

Abstract
PURPOSE
Cell-free DNA (cfDNA) and circulating tumor cell (CTC) based liquid biopsies have emerged as potential tools to predict responses to androgen receptor (AR)-directed therapy in metastatic prostate cancer. However, due to complex mechanisms and incomplete understanding of genomic events involved in metastatic prostate cancer resistance, current assays (e.g. CTC AR-V7) demonstrate low sensitivity and remain underutilized. The recent discovery of AR enhancer amplification in >80% of metastatic patients and its association with disease resistance presents an opportunity to improve upon current assays. We hypothesized that tracking AR/enhancer genomic alterations in plasma cfDNA would detect resistance with high sensitivity and specificity.
METHODS
We developed a targeted sequencing and analysis method as part of a new assay called Enhancer and neighboring loci of Androgen Receptor Sequencing (EnhanceAR-Seq). We applied EnhanceAR-Seq to plasma collected from 40 patients with metastatic prostate cancer treated with AR-directed therapy to monitor AR/enhancer genomic alterations and correlate these events with therapy resistance, progression-free survival (PFS) and overall survival (OS).
RESULTS
EnhanceAR-Seq identified genomic alterations in the AR/enhancer locus in 45% of cases, including a 40% rate of AR enhancer amplification. Patients with AR/enhancer alterations had significantly worse PFS and OS than those without (6-month PFS: 30% vs. 71%, P=0.0002; 6-month OS: 59% vs. 100%, P=0.0015). AR/enhancer alterations in plasma cfDNA detected 18 of 23 resistant cases (78%) and outperformed the CTC AR-V7 assay which was also run on a subset of patients.
CONCLUSION
cfDNA-based AR locus alterations, including of the enhancer, are strongly associated with resistance to AR-directed therapy and significantly worse survival. cfDNA analysis using EnhanceAR-Seq may enable more precise risk stratification and personalized therapeutic approaches for metastatic prostate cancer.
INTRODUCTION
Metastatic castration resistant prostate cancer (mCRPC) is the most aggressive form of prostate cancer1. Outcomes have improved significantly with the advent of androgen receptor (AR)-directed therapies such as abiraterone and enzalutamide2–4. Still, approximately 20-40% of patients exhibit primary resistance to these treatments and have significantly worse survival (median survival less than 6 months)5,6. Other patients develop secondary resistance to AR-directed therapy, responding well initially before eventually developing resistance7. There is thus an urgent need for molecular biomarkers that can detect resistance to AR-directed therapy early, especially primary resistance, which would enable clinicians to consider alternative treatments (i.e. chemotherapy, immunotherapy or systemic radiotherapy) and potentially improve patient survival.
The clinically validated circulating tumor cell (CTC) assay for detecting an aberrant AR splice variant (AR-V7), a predictive biomarker of resistance to AR-directed therapy, highlights the potential value of liquid biopsy analysis in mCRPC patients5,6,8. However, the reported sensitivity of this test for detecting AR-resistant mCRPC remains low at only ~30%6,8. Thus although indicated for clinical use, there is a need for more sensitive assays to detect resistance to AR-directed therapy.
Assessment of cell-free DNA (cfDNA) has recently emerged as a non-invasive means to assess relevant genomic alterations in multiple cancer types including prostate cancer9–16. cfDNA assessment of circulating tumor DNA has been shown to be sensitive for identifying tumor-specific somatic mutations with capability to even detect molecular residual disease10,11,13,16,17. In mCRPC, detection sensitivities have been shown to be high prior to treatment initiation and genomic alterations, including those that target the AR gene body, can be reliably measured9,12,15. Still, it remains to be seen if measuring these genomic alterations can reliably identify resistance to AR-directed therapy.
While AR is the key player in mCRPC treatment resistance, our understanding of the genomic alterations affecting AR is incomplete. To address this, recent large whole genome sequencing studies discovered a long-range non-coding enhancer upstream of AR that promotes AR expression and resistance to AR-directed therapies18–20. Indeed, the AR enhancer was found to be amplified in 81-87% of patients, the most frequent genomic alteration in mCRPC (11-17% more than AR gene body amplification)18,20. While studies have shown detection of AR gene body alterations in plasma cfDNA of mCRPC patients9,12,15, none of these tracked the AR enhancer. Here we present a liquid biopsy cfDNA technique to monitor genomic alterations that includes the AR enhancer called Enhancer and neighboring loci of Androgen Receptor Sequencing (EnhanceAR-Seq), and demonstrate the ability to detect resistance to AR-directed therapy with high sensitivity and specificity.
PATIENTS AND METHODS
Patient Enrollment
We prospectively enrolled 40 patients with metastatic prostate cancer treated with at least one month of standard-of-care AR-directed treatment (e.g. abiraterone or enzalutamide). All patients were maintained on standard androgen deprivation therapy (i.e. LHRH-R agonist or antagonist). Prior treatment with other systemic agents including chemotherapy was allowed. Patients with evidence of any active non-prostate malignancy other than localized skin cancer were excluded from the study. Eligible patients underwent blood collection for cfDNA analysis at the time of enrollment. All patients underwent continued clinical and laboratory follow-up as per the standard-of-care. Additionally, healthy adult blood donors (n=36) were recruited from the Washington University School of Medicine and the American Red Cross Blood Center in St. Louis, Missouri. All samples were collected with informed consent and institutional review board approval in accordance with the Declaration of Helsinki.
Sequencing and Analysis of Plasma cfDNA
We developed EnhanceAR-Seq as a targeted sequencing assay of plasma cfDNA to monitor genomic alterations in the AR gene and AR enhancer loci and other frequently altered genes9,18 in metastatic prostate cancer (Appendix Table A1). We performed EnhanceAR-Seq on plasma from all patients acquired at the time of enrollment and analyzed genomic alterations with respect to matched plasma-depleted whole blood and unmatched healthy donor samples (Fig 1; Appendix Tables A2–A8). In four patients, we also performed EnhanceAR-Seq on serial timepoints including at least one timepoint during AR-directed treatment.
Fig 1. Patient enrollment and sample collection.
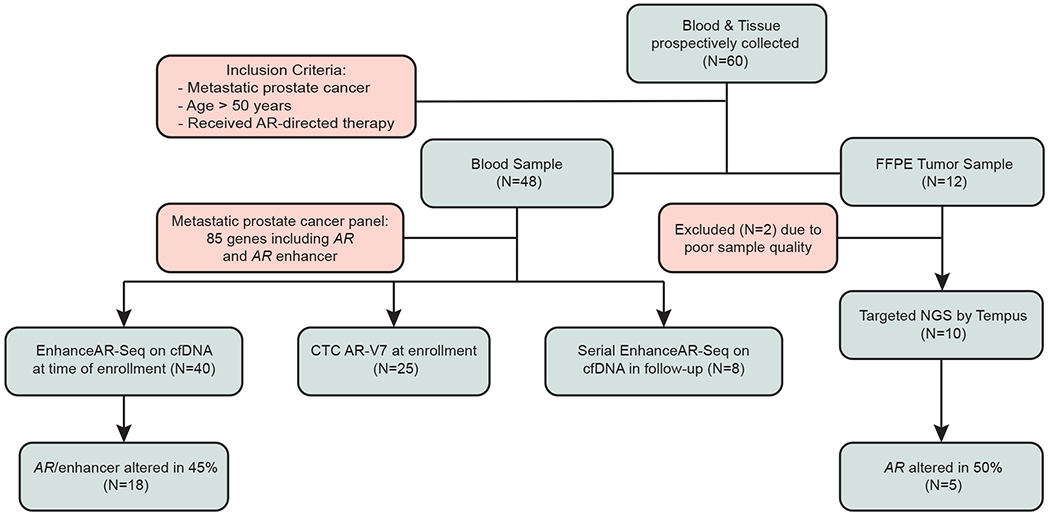
Patients with biopsy-proven metastatic prostate cancer treated with AR-directed therapy were enrolled onto the study and samples collected for tissue, cell-free DNA and CTC analyses. AR, androgen receptor; CTC, circulating tumor cell; EnhanceAR-Seq, Enhancer and neighboring loci of Androgen Receptor Sequencing; cfDNA, cell-free DNA; FFPE, formalin-fixed paraffin-embedded; NGS, next-generation sequencing.
Clinical Outcomes and Statistical Analysis
Resistance to AR-directed therapy was scored by a board-certified academic medical oncologist specializing in genitourinary cancers. Primary resistance was defined as PSA progression, change of therapy or death within 4 months of treatment initiation, or radiographic progression within 6 months. Secondary resistance was defined as PSA progression, change of therapy, radiographic progression or death outside of this timeframe. Associations between assay results and resistance to AR-directed therapy were assessed by Fisher’s Exact test. A progression-free survival (PFS) event was defined as the time to PSA progression by PCWG321 criteria or death, and an overall survival (OS) event was defined as the time to death. The Kaplan-Meier method and multivariate Cox proportional hazards models were used to analyze survival outcomes.
Additional methodological details are provided in the Appendix Methods.
RESULTS
Patient Characteristics
We prospectively enrolled 40 patients with metastatic prostate cancer treated with AR-directed therapy between November 2018 and November 2019 (Appendix Tables A2–A3). The median age was 69 years, ECOG performance status ranged between 0 and 2, and median follow-up time on study was 6.0 months. Among these patients, 11 were on their first line of systemic therapy, and the remaining 29 were on their second or greater line of systemic therapy for metastatic prostate cancer at the time of study enrollment.
EnhanceAR-Seq Detects Somatic Alterations in Plasma cfDNA
The most frequent genomic events detected in plasma cfDNA from our cohort were AR/enhancer alterations (most commonly copy number gain and tandem duplication) present in 18 patients (45%), including a 40% amplification rate in the AR enhancer region (Fig 2A; Appendix Tables A8–A9). Three patients (8%) were found to have independent AR enhancer amplification without AR gene body amplification, consistent with previous tissue-based results18,20 (Fig 2A; Appendix Fig A1). Other genes frequently found in cfDNA to be targeted by alterations included TP53 and PTEN which demonstrated copy number loss in 6 patients (15%) and COSMIC22-annotated nonsynonymous single nucleotide variants in 5 cases (13%) (Fig 2A; Appendix Tables A8 and A10). We also detected TMPRSS2-ERG gene fusion in 5 cases (13%) (Fig 2A; Appendix Table A9).
Fig 2. Genomic alterations in plasma cell-free DNA in metastatic prostate cancer including the AR/enhancer locus.
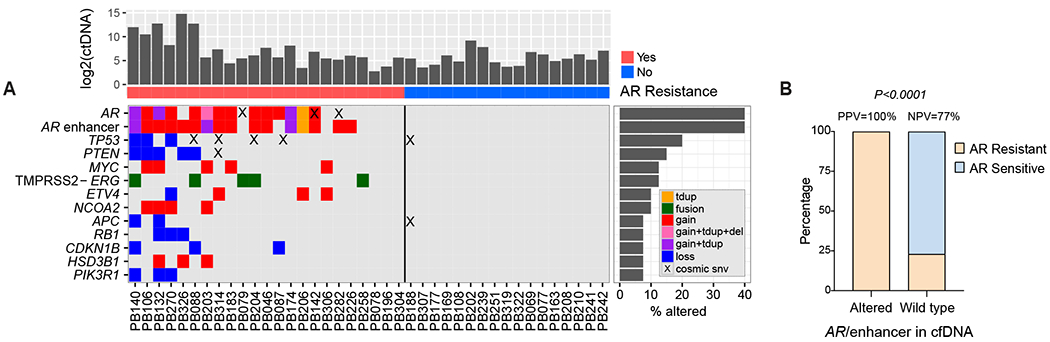
(A) Co-mutation plot based on cell-free DNA analysis of patients with metastatic prostate cancer treated with AR-directed therapy. Each column represents data from a single patient. Rates of queried genomic alterations are depicted by the bar graphs to the right. Only genes with >5% alteration rate (considering tandem duplications, fusions, deletions, copy number changes and COSMIC-indexed SNVs) are displayed. ctDNA levels are represented in the bar graph on top in log2 space. Resistance to AR-directed therapy is indicated below the bar graph as red (resistant) vs. blue (sensitive). (B) Proportion of patients with AR/enhancer genomically altered (N=18) or wild type (N=22) in cell-free DNA, who developed resistance (N=23) or not (N=17) to AR-directed therapy. P value was calculated by Fisher’s exact test. AR, androgen receptor; tdup, tandem duplication; del, deletion; SNV, single nucleotide variation; COSMIC, catalogue of somatic mutations in cancer; PPV, positive predictive value; NPV, negative predictive value.
Ten patients consented to additional tissue-based analyses using metastatic biopsy samples. These samples were analyzed by targeted NGS using the Tempus sequencing platform, which includes the AR gene body but not the enhancer23,24. Five patients had evidence of AR gene body alteration in tumor with four of those having the same genomic changes evident in plasma. Overall, genomic alterations in AR were 80% concordant between tissue and plasma (Appendix Fig A2; Appendix Table A11), consistent with work published by others25.
AR/Enhancer Alterations in cfDNA are Associated with Clinical Resistance
We observed the greatest concordance between genomic events and clinical resistance to AR-directed therapy for alterations in the AR locus including the enhancer (Fig 2). Alterations in the AR/enhancer locus predicted resistance with 78% sensitivity and 100% specificity (Fig 2B). There was a highly significant correlation between alterations detected in AR/enhancer in cfDNA and resistance to AR-directed therapy (P<0.0001). Interestingly, all three cases with AR enhancer amplification in cfDNA in the absence of AR gene body amplification progressed to resistance at a median of 5.3 months (range 0.6-8.0), indicative of improved sensitivity in identifying resistance when tracking the AR enhancer in addition to the gene body. The AR-V7 Nucleus Detect CTC assay was run at a median of 16 days from cfDNA analysis in 25 patients, including within 24 hours of cfDNA testing for 10 patients. AR-V7 was detected in CTCs from 2 patients (8%) and negative in the remaining 23 (Appendix Fig A3; Appendix Table A3).
AR/Enhancer Alterations in cfDNA Portend Poor Progression-Free Survival
PFS was significantly shorter among men with detected AR/enhancer alterations in plasma cfDNA (18 patients, 45%) compared to those without (22 patients, 55%) (HR 6.8; 95% CI, 2.5-18.6; P=0.0002) (Fig 3A). PFS remained significantly shorter with similar hazard ratio when restricting our analysis to just the AR enhancer region (HR 8.1; 95% CI 2.8-23.6; P=0.0001) (Appendix Fig A4A). cfDNA-detected alterations in the AR/enhancer locus or the AR enhancer alone remained highly significant by multivariate Cox proportional hazards regression, which included important baseline characteristics such as PSA concentration, circulating tumor DNA (ctDNA) level, number of lines of therapy received in the metastatic setting, prior enzalutamide vs. abiraterone treatment, metastatic disease burden and time since diagnosis (Appendix Tables A12–15). We also found that overall ctDNA levels and mutational burden did not correlate with clinical outcomes, nor were they significantly different between patients who developed AR-resistance vs. remained AR-sensitive (Appendix Fig A5; Appendix Table A16).
Fig 3. Progression-free and overall survival according to AR/enhancer alteration status in cell-free DNA.
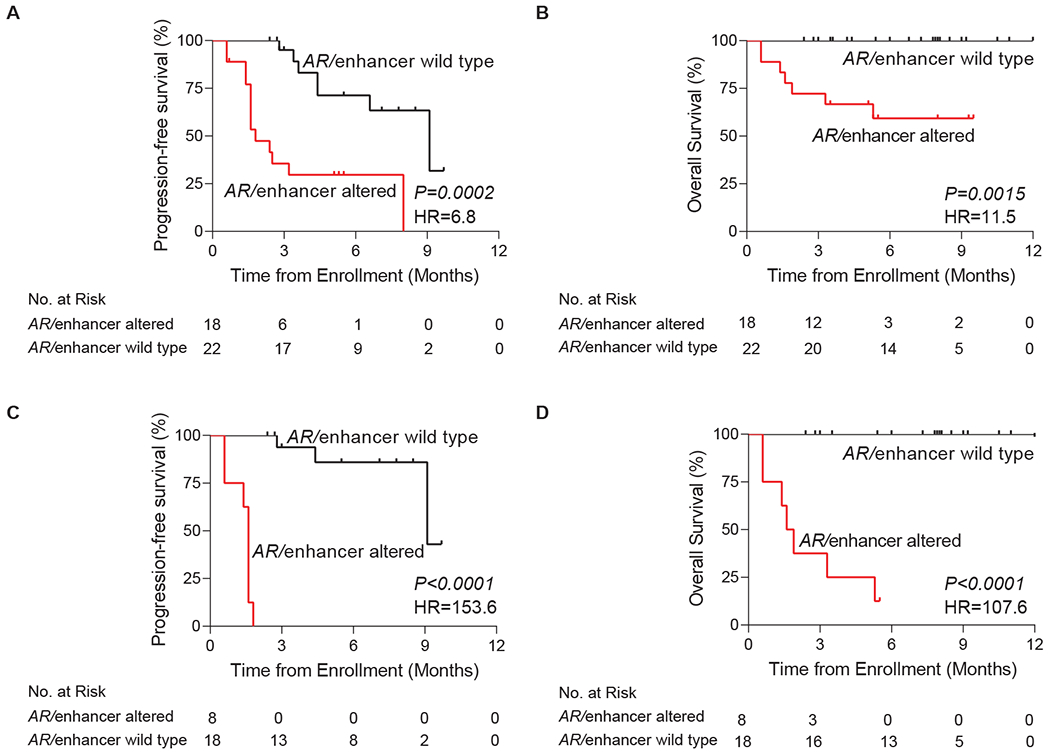
Panels A (PFS) and B (OS) represent the full 40 patient cohort while panels C (PFS) and D (OS) exclude patients with secondary resistance to AR-directed therapy. Kaplan-Meier analyses were performed from the time of sample collection (time of enrollment), stratified based on the genomic alteration status of AR/enhancer measured in cell-free DNA. P values were calculated by the log-rank test and hazard ratios by the Mantel-Haenszel method. PFS, progression-free survival; OS, overall survival; AR, androgen receptor; HR, hazard ratio.
AR/Enhancer Alterations in cfDNA Portend Poor Overall Survival
Although median follow-up of our cohort from time of enrollment was only 6.0 months, we performed a preliminary OS analysis. OS was significantly shorter among men with detected AR/enhancer alterations in plasma cfDNA compared to those without (HR 11.5; 95%CI 2.5-52.1; P=0.0015) (Fig 3B). OS remained significantly shorter with a high hazard ratio when ignoring AR gene body alterations and restricting our analysis to just the AR enhancer region (HR 16.4; 95% CI 3.5-77.2; P=0.0004) (Appendix Fig A4B).
AR/Enhancer Alterations in cfDNA in Primary versus Secondary Resistance
Our cohort included nine primary resistant and 14 secondary resistant cases. In all cases of primary resistance, patients experienced no response, while in cases of secondary resistance, patients experienced a temporary treatment response before ultimately progressing on AR-directed therapy. Notably, the previously published AR-V7 assay has only been shown to be capable of identifying primary resistance, albeit with limited sensitivity5,6. We thus decided to test EnhanceAR-Seq more exclusively in this space. Positive predictive value of cfDNA-derived AR/enhancer alterations for primary resistance was 100%, with every positive case progressing within 3 months and all but one dying within 6 months of study enrollment (Figs 3C and 3D). The sensitivity of our assay for detecting primary resistance was 89%, higher than the 71% we observed for secondary resistance, while specificity remained 100%.
We obtained serial samples in four patients with at least one timepoint being during AR-directed therapy (Fig 4). For patient PB078 (Fig 4A), EnhanceAR-Seq detected no evidence of AR/enhancer alterations at enrollment, and AR-V7 detection in CTCs was also negative. At 19 and ~45 weeks later, EnhanceAR-Seq revealed significantly elevated copy number amplification of both the AR gene body and enhancer, while the patient was actively developing resistance to enzalutamide followed by abiraterone. The CTC AR-V7 assay also became positive at ~45-weeks. Patients PB087 and PB203 similarly showed rapid increases in AR/enhancer copy number on enzalutamide and abiraterone, respectively, while AR-V7 testing remained negative (Figs 4B and 4C). Cell-free AR/enhancer amplification preceded rise in PSA and clinician-recognized resistance leading to therapy change. For patient PB140 (Fig 4D), AR/enhancer copy numbers increased more subtly on serial analysis, however in this case the baseline copy numbers for AR and its enhancer were already >8-fold elevated; reflective of this, the patient progressed rapidly 6 weeks after study enrollment and died from mCRPC at 22 weeks. These vignettes demonstrate the potential value of using cell-free DNA based AR/enhancer analysis as a precision modality to monitor treatment resistance in metastatic prostate cancer patients undergoing AR-directed therapy.
Fig 4. Serial timepoint liquid biopsy analyses of patients on AR-directed treatment.
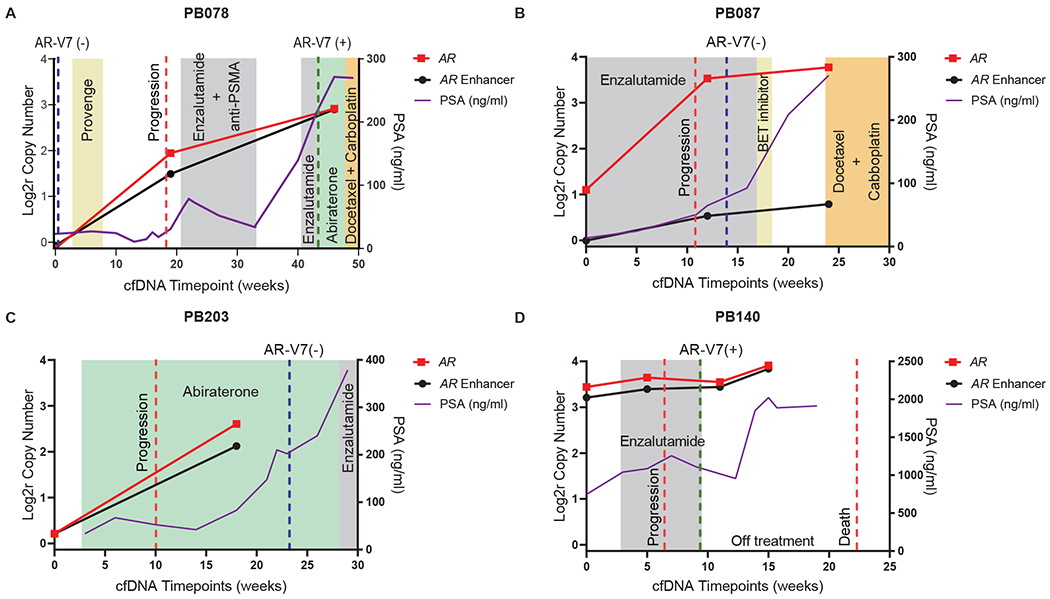
(A) The patient was negative for CTC AR-V7 and cell-free DNA AR/enhancer alteration at the time of enrollment. At week 19, shortly before receiving enzalutamide and anti-PSMA, he tested positive for amplifications in AR and its enhancer in cfDNA. The patient initially responded, then after a treatment break rapidly progressed on both enzalutamide and abiraterone. Repeat testing at this final timepoint (~45 weeks post-enrollment) was positive with further amplification observed in AR and its enhancer in cfDNA and AR-V7 detected in CTCs. (B-D) Clinical vignettes of three more mCRPC patients with serial cfDNA collected over time with at least one timepoint occurring during AR-directed therapy. AR and AR enhancer copy number ratios in cfDNA are shown over time in log2 space, and PSA concentrations in blood are shown in ng/mL. Treatments are indicated in colored boxes, time of progression or death as dashed red lines, and AR-V7 test results as dashed green lines (if positive) or blue lines (if negative). Weeks since study enrollment are shown on the X-axis. cfDNA, cell-free DNA; CTC, circulating tumor cell; AR, androgen receptor; Log2r, logarithm base 2 ratio; PSMA, prostate-specific membrane antigen; PSA, prostate-specific antigen; BET, bromodomain and extraterminal domain; mCRPC, metastatic castration resistant prostate cancer.
DISCUSSION
In this study, we developed and tested a cfDNA analysis method for assessing treatment resistance in metastatic prostate cancer, which we call EnhanceAR-Seq. Our results indicate that cfDNA analysis is a promising approach for detecting resistance to AR-directed therapy, with 100% positive predictive value and 78% sensitivity. Sensitivity increased to 89% when considering only primary resistant cases. EnhanceAR-Seq outperformed the CTC AR-V7 test utilized clinically which was performed for a subset of patients in our study. In available cases, we also performed tumor sequencing and observed 80% concordance between AR genomic alterations in tumor and plasma.
We also factored in baseline ctDNA level in multivariate Cox regression analyses to determine if it might be a confounding variable, which we found did not correlate with clinical outcomes and was not significantly different between AR-resistant and AR-sensitive patients. While other baseline differences between patients could have influenced our study’s outcomes, we accounted for them through four separate multivariate Cox regression analyses (Appendix Tables A12–A15) where we found that only AR/enhancer alterations including in the AR enhancer alone were highly significantly associated with resistance to AR-directed therapy (HR>10, P<0.005).
Within our cohort, nearly every patient with detectable alterations in AR or its enhancer in cell-free DNA developed resistance and progressed despite a relatively short follow-up period. AR/enhancer alterations were associated with significantly worse PFS and OS. In contrast, the Genomic Health CTC AR-V7 assay was positive in only 8% of tested cases and did not correlate significantly with outcomes. It is important to note, however, that larger studies have shown correlations of CTC AR-V7 detection with outcomes5,6,8, which may not have been evident here due to small cohort size, heterogeneous nature of our cohort, and CTC AR-V7 testing being performed in only 63% of our cohort. Still, the 8% positivity rate for the Genomic Health CTC AR-V7 assay in our cohort is not much different than the 10% positivity rate of this assay in high-risk mCRPC patients in the recently published PROPHECY trial26, suggesting our results may be in line with other prospective data.
Five cases of resistance to AR-directed treatment were not detected using our cfDNA assay. However, four of these represent secondary resistance to AR-directed therapy, where patients initially responded to treatment before eventually developing resistance. In this regard, we performed a serial timepoint analysis in a patient (PB078) where both EnhanceAR-Seq and CTC AR-V7 were negative at the initial responsive timepoint, but both assays became positive as the patient evolved resistance to enzalutamide followed by abiraterone. Serial timepoint analysis of two other patients (PB087 and PB203), including one who received abiraterone followed by enzalutamide, also demonstrated dramatically increasing AR/enhancer levels over time which anticipated clinical progression and rising PSA while on AR-directed treatment. In contrast, a fourth case (PB140) of primary resistance demonstrated high >8-fold amplification of AR and its enhancer at baseline which remained highly elevated on serial analysis. This correlated with rapid early progression on enzalutamide and death from mCRPC at 22 weeks. These data support the potential value of serial timepoint analysis, especially in the secondary resistance setting where AR/enhancer amplification may not be apparent at baseline. These clinical vignettes also suggest that our assay could potentially inform clinicians when to trial a different AR-directed treatment (when AR/enhancer copy numbers remain low) or switch to a different therapy-type altogether (when AR/enhancer copy numbers have risen high).
Resistant patients identified by AR/enhancer alterations may be completely distinct from those with AR-V7 messenger RNA splice variation27. Given assessment of different mechanisms of resistance, one at the DNA level (detected by EnhanceAR-Seq) and the other at the mRNA/protein level26 (detected by CTC-based assays), it may be valuable to run both methods to more comprehensively assess multiple mechanisms of resistance in certain cases. In our cohort, CTC AR-V7 results did not improve upon the sensitivity achieved with EnhanceAR-Seq, however we note that AR-V7 testing was done in only a subset of our patients.
To our knowledge, our assay is the first to monitor the AR enhancer in the cell-free compartment. In addition to showing that AR enhancer amplification can be detected in plasma cfDNA from patients with metastatic prostate cancer, we observed that 13% of resistant patients had AR enhancer amplification detectable in plasma cfDNA independent of gene body amplification. Although our cohort is small, the prevalence of independent AR enhancer amplification is consistent with prior studies18,20. Highlighting its clinical importance, AR enhancer amplification stratified patients by both resistance to AR-directed therapy and survival outcomes. All patients with independent AR enhancer amplification progressed to treatment resistance at a median of 5.3 months, highlighting the importance of monitoring the AR enhancer in addition to the gene body.
In addition to genomic alterations in AR and its enhancer, we assessed 84 other genes shown to be important in mCRPC9,18. In several cases, we observed multiple alterations involving different genes including TP53 and PTEN, consistent with prior work18. We also targeted a 13kb fusion hotspot in the TMPRSS2 intronic region, based on analysis of previously published WGS data in mCRPC18. This enabled us to identify a subset of TMPRSS2-ERG fusion events in our cohort. To monitor TMPRSS2-ERG fusions more comprehensively, we would have needed to target full lengths of TMPRSS2 and ERG gene bodies and introns, which would have required a much larger targeted space and limited our sequencing depth-of-coverage.
Limitations of our study include a short follow-up period, reducing our ability to assess long-term clinical outcomes such as PFS and OS. Despite this, hazard ratios for survival outcomes were high on Kaplan-Meier analysis. It is possible that with longer follow-up time, we would observe even greater predictive and prognostic value of measuring AR/enhancer alterations in cfDNA. Additionally, patients were enrolled while on different lines of therapy, leading to cohort heterogeneity, similar to clinical studies involving the CTC AR-V7 assay5,6,8. CTC AR-V7 testing was performed on only a subset of patients, which could have biased our ability to compare it to cfDNA analysis.
In conclusion, we developed a novel cfDNA assay, EnhanceAR-Seq, to detect genomic alterations in the AR locus including the enhancer. Our method effectively detected resistance to AR-directed therapy, and stratified patients based on progression-free and overall survival despite short follow-up time. Assay performance improved further when considering only primary resistant disease. Our results remained highly significant when accounting for baseline characteristics such as PSA concentration, ctDNA level and metastatic disease burden. Serial timepoint analysis in four patients demonstrated the potential value of using our assay to monitor for AR-resistance during treatment. While our cohort was relatively small, EnhanceAR-Seq applied to a single timepoint predicted resistance to AR-directed therapy with high sensitivity and specificity. Our results suggest that cfDNA analysis through EnhanceAR-Seq can help improve risk stratification and clinical decision-making for metastatic prostate cancer. Future clinical trials should be performed to validate our findings prior to clinical implementation.
Supplementary Material
CONTEXT.
KEY OBJECTIVE
Can we predict resistance to androgen receptor (AR)-directed therapy in metastatic prostate cancer patients by tracking genomic alterations in the AR enhancer in addition to the AR gene body (AR/enhancer) in plasma cell-free DNA (cfDNA)?
KNOWLEDGE GENERATED
We developed EnhanceAR-Seq to monitor AR/enhancer alterations via liquid biopsy and detected AR enhancer amplification in cfDNA of 40% of metastatic prostate cancer patients including 8% without AR gene body amplification. Patients with cfDNA-detected alterations in the AR enhancer or gene body ubiquitously exhibited resistance to AR-directed therapy and had significantly worse survival.
RELEVANCE
AR/enhancer alterations are the most frequent somatic event in metastatic prostate cancer, which we show are detectable in plasma cfDNA and predictive of resistance to AR-directed therapy and poor survival. Therefore, cfDNA liquid biopsy analysis of the AR/enhancer locus has the potential to improve risk stratification and help guide clinical decision-making for metastatic prostate cancer.
ACKNOWLEDGEMENTS
We are grateful to the patients and their families for study participation. We also thank Timothy Ley, MD for providing critical feedback on the manuscript.
SUPPORT
Supported by the Alvin J. Siteman Cancer Research Fund at Washington University in St Louis (A.A.C., C.A.M., R.K.P.), the National Cancer Institute of the National Institutes of Health under award number K08CA238711 (A.A.C.), the Cancer Research Foundation Young Investigator Award (A.A.C.), the American Cancer Society Institutional Research Grant under award number IRG-18-158-61 (H.X.D.), the Prostate Cancer Foundation Young Investigator Award (R.K.P.), the Sidney Kimmel Scholar Award (R.K.P.), and the Galen Hoskin and Dina Wolkoff Giving Fund (R.K.P.).
REFERENCES
- 1.Lowrance WT, Murad MH, Oh WK, et al. Castration-Resistant Prostate Cancer: AUA Guideline Amendment 2018. J Urol 2018;200:1264–1272. [DOI] [PubMed] [Google Scholar]
- 2.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–97. [DOI] [PubMed] [Google Scholar]
- 5.Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scher HI, Lu D, Schreiber NA, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol 2016;2:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandrasekar T, Yang JC, Gao AC, et al. Targeting molecular resistance in castration-resistant prostate cancer. BMC Med 2015;13:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scher HI, Graf RP, Schreiber NA, et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol 2018;4:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Annala M, Vandekerkhove G, Khalaf D, et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov 2018;8:444–457. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov 2017;7:1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin RI, Chen K, Usmani A, et al. Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Mol Diagn Ther 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conteduca V, Wetterskog D, Sharabiani MTA, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol 2017;28:1508–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corcoran RB, Chabner BA. Application of Cell-free DNA Analysis to Cancer Treatment. N Engl J Med 2018;379:1754–1765. [DOI] [PubMed] [Google Scholar]
- 14.Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandekerkhove G, Struss WJ, Annala M, et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur Urol 2019;75:667–675. [DOI] [PubMed] [Google Scholar]
- 16.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–238. [DOI] [PubMed] [Google Scholar]
- 17.Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quigley DA, Dang HX, Zhao SG, et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018;174:758–769 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeda DY, Spisak S, Seo JH, et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018;174:422–432 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viswanathan SR, Ha G, Hoff AM, et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018;174:433–447 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scher HI, Morris MJ, Stadler WM, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol 2016;34:1402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tate JG, Bamford S, Jubb HC, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019;47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beaubier N, Tell R, Huether R, et al. Clinical validation of the Tempus xO assay. Oncotarget 2018;9:25826–25832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beaubier N, Tell R, Lau D, et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 2019;10:2384–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wyatt AW, Annala M, Aggarwal R, et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armstrong AJ, Halabi S, Luo J, et al. Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J Clin Oncol 2019;37:1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho Y, Dehm SM. Androgen Receptor Rearrangement and Splicing Variants in Resistance to Endocrine Therapies in Prostate Cancer. Endocrinology 2017;158:1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.