

Abstract
Introduction
Microglial TYROBP (DAP12) is a network hub and driver in sporadic late-onset Alzheimer’s disease (AD). TYROBP is a cytoplasmic adaptor for TREM2 and other receptors, but little is known about its roles and actions in AD. Herein, we demonstrate that endogenous Tyrobp transcription is specifically increased in recruited microglia.
Methods and Results
Using a novel transgenic mouse overexpressing TYROBP in microglia, we observed a decrease of the amyloid burden and an increase of TAU phosphorylation stoichiometry when crossed with APP/PSEN1 or MAPTP301S mice, respectively. Characterization of these mice revealed Tyrobp-related modulation of Apoe transcription. We also showed that Tyrobp and Apoe mRNAs were increased in Trem2-null microglia recruited around either Aβ deposits or a cortical stab injury. Conversely, microglial Apoe transcription was dramatically diminished when Tyrobp was absent.
Conclusions
Our results provide evidence that TYROBP-APOE signaling does not require TREM2 and could be an initiating step in establishment of the Disease-Associated Microglia (DAM) phenotype.
Keywords: Tyrobp, Dap12, Apoe, Trem2, microglia, Alzheimer’s disease, amyloid, APP/PSEN1, tauopathy, PS19, RNAscope, DAM
1. NARRATIVE
1.1. Contextual background
Microglia play a sentinel role in the brain, capable of detecting a wide variety of environmental stimuli, including microbial pathogens, aggregated proteins (such as amyloid-β; Aβ) and cellular debris (such as membrane fragments). This sensing activity is an essential part of the host response and is broad in its scope, sometimes triggering homeostatic adjustment, while, at other times, activating a host defense response. Microglia are also of interest in neurodegenerative diseases due to proteinopathies, e.g. Alzheimer’s disease (AD), in which large genetic studies have reported increased disease risk linked to many loci associated with microglial genes implicated in clearance of Aβ peptides[1–9]. More recently, transcriptomic analyses have revealed distinct profiles and signatures for microglia associated with aging and aging-related diseases, indicating that a wide range of specific proteins in microglia underlies sensing, activation, and/or other cellular responses. Using RNA sequencing, Hickman et al. (2013) identified 100 transcripts highly enriched in microglia and coined the term “sensome” to describe this class of microglial transcripts[10]. Network analysis of this list identified a TYROBP (for tyrosine kinase binding protein; also known as DAP12, for DNAX activating protein-12)-centered pathway with 24 of these 100 genes interacting directly with TYROBP and 20 interacting indirectly with TYROBP. Concurrently, members of our team employed an integrative network-based approach and identified TYROBP as a key network driver in sporadic late-onset Alzheimer’s disease (AD)[11]. More recently, Keren-Shaul et al.[12] used single-cell RNA sequencing in mouse to define a specific microglial phenotype that they termed “Disease-Associated Microglia” (DAM). Tyrobp was one of the genes most robustly upregulated in the proposed earliest stage of transition of microglia from the basal “homeostatic state” into the DAM phenotype.
TYROBP is a transmembrane signaling polypeptide that contains an immunoreceptor phosphotyrosine-based activation motif (ITAM) in its cytoplasmic domain. TYROBP is expressed in microglia in the brain and serves as an adaptor for a variety of immune receptors, including two molecules closely linked to AD pathogenesis: TREM2 (triggering receptor expressed on myeloid cells 2) and CR3 (complement receptor 3). TREM2 is expressed at the plasma membrane of microglia in the brain and some mutations and polymorphisms of TREM2 are linked to autosomal dominant AD or sporadic late-onset AD[13]. Other TREM2 mutations can cause a polycystic leukoencephalopathy osteodystrophy also known by the eponym Nasu-Hakola disease[14]. Most TYROBP mutations represent loss-of-function mutations and also result in Nasu-Hakola disease[15]. Similarly, TYROBP genetic variants have been identified in early-onset AD[16]. TREM2 is (among other things) a microglial Aβ receptor promoting microglial phagocytosis and proliferation and is required in order for microglia to limit growth of Aβ deposits[17,18]. In addition to the influences of environmental factors on TREM2[19], this molecule is also essential for a full transition of homeostatic microglia to a DAM state. Keren-Shaul et al.[12] described a two-stage program for DAM transition with a Trem2-independent step (stage 1) where Tyrobp and other genes are upregulated, followed by a Trem2-dependent step during which both Tyrobp and Trem2 are upregulated (stage 2). Krasemann et al.[20] described a very similar microglial signature associated with neurodegenerative diseases, designated the “MGnD” phenotype, and showed that the transition from homeostatic to MGnD microglia was both TREM2- and apolipoprotein E (APOE)-dependent with a TREM2-APOE signaling pathway driving the transition from homeostatic microglia to MGnD. APOEε4, one of the APOE polymorphisms, is a major risk factor for late-onset AD, and emerging evidence suggests that APOE (mostly produced by astrocytes in a normal brain) can also bind to TREM2 [21,22]. This event defines an interesting and potentially disease-relevant pathway wherein extracellular APOE, as a ligand for TREM2, triggers upregulation of APOE in microglia.
When DAM and MGnD are compared, Keren-Shaul et al[12] also observed by single-cell RNA sequencing an apparent sequence of events whereby Tyrobp was upregulated prior to the upregulation of Trem2. For clarity, since DAM and MGnD microglia appear to share key features of the phenomena described here, we will refer only to DAM for the remainder of this report. However, insofar as we are aware, principles established here underpin both DAM and MGnD.
In light of the central role of TYROBP in the microglial sensome[10], its key role as adaptor for multiple microglial receptors[23], its upregulation in the early Trem2-independent DAM stage 1[12] and its upregulation in AD[11], we hypothesized that the upregulation of Tyrobp might be an early event that begins during the initial microglial response to the accumulation of Aβ deposits. Further, we propose that chronic sustained sensation of Aβ deposits by microglia might generate ongoing intracellular signals that influence the progression and pathogenesis of AD.
1.2. Study design and main results
We employed a number of strategies aimed at interrogation of the causes and consequences of TYROBP upregulation in microglia. Using dual RNA in situ hybridization and immunohistochemistry, we found that Tyrobp mRNA level is significantly increased when microglia are recruited, including in wild-type (WT) mice, in an APP/PSEN1 transgenic mouse model of cerebral Aβ amyloidosis, and in a MAPTP301S transgenic mouse model of tauopathy. To determine whether elevated TYROBP can modify microglial phenotype and AD pathogenesis, we generated a novel transgenic mouse, designated the Iba1Tyrobp mouse, wherein the Iba1 promoter was used to drive overexpression of a mouse Tyrobp transgene in microglia. We observed a reduced density of amyloid plaques and an apparent increase in the stoichiometry of TAU phosphorylation when Iba1Tyrobp mice were crossed with either APP/PSEN1 or MAPTP301S mice, respectively. In addition to the alteration of both APP/PSEN1 and MAPTP301S phenotypes, we observed that a constitutive increase in TYROBP influenced the transcription of Apoe and some associated genes. Finally, using two mouse models of cerebral Aβ amyloidosis and a mouse model of penetrating cortical stab injury, we showed that upregulation of Tyrobp and Apoe does not require Trem2, but that upregulation of microglial Apoe requires Tyrobp to reach normal levels.
1.3. Trem2-Tyrobp-Apoe choreography
TREM2, TYROBP and APOE are three microglial genes linked in a pathway contributing to the pathogenesis of AD and in the transition to DAM[12]. TYROBP was identified as a key driver in sporadic late-onset AD[11]. TREM2 binds to TYROBP, its intracellular adaptor, to initiate its signal transduction pathway, and naturally occurring loss-of-function mutations of either TYROBP or TREM2 can lead to Nasu-Hakola disease[15,24]. There is a general assumption among investigators in this research area that genetic deletion or overexpression of either TREM2 or TYROBP would result in identical phenotypes in disease models, but, until now, this has not been tested directly. We previously demonstrated amelioration of behavioral and electrophysiological deficits in APP/PSEN1 and MAPTP301S mice on a Tyrobp-null background, despite a concurrent absence of effect on amyloid pathology and an apparent increase in the stoichiometry of phosphorylated TAU vs total TAU[25–27]. Homozygous deletion of Trem2 can also lead to amelioration of both amyloidosis and tauopathy [28,29], but those effects vary according to the mouse model, and the age and level of deficiency at sacrifice [27,30,31]. Lee et al.[32] used bacterial artificial chromosome (BAC)-mediated transgenesis to overexpress the human TREM2 in the mouse genome and showed that TREM2 overexpression reduces amyloid accumulation in 5xFAD mice. Using a similar BAC system, Gratuze et al.[33] assessed the impact of TREM2R47H in MAPTP301S mice but no TREM2 overexpression was reported in that study.
The possible existence of an early TREM2-independent phase in conversion of microglia to DAM was described by Keren-Shaul et al.[12] but was not evident in studies by either Krasemann et al.[20] or Zhou et al.[34] Apoe has also been described as a participant in stage 1 of DAM with Tyrobp[12], and it has been suggested that APOE drives the DAM transition through a TREM2-APOE pathway[20]. Moreover, Apoe has been reported to influence both amyloidosis and tauopathy histological phenotypes in mouse models[35,36]. A complete elucidation of the choreography of the regulatory interactions among these genes and their cognate proteins therefore remains an area of intense interest. We would suggest that the discrepancies across the various analyses might be explained in part by the fact that DAM microglia are located in the immediate proximity of the plaques, and that neither bulk- nor single-cell- RNA sequencing can distinguish homeostatic vs DAM phenotypes since both techniques generate an average transcriptomic analysis from all microglia within a particular tissue sample. This formulation played a major role in prompting us to use dual RNA in situ hybridization and immunohistochemistry in the current study in which we sought to determine 1) the effects of transgenic overexpression of Tyrobp on amyloid and tau pathologies and 2) the relationship of the induction of Tyrobp to these pathologies and to the induction of Trem2 and Apoe.
While this manuscript was in preparation, Chen et al. (2020)[37] reported spatial transcriptomics and in situ sequencing in AppNL-G-F mice, employing this protocol due to considerations identical to those motivating our studies herein; i.e., in order to avoid any potential confound derived from averaging the transcriptomes in a tissue sample without providing for spatial resolution of isolated resident microglia vs plaque-associated recruited microglia. These investigators proposed the formulation of a plaque-induced gene (PIG) network within the microglia and astrocytes in immediate proximity to amyloid plaques; this PIG network was defined by differential expression of 57 genes[37]. Top genes highly upregulated in the proximity of the plaques and as early as 3 months of age were Tyrobp, Apoe and several complement-related genes. Notably, Tyrobp is a key regulator of the complement subnetwork[11], and C1q is down-regulated in APP/PSEN1 and MAPTP301S mice in the absence of Tyrobp[26,27]. The authors performed the same type of experiment in human AD brain slices and conrmed the enrichment of Tyrobp and several complement components (C1qA, C1qB, C1qC, and Clu). Of particular relevance to our data herein, Trem2 was not included in the human PIG network.
1.4. Study conclusions and perspectives
In addition to confirming that TYROBP overexpression in microglia is sufficient to alter both amyloidosis and tauopathy phenotypes, our data indicate that Tyrobp upregulation is an early marker of recruited microglia and can occur even in the brains of Trem2-deficient mice. Similarly, we observed that the increased Apoe mRNA level in microglia is Trem2-independent, whether in injury or AD-related mouse models. Finally, we observed that microglial Apoe mRNA level was greatly attenuated in plaque-associated microglia in Tyrobp-deficient mice. These data confirm the model proposed by Keren-Shaul et al.[12] in which Tyrobp and Apoe transcripts are increased first, and neither transcription event requires the presence of Trem2. Moreover, Meilandt et al.[38] recently reported that microglial APOE expression was not reduced, but, on the contrary, was increased in PS2APP;Trem2−/− mice when compared with microglial APOE expression in PS2APP; Trem2+/+ mice. In that same study, Meilandt and colleagues also analyzed the expression profiles of FACS-purified microglia from 5xFAD mice that expressed TREM2 normally and from FACS-purified microglia from 5xFAD mice deficient in Trem2. These investigators observed a two-fold reduction in Apoe in one dataset (GSE132508)[12,38] but no reduction at all in the other (GSE65067)[38,39]. However, Parhizkar et al.[40] reported that the absence of functional TREM2 reduces plaque-associated APOE. This is in line with what Krasemann et al.[20] proposed when they showed that genetic targeting of Trem2 suppresses the APOE pathway. Our observations and conclusions herein apparently differ from those of Parhizkar et al.[40] to the extent that, in our hands, microglial amyloid plaque sensing followed by upregulation of Tyrobp and Apoe are preserved in the absence of Trem2 and, as a consequence of the Trem2 deficiency, microglia recruitment into the proximity of amyloid plaques is reduced. This relationship points to the fact that the absence of functional TREM2 will block appearance of the full DAM phenotype and therefore the associated clearance of Aβ is reduced. Nevertheless, we propose a model wherein the sensing of amyloid plaques --which takes place upstream of amyloid plaque clearance-- involves Tyrobp and Apoe but not necessarily Trem2. Since APOE isoforms differentially influence AD age at onset and progression[41], future investigations will be aimed at elucidating whether and how various homozygous or heterozygous APOE isotype combinations affect TYROBP-APOE signaling, and/or microglial sensing and phenotyping switching[42].
TYROBP is a 113 amino acid polypeptide with a minimal extracellular region[43,44], making it unlikely that TYROBP is the sole player in a signal transduction pathway involving both the perception of the environment and the triggering of the switch from homeostatic phenotype to DAM. However, TYROBP is the adaptor for many receptors other than TREM2[23], and therefore, it is plausible and perhaps likely that other TYROBP receptors could play key roles in sensing the deposition of amyloid. For example, numerous SIGLEC proteins (sialic acid-binding immunoglobulin-type lectins) carry a positively charged residue in their transmembrane domain that participates in oligomerization of the SIGLEC with TYROBP. The primary SIGLEC ligand is a sialic acid that accumulates in many pathological conditions including cerebral Aβ amyloidosis[45,46]. Moreover, Siglec-H interacts with TYROBP, and its expression has been reported to be elevated in 5xFAD mice[32]. CD33 (SIGLEC-3) is also one of the most abundant SIGLECs in human brain, and genome-wide association studies (GWAS) implicated a polymorphism near CD33 as a genetic risk factor for AD[2,4,47]. CD33 and TREM2 both interact with TYROBP, either directly (TREM2) or via common intracellular signaling factors (CD33). Griciuc et al.[48] recently investigated crosstalk between CD33 and TREM2 and proposed that CD33 acts upstream of TREM2. They also showed that Cd33 and Tyrobp expression levels did not change in Trem2−/− versus WT microglia. This formulation provides evidence that CD33-TYROBP signaling could occur upstream of the recruitment and upregulation of TREM2.
Rather than the somewhat exclusively “Trem2-centric” view of DAM proposed in the existing AD microglia literature[20,34,40], we propose that Tyrobp can play a central role in an alternative and early pathway in the microglial sensome[10], even in the absence of any change in Trem2 levels. The data that we present here document the robust consequences of TYROBP overexpression in both APP/PSEN1 and MAPTP301S mice. We confirm here that upregulation in microglia of both Tyrobp and Apoe constitute interconnected events in microglia sensing of amyloid deposits, and that these events take place independently of Trem2.
2. CONSOLIDATED RESULTS AND STUDY DESIGN
With the inclusion of Tyrobp as a PIG gene[37] and because of our prior validation of its actions as a driver of AD[11,25–27], we hypothesized that constitutive overexpression of microglial Tyrobp via transgenesis would alter both amyloid and tau pathologies. In 4-month-old APP/PSEN1 mice, Tyrobp overexpression was associated with a 50% decreased in the amyloid burden, similar to what occurs with the upregulation of Trem2[32] (Fig. 3A–B). We confirmed this decrease by measuring levels of human Aβ42 and Aβ40 by ELISA in the cerebral cortices of these mice (Fig. 3C). An evaluation of plaque burden in 8-month-old mice in three different brain regions confirmed the magnitude and statistical significance associated with the reduction of amyloid burden (Fig. 3G–H). In MAPTP301S mice, we previously reported that deficiency of Tyrobp increased TAU phosphorylation and spread[27], and we were puzzled, therefore, to observe a similar increase in TAU phosphorylation in the MAPTP301S mice with overexpression of Tyrobp (Fig. 4A–C). MAPTP301S;Iba1Tyrobp microglia are also more reactive compared to MAPTP301S microglia, and reactive microglia have been reported to drive TAU pathology, so these two observations are compatible (Fig.4D–E, Supp Fig. 2B)[49,50]. This exacerbation of pathology under conditions of either down- or up-regulation of Tyrobp in MAPTP301S mice reveals the complexity of the microglial events underpinning tauopathy. Our formulation is that, for any particular microglial activation status, there exists some optimum level of Tyrobp expression, and that either elevation or deficiency of Tyrobp levels can be detrimental. These data also support a key role for microglial TYROBP in AD pathology progression, as we proposed in our previous reports on mice deficient in Tyrobp[25–27].
Figure 3: Transgene-derived Tyrobp upregulation decreases amyloid plaque load in APP/PSEN1 mice.

(A) Representative images of Thioflavine-S (ThioS) staining in APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. Scale bar = 500 μm. (B) Quantification of the number of ThioS-positive plaques per hemibrain in APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. N = 4–5 mice per genotype and sex with 3 slices per animal. (C) Human Aβ42 and Aβ40 concentrations measured by ELISA in the cortices of the same groups described in (B). (D) Representative images of double-label immunohistochemistry with anti-IBA1 and anti-6E10 antibodies in APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. Scale bar = 100 μm. (E) Quantification of the number of plaque-associated microglia in the 4 groups described in (B). N = 10–24 plaques from 4–5 mice per group. (F) RT-qPCR analyses of microglial gene mRNAs in the hippocampi of APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. N = 7–9 mice per group, females and males were pooled. (G) Representative images of ThioS staining in male APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 8 months of age. Scale bar = 500 μm. (H) Quantification of the ThioS immunoreactive area in male APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 8 months of age (somatomotor and visual areas of the cortex, and hippocampus were quantified). N = 3–4 mice per group. Error bars represent means ± SEM. Statistical analyses were performed using a Two-Way ANOVA followed by a Sidak post-hoc test for (B and E), a Kruskal-Wallis test for (C), a Student t-test for (F) and a Mann-Whitney test for (H), *p<0.05, **p<0.01, ***p<0.001.
Figure 4: Transgene-induced Tyrobp upregulation increases apparent stoichiometry of TAU phosphorylation and microglial activation in 4-month-old MAPTP301S mice.
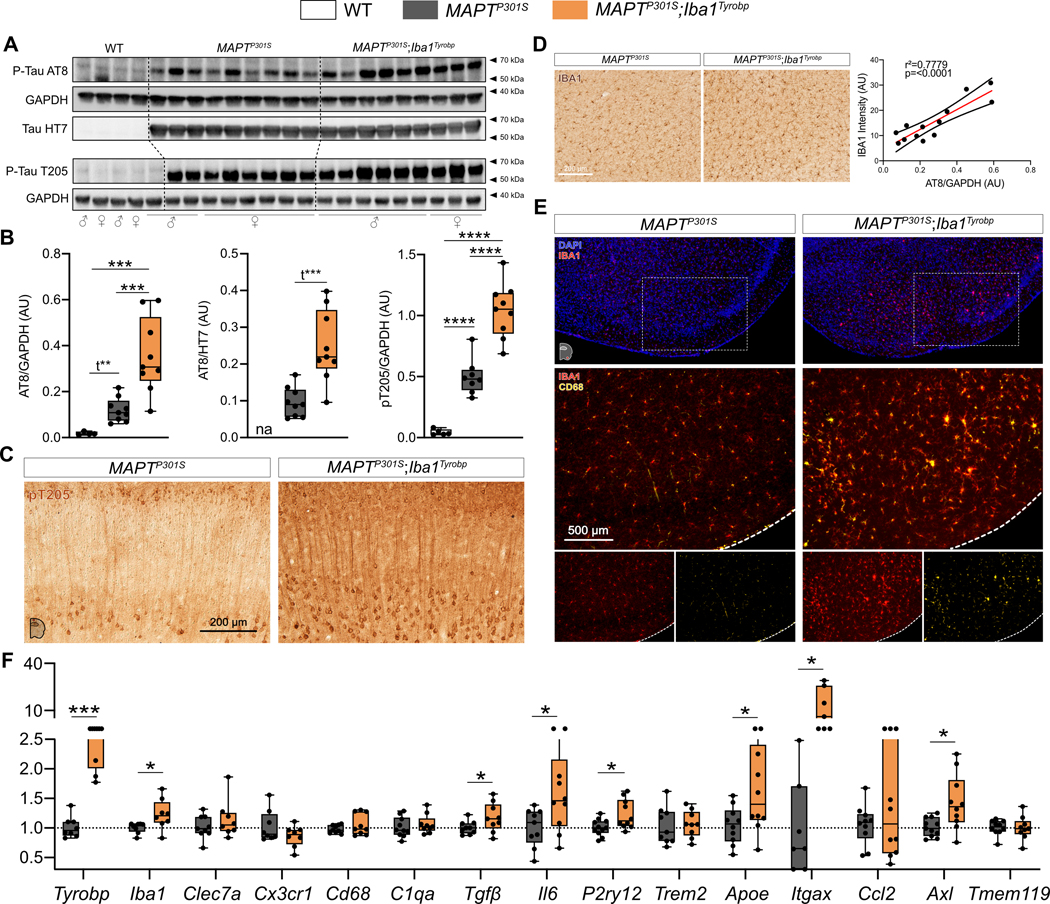
(A) Western blot analyses of phosphorylated-TAU on S202 or T205 epitopes (AT8 and pT205 antibodies) and total human TAU (HT7 antibody) in cortical homogenates from WT, MAPTP301S (PS19) and MAPTP301S;Iba1Tyrobp mice at 4 months-old. n = 4–9 mice per group. (B) Densitometric analyses of western blots presented in (A) standardized to GAPDH or HT7. (C) Representative images of DAB-immunohistochemistry with antibody pT205 in 4 month-old MAPTP301S and MAPTP301S;Iba1Tyrobp mice. Scale bar = 200 μm. (D) Left panel: representative images of anti-IBA1 immunohistochemistry on the same groups described in (C). Scale bar = 200 μm. Additional representative pictures are presented in Supplementary Figure 2. Right panel: western blot-AT8/GAPDH quantification plotted against anti-IBA1 immunoreactivity in the cortex. Linear regression with trend line (red line) and 95% confidence intervals (black lines) are indicated. (E) Representative images of double-label immunofluorescence with anti-IBA1 and anti-CD68 antibodies in the piriform cortex on the same groups described in (C). Scale bar = 500 μm. (F) RT-qPCR analyses of microglial gene mRNAs in the hippocampus of MAPTP301S and MAPTP301S;Iba1Tyrobp mice at 4 months of age. N = 7–11 per group. Error bars represent means ± SEM. Statistical analyses were performed using a One-Way ANOVA followed by a Tukey’s post-hoc test for (B) or a Student t-test for (B) when *t is indicated and (F), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Despite the obvious differences across APP/PSEN1 and MAPTP301S mouse models and the diverse consequences of Tyrobp upregulation in each of these mice, there are shared changes in Axl, Ccl2, Tgfβ and Il6 mRNAs in both APP/PSEN1 or MAPTP301S mice overexpressing TYROBP (Fig. 3F and 4F). These genes have been recently associated with Apoe in microglia, macrophages, and mononuclear phagocytes. AXL has been identified as a regulator of APOE[51], and accumulation of IL6 and CCL2 have been associated with APOE overexpression[52,53]. Similarly, reciprocal suppression of TGFβ and induction of APOE have been described in DAM microglia[20]. Apoe mRNA levels are indeed upregulated in MAPTP301S;Iba1Tyrobp mice as compared to MAPTP301S mice, but Apoe levels are unchanged in APP/PSEN1;Iba1Tyrobp mice as compared to APP/PSEN1 mice (Fig. 3F and 4F). However, in bulk RNA sequencing performed on hippocampi from male APP/PSEN1;Iba1Tyrobp vs APP/PSEN1 mice, we identified Apoe as a potential (activation z-score: 2.44; p-value overlap: 0.224) upstream regulator (Supp Fig. 3), suggesting the possible existence of a relationship between Tyrobp upregulation and Apoe. While a TREM2-APOE pathway has been described[20], it is interesting to note that our data indicate that the TYROBP-APOE relationship is detectable even in the absence of Trem2 upregulation (Fig. 3F and 4F).
To investigate further the interactions among Trem2, Tyrobp and Apoe in microglia, we used a penetrating cortical stab injury paradigm by introducing a small lesion via stereotactic surgery into one hemisphere of the mouse brain in order to induce a recruitment of microglia around the injury site32. Using this experimental paradigm in WT, Trem2−/− or Tyrobp−/− mice, we confirmed that microglia upregulate both Tyrobp mRNA and Apoe mRNA when recruited in close proximity to the injury (Fig. 5A–D), and these events are readily detectable even in the absence of Trem2. Interestingly, we failed to observe an upregulation of Apoe mRNA in microglia from Tyrobp−/− mice subjected to penetrating cortical stab injury (Fig. 5C). Because microglia are also recruited around Aβ plaques, we used two different mouse models of human cerebral Aβ-amyloidosis (i.e., TgCRND8 and APP/PSEN1 mouse lines) that were either WT or gene-targeted for Trem2 or Tyrobp, respectively. Similarly, we observed that both Tyrobp and Apoe mRNAs were upregulated in amyloid plaque-associated microglia even in the absence of Trem2 (Fig. 6A–B). Moreover, and as predicted with the stab-injury paradigm, we observed a substantial decrease in the induction of Apoe mRNA in plaque-associated microglia when Tyrobp was absent (Fig. 6D).
Figure 5: Increases of Tyrobp and Apoe mRNAs in microglia recruited to a site of stab injury are Trem2-independent.
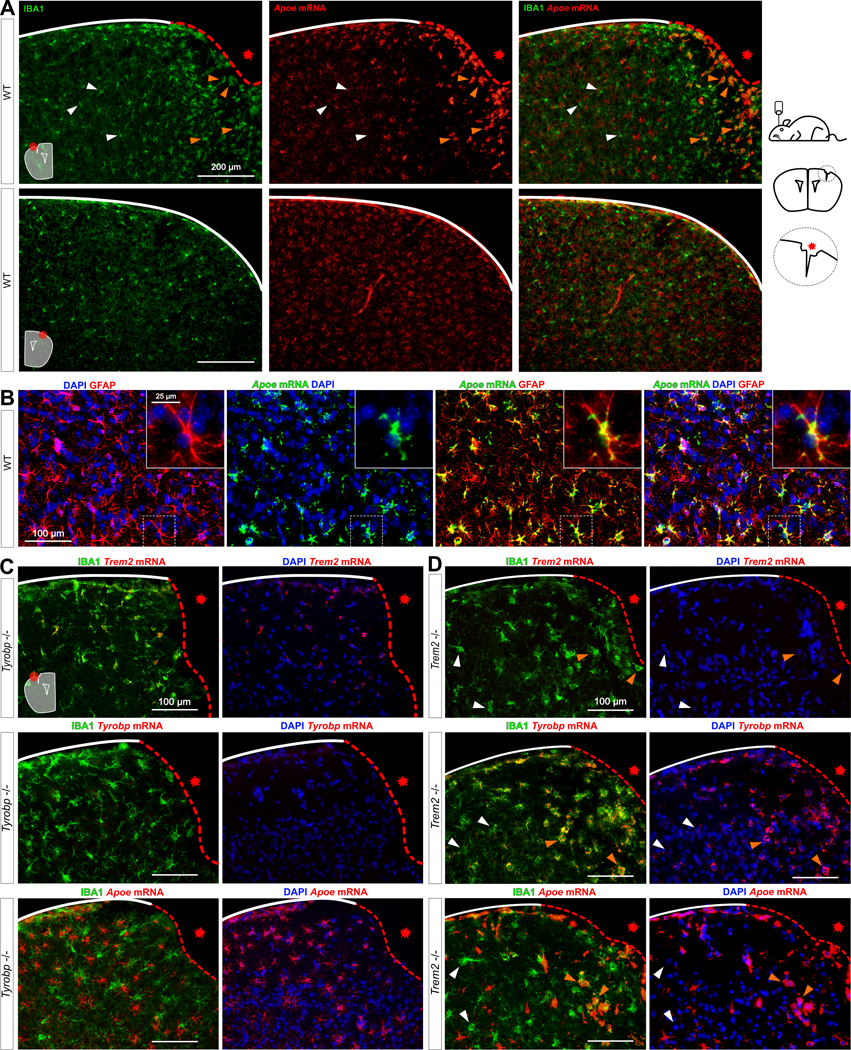
(A) Stab-injured WT mice were sacrificed 3 days after injury and dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe mRNA (red) and anti-IBA1 (green) respectively was performed. The injured ipsilateral area (red dotted line) is shown on the top row and the uninjured contralateral area is shown on the bottom row. Scale bar = 200 μm. (B) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe (green) and GFAP (red) in non-injured WT mice. (C-D) The same stab injury protocol was utilized in Tyrobp−/− (C) and Trem2−/− (D) mice. Anti-IBA1 staining and DAPI staining are shown in green and blue, respectively. Top row: Trem2 mRNA (red); middle row: Tyrobp mRNA (red); bottom row: Apoe mRNA (red). Mice were 4 months of age, and slice thickness = 10 μm. The red asterisk indicates the injured side. White and orange arrows indicate examples of non-recruited and recruited microglia, respectively.
Figure 6: Increases in Tyrobp and Apoe mRNAs in amyloid plaque-associated microglia are Trem2 independent.

(A) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Tyrobp mRNA (red), anti-IBA1 (green) and human Aβ (6E10 antibody) (blue) in TgCRND8 mice on WT (top row) or Trem2−/− (bottom row) background. Scale bar = 200 or 50 μm. (B) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe mRNA (red), anti-IBA1 (green) and human amyloid (6E10 antibody) (blue) in the same mice as in (A). Scale bar = 200 or 50 μm. (C) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Tyrobp mRNA (green), Apoe mRNA (red) and 6E10 (blue) in APP/PSEN1 mice. Scale bar = 50 μm. (D) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe mRNA (red), anti-IBA1 (green) and human Aβ (6E10 antibody) (blue) in APP/PSEN1 mice on a WT (top row) or Tyrobp-null (bottom row) background. Scale bar = 50 μm. Right panel: quantification of the number of plaque-associated microglia with upregulated Apoe mRNA in the same mice as in (D). N = 2–3 mice per group (A-D).
3. DETAILED METHODS AND RESULTS
3.1. Methods
3.2. Results
3.2.1. Tyrobp transcription is increased in recruited microglia
Microglia continuously sense changes in the brain environment and are recruited to sites of injury, microbial invasion, or where abnormal folding or modification of cellular constituents are detected, as with the accumulation of aggregated Aβ. We performed dual RNA in situ hybridization (RNAscope®) and immunohistochemistry for Tyrobp and IBA1 respectively in WT mice and observed increased levels of Tyrobp mRNA in areas exhibiting recruited microglia (Fig 1A). Using the same experimental approach in two independent mouse models of cerebral amyloidosis (APP/PSEN1[54] and 5xFAD[55]), we observed a similar pattern in that the Tyrobp mRNA level was extensively and selectively increased in microglia recruited in close proximity to amyloid plaques as compared to microglia that are more distant from the plaques (Fig. 1B–C). We similarly assayed Tyrobp mRNA and IBA1 protein in the MAPTP301S mice[56] (also known as PS19), a mutant tauopathy mouse model. We previously described an elevated number of anti-phosphorylated-TAU immunostained neurons in the piriform cortex of this mouse model (Fig. 1D)[27]. The changes that we observed in these mice were analogous to the changes observed around amyloid plaques in that we detected increased amounts of Tyrobp mRNA in microglia surrounding areas of aggregated protein pathology (Fig. 1D). We confirmed the increase of TYROBP at the protein level in microglia around amyloid plaques (Fig. 1E) as previously reported[57]. To discriminate between the role of TYROBP in activated vs recruited microglia, we isolated primary microglia from WT mice and exposed them to the gram-negative bacterial endotoxin lipopolysaccharide (LPS) to induce microglial activation[58], the status of which we established by quantifying the robust increase of Tnfα mRNA following LPS treatment. Interestingly, Tyrobp mRNA level was unchanged, suggesting that Tyrobp transcription may be increased only when microglia are both recruited and activated but not in resident microglia despite evidence that these residents are also activated (Fig. 1F).
Figure 1: Tyrobp mRNA is increased in recruited microglia.
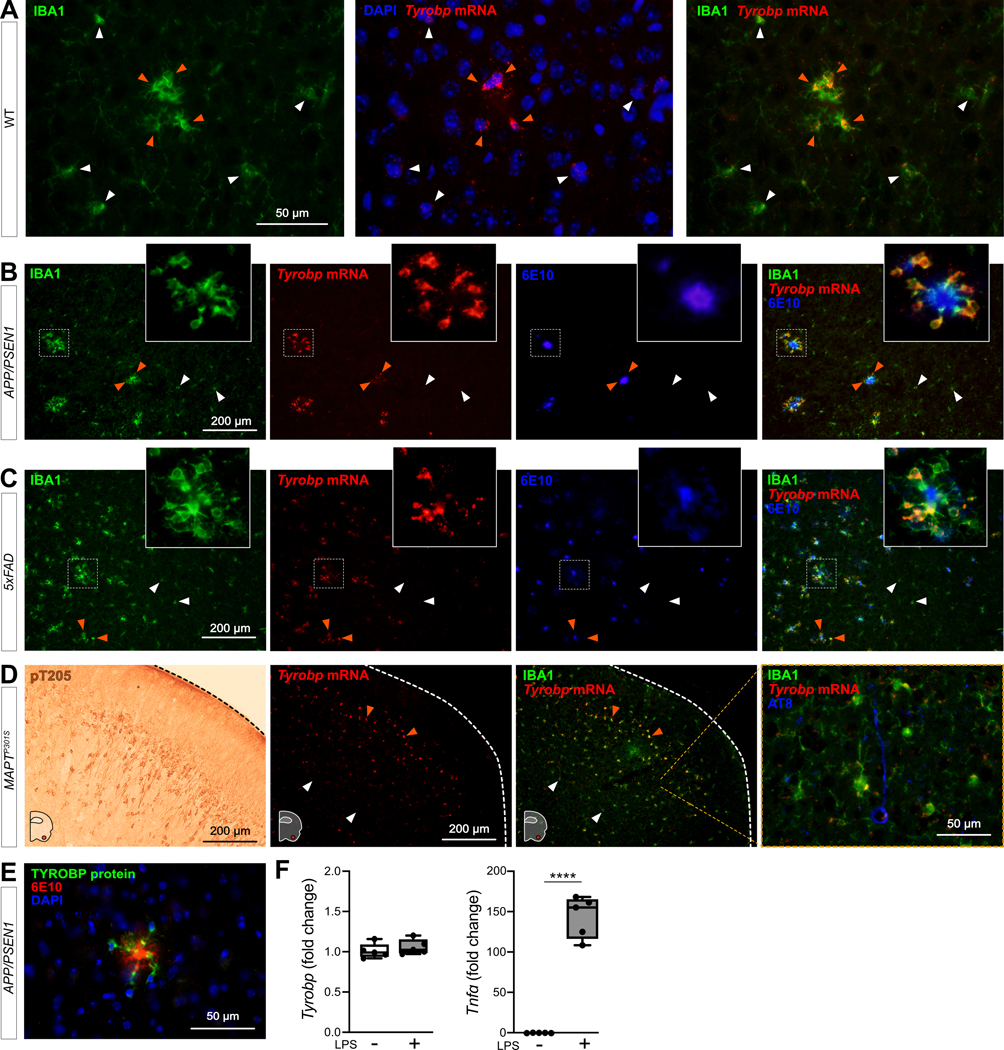
(A) Dual RNA fluorescent in situ hybridization (RNAscope®) and immunohistochemistry for Tyrobp mRNA (red) and IBA1 protein (green) respectively in WT mice (DAPI in blue). Scale bar = 50 μm. (B-C) Dual RNA in situ hybridization and immunohistochemistry for Tyrobp (red), IBA1 (Green) and Aβ (antibody 6E10) (blue) in APP/PSEN1 (B) and 5xFAD (C) mice. Scale bar = 200 μm. (D) Left panel: representative image of immunohistochemistry with antibody pT205 in the piriform cortex of MAPTP301S (PS19) mice. Scale bar = 200 μm. Right panels: dual RNA in situ hybridization and immunohistochemistry for Tyrobp (red), IBA1 (green) and pTau (antibody AT8) (blue) in the piriform cortex of MAPTP301S mice. Scale bars = 200 and 50 μm. (E) Co-immunohistochemistry for TYROBP (green) and human Aβ (antibody 6E10) (red) in APP/PSEN1 mice (DAPI in blue). Scale bar = 50μm. (F) RT-qPCR analyses of Tyrobp and TNFα mRNAs in WT primary microglia with and without LPS. Mice were either 4 (A) or 8 (B-E) months of age and were all WT for Tyrobp. White and orange arrows indicate examples of non-recruited and recruited microglia, respectively. Slice thickness = 10μm.
3.2.2. Microglia are normal in Iba1Tyrobp mice
To determine whether constitutive elevation of TYROBP via transgenesis may influence microglial phenotype and progression of AD pathology, we generated a novel transgenic mouse overexpressing Tyrobp selectively in microglia in the central nervous system. We used the mouse Tyrobp and Enhanced Green Fluorescent Protein (EGFP) sequences separated by an Internal Ribosome Entry Site (IRES) under the control of the mouse Iba1 regulatory sequences (Supp Fig. 1 [59]). Microinjections were performed in C57BL/6J mice and one line (B6.Cg-Tg(Iba1-Tyrobp-IRES-Egfp)34Mee/J) was selected for further use based on expression level of the transgene, now referred to Iba1Tyrobp. We first assessed the overexpression of Tyrobp mRNA by RT-qPCR and measured a ≈ 2.5-fold increase (Fig. 2A). Despite this elevated mRNA level, western blot analyses of protein extracts from the cortex did not reveal a significant overexpression at the protein level (Fig. 2B). Using combined RNA in situ hybridization and immunohistochemistry for Tyrobp and IBA1, respectively, in mice deficient in TYROBP (Tyrobp−/−), WT or overexpressing Tyrobp on the Iba1 promoter (Iba1Tyrobp), we confirmed the 2-fold increase in Tyrobp mRNA in Iba1Tyrobp mice compared to WT (Fig. 2C–D). We observed that only a subset of microglia was overexpressing Tyrobp mRNA in Iba1Tyrobp mice. This selectivity is likely due to the use of the Iba1 promoter in a WT background without extensive microglia activation, thereby also accounting for the lack of a significant increase of TYROBP at the protein level in the resting state. RNA sequencing of hippocampi from Iba1Tyrobp mice did not reveal any differentially expressed genes (DEGs) other than Aif1 (= Iba1), which is increased due to the inclusion of the first two exons in the transgenic vector (Supp Fig. 1). These data indicate that Iba1Tyrobp microglia do not display molecular and phenotypic changes in mice that are lacking in certain backgrounds of aggregated protein pathology.
Figure 2: Generation of Iba1Tyrobp mice.

(A) Hippocampi from 4-month old Tyrobp−/−, WT and Iba1Tyrobp mice were assayed for Tyrobp and Gfp mRNAs by RT-qPCR (n = 3–4 mice per group). (B) Representative western blot and quantification of TYROBP and GAPDH in the cortex of the same groups used in (A) (n = 2–6 mice per group). (C) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Tyrobp mRNA (red) and IBA1 (green) respectively (DAPI in blue) in Tyrobp−/−, WT and Iba1Tyrobp mice. Scale bar = 50 μm and slice thickness = 10μm. (D) Quantification of Tyrobp mRNA intensity from the experiment described in (C). n = 4, 17 and 17 slices per group (from N = 1 mouse per genotype) for Tyrobp−/−, WT and Iba1Tyrobp mice respectively. (E) Volcano plot representation of the whole hippocampal DEGs in Iba1Tyrobp vs WT mice (n = 4 4-month old males per genotype). Error bars represent means ± SEM. Statistical analyses were performed using a Student t-test (A) or a One-Way ANOVA followed by a Tukey’s post-hoc test (B, D), *p < 0.05, **p < 0.01, ****p < 0.0001. na = not applicable; ns = non-significant.
3.2.3. TYROBP overexpression in microglia decreases amyloid plaque load in APP/PSEN1 mice
To assess whether TYROBP overexpression in microglia modulates Aβ deposition in APP/PSEN1 mice, double-heterozygous APP/PSEN1;Iba1Tyrobp mice were generated and studied at 4 months of age. We measured a ≈50% decrease of the plaque density in the cerebral cortices of both male and female APP/PSEN1;Iba1Tyrobp mice compared to sex-matched APP/PSEN1 mice (Fig. 3A–B) in sections stained for amyloid plaques using Thioflavin S (ThioS). This observation was supported by measuring levels of human Aβ42 and Aβ40 by ELISA, both of which were apparently associated with a trend toward decreases in the cortex with TYROBP overexpression, mostly among male APP/PSEN1;Iba1Tyrobp mice (Fig. 3C). There was no genotype-dependent difference in the number of plaque-associated microglia (Fig. 3D–E), unlike what has been reported in 5xFAD mice in the presence of a transgenic increase in TREM2[32]. To evaluate microglial activation, we probed both groups with anti-IBA1 antibody and observed a weaker staining in APP/PSEN1;Iba1Tyrobp mice (Supp Fig. 2A). We next performed RT-qPCR on a group of microglial genes previously described in homeostatic or activated microglia. There was a significant increase of Axl and Ccl2 and a decrease of Cd68 and Tgfβ in brains of APP/PSEN1;Iba1Tyrobp mice (Fig. 3F). Finally, we observed that the decrease of amyloid plaques persisted in 8-month-old APP/PSEN1;Iba1Tyrobp mice as shown by the decreased percentage of ThioS positive areas in somatomotor, hippocampus, and visual areas (Fig. 3G–H).
3.2.4. TYROBP overexpression in MAPTP301S mice increases TAU phosphorylation and microglial activation
We previously reported that deletion of Tyrobp altered both mouse amyloidosis and tauopathy phenotypes and the microglial response to these pathologies[25–27]. In MAPTP301S;Iba1Tyrobp double heterozygous mice, western blot analyses using AT8 and T205 antibodies revealed increased levels of phosphorylated-TAU (pTAU) in the cortex of both male and female mice compared to MAPTP301S mice at 4 months of age, whereas total human TAU levels detected with the HT7 antibody were unchanged (Fig. 4A–B). Increased pTAU within brains from MAPTP301S;Iba1Tyrobp mice was further conrmed immunohistochemically (Fig. 4C). We also observed increased IBA1 intensity in MAPTP301S;Iba1Tyrobp compared to MAPTP301S mice and this IBA1 increase was correlated with the increased pTAU (Fig. 4D, Supp Fig. 2B). We confirmed an increased microglial activation state by double-label immunohistochemistry with anti-IBA1 and anti-CD68 in the piriform cortex (Fig. 4E). Using RT-qPCR, we measured increases of Tyrobp, P2ry12, Apoe, Axl, Itgax, Iba1, Tgfβ and Il6 mRNAs in MAPTP301S;Iba1Tyrobp mice compared to MAPTP301S mice (Fig. 4F).
3.2.5. Induction of microglial Tyrobp and Apoe is Trem2-independent in a model of cortical stab injury
To assess the interactions amongst Trem2, Tyrobp and Apoe in microglia, we used an injury paradigm by introducing a small penetrating cortical stab injury via stereotactic surgery into one hemisphere of the mouse brain in order to induce a recruitment of microglia around the injury site[60]. We first used injured- WT mice and combined RNA in situ hybridization and immunohistochemistry for Apoe and IBA1 respectively. In the intact hemisphere, most Apoe mRNA was not located in microglia but rather in astrocytes, the source of most APOE in brain. However, Apoe mRNA was dramatically increased in microglia recruited on the lesioned side (Fig. 5A–B). Following the same procedure in Tyrobp−/− mice, Apoe mRNA was not induced in microglia on either side (Fig. 5C), but strikingly, mRNA levels of Tyrobp and Apoe were highly upregulated in the recruited microglia of injured Trem2−/− mice (Fig. 5D, Supp Fig. 4). Taken together, these data indicate that Tyrobp upregulation in recruited microglia around the traumatic lesion is Trem2-independent. Moreover, the increase of Apoe transcripts in recruited microglia in the same mouse model of injury appears to be Tyrobp-dependent but Trem2-independent.
3.2.6. Induction of microglial Tyrobp and Apoe around amyloid plaques is Trem2-independent, and Apoe upregulation is dramatically decreased when Tyrobp is absent
In order to investigate further these interactions among Trem2, Tyrobp and Apoe in microglia in the presence of mutant human APP, we first performed dual RNA in situ hybridization and immunohistochemistry for Tyrobp, IBA1 and 6E10 in TgCRND8 mice[61] on either a WT or Trem2-null background. Despite reduced recruitment of microglia around plaques when Trem2 was deleted[57], Tyrobp mRNA was still increased in plaque-associated microglia (Fig. 6A) as was Apoe mRNA in plaque-associated microglia in the same TgCRND8;Trem2−/− mice (Fig. 6B). We confirmed that the plaques-associated microglia upregulating Tyrobp were the ones upregulating Apoe (Fig. 6C). We then assayed APP/PSEN1 mice that were either WT or deficient in Tyrobp and, while the expression of Apoe was not completely abolished by deletion of Tyrobp, we confirmed a substantial decrease in the induction of Apoe mRNA in plaque-associated microglia when Tyrobp was absent (Fig. 6D).
In summary, our results provide compelling evidence that: 1) upregulation of Tyrobp mRNA level is an early event occurring in recruited microglia; 2) TYROBP-APOE signaling in the microglial sensome is readily detectable even in the absence of Trem2. We propose that activation of the TYROBP-APOE pathway could be an early or even initiating step in the transformation of microglia from the homeostatic phenotype to the DAM phenotype (Figure 7).
Figure 7: Proposed ligand-induced Tyrobp signaling in recruited microglia.

Left panel: in response to penetrating stab injury or accumulation of amyloid-β (Aβ) deposits or misfolded tau, Tyrobp transcription is upregulated in microglia, thereby marking these cells as “recruited microglia”. Right panel: we observed that both microglial recruitment and Tyrobp upregulation occur in the absence of TREM2, indicating the existence of “sensing” receptors. Multiple alternative signaling pathways can be considered:
- Ligand signaling is initiated by apolipoprotein E, Aβ, debris, or other ligands at sensing receptors and leads to phosphorylation of the tyrosine residues in the cytoplasmic ITAM of TYROBP by SRC kinases and the recruitment of SYK. In turn, SYK signaling leads to up-regulated transcription of Tyrobp and Apoe. This series of events forms the basis for the phenotypic switch from homeostatic microglia to DAM.
- In mice lacking TREM2, microglial recruitment is retained, and transcription of both Tyrobp and Apoe is induced. Since these are constitutive TREM2 knockout mice, we are unable to exclude the possibility that some unknown sensor developed as compensation for the absence of TREM2. Another possibility is the existence of an unidentified sensing receptor that can upregulate TYROBP and APOE through a mechanism that does not require formation of TREM2/TYROBP complexes.
Abbreviations: TYROBP, tyrosine kinase binding protein; TREM2, triggering receptor expressed on myeloid cells-2; ITAM, immunoreceptor tyrosine-based activation motif; SYK, spleen tyrosine kinase; APOE, Apolipoprotein E; DAM, disease-associated microglia.
Supplementary Material
ACKNOWLEDGEMENTS
The study was supported by the National Institute on Aging (U01 AG046170 and R01 AG057907 to MEE, SG and BZ), the Alzheimer’s Disease Research Division of the BrightFocus Foundation (grant A2018253F to MA and grant A2016482F to JVHM), the Mount Sinai Alzheimer’s Disease Research Center (ADRC P50 AG005138 and P30 AG066514 to Mary Sano, with internal pilot grant awarded to MA). Figure 7 was created with BioRender.com.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
- [1].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013;45:1452–8. 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011;43:436–41. 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lambert J-C, Zelenika D, Hiltunen M, Chouraki V, Combarros O, Bullido MJ, et al. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol Aging 2011;32:756.e11–15. 10.1016/j.neurobiolaging.2010.11.022. [DOI] [PubMed] [Google Scholar]
- [4].Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 2011;43:429–35. 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jun G, Naj AC, Beecham GW, Wang L-S, Buros J, Gallins PJ, et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 2010;67:1473–84. 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009;41:1094–9. 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- [7].Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009;41:1088–93. 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019;51:404–13. 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019;51:414–30. 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013;16:1896–905. 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013;153:707–20. 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017;169:1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- [13].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117–27. 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhou S-L, Tan C-C, Hou X-H, Cao X-P, Tan L, Yu J-T. TREM2 Variants and Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. JAD 2019;68:1171–84. 10.3233/JAD-181038. [DOI] [PubMed] [Google Scholar]
- [15].Paloneva J, Kestilä M, Wu J, Salminen A, Böhling T, Ruotsalainen V, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet 2000;25:357–61. 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- [16].Pottier C, Ravenscroft TA, Brown PH, Finch NA, Baker M, Parsons M, et al. TYROBP genetic variants in early-onset Alzheimer’s disease. Neurobiol Aging 2016;48:222.e9–222.e15. 10.1016/j.neurobiolaging.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao Y, Wu X, Li X, Jiang L-L, Gui X, Liu Y, et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018;97:1023–1031.e7. 10.1016/j.neuron.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. The Journal of Cell Biology 2018;217:459–72. 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Montalvo V, Quigley L, Vistica BP, Boelte KC, Nugent LF, Takai T, et al. Environmental factors determine DAP12 deficiency to either enhance or suppress immunopathogenic processes. Immunology 2013;140:475–82. 10.1111/imm.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017;47:566–581.e9. 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mahley RW. Apolipoprotein E: Remarkable Protein Sheds Light on Cardiovascular and Neurological Diseases. Clin Chem 2017;63:14–20. 10.1373/clinchem.2016.255695. [DOI] [PubMed] [Google Scholar]
- [22].Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A 2006;103:5644–51. 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunological Reviews 2009;227:150–60. 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, et al. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiology of Aging 2017;53:194.e13–194.e22. 10.1016/j.neurobiolaging.2017.01.015. [DOI] [PubMed] [Google Scholar]
- [25].Haure-Mirande J-V, Audrain M, Fanutza T, Kim SH, Klein WL, Glabe C, et al. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol 2017;134:769–88. 10.1007/s00401-017-1737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haure-Mirande J-V, Wang M, Audrain M, Fanutza T, Kim SH, Heja S, et al. Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in cerebral Aβ amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Aβ burden. Mol Psychiatry 2018. 10.1038/s41380-018-0255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Audrain M, Haure-Mirande J-V, Wang M, Kim SH, Fanutza T, Chakrabarty P, et al. Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry 2018. 10.1038/s41380-018-0258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. The Journal of Experimental Medicine 2015;212:287–95. 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci USA 2017;114:11524–9. 10.1073/pnas.1710311114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener 2017;12:74. 10.1186/s13024-017-0216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, et al. Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc Natl Acad Sci USA 2018;115:10172–7. 10.1073/pnas.1811411115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee CYD, Daggett A, Gu X, Jiang L-L, Langfelder P, Li X, et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018;97:1032–1048.e5. 10.1016/j.neuron.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gratuze M, Leyns CEG, Sauerbeck AD, St-Pierre M-K, Xiong M, Kim N, et al. Impact of TREM2R47H variant on tau pathology–induced gliosis and neurodegeneration. Journal of Clinical Investigation 2020:10.1172/JCI138179. 10.1172/JCI138179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 2020;26:131–42. 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 2019. 10.1084/jem.20190980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ulrich JD, Ulland TK, Mahan TE, Nyström S, Nilsson KP, Song WM, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med 2018;215:1047–58. 10.1084/jem.20171265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen W-T, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020:S0092867420308151. 10.1016/j.cell.2020.06.038. [DOI] [PubMed] [Google Scholar]
- [38].Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee S-H, Srinivasan K, et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J Neurosci 2020:1871–19. 10.1523/JNEUROSCI.1871-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015;160:1061–71. 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nature Neuroscience 2019. 10.1038/s41593-018-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kloske CM, Wilcock DM. The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front Immunol 2020;11:754. 10.3389/fimmu.2020.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fitz NF, Wolfe CM, Playso BE, Biedrzycki RJ, Lu Y, Nam KN, et al. Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol Neurodegeneration 2020;15:41. 10.1186/s13024-020-g00394-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 1998;391:703–7. 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- [44].Tomasello E, Olcese L, Vély F, Geourgeon C, Bléry M, Moqrich A, et al. Gene structure, expression pattern, and biological activity of mouse killer cell activating receptor-associated protein (KARAP)/DAP-12. J Biol Chem 1998;273:34115–9. 10.1074/jbc.273.51.34115. [DOI] [PubMed] [Google Scholar]
- [45].Siddiqui SS, Matar R, Merheb M, Hodeify R, Vazhappilly CG, Marton J, et al. Siglecs in Brain Function and Neurological Disorders. Cells 2019;8. 10.3390/cells8101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Salminen A, Kaarniranta K. Siglec receptors and hiding plaques in Alzheimer’s disease. J Mol Med 2009;87:697–701. 10.1007/s00109-009-0472-1. [DOI] [PubMed] [Google Scholar]
- [47].Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet 2008;83:623–32. 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019;103:820–835.e7. 10.1016/j.neuron.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015;138:1738–55. 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 2015;18:1584–93. 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhao W, Fan J, Kulic I, Koh C, Clark A, Meuller J, et al. Axl receptor tyrosine kinase is a regulator of apolipoprotein E. Mol Brain 2020;13:66. 10.1186/s13041-020-00609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Levy O, Lavalette S, Hu SJ, Housset M, Raoul W, Eandi C, et al. APOE Isoforms Control Pathogenic Subretinal Inflammation in Age-Related Macular Degeneration. J Neurosci 2015;35:13568–76. 10.1523/JNEUROSCI.2468-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Levy O, Calippe B, Lavalette S, Hu SJ, Raoul W, Dominguez E, et al. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol Med 2015;7:211–26. 10.15252/emmm.201404524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 2004;13:159–70. 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- [55].Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006;26:10129–40. 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337–51. 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- [57].Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016;90:724–39. 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoogland ICM, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 2015;12:114. 10.1186/s12974-015-0332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tanaka KF, Matsui K, Sasaki T, Sano H, Sugio S, Fan K, et al. Expanding the Repertoire of Optogenetically Targeted Cells with an Enhanced Gene Expression System. Cell Reports 2012;2:397–406. 10.1016/j.celrep.2012.06.011. [DOI] [PubMed] [Google Scholar]
- [60].Clarke D, Penrose MA, Harvey AR, Rodger J, Bates KA. Low intensity rTMS has sex-dependent effects on the local response of glia following a penetrating cortical stab injury. Exp Neurol 2017;295:233–42. 10.1016/j.expneurol.2017.06.019. [DOI] [PubMed] [Google Scholar]
- [61].Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 2001;276:21562–70. 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- [62].Kang SS, Kurti A, Baker KE, Liu C-C, Colonna M, Ulrich JD, et al. Behavioral and transcriptomic analysis of Trem2-null mice: not all knockout mice are created equal. Human Molecular Genetics 2018;27:211–23. 10.1093/hmg/ddx366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Litvinchuk A, Wan Y-W, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018;100:1337–1353.e5. 10.1016/j.neuron.2018.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.