

Abstract
Objective:
We analyzed the longitudinal profile of Alzheimer’s disease (AD) cerebrospinal fluid (CSF) biomarkers in early Parkinson’s disease (PD) compared with healthy controls (HC) and tested baseline CSF biomarkers for prediction of clinical decline in PD.
Methods:
Amyloid-β (Aβ42), total tau (t-tau) and tau phosphorylated at the threonine 181 position (p-tau) were measured using the high-precision Roche Elecsys® electrochemiluminescence immunoassay in all available CSF samples from longitudinally studied PD (n=416) and HC (n=192) followed for up to three years in the Parkinson’s Progression Markers Initiative (PPMI). Longitudinal CSF and clinical data were analyzed with linear-mixed effects models.
Results:
We found PD patients had lower CSF t-tau (median=157.7pg/mL,range=80.9–467.0), p-tau (median=13.4pg/mL,range=8.0–40.1) and Aβ42 (median=846.2pg/mL,range=238.8–3707.0) than HC at baseline (CSF t-tau median=173.5pg/mL,range=82.0–580.8; p-tau median=15.4pg/mL,range=8.1–73.6; Aβ42 median=926.5pg/mL,range=239.1–3297.0;p<0.05–0.001) and a moderate-to-strong correlation among these biomarkers in both PD and HC (Rho=0.50–0.97; p<0.001). 31.5% of PD had pathologically-low levels of CSF Aβ42 baseline and these PD patients had lower p-tau levels (median=10.8pg/mL, range=8.0–32.8) compared to 27.7% of HC with pathologically-low CSF Aβ42 (CSF p-tau median=12.8pg/mL, range 8.2–73.6;p<0.03). In longitudinal CSF analysis, we found PD had greater decline in CSF Aβ42 (mean difference= −41.83 pg/ml; p=0.03) and CSF p-tau (mean difference= −0.38 pg/ml; p=0.03) at year three compared to HC. Baseline CSF Aβ42 values predicted small but measurable decline on cognitive, autonomic and motor function in early PD.
Interpretation:
Our data suggest baseline CSF AD biomarkers may have prognostic value in early PD and that the dynamic change of these markers, while modest over a three-year period, suggest biomarker profiles in PD may deviate from healthy aging.
Keywords: Parkinson’s disease, Alzheimer’s disease, biomarker, cerebrospinal fluid, tau, amyloid-beta, PPMI, alpha-synuclein
INTRODUCTION
There is significant clinical and pathological heterogeneity of Parkinson’s disease (PD), and while α-synuclein (aSyn) Lewy pathology and the associated synapse and neuronal loss is the hallmark of this disease, there is varying severity of mixed Alzheimer’s disease (AD) associated amyloid-beta (Aβ42) plaques and tau tangles found at autopsy in many PD patients. Indeed, approximately 30% of autopsy-confirmed PD have sufficient postmortem plaque and tangle pathology to meet neuropathologic criteria for a second diagnosis of AD, and these patients have a more rapid decline in cognition and overall survival than PD patients with minimal AD co-pathology1, 2. Thus, identifying markers of AD pathology during life may have important prognostic indications in PD to guide clinical trials for homogeneous patient selection.
Cerebrospinal fluid (CSF) analysis provides a mechanism to detect and measure protein species related to the accumulation of these pathological proteins over time in living patients; cross-sectional work finds CSF measures of AD pathology associate with cognitive performance3–6 and postmortem severity of AD co-pathology in PD7. Moreover, CSF tau and aSyn levels are highly correlated and, on average, lower in PD compared to controls8, 9; however, longitudinal AD CSF biomarker data in PD is rare10–14 and detailed longitudinal modeling of progressive changes in values are lacking.
One obstacle to longitudinal CSF studies is inter- and intra-assay variation15 which could reduce the sensitivity to detect changes between repeated measurements from an individual over time in longitudinal biomarker studies. The Roche Elecsys® analytical platform is fully-automated with high-reliability for measurement of AD CSF biomarkers16–18 and was previously validated with a reference measurement procedure approved by the Joint Committee for Traceability in Laboratory Medicine for CSF Aβ4219. PPMI20 is a unique multicenter international observational study collecting long term annual detailed harmonized clinical measures and biomarkers in a large cohort of newly diagnosed drug-naïve PD. We previously found CSF measurements of tau and Aβ42, as well as aSyn, related to cross-sectional and longitudinal clinical features in this cohort with follow-up up to one year8, 9, 12, 21.
Using the rich PPMI dataset with standardized longitudinal data for up to three years and the Elecsys® high-precision analytical platform, we evaluated the baseline and longitudinal progression of AD CSF biomarkers in PD and tested the relationship of these with clinical features.
METHODS
Sample
The sample consisted of participants in two of the cohorts of the PPMI multicenter prospective longitudinal observational study: early PD, drug-naïve at baseline, and HC20, with diagnostic criteria for enrollment as described previously8, 9. Those included for study (n=608) had at least one CSF sample at any timepoint available as of May 7th, 2018 (PD=416, HC=192). We did not include other PPMI cohort participants (symptomatic or asymptomatic individuals with PD-related genetic mutations, prodromal PD, or participants with parkinsonism but without evidence of dopaminergic deficit syndrome). CSF and clinical data were obtained from PPMI database for baseline, 6 months and annual follow up visits at years 1, 2 and 3. A subset of these participants were previously reported in a cross-sectional study of baseline CSF data (n=601)8, 9 or longitudinal CSF with only 1-year follow up (n=285)12 and using a different immunoassay platform (i.e. Innogenetics AlzBio3 Luminex platform).
All procedures were performed with prior approval from ethical standards committees at each participating institution and with informed consent from all study participants. The study is registered in clinicaltrials.gov as NCT01141023.
CSF Analysis
CSF collection, shipment and storage were performed using standard operating procedures at each institution as described in detail previously (please see biologics manual ppmi.info.org). CSF samples were shipped from the PPMI Biorepository Core laboratories to the University of Pennsylvania (Penn) Biomarker Research Laboratory for measurement of CSF amyloid-beta 1–42 (Aβ42), total-tau (t-tau) and tau phosphorylated at threonine 181 position (p-tau) using Elecsys® electrochemiluminescence immunoassays on the cobas e 601 analysis platform (Roche Diagnostics) as described16, 18. The analytical measurement range for the Aβ42 assay was 200 to 1700 pg/mL, the t-tau assay was 80 to 1300 pg/mL and the p-tau181 assay: 8 to 120 pg/mL. Roche extrapolated values above the upper technical limit from the calibration curve, 1700 pg/ml, in 96 measurements of Aβ42. Performance of this platform has been previously reported with intra- and inter-CV% <5%17–19. CSF total alpha-synuclein (aSyn) data from baseline visits were obtained from PPMI database and measured by BioLegend (San Diego, CA) using a commercially available sandwich immunoassay, as previously described8, 9, 12.
Clinical Data
Clinical data for each visit was obtained from the PPMI database as above and described in detail previously22. Variables included for analysis were demographics (age at baseline, age at symptoms onset, disease duration at visit, years of education and sex) and cognitive and motor testing scores. We chose continuous measures of cognitive functioning in several domains including: global functioning (Montreal Cognitive Assessment; MoCA), episodic memory (Hopkins Verbal Learning Test discrimination recognition score; HVLT), visuospatial functioning (Benton judgement of line orientation score; JOLO), language (semantic fluency; SF) and executive functioning (letter number sequencing; LNS). For motor functioning we used the Movement Disorders Society modified Unified Parkinson’s disease rating scale (MDS-UPDRS) part III total score and motor sub-scores for tremor and postural instability (PIGD), as previously defined9, as continuous variables. We also included the total score for the Scales for Outcomes in Parkinson’s Disease-Autonomic questionnaire (SCOPA-AUT) to capture non-motor/cognitive autonomic aspects of PD.
Genetic Data
Blood samples were analyzed for apolipoprotein ɛ (APOE) genotype at the PPMI genetics core as described8 and coded for analyses as the presence or absences of one or more ɛ4 alleles (i.e. dominant model).
Statistical Analyses
Data used in study were downloaded from PPMI database on May 7th 2018 and analyzed at the University of Iowa using SAS 9.4 Software (SAS Institute, Cary, NC) or at Penn using SPSS version 24.0 (IBM, Chicago, IL). We used a significance threshold of p<0.05 due to the hypothesis-driven approach for CSF-clinical correlations (please see results section for specifics).
Continuous demographic, clinical and biomarker data were compared between groups using Student’s t-test or Wilcoxon rank-sum test, as appropriate, and nominal variables compared with a chi-square or Fisher’s exact test. For non-parametric comparisons we calculated effect size r=z/√(N), where N is the total sample size23.
To test for associations of needle type used in CSF collection, we used univariate comparisons for measures of each analyte using the Kruskal-Wallis test within PD subjects. CSF needle was grouped by type coded in database: Quincke, Sprotte, or “other”.
Correlations between CSF biomarkers were computed using Spearman rank correlation and 95% confidence intervals (CI) obtained based on Fisher’s z transformation. To test biological associations of CSF biomarkers, we performed univariate subgroup analysis within PD and HC groups comparing patients with one or more copies of APOE ɛ4 allele compared to those without.
To characterize the AD CSF profile of PD and HC we applied a cut-point for amyloid-positivity established in AD16. To mitigate differences in pre-analytical factors between PPMI and AD cohorts that influence CSF Aβ42 levels24, we used the transformation formula from Shaw et al25 to convert Elecsys values to AlzBio3 equivalents [x= (CSF Aβ42 +251.55)/3.74] and applied the established cut-point of <250 pg/ml of AlzBio3 equivalent values16 to designate amyloid-positivity. A chi-square test was used to analyze proportional differences in amyloid-positivity among PD and HC at baseline. Within PD and HC, we compared demographics and CSF biomarker values between amyloid-positive and negative groups using univariate statistics.
For longitudinal analyses we focused on core AD CSF biomarkers (Aβ42, t-tau, and p-tau), rather than ratios of these analytes, to more directly test biomarker associations. To assess the difference in mean change from baseline for each AD CSF analyte between the PD and control groups, we used rank-based linear mixed models (LMM) with adjustment for age, sex and the baseline value of the CSF outcome. Akaike information criterion (AIC) was used in the determination to adjust for APOE and the model fit of including an interaction between time and group (i.e PD vs HC). We report the p-value from the rank-based LMM and mean estimates from a model based on the untransformed values for ease of interpretation. The model-based mean estimates of the change in CSF within PD and HC and their differences adjust for group specific differences in the baseline covariates (age, sex, baseline CSF, and APOE (if applicable)).
To test the associations between baseline CSF analyte levels and decline on clinical measures in PD, we used LMM or rank-based LMM with separate models for each baseline CSF measure as predictors for the dependent variable of change in each clinical measure (MoCA, HTLV, JLO, SF, LNS, UPDRS III total, tremor UPDRS subscore, PIGD UPDRS subscore and SCOPA-AUT) from baseline in PD subjects. All models adjusted for baseline age, gender, disease duration, and the baseline value of the clinical measure. AIC was used in the determination to adjust for APOE in the final models and to compare the model fit of including an interaction between time and baseline CSF. If AIC indicated the interaction did not provide better fit, the interaction term was removed. Using this approach, we found the optimal model structure for MoCA, LNS, UPDRS III and SCOPA-AUT was a linear time model with a random intercept and slope and an unstructured covariance structure. The optimal model for SF was a linear time model with a random intercept and an unstructured covariance structure. A non-linear time model had optimal fit for JLO. The final models for the clinical outcomes in LNS adjusted for APOE along with the MoCA models for Aβ42 and aSyn. Rank-based LMM were fit for Tremor, PIGD, and HVLT. We report the p-value from the rank-based LMM and effect estimates from a model based on the untransformed values for ease of interpretation.
RESULTS
Patient Demographics and Baseline Characteristics
PD and HC patient demographics are listed in Table 1. Similar to previous reports of this cohort at baseline8, 9, PD and HC groups did not differ in age, sex or APOE allele status.
Table 1.
Patient and Control Demographics and Baseline Characteristics
| Variables | PD N=416 | HC N=192 | P-value | |
|---|---|---|---|---|
| DEMOGRAPHIC | Age (y) | 61.7 (9.6) | 60.8 (11.3) | 0.3 |
| Sex | Male=272 (65.4%) Female=144 (34.6%) |
Male=123 (64.0%) Female =69 (35.9%) |
0.8 | |
| Education (y) | 15.5 (3.0) | 16.0 (2.9) | 0.06 | |
| APOE Ɛ4 Status | 0 alleles= 277 (73.3%) 1 allele= 92 (24.3%) 2 alleles= 9 (2.4%) Missing data=38 |
0 alleles= 129 (73.7%) 1 allele= 42 (24.0%) 2 alleles= 4 (2.3%) Missing data=17 |
>0.99 | |
| Age at Onset (y) | 59.7 (9.9) | NA | - | |
| Disease Duration (mo) | 6.7 (6.5) | NA | - | |
| MOTOR | UPDRS III Tremor | 0.5 (0.3) N=415 Missing data=1 |
0.03 (0.08) N=191 Missing data=1 |
<0.0001 |
| UPDRS III PIGD | 0.23 (0.22) N=415 Missing data=1 |
0.02 (0.09) N=191 Missing data=1 |
<0.0001 | |
| COGNITIVE | MOCA | 27.1 (2.3) N=413 Missing data=3 |
28.2 (1.1) N=192 |
<0.0001 |
| HVLT | 10 (−4,12) N=414 Missing data=2 |
11 (−4, 12) N=192 |
<0.001 | |
| SFT | 48.8 (11.6) N=415 Missing data=1 |
51.9 (11.3) N=192 |
<0.01 | |
| JLO | 13 (5,15) N=415 Missing data=1 |
14 (4, 15) N=192 |
0.06 | |
| LNS | 10.6 (2.6) N=415 Missing data=1 |
10.9 (2.6) N=192 |
0.1 | |
Data listed= mean (SD) for normally distributed variables or median (range) for non-normally distributed variables and frequency (%) for categorical variables. Missing Data noted in each cell where applicable.
Abbreviations: PD=Parkinson’s disease, HC=healthy controls, y= years, mo=months, APOE=apolipoprotein E, UPDRS= Unified Parkinson’s Disease Rating Scale, Tremor=Tremor sub score of UPDRS, PIGD= Postural instability and gait disturbance sub score of UPDRS, MOCA= Montreal Cognitive Assessment, HVLT= Hopkins Verbal Learning Test Discrimination Recognition Score , SFT= semantic fluency total score, JLO= Benton judgement of line orientation score, LNS= Letter-number sequencing score.
Cross-Sectional CSF Analysis
First, to test for pre-analytical factors that could influence CSF measurements on this platform, we performed univariate comparisons of needle type used in CSF collection cross-sectional data at each time point for CSF Aβ42, t-tau and p-tau. We did not find any association of needle type with biomarker values (data not shown) and needle-type did not have a significant effect on any of our subsequent CSF outcome models below.
Baseline levels of CSF Aβ42, t-tau, p-tau, aSyn and the ratio of p-tau/t-tau were lower in PD than HC (effect size=0.09–0.17, p<0.03–0.0001), while the ratios of t-tau/Aβ42, p-tau/Aβ42, t-tau/aSyn, p-tau/aSyn, Aβ42/aSyn were similar between groups (Figure 1). These group-level differences were similar across timepoints (Table 2); however, despite group-wise differences in these CSF biomarkers, there was individual-patient overlap in values between groups (Figure 1).
Figure 1. Group-wise comparison of baseline CSF biomarkers in PD and HC.
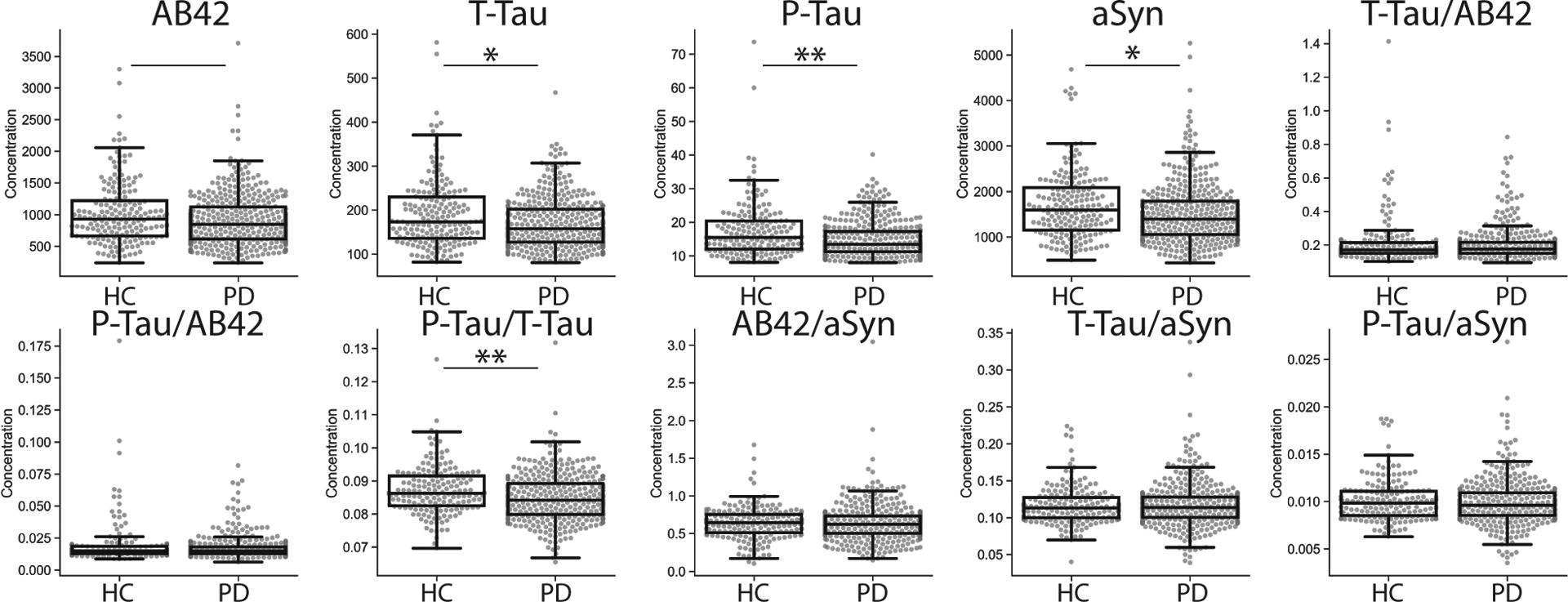
Solid line represents group-wise difference between PD and HC p<0.05, solid line plus single asterisk p<0.01 and solid line plus double-asterisk p<0.001.
Table 2.
Cross-Sectional Cerebrospinal Fluid Biomarker Data
| Analyte | Visit | PD (N=416) | HC (N=192) | Effect Size r | P-value |
|---|---|---|---|---|---|
| Aβ42 | Baseline | 846.15 (238.80 – 3707.00) Missing data=6 |
926.45 (239.10 – 3297.00) Missing data=4 |
0.09 | 0.02 |
| 6 Months | 849.70 (267.30 – 2888.00) Missing data=77 |
938.90 (372.90 – 3272.00) Missing data=35 |
0.14 | <0.002 | |
| Year 1 | 821.30 (249.50 – 2480.00) Missing data=90 |
1019.50 (312.00 –2678.00) Missing data=40 |
0.18 | <0.0001 | |
| Year 2 | 849.75 (260.30 – 3000.00) Missing data=112 |
955.75 (248.60 – 3551.00) Missing data=56 |
0.13 | <0.01 | |
| Year 3 | 855.25 (240.80 – 2396.00) Missing data=194 |
954.30 (282.00 – 2842.00) Missing data=79 |
0.12 | 0.03 | |
| t-tau | Baseline | 157.70 (80.93 – 467.00) Missing data=13 |
173.50 (81.96 – 580.80) Missing data=5 |
0.12 | <0.01 |
| 6 Months | 153.60 (80.64 – 387.50) Missing data=81 |
179.60 (82.64 – 551.50) Missing data=37 |
0.19 | <0.0001 | |
| Year 1 | 155.60 (82.24 – 388.70) Missing data=94 |
178.80 (82.36 – 600.10) Missing data=40 |
0.18 | <0.0001 | |
| Year 2 | 156.35 (80.88 – 463.60) Missing data=110 |
178.80 (85.92 – 619.70) Missing data=60 |
0.18 | <0.001 | |
| Year 3 | 160.45 (80.98 – 444.50) Missing data=190 |
173.60 (83.48 – 569.40) Missing data=79 |
0.16 | <0.01 | |
| p-tau | Baseline | 13.40 (8.01 – 40.13) Missing data=37 |
15.44 (8.08 – 73.61) Missing data=16 |
0.17 | 0.0001 |
| 6 Months | 13.34 (8.00 – 36.94) Missing data=106 |
15.69 (8.53 – 69.10) Missing data=42 |
0.20 | <0.0001 | |
| Year 1 | 13.41 (8.05 – 34.28) Missing data=124 |
15.87 (8.30 – 80.08) Missing data=48 |
0.21 | <0.0001 | |
| Year 2 | 13.39 (8.13 – 43.69) Missing data=136 |
15.59 (8.00 – 80.54) Missing data=66 |
0.21 | <0.0001 | |
| Year 3 | 13.31 (8.03 – 42.87) Missing data=205 |
15.31 (8.05 – 78.34) Missing data=90 |
0.22 | 0.0001 | |
| aSyn | Baseline | 1390.50 (432.40–5256.90) Missing data=2 |
1593.50 (488.60–4683.10) Missing data=2 |
0.13 | <0.002 |
| t-tau/Aβ42 | Baseline | 0.18 (0.10 – 0.84) Missing data=18 |
0.17 (0.10 – 1.41) Missing data=7 |
0.02 | 0.5 |
| p-tau/Aβ42 | Baseline | 0.01 (0.01 – 0.08) Missing data=42 |
0.01 (0.01 – 0.18) Missing data=18 |
0.01 | 0.8 |
| p-tau/t-tau | Baseline | 0.08 (0.07 – 0.13) Missing data=37 |
0.09 (0.07 – 0.13) Missing data=16 |
0.16 | <0.001 |
| Aβ42/aSyn | Baseline | 0.63 (0.15 – 3.04) Missing data=7 |
0.65 (0.10 – 1.68) Missing data=4 |
0.02 | 0.7 |
| t-tau/aSyn | Baseline | 0.11 (0.04 – 0.34) Missing data=14 |
0.11 (0.04 – 0.22) Missing data=5 |
0.02 | 0.5 |
| p-tau/aSyn | Baseline | 0.01 (0.00 – 0.03) Missing data=38 |
0.01 (0.01 – 0.02) Missing data=16 |
0.05 | 0.3 |
Data listed= median (range). Missing Data noted in each cell where applicable.
Abbreviations: PD=Parkinson’s disease, HC=healthy controls, Aβ42= Cerebrospinal fluid amyloid-beta 1–42, t-tau= cerebrospinal fluid total tau, p-tau= cerebrospinal fluid phosphorylated tau at threonine 181, aSyn= cerebrospinal fluid total alpha-synuclein.
Next, to test the association of AD CSF biomarkers with a known genetic marker of AD pathology26, we compared both PD and HC individuals with one or more copies of APOE ɛ4 genotype to those with no copies of APOE ɛ4 at baseline and found lower CSF Aβ42 in APOE ɛ4 carriers for both PD and HC (effect size=0.26–0.31, p<0.0001), while there was no difference between APOE genotype groups within PD or HC for t-tau or p-tau (Table 3). Interestingly, there was also lower baseline CSF aSyn in PD APOE ɛ4 carriers than non-carriers (effect size= 0.13, p=0.01), while CSF aSyn was similar between HC APOE groups (Table 3).
Table 3.
Baseline CSF Data by APOE Genotype
| CSF Analyte | PD | HC | ||||||
|---|---|---|---|---|---|---|---|---|
| APOE 4+ N=101 |
APOE 4− N=277 |
Effect Size r | p | APOE 4+ N=46 |
APOE 4− N=129 |
Effect Size r | p | |
| CSF Aβ42 | 697.1 (238.8 – 1795.0) Missing=1 |
912.8 (249.0 – 3707.0) Missing=4 |
0.26 | <0.0001 | 673.1 (239.1 – 1890.0) Missing=1 | 994.8 (336.1 – 3297.0) Missing=3 |
0.31 | <0.0001 |
| CSF T-Tau | 152.0 (85.0 – 349.8) Missing=5 |
159.9 (80.9 – 467.0) Missing=8 |
0.04 | 0.48 | 189.5 (93.3 – 554.5) Missing=2 |
168.6 (82.0 – 580.8) Missing=2 |
0.04 | 0.57 |
| CSF P-Tau | 13.3 (8.0 – 28.0) Missing=12 |
13.6 (8.0 – 40.1) Missing=22 |
0.03 | 0.56 | 17.0 (8.2 – 60.0) Missing=4 |
15.3 (8.1 – 73.6) Missing=10 |
0.05 | 0.52 |
| CSF aSyn | 1256.5 (432.4 – 3022.3) |
1432.7 (472.0 – 5256.9) Missing=2 |
0.13 | 0.01 | 1522.0 (488.6 – 4683.1) Missing=1 |
1662.6 (600.7 – 4271.3) Missing=1 |
0.07 | 0.36 |
Data listed= median (range). Number of missing data points is noted in each cell. Abbreviations: PD=Parkinson’s disease, HC=healthy controls, Aβ42= Cerebrospinal fluid amyloid-beta 1–42, t-tau= cerebrospinal fluid total tau, p-tau= cerebrospinal fluid phosphorylated tau at threonine 181, aSyn= cerebrospinal fluid total alpha-synuclein.
We found a moderate to strong correlation among AD CSF biomarkers (Aβ42 vs t-tau Rho=0.59, 95%CI (0.53–0.64), p<0.0001, n=583; Aβ42 vs p-tau Rho=0.51, 95%CI (0.45–0.57), p<0.0001, n=548; t-tau vs p-tau Rho=0.97, 95%CI (0.97–0.98), p<0.0001, n=555) and with AD CSF biomarkers and CSF aSyn (Aβ42 vs aSyn Rho=0.60, 95%CI (0.55–0.65), p<0.0001, n=597; t-tau vs aSyn Rho=0.80, 95%CI (0.77–0.83), p<0.0001, n=589; p-tau vs aSyn Rho=0.80, 95%CI (0.77–0.83), p<0.0001, n=554) in the total cohort at baseline (Figure 2).
Figure 2. Baseline correlation of CSF biomarkers in PD and HC.

Scatterplots depict individual patient datapoints for CSF values at baseline. Dashed-line represents fitted line with 95% confidence interval. Red=PD, Blue=HC.
Finally, we examined cross-sectional profiles of patients with presumed amyloid-positivity in PD and HC at baseline using an established cut-point in AD16. We found at baseline, relative equal frequencies of pathologically low CSF Aβ42 indicative of amyloidosis (+A) in PD (31.5%) and HC (27.7%) (Table 4) with no differences in demographics between PD+A and PD with normal CSF Aβ42 (-A) or HC+A and HC-A; however, there were lower CSF t-tau, p-tau and aSyn levels in PD+A vs PD-A (effect size=0.29–0.45, p<0.0001). In contrast, there was no difference in CSF p-tau between HC+A and HC-A, but CSF p-tau was lower in PD+A than HC+A (effect size= 0.19, p<0.03), suggesting a divergent interaction between AD CSF biomarkers in PD compared to controls (Table 4).
Table 4.
Baseline CSF AB Groups.
| PD-A | PD+A | HC-A | HC+A | |
|---|---|---|---|---|
| N (% total) | 281 (68.5%) | 129 (31.5%) | 136 (72.3%) | 52 (27.7%) |
| Sex F (%F) | 99 (35.2%) | 41 (31.8%) | 50 (36.8%) | 18 (34.6%) |
| Age at CSF | 61.47 (9.63) | 62.2 (9.6) | 60.7 (10.8) | 61.0 (13.0) |
| Disease Duration | 4.2 (0.4–35.8) | 4.6 (0.7–34.7) | NA | NA |
| CSF T-Tau | 169.50 (85.6–467.0) | 124.1*(80.9–339.2) Missing=12 |
183.0 (93.7–420.5) | 126.8ⴕ (81.96–580.8) Missing=3 |
| CSF P-Tau | 14.04 (8.2–40.1) Missing=3 |
10.82*,ф (8.0–32.8) Missing=33 |
15.6 (8.1–39.1) Missing=1 |
12.8 (8.2–73.6) Missing=13 |
| CSF aSyn | 1522.3 (606.1–5256.9) Missing=1 |
1026.6* (432.4–3638.3) | 1696.2 (733.8–4271.3) | 1131.9ⴕ (488.6–4683.1) |
Data listed= frequency (%) for categorical data, mean (standard deviation) for normally distributed data or median (range) for non-normally distributed data. Number of missing data points is noted in each cell. Abbreviations: PD-A=Parkinson’s disease with normal CSF Aβ42 levels; PD+A= PD with pathologically low CSF Aβ42 levels, HC-A=healthy controls with normal CSF Aβ42 levels, HC+A=HC with pathologically low CSF Aβ42 levels, F= female, t-tau= cerebrospinal fluid total tau, p-tau= cerebrospinal fluid phosphorylated tau at threonine 181, aSyn= cerebrospinal fluid total alpha-synuclein.
= p<0.0001 PD+A vs PD-A;
= p<0.0001 HC+A vs HC-A;
= p<0.03 PD+A vs HC+A.
Longitudinal Change in AD CSF Biomarkers
To further test the profile of AD CSF biomarkers longitudinally in PD vs HC, we performed linear-mixed model analysis to test the mean change from baseline at each time point between PD and HC. We did not find an interaction between group and time, suggesting the difference in change between PD and HC was largely constant over the three-year period (Figure 3). PD had greater decline in all three biomarkers over time; we found greater reduction in CSF Aβ42 (mean difference= −41.83 pg/ml, SE=18.94, p=0.03) and p-tau (mean difference= −0.38 pg/ml, SE=0.22, p=0.03), in PD compared to HC with a trend for CSF t-tau (mean difference= −3.7 pg/ml, SE=2.7, p=0.07) (Table 5). Examination of estimates of mean change at each time point in our models finds an increasingly negative mean change in CSF Aβ42 in PD compared to HC, where there is mild decline only at year three, and in PD more modest mean increases in CSF t-tau and p-tau seen only at year three compared to more consistent increases over time in HC (Table 5).
Figure 3. Individual patient data for median change in AD CSF biomarker measurements at each time point.
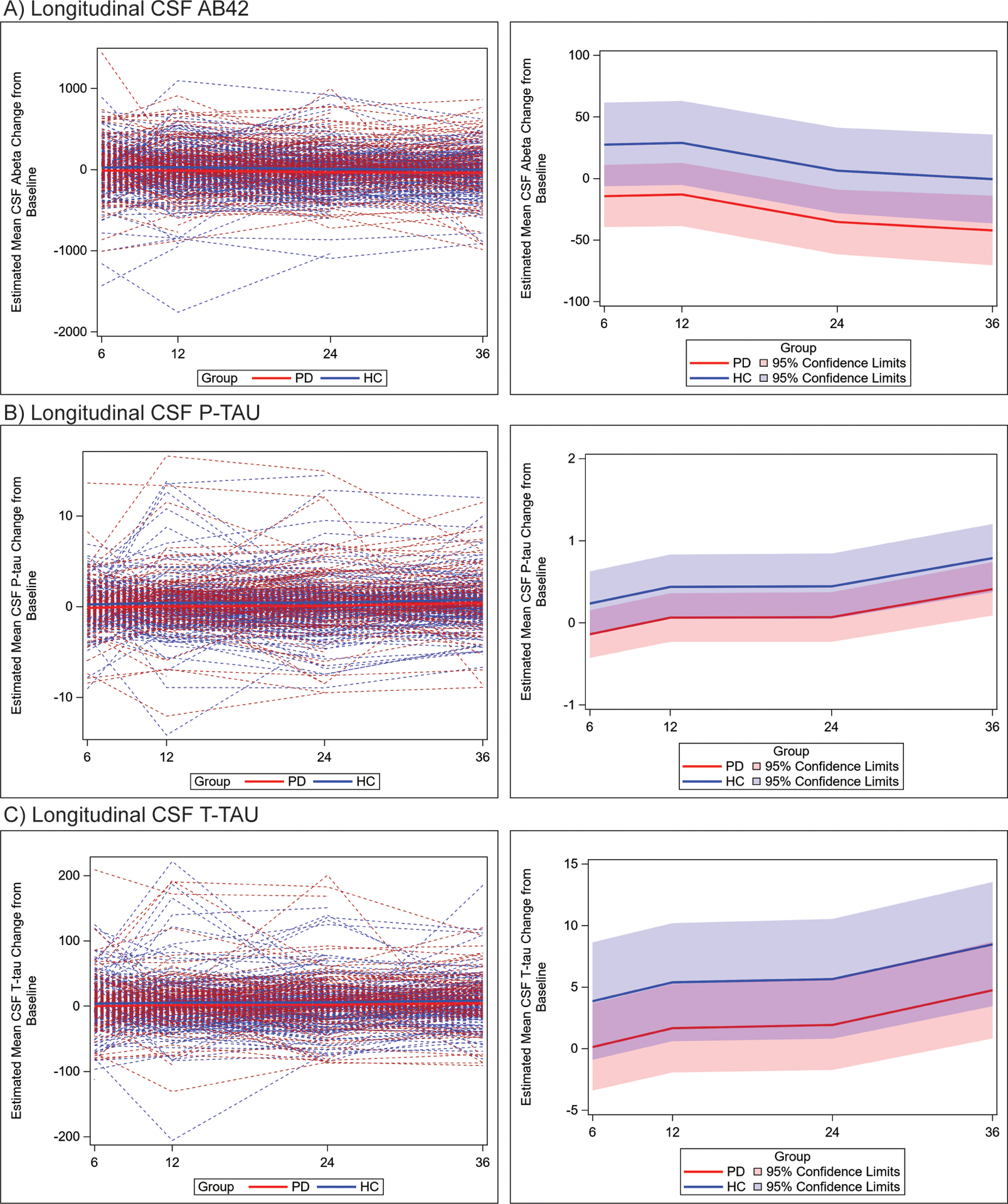
Spaghetti plot depicts individual-patient trajectories (left panels) and trend lines (right panels) depict mean values for PD (red) and HC (blue) and 95% CI for mean change in biomarker value at each time point using LMM adjusted for age, sex, and the baseline value of the CSF outcome for mean change in measurements for CSF Aβ42 (A) t-tau (B) and p-tau (C) at 6 months, 12 months, 24 months and 36-month time points. Across all time points we found a greater reduction in CSF Aβ42 (mean difference −41.83 pg/ml, SE=18.94, p=0.03) and p-tau (mean difference=−0.38 pg/ml, SE=0.22, p=0.03), in PD compared to HC with a non-significant trend for CSF t-tau (mean difference=−3.7 pg/ml, SE=2.7, p=0.07).
Table 5.
Mean Estimates of Change in AD CSF Biomarkers in PD and Healthy Controls.
| Variable | PD | HC | ||||||
|---|---|---|---|---|---|---|---|---|
| 6 Months | 1 Year | 2 Years | 3 Years | 6 Months | 1 Year | 2 Years | 3 Years | |
| AB42 | ||||||||
| Estimate (SE) | −14.29 (12.88) | −13.01 (13.08) | −35.39 (13.36)* | −42.27 (14.47)* | 27.53 (17.28) | 28.82 (17.37) | 6.44 (17.67)* | −0.44 (18.38)* |
| (95% CI) | (−39.56, 10.97) | (−38.67, 12.65) | (−61.60, −9.18) | (−70.65, −13.88) | (−6.38, 61.45) | (−5.26, 62.89) | (−28.23, 41.10) | (−36.50, 35.62) |
| P-TAU | ||||||||
| Estimate (SE) | −0.14 (0.15) | 0.06 (0.15) | 0.07 (0.15)* | 0.41 (0.16)* | 0.24 (0.20) | 0.44 (0.20) | 0.45 (0.20)* | 0.79 (0.21)* |
| (95% CI) | (−0.43, 0.15) | (−0.23, 0.36) | (−0.23, 0.37) | (0.09, 0.74) | (−0.15, 0.63) | (0.05, 0.83) | (0.05, 0.84) | (0.37, 1.20) |
| T-TAU | ||||||||
| Estimate (SE) | 0.13 (1.81) | 1.67 (1.83) | 1.93 (1.87)* | 4.75 (2.01)* | 3.87 (2.43) | 5.40 (2.44) | 5.66 (2.48)* | 8.49 (2.57)* |
| (95% CI) | (−3.41, 3.68) | (−1.93, 5.26) | (−1.73, 5.59) | (0.81, 8.69) | (−0.90, 8.63) | (0.61, 10.18) | (0.80, 10.53) | (3.45, 13.52) |
Estimates based on the raw values (not the ranks) from models adjusting for age, sex and baseline CSF outcome value. AIC criteria determined APOE included in AB42 model.
Denotes p<0.0001 for within-group comparison of estimates between time point and 6-month reference category.
Prediction of Longitudinal Cognitive, Motor and Autonomic Decline using Baseline AD CSF Biomarkers
We performed exploratory analyses based on previous postmortem27–30 and biomarker work3–5 to test predictive value of AD CSF biomarkers in PD. We hypothesized that AD CSF biomarkers would relate to overall cognitive decline, and more specifically in temporal-lobe mediated episodic memory and semantic fluency tasks. Moreover, we expected CSF aSyn would relate to decline on traditional-reported cognitive deficits in early PD22, 31, 32: spatial and executive/attention/working memory tasks. Further, we hypothesized CSF aSyn would relate to progression of classic PD features of motor impairment and autonomic instability. Finally, based on recent postmortem work27, we expected greater increase in motor postural instability to associate with lower CSF AB42.
We found greater baseline p-tau (β= −0.47 points per 10 pg/mL, 95%CI: (−0.91- −0.03), p<0.05) and lower CSF Aβ42 (Month 24 β=0.06 points per 100 pg/mL, 95%CI: (0.01–0.10), p=0.02; Month 36 β=0.09 points per 100 pg/mL, 95%CI: (0.03–0.15), p<0.01) predicted greater decline in global cognition (i.e. MOCA). We also found that both lower CSF baseline Aβ42 (β=0.04 points per 100 pg/mL, 95%CI: (0.0003–0.09), p<0.05) and aSyn (β=0.03 points per 100 pg/mL, 95%CI: (0.003–0.06), p=0.03) predicted greater decline in working memory (i.e. LNS). There was a non-significant trend for greater baseline CSF t-tau to be associated with longitudinal decline on semantic fluency (β= −0.57 points per 100 pg/mL, 95%CI: (−1.17– 0.03), p=0.06).
We found both lower baseline CSF Aβ42 and aSyn were associated with increased postural instability subscores (aSyn β= −0.004 points per 100 pg/mL, 95%CI: (−0.008- −0.0007), p<0.02; Aβ42 β=−0.007 points per 100 pg/mL, 95%CI: (−0.01- −0.001), p=0.04) and total UPDRS III motor scores (aSyn β=−0.10 points per 100 pg/mL, 95% CI: (−0.19- −0.003), p=0.04; Aβ42 β=−0.16 points per 100 pg/mL, 95% CI: (−0.30- −0.01), p=0.03). Finally, lower baseline CSF Aβ42 was also associated with an increase in autonomic symptoms on SCOPA-AUT (β=−0.12 points per 100 pg/mL, 95% CI: (−0.21- −0.02), p=0.02). We did not find other associations with baseline CSF biomarkers and longitudinal clinical measures (data not shown).
DISCUSSION
In this large-scale longitudinal study of well-characterized PD patients over a three-year period using a precise analytical platform (the Roche Elecsys® system) to measure AD CSF biomarker analytes we have several important findings. First, we find lower overall AD CSF biomarker values in PD vs HC (Figure 1, Table 2), with a moderate-to-strong correlation between markers in both PD and HC (Figure 2). 31.5% of PD had pathologically low CSF Aβ42 at baseline with relatively low CSF p-tau compared to HC with pathological CSF Aβ42 (Table 4). Moreover, we found modest but novel measurable group-level changes in AD CSF biomarkers over time in PD that were distinct from HC, with greater overall decline in CSF Aβ42 and p-tau in PD (Figure 3, Table 5). Finally, we find preliminary evidence for predictive value of CSF Aβ42 for global and domain-specific cognitive decline, motor and autonomic function in PD. These data have important implications for the interpretation of these emerging CSF biomarkers in PD.
Our group-wise comparisons at baseline (Figure 1, Table 2) using the high-precision immunoassay replicated previous findings of lower CSF levels of t-tau and p-tau on average in PD than HC and a strong correlation with CSF aSyn (Rho=0.8–0.9) (Figure 2)8–10. Similar to another study of early PD5, we found lower CSF Aβ42 in PD compared to HC and moderate correlations of CSF Aβ42 with CSF t-tau, p-tau and aSyn in both PD and HC (Figure 2). Moreover, low baseline CSF Aβ42 in this PD cohort was overall associated with lower baseline levels of CSF t-tau and p-tau, rather than higher levels of CSF tau as in preclinical and clinical AD cohorts16. Indeed, in our unique analysis applying an established AD cut point for CSF Aβ42, we found approximately one-third of PD had pathologically-low CSF Aβ42 (PD+A). Moreover, these patients had, on average, lower p-tau levels compared to HC with pathologically-low Aβ42 (HC+A; Table 4), suggesting the profiles of CSF Aβ42 and p-tau in PD may diverge from aging and AD. Interestingly, HC+A had lower CSF t-tau and aSyn compared to HC with normal CSF Aβ42m ( HC-A, Table 4) which is opposite than expected; however, there was heterogeneity in values with higher overall range in these analytes than seen in PD. Our observed frequency of 31% of early PD with positive AD CSF biomarker profile is similar to autopsy data in end-stage PD1, but lower than a previous study using a CSF p-tau/Aβ42 ratio to designate AD positive profile10. Our findings of low CSF p-tau in PD at baseline and follow-up suggest that a CSF p-tau cut-point established in AD cohorts may underestimate the frequency of AD co-pathology in PD. This is important to consider as biomarker classification strategies are being employed in AD and related neurodegenerative conditions33.
To further clarify the biological context of our findings, we tested the association of CSF Aβ42 with APOE Ɛ4 genotype, and similar to previous studies8, 10, 11, we found lower levels in APOE Ɛ4 carriers vs non-carries for both PD and HC groups (Table 3). These data suggest our measurements are related, at least in part, to amyloid-beta pathophysiology in PD. Interestingly, we also found lower CSF aSyn in APOE Ɛ4 carriers compared to non-carriers for PD but not HC; previous autopsy work finds an association of APOE Ɛ4 with pure aSyn neuropathology34 suggesting shared genetic risk for amyloidosis and aSyn aggregation that may be reflected in our CSF findings here. Interestingly, our clinical correlations, while preliminary, found similar associations of both CSF Aβ42 and aSyn with core clinical features of PD (see below), further suggesting these biomarkers may in part reflect similar underlying pathophysiological processes in PD.
Longitudinal analysis of CSF biomarkers in PD are rare10, 12, 14 with conflicting results. One study that included 30 sporadic PD patients found lower CSF Aβ42, t-tau and p-tau in PD compared to controls at baseline and 24-month follow-up13. Whereas in 62 PD patients of the BioFINDER study, on average there was an increase in CSF t-tau and p-tau at 24 months that was most pronounced in PD patients with longer disease duration14. In a large-scale prospective PD cohort with follow-up up to 8 years there was lower CSF Aβ42 in PD patients who developed cognitive impairment with more stable levels in PD without cognitive impairment11, but neither this study, the similarly-sized DATATOP study10, or other studies above modeled longitudinal change of CSF biomarkers over time.
Here, with the first automated high-precision measurements in PD and statistical modelling to account for demographic factors in the longitudinal change in biomarkers, we find modest but measurable group-wise changes in AD CSF biomarkers over a three-year period (Table 5, Figure 3). Importantly we find that the longitudinal profile in PD diverges from HC with greater overall decrease in CSF Aβ42 and lower overall increases in CSF t-tau and p-tau by year three. We previously reported a slight increase in CSF Aβ42 and CSF p-tau in the PPMI PD cohort at year one using the AlzBio3 assay and shorter follow-up12. There are several possibilities for discrepancies in the previous literature, including the size and demographic makeup of the patient population (e.g. stage/severity of disease), statistical approach and increased precision of the automated analytical platform in this study19. Moreover, there was large individual patient variability in this study (Figure 3) and our statistical modelling helped account for demographic and APOE status which could influence longitudinal measures of CSF analytes and obscure group-wise differences using traditional cross-sectional analyses used in previous work. Indeed, our observations in HC here are congruent with previous longitudinal CSF data in cognitively normal aged patients with mild decreases in CSF Aβ42 and increases in CSF t-tau and p-tau35, 36.
It is interesting to hypothesize the mechanism for our observations of decline in CSF Aβ42 in PD; as aforementioned, while low CSF Aβ42 has been linked to amyloid-beta pathophysiology in PD7, 37, low CSF Aβ42 may have independent associations with aSyn pathology7 and perhaps in some PD patients low CSF Aβ42 is reflective of mechanisms related to underlying aSyn pathology prior to, or in absence of, the accumulation of cerebral amyloidosis. We also found CSF t-tau and p-tau had divergent longitudinal profiles from HC, with minimal change until years 2–3, where there was mild overall increase in levels compared to the greater mean increases seen in HC (Table 5). Thus, the longitudinal profile of increasing CSF t-tau and p-tau with age may be partially suppressed in the context of PD. Other longitudinal studies with more advanced PD suggest highly correlated levels CSF tau and aSyn levels may eventually increase over time in more advanced disease14 and cross-sectional work finds greater CSF t-tau and p-tau levels in PDD compared to PD without dementia38. Moreover, both CSF aSyn and tau levels are elevated in AD39, suggesting increasing neurodegeneration may lead to increased CSF tau and aSyn. Thus, future work with molecular imaging and autopsy data are needed to establish CSF cut-points to accurately detect AD co-pathology in PD for prognosis and to elucidate the underlying pathophysiological changes contributing to patterns observed here.
Our longitudinal clinical correlation analyses provide further insight into the interpretation of these CSF markers in PD. While there are currently relative mild levels of overall cognitive impairment in the PPMI PD cohort even after 5 years21, 40, we found evidence for lower baseline CSF Aβ42 to predict global cognitive decline (i.e. change in MoCA score) in PD, similar to previous work3, 5, 11, 13, 21, 41–45. Moreover, we also found more modest associations of greater baseline CSF p-tau to predict decline in MoCA score in our PD cohort, similar to one study44 but not others13, 43. One possible interpretation is that despite the overall trend of declining CSF p-tau in the PD group, there is heterogeneity and some PD patients at risk for cognitive impairment have an early increase in p-tau levels. Future work with longer follow-up can elucidate potential biomarker-defined subgroups of PD. Nonetheless, these data suggest that baseline AD CSF profiles may have prognostic value for overall incipient cognitive decline in PD.
Cognitive impairment in PD is heterogeneous and although attention, working memory, executive abilities and visuospatial dysfunction are considered to be the core clinical features in the majority of initial PD cognitive deficits22, 31, 32, episodic memory loss and language dysfunction are not uncommon and previously linked to AD pathology28–30. Thus, we hypothesized domain-specific associations of AD CSF biomarkers for episodic memory and semantic fluency but surprisingly did not find an association. Instead, we found both lower CSF Aβ42 and CSF aSyn had predictive value of cognitive decline in working memory (i.e. a core cognitive feature of PD) and decline in motor UPDRS III total and PIGD sub-scores. Moreover, CSF Aβ42 alone predicted worsening of autonomic symptoms in PD. One study of early PD similarly found lower CSF Aβ42 related to postural instability scores46 and postmortem amyloid-pathology has been linked to postural instability in PD27; however our data also conflicts with some previous work that found associations of baseline AD CSF biomarkers with measures of memory impairment5 and findings of greater baseline CSF aSyn associated with cognitive and motor decline in PD42, 47. Moreover, another study of early PD did not find an association of CSF aSyn with cognitive or motor decline48 while, p-tau/t-tau and p-tau/Aβ42 ratios have been linked to motor decline in PD in one large-scale study10. Thus, there is a complex literature on baseline CSF biomarker prediction of progression in PD with varying methodologies and patient compositions which could contribute to these discrepancies, necessitating replication with follow-up capturing end-stage disease to fully discern predictive values of CSF biomarkers in PD. Here, the effect sizes of these changes were relatively small and statistical associations marginal so these findings remain preliminary in this early stage of PD; however the overall pattern of CSF Aβ42 clinical associations with core features of PD reinforce the possibility that this analyte may reflect biological processes integral to the pathophysiology of PD.
There are several limitations to acknowledge in this study. First, while this cohort represents a unique large-scale international coordinated multicenter effort to collect standardized longitudinal assessments, findings in this dataset from a research setting require replication in independent population-based cohorts to generalize findings. The Roche Elecsys® platform has advantages of high precision (%CV values<5%), linearity of dynamic range of measurements16–19 and standard operating procedures were used for harmonized methods of CSF collection across PPMI sites; we examined the effect of needle-type used during the LP procedure and found no significant association of needle-type with any of our AD CSF biomarkers, similar to other recent work in AD49, providing further critical data to optimize large-scale multicenter biomarker efforts needed to establish CSF biomarkers for use in clinical practice. While our predictive models were robust, the magnitude of change in our clinical and biomarker values were relatively modest, likely due to the early stage of disease and relative short duration of follow-up for longitudinal biomarker values that may take decades to show progression50. Finally, future work relating CSF biomarker profiles across the full natural history of PD to in vivo measures of pathology and autopsy data is needed to fully resolve the biological context of these analytes in PD. Nonetheless our unique large-scale longitudinal data suggest a distinct CSF AD biomarker profile in early PD with relatively greater decline in CSF Aβ42 and p-tau. Moreover, we find preliminary evidence of early predictive value of subtle changes in CSF biomarkers for cognition, motor and autonomic function in PD. Further follow-up of the PPMI cohort and other ongoing longitudinal PD studies5, 11, 14, 45 will be needed to determine predictive value for clinically relevant changes.
Supplementary Material
ACKNOWLEDGEMENTS
PPMI is sponsored by the Michael J. Fox Foundation for Parkinson’s Research (MJFF) and is co‐funded by MJFF, Abbvie, Allergan, Avid Radiopharmaceuticals, Biogen, BioLegend, Bristol‐Myers Squibb, Celgene, Denali, Eli Lilly & Co., F. Hoffman‐La Roche, Ltd., GE Healthcare, Genentech, GlaxoSmithKline, Lundbeck, Merck, MesoScale, Piramal, Prevail Therapeutics, Pfizer, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, Berily, Voyager Therapeutics. We are grateful to Roche for supplying all immunoassay reagents and supplies to the University of Pennsylvania Biomarker Research Laboratory to enable measurements of CSF biomarkers using the Elecsys® β-amyloid(1-42) CSF, the Elecsys® phosphotau (181P) CSF and Elecsys® total-tau CSF on a cobas e 601 analyzer (software version 05.02). DJI is supported by NIH NINDS (NS088341) and Penn Institute on Aging as well as NIA grant AG10124 (JQT). DJI and JF were responsible for generation of figures and we thank Nicholas Cullen and Claire Peterson for their assistance.
Potential Conflicts of Interest
Dr. Mollenhauer has received honoraria for consultancy from Roche, Biogen, UCB and Sun Pharma Advanced research Company. Dr. Kieburtz reports other from Clintrex Research Corp, other from Hoover Brown LLC, outside the submitted work; Dr. Galasko reports personal fees from Biogen, Inc, personal fees from vTv Pharmaceuticals, Inc, personal fees from Fujirebio, Inc, personal fees from Cognition therapeutics, outside the submitted work; .Dr. Simuni reports grants from Biogen, Roche, Neuroderm, Sanofi, Sun Pharma, Abbvie, IMPAX, Prevail, other from Acadia, Abbvie, Accorda , Adamas, Allergan, Amneal, Aptinyx, Denali, General Electric (GE) , Kyowa, Neuroderm, Neurocrine , Sanofi, Sinopia, Sunovion, Roche, Takeda, Voyager, US World Meds, during the conduct of the study; Dr. Tanner reports grants from Gateway LLC, grants from Roche/Genentech, grants and personal fees from Biogen Idec, personal fees from Acorda, personal fees from Adamas Therapeutics, personal fees from Amneal, personal fees from CNS Ratings, personal fees from Grey Matter LLC, personal fees from Northwestern University, personal fees from Partners, Harvard U, outside the submitted work; .Dr. Marek reports consulting from Michael J Fox, GE Heathcare, Takeda, Lundbeck, Neuron23, Roche, Neuroderm, o Invicro, outside the submitted work; PPMI is supported in part by Roche who manufacture the Elecsys® assays used in the study.
Footnotes
Data used in the preparation of this article were obtained from the PPMI database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org.
REFERENCES
- 1.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Annals of neurology. 2012. October;72(4):587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet neurology. 2017. January;16(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid {beta} 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010. September 21;75(12):1055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montine TJ, Shi M, Quinn JF, et al. CSF Abeta(42) and tau in Parkinson’s disease with cognitive impairment. Movement disorders : official journal of the Movement Disorder Society. 2010. November 15;25(15):2682–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alves G, Bronnick K, Aarsland D, et al. CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson’s disease: the Norwegian ParkWest study. Journal of neurology, neurosurgery, and psychiatry. 2010. October;81(10):1080–6. [DOI] [PubMed] [Google Scholar]
- 6.Compta Y, Marti MJ, Ibarretxe-Bilbao N, et al. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2009. November 15;24(15):2203–10. [DOI] [PubMed] [Google Scholar]
- 7.Irwin DJ, Xie SX, Coughlin D, et al. CSF tau and beta-amyloid predict cerebral synucleinopathy in autopsied Lewy body disorders. Neurology. 2018. March 20;90(12):e1038–e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang JH, Mollenhauer B, Coffey CS, et al. CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: the Parkinson’s Progression Markers Initiative study. Acta neuropathologica. 2016. June;131(6):935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang JH, Irwin DJ, Chen-Plotkin A, et al. Association of cerebrospinal fluid Aβ1–42, t-tau, p-tau181 and α-synuclein levels with clinical features of early drug naïve Parkinson’s disease patients. JAMA neurology. 2013;in press; doi: 10.1001/jamaneurol.2013.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Mattison HA, Liu C, et al. Longitudinal assessment of tau and amyloid beta in cerebrospinal fluid of Parkinson disease. Acta neuropathologica. 2013. November;126(5):671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lerche S, Wurster I, Roben B, et al. Parkinson’s disease: evolution of cognitive impairment and CSF Abeta1–42 profiles in a prospective longitudinal study. Journal of neurology, neurosurgery, and psychiatry. 2019. February;90(2):165–70. [DOI] [PubMed] [Google Scholar]
- 12.Mollenhauer B, Caspell-Garcia CJ, Coffey CS, et al. Longitudinal CSF biomarkers in patients with early Parkinson disease and healthy controls. Neurology. 2017. November 7;89(19):1959–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brockmann K, Schulte C, Deuschle C, et al. Neurodegenerative CSF markers in genetic and sporadic PD: Classification and prediction in a longitudinal study. Parkinsonism & related disorders. 2015. December;21(12):1427–34. [DOI] [PubMed] [Google Scholar]
- 14.Hall S, Surova Y, Ohrfelt A, et al. Longitudinal Measurements of Cerebrospinal Fluid Biomarkers in Parkinson’s Disease. Movement disorders : official journal of the Movement Disorder Society. 2016. June;31(6):898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta neuropathologica. 2011. May;121(5):597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw LM, Waligorska T, Fields L, et al. Derivation of cutoffs for the Elecsys((R)) amyloid beta (1–42) assay in Alzheimer’s disease. Alzheimers Dement (Amst). 2018;10:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rozga M, Bittner T, Hoglund K, Blennow K. Accuracy of cerebrospinal fluid Abeta1–42 measurements: evaluation of pre-analytical factors using a novel Elecsys immunosassay. Clinical chemistry and laboratory medicine : CCLM / FESCC. 2017. August 28;55(10):1545–54. [DOI] [PubMed] [Google Scholar]
- 18.Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-beta PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2018. November;14(11):1470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of beta-amyloid (1–42) in human cerebrospinal fluid. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2016. May;12(5):517–26. [DOI] [PubMed] [Google Scholar]
- 20.The Parkinson Progression Marker Initiative (PPMI). Progress in neurobiology. 2011. December;95(4):629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspell-Garcia C, Simuni T, Tosun-Turgut D, et al. Multiple modality biomarker prediction of cognitive impairment in prospectively followed de novo Parkinson disease. PloS one. 2017;12(5):e0175674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weintraub D, Simuni T, Caspell-Garcia C, et al. Cognitive performance and neuropsychiatric symptoms in early, untreated Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2015. June;30(7):919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fritz CO, Morris PE, Richler JJ. Effect size estimates: current use, calculations, and interpretation. J Exp Psychol Gen. 2012. February;141(1):2–18. [DOI] [PubMed] [Google Scholar]
- 24.Stewart T, Shi M, Mehrotra A, et al. Impact of Pre-Analytical Differences on Biomarkers in the ADNI and PPMI Studies: Implications in the Era of Classifying Disease Based on Biomarkers. J Alzheimers Dis. 2019;69(1):263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw LM, Hansson O, Manuilova E, et al. Method comparison study of the Elecsys(R) beta-Amyloid (1–42) CSF assay versus comparator assays and LC-MS/MS. Clinical biochemistry. 2019. October;72:7–14. [DOI] [PubMed] [Google Scholar]
- 26.Wider C, Ross OA, Nishioka K, et al. An evaluation of the impact of MAPT, SNCA and APOE on the burden of Alzheimer’s and Lewy body pathology. Journal of neurology, neurosurgery, and psychiatry. 2012. April;83(4):424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson’s disease. Brain : a journal of neurology. 2009. November;132(Pt 11):2947–57. [DOI] [PubMed] [Google Scholar]
- 28.Coughlin D, Xie SX, Liang M, et al. Cognitive and Pathological Influences of Tau Pathology in Lewy Body Disorders. Annals of neurology. 2019. February;85(2):259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraybill ML, Larson EB, Tsuang DW, et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology. 2005. June 28;64(12):2069–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peavy GM, Edland SD, Toole BM, Hansen LA, Galasko DR, Mayo AM. Phenotypic differences based on staging of Alzheimer’s neuropathology in autopsy-confirmed dementia with Lewy bodies. Parkinsonism Relat Disord. 2016. October;31:72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2007. September 15;22(12):1689–707; quiz 837. [DOI] [PubMed] [Google Scholar]
- 32.Litvan I, Goldman JG, Troster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Movement disorders : official journal of the Movement Disorder Society. 2012. March;27(3):349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jack CR Jr., Bennett DA, Blennow K, et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016. August 2;87(5):539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuang D, Leverenz JB, Lopez OL, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA neurology. 2013. February;70(2):223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta neuropathologica. 2013. November;126(5):659–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fagan AM, Mintun MA, Shah AR, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO molecular medicine. 2009. November;1(8–9):371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buongiorno M, Antonelli F, Compta Y, et al. Cross-Sectional and Longitudinal Cognitive Correlates of FDDNP PET and CSF Amyloid-beta and Tau in Parkinson’s Disease1. Journal of Alzheimer’s disease : JAD. 2017;55(3):1261–72. [DOI] [PubMed] [Google Scholar]
- 38.Hall S, Ohrfelt A, Constantinescu R, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Archives of neurology. 2012. November;69(11):1445–52. [DOI] [PubMed] [Google Scholar]
- 39.Shi M, Tang L, Toledo JB, et al. Cerebrospinal fluid alpha-synuclein contributes to the differential diagnosis of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2018. March 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marek K, Chowdhury S, Siderowf A, et al. The Parkinson’s progression markers initiative (PPMI) - establishing a PD biomarker cohort. Annals of clinical and translational neurology. 2018. December;5(12):1460–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrag A, Siddiqui UF, Anastasiou Z, Weintraub D, Schott JM. Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: a cohort study. Lancet Neurol. 2017. January;16(1):66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall S, Surova Y, Ohrfelt A, Zetterberg H, Lindqvist D, Hansson O. CSF biomarkers and clinical progression of Parkinson disease. Neurology. 2015. January 6;84(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terrelonge M Jr., Marder KS, Weintraub D, Alcalay RN. CSF beta-Amyloid 1–42 Predicts Progression to Cognitive Impairment in Newly Diagnosed Parkinson Disease. Journal of molecular neuroscience : MN. 2016. January;58(1):88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu C, Cholerton B, Shi M, et al. CSF tau and tau/Abeta42 predict cognitive decline in Parkinson’s disease. Parkinsonism & related disorders. 2015. March;21(3):271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alves G, Lange J, Blennow K, et al. CSF Abeta42 predicts early-onset dementia in Parkinson disease. Neurology. 2014. May 20;82(20):1784–90. [DOI] [PubMed] [Google Scholar]
- 46.Alves G, Pedersen KF, Bloem BR, et al. Cerebrospinal fluid amyloid-beta and phenotypic heterogeneity in de novo Parkinson’s disease. Journal of neurology, neurosurgery, and psychiatry. 2013. May;84(5):537–43. [DOI] [PubMed] [Google Scholar]
- 47.Stewart T, Liu C, Ginghina C, et al. Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. The American journal of pathology. 2014. April;184(4):966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forland MG, Ohrfelt A, Dalen I, et al. Evolution of cerebrospinal fluid total alpha-synuclein in Parkinson’s disease. Parkinsonism & related disorders. 2018. April;49:4–8. [DOI] [PubMed] [Google Scholar]
- 49.Rembach A, Evered LA, Li QX, et al. Alzheimer’s disease cerebrospinal fluid biomarkers are not influenced by gravity drip or aspiration extraction methodology. Alzheimer’s research & therapy. 2015. November 19;7(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jack CR Jr., Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet neurology. 2013. February;12(2):207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.