

Abstract
Therapy-related myeloid neoplasms (t-MNs) following treatment with alkylating agents are characterized by a del(5q), complex karyotypes, alterations of TP53, and a dismal prognosis. To decipher the molecular pathway(s) leading to the pathogenesis of del(5q) t-MN and the effect(s) of cytotoxic therapy on the marrow microenvironment, we developed a mouse model with loss of two key del(5q) genes, EGR1 and APC, in hematopoietic cells. We used the well-characterized drug, N-ethyl-N-nitrosurea (ENU) to demonstrate that alkylating agent exposure of stromal cells in the microenvironment increases the incidence of myeloid disease. In addition, loss of Trp53 with Egr1 and Apc was required to drive the development of a transplantable leukemia, and accompanied by the acquisition of somatic mutations in DNA damage response genes. ENU treatment of mesenchymal stromal cells induced cellular senescence, and led to the acquisition of a senescence-associated secretory phenotype, which may be a critical microenvironmental alteration in the pathogenesis of myeloid neoplasms.
Keywords: therapy-related myeloid neoplasms, del(5q), TP53, mesenchymal stromal cells, senescence
Introduction
Therapy-related myeloid neoplasms (t-MNs) comprise therapy-related acute myeloid leukemia (t-AML) and myelodysplastic syndrome (t-MDS), and are a late complication of cytotoxic therapy — chemotherapy and/or radiation therapy — used in the treatment of both malignant and non-malignant diseases (1,2). The genetic profile of t-MN is markedly skewed towards high-risk cytogenetic and molecular abnormalities, and complex karyotypes with deletions of the long arm of chromosome 5, del(5q), and TP53 mutation/loss are profoundly over-represented in t-MN as compared with de novo counterparts (3,4). A proximal minimally deleted region (MDR) in 5q31.2 (containing EGR1) was previously identified in t-MN, de novo AML and high-risk MDS, and a distal MDR in 5q33.1(containing mir145, RPS14 and CSNK1A1) was identified in myelodysplastic syndrome with an isolated del(5q) (previously referred to as the 5q- syndrome) (1,5). However, the deletion of 5q in all but rare patients encompasses both MDRs, spanning 5q14–5q33, resulting in loss of one allele for hundreds of genes. Although challenging to identify involved genes, we previously chose to examine two well-characterized del(5q) tumor suppressor genes, early growth response 1 (EGR1) (5q31.2) and adenomatous polyposis coli (APC) (5q22.2), and showed that they cooperate with TP53 (p53) loss to induce acute myeloid leukemia in mice (6). The low frequency of AML and long latency observed in this model, however, suggests that additional stochastic events, and/or mutations arising from environmental exposures may contribute to the etiology of disease.
Healthy individuals harbor clonal, somatic, gene mutations in white blood cells, and the frequency increases with age, leading to the expansion of some clones, a phenomenon known as “clonal hematopoiesis of indeterminant potential”, or CHIP (7,8). Recent studies have revealed that CHIP increases the risk for t-MN (9,10). The landmark finding of pre-existing TP53 mutations in hematopoietic cells originating prior to cytotoxic treatment of a primary malignant disease, and in the t-MNs that subsequently evolved, led to the proposal that chemotherapy and/or radiation may preferentially impair normal hematopoietic stem and progenitor cells (HSPCs), while providing a selective advantage to clones with pre-existing mutations (11). Follow-up studies showed that cytotoxic therapy results in the expansion of clones carrying mutations in DNA damage response genes, including TP53, PPM1D, ATM, BRCC3, SRCAP, and RAD21 (12); however, mutations in DNA repair genes, other than TP53 and PPM1D (11–13), are not present in leukemia samples isolated from t-MN patients. Interestingly, the DNA double-strand break response is abnormal in myeloblasts from t-MNs, even though somatic mutations of DNA repair genes do not appear to be the dominant mechanism responsible for this phenotype (14).
In addition to alterations in hematopoietic cells, there is compelling data suggesting that alterations to the bone marrow microenvironment also play a role in the pathogenesis of myeloid neoplasms (15–18). These alterations may include establishment of an inflammatory microenvironment (19) and/or age-associated changes of mesenchymal stromal cells (MSCs) (20). MSCs isolated from MDS and AML patients display several characteristic features of senescence, including TP53 pathway activation (21,22), increased senescence-associated β-galactosidase (SA-βgal) expression (22–24) and upregulation of senescence-associated secretory phenotype (SASP) factors, such as IL6 (25,26). Moreover, AMLs can induce a senescent phenotype in BM stromal cells, and targeting these senescent cells improves the survival of mice with leukemia (25), illustrating the importance of a senescent microenvironment in the pathophysiology of leukemia.
The complete compendium of genetic alterations and selective pressures leading to t-MN with a del(5q) remain unknown, especially with regard to the effects of cytotoxic therapy on the microenvironment. In the present study, we use a multi-faceted approach to model del(5q) t-MN in mice, and to define genetic alterations in HSPCs by whole exome and RNA sequencing, while simultaneously probing hematopoietic stem cell (HSC)-intrinsic and cell-extrinsic effects of alkylating-agent therapy. Our results pinpoint chemotherapy-induced alterations to bone marrow stromal cells, and genetic alterations to DNA repair genes in HSPCs as two factors critical for the pathogenesis of del(5q) t-MNs with aberrant TP53.
Results
Alkylating agent exposure increases the incidence of myeloid disease
A striking 40% of t-MN patients have a del(5q) (2), the majority (>95%) of which have haploinsufficient loss of both EGR1 (27) and APC (28). Moreover, ~ 75% of del(5q)t-MNs have loss and/or mutation of the TP53 gene (29). To model this, we previously showed that cell intrinsic loss of both Egr1 and Apc, or individual loss of either Egr1 or Apc, together with Trp53 knock-down, was not sufficient to promote AML; however, concordant loss of the three genes, Egr1, Apc and Trp53, modeled the development of del(5q) AML (6) (supplemental Fig. 1). In clinical settings, however, t-MN patients receiving chemotherapy have exposure of the entire body, including the bone marrow microenvironment and HSPCs. The relative contribution of drug treatment on the niche and/or HSPCs, and the interplay of genetics, the microenvironment and chemotherapy, in subsequent t-MN development is unclear.
To isolate these variables, we employed our del(5q) mouse model, with Egr1 and Apc haploinsufficiency and loss of Trp53, which spontaneously develops leukemia at a low frequency (<20%) (6). However, in this study we treated HSPCs and the niche separately with ENU, a proto-typical alkylating agent whose effects have been well-characterized in mice. We first assessed the outcome of ENU exposure of both the niche and hematopoietic cells. Egr1+/−, Apcdel/+ donor and WT recipient mice were treated with ENU. Egr1+/−, Apcdel/+ bone marrow cells were isolated two weeks after ENU treatment, transduced with Trp53 shRNA to knockdown expression of Trp53 (Trp53 KD), and transplanted into ENU-treated, lethally-irradiated WT recipient mice (Fig. 1A).
Fig. 1. Alkylating agent therapy increases the incidence of myeloid neoplasms.
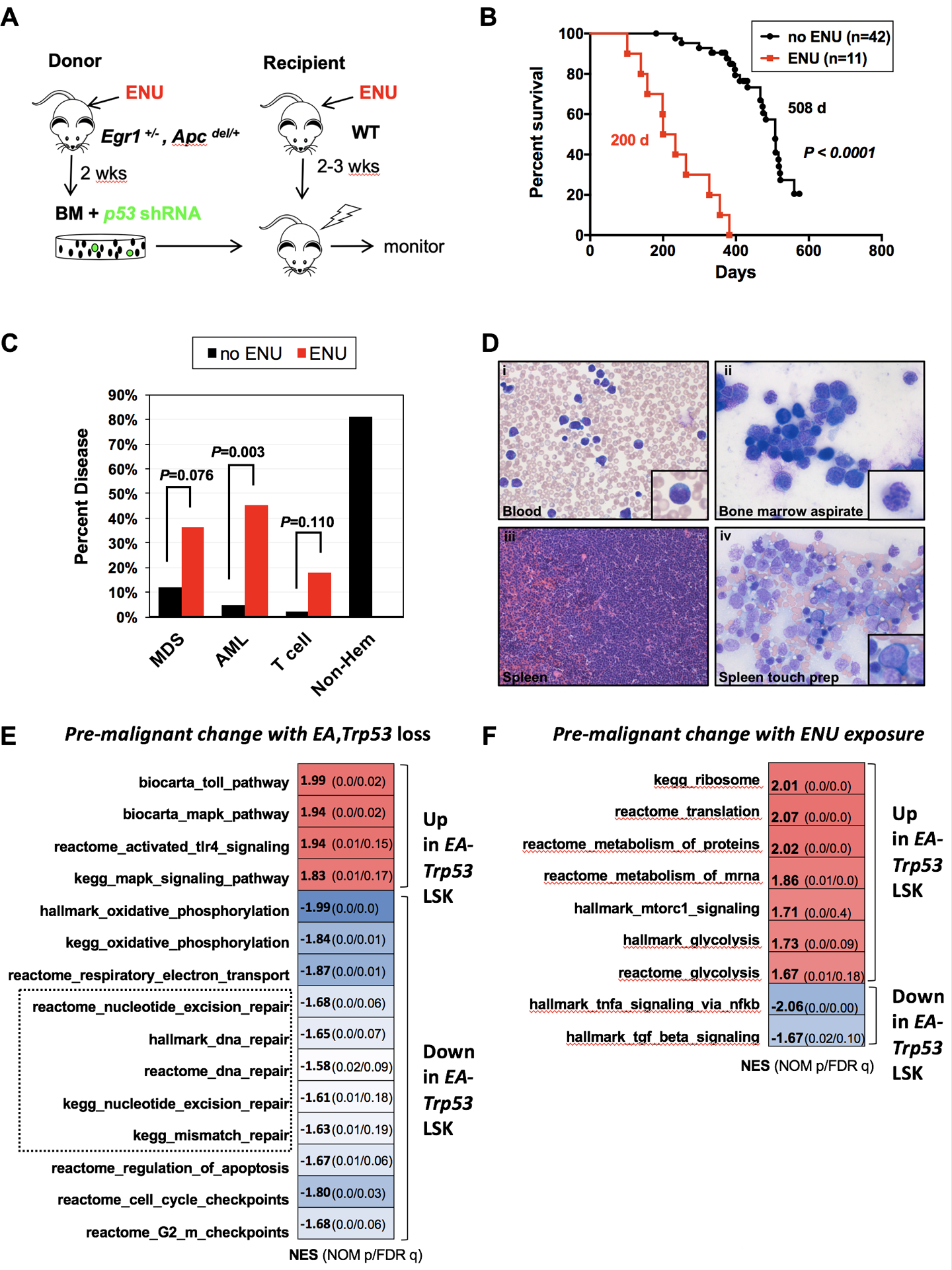
(A) Schematic of ENU-treatment and transplantation scheme used to develop a mouse model of t-MN. Egr1+/−, Apcdel/+ bone marrow cells were transduced with Trp53 shRNA, and transplanted into lethally-irradiated WT recipient mice. For the ENU cohort, donor mice were treated once with 100 mg/kg ENU 2 weeks before bone marrow harvest; recipients were treated once with 100 mg/kg ENU, 2–3 weeks before lethal irradiation and transplantation. (B) Kaplan-Meier survival curves of untreated and ENU-treated mice. Percentage survival (time to sacrifice) is plotted vs. time in days. In the absence of ENU treatment, the mice survive significantly longer (508 d vs. 200 d, P <0.001), with a small percentage developing MDS/AML, but the majority succumb to advanced age and non-hematological effects. (C) Histological classification of diseases that arose in the no-ENU and ENU-treated mice. There is a significantly increased frequency of AML in the ENU-treated group. Most of the no-ENU mice died due to age-related issues, rather than hematopoietic malignancies. Disease frequency was compared using Fisher exact test. (D) Images of the myeloid disease in ENU-treated mice were obtained using an Olympus BX41 microscope (Melville, NY) and a 50x/0.9 (oil) or 40x/0.9 objective, and processed with Adobe Photoshop (San Jose, CA). Peripheral blood smears, bone marrow smears, and spleen touch preparations were stained with Wright–Giemsa (500x magnification), and spleen sections were stained with H&E (200x magnification). Examples of a myeloblast (inset-i), dysplastic granulocyte (inset-ii), sheets of infiltrating blasts (iii), and a dysplastic erythroid precursor (inset-iv) are shown. (E, F) LSK+ (Lin-, Sca1+, Kit+) cells were sorted from 3 different mouse cohorts: recipients of WT, luc shRNA+ bone marrow (controls) (n=3), recipients of Egr1+/−, Apcdel/+, Trp53 shRNA+ cells (EA-Trp53), either untreated (n=3) or treated with ENU (n=3), ~ 70–90 d after transplant, prior to the onset of overt leukemia. (E). GSEA of WT control (n=3) compared to EA-Trp53 LSK+ samples (includes both the no-ENU and ENU-treated groups (n=6)) to identify pre-malignant changes as a consequence of Egr1, Apc, and Trp53 loss. (F) GSEA of EA-Trp53 LSK+ samples from the no-ENU (n=3) versus ENU-treated (n=3) group was used to identify genetic consequences of in vivo ENU exposure. Biological pathways/processes that were significantly enriched (FDR<20%, nominal p value <0.05) in two or more Molecular Signatures Database (MSigDB) gene sets are shown. Heat map of the normalized enrichment scores (NES) shows that DDR pathways (DNA repair, apoptosis, checkpoints) are downregulated due to loss of Egr1, Apc, and Trp53, with or without ENU treatment. (E), and energy production pathways, such as mTORC1, protein translation and glycolysis, are upregulated as a consequence of ENU exposure (F).
Compared to donor and recipient mice that did not receive ENU (no ENU), alkylating agent exposure significantly accelerated the onset of disease with a median survival of 200 d versus 508 d, P<0.0001 (Fig. 1B). In accordance with the Bethesda classification (30), the disease was classified as a myeloid leukemia when at least 20% blasts were observed in hematopoietic tissue, and as a myelodysplastic syndrome when anemia and morphologic dyspoiesis was observed. A significantly larger percentage of mice developed MDS (36% vs. 12%, P =0.076) or AML (45% vs. 5%, P = 0.003) in the ENU-treated group as compared to untreated mice (Fig. 1C). Eighty percent of non-ENU treated mice did not develop hematological disease (non-Hem) and were sacrificed due to age and poor health. The myeloid malignancies that developed in ENU-treated mice resembled human disease ranging from MDS - refractory anemia with excess blasts-1 or −2 (RAEB-1 or −2) to AML with myelodysplasia-related changes (Fig. 1D, supplemental Fig. 2A). Mice with myeloid neoplasms presented with splenomegaly and an effacement of the normal splenic architecture, as well as trilineage dysplasia similar to that observed in t-MN patients with a del(5q) (supplemental Figs. 2B, 3). Disease in mice with blast counts >20%, and classified as AML, was transplantable to secondary recipients and was, thus, no longer dependent on an ENU-exposed niche (supplemental Table 1).
To decipher the molecular pathways that are altered in the pre-malignant state, we sorted Egr1+/−, Apcdel/+, GFP+, Trp53-deficient lineage−, Sca-1+, Kit+ progenitors (EA-Trp53 LSKs) from both the no-ENU (n=3) and ENU-treated (n=3) groups prior to the development of overt leukemia (~80 d post-transplant) and performed RNA-seq. As a control, three independent LSK+ samples were obtained from WT recipients of WT bone marrow transduced with a non-specific (luciferase) shRNA. To decipher the effects of EA-Trp53 loss, we compared WT control (n=3) to EA-Trp53 LSK+ samples (includes both the no-ENU and ENU-treated groups (n=6)) (Fig. 1E). Gene set enrichment analysis (31) (GSEA) of the top enriched curated pathways revealed that cell intrinsic loss of Egr1, Apc, and Trp53 downregulates oxidative phosphorylation, DNA repair, apoptosis, and cell cycle checkpoints. Similar results were observed when we compared only the three ENU-exposed EA-Trp53 LSK+ samples to WT controls (supplemental Fig. 4).
To elucidate the effects of ENU, we compared EA-Trp53 cells with and without ENU-exposure (Fig. 1F). ENU exposure increased the expression of genes involved in energy production, such as mTOR signaling and protein translation (Fig. 1F); however, we cannot rule out that this simply reflects cells that are at a more advanced stage along the continuum of malignant transformation with higher metabolic activity, rather than a direct effect of ENU exposure. Interestingly, similar changes in gene expression were observed in patients who subsequently developed t-MN (32). Together, these results are consistent with a role for loss of genome-defense mechanisms in the development of myeloid neoplasms.
Incidence of AML increases when Egr1, Apc, and Trp53-deficient HSPCs as well as the bone marrow microenvironment are exposed to cytotoxic therapy.
We next separately tested the effects of alkylating agent therapy on HSPCs or the bone marrow microenvironment. To test the effect on HSPCs, we treated only bone marrow donors with ENU (2 wks prior to bone marrow isolation for Trp53 knockdown, Fig. 2A). To test the impact on the niche, we treated transplant recipients only (2–3 wks prior to transplant).
Fig. 2. Exposure of the bone marrow microenvironment to ENU plays a critical role in the development of MDS and AML.
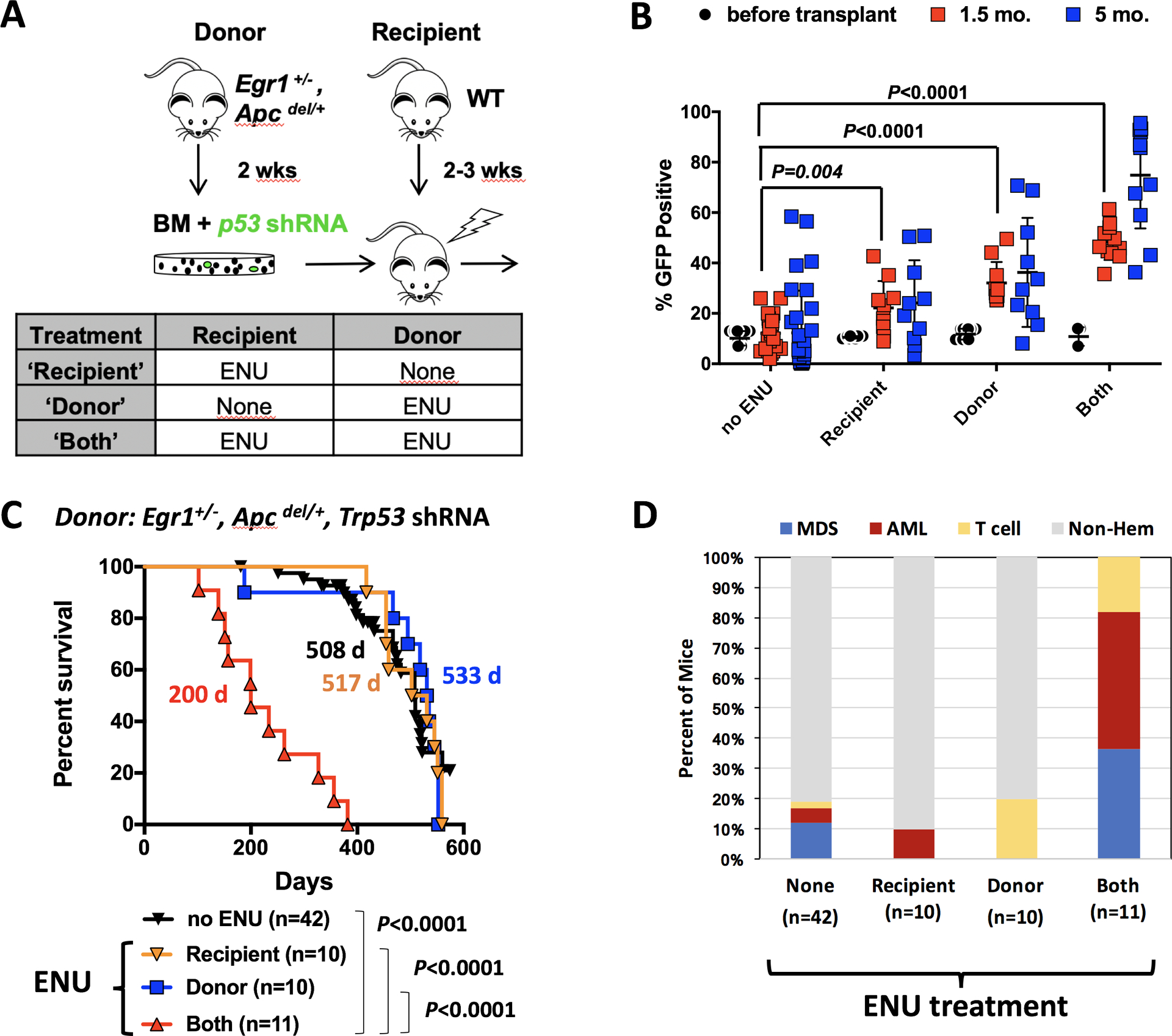
(A) Schematic of the transplantation schemes used to elucidate the effects of alkylating agents on HSPCs, with Trp53 knockdown (donors), and bone marrow microenvironment (WT-recipients). Donor mice received one injection of ENU (100 mg/kg) two weeks before bone marrow harvest. Recipient mice received one injection of ENU (100 mg/kg) 3 weeks before lethal irradiation and transplantion. (B) Percentage of GFP+ cells in the bone marrow at the time of transplantation, and in the blood of mice at 1.5 and 5 months post-transplantation. There was a significantly greater percentage of GFP+ cells in the blood of ENU-treated mice at 1.5 month and 5 months. The P-values from a two-tailed Student t test at 1.5 months are shown. At 5 months, only ‘donor’ and ‘both’ conditions showed a significant expansion of GFP+ cells (P<0.0001). ENU exposure of both donor and recipient creates a more optimal environment for the expansion of Trp53 shRNA, GFP+ cells. (C) Kaplan-Meier survival curves of WT recipients transplanted with Egr1+/−, Apcdel/+ bone marrow cells transduced with Trp53 shRNA. When both donor and recipient mice are exposed to ENU, survival time is significantly decreased (200 d). There was no statistical difference in survival between ENU-donor vs. no ENU (P=0.831) or ENU-recipient vs. no ENU (P=0.737), or ENU-donor vs. ENU-recipient cohorts (P=0.910). (D) Histologic classification of diseases arising in the mice.
Approximately 10% of Egr1+/−, Apcdel/+ bone marrow cells were transduced with the GFP-positive Trp53 shRNA (Trp53 shRNA-GFP+) prior to transplantation (Fig. 2B, % GFP+, black circles). This relatively low frequency allowed us to assess the effect of ENU on the competitive expansion of Trp53-deficient, Egr1+/−, Apcdel/+ bone marrow cells. ENU treatment of recipient or donor mice led to a modest expansion of Trp53 shRNA-GFP+ cells 1.5–5 months after transplant. However, the greatest expansion of Trp53 shRNA-GFP+ cells was observed when both donor and recipients were treated with ENU, suggesting that ENU exposure of both HSPCs and the microenvironment contributes to clonal expansion.
Even though ENU treatment of donor or recipient mice increased the expansion of Trp53 shRNA-GFP+ cells, it did not accelerate disease development (Fig. 2C; 517d or 533 d vs. 508d without ENU treatment), nor did it increase the frequency of myeloid neoplasms beyond what is observed without ENU treatment (Fig. 2D; compare “none” to “donor” or “recipient”). Only 1/10 recipient-treated mice developed AML, and none of the donor-treated mice developed a myeloid neoplasm (two died early from T lymphomas; the remainder were sacrificed (>500 d) due to age-related causes). In contrast, 82% of mice, in which both donor and recipient mice were exposed to ENU, developed MDS or AML. Thus, whereas exposure of HSPCs or the microenvironment alone to ENU promotes expansion of Trp53-shRNA+ cells, t-MN development is driven by the synergistic effects of chemotherapy exposure of pre-malignant hematopoietic cells, together with deleterious effects of cytotoxic therapy on the supporting microenvironment. Historically, it has been assumed that the effect of prior therapy in patients who develop t-MN was primarily on the HSPCs. To our knowledge, this is the first mouse model of del(5q) t-MN to directly show that chemotherapy-induced alterations to the BM microenvironment contribute to disease development.
Alkylating agent exposure induces senescence of mesenchymal stromal cells
In solid tumors, there is evidence that senescence of non-tumor cells promotes tumor growth and metastasis (33). Furthermore, cytotoxic treatment induces an acute inflammatory response in the BM microenvironment. We hypothesized that ENU induces a sustained effect via senescence of MSCs within the microenvironment and, thereby, alters the function of HSPCs. To evaluate this hypothesis, MSCs were treated for 24 hrs with increasing doses of ENU, and senescence was assessed seven days later. MSCs treated with ENU showed an increase in senescence-associated β-galactosidase (SA-β-gal) activity and a decline in DNA synthesis as determined by EdU incorporation (Fig. 3A, B). Senescence was further confirmed by elevated levels of mRNA encoding p21 (Cdkn1a), p16INK4a (Cdkn2a), and IL6 (Fig. 3C).
Fig. 3. ENU induces senescence of mesenchymal stromal cells.
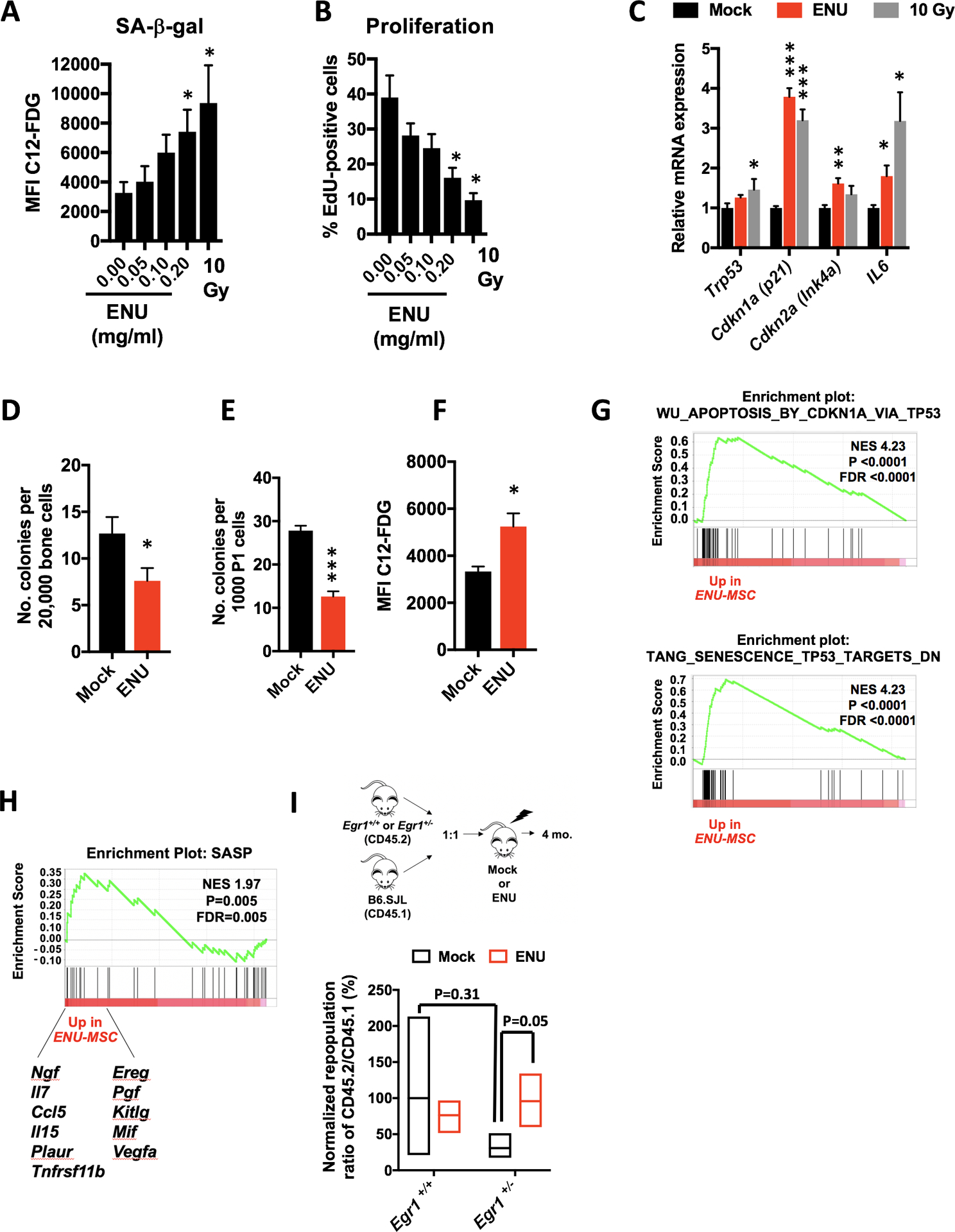
(A, B) MSCs were treated with varying doses of ENU for 24 hours in vitro. Seven days later, cells were either stained for SA-βgal or incubated 24 hrs with EdU, then fixed and stained. Five and three independent experiments were done for SA-βgal and EdU, respectively. A senescence-inducing dose of 10Gy was used as a positive control. (C) Quantitative PCR analysis of RNA isolated from control (10% ethanol) and ENU-treated (0.2mg/ml) MSCs at day 7. Values are presented as a ratio of target mRNA to 18S rRNA. N=3 independent experiments, each done in triplicate. (D-H) Mice were mock-treated (10% ethanol) or ENU-treated (100 mg/kg). One month later, mice were sacrificed and MSCs were isolated. (D, E) The colony-forming unit-fibroblast (CFU-F) assay was used to estimate the proliferative and clonogenic potential of MSCs. MSCs were isolated from collagenase-treated compact bones and either directly plated (D) or passaged once (E) before plating. N= 5 and 9 independent experiments for D and E, respectively. (F-H) Early passage MSCs (P1–2) were stained for SA-βgal or harvested for RNA. N=3 independent experiments. Significant differences between control (no ENU/mock) and experimental were calculated by two-tailed Student t test for panels A-F. * P<0.05, ** P<0.001, *** P<0.0001. (G, H) GSEA of MSCs from ENU-treated versus mock-treated mice shows an enrichment of the p53 pathway and specific SASP factors, shown below enrichment plot. (I) Equal numbers of Egr1+/+(WT) or Egr1+/− (CD45.2) and wild-type B6.SJL competitor cells (CD45.1) were transplanted into lethally-irradiated recipients that were mock- or ENU-treated, 3 weeks before transplantation. Ratios of CD45.2 (Egr1+/+ or Egr1+/−) vs. competitor (CD45.1) for short-term hematopoietic stem cells (ST-HSC; Lin- Sca1+ Kit+ CD48- CD150-) were calculated 4 months post-transplant. A repopulation ratio of CD45.2 (WT-Egr1+/+) to CD45.1 in mock-treated conditions was set to 1 (expressed as 100%). Without ENU, Egr1+/− ST-HSC showed an average repopulation frequency of 31%; with ENU treatment, repopulation increased to 95% (P=0.05 by two-tailed Student t test) suggesting that an ENU-treated microenvironment impacts stem cell fitness.
We next examined MSCs isolated from mice treated with one dose of ENU, which showed a decreased number of colony-forming unit-fibroblasts (CFU-F) (Fig. 3D, E) and an increase in SA-β-gal activity (Fig. 3F). Consistent with the finding that damage-initiated senescence strongly depends on TP53 (34), GSEA analysis of RNA-seq data revealed activation of the p53 pathway in MSCs isolated from ENU-treated mice, compared to mock-treated mice (Fig. 3G). Senescent cells express and secrete a variety of extracellular modulators collectively referred to as the senescence-associated secretory phenotype (SASP) (35). Enrichment of a SASP GSEA signature was observed in MSCs isolated from ENU-treated mice (Fig. 3H), providing a potential mechanism by which they contribute to the pathogenesis of leukemia.
To assess whether an ENU-treated microenvironment affects stem cell fitness in vivo, a competitive bone marrow transplant assay was performed as outlined in Fig. 3I. We used Egr1+/− donor cells, since Egr1 has been documented to play a role in stem cell quiescence (36). The repopulation ratio of CD45.2+ (Egr1+/+) to CD45.1+ short-term-HSC (ST-HSC) cells was normalized to 100% (mock conditions). In mock conditions, Egr1+/− ST-HSCs displayed a reduced normalized repopulation ratio of 31%. Although not statistically different (P=0.31), this trend may reflect stem cell exhaustion due to Egr1’s role in maintaining stem cell quiescence. With ENU treatment (red bars) , Egr1+/− ST-HSCs showed an increased repopulation of 95% (P=0.05) suggesting that an ENU-treated microenvironment may effect Egr1+/− stem cell fitness. Thus, alterations to the microenvironment may induce senescence of MSCs and trigger SASP, thereby creating a permissive environment that impacts stem cell fitness, and contributes to the pathogenesis of myeloid neoplasms.
Loss of Trp53, concurrent with two del(5q) genes, Egr1 and Apc, models high-risk MDS/AML.
TP53 mutations are associated with complex karyotypes, high-risk MDS, transformation to AML, and poor overall survival (3,4,37). We next compared disease development in mice transplanted with Egr1+/−, Apcdel/+ bone marrow transduced with luciferase control shRNA cells (EA-Luc, black) vs. Trp53 shRNA (EA-Trp53, red) (Fig. 4A). Both the donor and the recipient mice were treated with ENU. EA-Luc mice survived significantly longer (370 d vs. 200 d, P =0.0014) (Fig. 4A), and none developed AML (Fig. 4B). Most (93%, 13/14) developed MDS with marked proliferation of dysplastic erythroblasts, and some mild granulocytic and megakaryocytic dysplasia (supplemental Fig. 5). In contrast, the addition of Trp53 knockdown led to leukemia development; 45% (5/11) of EA-Trp53 mice (red) developed AML vs. none (0/14) of the EA-Luc mice (p =0.009) (Fig. 4B). Together, our data show that cell intrinsic loss of Egr1 and Apc is not sufficient to promote the development of overt leukemia, even when HSPCs and the microenvironment are exposed to ENU. However, concordant loss of Trp53 leads to the development of AML in almost half of the mice when both the HSPCs and microenvironment are exposed to ENU. The severity of disease appears to worsen with the additional loss of Apc, since E-Trp53 mice did not succumb to disease as quickly as did EA-Trp53 mice (369 d vs. 200 d, P =0.0177) (supplemental Figure 6A), and showed a trend towards more MDS and less AML compared to EA-Trp53 mice (supplemental Figure 6B). Thus, the EA-Trp53 mouse model mimics del(5q) MDS patients, where TP53 mutation and/or loss confers the highest risk for disease progression and death in patients.
Fig. 4. Following exposure to an alkylating agent, haploinsufficiency of both Egr1 and Apc promotes development of MDS; loss of these genes together with Trp53 promotes AML development.
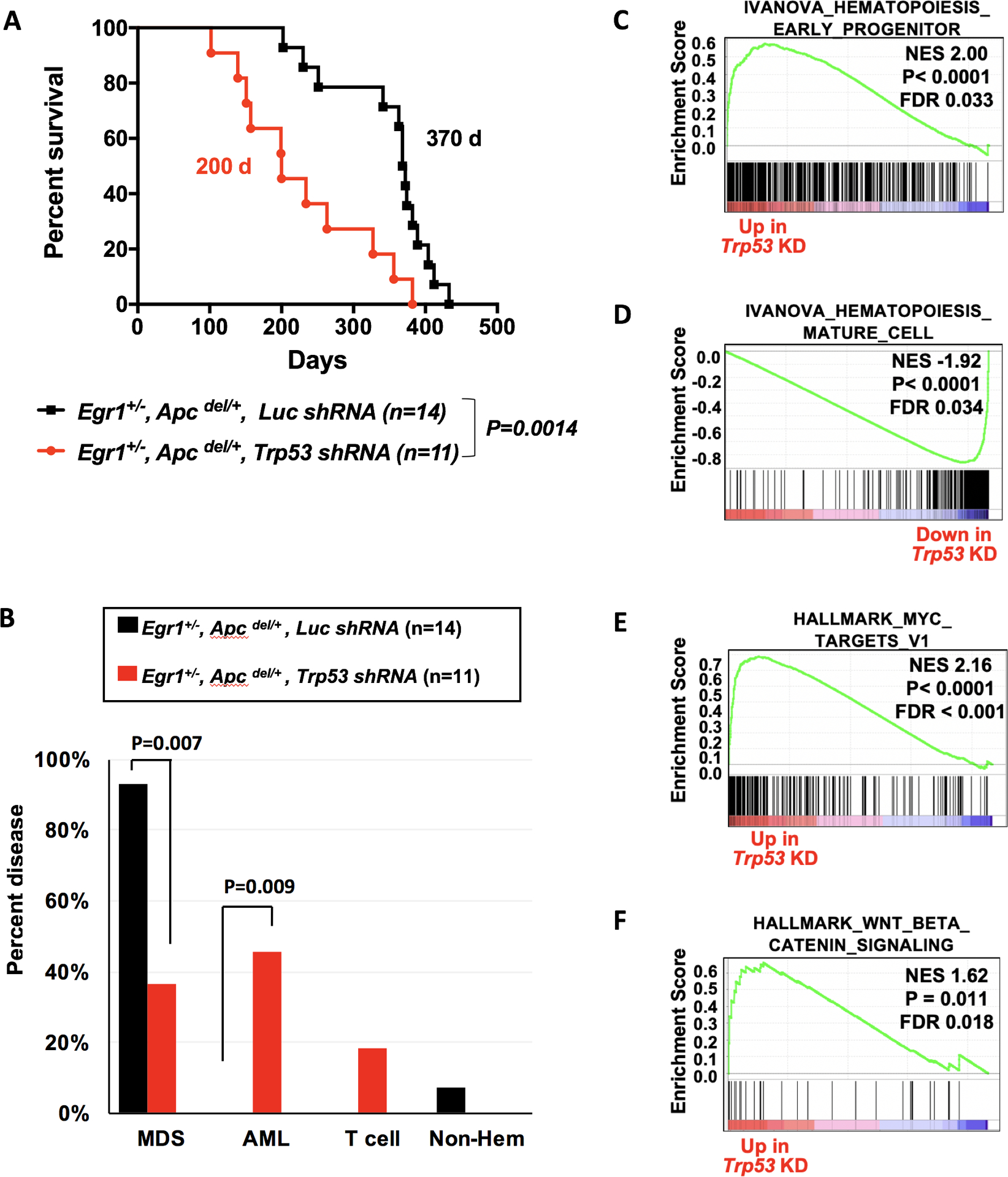
(A) Kaplan-Meier survival curves of WT recipients transplanted with Egr1+/−, Apcdel/+ bone marrow cells transduced with luc shRNA (EA-luc: black) or Trp53 shRNA (EA-Trp53: red). Both donor and recipient mice were treated with ENU. Disease development is significantly faster in EA-Trp53 mice compared to EA-luc mice (200d vs. 370 d, P=0.0014). (B) Histological classification of diseases shows that most EA-luc mice developed MDS, and none developed AML. (C-F) Mouse genes were collapsed to human gene names; differentially expressed genes in EA-Trp53 AML samples (n=6) vs. EA-luc MDS samples (n=6) were analyzed using the GSEA software. Features, such as a block in myeloid differentiation and upregulation of MYC target genes and WNT/β-catenin signaling recapitulate human t-MN.
Several lines of evidence, suggest that the EA-Trp53 AMLs model the progression to high-risk disease. Gene expression profiling of EA-Trp53 AML vs. EA-Luc MDS samples revealed a positive enrichment of the hematopoiesis early progenitor signature, and a negative enrichment of the hematopoiesis mature cell and the myeloid cell maturation signatures in AML samples (Fig. 4C, D), as well as an elevated (>20%) myeloblast count, modeling the transition from MDS to AML. A strong upregulation of MYC target genes and WNT/β-catenin signaling in EA-Trp53 AML samples (Fig. 4 E, F) modeled our previous expression profiling data of del(5q) t-MN patients, which showed a distinctive upregulation of the MYC oncogene, and deregulated WNT signaling (38,39). In EA-Trp53 samples, compared to EA-Luc MDS, there were a greater number of mice with a clonal chromosomal aberration (75% (6/8) vs. 28% (2/7), P=0.13), and on average a greater number of aberrations (7 vs. 1.6, P=0.22) (supplemental Table 2); however, the sample size was small and these differences did not reach statistical significance. Overall, our mouse model provides a tractable system to understand the molecular events defining del(5q) t-MNs with TP53 deficiency (through 17p loss or TP53 mutation) and, potentially, to test novel therapies.
Although AML samples were transplantable to secondary recipients, MDS samples (with or without Trp53 shRNA) were not (supplemental Table 1). The inability to transplant MDS is similar to the observation that transplantation of CD34+ cells from patients with MDS does not result in engraftment or propagation in murine xenograft models, unless MDS-derived MSCs, but not MSCs from age-matched healthy controls, are co-injected (16), supporting a contribution of the BM microenvironment to the pathogenesis of MDS. In our EA-luc model, we noted that development of MDS was particularly sensitive to ENU’s effect on the microenvironment (Fig. 5A). ENU-treatment of donor Egr1, Apcdel/+ mice-exposing the HSPCs to ENU- was not sufficient to promote the development of MDS (Fig. 5B). However, when WT recipient mice were treated with ENU, EA-luc mice developed MDS at the same penetrance and latency as when both donor and recipient were treated with ENU (80% MDS, vs. 93% MDS, P=0.55; 390 d vs. 374 d median MDS survival, P= 0.271; ENU-recipient vs. ENU-both, respectively) (Fig. 5B, C). Since an expansion of erythroblasts in conjunction with morphologic dyspoiesis was observed, this data is consistent with a neoplastic rather than reactive process. These data suggests that ENU treatment of the stromal microenvironment influences disease progression of Egr1, Apcdel/+ HSPCs resulting in MDS. Thus, chemotherapy may, in some instances, be a cell-extrinsic factor driving emergence of MDS clones.
Fig. 5. ENU exposure of the BM microenvironment is a major force driving MDS development in mice transplanted with Egr1 and Apc haploinsufficient HSPCs.
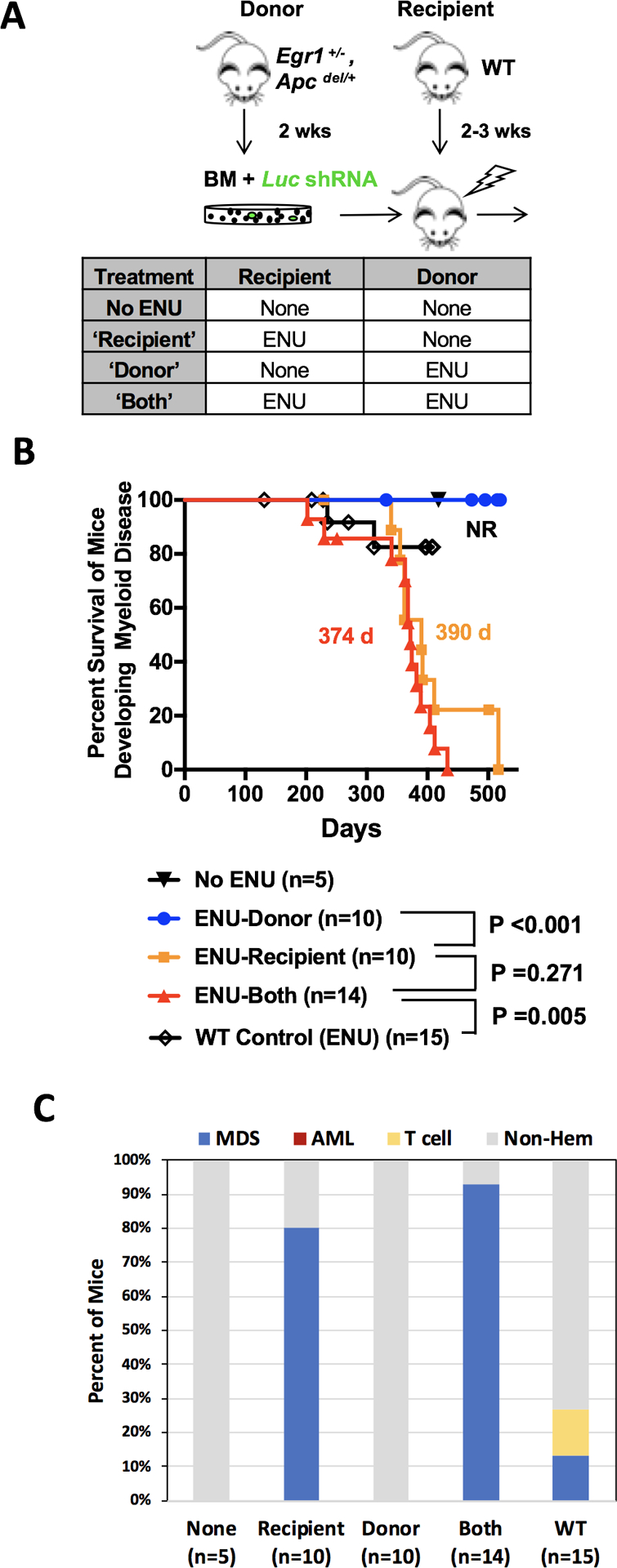
(A) Schematic of the transplantation schemes used to elucidate the effects of alkylating agents on HSPCs (donor) vs. the microenvironment (recipient) in the absence of Trp53 knockdown. WT recipients were transplanted with Egr1+/−, Apcdel/+ bone marrow cells transduced with Luc shRNA (control) under 4 different ENU conditions. Donor mice received one injection of ENU (100 mg/kg) 2 weeks before bone marrow harvest. Recipient mice received one injection of ENU (100 mg/kg) 3 weeks before lethal irradiation and transplantation. (B) Kaplan Meier survival curves of mice developing myeloid disease. Survival time is similar when when only recipient mice are exposed (390 d) versus when both donor and recipient mice are exposed to ENU (374 d) (P= 0.271). As a control, WT bone marrow was transplanted into WT recipients, and then treated with ENU; median survival was not reached as only 2/15 (13%) of mice developed MDS. (C) Histologic classification of diseases arising in the mice. The percent of mice that developed MDS in ENU-recipient (80%) and ENU-both (93%) conditions was similar (P=0.55 by Fisher exact test).
DNA damage response genes are somatically mutated in AMLs modeling del(5q) t-MN.
To identify whether loss of Egr1, Apc, and Trp53 was sufficient to drive AML transformation, or whether additional mutations were required, we performed whole exome sequencing (WES) on seven EA-Luc MDS and eight EA-Trp53 AML samples. All seven MDS and five of eight AML samples were derived from ENU-both conditions; two AMLs were derived without ENU (1586 and 4525) and one AML (7914) after ENU treatment of the recipient only. The median number of deleterious mutations per mouse was eight (range: 0–24) for the EA-Luc MDS samples, and 12.5 (range: 1–40) for the EA-Trp53 AML samples (Fig. 6A). Indel mutations were detected only in the EA-Trp53 AML group, and ranged from 0–3 per sample. The median variant allele frequency (VAF) was similar for the EA-Luc MDS and EA, Trp53-AML groups (27%, range 20–55%; and 30%, range 20–64%, respectively). A complete list of mutated genes, with a GERP score >2 and considered to be deleterious, is found in supplemental Table 3.
Fig. 6. Enrichment of DNA repair gene mutations in myeloid neoplasms with Trp53 knockdown.
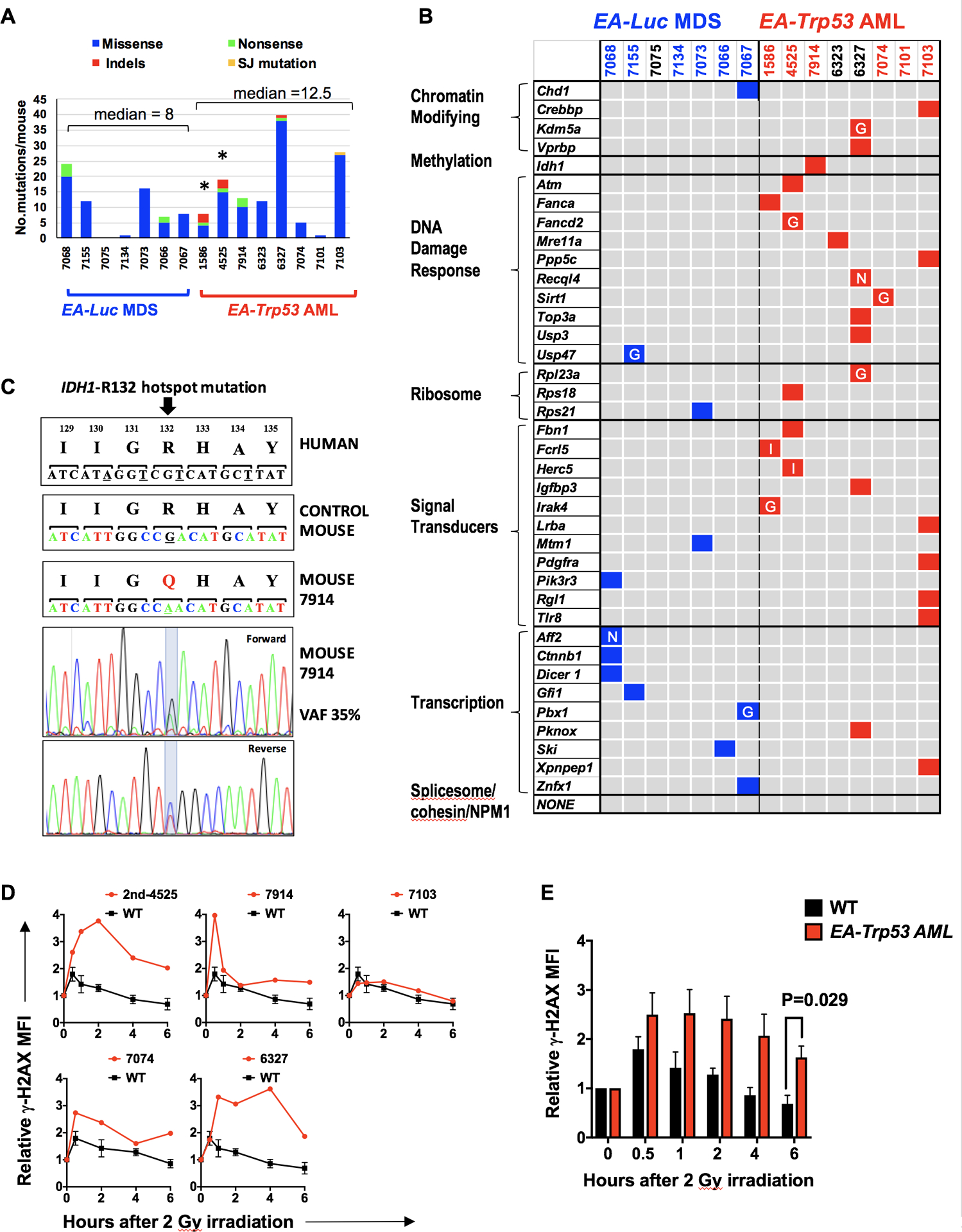
(A) Summary of non-synonymous mutations in 7 EA-Luc MDS and 8 EA, Trp53-AML samples. Both donor and recipient mice were treated with ENU with the exception of 3 mice: 1586 and 4525 (no ENU); 7914 (ENU recipient alone). All EA, Trp53 mice had a >20% blast count, except 6327 which had 18% blasts. The median number of mutations was not statistically different in the EA-Luc MDS vs. the EA-Trp53 AML group (8 vs. 12.5). Two EA, Trp53-AMLs (1586, 4525), which arose without ENU treatment (*) had a median of 13.5 mutations. (B) Since there were no genes that were mutated in multiple mice, mutations were classified into 10 gene categories that are commonly mutated (somatic and inherited) in patients with MDS or AML. Colored boxes identify genes that are mutated in each mouse. Nonsense (N) and indel (I) mutations are indicated. Most missense mutations were considered to be pathogenic by at least two of the following algorithms: GERP, SIFT and PolyPhenv2 (supplemental Table 4). Missense mutations considered pathogenic by GERP score alone are indicated by the letter “G”. With the exception of 7101, all EA, Trp53-AMLs harbor a mutation in a gene within the gene ontology term ‘DNA repair’. Bone marrow samples from mice identified in blue and red numbering were used for RNA-seq analysis. (C) The normal nucleotide and amino acid sequence and position (human) of the IDH1 gene are shown in the upper panels. The amino acid sequence is conserved in this region. Mouse 7914 had a mutation at the R132 position, which is a hotspot mutation in humans. The chromatograms with the mutation are shown. (D) γH2AX levels were measured in CD117 (Kit) + (4525–2nd, 7914) or CD11b+ (7074, 6327, 7103, 7098) cells at baseline, and up to 6 hours post 2Gy irradiation (4525–2nd and 7914 expressed Kit, but not CD11b). We previously showed that leukemia cells from 1586 and 4525 have an aberrant DNA damage response (6). AML that arose in a secondary transplant using 4525 donor cells (4525–2nd sample) also showed a defective DNA repair response. The mean fluorescent intensity measured using flow cytometry, was normalized to the 0 hr time point for each sample, to compare the induction and resolution kinetics among samples. Kinetics of each AML sample is shown in comparison to the average kinetics of CD11b+ wild-type cells. We could not assess the DNA damage response in 7101 (no DDR mutation), since the irradiation did not induce a γH2AX signal at any timepoint. (E) The mean γH2AX levels for WT (n=3) and EA, Trp53-AML (n=5) samples ± standard error of the mean are shown. An independent, two-tailed t-test was performed for each time point. The difference was statistically significant at the 6 hour time point.
Mutated genes were categorized into the ten functional categories of genes that are recurrently mutated in MDS and AML: genes encoding chromatin modifiers, DNA methylation-regulators, DNA repair proteins, ribosomal proteins, signal transducers, transcription factors, tumor suppressors, cohesin and splicesome components, and NPM1 (40,41) (Fig. 6B). There were two nonsense mutations (Recql4, Aff2), and two indel mutations (Fcrl5, Herc5); the remaining missense mutations were considered to be deleterious by the GERP, Sorting Tolerant from Intolerant (SIFT) and/or Polymorphism Phenotyping v2 (PolyPhen-2) algorithms (Fig. 6 B, supplemental Table 4). Interestingly, six similar missense gene mutations (Idh1, Atm, Lrba, Pdgfra, Pik3r3, and Dicer) were identified in human patients using the Clinvar database.
Within the ten categories listed above, there was a significantly greater number of genes mutated in the EA, Trp53-AML vs. EA-Luc MDS group (26 vs. 12, P=0.0026 by Fisher Exact test). Ribosomal and signaling genes were mutated in both groups, with signaling gene mutations occurring more frequently in EA-Trp53 AML samples (P=0.0089). Genes encoding transcription factors were mutated more frequently in EA-Luc MDS samples (P=0.057). The chromatin modifying (Crebbp, Kdm5a and Vprbp) and DNA methylation (Idh1) genes were mutated in EA, Trp53-AMLs. Mutations in genes encoding cohesin or splicesome components or Npm1, which are not enriched in the del(5q) subgroup of t-MN (1), were not detected.
Investigation of the mutated genes with the Molecular Signatures Database revealed that there was a significant enrichment of genes within the Gene Ontology (GO)_DNA repair (P= 1.18 × 10−8) collection and include Atm, Fanca, Fancd2, Mre11a, Ppp5c, Recql4, Sirt1, Top3a, Usp3, and Usp47 (Supplemental Table 5). Remarkably, a significantly larger number of DNA damage response (DDR) gene mutations were in the EA-Trp53 AML vs. the EA-Luc MDS group (P= 0.0003, Fisher Exact Test). Additionally, Idh1 mutations have been linked to altered DNA repair (42), and mouse 7914 had a somatic Idh1 mutation that corresponded with the IDH1 mutation involving arginine 132 (R132 “hotspot”) frequently observed in human malignancies (Fig. 6C). Mutations were confirmed by Sanger sequencing (supplemental Fig. 7). Fig. 6B illustrates that each EA-Trp53 AML had at least one mutation in a DNA repair gene (with the exception of mouse 7101). Somatic mutations in IDH1 and ATM, and germline mutations in Fanconi anemia genes have been observed in t-MN patients (29,43) (Supplemental Table 6). In AML samples from patients, curated by the catalogue of somatic mutations in cancer (COSMIC) database, there are codon-altering mutations in IDH1, ATM, FANCA, MRE11, RECQL5, USP3 and USP47; FANCD2, PPP5C, TOP3A are mutated only in lymphoid neoplasms (Supplemental Table 6). These findings support the hypothesis that mutation of the DDR genes identified in EA-Trp53 AMLs contributes to the development of myeloid disease.
To test for a defect in the DNA damage response, we measured the induction and resolution of phosphorylated histone H2AX (γH2AX), a well-established marker for DSBs, after 2Gy of irradiation, and showed that cells from 4/5 EA-Trp53 AMLs tested have an aberrant double-strand break response (as measured by resolution of γH2AX fluorescence), similar to our previously published results (6) (Fig. 6D). Cells from one leukemia mouse (7103) had a normal γH2AX response even though they had a mutation in the Ppp5c gene, which encodes a serine/threonine phosphatase involved in multiple cellular processes, including the DNA damage response. At 6 hours post irradiation, the mean fluorescence intensity of γH2AX was ~2.4 fold higher in EA-Trp53 AML samples suggesting that AML cells are unable to resolve DSBs to the same extent as WT cells (Fig. 6E). Thus, haploinsufficiency of two critical del(5q) genes, Egr1 and Apc, with concurrent Trp53 loss is sufficient to initiate disease; however, acquisition of a DDR gene mutation, and ensuing abnormal DNA repair response, appears to be critical for transformation to AML.
Discussion
In the present study, we addressed the long-standing question of the role of cytotoxic therapy in the pathogenesis of t-MN, and why prior cytotoxic therapy strongly selects for TP53 loss and/or mutation in conjunction with a del(5q). We showed that loss of Trp53 in HSPCs in conjunction with two key tumor suppressor del(5q) genes, Egr1 and Apc, was required for the development of AML; the development of AML was associated with the acquisition of deleterious mutations in DNA repair genes. Modeling the consequence of a cytotoxic-exposed microenvironment, our data suggests that chemotherapy-induced alterations to the microenvironment should be considered one of the predisposing risk factors for the development of t-MN. Although the seminal study by Wong et al. demonstrated that selection of cells with TP53 mutations/loss occurs after exposure to DNA-damaging chemotherapeutics (11), this study did not distinguish the effect of chemotherapy on HSPCs versus the microenvironment. Our in vivo model shows the unexpected finding that a chemotherapy-exposed microenvironment promotes this expansion. Moreover, we introduce the concept that senescent phenotype within the bone marrow microenvironment, induced by prior cytotoxic therapy, may contribute to the pathogenesis of t-MN.
Jacoby et al. previously demonstrated that myeloblasts from the majority of t-MN patients are characterized by deregulation of the DNA damage response, even though few somatically mutated DDR genes were identified (14). It is striking that in our model of del(5q) t-MN, leukemia development in mice selected for mutations of DDR genes, impairing the DNA damage response, similar to t-MN patients. The results of our studies suggest that TP53 loss on its own does not lead to an impaired DNA damage response in t-MN, since nearly every mouse AML with Trp53 loss had acquired a mutation of a DDR gene. Although t-MN patients do not generally exhibit somatically mutated DDR genes (1), inherited and acquired mutations, epigenetic modifications, and/or chromosomal loss of DNA repair gene(s) may all contribute to an aberrant DNA damage response in patients. The finding that the AML developing in mice selected for DDR gene mutations in a model of t-MN lends support to the idea that therapeutic approaches exploiting a dysfunctional double-strand break response should be considered for t-MN.
A new paradigm in leukemogenesis is that alterations of the microenvironment contribute to the pathogenesis of myeloid malignancies (15,17,18,44). This may be due to genetic alterations, as is the case for the Shwachman-Bodian-Diamond syndrome gene, remodeling of the niche by malignant myeloid cells (16,25), or environmental exposures such as cytotoxic therapy, as suggested by this study. We hypothesize that MSCs in the BM microenvironment are key players, since recipient mice were lethally irradiated, leading to cell death of immune cells but not the MSCs, which are typically radioresistant. Although chemotherapy is known to transiently increase pro-inflammatory cytokines (45), we waited two weeks after ENU-administration before transplanting mice. Since ENU-treatment of recipient mice appeared to “set the stage” for malignant transformation, only observed months later, we hypothesized that chemotherapy must have long-term consequences on BM stromal cells, such as establishing a senescent phenotype. In support of this, senescence of non-tumor cells, following doxorubicin treatment, was shown to play a key role in cancer relapse and spread to other tissues in a mouse model of breast cancer (33). Our study expands on this idea and suggests that cytotoxic therapy for a prior malignancy may have long-term effects on the microenvironment, possibly through the induction of senescence, creating an inflammatory environment and favorable conditions for the development of t-MN. Consistent with our hypothesis, it was recently shown that MSCs from t-MN patients had a higher senescent rate as compared to MSCs isolated from healthy controls, patients with primary MDS, and patients with two unrelated cancers, without prior cytotoxic exposure (46).
In Fig. 7, we propose that there are several pathways that lead to t-MN. Various predisposing factors may influence whether a patient develops t-MN (left panel) (1). The presence of a germline mutation in genes involved in DNA repair and/or DNA damage‐sensing pathways, such as BRCA1, BRCA2, CHEK2, TP53 and Fanconi anemia genes, has been identified in some patients with t-MN, and may lead to ineffective DNA repair following cytotoxic therapy and the acquisition of somatic mutations (43,47–49). Individuals who have CHIP, or small pre-existing, age-related resilient clones, may be at increased risk of developing t-MN following cytotoxic therapy (9–13). Finally, it is also possible that the bone marrow microenvironment is damaged prior to cytotoxic therapy, due to aging, infection, chronic inflammation, or inherited mutations (50). No recurrent somatic mutations or karyotypic abnormalities have been found in MDS-derived MSCs (51). Rather, it has been proposed that malignant stromal alterations may be of an epigenetic nature (52); however, at present it not known if alterations precede disease or are induced by malignancy, and how chemotherapy may impact these epigenetic changes.
Fig. 7. Model illustrating how multiple, converging pathways may lead to t-MN.
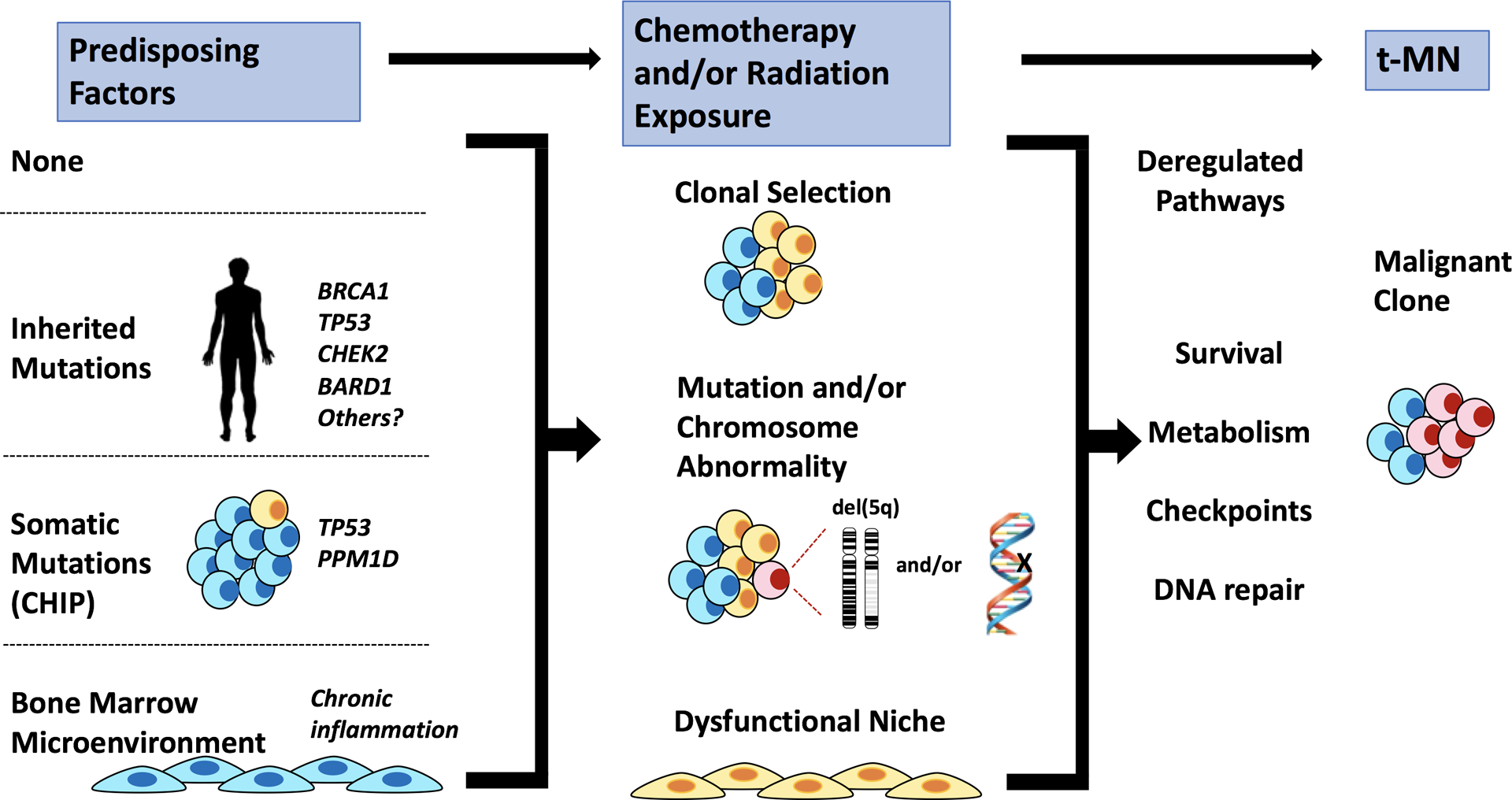
The left panel illustrates various predisposing factors that may influence whether a patient develops t-MN. A patient may have no predisposing factors, have an inherited mutation (predisposing them to aberrant DNA repair), have a pre-existing somatic mutation (CHIP), or have an aberrant bone marrow microenvironment (due to aging and/or chronic inflammation). The middle panel illustrates how multiple cycles of cytotoxic therapy may promote clonal expansion of cells with pre-existing and/or newly acquired mutations, and alter the BM microenvironment, possibly via therapy-induced senescence. Together, this creates permissive conditions for malignant transformation of hematopoietic cells. The right panel illustrates how these changes converge to deregulate the DNA damage response and cell cycle checkpoint, as well as enhance metabolism and survival, ultimately leading to t-MN.
Our model suggests that multiple cycles of cytotoxic therapy may create conditions that promote clonal expansion of cells with pre-existing and/or newly-acquired mutations and/or chromosomal aberrations. Cytotoxic therapy may also alter the BM microenvironment, possibly through therapy-induced senescence, to help promote malignant transformation in t-MN. Consistent with this idea, senescence-associated changes have been observed in MSCs isolated from MDS and AML patients (20,25). The pathways illustrated in Fig. 7 are not mutually exclusive, and one or multiple pathways may ultimately lead to decreased DDR, deficient checkpoint responses, and alterations to metabolism, collectively contributing to increased survival and expansion of the t-MN clone. Our results suggest that niche-based therapeutic approaches, most likely in conjunction with approaches targeting the hematopoietic cells, should be considered. Thus, elucidating the effects of cytotoxic therapy on the bone marrow microenvironment and on the HSPCs, as well as the contribution of an aberrant DNA damage response may provide insights relevant to the development of targeted therapies for t-MN.
Methods
Mice
All mouse studies were approved by the University of Chicago’s Institutional Animal Care and Use Committee (IACUC), and mice were housed in a facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Egr1+/−, Mx1-Cre−Apc fl/+ (Egr1+/−) and Egr1+/−, Mx1-Cre+Apc fl/+ (Egr1+/−, Apc del/+), mice were generated by crossing Mx1-Cre transgenic mice with Egr1−/−, Apcfl/fl mice (C57Bl/6 genetic background) as described previously (6).
Mouse model of MDS/AML
Total bone marrow cells, isolated from 10–12 week old Egr1+/− or Egr1+/−, Apc del/+ mice, were transduced with a Luc (control) or Trp53-specific shRNA (p53.1224) (6) and transplanted, via retro-orbital injection, into lethally irradiated (8.6 Gy) WT mice (B6.SJL/Cd45.1). Where indicated, donor mice were injected once with N-ethyl-N-nitrosurea (ENU) (100mg/kg) 2 weeks after polyinosinic:polycytidylic acid (pI-pC) injections to induce the deletion of the floxed Apc allele, and recipient mice were injected once with ENU (100mg/kg) two weeks before transplantation. In ENU ‘both’ conditions, Mx1-Cre−Apc fl/+ mice were used as WT recipient mice, instead of B6.SJL/Cd45.1 mice. Peripheral blood, histology, spectral karyotyping, and flow cytometric analyses were performed as described previously (6). To ensure that we were not biasing the disease phenotype, animals were euthanized when they displayed at least four of the following symptoms: poor grooming, abnormal hunched posture, decreased activity, listlessness or lethargy, a body conditioning score of 2 (lean), anemia (Hb <8 g/dL), leukocytosis (> 25K/μL) or thrombocytopenia (< 500 K/μL), and enlarged spleen, thymus or masses (by palpation). Survival times (time to sacrifice) were estimated by the Kaplan Meier method and compared between groups via log rank test.
Whole Exome and RNA Sequencing
Exome libraries were generated using the SureSelectXT Mouse All Exon kit (Agilent, Santa Clara, CA) following the manufacturer’s directions. 100 bp paired-end sequencing was performed on a HiSeq 4000 sequencing system. Reads were assessed for quality (Short Read and FastQC), overlapping reads were merged using FLASH (ver 1.2.11) and aligned to mm9 using BWA (ver 0.7.12) (53). SAMtools v0.1.18 (53) was used to remove duplicates, and GATK was used for local realignment and score recalibration (54). Mutation calling was performed with reads with mapping qualities ≥ 30, requiring ≥ 8X coverage in both tumor and normal DNA (Egr1+/−, Apc fl/+) requiring a variant read on each strand, and a minimum variant allele frequency of 20%. Indels identified using VarScan v2.3.4 (55), and single nucleotide mutations identified using both MuTect v1.1.4 (56) and VarScan were retained. Known single nucleotide polymorphisms (SNPs) for the C57Bl/6 strain were removed. Only mutations with a Genomic Evolutionary Rate Profiling (GERP) score (57) >2 predicted to have deleterious consequences were included. All gene mutations listed in Fig. 5B were validated by Sanger Sequencing. RNA sequencing analysis was performed as described, with the exception that reads were aligned to mm10 and the MSC dataset was pseudoaligned with Kallisto(58). Sequencing data is available on the Gene Expression Omnibus database (GSE135866) and Sequence Read Archive (PRJNA597332).
MSC senescence assays
To isolate MSCs, femur, tibia, and humerus bones were flushed to remove hematopoietic cells, treated with collagenase, and cultured using MesenCult™ (Mouse) with MesenPure™ in 5% O2 according to the manufacturer’s recommendations (Stem Cell Technologies, Vancouver, BC). To measure senescence-associated β-galactosidase activity (SA-βgal) that is detectable at pH6 (due to increased lysosomal biogenesis in senescent cells), the internal pH of lysosomes was increased to ~pH 6 using bafilomycin A1. Cells were then incubated with 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG) (Cayman Chemicals, Ann Arbor, MI), a β-galactosidase substrate that becomes fluorescent after cleavage by the enzyme. SA-βgal activity was estimated using flow cytometry. Cell proliferation was assayed with the EZClick™ EdU proliferation kit (BioVision Inc., San Francisco, CA). For colony-forming units-fibroblastic (CFU-F) assays, either 2×104 directly isolated MSCs (post collagenase) or 2×103 first passage MSCs were plated per well of a 6-well plate, and stained with 0.5% crystal violet after 14 days to enumerate colonies. Quantitative PCR was performed as previously described (59). Primer sequences are provided in supplemental Table 7. For RNA-seq analysis, MSCs were isolated from mock- or ENU-treated mice, ~1 month post treatment, and expanded in vitro to passage 1 or 2.
Supplementary Material
Statement of Significance.
This study challenges the historic view that prior cytotoxic therapy targets only hematopoietic cells, and shows that chemotherapy-induced alterations to the microenvironment contribute to myeloid neoplasms in a model of del(5q) t-MN. The DNA damage response in hematopoietic cells and senescence of stromal cells are identified as potential therapeutic targets.
Acknowledgments
We would like to thank the Le Beau and McNerney laboratory members for the insightful discussions about this research project, and the University of Chicago Biostatistics Core Facility for providing statistical support.
Financial Support: This work was supported by a Public Health Service, National Institutes of Health, National Cancer Institute grant (RO1 CA190372), and a grant from the Edward P. Evans Foundation (M.M.L.B.), as well as the Genomics Core Facility and the Cytometry and Antibody Technology Facility of the University of Chicago Medicine Comprehensive Cancer Center (P30 CA014599). M.E.M. is supported by R01 CA231880, the American Cancer Society Research Scholar Award, and the Brinson Foundation.
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
References
- 1.McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer 2017;17(9):513–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 2003;102(1):43–52 [DOI] [PubMed] [Google Scholar]
- 3.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2001;19(5):1405–13 [DOI] [PubMed] [Google Scholar]
- 4.Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125(9):1367–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV, et al. Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30(12):1343–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoddart A, Fernald AA, Wang J, Davis EM, Karrison T, Anastasi J, et al. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood 2014;123(7):1069–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine 2014;371(26):2488–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine 2014;20(12):1472–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol 2017;18(1):112–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol 2017;18(1):100–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015;518(7540):552–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong TN, Miller CA, Jotte MRM, Bagegni N, Baty JD, Schmidt AP, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun 2018;9(1):455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. The New England journal of medicine 2017;376(6):536–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby MA, De Jesus Pizarro RE, Shao J, Koboldt DC, Fulton RS, Zhou G, et al. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2014;28(6):1242–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014;506(7487):240–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014;14(6):824–37 [DOI] [PubMed] [Google Scholar]
- 17.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010;464(7290):852–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoddart A, Wang J, Hu C, Fernald AA, Davis EM, Cheng JX, et al. Inhibition of WNT signaling in the bone marrow niche prevents the development of MDS in the Apcdel/+ MDS mouse model. Blood 2017;129(22):2959–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016;19(5):613–27 [DOI] [PubMed] [Google Scholar]
- 20.Mattiucci D, Maurizi G, Leoni P, Poloni A. Aging- and Senescence-associated Changes of Mesenchymal Stromal Cells in Myelodysplastic Syndromes. Cell transplantation 2018;27(5):754–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fei C, Zhao Y, Guo J, Gu S, Li X, Chang C. Senescence of bone marrow mesenchymal stromal cells is accompanied by activation of p53/p21 pathway in myelodysplastic syndromes. European journal of haematology 2014;93(6):476–86 [DOI] [PubMed] [Google Scholar]
- 22.Kornblau SM, Ruvolo PP, Wang RY, Battula VL, Shpall EJ, Ruvolo VR, et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica 2018;103(5):810–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geyh S, Oz S, Cadeddu RP, Frobel J, Bruckner B, Kundgen A, et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013;27(9):1841–51 [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Wu D, Fei C, Guo J, Gu S, Zhu Y, et al. Down-regulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica 2015;100(2):194–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdul-Aziz AM, Sun Y, Hellmich C, Marlein CR, Mistry J, Forde E, et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019;133(5):446–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calkoen FG, Vervat C, van Pel M, de Haas V, Vijfhuizen LS, Eising E, et al. Despite differential gene expression profiles pediatric MDS derived mesenchymal stromal cells display functionality in vitro. Stem cell research 2015;14(2):198–210 [DOI] [PubMed] [Google Scholar]
- 27.Joslin JM, Fernald AA, Tennant TR, Davis EM, Kogan SC, Anastasi J, et al. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood 2007;110(2):719–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Fernald AA, Anastasi J, Le Beau MM, Qian Z. Haploinsufficiency of Apc leads to ineffective hematopoiesis. Blood 2010;115(17):3481–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica 2013;98(6):908–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 2002;100(1):238–45 [DOI] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L, Li M, Sun C, Francisco L, Chakraborty S, Sabado M, et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell 2011;20(5):591–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer discovery 2017;7(2):165–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature reviews Molecular cell biology 2007;8(9):729–40 [DOI] [PubMed] [Google Scholar]
- 35.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual review of pathology 2010;5:99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min IM, Pietramaggiori G, Kim FS, Passegue E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2008;2(4):380–91 [DOI] [PubMed] [Google Scholar]
- 37.Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res 2015;39(3):348–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM. Expression profiling of CD34+ hematopoietic stem/ progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci U S A 2002;99(23):14925–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoddart A, Nakitandwe J, Chen SC, Downing JR, Le Beau MM. Haploinsufficient loss of multiple 5q genes may fine-tune Wnt signaling in del(5q) therapy-related myeloid neoplasms. Blood 2015;126(26):2899–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014;28(2):241–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine 2013;368(22):2059–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016;30(2):337–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voso MT, Fabiani E, Zang Z, Fianchi L, Falconi G, Padella A, et al. Fanconi anemia gene variants in therapy-related myeloid neoplasms. Blood Cancer J 2015;5:e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007;129(6):1097–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith LB, Leo MC, Anderson C, Wright TJ, Weymann KB, Wood LJ. The role of IL-1beta and TNF-alpha signaling in the genesis of cancer treatment related symptoms (CTRS): a study using cytokine receptor-deficient mice. Brain, behavior, and immunity 2014;38:66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kutyna MM, Wee A, Paton S, Cakouros D, Arthur A, Chhetri R, et al. Aberrant Bone Marrow Microenvironment in Therapy Related Myeloid Neoplasm (t-MN) Blood 2019;134(Supplement 1):1694 [Google Scholar]
- 47.Schulz E, Valentin A, Ulz P, Beham-Schmid C, Lind K, Rupp V, et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J Med Genet 2012;49(7):422–8 [DOI] [PubMed] [Google Scholar]
- 48.Link DC, Schuettpelz LG, Shen D, Wang J, Walter MJ, Kulkarni S, et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA 2011;305(15):1568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Churpek JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer 2016;122(2):304–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inflammation Lepperdinger G. and mesenchymal stem cell aging. Current opinion in immunology 2011;23(4):518–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von der Heide EK, Neumann M, Vosberg S, James AR, Schroeder MP, Ortiz-Tanchez J, et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia 2017;31(5):1069–78 [DOI] [PubMed] [Google Scholar]
- 52.Bhagat TD, Chen S, Bartenstein M, Barlowe AT, Von Ahrens D, Choudhary GS, et al. Epigenetically Aberrant Stroma in MDS Propagates Disease via Wnt/beta-Catenin Activation. Cancer research 2017;77(18):4846–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22(3):568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31(3):213–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005;15(7):901–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.An N, Khan S, Imgruet MK, Gurbuxani SK, Konecki SN, Burgess MR, et al. Gene dosage effect of CUX1 in a murine model disrupts HSC homeostasis and controls the severity and mortality of MDS. Blood 2018;131(24):2682–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stoddart A, Qian Z, Fernald AA, Bergerson RJ, Wang J, Karrison T, et al. Retroviral insertional mutagenesis identifies the del(5q) genes, CXXC5, TIFAB and ETF1, as well as the Wnt pathway, as potential targets in del(5q) myeloid neoplasms. Haematologica 2016;101(6):e232–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.