

Abstract
Metastatic breast cancer (MBC) is an extremely recalcitrant disease capable of bypassing current targeted therapies via engagement of several growth promoting pathways. SH2 containing protein tyrosine phosphatase-2 (SHP2) is an oncogenic phosphatase known to facilitate growth and survival signaling downstream of numerous receptor inputs. Herein, we used inducible genetic depletion and two distinct pharmacological inhibitors to investigate the therapeutic potential of targeting SHP2 in MBC. Cells that acquired resistance to the ErbB kinase inhibitor, neratinib, displayed increased phosphorylation of SHP2 at the Y542 activation site. Additionally, higher levels of SHP2 phosphorylation, but not expression, were associated with decreased survival of breast cancer patients. Pharmacological inhibition of SHP2 activity blocked ERK1/2 and AKT signaling generated from exogenous stimulation with FGF2, PDGF and hGF and readily prevented MBC cell growth induced by these factors. SHP2 was also phosphorylated upon engagement of the extracellular matrix (ECM) via focal adhesion kinase. Consistent with the potential of SHP2-targeted compounds as therapeutic agents, the growth inhibitory property of SHP2 blockade was enhanced in ECM-rich 3D culture environments. In vivo blockade of SHP2 in the adjuvant setting decreased pulmonary metastasis and extended the survival of systemic tumor-bearing mice. Finally, inhibition of SHP2 in combination with FGFR-targeted kinase inhibitors synergistically blocked the growth of MBC cells. Overall, our findings support the conclusion that SHP2 constitutes a shared signaling node allowing MBC cells to simultaneously engage a diversity of growth and survival pathways, including those derived from the ECM.
Introduction
Stage IV, metastatic breast cancer (MBC) is a major clinical challenge, with five year survival rates currently at ~23% [1]. Tyrosine kinase inhibitors targeting human epidermal growth factor receptor 2 (HER2), such as lapatinib and neratinib, can provide therapeutic benefit to HER2+ patients. However, inherent and acquired drug resistance inevitably occur in the metastatic setting, leading to further disease progression [2–6]. Given the relationship between drug resistance and metastasis, there exists the possibility that the underlying drivers of these processes may be shared. Identification and pharmacological inhibition of the shared targets of drug resistance and metastasis remains a major barrier to the development of effective therapeutics for the treatment of late-stage disease.
Multiple experimental studies, including findings from our lab, describe a role for fibroblast growth factor receptor 1 (FGFR1) in acquired and inherent resistance to HER2-targeted therapeutics [7–10]. The locus encoding FGFR1 is amplified in 14% of breast cancer patients, and FGFR1 expression can be further upregulated through the process of epithelial-mesenchymal transition (EMT), a key driver of both drug resistance and metastasis [11, 12]. In addition to FGFR, multiple other receptor tyrosine kinases (RTKs) have been linked to both drug resistance and metastasis, including PDGFR, VEGFR, EGFR, and cMET [13–15]. Finally, active signaling events generated through integrin-mediated sensing of the extracellular matrix (ECM) can function independently and in conjunction with RTKs to support the growth and survival of MBC cells in the presence of targeted therapies and chemotherapies [16, 17]. Therefore, we sought to identify signaling nodes shared between multiple RTKs and ECM signaling events as targeting these nodes may hold the key to the development of MBC therapies.
Src homology region 2 (SH2)-containing protein tyrosine phosphatase-2 (SHP2) is an oncogenic tyrosine phosphatase involved in downstream signaling of several RTKs, and was therefore chosen for analysis as a shared signaling node [18–21]. SHP2 exists in an inactivated form due to an intramolecular allosteric interaction between the N-terminal SH2 domain and the C-terminal phosphatase domain. Phosphorylation at Y542 releases this inhibition and the molecule unfolds, becoming enzymatically active [22, 23]. The phosphatase activity of SHP2 is clearly required for critical cancer-associated signaling pathways such as RAS and PI3K, but the biochemical mechanisms of how SHP2 supports oncogenic signaling still remain to be definitively determined [24, 25].
Two types of small molecule inhibitors of SHP2 have recently emerged. SHP099 is an allosteric inhibitor of SHP2, and we have recently developed 11a-1, as an active-site inhibitor of SHP2 [26–28]. Genetic deletion and pharmacological inhibition of SHP2 have been reported to block the growth of primary tumors in breast cancer xenograft and genetically engineered mouse models [29–32]. Given recent findings around the function of SHP2, and the established pharmacological tools to therapeutically inhibit its activity, we sought to address the hypothesis that SHP2 will be an effective therapeutic target for drug-resistant MBC [33].
Herein, we demonstrate that SHP2 becomes phosphorylated through ECM signaling, correlating with enhanced efficacy of SHP2 inhibitors in 3D growth environments as compared to traditional 2D culture. In vivo, SHP2 inhibition blocked proliferation of MBC cells and delayed pulmonary metastatic progression, a result that could be enhanced by combination with FGFR-targeted kinase inhibitors. Overall, we posit that combined inhibition of FGFR and SHP2 may be an effective treatment strategy for MBC.
Results
SHP2 facilitates in vitro growth of metastatic breast cancer cells
We used three independent doxycycline-inducible shRNAs targeting PTPN11 to manipulate SHP2 expression in two MBC cell lines. The 4T1 and D2.A1 cells are murine models of stage IV breast cancer, capable of producing systemic metastases following orthotopic engraftment of mammary fat pad tumors. Similar to human disease, previous studies from our lab and others indicate that the metastatic phenotypes in these cells are driven by a variety of RTK and ECM signaling events [12, 34–36]. Upon stable transduction with inducible shRNA expression constructs, these cells were transiently treated with doxycycline and GFP+ cells were isolated by FACS (Supplementary Fig. 1a–f). Using this approach we were able to deplete SHP2 levels by 79.8% in the 4T1 cells and 89.5% in the D2.A1 cells upon addition of doxycycline (Fig. 1a–c and Supplementary Fig. 1g and 1h). We next utilized these depletion systems to perform growth assays under 2D and 3D culture conditions. As shown in figure 1d, cells were either cultured directly on tissue culture polystyrene for 2D culture conditions, or cells were placed at the interface of two semi-solid layers of ECM matrix for 3D culture conditions (Fig. 1d). Depletion of SHP2 significantly inhibited the growth of the 4T1 cells under both 2D and 3D culture conditions, but the growth inhibitory effects of SHP2 depletion were only observed when D2.A1 cells were cultured under 3D conditions (Fig. 1e–g). The specific growth inhibitory effects of SHP2 depletion under 3D culture conditions led us to further investigate the functions of SHP2 in ECM signaling.
Figure. 1.
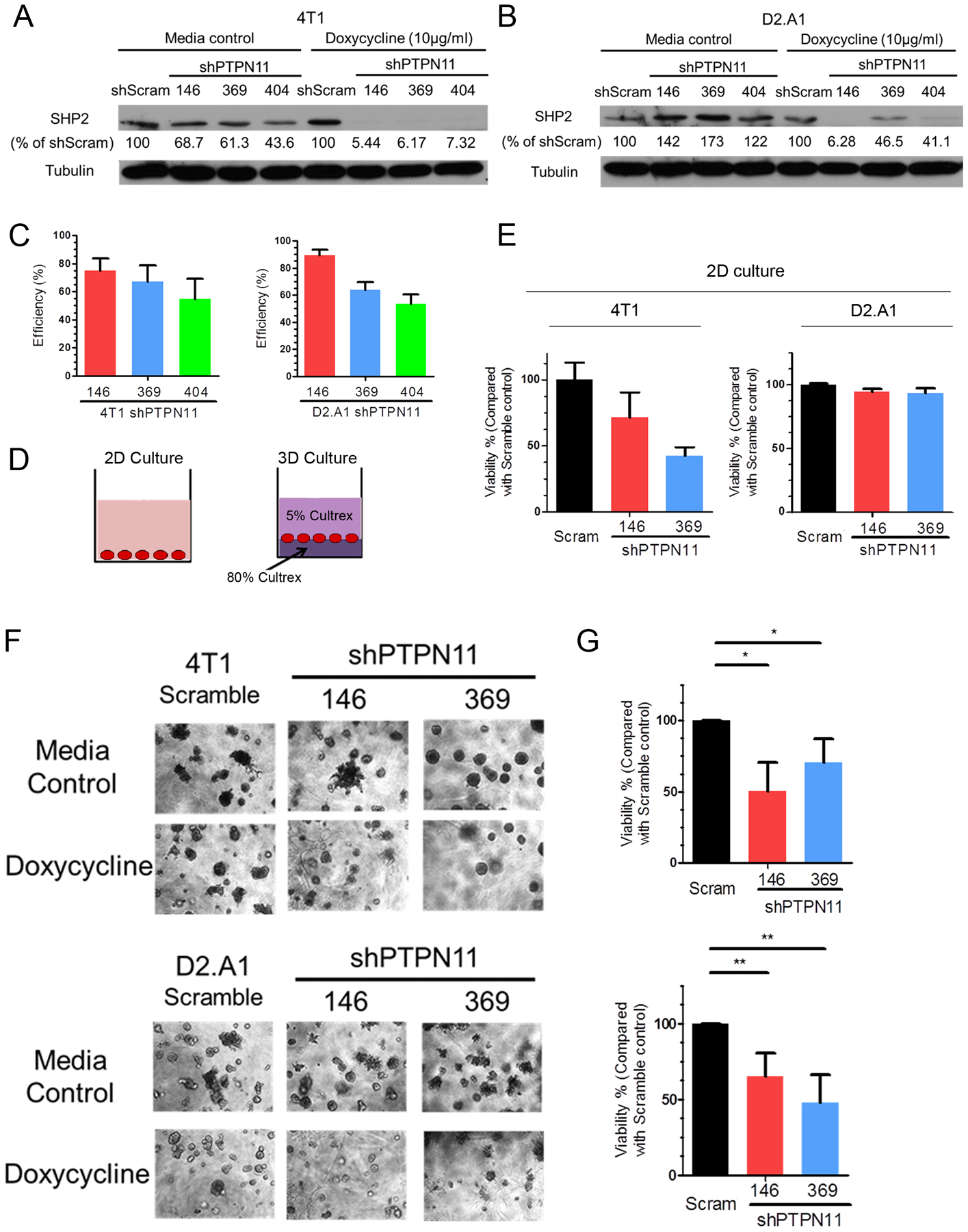
Depletion of SHP2 inhibits the growth of metastatic breast cancer cells. a, b Immunoblot analyses with quantification for SHP2 in 4T1 and D2.A1 cells stably expressing three independent doxycycline-inducible shRNA sequences targeting PTPN11 with and without doxycycline induction, compared with a scrambled (scram) shRNA control. c Quantification of SHP2 depletion for each shRNA construct in 4T1 and D2.A1 cells (n=3). Depletion efficiency was defined as the percentage of SHP2 protein decrease normalized to scramble controls upon doxycycline induction. d Schematic representation of 2D and 3D growth conditions. e 2D growth assays showing differential cell viability of 4T1 and D2.A1 cells upon doxycycline-induced depletion of SHP2. Data are the mean ± s.e.m. of two biological replicates completed in triplicate. f Representative photos showing 3D morphologies of 4T1 and D2.A1 cells upon depletion of SHP2. g Quantification of differential cell viability of 4T1 and D2.A1 cells upon depletion of SHP2. Data are shown as mean ± s.e.m. (n = 3 for 4T1; *p < 0.05; n = 4 for D2.A1, **p<0.01)
Pharmacologic targeting of SHP2 decreases pulmonary metastasis
To further identify whether targeting SHP2 can inhibit metastatic tumor growth we implemented an adjuvant treatment protocol in conjunction with the 4T1 orthotopic model of metastasis (Fig. 2a). This approach recapitulates a surgical intervention to remove the primary tumor followed by pharmacological inhibition of residual systemic disease [37]. Following surgical resection of the primary tumor, systemic tumor bearing animals were treated with the allosteric SHP2 inhibitor, SHP099, or vehicle via oral gavage for a period of 11 days. This treatment led to a significant reduction in outgrowth of pulmonary metastases as determined by bioluminescent imaging, ex vivo quantification of pulmonary metastatic nodules, H&E staining of histological sections, and overall pulmonary weight (Fig. 2b–f).
Figure 2.
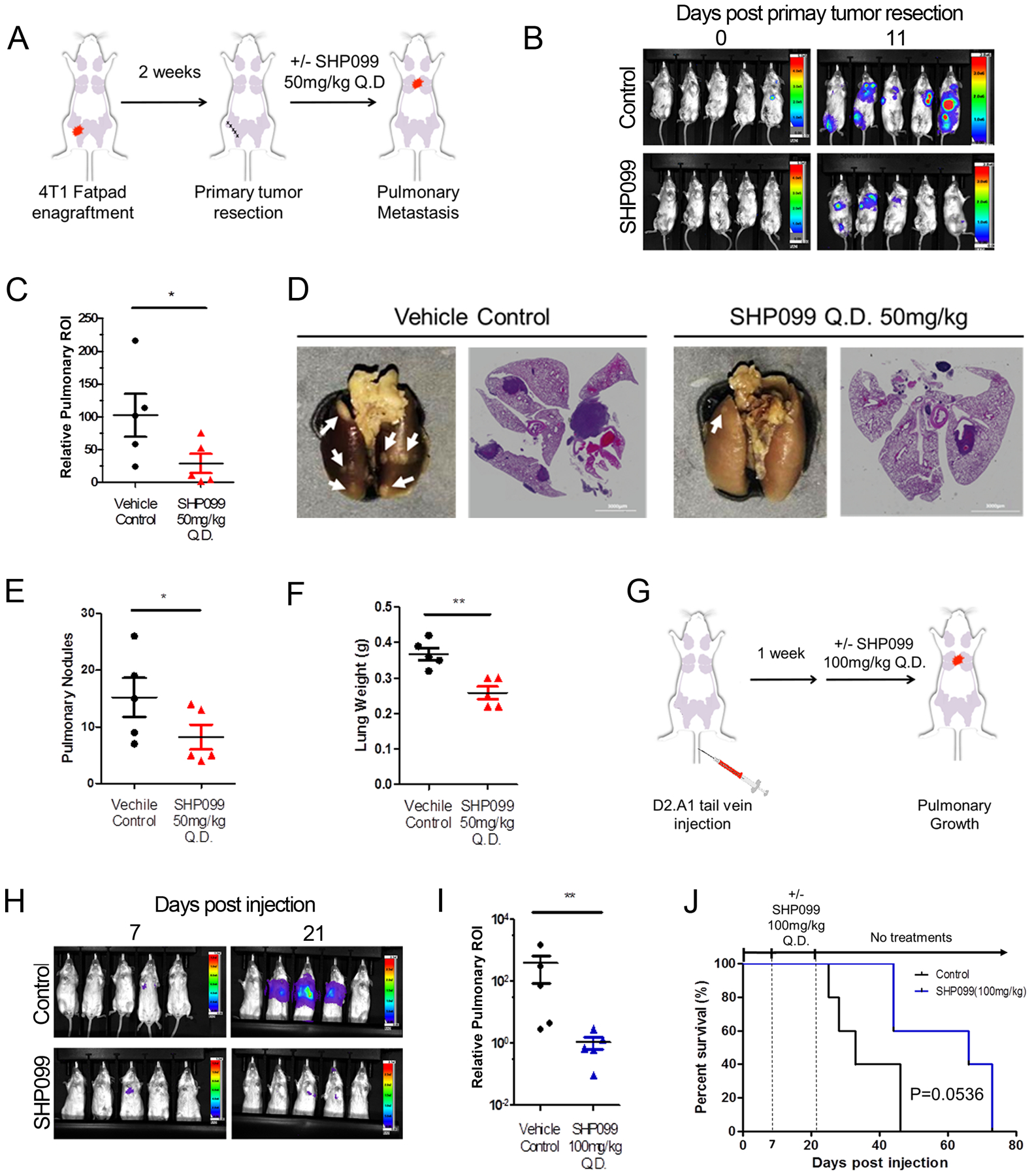
Pharmacological inhibition of SHP2 blocks in vivo pulmonary metastasis. a Schematic representation of the 4T1 post-surgical model of metastasis. b Representative bioluminescent images taken at Day 0 and Day 11 of SHP099 treatment. c Bioluminescent values of pulmonary regions of interest (ROI) were quantified as a measure of metastasis. Data are the ratio of luminescence values at Day 11 of treatment compared to Day 0. (*p<0.05 for n=5 mice per group). d Representative photos of pulmonary metastases (arrow heads) and H&E staining of pulmonary histological sections in control and SHP099 treated mice. e, f Plots comparing numbers of pulmonary nodules and lung weights from control and SHP099 treated mice (*p<0.05, **p<0.01, n=5 mice per group). g Schematic of the D2.A1 model of pulmonary tumor growth. h Representative images of pulmonary growth monitored by bioluminescence at Day 7 and Day 21 post injection. i Bioluminescent values from pulmonary ROI quantified as the ratio of day 21 to day 7 following tumor cell injection. (**p<0.01, n=5 mice per group). j Kaplan-meier analyses of control and SHP099 treated mice, bearing D2.A1 pulmonary tumors, resulting in the indicated P value (n=5 mice per group).
To further determine the efficacy of SHP099 in the pulmonary microenvironment, we inoculated mice with D2.A1 cells via a tail vein injection (Fig. 2g). One week after tail vein engraftment, mice were again treated with SHP099 and pulmonary tumor growth was quantified via bioluminescence. Consistent with what we observed in the 4T1 model, 14 days of SHP099 therapy resulted in a significant decrease in pulmonary tumor burden (Fig. 2h and 2i). Finally, this 14-day SHP099 treatment period also extended the subsequent survival of mice as compared with the vehicle control group (Fig. 2j).
Three dimensional culture enhances growth inhibition by SHP2 blockade
Genetic depletion of SHP2 inhibited cell growth to a greater extent under 3D culture conditions as compared to 2D culture (Fig. 1e–g). Therefore, we sought to define the impact of extracellular matrix signaling on SHP2. To pharmacologically recapitulate our results from figure 1, we treated 4T1 cells with two small molecule inhibitors of SHP2 under the 2D and 3D culture conditions described in figure 1d. Treatment with effective concentrations of either 11a-1 or SHP099 under serum containing conditions failed to significantly inhibit the growth of 4T1 cells in 2D culture (Fig. 3a). In contrast, use of these SHP2 inhibitors at the same concentrations significantly decreased the growth of 4T1 cells in 3D culture (Fig. 3b). Similar results were also observed in several other MBC cell lines (Supplementary Fig. 2).
Figure 3.
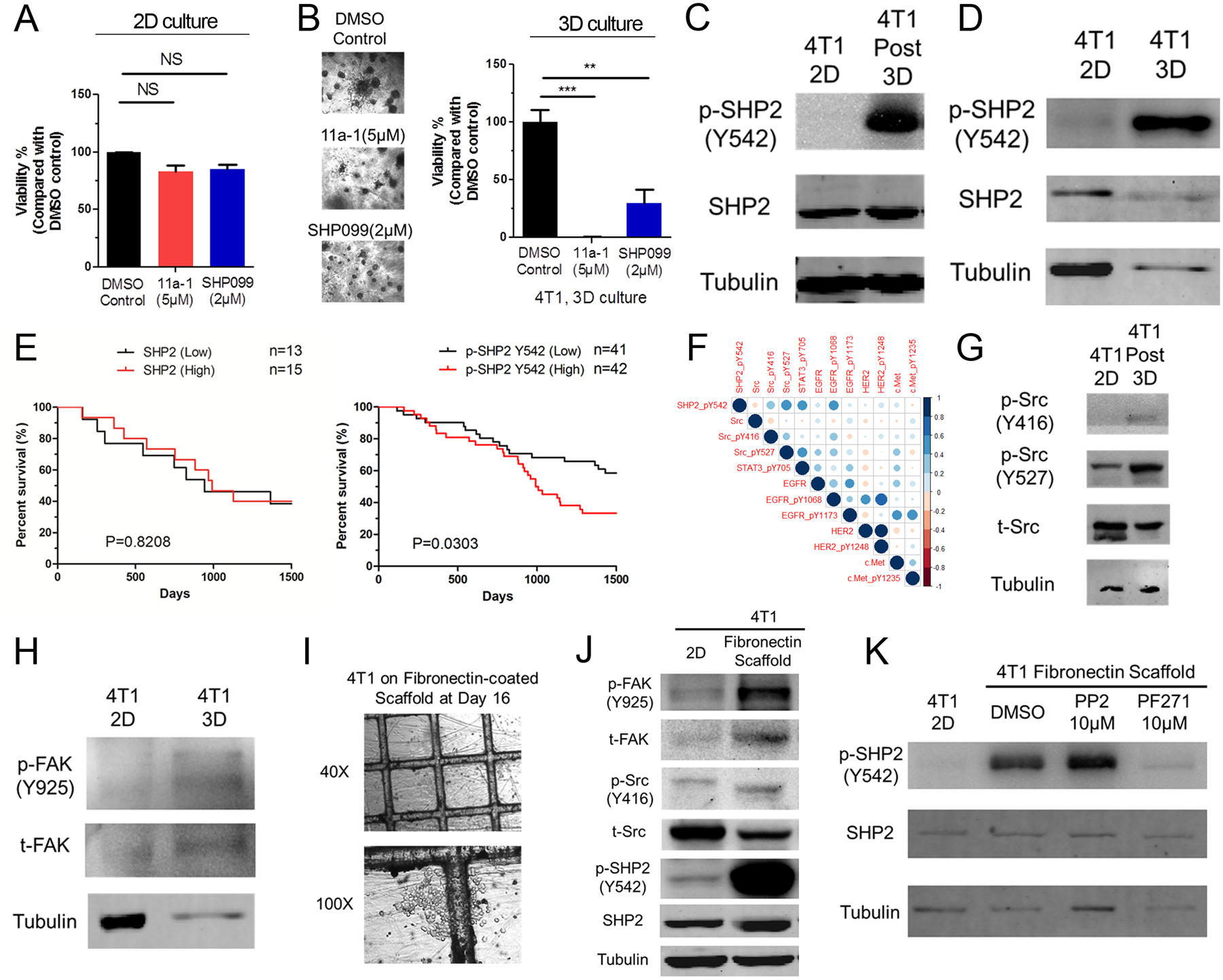
The extracellular matrix promotes SHP2 phosphorylation. a Cell viability of 4T1 cells cultured in 2D for 6 days in the absence or presence of the indicated concentrations of 11a-1 and SHP099. b Representative photomicrographs and quantification of 4T1 cell viability after 16 days in 3D culture in the absence or presence of the indicated concentrations of 11a-1 and SHP099. For panels A and B data are normalized to untreated controls and are the mean ± s.e.m. of three experiments where **p<0.01 and ***p<0.001. c, d Immunoblot analyses showing differential phosphorylation of SHP2 at Y542 in 4T1 cells following 3D culture (C) or in 4T1 cells lysed directly from gel-based 3D culture (D). e Kaplan-Meier analyses of patients from the TCGA breast cancer dataset separated into two groups based on the median mRNA expression value of SHP2 (left) or median phosphorylation levels of SHP2 at Y542 (right). Overall survival was analyzed by a log-rank test resulting in the indicated p-values. f RPPA data from the TCGA breast cancer dataset were analyzed for correlation of total expression levels and post translational modifications of the indicated proteins in relation to SHP2-Y542 phosphorylation. The heat map indicates the Pearson correlation coefficient and the size of the circle is representative of the value. g Immunoblot analyses showing differential phosphorylation of Src in 4T1 cells following culture under gel-based 3D conditions as compared to 2D culture. h Immunoblot analyses showing differential phosphorylation of FAK at Y925 in 4T1 cells isolated directly from gel-based 3D cultures as compared to 2D culture. i Representative photomicrographs showing 4T1 cells cultured on FN-coated tessellated scaffolds. j Immunoblot analyses showing differential phosphorylation of FAK, Src and SHP2 in 4T1 cells cultured on FN-coated scaffolds as compared to 2D culture. k 4T1 cells were cultured on FN-coated scaffolds for 16 days and treated with the indicated concentrations of a Src inhibitor (PP2) or a FAK inhibitor (PF271) for the last 24 hours before harvesting cells for immunoblot analyses of SHP2 phosphorylation at Y542. All immunoblots shown are representative of at least 3 independent experiments.
Given this enhanced reliance of cells on SHP2 for growth in 3D culture we next assessed changes in SHP2 phosphorylation upon cellular engagement of the ECM [22]. Phosphorylation of SHP2 at Y542 was clearly enhanced when 4T1 cells were cultured under 3D conditions (Fig. 3c and 3d). We also observed induction of SHP2 Y542 phosphorylation in D2.A1 cells upon 3D culture and in 4T1 ex vivo lysates derived from lung metastases (Supplementary Fig. 3a and 3b). Consistent with these data, analysis of the TCGA dataset indicated differential expression levels of SHP2 were not predictive of patient survival, but elevated phosphorylation of SHP2 at Y542 was associated with decreased breast cancer patient survival (Fig. 3e). Further analysis of the TCGA dataset demonstrated a positive correlation between SHP2 Y542 phosphorylation and that of EGFR, STAT3 and Src phosphorylation (Fig. 3f). As Src is a key enzyme in ECM signaling, we focused on characterizing the relationship between Src and SHP2. Similar to SHP2, phosphorylation of Src at Y416 was increased in 4T1 cells following 3D culture (Fig. 3g). We also noted that 3D culture conditions enhanced phosphorylation of focal adhesion kinase (FAK) (Fig. 3h). Therefore, we next investigated whether Src and FAK contributed to SHP2 phosphorylation. To do this we utilized our recently developed tessellated scaffold system [38]. In the gel-based 3D culture system described previously ECM proteins are largely in a globular form. However, our tessellated system allows for creation of fibular networks of ECM matrixes that are more characteristic of the in vivo condition. Additionally, the absence of gel allows for more efficient cell recovery, making the system more suitable for signaling analyses (Fig. 3i). Using this culture system, we found that phosphorylation of SHP2 at Y542 was elevated upon engagement of fibrillar fibronectin. This event could be ablated by addition of the FAK inhibitors PF-562271 (PF271) or defactinib, but not by the Src inhibitor PP2 (Fig. 3j, 3k and Supplementary Fig. 3e). We similarly observed FAK-dependent phosphorylation of SHP2 in the D2.A1 cell line when cultured on fibrillar fibronectin (Supplementary Fig. 3c–d and 3f–3g). In summary, these data suggest that phosphorylation of SHP2 at Y542 is associated with disease progression and that this event can be mediated by the ECM in a FAK-dependent manner.
Inhibition of SHP2 targets the growth of drug resistant HER2+ breast cancer cells
We have previously reported that acquisition of resistance to lapatinib in HER2-transformed mammary epithelial cells (HME2) results in a mesenchymal phenotype that includes upregulation of FGFR1 and several ECM proteins, including fibronectin [10, 38]. Addition of SHP2 inhibitors to a low dose of the pan-ErbB inhibitor, neratinib, could significantly reduce the growth of the parental HER2-transformed cells (Supplementary Fig. 4). However, given our results in Figure 3, we hypothesized that SHP2 also facilitates resistance to ErbB-targeted kinase inhibitors by facilitating bypass ECM and alternate RTK signaling. Therefore we first validated that our lapatinib resistant (LAPR) cells were independent of all ErbB signaling by demonstrating they were similarly resistant to neratinib (Fig. 4a). The LAPR cells also displayed elevated phosphorylation of SHP2 at Y542 compared to the drug sensitive parental cell line (Fig. 4b). Furthermore, pharmacological inhibition of SHP2 preferentially inhibited the growth of the LAPR cells as compared to the parental cells (Fig. 4c and 4d). Next, we focused on signaling recovery events following ErbB blockade. The addition of neratinib clearly prevented phosphorylation of HER2 in both the parental and LAPR cells, but downstream phosphorylation of AKT and ERK1/2 recovered quicker in LAPR cells compared to parental cells (Fig. 4e). These results are consistent with acquisition of bypass drivers of cell growth. To elucidate the mediators of ErbB bypass signaling we exposed LAPR cells to neratinib for 1 hour, the drug was washed-off, and cells were subsequently allowed to recover in the presence or absence of compounds targeting SHP2 or FGFR for an additional 12 hours (Fig. 4f). Following this transient treatment with neratinib recovery of AKT and ERK1/2 phosphorylation were delayed in the presence of SHP2 or FGFR inhibitors (Fig. 4f).
Figure 4.
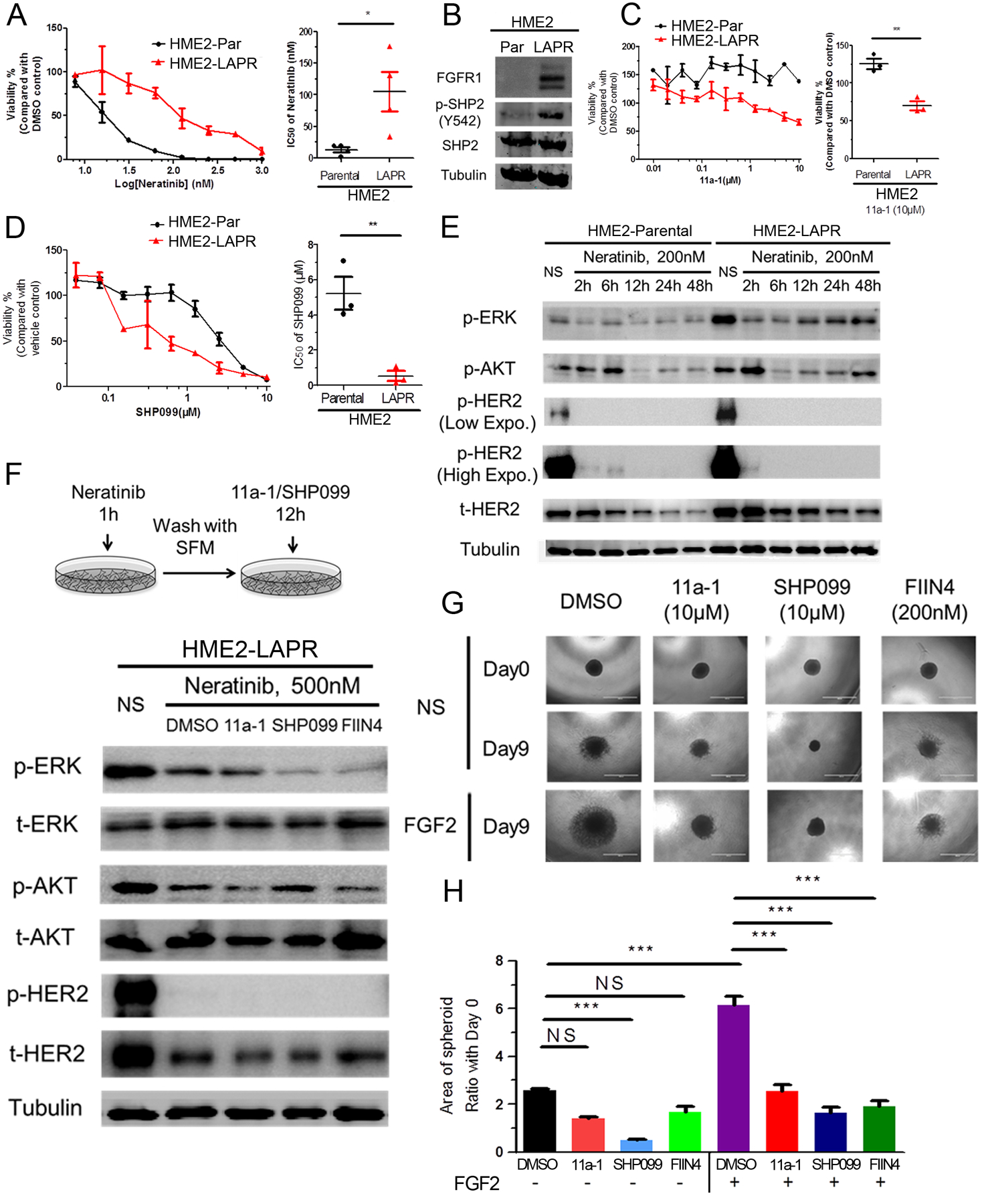
Drug resistant breast cancer cells can be targeted by inhibition of SHP2. a Representative dose response of HME2 parental (HME2-Par) and lapatinib resistant (HME2-LAPR) cells treated with neratinib for 6 days. (right) The IC50 values for each independent experiment were calculated (n=4) and analyzed using a two-tail student t-test where *p<0.05. b Immunoblot analyzes showing differential expression of FGFR1 and phosphorylation of SHP2 at Y542 in HME2-LAPR cells compared to their HME2-Par counterparts. c Representative dose response upon 6 days treatment with 11a-1. (right) Cell viability upon treatment with 10 μM 11a-1 for each independent experiment was calculated (n=4) and analyzed using a two-tail student t-test where **p<0.01. d Representative dose response curves of HME2 parental (HME2-Par) and HME2-LAPR cells treated with SHP-099 for 6 days. (right) The IC50 values for each independent experiment were calculated (n=3) and analyzed using a two-tail student t-test where **p<0.01. e Immunoblotting showing phosphorylation of AKT, ERK1/2, and HER2 in HME2-Par and HME2-LAPR cells upon the indicated neratinib treatments. f (top) Schematic representation of the signaling recovery assays. HME2-LAPR cells were treated with neratinib for 1 hour, the drug was removed, and the cells were allowed to recover in serum free media in the presence or absence of SHP2 or FGFR inhibitors for 12 hours. DMSO was used as a vehicle control. (bottom) Recovery of AKT, ERK1/2, and HER2 phosphorylation were analyzed by immunoblot. g, h HME2-LAPR spheres were formed in a round bottom plate and then plated onto a bed of gel matrix in the presence of the indicated concentrations of SHP2 or FGFR inhibitors. The growth of spheroids was further induced by addition of exogenous FGF2 (20ng/ml). The area of the sphere 9 days after placement on the ECM was measured, and these values were normalized to the initial sphere size (Day 0). Data are the mean ± s.e.m. (n = 3) were ***p<0.001.
To further define the role of FGFR:SHP2 signaling in promoting the growth of drug resistant cells, we used a 3D tumor sphere assay. We recently described this assay, which consists of forming a large multicellular sphere of tumor cells using a non-adherent round bottom plate, followed by transferring the sphere to a bed of ECM to allow for tumor growth and invasion [39]. In these ECM-rich culture conditions, the growth of LAPR spheres was significantly increased by addition of exogenous FGF2. This FGF2-driven growth was abolished by inhibition of SHP2 or, as a control, direct targeting of FGFR kinase activity (Fig. 4g and 4h). These results indicate that in the presence of the ECM, SHP2 activity is required for FGFR signaling to facilitate HER2-independent cell growth and resistance to ErbB-targeted compounds.
SHP2 facilitates signaling from multiple receptor tyrosine kinases
We next sought to investigate whether SHP2 facilitates proliferative signaling from additional RTKs that are also known to be involved in drug resistance and metastasis. We first detected the expression levels of multiple RTKs in several MBC cell lines and determined the ability of their cognate ligands to induce SHP2 phosphorylation (Supplementary Fig. 5). In the metastatic D2.A1 cells, transient addition of FGF2, platelet-derived growth factor (PDGF) and hepatocyte growth factor (hGF) could induce SHP2 phosphorylation at Y542 and downstream ERK1/2 phosphorylation (Fig. 5a). FGF2 induced the growth of D2.A1 cells under both 2D and 3D conditions while hGF and PDGF were only growth promoting under 2D and 3D culture conditions, respectively (Fig. 5b and 5c). Depletion of SHP2 significantly blocked cell growth induced by these specific growth factors (Fig. 5d and 5e). These results could also be recapitulated using SHP2-targeted inhibitors (Supplementary Fig. 6). To illustrate the downstream signaling events facilitated by SHP2, we performed ligand-induced signaling assays in the presence of SHP099. Phosphorylation of AKT and ERK1/2 induced by FGF2, PDGF and hGF could all be reduced by pretreatment with SHP099 (Fig. 5f–h). Finally, unlike what we observed upon ECM-mediated signaling, growth factor-mediated phosphorylation of SHP2 could be readily blocked by the addition of PP2 (Fig. 5i).
Figure 5.
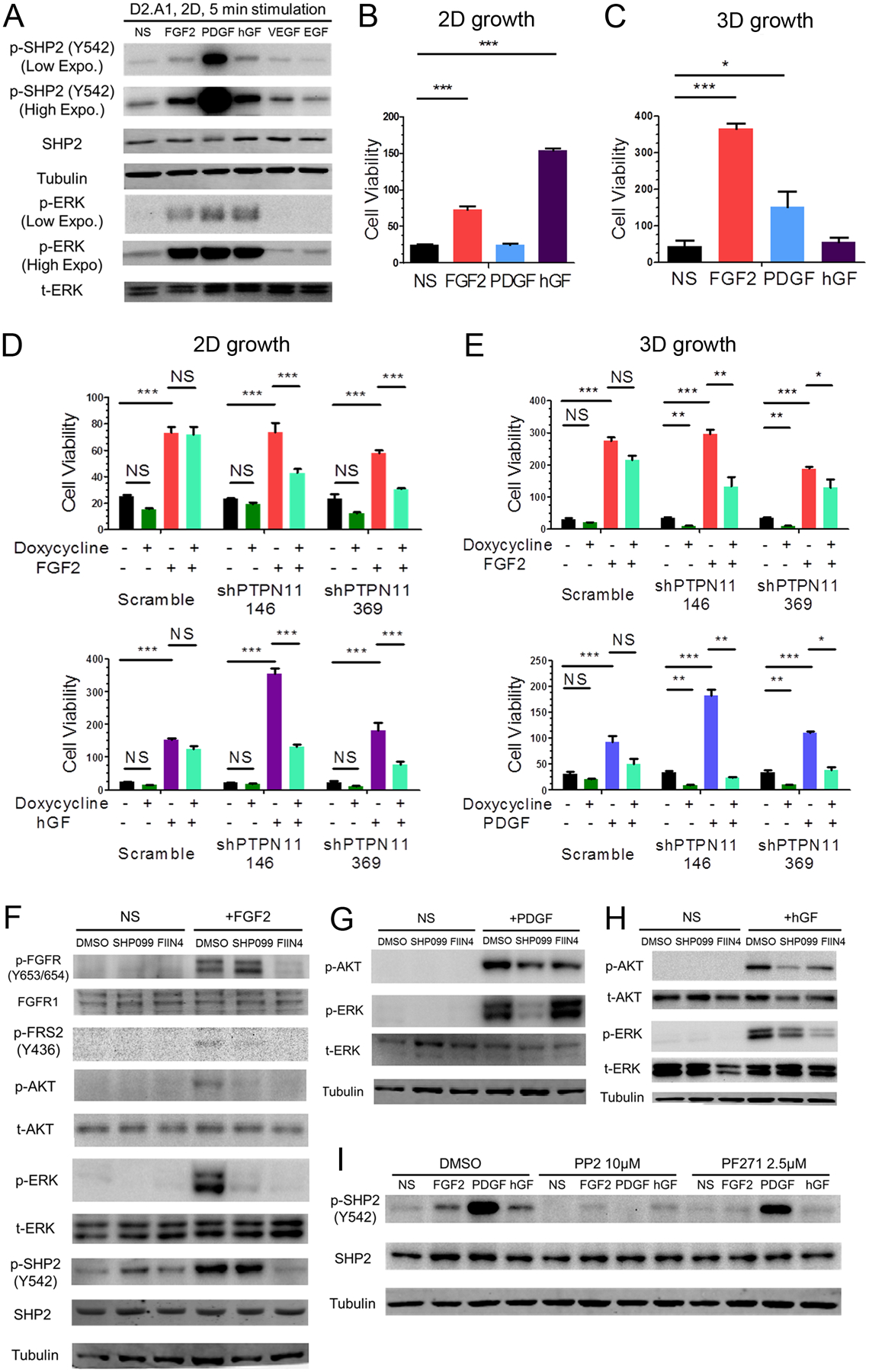
SHP2 facilitates signaling in response to several growth factors. a Immunoblot analyses showing the phosphorylation of SHP2 and ERK1/2 (ERK) induced by the indicated growth factors in D2.A1 cells. b, c D2.A1 cell growth was induced by addition of exogenous FGF2, PDGF and hGF in 2D (B) or 3D (C) culture for 6 days. d,e D2.A1 cells expressing doxycycline inducible shRNAs targeting PTPN11 were treated with doxycycline in 2D culture (D) or 3D culture (E) in the presence of FGF2, hGF, or PDGF as indicated. For panels B-E cell viability was quantified as a relative bioluminescence ratio normalized to day 0. Data are the mean ±s.e.m. for at least three independent assays resulting in *p<0.05, **p<0.01, ***p<0.001, or no significance (NS) as determined by a two-tail Student’s t-test. f-h D2.A1 cells were pre-treated with SHP099 or FIIN4 for 24 hours in serum-free media and cells were subsequently induced for 5 minutes with FGF2, PDGF, or hGF as indicated. Immunoblot analyses were used to detect phosphorylation of FGFR, FRS2, ERK1/2, AKT, and SHP2. i D2.A1 cells were pre-treated with PP2 or PF271 for 24 hours in serum-free media, and cells were then induced for 5 minutes with FGF2, PDGF, or hGF. Immunoblot analyses were used to detect phosphorylation of SHP2. All immunoblots are representative of at least 3 independent experiments.
Combined targeting of SHP2 and FGFR improves therapeutic inhibition of metastasis
Our results thus far indicated that SHP2 is a shared node of both RTK and ECM signaling, suggesting there may be therapeutic potential for the use of SHP2 inhibitors in combination with targeted inhibition of a specific RTK. Therefore, we evaluated SHP099 in combination with the FGFR-targeted kinase inhibitor, FIIN4. Using concentrations of both compounds previously determined to be below effective dosages against the 4T1 cells resulted in complete blockade of cell growth when used in combination (Fig. 6a–c). Similar results were obtained with 11a-1 in 4T1 and other MBC cells (Supplementary Fig. 7). The synergy between SHP2 blockade and FGFR targeting was defined by establishing a combination index (CI) using COMPUSYN 1.0 for the 4T1 and other MBC cell lines (Supplementary Fig. 8). We also extended this combined therapeutic approach in vivo by treating late stage 4T1 metastases with SHP099:FIIN4 combination therapy. As opposed to our approach in Figure 2, here mice were left untreated for 5 additional days following primary tumor removal to allow metastatic outgrowth to progress. Using this approach, we found that only the combination of SHP099 and FIIN4 was able to significantly delay late-stage metastatic progression and extend animal survival as compared to control (Fig. 6d–f). No significant weight loss was observed in the combination group, suggesting that this therapeutic protocol does not result in unacceptable toxicity (Supplementary Fig. 9). These data demonstrate the efficacy and feasibility of combining SHP2 inhibitory compounds with FGFR-targeted kinase inhibitors for the treatment of advanced breast cancer.
Figure 6.
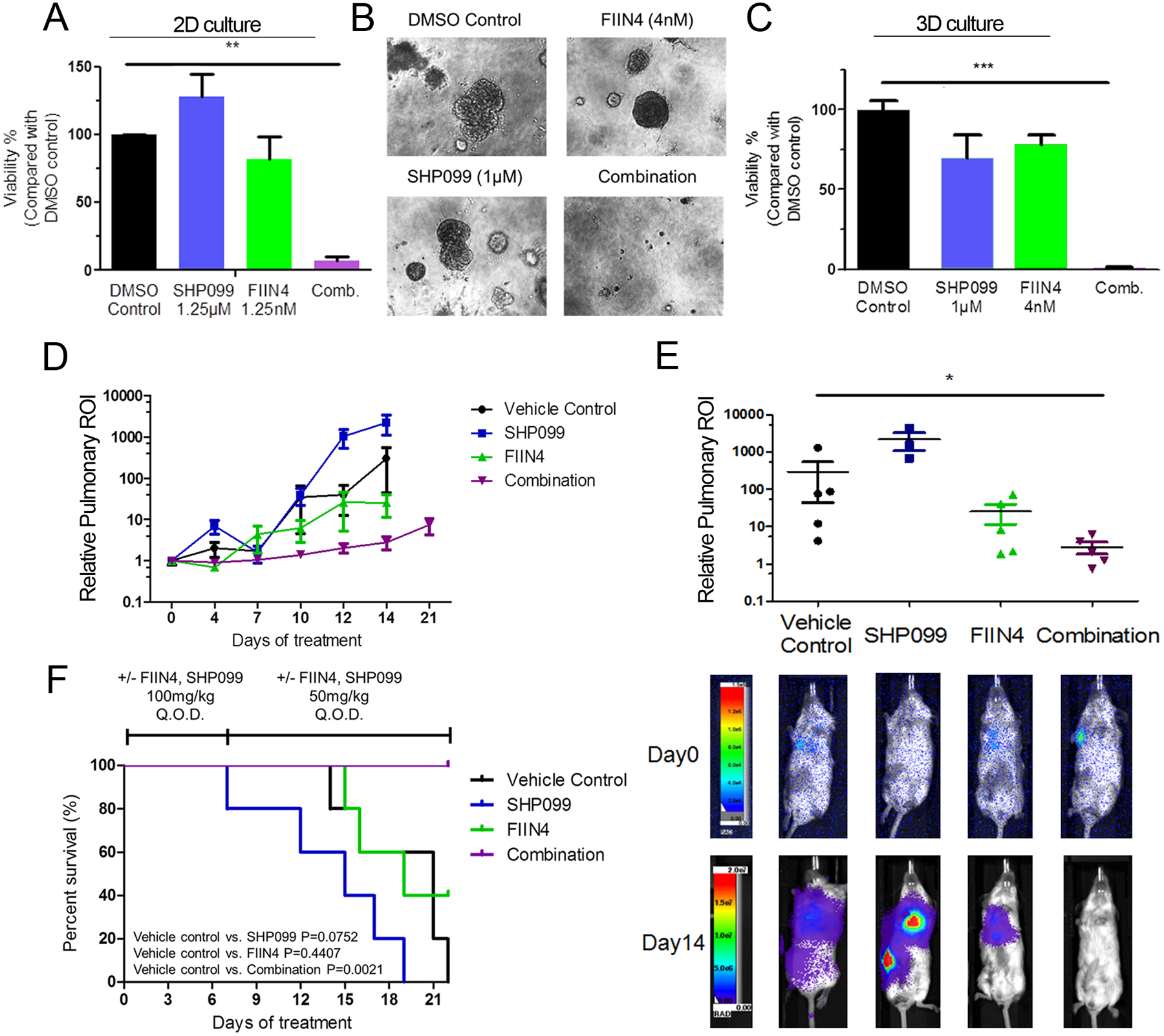
Inhibition of SHP2 synergizes with blockade of FGFR in vitro and in vivo. a 4T1 cells were seeded in 2D culture, and treated with the indicated concentrations of SHP099, FIIN4 alone, or both compounds for 6 days. b, c Single cell suspensions of 4T1 cells were plated in 3D matrices and treated with the indicated compounds alone or in combination for 12 days. Media containing DMSO was used as a vehicle control. Data in panels A and C data are bioluminescence values normalized to DMSO controls and are the mean ± s.e.m. of at least 3 independent experiments where **p<0.01, ***p<0.001 as determined by a two-tail Student’s t-test. d Bioluminescence values for pulmonary regions of interest (ROI) from mice bearing 4T1 metastases normalized to values at the initiation of treatment. Data are the mean ±s.e.m. of 5 mice per treatment group. e (bottom) Representative bioluminescent images of metastasis and (top) quantified pulmonary ROI values from mice bearing 4T1 metastases 14 days after treatment initiation. Data are the mean ±s.e.m. of 5 mice per group resulting in *p<0.05 as determined by a two-tail Mann Whitney test. f Differential survival analyses of tumor bearing mice in each treatment group, resulting in the indicated P values as determined by a log-rank test.
Discussion
Drug-resistant progression of MBC can occur due to signaling events generated from multiple RTKs and can be further influenced by interactions with the ECM. This diversity in upstream inputs, and the downstream signaling pathways for which they activate, emphasizes the importance of targeting shared signaling nodes in the design of anti-metastatic therapeutics. Although recent findings have begun to illustrate the oncogenic roles of SHP2 in breast cancer, mechanistic understanding of SHP2 signaling and the therapeutic value of targeting SHP2 in drug resistant MBC was still limited [30–32]. Herein, we demonstrate that SHP2 is indeed a shared signaling node that facilitates signaling from multiple RTKs as well as the ECM. Our work is supported by several other studies that clearly demonstrate SHP2 participates in RTK-mediated signaling [40–44].
Using an inducible shRNA approach, we demonstrate that depletion of SHP2 specifically inhibited the ability of D2.A1 cells to grow under 3D culture conditions. These data are consistent with previous findings from our group and others that establish the requirement of β1 integrin:FAK signaling for D2.A1 cell growth in 3D culture [45, 46]. Consistent with these findings, culture of several additional MBC cell types under 3D conditions consistently induced SHP2 phosphorylation. Use of our recently-established tessellated scaffold culture system allowed for evaluation of specific ECM components, where we determined that fibronectin-mediated SHP2 phosphorylation occurs in a FAK-dependent manner. In contrast, SHP2 phosphorylation induced by various growth factors was reduced by the Src inhibitor PP2. These findings may serve to explain the clinical limitations observed in targeting either Src or FAK and suggest novel mechanisms by which ECM components interface with growth factor receptor signaling to support cell growth and survival (Fig. 7). Continued elucidation of upstream mechanisms of SHP2 phosphorylation remain important topics of research, but our findings herein clearly indicate that directed pharmacological targeting of SHP2 phosphatase activity is highly effective at inhibiting cell growth under 3D culture conditions. This is an unusual observation, as 3D culture typically diminishes cellular responses to targeted small molecule inhibitors. The ability of SHP2 inhibition to ablate matrix-mediated growth and survival signals indicate that SHP2 inhibitory compounds might synergize with RTK inhibitors or other therapeutics that particular tumors have an initial sensitivity to. We demonstrate this concept here using cell lines that we have previously demonstrated to initially respond to targeted inhibition of FGFR [10, 12, 35]. This concept is consistent with findings from other recent studies linking SHP2 function to acquisition of resistance of RAS mutant cancers to inhibition of MEK/ERK signaling [29, 33].
Figure 7.
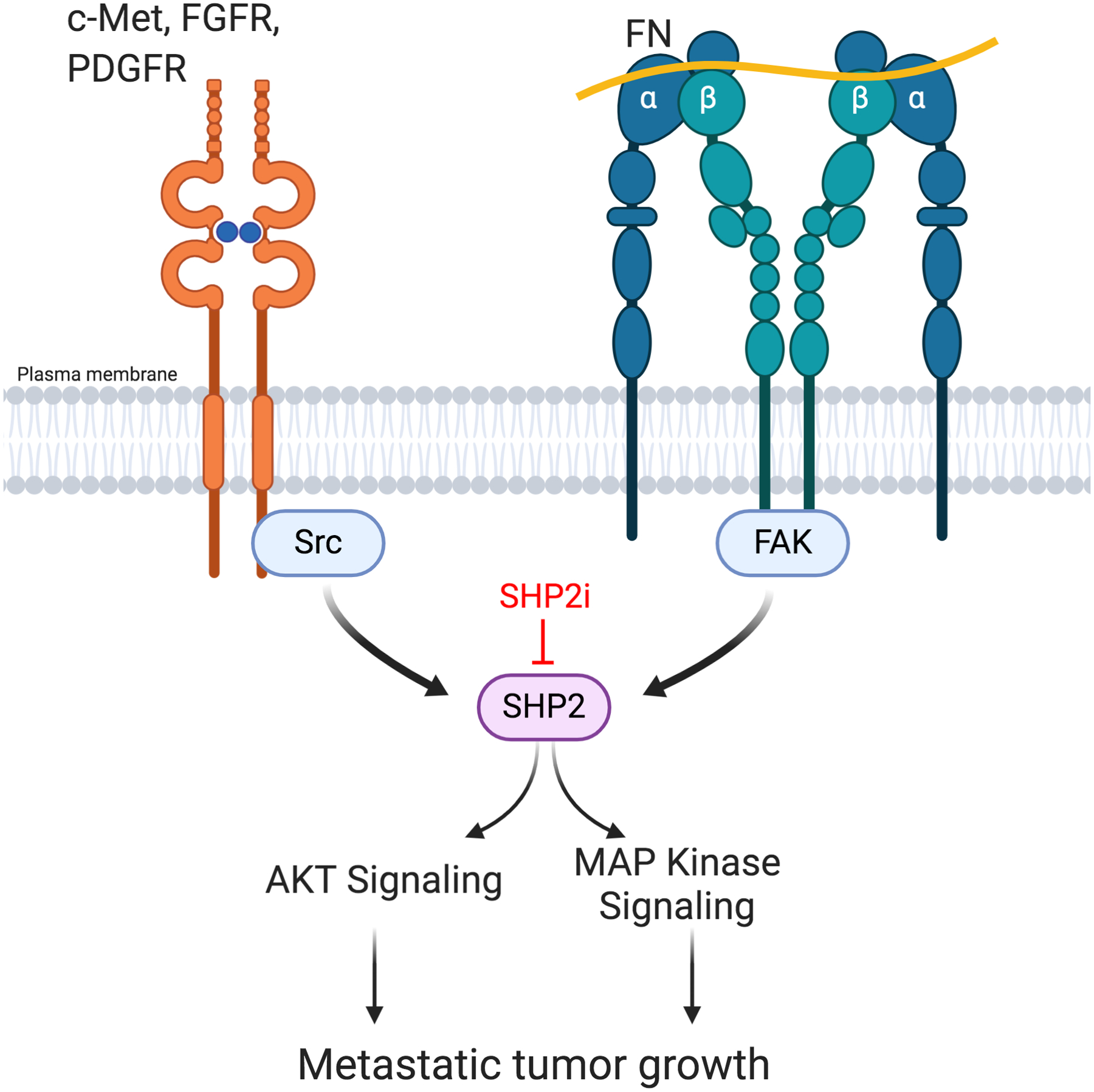
SHP2 is a shared node for ECM and RTK signaling. Activation of FAK via integrin engagement of ECM components drives SHP2 phosphorylation. Growth factor receptor signaling additionally contributes to phosphorylation of SHP2 via Src and FAK. SHP2 activity contributes to various downstream signaling pathways that facilitate metastatic tumor growth in the presence of currently used targeted therapies. Targeted inhibition of SHP2 (SHP2i), enhances responses to existing therapeutics. Figure was created using BioRender.
The importance of ECM-mediated phosphorylation of SHP2 is further emphasized by our findings indicating differential patient survival based on this biomarker. Clearly, SHP2 phosphorylation status, not mRNA expression levels, is the key determinant in patient prognosis. Current biomarkers that drive therapeutic indications are largely focused on determining genetic mutations or expression level changes of enzymes that are to be directly targeted with antibodies or small molecules. Targeting of signaling nodes, like SHP2, will require continued understanding of upstream inputs and downstream consequences and how they can be best utilized to identify the optimal patient population that will benefit from the addition of SHP2 therapy to other current standards of care.
Pharmacologic targeting of SHP2 remains an active area of research and discovery in the field of medicinal chemistry. Here we utilize a combination of genetic and pharmacological approaches to confidently elucidate the oncogenic functions of SHP2 in MBC. Both the SHP2 active site directed inhibitor 11a-1 and the allosteric inhibitor SHP099 phenocopied genetic depletion of SHP2, indicating that the observed pharmacologic effects are unlikely to be nonspecific. We did observe differences in outcomes between active site inhibition via 11a-1 versus allosteric inhibition via SHP099 in several of our assays. However, our studies fail to offer clear advantages of active site versus allosteric inhibition of SHP2. Several clinical trials are currently underway evaluating toxicity of allosteric inhibitors of SHP2 in various solid tumors (NCT03565003, NCT03634982). Further mechanistic studies comparing these modes of inhibition are required to better define their impact on SHP2 function and therapeutic outcome.
It is worthwhile to point out that our in vivo studies utilize two immune-competent mouse models. In addition to being expressed in epithelial cells, SHP2 phosphatase activity is also critical to T-cell function where it acts in a more traditional sense to facilitate immunoreceptor tyrosine-based inhibition motif (ITIM) signaling downstream of checkpoint receptors such as PD-1 and CTLA4 [47]. Given the importance of SHP2 in facilitating immune checkpoint signaling in cytolytic T-cells, our in vivo data using systemic administration of small molecule inhibitors could also reflect an enhancement of anti-tumor immunity [47, 48]. Indeed, recent studies suggest that systemic inhibition of FGFR increases recruitment of T-cells into tumors, enhancing response to antibody-mediated checkpoint blockade [49, 50]. Studies to further delineate the role of SHP2 inhibition in driving anti-metastatic immunity through increased T-cell recruitment and checkpoint inhibition are currently underway in our laboratory.
In summary, we show that inputs from ECM and RTK signaling that contribute to drug resistance and metastasis are facilitated by SHP2 (Fig. 7). Our study pioneers the role of SHP2 in ECM signaling and presents pharmacological inhibition of SHP2 as a robust means to overcome matrix-mediated drug resistance. We also highlight the correlation of SHP2 Y542 phosphorylation status, not mRNA expression levels, to clinical prognosis in breast cancer patients. Finally, we elucidate a synergistic combination of SHP2 and FGFR inhibition in targeting MBC. Given the recent FDA approval of FGFR inhibitors and the current clinical evaluations of SHP2 targeted therapies, our studies strongly support development of this combined therapy for the treatment of MBC.
Materials and Methods
Cell lines and culture conditions
The names and culture conditions of the cell lines in this study are listed in Supplementary Table 1. Luciferase expressing, 4T1, HMLE cells transformed by HER2 overexpression (HME2), their lapatinib resistant counterparts (LAPR), and the D2.A1 cells were constructed as we previously described [10, 12]. The other cell lines were purchased from ATCC. All cell lines were authenticated via the IDEXX IMPACT III CellCheck in December of 2018. All cell lines are regularly tested for mycoplasma contamination via PCR.
Depletion of SHP2 with Dox-inducible shRNA
Dox-inducible eGFP-shRNA constructs with were purchased from Dharmacon. The targeting sequences are listed in Supplementary Table 2. The same vector with a scrambled non-targeting control shRNA was also purchased from Dharmacon. Lentiviral particles were produced in HEK-293 cells and were used to transduce 4T1 and D2.A1 cells, and stable integration was selected for using 5 μg/ml puromycin (Fisher). The stable cell lines were induced with doxycycline (Fisher) at 10 μg/ml for 3 days, and sorted for GFP positivity.
Cell viability assays
The manufactures of the compounds used in in vitro assays are listed in Supplementary Table 3. 11a-1 was synthesized as previously described [27]. In 2D growth assays, the cells were seeded in 96-well plates with the indicated drug concentrations and cultured for 6 days. In 3D growth assays, 96-well plates were coated with 80% Cultrex (Sigma), which is a growth-factor-reduced 3D culture hydrogel matrix. Single cell suspensions were seeded on top of these hydrogel matrices in media containing 5% Cultrex and 10% FBS. In separate experiments 1×104 tumor cells were formed into a macroscopic sphere via a 72 hour plating in a non-adherent round bottom dish (Corning). These tumor spheres were similarly transferred to Cultrex matrices and tumor sphere growth and invasion were quantified by ImageJ 1.52a (NIH). Where indicated small molecules and growth factors were included upon seeding the tumor spheres onto the Cultrex and were refreshed every 3–4 days thereafter. The growth of luciferase-constructed cell lines was monitored by bioluminescent readings after addition of luciferin. Cell viability was also obtained using CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the manufacturer’s instructions at the end points of assays.
Immunoblotting
Immunoblotting of cells in 2D and 3D culture environments was executed as previously described [45, 51]. In the ligand-induced signaling assays, the cells were pretreated with inhibitors in serum-free media for 24 hours prior to stimulation with FGF2 (20ng/ml), PDGF (100ng/ml), hGF (50ng/ml), VEGF (100ng/ml) and EGF (50ng/ml). In the assays with tessellated scaffold culture system, the FN-coated scaffolds were prepared as previously described [38]. The cells were grown on scaffolds for 16 days, and treated with inhibitors for 24 hours before harvesting. Primary and secondary antibodies used in this study are listed in Supplementary Table 4. Immunoblot results were collected and recorded using X-ray films, ChemiDoc Gel Imaging System (Bio-Rad) and LI-COR imaging (LI-COR Biosciences). Quantification of the blots was performed with ImageJ 1.52a (NIH).
Animal care and in vivo metastatic models
For the 4T1 spontaneous metastasis model, 50k cells were engrafted onto the mammary fat pad via an intraductal injection. For the D2.A1 model, 1×106 cells were injected via the lateral tail vein [12, 35]. All in vivo studies were conducted in 4–6 week old, female BALB/cJ mice purchased from Jackson Laboratories. All small molecule inhibitors were administered via oral gavage at the indicated concentrations and frequencies. Small molecule manufactures and gavage formulations are listed as Supplementary Table 3. Metastasis was monitored using bioluminescent imaging after intraperitoneal injection of luciferin using an AMI HT (Spectral Instruments). The lungs were fixed overnight by 10% formaldehyde (Fisher) immediately after sacrificing the mice and stored in 75% ethanol. Paraffin sectioning at 5 μm thickness and H&E staining were performed by AML Laboratories, Inc. (Jacksonville, FL). The photos of lung sections were acquired by Cytation 5 cell imaging multi-mode reader with Gen5 software (BioTek Instruments, Inc.). All in vivo assays were conducted under IACUC approval from Purdue University. No randomization or blinding was done.
Drug combination study and data interpretation
The in vitro drug combination study was designed as described in the user guide of COMPUSYN 1.0 (ComboSyn Inc.; [52]). The small molecules FIIN4 and SHP099 were combined at a constant ratio.
Clinical dataset analysis and code availability
For clinical outcomes, mRNA and Reverse Phase Protein Array (RPPA) raw data from Breast Invasive Carcinoma (BRCA) cohort of the Cancer Genome Atlas (TCGA) were downloaded from FireBrowse (www.firebrowse.org, Broad Institute of MIT and Harvard, MA). The RPPA data were first input into R, and the correlation plot of different genes was generated using the “corrplot” R package [53]. To analyze patient survival, we set 1500 days as the threshold of survival and then selected data from non-living patients. The patients were separated into two groups based on median expression levels. Finally, the survival curves were created by GraphPad Prism 5.0, and analyzed via a log-rank test. The detailed step-by-step methods and the R scripts for the correlation plot and data cleaning are included in the Supplementary Information.
Statistical analysis
A two tailed student’s t-test was used for comparing differences between two groups of measurements in in vitro assays. Error bars show the standard error of the mean. IC50 values were calculated using GraphPad Prism 5.0 software via curve fit. Group measurements of the in vivo assays were compared with a Mann-Whitney non parametric test. Survival analysis was performed with GraphPad Prism 5.0 software, and the distributions of survival were compared by a log-rank test. No exclusion criteria were used in these studies, all statistical tests were appropriate as the groups being compared met the assumptions of the test and had similar variance.
Supplementary Material
Acknowledgements
This work was supported by National Cancer Institute (R01CA207751), Metavivor foundation Translation Research Award, and American Cancer Society Research Scholar Grant (RSG-CSM-130259) to MKW and National Cancer Institute (RO1CA207288) to ZYZ. We gratefully acknowledge the SIRG Graduate Research Assistantships Award (P30CA023168) and the support from the Purdue University Center for Cancer Research. We thank Dr. Jill E. Hutchcroft, Dr. Wen-Hung Wang and Flow Cytometry and Cell Separation Core Facility at Bindley Bioscience Center, Purdue University for technical support. We also thank Saeed Salehin Akhand, Hang Lin, Juan Sebastian Paez and other members at Wendt Lab for technical support.
Footnotes
Conflict of interest
ZYZ is a co-founder and serves on the scientific advisory board of Tyligand Bioscience.
References
- 1.Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the number of women living with metastatic breast cancer in the United States. Cancer Epidemiology and Prevention Biomarkers 2017; 26: 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer research 2006; 66: 1630–1639. [DOI] [PubMed] [Google Scholar]
- 3.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. New England Journal of Medicine 2006; 355: 2733–2743. [DOI] [PubMed] [Google Scholar]
- 4.Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol 2010; 28: 1301–1307. [DOI] [PubMed] [Google Scholar]
- 5.Martin M, Holmes FA, Ejlertsen B, Delaloge S, Moy B, Iwata H et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 2017; 18: 1688–1700. [DOI] [PubMed] [Google Scholar]
- 6.Hanker AB, Brewer MR, Sheehan JH, Koch JP, Sliwoski GR, Nagy R et al. An acquired HER2 T798I gatekeeper mutation induces resistance to Neratinib in a patient with HER2 mutant–driven breast cancer. Cancer discovery 2017; 7: 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clinical Cancer Research 2007; 13: 4909–4919. [DOI] [PubMed] [Google Scholar]
- 8.Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell reports 2015; 11: 390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shattuck DL, Miller JK, Carraway KL, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer research 2008; 68: 1471–1477. [DOI] [PubMed] [Google Scholar]
- 10.Brown WS, Akhand SS, Wendt MK. FGFR signaling maintains a drug persistent cell population following epithelial-mesenchymal transition. Oncotarget 2016; 7: 83424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clinical Cancer Research 2016; 22: 259–267. [DOI] [PubMed] [Google Scholar]
- 12.Brown WS, Tan L, Smith A, Gray NS, Wendt MK. Covalent targeting of fibroblast growth factor receptor inhibits metastatic breast cancer. Molecular cancer therapeutics 2016; 15: 2096–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minuti G, Cappuzzo F, Duchnowska R, Jassem J, Fabi A, O’Brien T et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. British journal of cancer 2012; 107: 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander PB, Chen R, Gong C, Yuan L, Jasper JS, Ding Y et al. Distinct receptor tyrosine kinase subsets mediate anti-HER2 drug resistance in breast cancer. Journal of Biological Chemistry 2017; 292: 748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P et al. Autocrine PDGFR signaling promotes mammary cancer metastasis. The Journal of clinical investigation 2006; 116: 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li D-M, Feng Y-M. Signaling mechanism of cell adhesion molecules in breast cancer metastasis: potential therapeutic targets. Breast cancer research and treatment 2011; 128: 7. [DOI] [PubMed] [Google Scholar]
- 17.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature Reviews Cancer 2010; 10: 116. [DOI] [PubMed] [Google Scholar]
- 18.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Molecular and cellular biology 2003; 23: 7875–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nature communications 2015; 6: 8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, Yu W-M, Zhang J, Chan RJ, Loh ML, Zhang Z et al. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia 2017; 31: 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ran H, Tsutsumi R, Araki T, Neel BG. Sticking it to cancer with molecular glue for SHP2. Cancer cell 2016; 30: 194–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. Journal of Biological Chemistry 2003; 278: 41677–41684. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Lu S, Ouyang M, Lin L-J, Zhuo Y, Liu B et al. Antagonism between binding site affinity and conformational dynamics tunes alternative cis-interactions within Shp2. Nature communications 2013; 4: 2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dance M, Montagner A, Salles J-P, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cellular signalling 2008; 20: 453–459. [DOI] [PubMed] [Google Scholar]
- 25.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R et al. Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Molecular and cellular biology 2002; 22: 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016; 535: 148. [DOI] [PubMed] [Google Scholar]
- 27.Zeng L-F, Zhang R-Y, Yu Z-H, Li S, Wu L, Gunawan AM et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. Journal of medicinal chemistry 2014; 57: 6594–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang R-Y, Yu Z-H, Zeng L, Zhang S, Bai Y, Miao J et al. SHP2 phosphatase as a novel therapeutic target for melanoma treatment. Oncotarget 2016; 7: 73817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov 2018; 8: 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Martin E, Matalkah F, Shah N, Ivanov A, Ruppert JM et al. Conditional knockout of SHP2 in ErbB2 transgenic mice or inhibition in HER2-amplified breast cancer cell lines blocks oncogene expression and tumorigenesis. Oncogene 2019; 38: 2275–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lan L, Holland JD, Qi J, Grosskopf S, Vogel R, Györffy B et al. Shp2 signaling suppresses senescence in PyMT‐induced mammary gland cancer in mice. The EMBO journal 2015; 34: 1493–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nature medicine 2012; 18: 529. [DOI] [PubMed] [Google Scholar]
- 33.Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA et al. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep 2019; 26: 65–78 e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou B, Gibson-Corley KN, Herndon ME, Sun Y, Gustafson-Wagner E, Teoh-Fitzgerald M et al. Integrin α3β1 can function to promote spontaneous metastasis and lung colonization of invasive breast carcinoma. Molecular Cancer Research 2014; 12: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wendt MK, Taylor MA, Schiemann BJ, Sossey-Alaoui K, Schiemann WP. Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor β1 signaling in metastatic breast cancers. Breast cancer research 2014; 16: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibue T, Weinberg RA. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proceedings of the National Academy of Sciences 2009; 106: 10290–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pulaski BA, Ostrand‐Rosenberg S. Mouse 4T1 breast tumor model. Current protocols in immunology 2000; 39: 20.22. 21–20.22. 16. [DOI] [PubMed] [Google Scholar]
- 38.Shinde A, Libring S, Alpsoy A, Abdullah A, Schaber JA, Solorio L et al. Autocrine fibronectin inhibits breast cancer metastasis. Molecular Cancer Research 2018; 16: 1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ali R, Brown W, Purdy SC, Davisson VJ, Wendt MK. Biased signaling downstream of epidermal growth factor receptor regulates proliferative versus apoptotic response to ligand. Cell death & disease 2018; 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montagner A, Yart A, Dance M, Perret B, Salles J-P, Raynal P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. Journal of Biological Chemistry 2005; 280: 5350–5360. [DOI] [PubMed] [Google Scholar]
- 41.Koyama T, Nakaoka Y, Fujio Y, Hirota H, Nishida K, Sugiyama S et al. Interaction of scaffolding adaptor protein Gab1 with tyrosine phosphatase SHP2 negatively regulates IGF-I-dependent myogenic differentiation via the ERK1/2 signaling pathway. Journal of Biological Chemistry 2008; 283: 24234–24244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hadari Y, Kouhara H, Lax I, Schlessinger J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Molecular and cellular biology 1998; 18: 3966–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gotoh N, Ito M, Yamamoto S, Yoshino I, Song N, Wang Y et al. Tyrosine phosphorylation sites on FRS2α responsible for Shp2 recruitment are critical for induction of lens and retina. Proceedings of the National Academy of Sciences 2004; 101: 17144–17149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunnick JM, Dorsey JF, Munoz-Antonia T, Mei L, Wu J. Requirement of SHP2 binding to Grb2-associated binder-1 for mitogen-activated protein kinase activation in response to lysophosphatidic acid and epidermal growth factor. Journal of Biological Chemistry 2000; 275: 13842–13848. [DOI] [PubMed] [Google Scholar]
- 45.Wendt MK, Taylor MA, Schiemann BJ, Schiemann WP. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Molecular biology of the cell 2011; 22: 2423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS et al. Metastatic growth from dormant cells induced by a col-I–enriched fibrotic environment. Cancer research 2010; 70: 5706–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharmaceutica Sinica B 2019; 9: 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. Journal of Experimental Medicine 2012; 209: 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palakurthi S, Kuraguchi M, Zacharek SJ, Zudaire E, Huang W, Bonal DM et al. The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunol Res 2019; 7: 1457–1471. [DOI] [PubMed] [Google Scholar]
- 50.Ye T, Wei X, Yin T, Xia Y, Li D, Shao B et al. Inhibition of FGFR signaling by PD173074 improves antitumor immunity and impairs breast cancer metastasis. Breast Cancer Res Treat 2014; 143: 435–446. [DOI] [PubMed] [Google Scholar]
- 51.Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-β signaling and metastasis. Breast cancer research 2009; 11: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 2010; 70: 440–446. [DOI] [PubMed] [Google Scholar]
- 53.Wei T, Simko V, Levy M, Xie Y, Jin Y, Zemla J. Package ‘corrplot’. Statistician 2017; 56: e24. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.