term risk for life-threatening complications were not eligible. Other exclusion criteria are described in the study protocol (Data Supplement).
Originally, it was planned to enroll approximately 10 patients with ATC, reflecting the low prevalence of this disease. This was then increased to approximately 40 patients. As data are limited on the expected efficacy of a PD-1 inhibitor in patients with ATC, 10% was selected as the boundary for moderate efficacy. With an observed ORR of 20% at a sample size of 40, the 90% credible interval (based on a minimally informative prior β [0.25 to 1]) for the true ORR would be (10.8%–30.9%), giving a lower bound greater than the estimated moderate efficacy.
Treatment Plan
Patients in the phase II ATC treatment group received spartalizumab 400 mg, administered intravenously (IV), once every 4 weeks. Treatment was administered until unacceptable toxicity, progressive disease as per irRC, or patient/physician decision. If more than two consecutive doses were missed because of toxicity, treatment was discontinued. Delays of up to 7 days were permitted; dose modifications were not allowed.
Safety and Response Assessments
Regular safety assessments were performed, including physical examination, ECOG performance status, and laboratory parameters. Adverse events (AEs), defined by the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03, were assessed at every visit.
Tumor response was evaluated by local investigator’s assessment of imaging data using RECIST v1.1 and irRC. Tumor assessments were performed at baseline, and subsequent assessments were performed every 8 weeks up to 40 weeks, then every 12 weeks until disease progression per irRC, withdrawal of consent, or loss to follow-up.
Pharmacodynamic and Biomarker Analyses
At screening, an archival tumor sample and/or a newly obtained pretreatment tumor biopsy was collected from each patient, and an additional tumor biopsy was obtained during treatment.
Biomarker assessments included baseline PD-L1 levels in tumor cells, assessed centrally by immunohistochemistry using a diagnostic developed for non–small-cell lung cancer (Dako PD-L1 IHC 22C3 pharmaDx, performed by HistoGeneX, Wilrijk, Belgium), baseline CD8+ levels assessed by immunohistochemistry, expressed as CD8+ lymphocyte staining as a percentage of the total sample area, and baseline BRAF status, assessed by Cobas 4800 BRAF V600 mutation test (performed by HistoGeneX, Wilrijk, Belgium).
Patient samples were also analyzed by next-generation sequencing (Foundation Medicine, Cambridge, Massachusetts) to assess mutations affecting cancer-related genes and tumor mutation burden,17 and by RNA sequencing (RNAseq) to evaluate gene expression patterns, including interferon gamma (IFNγ) signature.
Statistical Methods
Detailed statistical methods are summarized in the Data Supplement. ORRs are summarized with accompanying 95% exact binomial CIs. Associations between ORR and potential biomarkers were assessed using exact Fisher’s tests; Spearman correlation coefficient (95% CI) was estimated for the correlation between efficacy and IFNγ signature. Median survival (95% CI) was estimated using Kaplan-Meier method.
RESULTS
Patient Characteristics and Disposition
During the phase I part of the study, a patient with ATC treated with spartalizumab 3 mg/kg once every 2 weeks experienced a mixed response, with significant reductions in two target lesions and a best overall response of stable disease, before developing progressive disease at study Day 222. On the basis of this patient and the associated biomarker data, which identified an increase in CD8 between the baseline and on-treatment biopsy, a phase II cohort was opened for patients with ATC.
As of February 1, 2019, 42 patients with ATC had been treated with 400 mg spartalizumab IV once every 4 weeks in the phase II part of the study. Patient demographics and baseline characteristics are shown in Table 1. Tumor tissue was available for central pathology review for 40 patients (95%), with 38 patients (90%) confirmed to have ATC. For the two remaining patients, the diagnosis was high-grade malignant neoplasm with chondroblastic differentiation and Hürthle cell carcinoma. Median age was 62.5 years (range, 46–83 years), and all patients had a performance status of 0 or 1. Sixty percent of patients had received prior antineoplastic therapy, with common prior therapies including doxorubicin in 13 (31%), paclitaxel in 12 (29%), and lenvatinib in 4 patients (10%). Seventy-one percent of patients also received prior radiotherapy, and 67% had undergone thyroidectomy/neck dissection. Most patients had metastatic disease at study entry, with lungs (83%) and lymph nodes (48%) as the most common sites of metastasis.
TABLE 1.
Baseline Patient Demographics and Characteristics
| Characteristic | Spartalizumab 400 mg Every 4 Weeks (N = 42) |
|---|---|
| Central pathology review | |
| ATC | 38 (90.5) |
| Other | 2 (4.8) |
| Missing | 2 (4.8) |
| Age, years, median (range) | 62.5 (46–83) |
| Sex, male | 23 (54.8) |
| ECOG PS | |
| 0 | 17 (40.5) |
| 1 | 25 (59.5) |
| Prior treatment regimens | |
| 0 | 17 (40.5) |
| 1 | 20 (47.6) |
| ≥ 2 | 5 (11.9) |
| Prior radiation | |
| Yes | 30 (71.4) |
| Prior thyroidectomy and/or neck dissection | |
| Yes | 28 (66.7) |
| Metastatic sites at study entry | |
| Lung | 35 (83.3) |
| Lymph node | 20 (47.6) |
| Bone | 5 (11.9) |
| Liver | 3 (7.1) |
| BRAF V600 mutation by Cobas 4800 | |
| Mutant | 12 (28.6) |
| Nonmutant | 26 (61.9) |
| Missing | 4 (9.5) |
NOTE. Data are presented as No. (%) unless otherwise noted.
Abbreviations: ATC, anaplastic thyroid carcinoma; ECOG PS, Eastern Cooperative Oncology Group performance status.
As of February 1, 2019, treatment was ongoing for 7 patients (17%); 35 patients (83%) discontinued from the study because of progressive disease (57%), death (21%; due to study indication [n = 8] and shock unrelated to study treatment [n = 1]), AEs (2%), or patient/guardian decision (2%).
Safety and Tolerability
AEs, regardless of relationship to study treatment, were reported in 41 patients (98%) and grade 3/4 AEs regardless of relationship to study treatment were reported in 29 patients (69%; Data Supplement). AEs attributed to study treatment were experienced by 19 patients (45%; Table 2). The most frequent were diarrhea, pruritus, fatigue, and pyrexia. Grade 3/4 AEs attributed to study treatment were reported in four patients (10%): anemia, rash, and metastases to the CNS. Ten patients experienced potential immune-related AEs of special interest, suspected to be treatment related. These were mostly grade 1 or 2, including diarrhea, pruritus (five patients each; 12%), pruritus generalized (one patient; 2%), rash (two patients; 5%; one grade 3), rash macular, increased blood thyroid-stimulating hormone, and hypothyroidism (one patient each; 2%). Additional AEs of potential immune-related etiology regardless of relationship to treatment are summarized in the Data Supplement. Metastasis to the CNS was the only serious AE suspected to be related to study treatment. This patient was enrolled with no knowledge of brain metastases but reported vertigo, vision loss, and vomiting before study enrollment; inflammation induced by treatment with spartalizumab was proposed by the investigator as a possible mechanism for the serious AE. Dose interruptions due to AEs occurred in three patients. Discontinuation of study treatment was reported in one patient who developed grade 4 obstructive airway disorder, not suspected to be related to treatment. A total of 11 patients died during the treatment period; deaths were attributed to hemorrhage due to disease (n = 1) and disease progression (n = 10).
TABLE 2.
Adverse Events (any grade, occurring in ≥ 3% of patients) Suspected to Be Related to Study Treatment
| Preferred Term | Spartalizumab 400 mg Every 4 Weeks (N = 42) |
|
|---|---|---|
| All | Grade 3/4 | |
| Total | 19 (45.2) | 4 (9.5) |
| Diarrhea | 5 (11.9) | 0 |
| Pruritus | 5 (11.9) | 0 |
| Fatigue | 3 (7.1) | 0 |
| Pyrexia | 3 (7.1) | 0 |
| Anemia | 2 (4.8) | 2 (4.8) |
| Asthenia | 2 (4.8) | 0 |
| Myalgia | 2 (4.8) | 0 |
| Rash | 2 (4.8) | 1 (2.4) |
NOTE. Data presented as No. (%).
Efficacy
As expected for this aggressive disease, the median duration of spartalizumab treatment was short at 8 weeks (range, 1.7–113.6 weeks). Twenty-four patients discontinued treatment for progressive disease, eight for death due to study indication, and one for an AE not suspected to be related to treatment. Eleven patients (26%) received treatment of ≥ 50 weeks (Fig 1A). Best overall response was evaluated by both RECIST v1.1 and irRC (Data Supplement). The ORR according to RECIST v1.1 was 19% (95% CI, 8.6% to 34.1%; n = 8/42), including three patients with a complete response (CR; 7%) and five patients with a partial response (PR; 12%). All responding patients had ATC confirmed by central pathology review. Duration of response ranged from 16.7 weeks to 1.6 years (ongoing at data cutoff). Median duration of response has not yet been reached, and six of eight responding patients were still receiving treatment with ongoing responses at the data cutoff. By irRC, the ORR was 24% (95% CI, 12.1% to 39.5%), including three patients with a CR and seven patients with a PR. All patients with a confirmed response by RECIST v1.1 or irRC had ATC confirmed by central pathology analysis.
FIG 1.
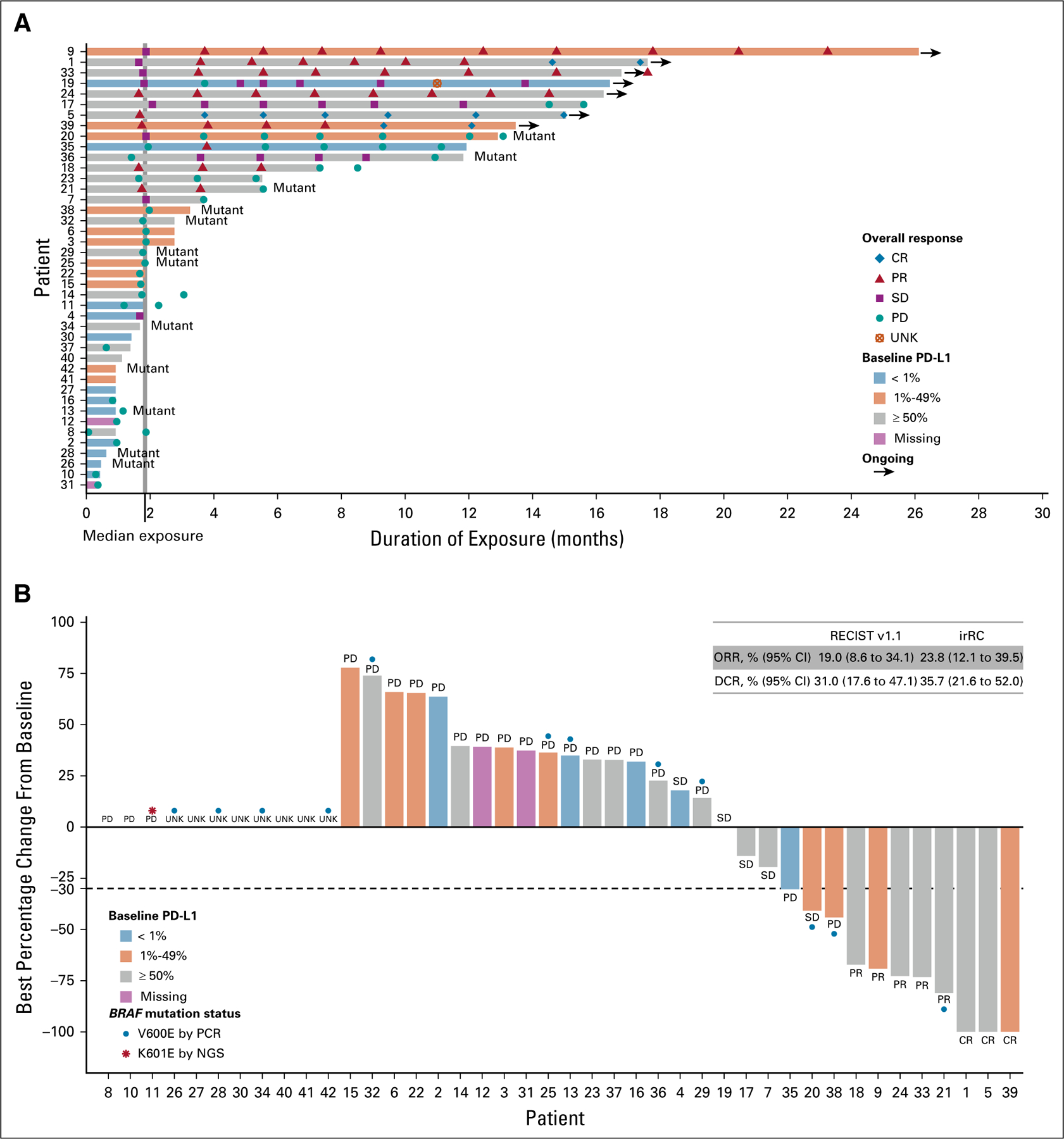
Duration of exposure to spartalizumab and percentage change from baseline in sum of diameters of target lesions, by programmed death-ligand 1 (PD-L1) expression at baseline. (A) Duration of exposure to spartalizumab. Mutant denotes BRAF mutation. (B) Best percentage change from baseline in sum of diameters of target lesions. Thirty-one patients were evaluable for best percentage change; 11 patients were not evaluable because of discontinuation or death before first postbaseline assessment (n = 8) or missing postbaseline assessment (n = 3). Best overall response by RECIST v1.1 is indicated. Where available, BRAF mutation status is indicated. Patient number refers to number in Data Supplement. CR, complete response; DCR, disease control rate [CR + PR + SD]; irRC, immune-related response criteria; NGS, next-generation sequencing; ORR, overall response rate [CR + PR]; PCR, polymerase chain reaction; PD, progressive disease; PR, partial response; SD, stable disease; UNK, unknown.
The extent of disease reduction and change over time is shown in Fig 1B and the Data Supplement. Of 31 patients with evaluable postbaseline sum of target lesions measurements, 13 had some reduction from baseline. Eleven patients were not evaluable for best percentage change from baseline, because of discontinuation or death before first postbaseline assessment (n = 8) or missing postbaseline assessments (n = 3).
Median PFS (Data Supplement) was 1.7 months by both RECIST v1.1 and irRC (95% CI, 1.2 to 1.9 months and 1.2 to 2.0 months, respectively). At 1 year, the PFS rate was 17% and 22% by RECIST v1.1 and irRC, respectively. Median OS was 5.9 months (95% CI, 2.4 months to not reached), with 40% of patients alive at 1 year (Fig 1A).
Biomarker Analyses
Patient tumor biopsies were analyzed by immunohistochemistry for baseline (before therapy) expression of PD-L1 and CD8+ lymphocytes, RNA sequencing to characterize gene expression, tumor mutational burden (TMB), and mutations affecting genes related to cancer by DNA sequencing. The outcome of these analyses is summarized per patient in the Data Supplement. Commonly mutated genes included TP53, TERT, BRAF, PTEN, and CDKN2A/B. TMB was evaluable in 27 patients, and median TMB was low at 3.78 mutations/Mb (range, 0–13.87 mutations/Mb). Six of 10 patients with RECIST/irRC responses had TMB analyses; only two had TMB > 5 mutations/Mb. One responder, classified as microsatellite stable by Foundation Medicine testing with a TMB in the intermediate range (14 mutations/Mb, the highest TMB in the cohort), had a frameshift mutation in the DNA mismatch repair gene MSH6 that contributes to tumor genomic instability.18 Of the other five RECIST/irRC responders who had mutational analysis, none had detected mutations affecting mismatch repair proteins.
Tumor samples were analyzed for baseline PD-L1 expression by immunohistochemistry; data were available for 40 patients. The majority (n = 28/40; 70%) of tumor biopsies were positive for PD-L1 expression (≥ 1% positive in tumor cells), and the confirmed RECIST response rate was 29% (95% CI, 13.2 to 48.7; n = 8/28) in patients with PD-L1–positive disease. The response rate was highest (ORR, 35%; 95% CI, 14.2 to 61.7) in the subset of patients with PD-L1 ≥ 50%; no RECIST responses were observed in patients with PD-L1 expression < 1% (0/12 patients; Data Supplement). The difference in response rates between PD-L1–positive and –negative groups reached statistical significance at level 0.1 (Fisher’s exact two-sided test, P = .079). One patient with PD-L1 < 1% had an irRC PR. Survival rates were also higher in patients with baseline tumor samples positive for PD-L1 expression. Although median PFS remained < 2 months for all PD-L1 subsets, 1-year PFS rates were notably increased in PD-L1–positive patients, with 0%, 20%, and 29%, respectively for PD-L1 < 1%, 1%–49%, and ≥ 50%. OS also correlated with PD-L1 status, with a median OS of 1.6 months in patients with PD-L1 < 1%, compared with not yet reached in PD-L1–positive patients (Fig 2).
FIG 2.
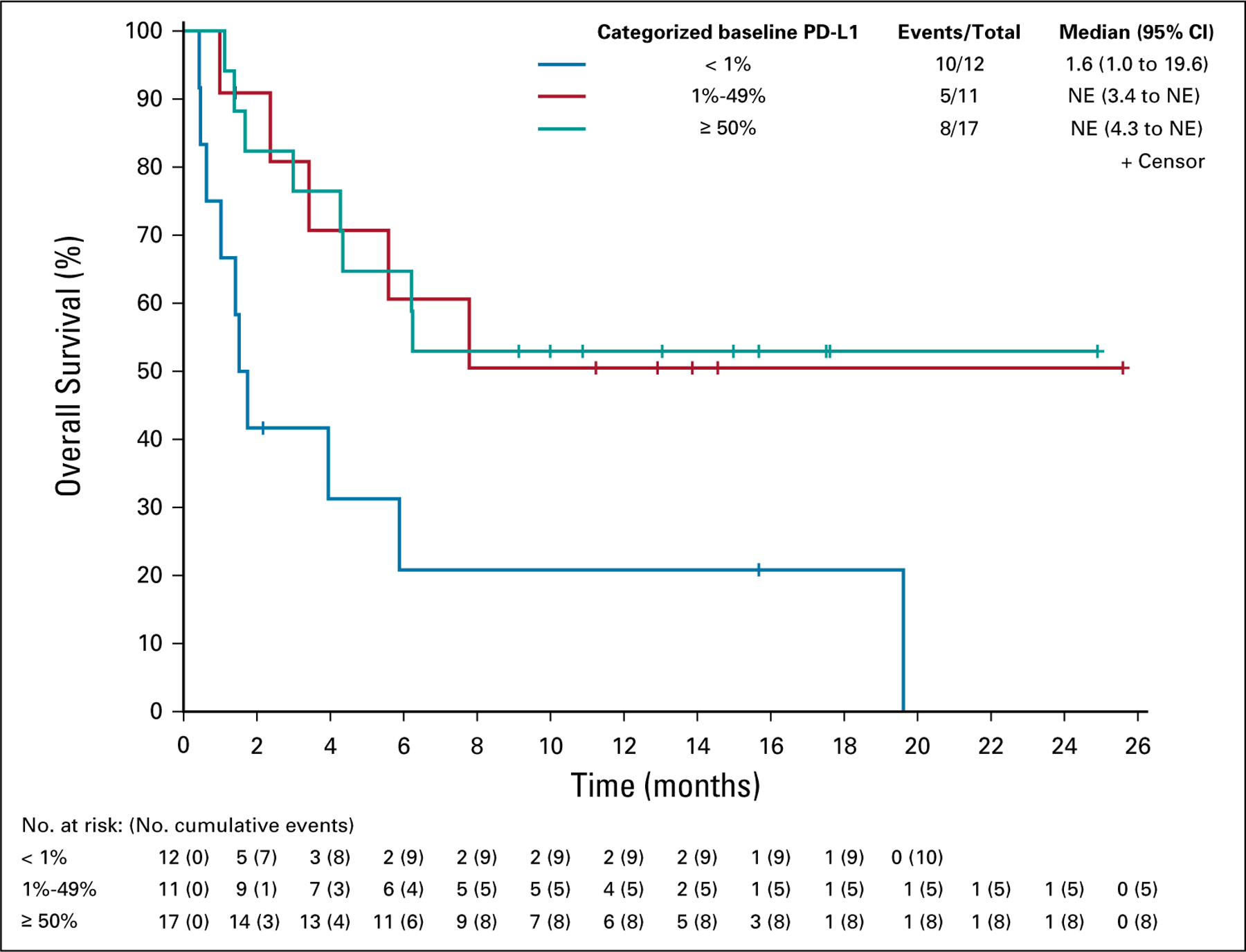
Overall survival by PD-L1 expression at baseline. NE, not estimable; PD-L1, programmed death-ligand 1.
Baseline CD8 expression by immunohistochemistry was available for 37 patients. Forty-three percent (n = 16/37) of tumors had ≥ 1% CD8+ staining. There was a weak correlation between CD8+ lymphocyte infiltration and PD-L1 expression (Spearman coefficient, 0.24; 95% CI, −0.1 to 0.7). The ORR was numerically higher in patients with tumors containing CD8 ≥ 1% compared with patients with CD8 expression < 1% (25% [95% CI, 7.3% to 52.4%] v 14% [95% CI, 3.0% to 36.3%]; Fisher’s exact two-sided test, P = .437; Data Supplement). There were 14 patients with tumors positive for both PD-L1 and CD8; the ORR in this subgroup was 29% (95% CI, 8.4% to 58.1%).
Tumor samples were analyzed for BRAF mutation status, available for 38 patients; of these, a third had BRAF mutation (n = 12/38; 32%). The ORR was lower in patients with BRAF mutation than in those without (8% [95% CI, 0.2% to 38.5%] v 23% [95% CI, 9.0% to 43.6%]; Fisher’s exact two-sided test, P = .395; Data Supplement). Nine of 12 patients with a BRAF mutation by polymerase chain reaction had a PD-L1 of ≥ 1%.
Baseline tumor samples were also evaluated for gene expression by RNAseq. In data available from 18 patients, a correlation was observed between the best percentage change from baseline in sum of diameters of target lesions and IFNγ signature (Fig 3; Spearman coefficient, −0.67; 95% CI, −0.9 to −0.3).
FIG 3.
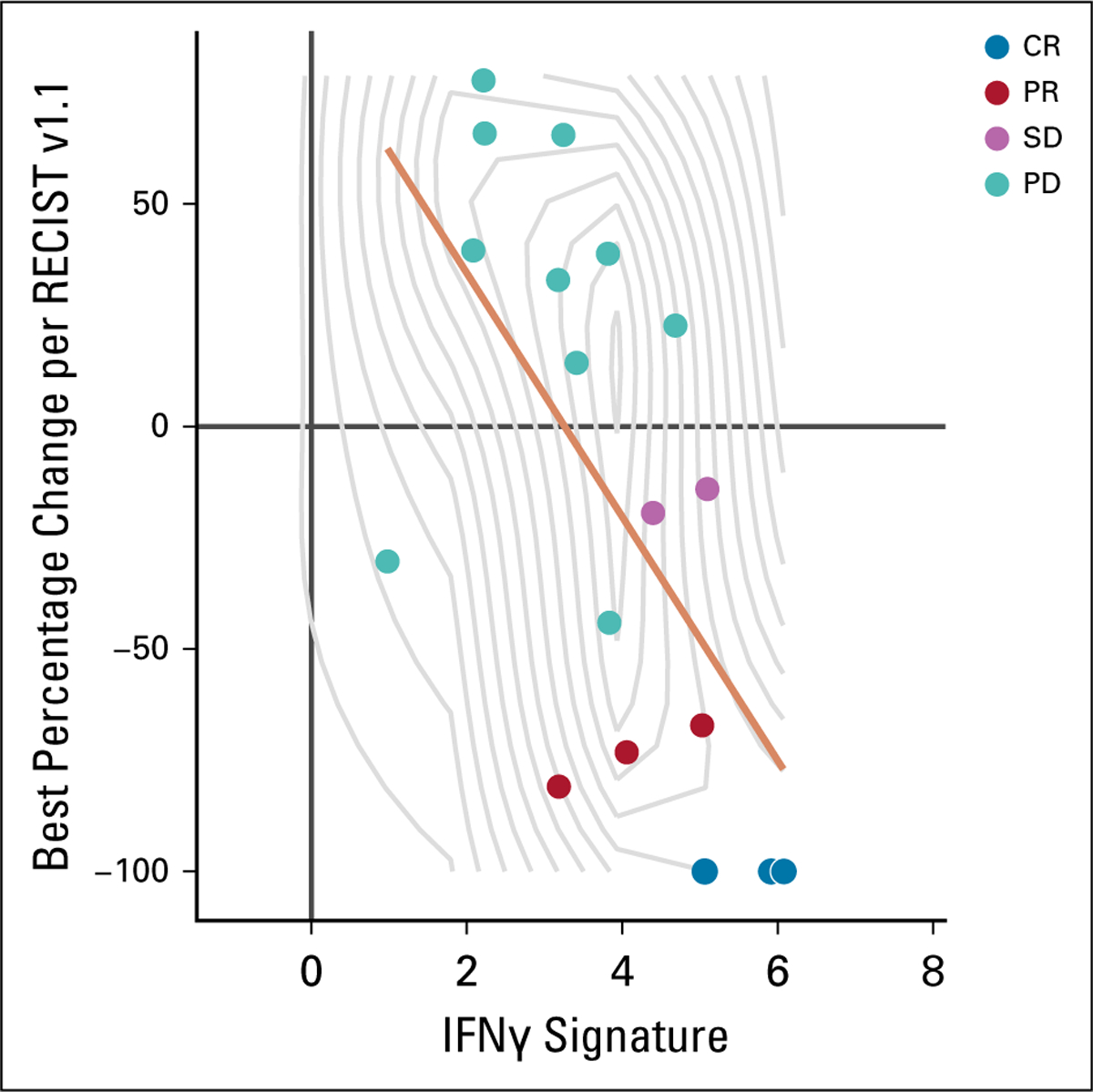
Interferon g (IFNγ) signature, by best percentage change from baseline in sum of diameters of target lesions. Best overall response by RECIST v1.1 is indicated. Data were available for 18 patients. Spearman coefficient, −0.67; 95% CI, −0.9 to −0.3. CR, complete response; PD, progressive disease; PD-L1, programmed death-ligand 1; PR, partial response; SD, stable disease.
DISCUSSION
Spartalizumab was well tolerated in this patient population, and toxicities were consistent with those seen with other PD-1–targeting monoclonal antibodies.19,20 Treatment with spartalizumab achieved durable responses in a subset of patients, with three complete and five partial responses to treatment, including seven responses in patients without detectable BRAF mutation. Of eight patients (19%) with a response according to RECIST criteria, six were ongoing at the time of data cutoff, lasting more than a year after starting therapy. Additional responses to treatment were observed by irRC, with an ORR of 24%. ATC diagnosis was confirmed by central pathology review in 95% of 40 evaluable patients; all responding patients had ATC. This evidence of clinical benefit in ATC, a disease with an especially grave prognosis, is striking.
Response to treatment with spartalizumab was particularly notable in patients with evidence of an inflamed tumor microenvironment at baseline, as suggested by higher PD-L1 expression, presence of CD8+ lymphocytes, and expression of genes involved in IFNγ signaling. An association was also seen between PD-L1 expression at baseline and survival, with median OS not yet reached in the subset of patients with PD-L1 ≥ 1% expression at baseline. These findings are consistent with the mechanism of action of spartalizumab and with data reported for inhibitors of PD-1 in other indications.11
The mechanism underlying the immune activation seen in ATC is unclear. Response to PD-1 blockade is associated with higher levels of novel antigens created by chronic exposure to mutagens, such as tobacco smoke or UV light, or by defects in DNA repair, such as is found in microsatellite-unstable colorectal cancer.21 A subset of these neoantigens are immunogenic and provide targets for recognition by the immune system and disease control. Overall, TMB was low across the analyzed patients, with only one patient with TMB > 10 mutations/Mb; these findings are consistent with published reports.5,6 Patient responses despite a low or intermediate TMB have been observed in other cancers, including renal cell carcinoma, where a high prevalence of insertions and deletions has been postulated as a mechanism for neoantigen formation,22 and Merkel cell polyomavirus–positive tumors, where the presence of viral antigens may explain checkpoint activity.23,24 Additional investigation will be necessary to characterize the mechanism for the efficacy observed with checkpoint inhibition in ATC. The rapid disease progression and very short OS in spartalizumab nonresponders highlight an important unmet need to build on PD-1–based immunotherapy to further improve treatment of patients with ATC.
In summary, spartalizumab demonstrated promising clinical activity and a good safety profile in a patient population with aggressive incurable disease and short life expectancy. Targeting PD-1/PD-L1 may provide a much-needed treatment option for patients with PD-L1–positive advanced ATC, including the BRAF wild-type population.
Supplementary Material
ACKNOWLEDGMENT
We thank the patients who participated in the trial and their families and thank the physicians, nurses, research coordinators, and other staff at each site who assisted with the study. We also thank Jennifer Mataraza, Victor Antona, Antonio Silva, and Emilie Bossuge for their contributions to this study. Editorial assistance was provided by Laura Hilditch, PhD, and was funded by Novartis Pharmaceuticals.
Financial support: Scott Cameron
SUPPORT
Supported by Novartis Pharmaceuticals.
Footnotes
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Jaume Capdevila
Consulting or Advisory Role: Bayer, Eisai, Sanofi, Exelixis, Novartis, Ipsen, Pfizer, Merck Serono, Advanced Accelerator Applications
Speakers’ Bureau: Bayer, Eisai, Sanofi, Novartis, Ipsen, Pfizer, Merck Serono
Research Funding: Eisai (Inst), AstraZeneca (Inst), Advanced Accelerator Applications (Inst), Novartis (Inst), Ipsen (Inst), Bayer (Inst), Pfizer (Inst)
Travel, Accommodations, Expenses: Pfizer, Ipsen, Eisai
Lori J. Wirth
Consulting or Advisory Role: Merck, Loxo, Blueprint Medicines, Eisai, Ayala Pharmaceuticals, CUE Biopharma, Genentech, Rakuten Medical, Lilly, Bayer, Exelexis
Thomas Ernst
Consulting or Advisory Role: Novartis, Bristol-Myers Squibb, Pfizer, Incyte
Santiago Ponce Aix
Consulting or Advisory Role: Roche, Bristol-Myers Squibb, Merck
Speakers’ Bureau: Bristol-Myers Squibb, Merck, Roche
Travel, Accommodations, Expenses: AstraZeneca, Roche, Merck
Chia-Chi Lin
Honoraria: Novartis, Roche, Daiichi Sankyo
Consulting or Advisory Role: Novartis, Boehringer Ingelheim, Blueprint Medicines
Travel, Accommodations, Expenses: Lilly, Daiichi Sankyo, BeiGene, Novartis, Daiichi Sankyo
Rodryg Ramlau
Consulting or Advisory Role: BMS, Boehringer Ingelheim, MSD Oncology, Merck, Roche, Novartis, Takeda, AstraZeneca, Pfizer
Travel, Accommodations, Expenses: BMS, Boehringer Ingelheim, Roche, AstraZeneca
Marcus O. Butler
Honoraria: Roche, Merck, Bristol-Myers Squibb, Novartis
Consulting or Advisory Role: Merck, Bristol-Myers Squibb, Novartis, Immunovaccine, Immunocore, Adaptimmune, EMD Serono, GlaxoSmithKline, Genzyme, Sanofi Canada, LaRoche Possey
Research Funding: Merck, Takara Bio
Expert Testimony: Merck
Jean-Pierre Delord Consulting or Advisory Role: Novartis, Roche/Genentech, Bristol-Myers Squibb, MSD Oncology
Research Funding: Genentech (Inst), Bristol-Myers Squibb (Inst), MSD Oncology (Inst)
Hans Gelderblom
Patents, Royalties, Other Intellectual Property: Amgen, Boehringer-Ingelheim, Pfizer, Novartis, Pharmamar, Daiichi, Five Prime (Inst)
Paolo A. Ascierto
Stock and Other Ownership Interests: PrimeVax
Consulting or Advisory Role: Bristol-Myers Squibb, Roche/Genentech, Merck Sharp & Dohme, Novartis, Amgen, Array BioPharma, Merck Serono, Pierre Fabre, Newlink Genetics, Genmab, Incyte, MedImmune, AstraZeneca, Syndax, Sun Pharma, Sanofi, Idera, Ultimovacs, Sandoz, Immunocore, 4SC, Alkermes, Italfarmaco
Research Funding: Bristol-Myers Squibb (Inst), Roche/Genentech (Inst), Array BioPharma (Inst)
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Patrick M. Forde
Consulting or Advisory Role: AstraZeneca/MedImmune, Bristol-Myers Squibb, Janssen, AbbVie
Research Funding: Bristol-Myers Squibb (Inst), AstraZeneca/MedImmune (Inst), Kyowa Hakko Kirin (Inst), Novartis (Inst), Corvus Pharmaceuticals
Anna Wrona
Speakers’ Bureau: BMS, Roche, Pfizer
Travel, Accommodations, Expenses: BMS, Roche, Pfizer
Armando Santoro
Consulting or Advisory Role: Bristol-Myers Squibb, SERVIER, Gilead Sciences, Pfizer, Eisai, Bayer, MSD, Sanofi, ARQULE
Speakers’ Bureau: Takeda, Roche, AbbVie, Amgen, Celgene, AstraZeneca, ArQule, Eli Lilly, Sandoz, Novartis, BMS, Servier, Gilead, Pfizer, Eisai, Bayer, MSD
Peter M. Sadow
Consulting or Advisory Role: Novartis, Interpace Diagnostics, Veracyte
Sebastian Szpakowski
Employment: Novartis, Compass Therapeutics (I)
Stock and Other Ownership Interests: Novartis, Compass Therapeutics (I), Sarepta Therapeutics (I), Alexion Pharmaceuticals (I)
Travel, Accommodations, Expenses: Novartis, Compass Therapeutics (I)
Hongqian Wu
Employment: Novartis Pharmaceuticals
Stock and Other Ownership Interests: Biohaven Pharmaceuticals (I), Nektar, One Medical (I), Amarin
Travel, Accommodations, Expenses: Novartis Pharmaceuticals
Geraldine Bostel
Employment: Novartis Pharmaceuticals, Novartis Pharmaceuticals (I)
Stock and Other Ownership Interests: Novartis Pharmaceuticals, Novartis Pharmaceuticals (I)
Jason Faris
Employment: Novartis
Stock and Other Ownership Interests: Novartis
Scott Cameron
Employment: Novartis
Leadership: Novartis
Stock and Other Ownership Interests: Novartis
Patents, Royalties, Other Intellectual Property: Patents related to drugs being developed by Novartis
Travel, Accommodations, Expenses: Novartis
Andreea Varga
Research Funding: AstraZeneca (Inst), Novartis (Inst), Pfizer (Inst), Roche (Inst), Sanofi (Inst), Bristol-Myers Squibb (Inst), Boehringer Ingelheim (Inst), Merck (Inst), Janssen-Cilag (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Roche, Boehringer Ingelheim
Matthew Taylor
Consulting or Advisory Role: Bristol-Myers Squibb, Eisai, Array BioPharma, Loxo, Bayer, ArQule, Blueprint Medicines, Novartis, Sanofi/Genzyme
Speakers’ Bureau: Bristol-Myers Squibb, Eisai, Merck
No other potential conflicts of interest were reported.
ASSOCIATED CONTENT
PRIOR PRESENTATION
Presented in part at the American Society of Clinical Oncology Annual Meeting, Chicago, IL, June 1–5, 2018.
CLINICAL TRIAL INFORMATION
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST AND DATA AVAILABILITY STATEMENT
Disclosures provided by the authors and data availability statement (if applicable) are available with this article at DOI https://doi.org/10.1200/JCO.19.02727.
REFERENCES
- 1.Haddad RI, Nasr C, Bischoff L, et al. : NCCN guidelines insights: Thyroid carcinoma, version 2.2018. J Natl Compr Canc Netw 16:1429–1440, 2018 [DOI] [PubMed] [Google Scholar]
- 2.Smallridge RC, Ain KB, Asa SL, et al. : American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid 22: 1104–1139, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Smallridge RC, Copland JA: Anaplastic thyroid carcinoma: Pathogenesis and emerging therapies. Clin Oncol (R Coll Radiol) 22:486–497, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiedje V, Stuschke M, Weber F, et al. : Anaplastic thyroid carcinoma: Review of treatment protocols. Endocr Relat Cancer 25:R153–R161, 2018 [DOI] [PubMed] [Google Scholar]
- 5.Landa I, Ibrahimpasic T, Boucai L, et al. : Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 126: 1052–1066, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pozdeyev N, Gay LM, Sokol ES, et al. : Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 24:3059–3068, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunstman JW, Juhlin CC, Goh G, et al. : Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet 24:2318–2329, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subbiah V, Kreitman RJ, Wainberg ZA, et al. : Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol 36:7–13, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robert C, Schachter J, Long GV, et al. : Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372:2521–2532, 2015 [DOI] [PubMed] [Google Scholar]
- 10.Borghaei H, Paz-Ares L, Horn L, et al. : Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 373:1627–1639, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khunger M, Hernandez AV, Pasupuleti V, et al. : Programmed cell death 1 (PD-1) ligand (PD-L1) expression in solid tumors as a predictive biomarker of benefit from PD-1/PD-L1 axis inhibitors: A systematic review and meta-analysis. JCO Precis Oncol doi: 10.1200/PO.16.00030 [DOI] [PubMed] [Google Scholar]
- 12.Ahn S, Kim TH, Kim SW, et al. : Comprehensive screening for PD-L1 expression in thyroid cancer. Endocr Relat Cancer 24:97–106, 2017 [DOI] [PubMed] [Google Scholar]
- 13.Zwaenepoel K, Jacobs J, De Meulenaere A, et al. : CD70 and PD-L1 in anaplastic thyroid cancer - promising targets for immunotherapy. Histopathology 71: 357–365, 2017 [DOI] [PubMed] [Google Scholar]
- 14.Kollipara R, Schneider B, Radovich M, et al. : Exceptional response with immunotherapy in a patient with anaplastic thyroid cancer. Oncologist 22:1149–1151, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyer PC, Dadu R, Gule-Monroe M, et al. : Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma. J Immunother Cancer 6:68, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naing A, Gainor JF, Gelderblom H, et al. : A first-in-human phase 1 dose escalation study of spartalizumab (PDR001), an anti-PD-1 antibody, in patients with advanced solid tumors. J Immunother Cancer 8:e000530, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023–1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perucho M: Cancer of the microsatellite mutator phenotype. Biol Chem 377:675–684, 1996 [PubMed] [Google Scholar]
- 19.Khoja L, Butler MO, Kang SP, et al. : Pembrolizumab. J Immunother Cancer 3:36, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brahmer JR, Hammers H, Lipson EJ: Nivolumab: Targeting PD-1 to bolster antitumor immunity. Future Oncol 11:1307–1326, 2015 [DOI] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415–421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turajlic S, Litchfield K, Xu H, et al. : Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol 18:1009–1021, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Maleki Vareki S: High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 6:157, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufman HL, Russell JS, Hamid O, et al. : Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer 6:7, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.