

Abstract
Osteoarthritis (OA) is a debilitating and painful disease characterized by the progressive loss of articular cartilage. Post-traumatic osteoarthritis (PTOA) is an injury-induced type of OA that persists in an asymptomatic phase for years before it becomes diagnosed in ~50% of injured individuals. Although PTOA is not classified as an inflammatory disease, it has been suggested that inflammation could be a major driver of PTOA development. Here we examined whether a state of systemic inflammation induced by lipopolysaccharide (LPS) administration 5-days before injury would modulate PTOA outcomes. RNA-seq analysis at 1-day post-injury followed by micro-computed tomography (μCT) and histology characterization at 6 weeks post-injury revealed that LPS administration causes more severe PTOA phenotypes. These phenotypes included significantly higher loss of cartilage and subchondral bone volume. Gene expression analysis showed that LPS alone induced a large cohort of inflammatory genes previously shown to be elevated in synovial M1 macrophages of rheumatoid arthritis (RA) patients, suggesting that systemic LPS produces synovitis. This synovitis was sufficient to promote PTOA in MRL/MpJ mice, a strain previously shown to be resistant to PTOA. The synovium of LPS-treated injured joints displayed an increase in cellularity, and immunohistological examination confirmed that this increase was in part attributable to an elevation in type 1 macrophages. LPS induced the expression of Tlr7 and Tlr8 in both injured and uninjured joints, genes known to be elevated in RA. We conclude that inflammation before injury is an important risk factor for the development of PTOA and that correlating patient serum endotoxin levels or their state of systemic inflammation with PTOA progression may help develop new, effective treatments to lower the rate of PTOA in injured individuals.
Keywords: ACL, BONE, INFLAMMATION, LPS, MRL/MpJ, OSTEOARTHRITIS, RNA SEQUENCING, μCT
Introduction
Post-traumatic osteoarthritis (PTOA) develops in approximately half of patients who have experienced an articular trauma such as a tibial plateau fracture, meniscus tear, or an anterior cruciate ligament (ACL) rupture.(1–3) Within seconds of the traumatic event, cell necrosis, collagen rupture, swelling of the cartilage and synovium, hemarthrosis, and loss of glycosaminoglycans occur,(4) events likely to contribute to PTOA initiation. The healing process initiates during the acute phase, which is characterized by significant apoptosis, leukocyte infiltration, matrix degradation, loss of lubrication, and overexpression of inflammatory mediators.(5,6) For ~50% of these patients who undergo ACL reconstruction, the injury repairs and after surgery the joint never develops PTOA, but the rest persist in an asymptomatic pre-PTOA phase. This degenerative phase can last as little as a couple of years or can linger for decades before entering the chronic phase that eventually leads to a PTOA diagnosis.(5) Although osteoarthritis is currently not classified as an inflammatory disease, many reports have described a state of elevated joint inflammation post-injury as strongly correlating with progression to the disease state.(7) Importantly, studies examining both inflammation and joint injury have not looked at how a brief burst of systemic inflammation shortly before an injury could influence the outcomes of PTOA. A more detailed understanding of how elevated inflammation before injury contributes to PTOA development could lead to the identification of prognostic inflammatory markers during the acute phase as well as the development of new prophylactics for blocking unfavorable immune components that promote cartilage degeneration.
The endotoxin lipopolysaccharide (LPS) is secreted by gram-negative bacteria, such as Escherichia coli, and in healthy individuals LPS is released into circulation by the gut biome to provide a beneficial low-level immune activation.(8) During a gram-negative bacteria infection, circulating levels of LPS increase, which helps the immune system clear the bacteria, but this elevated immune activation may also negatively influence physiological processes sensitive to levels of inflammatory cytokines. For example, it has been previously shown that mice with persistent high levels of circulating LPS have a significantly lower bone mass than mice with low levels of LPS.(9–12) Furthermore, focal LPS administration to joints has been shown to induce synovitis in horses and has been used as a model to evaluate potential treatments for acute synovitis.(13–17) Studies have been conducted on the differences between LPS levels in serum of germ-free mice and specific pathogen-free mice; this experiment had LPS assay inhibitors, therefore they did not show any difference between circulating LPS levels in germ-free mice and specific pathogen-free mice.(18) No study to date has investigated the effect of systemic LPS-induced inflammation before ACL rupture on the outcomes of PTOA in strains of mice that have been shown to be either sensitive (C57Bl/6J) or resistant (MRL/MpJ) to PTOA.(5) In prior work, C57Bl/6J male mice have been shown to develop PTOA post-injury, whereas Murphy Roths Large (MRL/MpJ) male mice retained a relatively normal joint morphology after being subjected to several models of PTOA, including tibial compression overload,(5) intraarticular fracture,(19) and destabilization of the medial meniscus.(20) MRL/MpJ have been suggested to be resistant to PTOA due to their ability to resolve inflammation faster than C57Bl/6J and also to repair damaged cartilage; these two characteristics make them virtually resistant to PTOA.(5,21) Previously published data had identified differences in the resolution of acute inflammation between sexes where lower monocyte and neutrophil levels were recorded for females, but no study to date has addressed how sex differences in inflammation and injury would affect PTOA outcomes.(22)
Here we show that a single systemic inflammation-inducing dose of LPS administered 5 days before a noninvasive ACL injury is sufficient to modify outcomes by producing a more severe PTOA phenotype.(23) LPS timeline was chosen based on clinical parameters demonstrated for LPS challenges in mice, where fever subsided ~3 days post-injection and locomotive activity returned to normal 5 to 6 days after an intraperitoneal LPS injection at the same concentration as the one used in this study on C57Bl/6J mice.(24,25) Molecular characterization of the joint identified significant downregulation of bone and cartilage matrix proteins during the acute phase and an increase in the number of type 1 macrophages in the synovium, 6 weeks after injury, suggesting that systemic inflammation when combined with ACL rupture initiates synovitis, suppressing cartilage and bone repair. More interestingly, the MRL/MpJ strain of mice also developed PTOA in response to LPS, suggesting that these mice are not able to block the negative effects of this inflammatory process. These results have impactful clinical ramifications and suggest that knowledge of systemic inflammation in the patient at the time of injury may be useful to predict future clinical risks. Furthermore, identifying and prophylactically blocking immune components involved in this process may guide the discovery of new therapies for the prevention of PTOA.
Materials and Methods
Experimental animals and ACL injury model
Male and female C57Bl/6J mice were purchased (Jackson Laboratory, Bar Harbor, ME, USA; stock no. 000664) at 4 weeks of age and kept under normal cage conditions for 6 weeks. MRL/MpJ strains were bred in house. Five days before injury, at 10 weeks of age, cohorts of mice (RNASeq: n ≥ 4 per group C57Bl/6J, histology and micro-computed tomography [μCT]: n ≥ 4 per group C57Bl/6J male and female; histology and μCT: n = 3 per group MRL/MpJ female, n = 4 per group MRL/MpJ male) were separated into two groups (vehicle [VEH] and lipopolysaccharide [LPS]) for both males and females. The LPS group received an intraperitoneal (ip) injection of LPS (1 mg/kg), whereas the vehicle (VEH) group received an ip injection of saline of equivalent volume. On the day of injury, both groups were subjected to ACL rupture using tibial compressive (TC) loading. TC loading is a noninvasive single dynamic tibial compressive overload using an electromagnetic material testing system (ElectroForce 3200, TA Instruments, New Castle, DE, USA) as previously described.(26–28) Cohorts were placed under anesthesia using isoflourane before injury.(29) ACL injury was performed by placing the mouse in the system and applying enough force (C57Bl/6J: 12 N-14 N; MRL/MpJ 15 N-18 N) to rupture the ACL; the uninjured group was placed in the system and received a non-injury inducing force (2 N-3 N). After injury mice cohorts were given saline (0.05 ml) and buprenorphine (0.05 mg/kg) and returned to normal cage activity as previously described.(5,23,27,28) All animal experiments were approved by the Lawrence Livermore National Laboratory and University of California, Davis Institutional Animal Care and Use Committee and conformed to the Guide for the care and use of laboratory animals.
Micro-computed tomography
C57Bl/6J injured (right leg), injured-contralateral (left leg), and uninjured joints (right and left legs) were collected 6 weeks post-injury for female and male VEH and LPS groups. Samples were dissected and fixed for 72 hours at 4°C using 10% neutral buffer formalin; samples were stored in 70% ethanol at 4°C until scanned. Whole knees were scanned using a Scanco μCT 35 (Bruttisellen, Switzerland) according to the rodent bone structure analysis guidelines (X-ray tube potential = 55 kVp, intensity = 114 mA, 10 μm isotropic nominal voxel size, integration time = 900 ms).(23) Trabecular bone in the distal femoral epiphysis was analyzed by manually drawing contours on 2D transverse slides. The distal femoral epiphysis was designated as the region of trabecular bone enclosed by the growth plate and subchondral cortical bone plate. We quantified trabecular bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp).(30) Mineralized osteophyte volume in injured and contralateral joints was quantified by drawing contours around all heterotopic mineralized tissue attached to the distal femur and proximal tibia as well as the whole fabellae, menisci, and patella. Total mineralized osteophyte volume was then determined as the volumetric difference in mineralized tissue between injured and uninjured joints.
Histological assessment of articular cartilage and joint degeneration
VEH and LPS-treated injured, contralateral, and uninjured joints were dissected 6 weeks post-injury, fixed, dehydrated, paraffin embedded, and sectioned as previously described.(27) The cartilage was visualized in sagittal 6-μm paraffin serial using Safranin-O (0.1%, Sigma, St. Louis, MO, USA; S8884) and Fast Green (0.05%, Sigma; F7252) as previously described.(28) OA severity was evaluated using a modified Osteoarthritis Research Society International (OARSI) scoring scale as previously described.(31) Cartilage scoring began ~0.4 mm out from the start of synovium to the articular cartilage. Blinded slides were evaluated by seven scientists (six with and one without expertise in OA) utilizing modified (sagittal) OARSI scoring parameters because of the severe phenotype caused by TC loading-destabilization that promotes mechanical-induced tibial degeneration on injured joints.(27,31) Modified scores are: (0) for intact cartilage staining with strong red staining on the femoral condyle and tibia, (1) minor fibrillation without cartilage loss, (2) clefts below the superficial zone, (3) cartilage thinning on the femoral condyle and tibia, (4) lack of staining on the femoral condyle and tibia, (5) staining present on 90% of the entire femoral condyle with tibial degeneration, (6) staining present on more than 80% of the femoral condyle with tibial degeneration, (7) staining present on 75% of the femoral condyle with tibial degeneration, (8) staining present on more than 50% of the femora condyle with tibial degeneration, (9) staining present in 25% of the femoral condyle with tibial degeneration, (10) staining present in less than 10% of the femoral condyle with tibial degeneration.
Statistical analysis
Data expressed using box and whisker plots with median and interquartile range. Statistical analysis performed using Prism 8. Statistical comparisons were performed using 2-way ANOVA. For all tests, p < 0.05 was considered statistically significant.
Immunofluorescent staining
Six-micrometer sections from injured samples from both treatment groups of C57Bl/6J were used. Unitrieve was used as an antigen retrieval method for 30 minutes at 65°C.(32) Primary antibodies: Anti-F4/80 (Abcam, Cambridge, MA, USA; ab16911 [1:50]), Anti-CD206 (Abcam, ab64693 [1:500]), Anti-iNOS (Abcam, ab15323 [1:100]), CYTL1 (Proteintech, Chicago, IL, USA; 15856–1-AP [1:50]), and CHAD (Invitrogen, Carlsbad, CA, USA; PA5–53761 [1:300]) were used and incubated overnight at room temperature in a dark, humid chamber. Negative control slides were incubated with secondary antibody only. Stained slides were mounted with Prolong Gold with DAPI (Molecular Probes, Eugene, OR, USA). Slides were imaged using a Leica (Buffalo Grove, IL, USA) DM5000 microscope. ImagePro Plus V7.0 Software and a QIClick CCD camera (QImaging, Surrey, BC, Canada) were used for imaging and photo editing.
RNA sequencing and data analysis
C57Bl/6J injured and uninjured joints from VEH and LPS-treated male and female mice were collected 24 hours after injury (n ≥ 4). Joints were dissected and cut at the edges of the joint region with small traces of soft tissue to preserve the articular integrity. RNA was isolated and sequenced as previously described.(5) RNA-seq data quality was checked using FastQC software (version 0.11.5). Sequence reads were aligned to the mouse reference genome (mm10) using STAR (version 2.6). After read mapping, featureCounts from Rsubread package (version 1.30.5) was used to perform read summarization to generate gene-wise read counts. RUVseq (version 1.16.0) was used to identify factors of unwanted variation. Differentially expressed genes were identified using edgeR (version 3.22.3), adjusting for unwanted variation. Genes with false discovery rate (FDR)-corrected p value less than 0.05 and fold change greater than 1.25 were considered as differentially expressed genes (DEGs). Heatmaps were generated using heatmap.2 function in R package gplots.
Results
LPS administration causes a more severe PTOA phenotype
Using a tibial compression PTOA mouse model,(33) we examined whether a single dose of LPS (1 mg/kg) would impact OA outcomes at 6 weeks post-injury. C57Bl/6J male and female VEH uninjured joints looked similar (Fig. 1A, E). Joints of VEH injured female mice displayed more cartilage erosion than the VEH injured male mice (Fig. 1B, F), particularly on the medial side (Fig. 1b, f; arrow, asterisk); VEH injured females showed increased synovitis compared with the VEH injured male mice (Fig. 1bb, ff, asterisk). Although the cartilage of male LPS-treated uninjured joints was comparable to male VEH-treated contralateral and uninjured groups (Fig. 1A, C), significant differences were observed in the LPS-treated male injured joints (Fig. 1B, D). LPS treatment led to significant erosion of both cartilage and bone on the femoral and tibial posterior side (Fig. 1B, D). There was a clear reduction in the amount of subchondral bone visualized in the LPS-treated femurs relative to VEH-treated femurs (Fig. 1B, D, F, H, asterisks). VEH-treated injured joints retained significantly more articular cartilage integrity throughout the joint compared with LPS-treated injured joints, which displayed significant thinning of the femoral articular cartilage (Fig. 1b, d, f, h, arrow, asterisks). LPS-treated injured joints also displayed significantly hyperplastic synovium (Fig. 1 bb, dd, ff, hh) indicative of synovitis and elevated inflammation occurring in LPS-treated but not VEH-treated injured joints. The PTOA phenotype was more severe in females than males treated with or without LPS (Fig. 1D, H); however, it was only in the injured LPS-treated females that there was a lack of proteoglycan staining in the growth plate, when comparing it to the female LPS-treated contralateral (Fig. 1G, H). When comparing OARSI scores, LPS-treated injured joints had significantly more severe cartilage loss than VEH-treated injured joints, independent of sex (Fig. 1I). Lastly, we found a significant difference between the male and female injured joints, independent of treatment.
Fig 1.
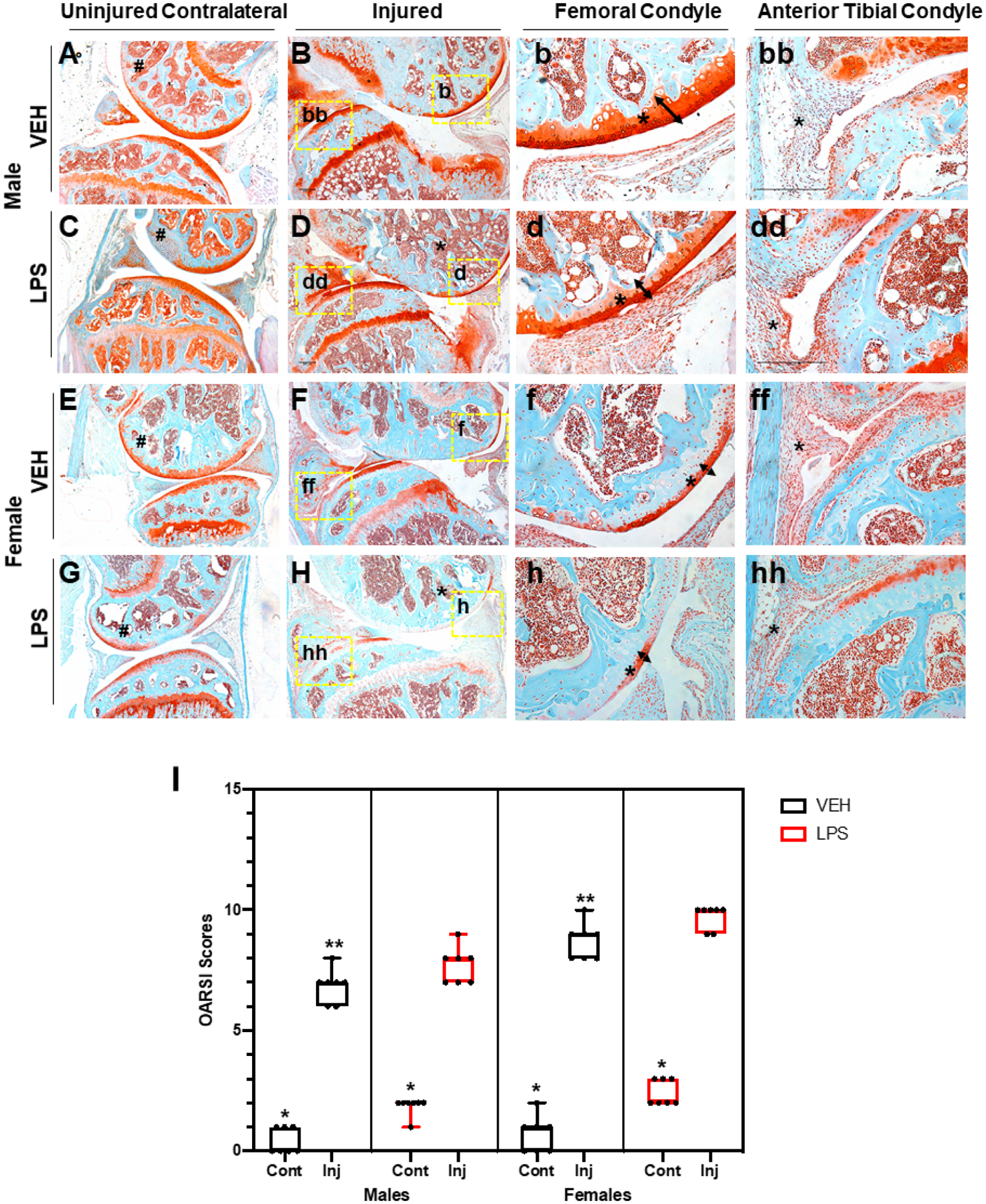
Histological assessment of LPS-treated tibial compression injured joints. (A) VEH male uninjured contralateral joint shows in red the cartilage and in blue the bone both intact. (B) VEH male injured joint shows degradation of cartilage on the femoral condyle and tibial resorption. (b) VEH male injured shows intact cartilage through the entire femoral condyle shown by the tibial. (bb) VEH male injured tibial condyle shows some cellular infiltration. (C) LPS male uninjured contralateral joints show proteoglycan degradation that results in lack of staining on the femoral condyle. (D) LPS male injured joint shows thinning of the cartilage and less calcified matrix on the femur. (d) LPS male injured shows cartilage thinning on the femoral condyle. (dd) LPS male shows an increase in cellular infiltration on the anterior tibial condyle. (E) VEH female uninjured contralateral shows strong cartilage staining. (F) VEH female injured shows lack of cartilage staining. (f) VEH female injured shows thinner cartilage on the femoral condyle. (ff) VEH female injured joints show synovium thickening and cellular infiltration on the anterior tibial condyle. (G) LPS female uninjured contralateral joints show lack of staining on the femoral condyle. (H) LPS female injured joints show virtually no cartilage staining. (h) LPS female injured shows almost no staining on the femoral condyle. (hh) LPS female injured shows cellular infiltration and little cartilage staining. (I) OARSI scoring shows statistically significant differences between sexes and treatment types when comparing injured joints. Uninjured joints were not found to be statistically significant independent of treatment and sex. *Statistically significantly different than injured joint of the same sex and treatment (p-value < 0.05). **Statistically significantly different than the LPS injured of the same sex (p-values < 0.05).
LPS administration reduces subchondral bone and osteophyte volume in injured joints
Because several studies have shown that LPS increases bone resorption and decreases bone mass,(34,35) we next examined loss of trabecular bone volume and osteophyte volume in LPS- and VEH-treated mice by quantifying the gain in osteophyte volume and the loss in subchondral trabecular bone at 6 weeks post-injury through the use of μCT (Fig. 2). VEH male injured femurs had significantly less subchondral bone volume (BV/TV) by ~4% and ~ 6.6% when compared with uninjured and contralateral, respectively. VEH females had significantly less BV/TV with ~3.3% less in injured relative to contralateral. LPS female injured joints had significantly less BV/TV than the uninjured by ~2.7% and contralateral by ~3.3%; injured LPS males had ~9.3% and ~ 9.5% significantly less BV/TV than uninjured and contralateral, respectively. In all groups examined, the percent loss of BV/TV in females was significantly less than in males (Fig. 2A). VEH-treated females had ~6.5%, ~5.2%, and ~8.4% less BV/TV than VEH males on the uninjured, injured, and contralateral joints, respectively; all were statistically significant. LPS-treated females had ~9.28%, ~5.22%, and ~9.49% less BV/TV than VEH males on the uninjured, injured, and contralateral joints, respectively; injured and contralateral were statistically significant. However, LPS treatment promoted a significant loss of BV/TV in injured joints when compared with VEH-treated joint, independent of sex. The BV/TV loss between injured VEH and injured LPS was statistically significant and was ~2.8% in females and ~2.7% in males, respectively (Fig. 2A). The LPS-related changes between sexes were not statistically significant.
Fig 2.
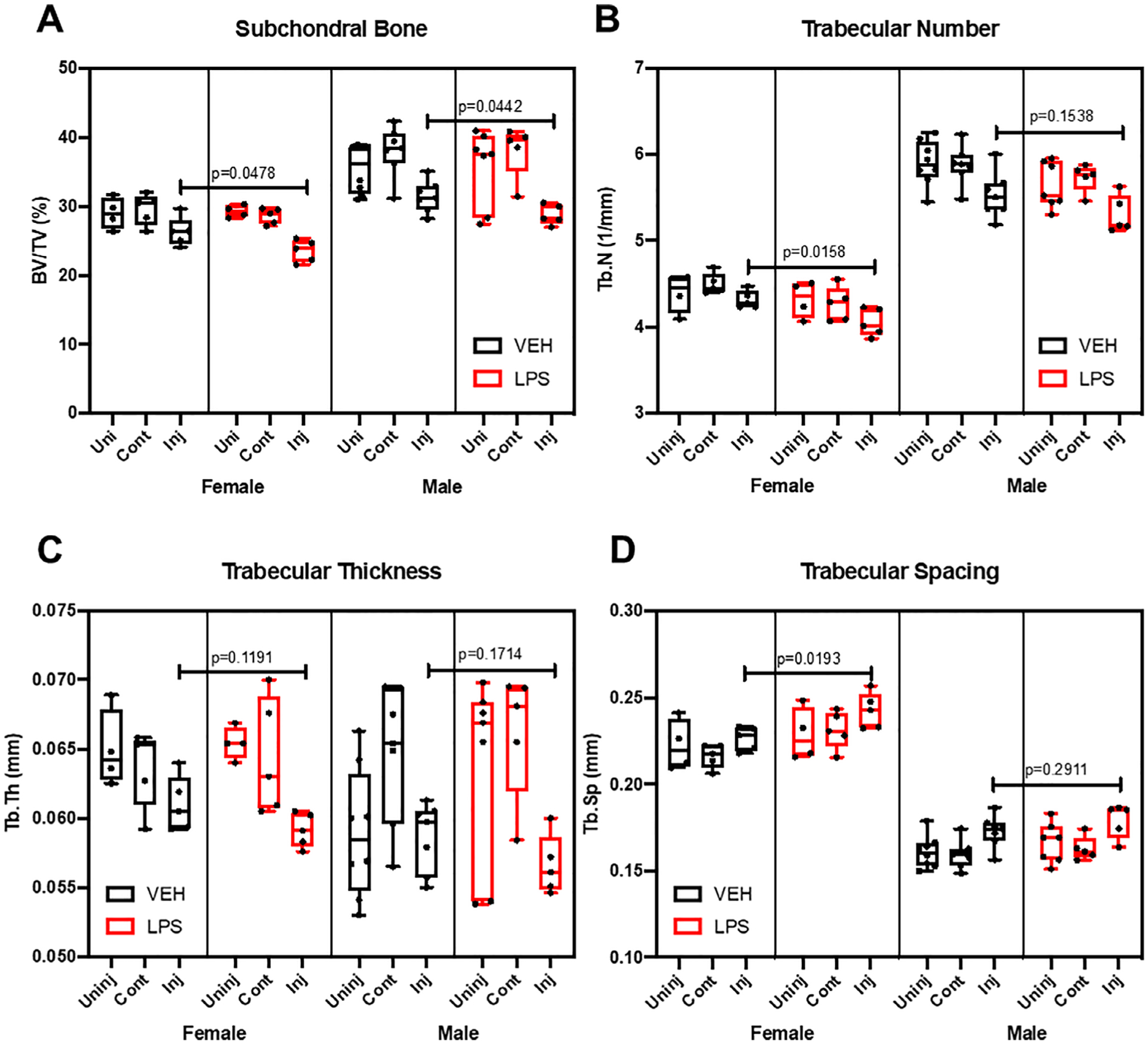
Bone microstructure assessment using micro-CT. (A) Subchondral bone volume fraction (BV/TV) is significantly lower in the LPS injured compared with the VEH. (B) Trabecular number (Tb.N) was lower in the LPS-treated injured female compared with the VEH injured female. (C) Trabecular thickness (Tb.Th) did not change with LPS treatment. (D) Trabecular spacing (Tb.Sp) was significantly higher in the LPS-treated female injured samples compared with the injured VEH females. Statistics were performed using two-way ANOVA and Student’s t test. *p < 0.05.
Other bone parameters were consistent with a bone loss phenotype in the distal femoral epiphysis of injured groups (Fig. 2). LPS treatment significantly reduced trabecular number (Tb.N) in injured females by ~6%; injured LPS males had ~4.2% less Tb. N, which was not statistically significant (Fig. 2B). Tb.N was significantly lower in females than males on both VEH and LPS. Trabecular thickness (Tb.Th) did not change in the injured joints in response to LPS administration (Fig. 2C). VEH uninjured females had significantly higher trabecular thickness than VEH males by ~9.2%. Trabecular spacing (Tb.Sp) increased on the LPS injured females by ~6.6% but was not significant on the males.
Osteophyte volume was significantly higher in male than in female VEH injured joints (Fig. 3). LPS administration induced a reduction in the amount of ectopic bone formed in females and males; this reduction was statistically significant in females only (Fig. 3). These findings suggest that LPS treatment enhances the injury-mediated catabolic activity in the subchondral bone, resulting in higher subchondral bone loss, but it slightly protects the injured joint from osteophyte formation.
Fig 3.
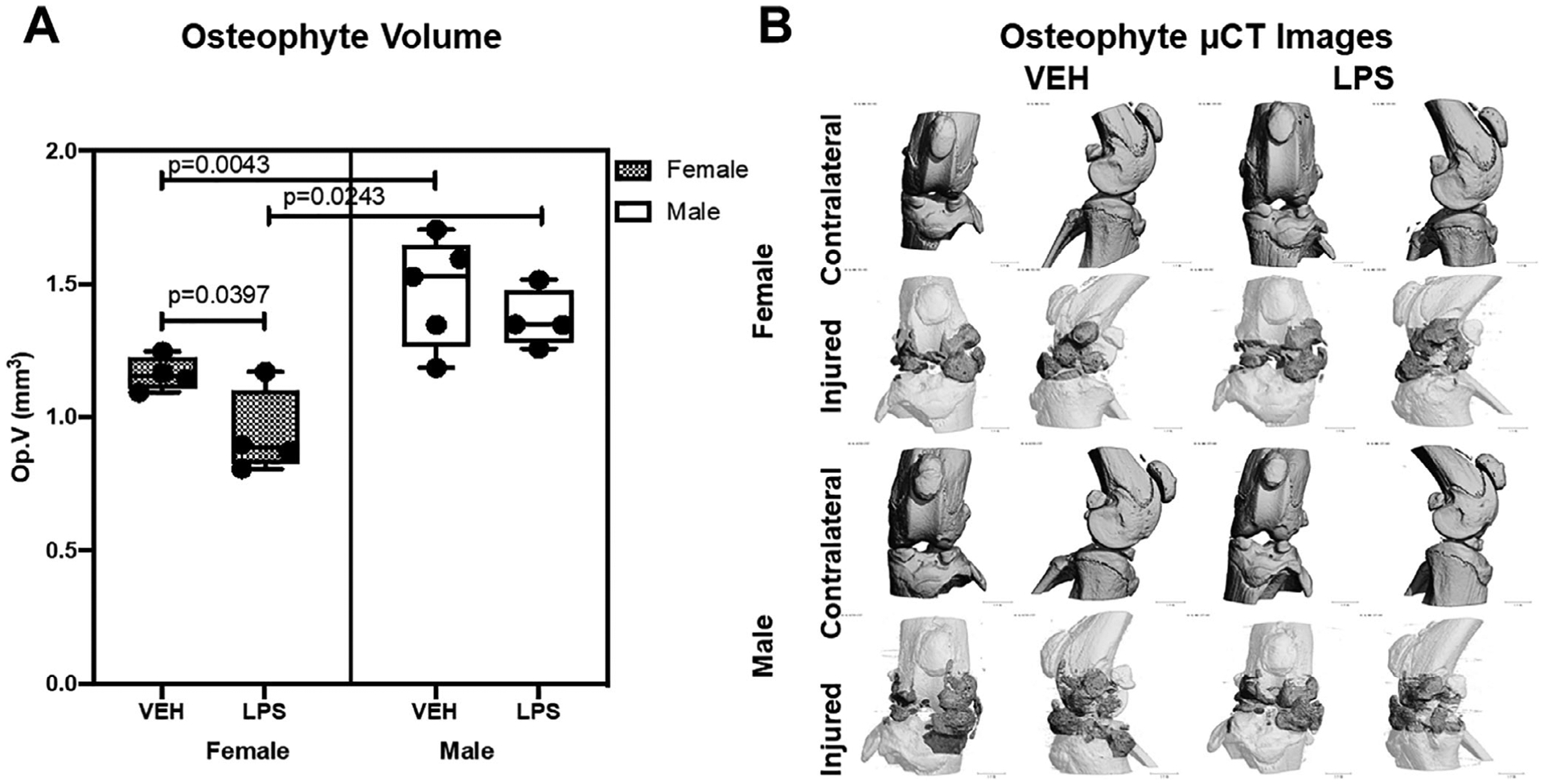
Osteophyte representation using micro-computed tomography. (A) There was significantly lower osteophyte volume on the VEH females compared with the males. There was significantly lower osteophyte volume on the LPS females than the VEH females. (B) Micro-computed tomography reconstructions of representative joints from all groups. For injured joints, dark gray regions show osteophyte growth compared with the native bone (white).
Cellular infiltration by type 1 macrophages is elevated in LPS-treated groups
Histological analysis showed that the anterior tibial condyle presented cellular infiltration on the LPS injured joints (Fig. 1D, dd). To determine whether LPS alters the composition of M1 and M2 macrophages in the injured joint, we used M1/M2-specific antibody markers to distinguish these subtypes. Immunohistochemical examination with macrophage markers determined that the increase in cellularity correlated with a larger amount of M1 macrophages present in the LPS injured joints than in the VEH injured joints, indicative of a higher level of inflammation (Fig. 4 bbb). In contrast, we observed a slight decrease in the abundance of M2 macrophages in LPS relative to VEH injured joints, suggesting that the joints of LPS treated animals lack sufficient anti-inflammatory macrophages. The change in M1/M2 ratio in the joint may influence the repair process and modulate PTOA phenotypes towards a more severe outcome (Fig. 4B, D).
Fig 4.
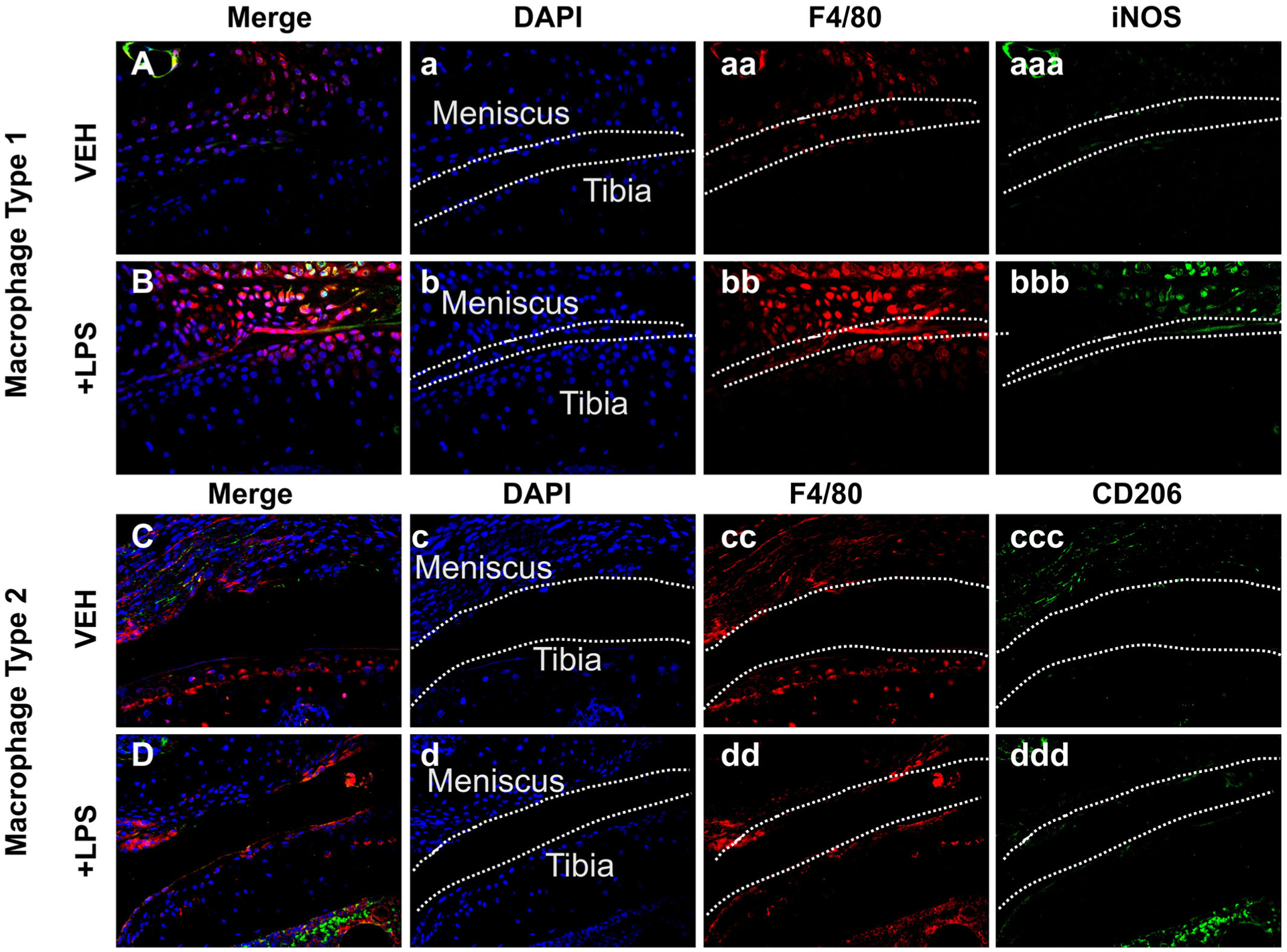
Fluorescent immunohistochemistry analysis of injured joints 6 weeks post-injury. (A, B) Fluorescent immunohistochemistry (IHC) of macrophage type 1 markers F4/80 and iNOS. There is a higher amount of M1 on the LPS sample. (C, D) Fluorescent IHC of macrophage type 2 markers F4/80 and CD205. There is a similar amount of M2 on VEH and LPS. Dashed line in white represents the surface of the anterior tibial condyle for all images.
Systemic LPS treatment induces inflammatory genes in the joint
Global gene expression analyses of male cohorts at 1 day post-injury identified 590 genes upregulated in LPS uninjured compared with VEH uninjured and 467 genes upregulated in LPS injured compared with VEH injured. Seventy-three genes associated with inflammation and immune responses were significantly upregulated in the LPS-treated samples (both injured and uninjured) when compared with the VEH uninjured controls. Genes associated with inflammation included transcripts such as toll-like receptors 5, 7, and 8 (Tlr5, Tlr7, Tlr8)(36,37) (Fig. 5A). We found that genes associated with the innate immune system, specifically monocyte infiltration like Ccl6 and Ccr2, were upregulated in the LPS-treated joints.(38–40) Cd80, a gene associated with T-cell activation, was also upregulated in the LPS-treated samples.(41) These data suggested that both innate and adaptive immune responses are being activated in LPS-treated joints when compared with the VEH uninjured. We also found 343 genes downregulated in LPS uninjured compared with VEH uninjured and 402 genes downregulated in LPS injured compared with VEH injured in male joints. There were 78 downregulated genes that were exclusive to male LPS injured when compared with all other groups, suggesting that these genes may correlate with the more severe PTOA phenotype. Several genes associated with bone and cartilage formation, such as Col9a1,(42) Col10a1,(43) Chad,(44) Notum,(45) and Sost,(28) were also downregulated in LPS injured samples (Fig. 5C). In addition, we found several genes whose knockouts produce OA phenotypes, such as Ucma,(46) and Cytl1,(47) were also downregulated. Transcriptional downregulation of Chad and Cytl1 was also confirmed at the protein level, where diminished expression of these proteins was detected in the articular cartilage of LPS injured (Fig. 5E, G) relative to the PBS injured (Fig. 5D, F). These early markers of lower cartilage and bone formation could be an indicator of the more severe PTOA phenotype in LPS injured samples.
Fig 5.
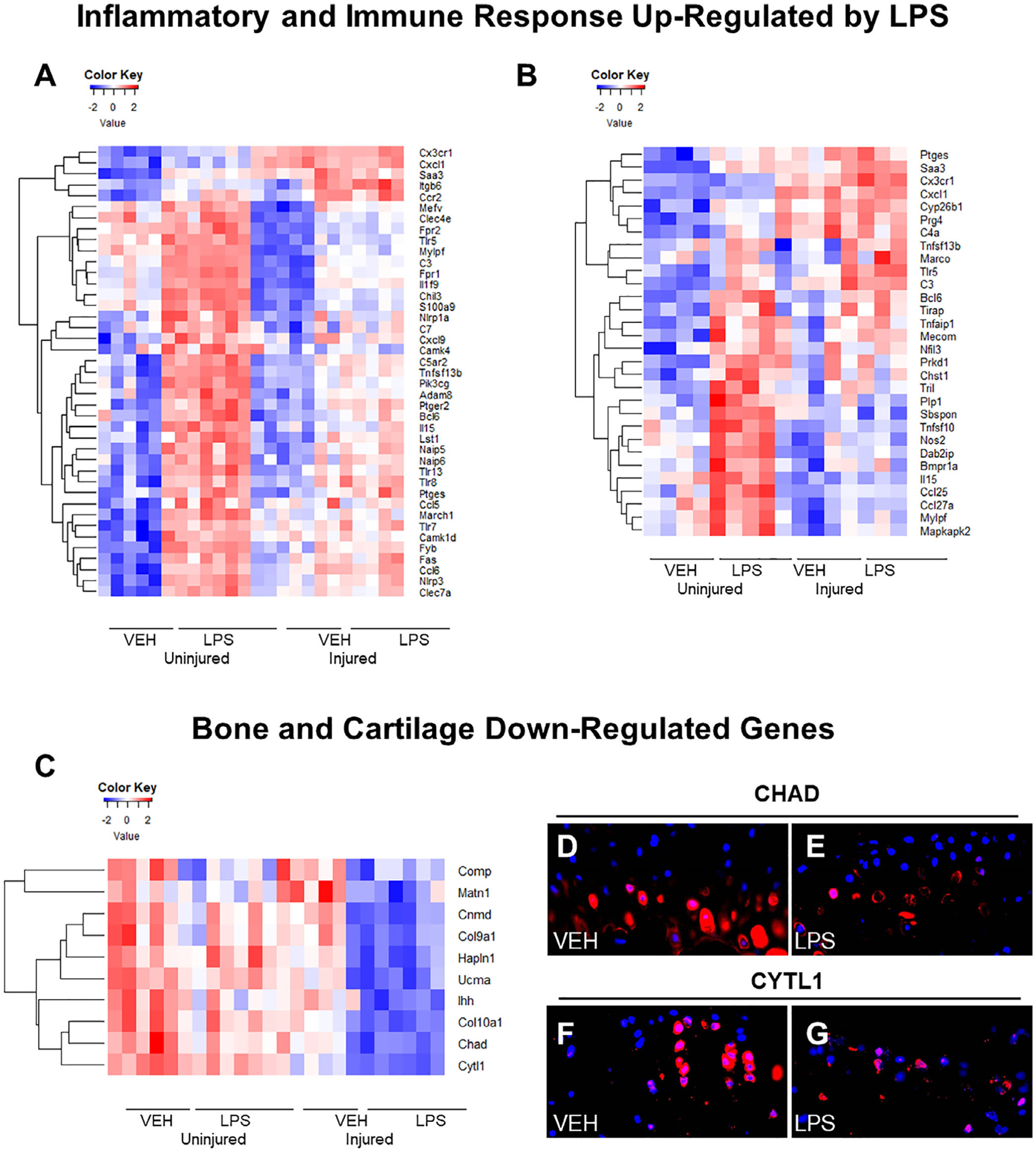
Gene expression changes caused by LPS and injury 24 hours post-injury. (A) Inflammatory/immune system genes upregulated in response to LPS treatment and injury in males. (B) Immune/inflammatory response genes upregulated in response to LPS treatment and injury in females. (C) Bone and cartilage formation genes downregulated in response to LPS treatment and injury in males. (D) Injured C57Bl/6J VEH male CYTL1 immunohistochemistry expression. (E) Injured C57Bl/6J LPS male CYTL1 immunohistochemistry expression is lower than VEH. (F) Injured C57Bl/6J VEH CHAD immunohistochemistry expression. (G) Injured C57Bl/6J LPS CHAD immunohistochemistry expression was lower than VEH.
In females, 1426 genes were upregulated in LPS uninjured compared with VEH uninjured, and 79 genes were upregulated in LPS injured compared with VEH injured. C57Bl/6J female uninjured LPS joints showed similarities to the corresponding males as reflected in the upregulation of inflammatory and immune response genes such as Tlr5, Bcl6,(48) Cxcl1,(49) Il15,(50) Prkca,(51) Ptges,(52) and Saa3(53) (Fig. 5A, B). A higher level of upregulated inflammatory genes was observed in the injured LPS female cohort relative to the LPS injured male cohort, including Tlr5, Saa3, Bcl6, Cx3xr1,(54) and Marco(55) (Fig. 5A, B). We also found 1815 genes downregulated in LPS uninjured compared with VEH uninjured and 307 genes downregulated in LPS injured compared with VEH injured. There were 117 genes in common between the downregulated injured males, a number of them being associated with B-cell differentiation and activation. Downregulated genes that were present in injured LPS females only were mainly associated with adaptive immunity activity.
LPS administration causes PTOA in MRL/MpJ mice
All uninjured MRL/MpJ joints showed strong cartilage staining throughout the entire joint (Fig. 6A, C, E, G), suggesting that LPS itself did not produce OA phenotypes. Injured LPS males displayed a reduction in staining on the femoral condyle cartilage and had visible clefts compared with the VEH (Fig. 6b, d, arrow, asterisk). There was a lack of staining and more cellularity in the male injured LPS compared with the VEH injured males on the anterior tibial condyle (Fig. 6 bb, dd, asterisk). VEH injured females showed less staining than the uninjured VEH MRL/MpJ, suggesting proteoglycan loss (Fig. 6E, F). VEH injured MRL/MpJ females showed a thinning of the femoral condyle (Fig. 6f, arrow, asterisk); there was cellularity and a reduction in staining on the anterior tibial condyle (Fig. 6ff, asterisk). The LPS injured MRL/MpJ females (Fig. 6H) also showed a thinning of the femoral condyle cartilage compared with VEH (Fig. 6f, h, arrow, asterisk) and a hyperplastic synovium (Fig. 6hh, asterisk). LPS had a catabolic effect on the cartilage of both female and male MRL/MpJ injured joints. Uninjured VEH MRL/MpJ joints did not show statistically significant differences when compared with the injured VEH of either sex (Fig. 6I), and this was consistent with prior reports.(5) OARSI score was significantly higher in the LPS injured compared with the LPS uninjured and VEH injured. These data suggest that LPS administration to male mice produces a significant PTOA phenotype in a mouse strain previously shown to be resistant to PTOA.(5)
Fig 6.
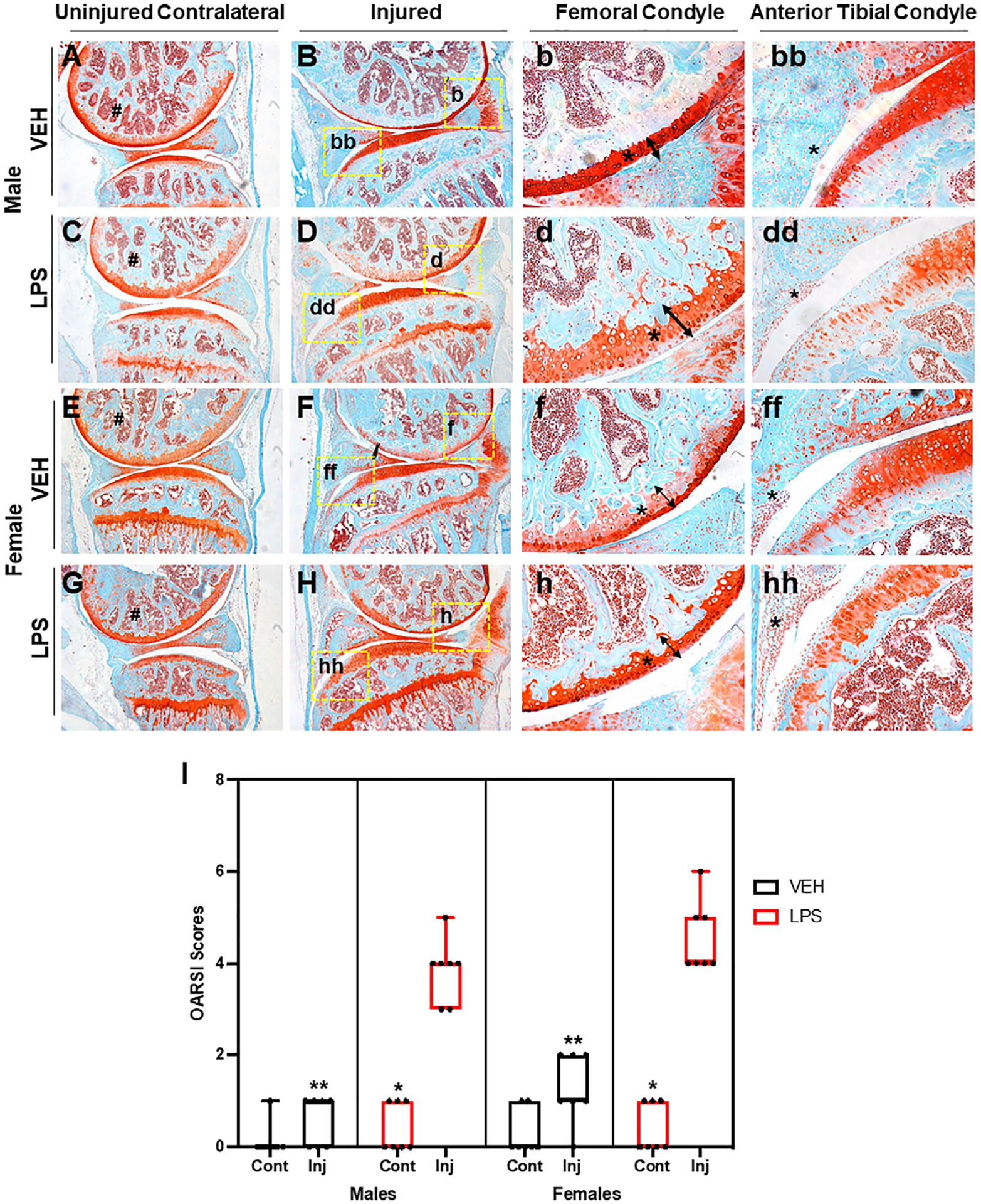
LPS induces PTOA on MRL/MpJ mice. (A, B) VEH uninjured and injured MRL/MpJ male mice. There is strong staining through the femoral condyle on both. (C, D) LPS uninjured MRL/MpJ male shows strong staining through the femoral condyle and tibia. LPS injured MRL/MpJ male shows fibrillations, clefts, and lack of staining on areas of the tibia and femoral condyle. (E, F) VEH uninjured MRL/MpJ female shows strong staining through the femoral condyle. VEH injured MRL/MpJ female shows clefts, cartilage thinning, and fibrillations on the femoral and tibial condyle. (G, H) LPS uninjured MRL/MpJ female shows strong cartilage staining through the femoral and tibial condyle. LPS injured MRL/MpJ female shows cartilage thinning and lack of staining on the femoral condyle with cellular infiltration in the anterior tibial condyle. (I) OARSI scoring did not show significant differences when comparing injured and uninjured VEH independent of sex. OARSI scores were significantly higher on LPS injured joints when compared with LPS uninjured and VEH injured independent of sex. There were no significant differences in the sexes when comparing different treatment. *Statistically significantly different than injured joint of the same sex and treatment (p-values < 0.05). **Statistically significantly different than the LPS injured of the same sex (p-values < 0.05).
Discussion
The effects of inflammation after injury have been shown to increase the likelihood of PTOA development.(56,57) Previous studies with germ-free and specific pathogen-free mice using destabilization of the medial meniscus (DMM) surgery have shown no differences between LPS levels in serum; however, they had LPS inhibitors that might have modified activity.(18) However, the effects of LPS-induced systemic inflammation before joint injury on the development of PTOA has not been previously examined. This study provides insights into how unresolved systemic inflammation at the time of injury accelerates the progression to PTOA after an ACL injury in mice. We were able to show how a single LPS injection 5 days before injury can cause significant changes to PTOA phenotypes by increasing the amount of cartilage degradation, reducing the subchondral bone volume fraction and trabecular bone microstructure, and reducing osteophyte volume. These effects were significantly greater in the female LPS injured mice than in the corresponding males, although similar trends were found in both sexes. These results are consistent with different responses to acute inflammation on females when compared with males. Immunohistochemistry analysis showed the presence of activated inflammatory macrophages 6 weeks post-injury; this indicates that the inflammation is not resolved on LPS-treated samples.
Male RNA sequencing data showed that systemic LPS significantly upregulated the expression of genes associated with inflammation in the joints of uninjured mice. At 6 days post-LPS injection, data indicated 73 of these immune and inflammatory response genes were significantly upregulated in both injured and uninjured LPS samples when compared with the VEH uninjured. More striking was the discovery that members of the Toll-like receptor (TLR) family of genes were activated in both LPS-treated samples and, in particular, Tlr5, Tlr7, and Tlr8. Three endosomal TLRs have been previously shown to be activated in the synovium and synovial fluid macrophages of RA patients.(37,58,59) This correlation would indicate that systemic LPS induces mild RA-like state in the joints of uninjured mice (Fig. 7) and that this transient RA-like state is sufficient to exacerbate cartilage degeneration in response to a joint injury, potentially through Tlr5, Tlr7, and Tlr8 expressing M1 macrophages (Fig. 7). Evidence in support of this correlation is provided by an RA and OA study comparing the synovial tissue where they found Tlr5 to be elevated in the lining and sublining macrophages of RA and OA patients.(60) An OA study in Tlr7-deficient mice showed Tlr7−/− mice develop a milder form of arthritis compared with control groups in the murine collagen induced arthritis (CIA) model.(36) Furthermore, overexpression of human Tlr8 (huTLR8) in mice promoted spontaneous and induced arthritis and the levels of huTLR8 correlated with proinflammatory cytokines in the joints of these animals.(36) These studies highlight the potential role for these endosomal Tlrs in the maintenance of inflammation in rheumatoid arthritis. Activation of these Tlr5/7/8 by LPS might have contributed to the enhanced PTOA phenotype observed in LPS-treated animals. Tlr7 and Tlr8 were activated in males only, whereas Tlr5 was activated in females as well. Additionally in females, the expression of Saa3, which has previously been shown to be upregulated in rheumatoid arthritis, could also promote the more severe PTOA phenotype and has been suggested to induce transcription of matrix metalloproteinases.(61) LPS upregulates different genes associated with inflammation in males and females that both lead to an accelerated PTOA phenotype. In healthy individuals, plasma endotoxin levels are less than 0.05 EU/mL, but a study of patients with active urinary tract infections (UTI) showed significantly higher levels of plasma endotoxins. This study concluded that patients with gram-negative bacterial UTIs present symptoms of systemic inflammatory responses due to high levels of plasma endotoxin(62) and would suggest that such individuals may be at significantly higher risk of PTOA in the event of a joint injury.
Fig 7.
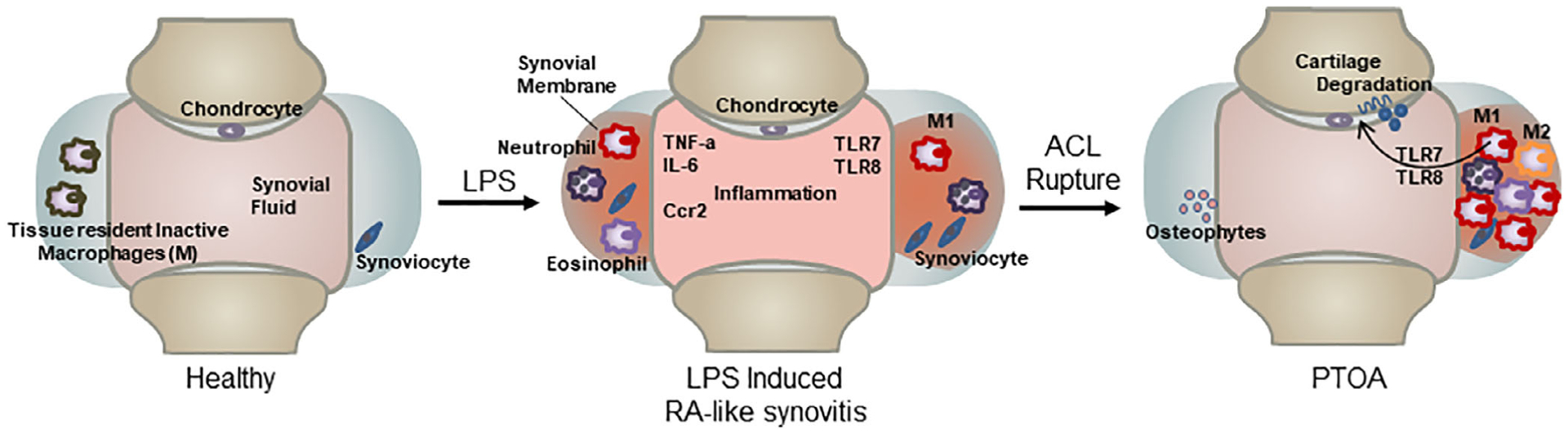
LPS enhances the vulnerability to PTOA development. Healthy individuals exposed to systemic inflammation, like LPS, will develop symptoms similar to those of rheumatoid arthritis patients. In the joint, it will cause synovitis, which has an increase of macrophages type 1, eosinophils, neutrophils, and inflammatory cytokines and chemokines. An injury that happens at the time of elevated systemic inflammation will increase vulnerability and accelerate the development of PTOA.
The histological analysis of the injured joints and the RNAseq data both point to the M1 macrophage as the source of the joint inflammation (Fig. 7), and our data is backed by publications showing that M1 macrophages express higher mRNA levels of Tlr5, Tlr7, and Tlr8 than M2 macrophages,(63) while TLR5/TLR7/TLR8 expression is significantly higher in the macrophages within synovial tissue of RA patients when compared with normal. Future studies will have to also survey the contribution of other immune cell types to the development of PTOA since genes associated with B-cell activation, such as Blk,(64) Blnk,(65) Cd22,(66) Cd79a,(67) and Cd79b,(68) were downregulated in the injured joints tread with LPS in both male and female cohorts. The role of B cells has not yet been investigated in the context of osteoarthritis and could represent potential therapeutic candidates for PTOA research. Also, the fact that the super-healer mice MRL/MpJ developed PTOA after LPS administration highlights that these mice are not resistant from inflammatory-mediated PTOA risks.
In our model system, PTOA develops in response to joint injury in both VEH- and LPS-treated joints; however, patients receive reconstructive surgery post-ACL rupture, and only 50% of these patients develop PTOA. Future experiments will have to address two key questions: (i) Do injured individuals with high plasma endotoxin levels develop PTOA at higher frequency than individuals with normal plasma endotoxin levels? (ii) Do anti-inflammatory drugs administered immediately post-joint injury lower the risk of developing PTOA? Furthermore, genes associated with bone and cartilage metabolism were significantly downregulated in male LPS injured samples relative to all other groups, suggesting that LPS + injury may have a significant bone and cartilage catabolic effect in males, whereas inflammation resolution might lead to an accelerated PTOA phenotype in females. Genes downregulated in males included Comp,(69) a noncollagenous extracellular matrix encoding gene; Hapln1,(70) Chad, Col9a1, and Col10a1, which encode cartilage matrix proteins; Cnmd, which is known to promote chondrocyte growth;(71,72) and Sost, which has been previously shown to have a protective role in the development of PTOA after injury.(28) LPS alone kept the inflammatory pathways elevated in males and females, while the addition of injury inhibited both bone and cartilage formation, influencing skeletal metabolism.
Our study uniquely examined how unresolved inflammation affects PTOA progression in a noninvasive ACL rupture model in mice. This noninvasive model closely mimics human ACL rupture from a single high impact event that leads to PTOA development. Although we recognize that patients receive reconstructive surgery and that our model remains unstabilized throughout, the ability to track molecular and morphological changes in the joint during PTOA progression will capture unknown events that cannot be perceived in humans because most patient samples are from individuals at advanced stages of the disease. The findings that a pre-existing systemic inflammation state produces RA-like molecular phenotypes in the uninjured joint and accelerates the progression to PTOA after injury may guide future clinical risk assessments of patients. Most striking is the revelation that unresolved inflammation in individuals with a mild autoimmune disease or suffering from a gram-negative-mediated infection such as UTI at the time of injury may be at significantly higher risk of developing severe PTOA phenotypes. In future experiments, this model will allow us to study whether blocking immune-derived molecules such as Tlr5, Tlr7, and Tlr8 immediately post-injury would prevent unwarranted PTOA phenotypes such as cartilage degeneration.
Acknowledgments
This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344.
MEM was supported by the Eugene Cota Robles Fellowship. AS, DKM, NRH, and GGL were supported by Lawrence Livermore National Laboratory LDRD grant 16-ERD-007. GGL and BAC were supported by the Department of Defense grant PR180268P1. BAC was supported by the National Institutes of Health grant R01 AR075013.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
References
- 1.Carbone A, Rodeo S. Review of current understanding of post-traumatic osteoarthritis resulting from sports injuries. J Orthop Res. 2017;35(3):397–405. [DOI] [PubMed] [Google Scholar]
- 2.Daniel DM, Stone ML, Dobson BE, Fithian DC, Rossman DJ, Kaufman KR. Fate of the ACL-injured patient. A prospective outcome study. Am J Sports Med. 1994;22(5):632–44. [DOI] [PubMed] [Google Scholar]
- 3.Kessler MA, Behrend H, Henz S, Stutz G, Rukavina A, Kuster MS. Function, osteoarthritis and activity after ACL-rupture: 11 years follow-up results of conservative versus reconstructive treatment. Knee Surg Sports Traumatol Arthrosc. 2008;16(5):442–8. [DOI] [PubMed] [Google Scholar]
- 4.Tiderius CJ, Olsson LE, Nyquist F, Dahlberg L. Cartilage glycosaminoglycan loss in the acute phase after an anterior cruciate ligament injury: delayed gadolinium-enhanced magnetic resonance imaging of cartilage and synovial fluid analysis. Arthritis Rheum. 2005;52(1): 120–7. [DOI] [PubMed] [Google Scholar]
- 5.Sebastian A, Chang JC, Mendez ME, et al. Comparative transcriptomics identifies novel genes and pathways involved in post-traumatic osteoarthritis development and progression. Int J Mol Sci. 2018;19(9):2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DD, Chubinskaya S, Guilak F, et al. Post-traumatic osteoarthritis: improved understanding and opportunities for early intervention. J Orthop Res. 2011;29(6):802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bigoni M, Sacerdote P, Turati M, et al. Acute and late changes in intraarticular cytokine levels following anterior cruciate ligament injury. J Orthop Res. 2013;31(2):315–21. [DOI] [PubMed] [Google Scholar]
- 8.Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14(7): 676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goel PN, Egol AJ, Moharrer Y, Brandfield-Harvey B, Ahn J, Ashley JW. Notch signaling inhibition protects against LPS mediated osteolysis. Biochem Biophys Res Commun. 2019;515(4):538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol. 2009;201(3):309–20. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. 2008;29(4):403–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11(3):234–50. [DOI] [PubMed] [Google Scholar]
- 13.Todhunter PG, Kincaid SA, Todhunter RJ, et al. Immunohistochemical analysis of an equine model of synovitis-induced arthritis. Am J Vet Res. 1996;57(7):1080–93. [PubMed] [Google Scholar]
- 14.Firth EC, Wensing T, Seuren F. An induced synovitis disease model in ponies. Cornell Vet. 1987;77(2):107–18. [PubMed] [Google Scholar]
- 15.Palmer JL, Bertone AL. Experimentally-induced synovitis as a model for acute synovitis in the horse. Equine Vet J. 1994;26(6):492–5. [DOI] [PubMed] [Google Scholar]
- 16.Hawkins DL, MacKay RJ, Gum GG, Colahan PT, Meyer JC. Effects of intra-articularly administered endotoxin on clinical signs of disease and synovial fluid tumor necrosis factor, interleukin 6, and prostaglandin E2 values in horses. Am J Vet Res. 1993;54(3):379–86. [PubMed] [Google Scholar]
- 17.Santos LCP, de Moraes AN, Saito ME. Effects of intraarticular ropivacaine and morphine on lipopolysaccharide-induced synovitis in horses. Vet Anaesth Analg. 2009;36(3):280–6. [DOI] [PubMed] [Google Scholar]
- 18.Ulici V, Kelley KL, Azcarate-Peril MA, et al. Osteoarthritis induced by destabilization of the medial meniscus is reduced in germ-free mice. Osteoarthr Cartil. 2018;26(8):1098–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furman BD, Huebner JL, Seifer DR, Kraus VB, Guilak F, Olson SA. MRL/MpJ mouse shows reduced intra-articular and systemic inflammation following articular fracture Poster presented at: 55th Annual Meeting of the Orthopaedic Research Society; Las Vegas, NV; February 22–25, 2009. Poster no. 1120. [Google Scholar]
- 20.Ward BD, Furman BD, Huebner JL, Kraus VB, Guilak F, Olson SA. Absence of posttraumatic arthritis following intraarticular fracture in the MRL/MpJ mouse. Arthritis Rheum. 2008;58(3):744–53. [DOI] [PubMed] [Google Scholar]
- 21.Deng Z, Gao X, Sun X, et al. Characterization of articular cartilage homeostasis and the mechanism of superior cartilage regeneration of MRL/MpJ mice. FASEB J. 2019;33(8):8809–21. [DOI] [PubMed] [Google Scholar]
- 22.Kay E, Gomez-Garcia L, Woodfin A, Scotland RS, Whiteford JR. Sexual dimorphisms in leukocyte trafficking in a mouse peritonitis model. J Leukoc Biol. 2015;98(5):805–17. [DOI] [PubMed] [Google Scholar]
- 23.Lockwood KA, Chu BT, Anderson MJ, Haudenschild DR, Christiansen BA. Comparison of loading rate-dependent injury modes in a murine model of post-traumatic osteoarthritis. J Orthop Res. 2014;32(1):79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furube E, Kawai S, Inagaki H, Takagi S, Miyata S. Brain region-dependent heterogeneity and dose-dependent difference in transient microglia population increase during lipopolysaccharide-induced inflammation. Sci Rep. 2018;8(1):2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozak W, Conn CA, Kluger MJ. Lipopolysaccharide induces fever and depresses locomotor activity in unrestrained mice. Am J Physiol. 1994;266(1) Pt 2:R125–35. [DOI] [PubMed] [Google Scholar]
- 26.Christiansen BA, Guilak F, Lockwood KA, et al. Non-invasive mouse models of post-traumatic osteoarthritis. Osteoarthr Cartil. 2015;23(10):1627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang JC, Sebastian A, Murugesh DK, et al. Global molecular changes in a tibial compression induced ACL rupture model of post-traumatic osteoarthritis: global molecular changes after ACL injury. J Orthop Res. 2017;35(3):474–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang JC, Christiansen BA, Murugesh DK, et al. SOST/Sclerostin improves posttraumatic osteoarthritis and inhibits MMP2/3 expression after injury: SOST overexpression improves PTOA outcomes. J Bone Miner Res. 2018;33(6):1105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prys-Roberts C. Isoflurane. Br J Anaesth. 1981;53(12):1243–5. [DOI] [PubMed] [Google Scholar]
- 30.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 31.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr Cartil. 2010;18 (Suppl 3):S17–23. [DOI] [PubMed] [Google Scholar]
- 32.Yee CS, Xie L, Hatsell S, et al. Sclerostin antibody treatment improves fracture outcomes in a type I diabetic mouse model. Bone. 2016;82: 122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JHN, Haudenschild DR. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthr Cartil. 2012;20(7):773–82. [DOI] [PubMed] [Google Scholar]
- 34.Teramachi J, Inagaki Y, Shinohara H, et al. PKR regulates LPS-induced osteoclast formation and bone destruction in vitro and in vivo. Oral Dis. 2017;23(2):181–8. [DOI] [PubMed] [Google Scholar]
- 35.Souza PPC, Lerner UH. The role of cytokines in inflammatory bone loss. Immunol Invest. 2013;42(7):555–622. [DOI] [PubMed] [Google Scholar]
- 36.Guiducci C, Gong M, Cepika A-M, et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J Exp Med. 2013;210(13):2903–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim S-J, Chen Z, Chamberlain ND, et al. Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in rheumatoid arthritis and experimental arthritis. J Immunol. 2014; 193(8):3902–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coelho AL, Schaller MA, Benjamim CF, Orlofsky AZ, Hogaboam CM, Kunkel SL. The chemokine CCL6 promotes innate immunity via immune cell activation and recruitment. J Immunol. 2007;179(8):5474–82. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Khairallah C, Sheridan BS, van der Velden AWM, Bliska JB. CCR2+ inflammatory monocytes are recruited to Yersinia pseudotuberculosis pyogranulomas and dictate adaptive responses at the expense of innate immunity during oral infection. Infect Immun. 2018;86(3):e00782–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaFleur AM, Lukacs NW, Kunkel SL, Matsukawa A. Role of CC chemokine CCL6/C10 as a monocyte chemoattractant in a murine acute peritonitis. Mediators Inflamm. 2004;13(5–6):349–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podojil JR, Kohm AP, Miller SD. CD4+ T cell expressed CD80 regulates central nervous system effector function and survival during experimental autoimmune encephalomyelitis. J Immunol. 2006;177(5): 2948–58. [DOI] [PubMed] [Google Scholar]
- 42.Dreier R, Opolka A, Grifka J, Bruckner P, Grässel S. Collagen IX-deficiency seriously compromises growth cartilage development in mice. Matrix Biol. 2008;27(4):319–29. [DOI] [PubMed] [Google Scholar]
- 43.Gu J, Lu Y, Li F, et al. Identification and characterization of the novel Col10a1 regulatory mechanism during chondrocyte hypertrophic differentiation. Cell Death Dis. 2014;5:e1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Batista MA, Nia HT, Önnerfjord P, et al. Nanomechanical phenotype of chondroadherin-null murine articular cartilage. Matrix Biol. 2014; 38:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kakugawa S, Langton PF, Zebisch M, et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature. 2015;519(7542): 187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viegas CSB, Costa RM, Santos L, et al. Gla-rich protein function as an anti-inflammatory agent in monocytes/macrophages: implications for calcification-related chronic inflammatory diseases. PloS One. 2017;12(5):e0177829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeon J, Oh H, Lee G, et al. Cytokine-like 1 knock-out mice (Cytl1−/−) show normal cartilage and bone development but exhibit augmented osteoarthritic cartilage destruction. J Biol Chem. 2011;286(31):27206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang H, Green MR. Epigenetic programing of B-cell lymphoma by BCL6 and its genetic deregulation. Front Cell Dev Biol. 2019;7:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva RL, Lopes AH, Guimarães RM, Cunha TM. CXCL1/CXCR2 signaling in pathological pain: role in peripheral and central sensitization. Neurobiol Dis. 2017;105:109–16. [DOI] [PubMed] [Google Scholar]
- 50.Grabstein KH, Eisenman J, Shanebeck K, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science. 1994;264(5161):965–8. [DOI] [PubMed] [Google Scholar]
- 51.Barton A Association of protein kinase C alpha (PRKCA) gene with multiple sclerosis in a UK population. Brain. 2004;127(8):1717–22. [DOI] [PubMed] [Google Scholar]
- 52.Abraham JE, Harrington P, Driver KE, et al. Common polymorphisms in the prostaglandin pathway genes and their association with breast cancer susceptibility and survival. Clin Cancer Res. 2009;15(6):2181–91. [DOI] [PubMed] [Google Scholar]
- 53.Larson MA, Wei SH, Weber A, Weber AT, McDonald TL. Induction of human mammary-associated serum amyloid A3 expression by prolactin or lipopolysaccharide. Biochem Biophys Res Commun. 2003; 301(4):1030–7. [DOI] [PubMed] [Google Scholar]
- 54.Lee M, Lee Y, Song J, Lee J, Chang S-Y. Tissue-specific role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Netw. 2018;18(1):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaur P, Myles A, Misra R, Aggarwal A. Intermediate monocytes are increased in enthesitis-related arthritis, a category of juvenile idiopathic arthritis. Clin Exp Immunol. 2017;187(2):234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lieberthal J, Sambamurthy N, Scanzello CR. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthr Cartil. 2015;23(11):1825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adamopoulos IE. Inflammation in bone physiology and pathology. Curr Opin Rheumatol. 2018;30(1):59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Culemann S, Grüneboom A, Nicolás-Ávila JÁ, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature. 2019;572(7771):670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chamberlain ND, Kim S, Vila OM, et al. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFα in monocytes. Ann Rheum Dis. 2013;72(3):418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chamberlain ND, Vila OM, Volin MV, et al. TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-α levels. J Immunol. 2012;189(1):475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vallon R, Freuler F, Desta-Tsedu N, et al. A (apoSAA) expression is upregulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J Immunol. 2001;166(4):2801–7. [DOI] [PubMed] [Google Scholar]
- 62.Goto T, Makinose S, Ohi Y. Plasma endotoxin concentrations in patients with urinary tract infections. Int J Urol. 1995;2(4):238–42. [DOI] [PubMed] [Google Scholar]
- 63.Liang D, Leung RK-K, Guan W, Au WW. Involvement of gut microbiome in human health and disease: brief overview, knowledge gaps and research opportunities. Gut Pathog. 2018;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Texido G, Su IH, Mecklenbräuker I, et al. The B-cell-specific Src-family kinase Blk is dispensable for B-cell development and activation. Mol Cell Biol. 2000;20(4):1227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu C, Turck CW, Kurosaki T, Chan AC. BLNK: a central linker protein in B cell activation. Immunity. 1998;9(1):93–103. [DOI] [PubMed] [Google Scholar]
- 66.Clark EA, Giltiay NV. CD22: a regulator of innate and adaptive B cell responses and autoimmunity. Front Immunol. 2018;9:2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luger D, Yang Y-A, Raviv A, et al. Expression of the B-cell receptor component CD79a on immature myeloid cells contributes to their tumor promoting effects. PloS One. 2013;8(10):e76115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCarron KF, Hammel JP, Hsi ED. Usefulness of CD79b expression in the diagnosis of B-cell chronic lymphoproliferative disorders. Am J Clin Pathol. 2000;113(6):805–13. [DOI] [PubMed] [Google Scholar]
- 69.Sengul A, Mohan S, Kesavan C. Bone response to loading in mice with targeted disruption of the cartilage oligomeric matrix protein gene. Physiol Res. 2012;61(6):637–41. [DOI] [PubMed] [Google Scholar]
- 70.Urano T, Narusawa K, Shiraki M, et al. Single-nucleotide polymorphism in the hyaluronan and proteoglycan link protein 1 (HAPLN1) gene is associated with spinal osteophyte formation and disc degeneration in Japanese women. Eur Spine J. 2011;20(4):572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kanbe K, Yang X, Wei L, Sun C, Chen Q. Pericellular matrilins regulate activation of chondrocytes by cyclic load-induced matrix deformation. J Bone Miner Res. 2007;22(2):318–28. [DOI] [PubMed] [Google Scholar]
- 72.Amano K, Densmore MJ, Lanske B. Conditional deletion of Indian hedgehog in limb mesenchyme results in complete loss of growth plate formation but allows mature osteoblast differentiation. J Bone Miner Res. 2015;30(12):2262–72. [DOI] [PubMed] [Google Scholar]