

Abstract
Smallpox, a contagious and deadly disease caused by variola virus, was eradicated by a strategy that included vaccination with vaccinia virus, a live-virus vaccine. Because the threat of bioterrorism with smallpox persists; infections with zoonotic poxvirus infections like monkeypox continue; and there may be a time when an alternative vaccine platform is needed, recombinant-subunit vaccine strategies for poxviruses have been pursued. Our prior work focused on understanding the immune responses generated to vaccine-formulations containing the virus protein L1. In this work, we examine vaccine-formulations with additional key protein targets: A33 and B5 (components of the extracellular virus) and another protein on the mature virus (A27) adjuvanted with aluminum hydroxide (AH) with and without CpG- oligonucleotide. Each vaccine was formulated to allow either adsorption or non-adsorption of the protein (and CpG) to AH. Mice given a prime and single boost produced long-lasting antibody responses. A second boost (given ~5-months after the first) further increased antibody titers. Similar to our prior findings with L1 vaccine-formulations, the most protective A33 vaccine-formulations included CpG, resulted in the generation of IgG2a-antibody responses. Unlike the prior findings with L1 (where formulations that adsorbed both the protein and the CpG to AH resulted in 100% survival after challenge and minimal weight loss), the AH-adsorption status of A33 and CpG did not play as important a role, since both AH-adsorbed and non-adsorbed groups lost weight after challenge and had similar survival. Vaccination with B5-formulations gave different results. While CpG-containing formulations were the only ones that generated IgG2a-antibody responses, the vaccine-formulation that adsorbed B5 to AH (without CpG) was as equally effective in protecting mice after challenge. These results indicate that the mechanism of how antibodies against A33 and B5 protect differ. The data also show the complexity of designing optimized vaccine-formulations containing multiple adjuvants and recombinant protein-based antigens.
Keywords: smallpox, variola virus; monkeypox, vaccinia virus; adjuvants, aluminum hydroxide; CpG oligonucleotide; antibody responses; mice
Introduction
Smallpox was successfully eradicated in the last century using a live virus vaccine [1]. Once eradicated, routine vaccination with vaccinia virus (VACV) was no longer carried out and production of the vaccine ended. Because smallpox vaccination also protected against zoonotic poxviruses, the number of cases of monkeypox has increased significantly in Africa [2–4]. Due to concerns of bioterrorism with variola virus, there was a need to replenish vaccine stockpiles as a countermeasure [5]. This was a challenge because pharmaceutical companies had produced the historical smallpox vaccine in lymph and skin of inoculated animals. To produce new stocks of vaccine, a cell culture-based vaccine was developed [6, 7]. Since side effects and rare life-threatening complications were still seen with the cell culture-based vaccine [8], safer smallpox vaccines were pursued. The U.S. chose to move forward with the more attenuated live virus vaccine, modified vaccinia Ankara (MVA) [9, 10]. This virus is safer because it does not generate infectious progeny in human cells. However, to generate similar immune responses to the historical VACV vaccine, a dose 1000-times greater than the historical vaccine is needed. Also, two doses are required. While MVA was chosen as the next generation smallpox vaccine that could also combat monkeypox [10, 11], recombinant subunit vaccine strategies had also been pursued and may someday be used as a future generation smallpox vaccine strategy.
Since poxviruses generate two forms of infectious progeny virus, subunit vaccines that generate protective antibody responses typically target key viral proteins on the membrane of the mature virus (MV) (e.g., L1, A27) and proteins on the membrane of the extracellular virus (EV) (e.g., A33 and B5) [12–31]. It is believed that antibodies to these two forms of infectious virions are important for both the initial control of incoming virus and control of viral spread in the infected host.
Our previous biophysical study showed that binding of the L1 protein to the adjuvant aluminum hydroxide (along with the TLR-9 adjuvant, CpG), was critical in generating a protective antibody response [29]. Here we studied the immunogenicity of the other key targets of a poxvirus recombinant subunit vaccine (non-tagged, smallpox homologs: A27V, A33V, B5V) with two adjuvants, aluminum hydroxide (Alhydrogel, AH) and CpG oligonucleotide after a prime and boost, as well as after a second boost vaccination ~5 months after the initial prime vaccination. We also examined the ability of these single subunit protein vaccine formulations to protect mice from lethal challenge with VACV.
Material and methods
Generation and purification of variola virus A33, B5, A27 proteins produced by baculovirus infected insect larvae
With WHO approval and safeguards in place, the ectodomains of the variola Bangladesh 1975 A27-homolog (amino acids 1 to 110; gene bank accession number AAA60882.1), variola India 3 Major 1967 A33-homolog (amino acids 58 to 184; gene bank accession number NP_042184.1), and variola Bangladesh 1975 B5-homolog (amino acids 20 to 275; gene bank accession number AAA60915.1) were synthetically generated and individually expressed using recombinant baculovirus technology as previously described for L1V [29]. Insect larvae were infected and the non-tagged proteins were isolated and purified to 99% homogeneity using an ion-exchange capture step, affinity (Blue sepharose) and orthogonal ion exchange chromatographic methods, with buffer exchange over G-25 for final formulation in histidine buffer. Purity of each protein can be seen by silver stain (see Fig. 2). To distinguish these proteins from previously used Histidine-tagged VACV proteins [19, 26], the proteins will be referred to as A27V, A33V and B5V. This study was not designed to provide information on protein dose selection. A protein antigen dose of 2 μg or 10 μg was used to match prior studies that used similar antigens [14, 19, 21, 29].
Figure 2.
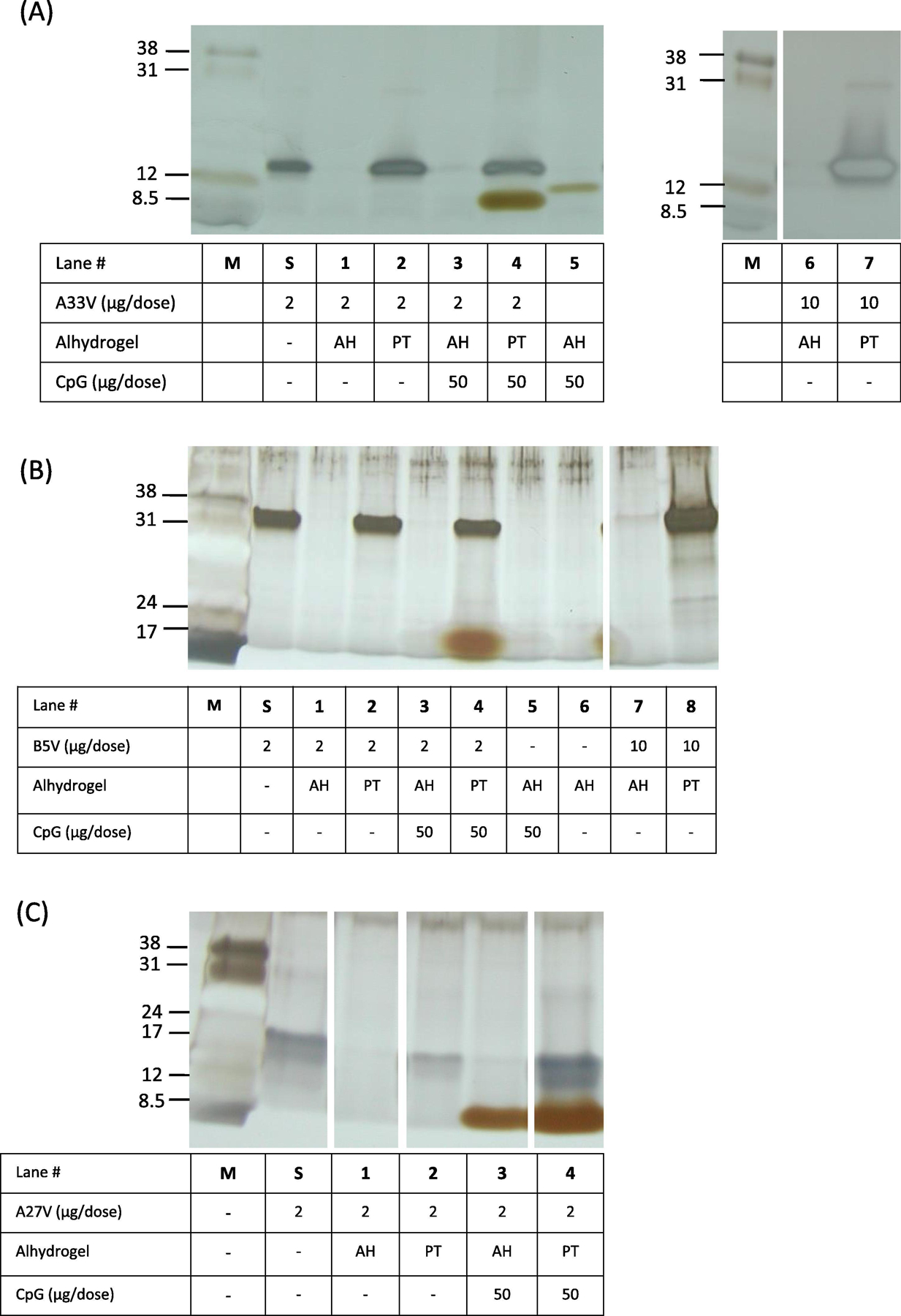
Characterization of vaccine formulations by silver-staining of SDS-PAGE. After vaccine formulations were made, the tube was centrifuged to pellet the solid aluminum hydroxide and the supernatants (5 μl) from each vaccine formulation were processed and loaded in each lane. Lane “M” is low-range molecular weight markers (Amersham) with the indicated apparent molecular weights in kDa. Lanes “S” is the starting amount of protein at a concentration of 2 μg/50 μl of A33V, B5V, A27 in histidine buffer. A. A33V formulations. PT = 40 mM KH2PO4. B. B5V formulations. PT = 40 mM KH2PO4. C. A27V formulations. PT = 100 mM KH2PO4. Note, the band in panel A, lane 5, is spill over from unrelated adjacent lane to the right of lane 5. AH, Alhydrogel; PT, phosphate treated Alhydrogel at the indicated concentration.
Vaccine formulations
All vaccine formulations contained Alhydrogel (AH) in a histidine buffer (10 mM histidine buffer with 150 mM NaCl, pH 7). Phosphate-treated Alhydrogel (PTAH) contained the indicated amount of phosphate buffer (see text) in the histidine buffer. The CpG adjuvant, CpG-10104 (5’-TCGTCGTTTCGTCGTTTTGTCGTT-3’; Coley Pharmaceutical) was used in the formulation containing Alhydrogel (AH) or phosphate-treated Alhydrogel (PTAH). Unless indicated, each vaccine formulation containing purified protein antigen at 2 μg or 10 μg, aluminum ion at 2 mg/mL (100 μg aluminum), with or without 50 μg CpG in a total of 50 μl volume were used per mouse [29]. Formulations were based on the biophysical characterization and adjuvant binding studies for each protein including addition of phosphate buffer ions which altered the surface charge of AH adjuvant, which in turn affected the interaction with each protein antigen with AH (Companion manuscript in preparation).
Vaccine preparation
The vaccines (50 μL/dose/mouse) were prepared on the day of vaccination. As previously described [19, 26, 29], Alhydrogel (100 μg/dose per mouse based on aluminum ion) was incubated in histidine buffer with or without phosphate buffer at 5 °C for 1 hr. Proteins (A33V, B5V, L1V, A27V) in histidine buffer were then added at 2 μg/dose or 10 μg/dose per mouse. CpG (50 μg/dose) was then added and formulations with or without CpG were incubated at 5 °C for an additional ~5 h. The adsorption status of proteins (and CpG) to AH and PTAH were determined by SDS-PAGE (4–15% gradient Mini-Protean TGX gel) under reducing and denaturing conditions. Protein bands and CpG bands were visualized in the gels by silver staining (Invitrogen Silverquest Staining Kit) following the manufacturer’s instructions.
Mouse vaccination and challenge
Female BALB/c mice (Charles River Laboratories) were housed in the ABSL2 facility at the University of Pennsylvania and worked with in accordance with institutional guidelines. Only female mice were used to compare results with our prior work. After a one-week acclimation period and as previously described [19, 26, 29], the six-week-old mice were randomly divided into 5 mice (for longer-term) or 10 mice per group (for short-term) for vaccination with various formulations. Briefly, for ease of handling, mice were anesthetized with ketamine/xylazine and intramuscularly vaccinated in the gastrocnemius muscle. The effect anesthetizing mice had on immune responses was not investigated. For short-term studies (Fig. 1), mice were vaccinated (primed) and then revaccinated (boosted) two weeks later. Mice were bled 3 weeks after the boost. For longer-term vaccination studies, mice were similarly, primed and then boosted 2 weeks after priming. Then at 21-weeks after priming, mice received a second boost vaccination (Fig. 1). These mice were bled at ~5 months after the first boost and then 3 weeks after the second boost. For both short-term and longer-term studies, 3-weeks after the boost vaccination, anesthetized mice were intranasally challenged with an indicated lethal dose of VACV (strain WR) in 20 μl PBS. Mice were monitored and weighed daily. Mice that reached end-point criteria and/or had 30% weight loss were humanely euthanized.
Figure 1.
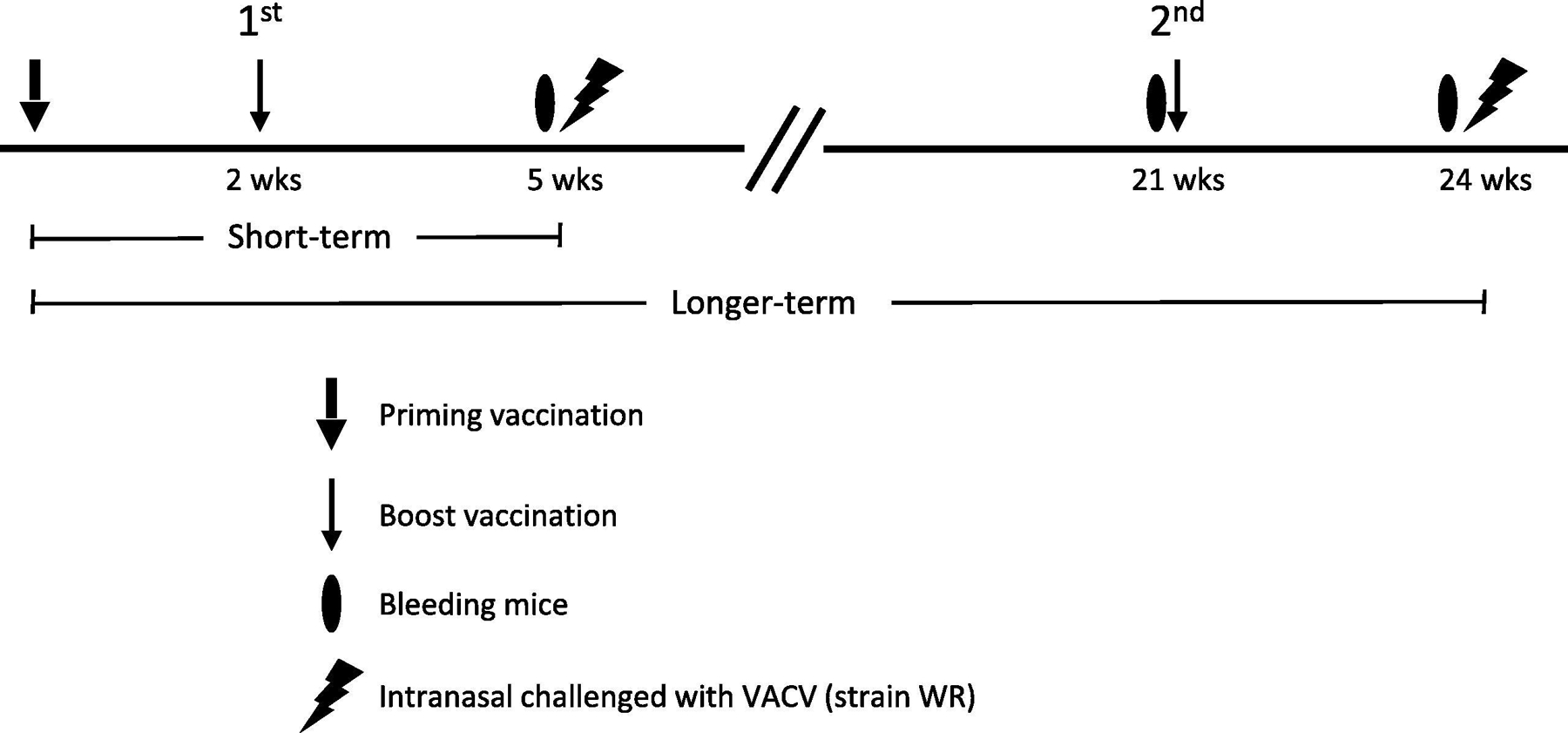
Timeline for in vivo study of short-term and longer-term subunit protein vaccinations, bleedings, and challenges.
Determination of antibody responses by ELISA
Equal volumes of sera from individual mice from each group were mixed and heat inactivated at 56 °C for 30 min. Total IgG and subclasses IgG1 and IgG2a were assayed by direct ELISA as previously described [19, 26, 29]. Briefly, 96-well ELISA plates were pre-coated with 0.5 μg/ml purified A33V or B5V or L1V or A27 proteins in PBS, and incubated at 4 °C overnight with two-fold dilutions of pooled serum samples starting at a 1:400 dilution. Wells were then probed with either HRP-conjugated Rat anti-mouse IgG (Abcam ab6728) diluted 1:2000, or IgG1 (BD Pharmingen 559626) and anti-IgG2a (BD Pharmingen 553391) each diluted at 1:1000. All samples were tested in triplicate. For each protein ELISA figure shown, samples were run at the same time to allow direct comparisons. The absorbance and antibody titers were determined as previously described [19, 26, 29].
Statistical analysis
Data are reported per group and mean ± standard error of the mean (SEM). The significant differences of the percentage weight loss between groups were analyzed from day 4 to day 12. Area-under-the-curve value was calculated for each set of percentage weight loss and ELISA data. Area-under-the-curve values were compared between groups by a One-way ANOVA statistical test using GraphPad Prism version 6 software. The p values < 0.05 were considered significant.
Results
Expression and purification of variola virus A33, B5 and A27 homolog proteins
We previously showed that a vaccine formulation of the pox protein L1V bound to aluminum hydroxide along with CpG induced a protective humoral immune response that was not found with formulations that lacked CpG or did not adsorb L1V to aluminum hydroxide [29]. In the work reported here, we individually studied the other key variola virus envelope protein homologs from the extracellular virus (A33 and B5) and mature virus (A27). Each of the protein antigens used was untagged and highly purified. While the expressed protein sequence of VACV L1 and variola L1 are identical, there were amino acid differences between the expressed ectodomains of VACV (strain WR) and variola proteins. Comparison of these VACV proteins to the VARV proteins reveals 10 amino acid differences in the 126 amino acid ectodomain of A33, 21 amino acid differences in the 255 amino acid ectodomain of B5, and 2 amino acid differences in in the110 amino acids of A27. Similar to L1V [29], all recombinant protein antigens (A33V, B5V and A27V) reacted with available anti-VACV A33, B5, A27 polyclonal and monoclonal antibodies (data not shown).
Adsorption state of adjuvanted A33V, B5V, A27V with or without CpG in Alhydrogel in the presence or absence of phosphate buffer
Previously, we found that phosphate buffer (20 mM) treated Alhydrogel (PTAH) adjuvant resulted in no adsorption of L1V antigen and/or CpG adjuvant to PTAH, and this impacted immune responses and protection [29]. In this study we investigated single additional recombinant protein antigens (A33V, B5V, A27V) adjuvanted with either AH or PTAH, with or without CpG. Biophysical studies of these proteins with Alhydrogel revealed the 40 mM phosphate buffer was needed to prevent adsorption of A33V and B5V to Alhydrogel, while 100 mM of phosphate buffer was needed for A27 (companion manuscript in preparation).
Both the A33V and B5V proteins at 2 μg/dose showed a similar AH adsorption pattern as we previously found with L1V [29]. As shown in Fig. 2, A33V with or without CpG was completely adsorbed to AH resulting in no bands visually observed in the gel (Fig. 2A, lanes 1 and 3). A33V with or without CpG was not adsorbed to PTAH (Fig. 2A, lanes 2 and 4) resulting in clearly observed bands in the gel. Similar experiments with B5V with or without CpG also showed both to be completely adsorbed to AH (Fig. 2B, lanes 1 and 3), and not adsorbed to PTAH (Fig. 2B, lanes 2 and 4). A higher dose of A33V and B5V (10 μg/dose) had similar results with AH able to adsorb, and PTAH not adsorbing, all of the protein (Fig. 2A, lanes 6 and 7, 2B, lanes 7 and 8). A higher concentration of phosphate buffer (100 mM) was needed to prevent A27V from being adsorbed to Alhydrogel (Fig. 2C, lane 4). Interestingly, unlike the other formulations with AH, some CpG remained unbound to AH (Fig. 2C, lane 3) in the presence of A27V.
A33V/CpG adsorbed to Alhydrogel (AH) induced higher antibody levels compared to protein non-adsorbed to phosphate treated Alhydrogel (PTAH)
After a prime and boost, antibody responses were measured 3 weeks after the boost. Our previously published data with L1V showed that antibody responses induced by L1V adsorbed to AH gave significantly higher antibody responses than the non-adsorbed formulation [29]. In this study, we similarly found that A33V adsorbed to AH (A33/AH) produced significantly higher IgG and IgG1 antibody responses than the non-adsorbed A33/PTAH formulation (Fig.3A (total IgG) and 3B (IgG1), closed circle, dashed line vs. open circle, dashed line; p= 0.01 and p= 0.02, respectively). Neither formulation without CpG produced measurable complement-fixing isotype antibody, IgG2a (Fig. 3C). When CpG was included in the formulation with A33V/AH (A33/AH/CpG), higher total IgG and IgG2a antibody titers were generated than the formulation not containing CpG (Fig. 3A, closed circle, solid line vs. closed circle, dashed line, IgG, p= 0.01 and Fig. 3C, IgG2a, p=0.0001). Comparing adsorbed to non-adsorbed A33V in the presence of CpG (A33/AH/CpG vs. A33/PTAH/CpG), adsorbed A33/CpG gives higher total IgG, IgG1, and IgG2a responses than non-adsorbed A33/CpG (Fig. 3 closed circle, solid line vs. open circle solid line; p= 0.002, p= 0.01 p= 0.002, respectively). Increasing the amount of A33V to 10 μg/dose of A33V adsorbed to AH (Fig. 3, closed triangle, dashed line) or non-adsorbed in PTAH (Fig. 3, open triangle, dashed line) did not drastically change total IgG titers compared to the lower 2 μg dose of A33V (p= 0.67) and decreased the amount of IgG1 (p= 0.007). The higher dose of A33V in the absence of CpG still did not form measurable amounts of IgG2a (Fig. 3C). In summary, these results indicate that increasing the amount of A33V in the formulation will not produce a higher antibody response or result in an isotype switch. However, CpG in the vaccine formulation is important for enhancing total immune responses and inducing antibody isotype IgG2a (Fig.3C). Adsorption of A33V and CpG to AH gave the highest overall antibody responses.
Figure 3.
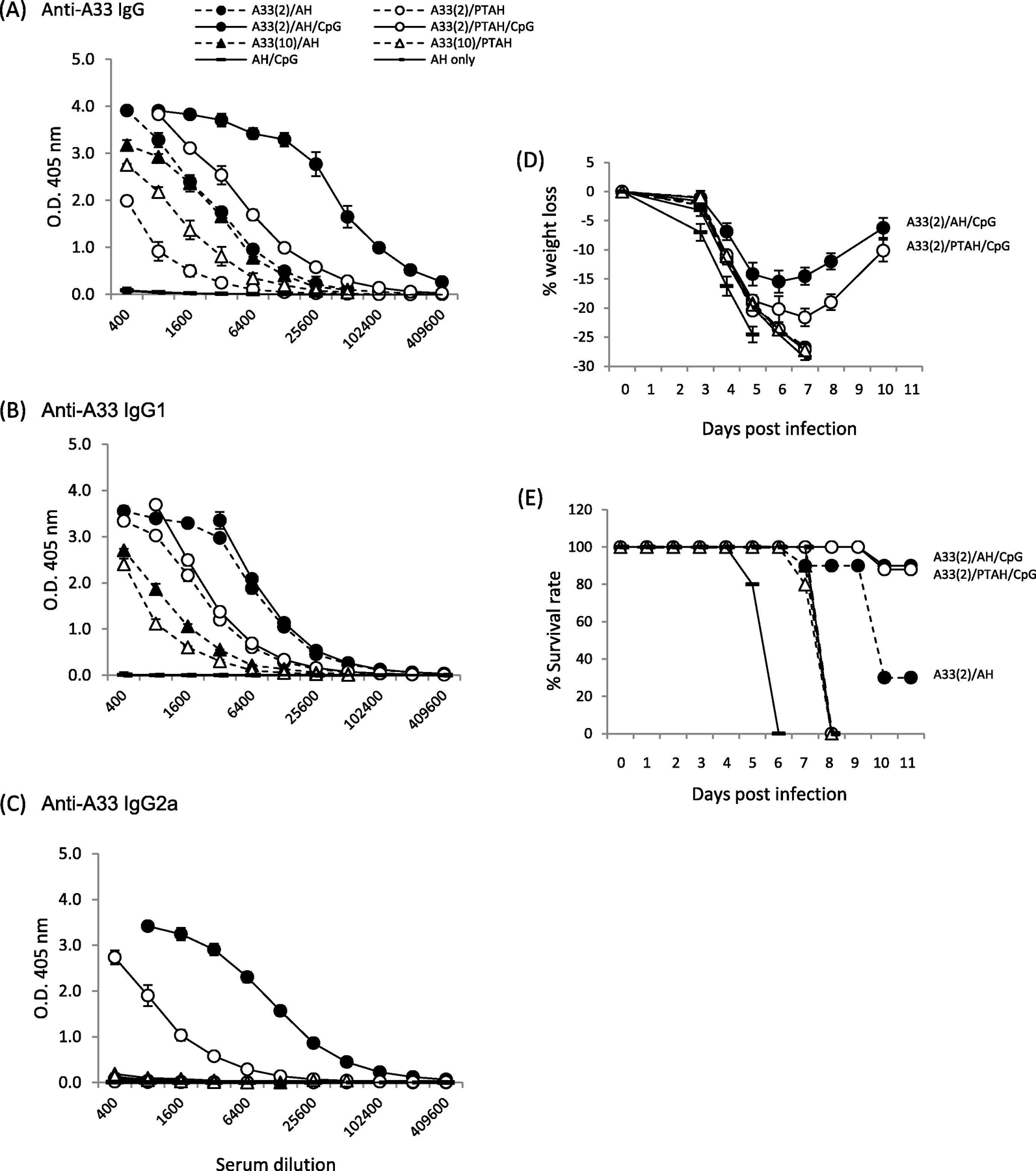
Evaluation of antibody responses after short-term vaccination of groups of mice with A33V formulations and pathogenesis after VACV challenge. A to C. Sera samples were obtained 3 weeks after one boost vaccination and antibody responses were measured by ELISA. (A) total IgG; (B) isotype IgG1; (C) isotype IgG2a. Mice (10 mice/group) were then challenged intranasally with VACV 1.7×105 pfu per mouse and followed for (D) weight loss and (E) survival rate. Solid symbols represent vaccines formulated in Alhydrogel (AH). Open symbols represent vaccines formulated in phosphate treated Alhydrogel (PTAH). Solid lines represent vaccines formulated with CpG. Dashed lines represent vaccines formulated without CpG.
Formulations with B5V behave differently than those with A33V in inducing antibody levels with or without adjuvant CpG
Given our prior results with L1V [29] and the results found with A33V, we expected B5V to behave similarly. However, as shown in Fig. 4, some antibody level differences between formulations with B5V were less distinct. There were obvious differences with the formulation with 2 μg of protein non-adsorbed B5V (B5/PTAH), which gave the lowest total IgG responses compared to adsorbed B5V at the same dose (B5/AH) giving a higher IgG responses (Fig.4A open circle, dashed line vs. closed circle, dashed line; p= 0.03). Inclusion of CpG in both formulations increases the total IgG levels to the same levels (Fig. 4A, closed circle, solid line vs. open circle, solid line; p= 0.3). Interestingly, increasing the amount of B5V in the formulation to 10 μg results in total IgG amounts at the same level of the formulations with CpG (Fig. 4A, closed circle, solid line vs. closed triangle, dashed line (p= 0.50) and open circle, solid line vs. open triangle, dashed line (p= 0.46)). Some of these trends are seen in the IgG1 response, although there is a clearer difference between adsorbed and non-adsorbed formulations with CpG (Fig. 4B. open circle, solid line vs. closed circle, solid line, p= 0.02). Again, unlike A33V, the 10 μg/dose of B5V non-adsorbed in PTAH drastically increased IgG1 levels compared to the lower 2 μg dose of B5V (Fig. 4B, open triangles, dashed line vs. open circle, dashed line, p= 0.03). However, 10 μg and 2 μg/dose of B5V adsorbed to AH produced the same levels of IgG1 (Fig. 4B, solid circle, dashed line vs. solid triangle, dashed line, p= 0.75). In summary, these results showed that the amount of B5V proteins and adsorption of protein to AH are important for stimulating a Th2 immune response and IgG1 production. While the development of isoclass switch to IgG2a is dependent on the presence of CpG (Fig. 4C), the adsorption state of the B5V protein and CpG appears less important than was seen with L1V [29] and A33V (Fig. 3C). In Fig. 4C, the highest IgG2a response is seen with B5/AH/CpG (Fig. 4C, closed circle, solid line vs. open circle, solid line; p=0.03), but the non-adsorbed formulation with phosphate buffer still produces high levels of IgG2a.
Figure 4.
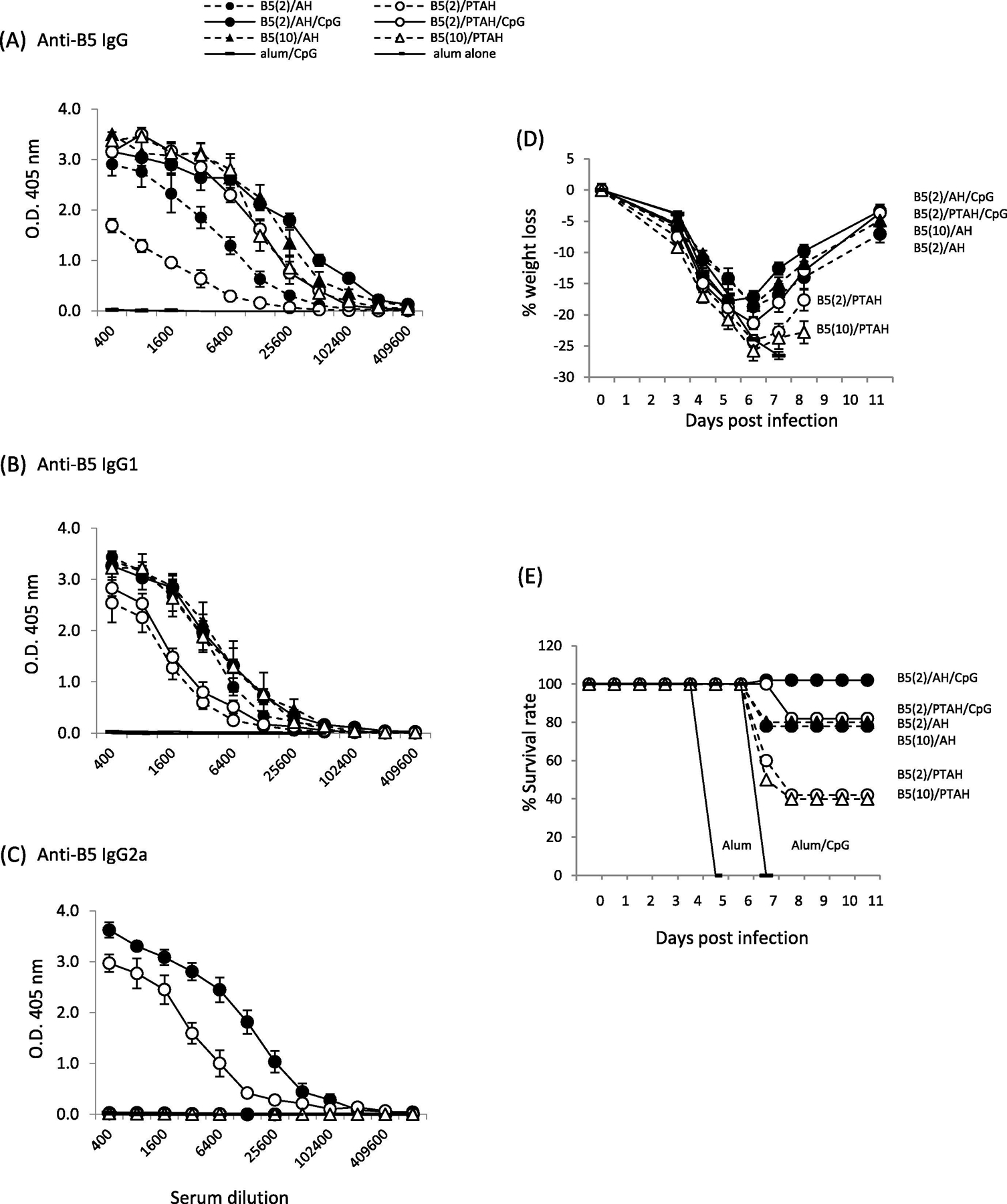
Evaluation of antibody responses after short-term vaccination of groups of mice with B5V formulations and pathogenesis after VACV challenge. A to C. Sera samples were obtained 3 weeks after one boost vaccination and antibody responses were measured by ELISA. (A) total IgG; (B) isotype IgG1; (C) isotype IgG2a. Mice (10 mice/group) were then challenged intranasally with VACV 3.7×105 pfu per mouse and followed for (D) weight loss and (E) survival rate. Solid symbols represent vaccines formulated in Alhydrogel (AH). Open symbols represent vaccines formulated in phosphate treated Alhydrogel (PTAH). Solid lines represent vaccines formulated with CpG. Dashed lines represent vaccines formulated without CpG.
Formulations with A27V induce high antibody levels when the adjuvant includes CpG
For A27V protein formulations there appeared to be predictable antibody responses (Fig. 5). Highest antibody responses were with A27V formulations that included CpG. The AH-adsorption status did not matter (Fig. 5A, B, and C, closed circle solid line vs. open circle solid line; p =0.84 (IgG), p=0.71 (IgG1) and p= 0.89 (IgG2a)). The A27V formulations that were adjuvanted with Alhydrogel alone gave lower antibody responses when compared to formulations without CpG (Fig. 5A & B, solid circle solid lines vs. solid circle dashed line; p =0.03 (IgG), p=0.01 (IgG1) and open circle solid lines vs. open circle dashed line; p= 0.028 (IgG), p=0.024 (IgG1)), The only A27V formulations that generated an IgG2a response were those with CpG (Fig. 5C). Unlike that found with L1V [29] or reported here for A33V and B5V, the adsorption status of A27V along with CpG did not alter the IgG2a response (Fig. 5C, closed circle, solid line vs. open circle, solid line; p= 0.89).
Figure 5.
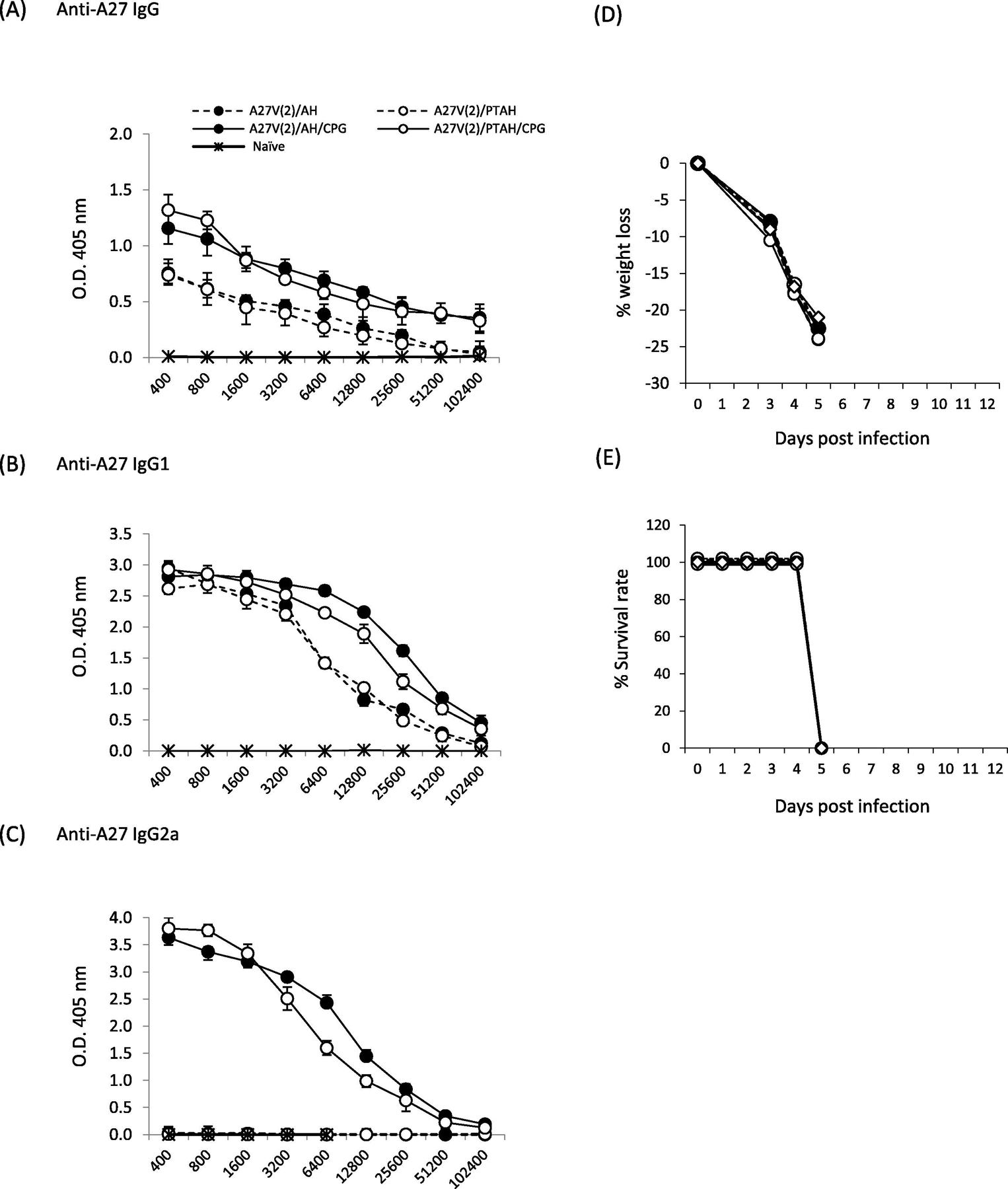
Evaluation of antibody responses after short-term vaccination of groups of mice with A27V formulations and pathogenesis after VACV challenge. A to C. Sera samples were obtained 3 weeks after one boost vaccination and antibody responses were measured by ELISA. (A) total IgG; (B) isotype IgG1; (C) isotype IgG2a. Mice (5 mice/group) were then challenged intranasally with VACV 4.0×105 pfu per mouse and followed for (D) weight loss and (E) survival rate. Solid symbols represent vaccines formulated in Alhydrogel (AH). Open symbols represent vaccines formulated in phosphate treated Alhydrogel (PTAH). Solid lines represent vaccines formulated with CpG. Dashed lines represent vaccines formulated without CpG.
Protection of vaccinated mice with formulations containing A33V, B5V, or A27V against intranasal lethal dose VACV challenge 3-weeks after a single boost
To determine which formulations could protect mice from intranasal VACV challenge 3-weeks after a single boost vaccination (Fig. 1), we followed weight loss and survival after challenge. For vaccines formulated with A33V (Fig. 3D and E), control mice vaccinated with AH alone or AH/CpG all died or needed to be humanely sacrificed. Groups of mice vaccinated with formulations that did not contain CpG (A33V (2 μg or 10 μg)) formulated with AH or PTAH all lost significant amounts of weight and succumb to infection. Groups of mice vaccinated with formulations that contained CpG resulted in 9 of 10 mice surviving (Fig. 3E, solid lines). But the group that was vaccinated with the formulation that adsorbed the A33V and CpG lost less weight than the group formulated with PTAH (Fig. 3D, closed circle, solid line vs. open circle, solid line; p=0.0078). These data indicate that formulations with CpG that generate antibody subtype IgG2a are protective. Mice vaccinated with the formulation with A33V/CpG adsorbed to AH was slightly better at protecting against weight loss than the A33V/CpG/PTAH formulation.
In contrast with the findings with A33V, a number of non-CpG containing formulations with B5V conferred some level of protection (Fig. 4D and E). Groups of mice vaccinated with either B5V (2 μg or 10 μg) adsorbed to AH lost similar amounts of weight and had similar levels of survival compared to the CpG-containing formulations (both adsorbed and non-adsorbed). These four groups lost 15 to 20% weight and had 80 to 100% survival. Less protective were non-adsorbed formulations B5V (2 μg)/PTAH and B5V (10 μg)/PTAH. These groups lost 25% of their weight and only 4 of 10 mice from each group survived challenge. While the differences in antibody response to many of the formulations can explain some of these findings (e.g., the formulation that had the least relative protection (B5V (2 μg)/PTAH) generated the lowest overall anti-B5V antibody responses), the similar levels of protection and weight loss of many of the formulations may indicate the important role antibody generated to B5 has in the protection from lethal challenge.
Despite high antibody responses to A27V in all formulations (Fig. 5), none of the vaccines protected mice from weight loss or survival (Fig. 5D &E). The lack of protection generated by A27V vaccines is likely due to the inability of these antibodies to neutralize MV in plaque reduction assays (data not shown). We had similar findings when the histidine-tagged VACV A27 protein was used in vaccine formulations [19].
Longer-term antibody responses to vaccine formulations
To understand the longer-term immune responses to these vaccines with adsorbed and non-adsorbed proteins, antibody responses were also examined ~6 months after the initial prime with one or two boosts (Fig. 1). Our prior work showed poorer protection of protein vaccines formulated without CpG [19, 26, 29]. For these studies, we examined the longer term antibody responses of various adjuvanted formulations with each of the four recombinant protein antigens including A33V, B5V, A27 (described in this work) as well as L1V described previously [29].
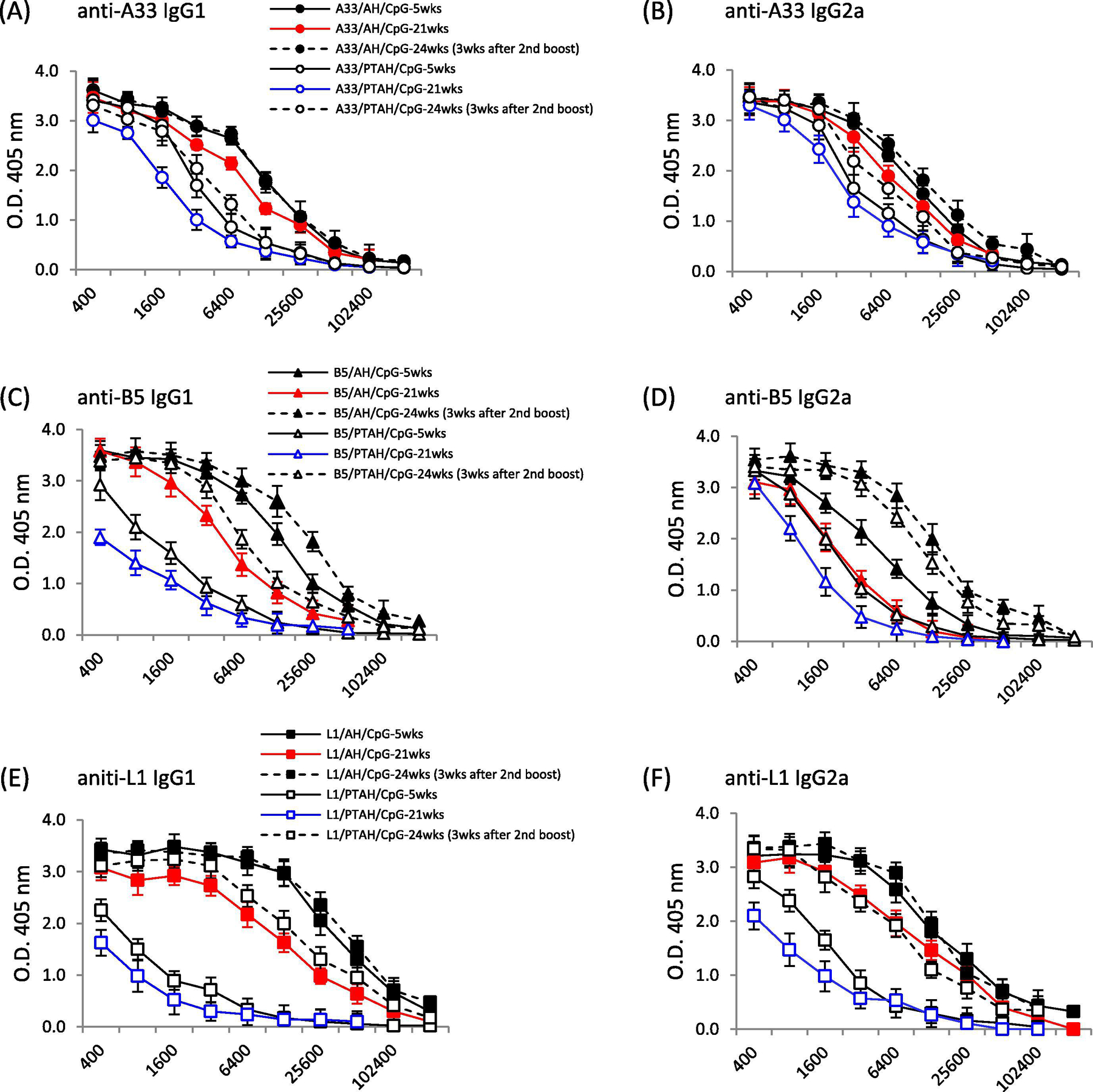
As shown in Fig. 6A and B, IgG1 and IgG2a antibody levels generated by A33V/AH/CpG at 21-weeks (19-weeks after the first boost (red circles, red line)) remained remarkedly high. The antibody levels were just slightly lower than the antibody response 3-weeks after the first boost (black solid circle, solid line vs. red circles, red line; p=0.06). The second boost brings IgG1 and IgG2a levels back to this same level as this earlier time point (solid circle, dashed line vs. solid circle, solid line; p=0.81). The A33V formulations that did not adsorb A33V (A33V/PTAH/CpG) had lower responses (open circles), but showed the same trends. IgG1 and IgG2a antibody levels remained relatively high at 21-weeks (19-weeks after the first boost (blue circles, blue line)) and were increased after a second boost.
Figure 6.
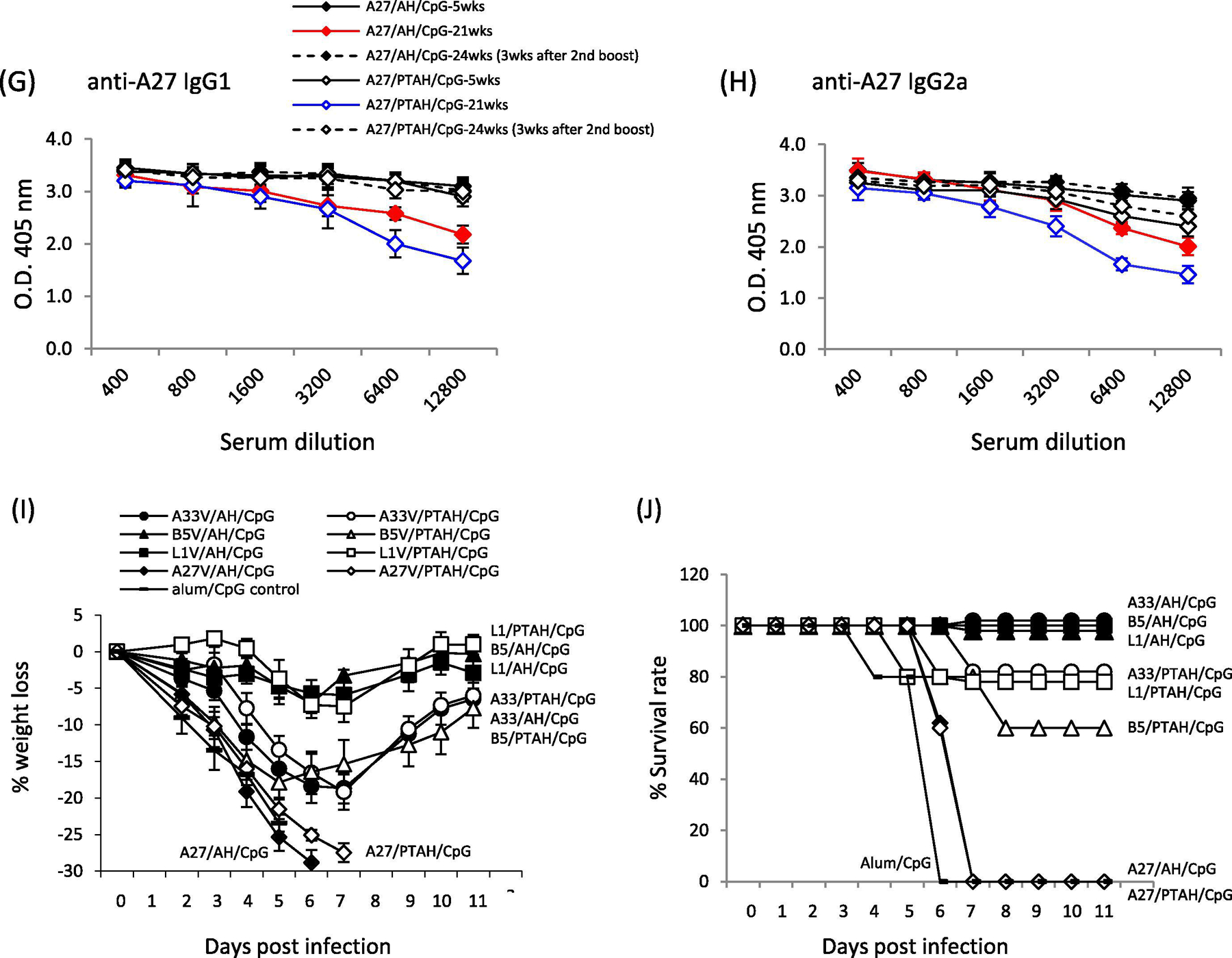
Comparison longer-term antibody response after one or two boost vaccinations and pathogenesis after VACV challenge. A to H. Sera samples were obtained 3 weeks and 19 weeks after first boost as well as 3-weeks after second boost. Antibody responses were measured by ELISA. Shown are responses to A33V (A) IgG1 (A) and (B) IgG2, B5V (C) IgG1 and (D) IgG2, L1V (E) IgG1 and (F) IgG2, A27V (G) IgG1 and (H) IgG2a. Solid symbols represent vaccines formulated in Alhydrogel (AH). Open symbols represent vaccines formulated in phosphate treated Alhydrogel (PTAH). Solid lines represent the groups that received a single boost 3-weeks after the initial prime vaccination. Dashed lines represent the groups that received a second boost 21-weeks after the initial prime vaccination. Red line shows antibody responses to vaccine formulations with proteins and CpG adsorbed to AH (AH) 19 weeks after the first boost. Blue line shows proteins and CpG not adsorbed to AH (PTAH) 19 weeks after the first boost. Groups of mice (5 mice/group) that received a second boost vaccination were then challenged intranasally with VACV 4.4×105 pfu per mouse and followed for (I) weight loss and (J) survival rate.
The formulations containing B5V followed similar trends to that seen with A33V. As shown in Fig. 6C, IgG1 antibody levels generated by B5V/AH/CpG at 21-weeks (19-weeks after the first boost (red triangles, red line)) still remained high. But the antibody levels were lower than the antibody response 3-weeks after the first boost (black solid triangle, solid line vs. red triangle, red line; p= 0.01). The second boost brings IgG1 levels to a higher level than the earlier time point (solid triangle, dashed line vs. solid triangle, solid line; p= 0.04). The B5V formulations that did not adsorb B5V (B5V/PTAH/CpG) had lower responses (open triangles), but showed the same trends. IgG1 antibody levels remained relatively high at 21-weeks (19-weeks after the first boost (blue triangles, blue line)) and were increased after a second boost (open triangle, dashed line vs. blue triangles, blue line; p= 0.01). For adsorbed and non-adsorbed B5V formulations, the IgG2a levels at 21-weeks (19-weeks after the first boost) were lower than 3 weeks after the first boost, but each boosted to a level higher than the level 3-weeks after boost vaccination (closed triangles, dashed line vs. red triangles, red triangles, red line (p=0.03), open triangles, dashed line vs. blue triangles, blue line (p=0.02)), Also, the second boost produced significant higher IgG2a titers than the first boost (closed triangles, dashed line vs. closed triangles, solid line (p= 0.03), open triangles, dashed line vs. open triangles, solid line (p= 0.02)). Interestingly, the adsorption status B5V/CpG after the second boost was less critical since non-adsorbed B5V/CpG generated similar high levels of IgG2a antibody to those produced by adsorbed B5V/CpG (closed triangle, dashed line vs. open triangle, dashed line; p=0.08)
We also examined longer-term responses to adsorbed and non-adsorbed L1V formulations with CpG. Similar to findings with A33V, as shown in Fig. 6E and F, IgG1 and IgG2a antibody levels generated by L1V/AH/CpG at 21-weeks (19-weeks after the first boost (red squares, red line)) remained very high. The IgG2a antibody levels were just slightly lower than the antibody response 3-weeks after the first boost (red squares, red line vs. solid squares, solid line; p=0.1). The second boost brings IgG1 and IgG2a levels back to the same level as this earlier time point (solid square, dashed line vs. solid square, solid line). The L1V formulations that did not adsorb L1V (L1V/PTAH/CpG) had lower responses (open squares), but showed the same trends. IgG1 and IgG2a antibody levels at 21-weeks (19-weeks after the first boost), remained similar to the antibody response 3 weeks after the first boost (blue squares, blue line vs. open black squares, solid black line; p= 0.09 for IgG1, p= 0.078 for IgG2a). However, the second boost resulted in high IgG1 and IgG2a levels like the adsorbed L1V formulation (L1V/AH/CpG) at 21-weeks (19-weeks after the first boost (open squares, dashed line vs. red squares, red line; IgG1, p= 0.2; IgG2a, p= 0.19)). Thus, a second boost of the non-adsorbed formulation can result in higher anti-L1 antibody responses and additional class-switched IgG2a antibody (open square, solid line vs. open square, dashed line in IgG1, p=0.002, IgG2a, p=0.01).
All formulations with A27V protein produced very high IgG1 and IgG2a antibody titers that were high even at 21-weeks (19-weeks after the first boost (Fig. 6G and H)). After the second boost, the IgG1 and G2a titers only slightly increased.
Vaccine formulations containing A33V, B5V, or L1V and CpG provide longer-term protection of mice against intranasal lethal dose VACV challenge 3-weeks after a second boost
We next determined if the immune responses after a second boost vaccination could protect mice against VACV infection six months after the initial priming vaccination. Boosted mice were challenged after they received a second boost, 3-weeks prior to challenge (Fig. 1). As shown on Fig. 6I and J, all groups except for those vaccinated with either formulation of A27V showed evidence of protection. The groups that had the least weight loss and 80 to 100% survival were the adsorbed and non-adsorbed formulations of L1V (open and closed squares) and the adsorbed formulation of B5V (closed triangles). The other groups (adsorbed A33V (closed circles), 100% survival; non-adsorbed A33V (open circles), 80% survival; and non-adsorbed B5V, 60% survival) all had significantly more weight loss (~20%) compared to the more protective formulations (e.g., B5V/PTAH/CpG vs. L1V/PTAH/CpG; p= 0.001). Formulations with A27V induced high antibody immune responses that poorly protect mice against virus infection. This was also found when the histidine-tagged VACV A27 protein was used as a subunit [19, 32].
Discussion
Recombinant protein subunit vaccines can have advantages over live virus vaccines. The most obvious is that they can have a better safety profile. However, live virus vaccines are considered the gold standard because of the breadth and longevity of the protective immune response they generate. Nevertheless, for the first time, a protein-based subunit vaccine used to prevent recurrent varicella zoster provided levels of protection better than those reported with a live attenuated virus vaccine [33, 34]. The new herpes zoster subunit vaccine (HZ/su, Shingrix) is based on a recombinant expressed glycoprotein E (gE) and the AS01B adjuvant (containing MPL and QS-21 adjuvants in a liposomal formulation). Like the pox protein antigens and multiple adjuvants we studied in this report in mice, the gE protein along with adjuvant AS01B induce a potent immune response in humans. Studies in humans showed that HZ/su elicits both high VZV gE-specific humoral and cell-mediated response. The overall efficacy against herpes zoster was 97.2%, and between 96.6% and 97.9% for all age groups [33]. In adults 70 years or older, the HZ/su efficacy against herpes zoster was around 90.0% after a 2-dose vaccination and reduced the postherpetic neuralgia among adults 70 years of age or older [34]. While not directly compared to the live attenuated VZV vaccine, the efficacy of HZ/su was better than historical data with the live vaccine. Thus, the right recombinant protein antigen/adjuvant combination could provide a safer and more effective alternative to a live virus vaccine.
Finding an equally efficient safe vaccine alternative to live-virus smallpox vaccine remains important, especially should the day comes when society and/or regulatory agencies no longer accept the safety vs efficacy profile of live virus vaccines. Furthermore, the threat of bioterrorism with an agent like variola virus remains present. Also, zoonotic infections from monkeypox occurs in Africa with known importations into non-endemic areas of the globe including the United States [35, 36] and elsewhere around the world [37, 38]. The long term stockpiling of a subunit vaccine may be easier than a live virus vaccine both in terms of vaccine stability and ease of large scale manufacturing. Since adjuvants can boost the effectiveness of subunit vaccines, the selection of subunit antigens and adjuvants, both in terms of in vivo immunopotentiation as well as their in vitro pharmaceutical properties, is a key aspect of their clinical development. Developing such vaccine formulations includes determining the nature of the interactions (or non-interactions) between the antigen-adjuvant components to optimize both physicochemical storage stability and in vivo potency. Despite the long history of vaccine development using aluminum adjuvants, much is still to be learned in terms of formulation development to optimize their use in combination with various subunit antigens [39, 40] and with newer adjuvants [41].
In our previous work, we studied antibody responses and protection conferred by subunit vaccines containing a combination of VACV proteins A33, B5, L1, and A27 adjuvanted with CpG and aluminum hydroxide in mice and non-human primates [19, 26]. These proteins were histidine-tagged. To begin to generate non-tagged antigens that could be used in humans, we moved to variola virus homologs and initially studied L1V [29]. L1 is a key viral envelope protein on the surface of MV. It is required for VACV cell entry [42]. In our prior work with L1V, we found clear correlation between the vaccine formulation and protection. L1V and CpG adsorbed to Alhydrogel resulted in the least weight loss after challenge and 100% survival when compared to the non-adsorbed formulation. In the work described here, we studied other viral components which could be used in a multi-subunit smallpox vaccine.
A33 is an EV-specific protein that is required for efficient cell-to-cell spread [43, 44]. For A33V, the status of the adsorption of protein to AH and the presence or absence of CpG resulted in antibody levels (Fig. 3) similar to those found with L1V. Like responses to L1V, the presence of CpG resulted not only in the highest total antibody response, but also resulted in the production of an IgG2a antibody response. While compared to the clearer differences in weight loss between L1V/AH/CpG and L1V/PTAH/CpG, there was less of a difference in weight loss of adsorbed and non-adsorbed A33V/CpG, although there were statistically significant differences between two groups. Both formulations also resulted in similar high survival levels after challenge (Fig. 3E). Thus, the ability of both adsorbed and non-adsorbed A33V/CpG to generate an IgG2a isotype response to A33 seems important. Mouse IgG2a is complement fixing and functions in antibody-dependent cellular cytotoxicity (ADCC). It has been shown that enhanced neutralization of EV is through complement fixing antibody [45] and thus our results with A33V make biological sense. The longer-term challenge experiment shows the exceptionally high antibody levels are present nearly 5 months after the last boost vaccination (Fig. 6A & B). Weight loss and survival of groups vaccinated with adsorbed and non-adsorbed A33V/CpG were similar (Fig. 6I & J, circles). While the experiment was not designed to examine the longer-term protection after a single boost vaccination, the antibody levels indicate that some protection would have been expected months after the first boost vaccination.
B5 is another key EV glycoprotein that is involved in EV formation and is involved in cell-to-cell and long-range spread of the virus in vivo [46]. While the only A33V vaccine formulations that conferred protection 3 weeks after the boost included CpG (Fig. 3E), our findings of antibody responses and survival with B5V vaccine formulations were different. Similar to results with A33V, B5V formulations that contained CpG were the only ones that generated IgG2a isotype responses (Fig. 4C). But opposed to A33V, formulations without CpG but with B5V adsorbed to AH had similar levels of protection as the CpG containing formulations as measured by weight loss and survival after challenge (Fig. 4E & F). This indicates that while IgG2a antibody responses can be important in neutralizing EV [45], the antibody response generated to the protein (either at 2 μg/dose or 10 μg/dose) adsorbed to AH generated a protective response to either neutralize EV [47, 48] and/or prevent efficient cell-to-cell spread [44, 46]. It also reveals that anti-B5 antibodies can protect in a manner different than antibodies to A33. The data also indicates a role for adsorption of B5V to AH to generate a protective immune response that was previously seen with L1V. Similar to results with A33V, the longer-term challenge experiment shows that there were measurable antibody levels present nearly 5 months after the last boost vaccination (Fig. 6C & D). Weight loss and survival of groups vaccinated with adsorbed B5V/CpG were better than the non-adsorbed formulation (Fig. 6I & J, closed triangles compared to open triangles). While the experiment was not designed to examine longer term protection after a single boost vaccination, based on the pre-second boost antibody levels and weight loss and survival after the second boost, some protection might be expected for the adsorbed B5V/CpG, but not the non-adsorbed B5V/CpG.
The experiment that examined longer-term antibody responses to L1V/CpG formulations confirm our prior findings of antibody responses 3-weeks after the initial boost [29]. That is, higher IgG1 and IgG2a responses to adsorbed L1V/CpG than non-adsorbed L1V/CpG (Fig. 6E & F, solid square, solid line vs. open square, solid line). These antibody levels are slightly lower ~5 months after the first boost (Fig. 6E & F, red square/line vs. blue square/line). A second boost results in high antibody levels for both formulations (solid and open squares, dashed line). These antibody levels result in little weight loss and 100% survival of challenged mice.
In conclusion, similar to our work with L1V, A33V adsorption to aluminum hydroxide along with CpG maximized antibody responses and protected mice against lethal dose VACV infection 3-weeks after a boost vaccination. While B5V adsorption to aluminum hydroxide (along with CpG) also maximized antibody responses, the presence of CpG was less critical for protection of mice against lethal dose VACV infection 3-weeks after the boost vaccination. While formulations with A27V induced high antibody responses, none of the formulations resulted in protection after challenge. The individual protein vaccines adsorbed and non-adsorbed to Alhydrogel induced long-lasting antibody immune responses, which rebounded to higher antibody titers after a second boost. After the second boost, the best protection was conferred by formulations with adsorbed B5V or L1V as well as non-adsorbed L1V.
Acknowledgments
This work was funded by U01 AI077913 as well as prior support from the NIAID Middle Atlantic Regional Center of Excellence (MARCE) grant U54 AI057168.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. Smallpox and its eradication. 1st ed. Geneva: World Health Organization; 1988. [Google Scholar]
- [2].Rimoin AW, Mulembakani PM, Johnston SC, Lloyd Smith JO, Kisalu NK, Kinkela TL, et al. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. Proc Natl Acad Sci U S A. 2010;107:16262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoff NA, Doshi RH, Colwell B, Kebela-Illunga B, Mukadi P, Mossoko M, et al. Evolution of a Disease Surveillance System: An Increase in Reporting of Human Monkeypox Disease in the Democratic Republic of the Congo, 2001–2013. Int J Trop Dis Health. 2017;25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reynolds MG, Doty JB, McCollum AM, Olson VA, Nakazawa Y. Monkeypox reemergence in Africa: a call to expand the concept and practice of One Health. Expert Rev Anti Infect Ther. 2019;17:129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wiser I, Balicer RD, Cohen D. An update on smallpox vaccine candidates and their role in bioterrorism related vaccination strategies. Vaccine. 2007;25:976–84. [DOI] [PubMed] [Google Scholar]
- [6].Weltzin R, Liu J, Pugachev KV, Myers GA, Coughlin B, Blum PS, et al. Clonal vaccinia virus grown in cell culture as a new smallpox vaccine. Nat Med. 2003;9:1125–30. Epub 2003 Aug 17. [DOI] [PubMed] [Google Scholar]
- [7].Greenberg RN, Kennedy JS. ACAM2000: a newly licensed cell culture-based live vaccinia smallpox vaccine. Expert Opin Investig Drugs. 2008;17:555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].CDC. Progressive vaccinia in a military smallpox vaccinee -- United States, 2009. MMWR Morb Mortal Wkly Rep. 2009;58:532–6. [PubMed] [Google Scholar]
- [9].Kennedy JS, Greenberg RN. IMVAMUNE: modified vaccinia Ankara strain as an attenuated smallpox vaccine. Expert Rev Vaccines. 2009;8:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pittman PR, Hahn M, Lee HS, Koca C, Samy N, Schmidt D, et al. Phase 3 Efficacy Trial of Modified Vaccinia Ankara as a Vaccine against Smallpox. N Engl J Med. 2019;381:1897–908. [DOI] [PubMed] [Google Scholar]
- [11].Earl PL, Americo JL, Wyatt LS, Eller LA, Whitbeck JC, Cohen GH, et al. Immunogenicity of a highly attenuated MVA smallpox vaccine and protection against monkeypox. Nature. 2004;428:182–5. [DOI] [PubMed] [Google Scholar]
- [12].Hooper JW, Custer DM, Schmaljohn CS, Schmaljohn AL. DNA vaccination with vaccinia virus L1R and A33R genes protects mice against a lethal poxvirus challenge. Virology. 2000;266:329–39. [DOI] [PubMed] [Google Scholar]
- [13].Hooper JW, Custer DM, Thompson E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology. 2003;306:181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fogg C, Lustig S, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J Virol. 2004;78:10230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hooper JW, Thompson E, Wilhelmsen C, Zimmerman M, Ichou MA, Steffen SE, et al. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J Virol. 2004;78:4433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pulford DJ, Gates A, Bridge SH, Robinson JH, Ulaeto D. Differential efficacy of vaccinia virus envelope proteins administered by DNA immunisation in protection of BALB/c mice from a lethal intranasal poxvirus challenge. Vaccine. 2004;22:3358–66. [DOI] [PubMed] [Google Scholar]
- [17].Fang M, Cheng H, Dai Z, Bu Z, Sigal LJ. Immunization with a single extracellular enveloped virus protein produced in bacteria provides partial protection from a lethal orthopoxvirus infection in a natural host. Virology. 2006;345:231–43. [DOI] [PubMed] [Google Scholar]
- [18].Heraud JM, Edghill-Smith Y, Ayala V, Kalisz I, Parrino J, Kalyanaraman VS, et al. Subunit recombinant vaccine protects against monkeypox. J Immunol. 2006;177:2552–64. [DOI] [PubMed] [Google Scholar]
- [19].Xiao Y, Aldaz-Carroll L, Ortiz AM, Whitbeck JC, Alexander E, Lou H, et al. A protein-based smallpox vaccine protects mice from vaccinia and ectromelia virus challenges when given as a prime and single boost. Vaccine. 2007;25:1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Golovkin M, Spitsin S, Andrianov V, Smirnov Y, Xiao Y, Pogrebnyak N, et al. Smallpox subunit vaccine produced in planta confers protection in mice. Proc Natl Acad Sci U S A. 2007;104:6864–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fogg CN, Americo JL, Lustig S, Huggins JW, Smith SK, Damon I, et al. Adjuvant-enhanced antibody responses to recombinant proteins correlates with protection of mice and monkeys to orthopoxvirus challenges. Vaccine. 2007;25:2787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thornburg NJ, Ray CA, Collier ML, Liao HX, Pickup DJ, Johnston RE. Vaccination with Venezuelan equine encephalitis replicons encoding cowpox virus structural proteins protects mice from intranasal cowpox virus challenge. Virology. 2007;362:441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Berhanu A, Wilson RL, Kirkwood-Watts DL, King DS, Warren TK, Lund SA, et al. Vaccination of BALB/c mice with Escherichia coli-expressed vaccinia virus proteins A27L, B5R, and D8L protects mice from lethal vaccinia virus challenge. J Virol. 2008;82:3517–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kaufman DR, Goudsmit J, Holterman L, Ewald BA, Denholtz M, Devoy C, et al. Differential antigen requirements for protection against systemic and intranasal vaccinia virus challenges in mice. J Virol. 2008;82:6829–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hooper JW, Ferro AM, Golden JW, Silvera P, Dudek J, Alterson K, et al. Molecular smallpox vaccine delivered by alphavirus replicons elicits protective immunity in mice and non-human primates. Vaccine. 2009;28:494–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Buchman GW, Cohen ME, Xiao Y, Richardson-Harman N, Silvera P, DeTolla LJ, et al. A protein-based smallpox vaccine protects non-human primates from a lethal monkeypox virus challenge. Vaccine. 2010;28:6627–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rudraraju R, Ramsay AJ. Single-shot immunization with recombinant adenovirus encoding vaccinia virus glycoprotein A27L is protective against a virulent respiratory poxvirus infection. Vaccine. 2010;28:4997–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Clark KM, Johnson JB, Kock ND, Mizel SB, Parks GD. Parainfluenza virus 5-based vaccine vectors expressing vaccinia virus (VACV) antigens provide long-term protection in mice from lethal intranasal VACV challenge. Virology. 2011;419:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xiao Y, Zeng Y, Alexander E, Mehta S, Joshi SB, Buchman GW, et al. Adsorption of recombinant poxvirus L1-protein to aluminum hydroxide/CpG vaccine adjuvants enhances immune responses and protection of mice from vaccinia virus challenge. Vaccine. 2013;31:319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Paran N, Lustig S, Zvi A, Erez N, Israely T, Melamed S, et al. Active vaccination with vaccinia virus A33 protects mice against lethal vaccinia and ectromelia viruses but not against cowpoxvirus; elucidation of the specific adaptive immune response. Virol J. 2013;10:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Reeman S, Gates AJ, Pulford DJ, Krieg A, Ulaeto DO. Protection of Mice from Lethal Vaccinia Virus Infection by Vaccinia Virus Protein Subunits with a CpG Adjuvant. Viruses. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fogg CN, Americo JL, Earl PL, Resch W, Aldaz-Carroll L, Eisenberg RJ, et al. Disparity between levels of in vitro neutralization of vaccinia virus by antibody to the A27 protein and protection of mice against intranasal challenge. J Virol. 2008;82:8022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lal H, Cunningham AL, Godeaux O, Chlibek R, Diez-Domingo J, Hwang SJ, et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med. 2015;372:2087–96. [DOI] [PubMed] [Google Scholar]
- [34].Cunningham AL, Lal H, Kovac M, Chlibek R, Hwang SJ, Diez-Domingo J, et al. Efficacy of the Herpes Zoster Subunit Vaccine in Adults 70 Years of Age or Older. N Engl J Med. 2016;375:1019–32. [DOI] [PubMed] [Google Scholar]
- [35].Centers for Disease C, Prevention. Update: multistate outbreak of monkeypox--Illinois, Indiana, Kansas, Missouri, Ohio, and Wisconsin, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:642–6. [PubMed] [Google Scholar]
- [36].Reed KD, Melski JW, Graham MB, Regnery RL, Sotir MJ, Wegner MV, et al. The detection of monkeypox in humans in the Western Hemisphere. N Engl J Med. 2004;350:342–50. [DOI] [PubMed] [Google Scholar]
- [37].Vaughan A, Aarons E, Astbury J, Balasegaram S, Beadsworth M, Beck CR, et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Euro Surveill. 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ng OT, Lee V, Marimuthu K, Vasoo S, Chan G, Lin RTP, et al. A case of imported Monkeypox in Singapore. Lancet Infect Dis. 2019;19:1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hem SL, HogenEsch H, Middaugh CR, Volkin DB. Preformulation studies--The next advance in aluminum adjuvant-containing vaccines. Vaccine. 2010;28:4868–70. [DOI] [PubMed] [Google Scholar]
- [40].HogenEsch H, O’Hagan DT, Fox CB. Optimizing the utilization of aluminum adjuvants in vaccines: you might just get what you want. NPJ Vaccines. 2018;3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shi S, Zhu H, Xia X, Liang Z, Ma X, Sun B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine. 2019;37:3167–78. [DOI] [PubMed] [Google Scholar]
- [42].Bisht H, Weisberg AS, Moss B. Vaccinia virus l1 protein is required for cell entry and membrane fusion. J Virol. 2008;82:8687–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roper RL, Wolffe EJ, Weisberg A, Moss B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J Virol. 1998;72:4192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Doceul V, Hollinshead M, van der Linden L, Smith GL. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science. 2010;327:873–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Benhnia MR, Maybeno M, Blum D, Aguilar-Sino R, Matho M, Meng X, et al. Unusual features of vaccinia virus extracellular virion form neutralization resistance revealed in human antibody responses to the smallpox vaccine. J Virol. 2013;87:1569–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Doceul V, Hollinshead M, Breiman A, Laval K, Smith GL. Protein B5 is required on extracellular enveloped vaccinia virus for repulsion of superinfecting virions. J Gen Virol. 2012;93:1876–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bell E, Shamim M, Whitbeck JC, Sfyroera G, Lambris JD, Isaacs SN. Antibodies against the extracellular enveloped virus B5R protein are mainly responsible for the EEV neutralizing capacity of vaccinia immune globulin. Virology. 2004;325:425–31. [DOI] [PubMed] [Google Scholar]
- [48].Putz MM, Midgley CM, Law M, Smith GL. Quantification of antibody responses against multiple antigens of the two infectious forms of Vaccinia virus provides a benchmark for smallpox vaccination. Nat Med. 2006;12:1310–5. [DOI] [PubMed] [Google Scholar]