

Abstract
Long-range oncogenic enhancers play an important role in cancer. Yet, whether similar regulation of tumor suppressor genes is relevant remains unclear. Loss of expression of PTEN is associated with the pathogenesis of various cancers, including T-cell leukemia (T-ALL). Here, we identify a highly conserved distal enhancer (PE) that interacts with the PTEN promoter in multiple hematopoietic populations, including T-cells, and acts as a hub of relevant transcription factors in T-ALL. Consistently, loss of PE leads to reduced PTEN levels in T-ALL cells. Moreover, PE-null mice show reduced Pten levels in thymocytes and accelerated development of NOTCH1-induced T-ALL. Furthermore, secondary loss of PE in established leukemias leads to accelerated progression and a gene expression signature driven by Pten loss. Finally, we uncovered recurrent deletions encompassing PE in T-ALL, which are associated with decreased PTEN levels. Altogether, our results identify PE as the first long-range tumor suppressor enhancer directly implicated in cancer.
Keywords: PTEN, enhancer, T-cell acute lymphoblastic leukemia, T-ALL, NOTCH1
Introduction
Enhancers are distal DNA regulatory regions that act in cis, and in orientation-independent manner, to regulate gene expression through three-dimensional physical interaction with gene promoters (1–4). In recent years, enhancer malfunction has been shown to play a key role in cancer (5). Specifically, cancer cells can use enhancers via epigenetic activation or via non-coding mutations that create enhancers de novo, increasing the expression of a variety of oncogenes. In addition, we and others have described numerous tissue-specific oncogenic enhancers as recurrently duplicated in cancer (5,6). In this setting, MYC is the best example to date of an oncogene regulated by a plethora of enhancer regions, all of which can be dysregulated in different cancer types (7). However, whether long-range enhancer regions might control the expression of tumor suppressor genes (TSGs), and whether these might also play a role in cancer development, remains largely unknown. T-cell acute lymphoblastic leukemia (T-ALL) is a NOTCH1-driven aggressive hematologic malignancy that presents in both pediatric and adult patients, and requires treatment with intensified chemotherapy (8). Importantly, 15–20% of T-ALL patients show loss of expression of the PTEN tumor suppressor gene (9,10), which leads to resistance to anti-NOTCH1 therapies with gamma-secretase inhibitors (GSIs) in T-ALL in vivo (11). However, not all of the T-ALL cases with loss of PTEN expression can be explained by prototypical mutations or deletions in its coding region, by defective splicing or by epigenetic silencing (10), highlighting our incomplete understanding of the mechanisms mediating loss of expression of PTEN. In this context, we hypothesized that PTEN expression in T-ALL might be controlled by yet-unidentified enhancer regions, and that loss of these putative enhancers might help explain the lack of PTEN expression in certain T-ALL cases. To test this hypothesis, we integrated chromosome conformation and epigenetic profiling analyses and identified a bona-fide enhancer of PTEN, whose functionality we further dissected using CRISPR/Cas9-mutant T-ALL cell lines and novel mouse models in vivo.
Results
Identification of PE, a PTEN enhancer in T-ALL
To reveal the global pattern of chromosomal interactions of the PTEN promoter in the leukemia genome and identify potential regions that might act as enhancers of PTEN, we performed chromosome conformation capture 4C-seq experiments (12,13), in three independent T-ALL cell lines (DND41, HPB-ALL and JURKAT) using the PTEN promoter as the viewpoint. These analyses revealed that the PTEN promoter interacts at high frequency with several regions along most of its topologically associating domain (TAD) (14) (Fig. 1A, upper tracks). Among these, we identified a region ~550Kb downstream of PTEN (hereby named PE, for PTEN enhancer), located in an intronic sequence of the neighboring RNLS gene, that shows high levels of interaction with the PTEN promoter. Moreover, epigenetic profiling analyses revealed concomitant bona fide enhancer marks in this region, including high levels of H3K27ac and H3K4me1, together with binding of CTCF, BRD4 and ZNF143 (Fig. 1A, middle tracks). Importantly, the CTCF binding sites in the PTEN promoter and the PE enhancer are located in convergent orientation (Fig. 1A, CTCF track), suggesting that cohesin-mediated loops might be responsible for this interaction (15–18). In addition, GRO-seq analyses of T-ALL cells (19) uncovered bi-directional transcription from this region (Fig. 1A, lower tracks), which has also been suggested to mark active enhancer regulatory elements (20,21). Finally, analyses of publicly available H3K27ac Hi-ChIP data in CUTLL1 T-ALL cells (19) revealed that, among the different interacting regions with the PTEN promoter, the interaction with the PE enhancer is the only one detected at FDR <1E-15 (Fig. 1A, top). Indeed, the PTEN-PE interaction is detected at an extremely low FDR value <1E-49 (Fig. 1A, top), overall suggesting a prominent role for PE in the regulation of PTEN.
Figure 1.
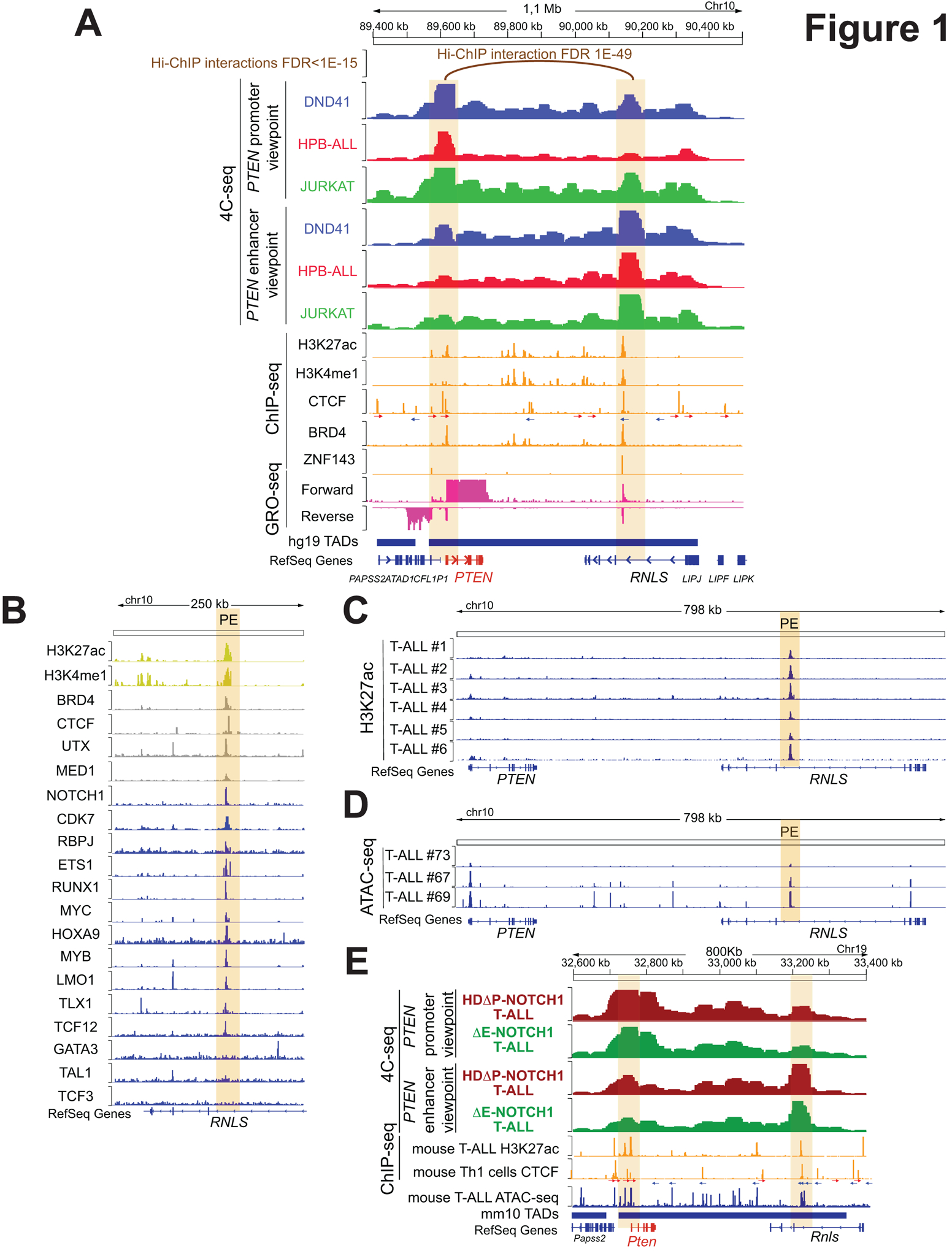
Identification of PE, a PTEN enhancer in T-ALL. A, H3K27ac Hi-ChIP, 4C-seq, ChIP-seq, GRO-seq and ATAC-seq tracks in human T-ALL cells. Top track shows H3K27ac Hi-ChIP interactions with the PTEN promoter in CUTTl1 T-ALL cells at FDR <1E-15. Upper tracks show 4C-seq data in DND41 (blue), HPB- ALL (red) or JURKAT (green) T-ALL cells, using either the PTEN promoter or the PE enhancer as the viewpoints. 4C signal is merged across three independent replicates per condition. Middle tracks show ChIP-seq analyses in different T-ALL cell lines for the presence of epigenetic marks or enhancer-associated factors (orange). CTCF motifs are indicated by arrows (red arrow: forward core motif, blue arrow: reverse core motif). Lower tracks show GRO-seq data from CUTTL1 cells (pink). Bottom track shows the PTEN TAD (hg19). The PTEN promoter and the PE enhancer are highlighted by orange columns. B, Analysis of epigenetic marks (yellow), epigenetic factos (gray) and transcription factor (blue) PE occupancy by ChIP-seq in human T-ALL cells. PE enhancer is highlighted by an orange column. C, H3K27ac mark by ChIPmentation around the PE enhancer (highlighted in orange) in 6 independent human primary T-ALLs. D, ATAC-seq profile around the PE enhancer (highlighted in orange) in 3 independent human primary T-ALLs (GSE124223). E, 4C-seq, ChIP-seq and ATAC-seq tracks in mouse T-ALL cells. Upper tracks show 4C-seq data from NOTCH1-induced mouse primary T-ALLs driven by either a NOTCH1-HDΔP construct (brown) or a NOTCH1-ΔE construct (green), using either the Pten promoter or the PE enhancer as the viewpoints. 4C signal is merged across three independent replicates per condition. Middle tracks show ChIP-seq (orange) of H3K27ac mark in mouse T-ALL cells and CTCF binding in mouse Th1 cells. CTCF motifs are indicated by arrows (red arrow: forward core motif, blue arrow: reverse core motif). Lower tracks show ATAC-seq data from a mouse primary T-ALL (blue), as well as the track showing the Pten TAD (mm10). The Pten promoter and the PE enhancer are highlighted by orange columns.
Following up on these findings, we next performed reciprocal 4C-seq analyses using this putative PE enhancer region as the viewpoint. Notably, these analyses confirmed its interaction with the PTEN promoter in all three cell lines (Fig. 1A, upper tracks) regardless of PTEN mutational status, as JURKAT cells (PTEN-null (9)) show similar levels of interaction as compared to HPB-ALL and DND41 cells (PTEN-positive (9)). This is also in line with ChIP-seq data showing H3K27ac in the PE enhancer both in PTEN-positive and PTEN-null cell lines (Supplementary Fig. S1A). Further inspection of ENCODE data (22) in this area in DND41 T-ALL cells revealed that the PE region is a ~10kb super-enhancer with higher H3K27ac levels along a ~4kb core sequence. However, reduced levels of H3K27ac are seen in the GM12878 B-lymphoblastoid cells or primary CD20+ B cells (Supplementary Fig. S1B), while greatly decreased or completely absent levels of H3K27ac were observed in a variety of solid tumor cell lines, primary CD14+ monocytes or in other primary cells derived from a variety of nonhematopoietic tissues (Supplementary Fig. S1B). Moreover, H3K27ac was also absent in PTEN-positive prostate (23) and thyroid (24) cancer cells (Supplementary Fig. S1C), which is of particular relevance given the well-described role of PTEN loss in the development of both prostate and thyroid cancer (25–27). Overall, these data suggest that PE might be an hematopoietic-specific enhancer prominently active in the lymphoid compartment.
Next, to investigate what transcription factors might regulate PE, we mined publicly available ChIP-seq data in T-ALL cells and identified a wide range of key T-cell transcription factors that bind to the PE enhancer, including MED1, UTX, NOTCH1, RBPJ, ETS1, RUNX1, MYC, HOXA9, MYB, LMO1, TLX1, TCF12 and CDK7 (Fig. 1B). Of note, the co-binding of NOTCH1 and CDK7 to enhancer regions has been previously reported in T-ALL (19). Indeed, some of these NOTCH1/CDK7-bound enhancers are GSI-sensitive while the rest of them are GSI-insensitive but sensitive to CDK7 inhibition (19). In the case of PE, analyses of CUTLL1 T-ALL cells treated with either a GSI or the THZ1 CDK7 inhibitor revealed lower H3K27ac levels only after THZ1 treatment, highlighting that PE likely belongs to the GSI-insensitive but CDK7-sensitive group of enhancers (Supplementary Fig. S1D). On the other hand, we detected minimal or no binding of TAL1 or TCF3, and we also failed to detect significant binding of GATA3 binding (Fig. 1B), which we previously showed to be a critical pioneering factor for the N-Me enhancer (1), underscoring that other pioneering factors might be more relevant in the case of PE. Finally, we also performed epigenetic profiling in human primary T-ALLs. These analyses revealed a robust H3K27ac mark (Fig. 1C) together with open chromatin via ATAC-seq (Fig. 1D). Interestingly, even if both marks show variable intensity in the PE region, they are present in all of the samples tested and correlate with the H3K27ac and ATAC-seq signal observed in the PTEN promoter (Fig. 1C–D). Consistently, inspection of Hi-C data from human normal T-cells and primary T-ALLs (19) revealed a clear interaction between the PTEN promoter and PE in both normal and transformed T-cells (Supplementary Fig. S1E–F). Overall, these results suggest that PE is a very relevant enhancer hub in human T-ALL controlled by inputs from a plethora of important T-cell transcription factors and highlight a relevant role for CDK7 in maintaining its activity.
Interestingly, multispecies DNA sequence alignment of the PE sequence revealed remarkable conservation of this region in mice and other mammals, with its central sequence also being conserved in reptiles but not in birds, fish or amphibians (Supplementary Fig. S2). Thus, we then performed 4C-seq analyses in NOTCH1-induced mouse primary T-ALLs (either driven by a potent ΔE-NOTCH1 construct or a weaker HDΔP-NOTCH1 construct) to uncover the global pattern of chromosomal interactions of the mouse Pten promoter in the leukemia genome. These experiments revealed that the mouse Pten promoter shows more promiscuous interactions inside its own TAD as compared to the human PTEN promoter (Fig. 1E). However, and most notably, among these contacts we observed again high levels of interaction with the PE homologous region in mice, which also shows high levels of H3K27ac (Fig. 1E). In addition, reciprocal 4C-seq analyses using the mouse PE enhancer as the viewpoint confirmed its interaction with the Pten promoter. Finally, and similar to the results observed in human cells, ChIP-seq in mouse Th1 cells showed CTCF binding in both the Pten promoter and PE enhancer with CTCF binding sites in convergent orientation, while ATAC-seq analyses from mouse T-ALL cells revealed that the PE enhancer is enriched for open chromatin in mouse leukemias (Fig. 1E). Overall, our results identify the PE enhancer as a long-range regulatory region that might be involved in the control of PTEN in T-ALL.
Functional characterization of the PE enhancer
To functionally characterize the potential role of this regulatory region in the control of PTEN expression, we first performed reporter luciferase assays to test the transactivation capability of PE. These experiments showed strong, orientation-independent activation of reporter constructs containing the core ~4Kb sequence of both human and mouse PE (Fig. 2A). Next, we hypothesized that PE loss in T-ALL cells might lead to reduced levels of PTEN. To test this, we generated DND41 cells with CRISPR/Cas9-induced heterozygous loss of PE (Fig. 2B). Notably, qRT-PCR analyses showed reduced PTEN levels in two independent clones with heterozygous loss of PE (Fig. 2C). Consistently, intracellular FACS staining revealed reduced levels of PTEN protein expression in both clones (Fig. 2D). This effect was further confirmed by western blot analysis, which showed reduced levels of PTEN together with a concomitant activation of AKT (Fig. 2E), consistent with the well-known role of PTEN as a negative regulator of the PI3K pathway (28). Finally, and in line with the well-described role of PTEN as a tumor suppressor gene, both clones of DND41 cells with heterozygous loss of PE displayed a proliferative advantage as compared to non-targeted DND41 control cells (Fig. 2F). Together, these results demonstrate that PE behaves as a bona-fide enhancer of PTEN.
Figure 2.
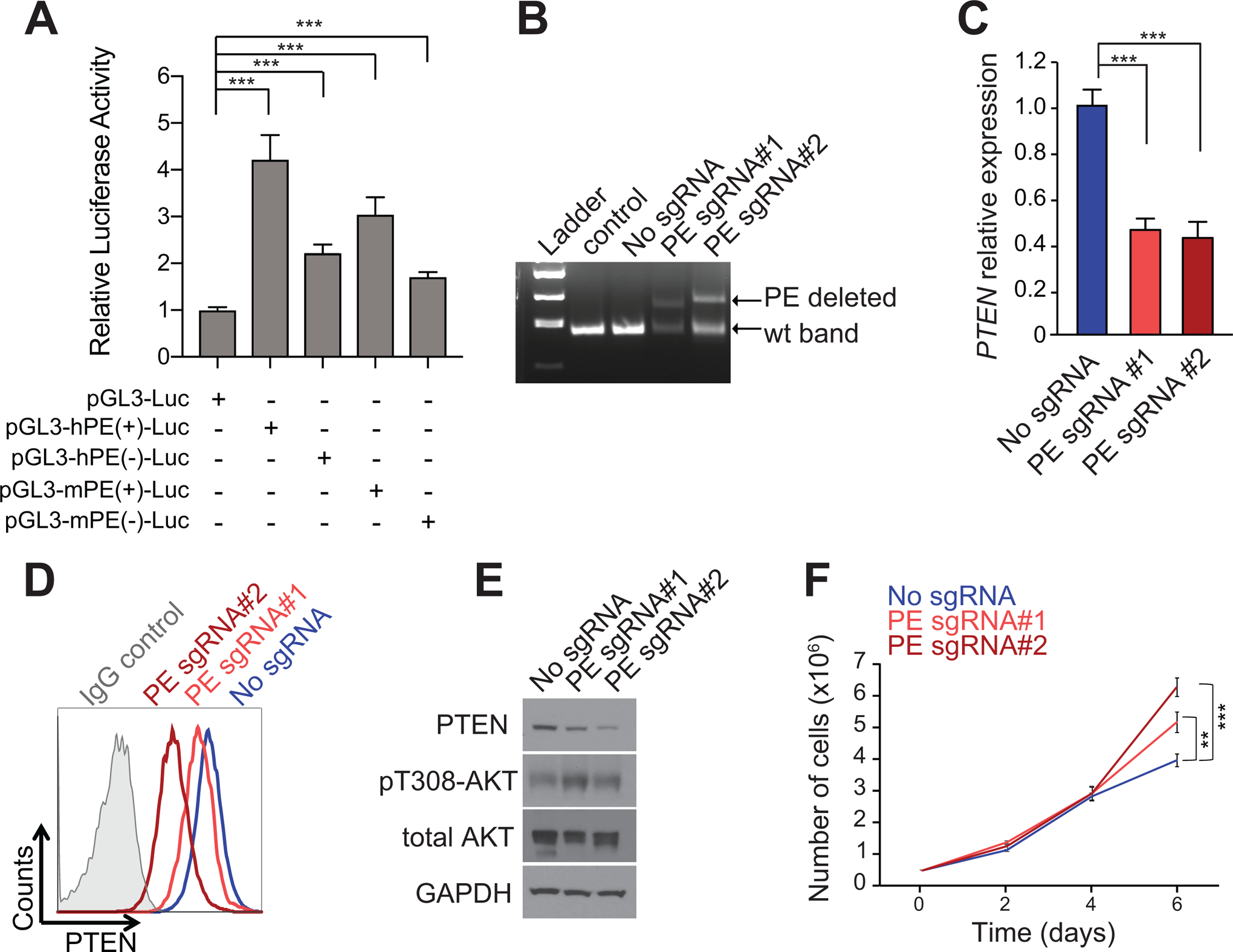
Functional characterization of the PE enhancer. A, Luciferase reporter activity in JURKAT T-ALL cells of a pGL3 promoter empty construct (pGL3-Luc), a pGL3 promoter plus the human PE enhancer in the forward (hPE(+)-Luc) or reverse (hPE(−)-Luc) orientation, or a pGL3 promoter plus the mouse PE enhancer in the forward (mPE(+)-Luc) or reverse (mPE(−)-Luc) orientation. Data from three independent electroporation replicates are shown. B, Genotyping of DND41 single cell colonies harboring a heterozygous deletion for the PE enhancer. DND41 cells not electroporated (control) or electroporated with Cas9 but without gRNAs (no sgRNA) are shown as controls. C-E, PTEN mRNA expression levels (C) and PTEN protein expression levels via intracellular FACS staining (D) or via western blot analysis, together with AKT activation (E) in DND41 control cells or two independent DND41 single-cell colonies with PE heterozygous deletion. F, Proliferation curve of DND41 control cells or two independent DND41 single-cell colonies with PE heterozygous deletion. **P < 0.01; and ***P < 0.005 values calculated using two-tailed Student’s t-test.
Germline loss of PE leads to reduced Pten levels during thymocyte development
Next, and to test the relevance of the PE enhancer in T-cell development and transformation, we genetically engineered mice with PE deletion using CRISPR/Cas9. To this end, C57BL/6J mouse zygotes were injected with Cas9 protein complexed with synthetic sgRNAs which targeted two sites flanking the ~4Kb core PE enhancer region with high levels of H3K27ac in the mouse (Supplementary Fig. S3A). Importantly, we obtained three different founders that showed specific deletion of the PE enhancer, with only 1–3bp differences in the size of the deleted fragment (Supplementary Fig. S3A–B). PE-null mice show normal postnatal development without any apparent abnormality in behavior or body size, they are fertile and are born with the expected Mendelian ratios (Supplementary Table S1). However, analyses of thymi from ~6-week-old mice revealed a marked reduction in Pten mRNA and protein expression levels in PE knockout mice from the three independent strains, as compared to littermate controls (Fig. 3A–C), while Pten expression remained unchanged across a comprehensive list of non-thymic tissues (Supplementary Fig. S4A). Of note, PTEN levels seem to be partially reduced in PE heterozygous mice, suggesting a dose-dependent effect for the PE enhancer (Fig. 3A–C). In addition, we did not detect any decrease in Rnls mRNA levels in these same samples, suggesting that the deletion of the PE enhancer does not affect Rnls expression, which is also consistent with the lack of interaction between the PE enhancer and the Rnls promoter in our previous 4C-seq analyses (Fig. 1E and Supplementary Fig. S4B). Finally, immunohistochemical analyses of these thymi revealed a generally preserved architecture and confirmed reduced PTEN levels in PE heterozygous mice, which were further and drastically reduced in PE knockout mice (Fig. 3D and E).
Figure 3.
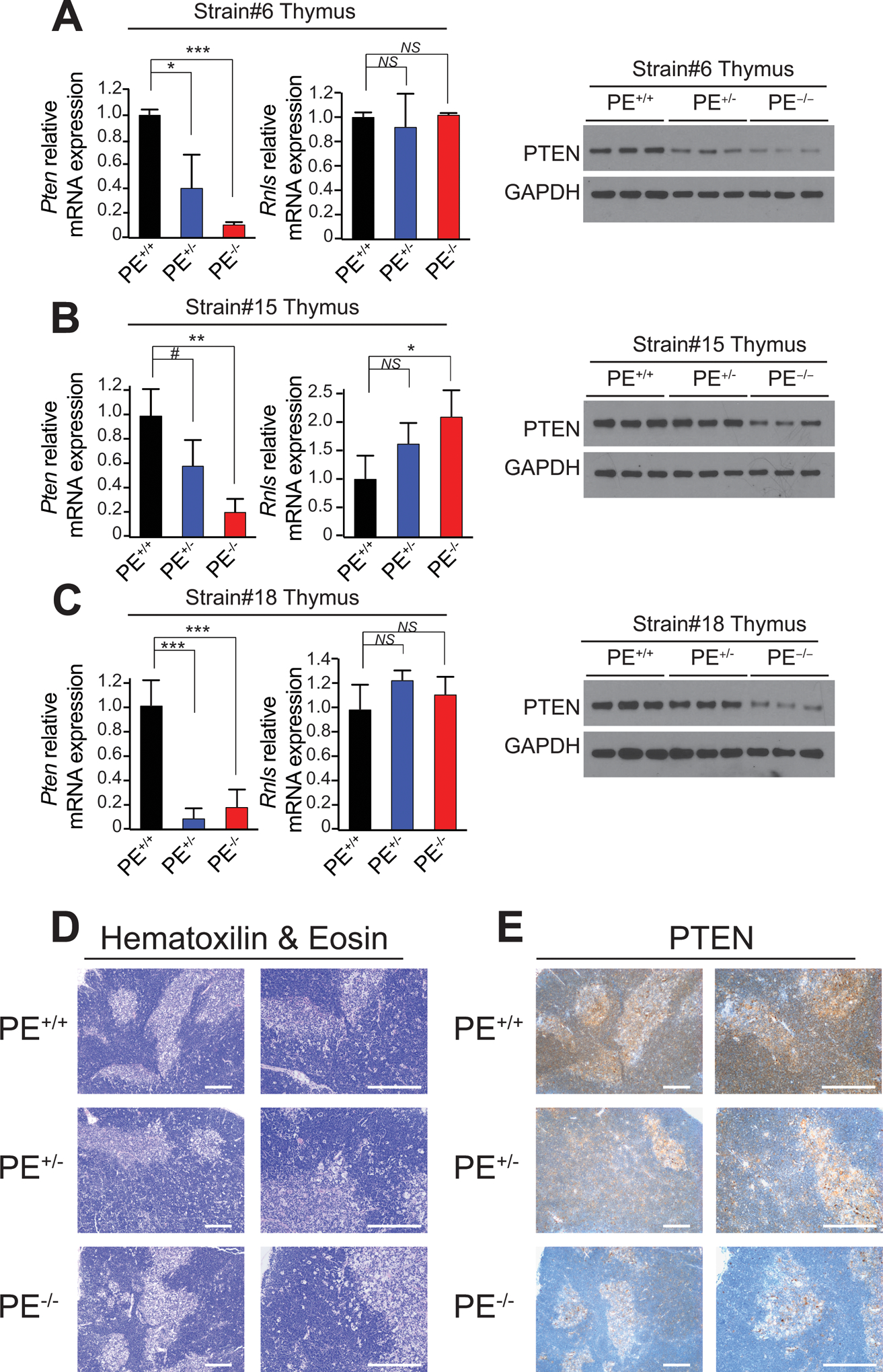
PE-deficient mice show reduced Pten levels in the thymus. A-C, Pten and/or Rnls mRNA and protein levels in thymi from PE+/+, PE+/− and PE−/− mice (n = 3 per genotype) from strain #6 (A), #15 (B) or #18 (C). #P < 0.1; *P < 0.05; **P < 0.01; and ***P < 0.005 values calculated using two-tailed Student’s t-test. NS = not significant. D-E, Hematoxilin & Eosin (D) and PTEN (E) staining in thymi from PE+/+, PE+/− and PE−/− mice (strain #6). Scale bars = 50μm.
Following up on these results, we next performed detailed immunophenotypic analyses of thymic populations in ~6-week-old mice with different PE allelic dosages. These experiments revealed that mice lacking PE show normal thymus weight and display a mostly normal thymocyte development across the different strains, with no consistent changes in CD4−CD8−-double negative, CD4+CD8+-double positive or single positive CD4+ and CD8+ cells as compared to wild-type control mice (Fig. 4A–C and Supplementary Fig. S5A and S5B), and further analyses also failed to show noticeable differences in the spleens from these mice (Supplementary Fig. S5C and S5D). Of note, this is consistent with the phenotypes previously reported in T-cell specific Pten knockout mice (29). Given the drastic reduction in PTEN levels observed in total thymus lysates from PE knockout mice (Fig. 3), we next performed single-cell RNA-seq analyses in thymi from PE wild-type or knockout mice, in order to dissect the specific stage/s of T-cell development in which the loss of PE might result in reduced levels of Pten. These analyses revealed that Pten levels in PE knockout mice are significantly reduced at the CD4−CD8−-double negative (DN), CD8+-immature single positive (ISP) and CD4+CD8+-double positive (DP) stages of thymocyte development (Fig. 4D–F). Moreover, a statistically not-significant trend of decreased Pten levels was also observed in CD4+ and CD8+-single positive mature cells (Fig. 4D–F). Interestingly, Rnls levels were not detectable by scRNA-seq in the thymus, and other neighboring genes contained in the TADs either upstream or downstream of the Pten TAD showed no differences or were also undetectable by sc-RNAseq (Supplementary Fig. S6), underscoring that PE might be primarily controlling the expression of Pten. We next investigated PE chromatin status throughout mouse hematopoietic development by mining ATAC-seq data available from the Immgen consortium (30). These data showed open chromatin already present in LT-HSCs, which is maintained throughout the development of different bone marrow and thymocyte populations (Supplementary Fig. S7A). Interestingly, even if the ATAC-seq signal is variable between DN-ISP-DP-SP stages, we detected significant differences in Pten levels in DN-ISP-DP cells only (Fig. 4F). In addition, analyses of tagHi-C data from different early mouse hematopoietic populations (31) confirmed the interaction Pten promoter-PE (Supplementary Fig. S7B). Similarly, epigenetic profiling of different human hematological and thymic populations along the T-cell differentiation trajectory showed open chromatin and H3K27ac mark in PE across the different populations (Supplementary Fig. S8A–B) (32). Consistently, analyses of publicly available capture-HiC data in human hematological populations (33) showed strong interaction between the PTEN promoter and the PE enhancer in fetal thymus, total CD4 cells, total CD8 cells, total B cells and megakaryocytes, with lower interactions detected in neutrophils, erythroblasts, monocytes or endothelial precursors (Supplementary Fig. S9).
Figure 4.
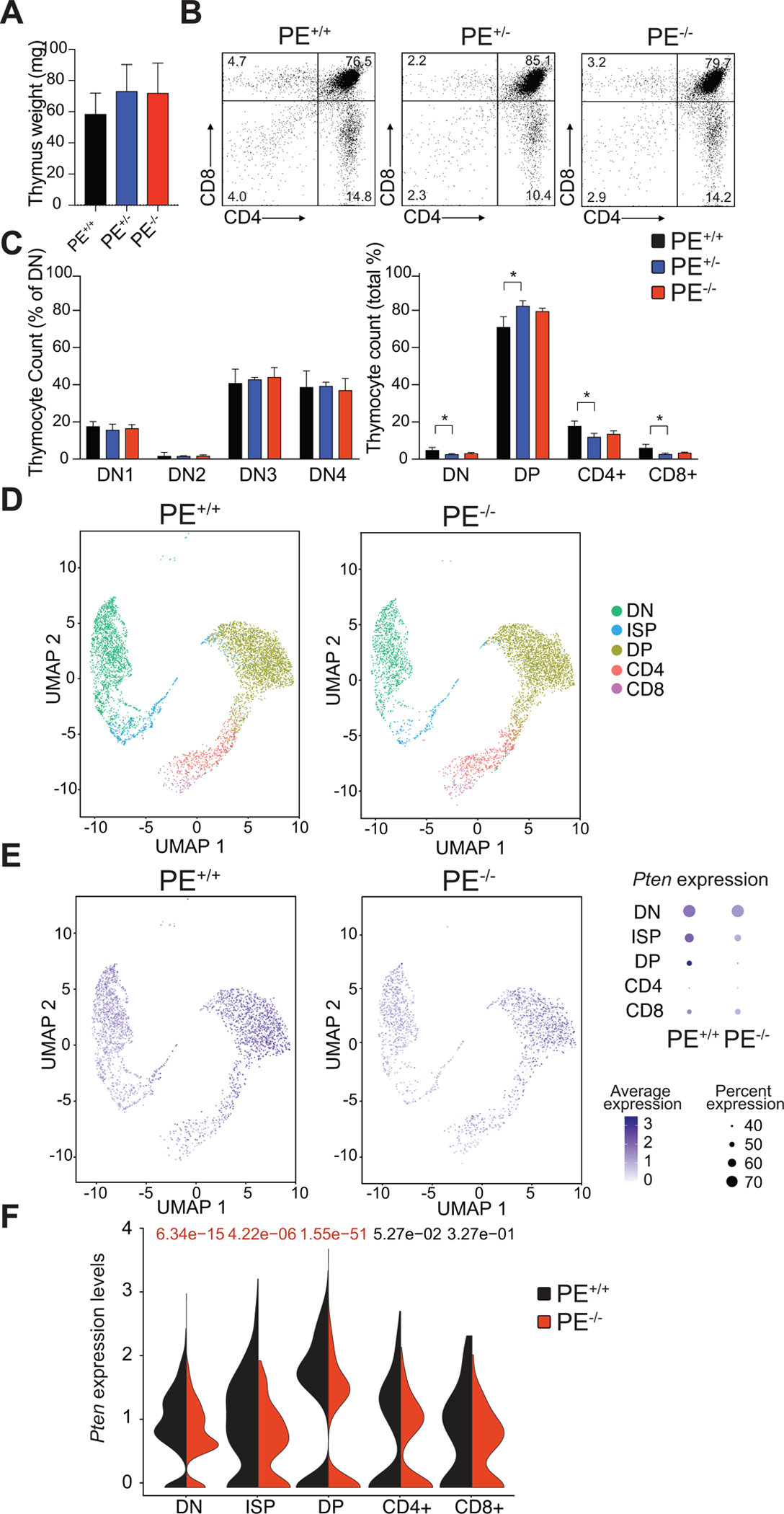
Effects of PE loss in thymic T-cell development. A, Thymus weight in 6-week-old wild- type (PE+/+), PE heterozygous knockout (PE+/−) and PE homozygous knockout (PE−/−) mice (n = 3 per genotype). B, Representative flow cytometry plots of thymocyte populations stained with antibodies to CD4 and CD8 in PE wild-type, PE heterozygous knockout and PE homozygous knockout 6-week-old mice. Percentage populations are indicated in each quadrant. C, Quantification of intrathymic T-cell populations in PE wild-type, PE heterozygous knockout and PE-null mice in relative numbers. Significance was calculated using two-tailed Student’s t-test (*P< 0.05; all other comparisons not significant). D, Single-cell RNA-seq analysis of a 1:1 mix of total thymocytes and CD4−CD3− thymocytes from 6-week-old PE wild-type and PE homozygous knockout mice. UMAP embeddings show the cells annotated to each thymic population. E, UMAP embeddings as in D representing single-cell Pten expression. Dot plots (right) represent the expression of Pten. Size of the dots is proportional to the percentage of cells expressing Pten in each population; color of the dots represents Pten average expression. F, Violin plots represent the expression of Pten in the different thymic populations. Significance was calculated using the Wilcoxon rank-sum test (significant values shown in red).
Overall, our results demonstrate that deletion of the PE enhancer leads to reduced expression of Pten throughout thymic T-cell development in mice, and highlight that the PE enhancer might also contribute to regulate PTEN expression in other hematopoietic populations.
Loss of PE results in acceleration of NOTCH1-induced T-ALL
Given the fundamental importance of NOTCH1 in T-ALL pathogenesis (34), where ~60% of T-ALL patients show activating mutations of NOTCH1 (8), and given that ~20% of T-ALL patients show loss of PTEN, which might co-occur with NOTCH1 mutations (9), we hypothesized that reduced levels of PTEN due to loss of the PE enhancer might accelerate NOTCH1-induced leukemogenesis. To test this hypothesis, we retrovirally expressed a constitutively active oncogenic mutant form of the NOTCH1 receptor (lacking its extracellular portion, but still tethered to the membrane and requiring gamma-secretase cleavage) with concomitant expression of the GFP marker (ΔE-NOTCH1-GFP) (6) in bone marrow progenitors obtained from PE wild-type, heterozygous or homozygous knockout mice, followed by transplantation into lethally irradiated recipient mice (Fig. 5A). Enforced expression of constitutively active NOTCH1 leads to the exclusive development of T-cell lineage leukemia with CD4+CD8+-double positive features, accompanied by acute leukemic infiltration of the spleen, in line with the critical role of NOTCH1 in T-cell specification (34,35). Notably, mice typically develop a CD4+CD8+-double positive wave of preleukemic cells in peripheral blood only 3 weeks after transplantation of NOTCH1-infected cells, and this preleukemic double-positive wave is known to be a good predictor of mouse survival (6). Indeed, our results showed a significantly higher number of double positive cells in mice transplanted with either PE heterozygous or PE homozygous knockout NOTCH1-infected bone marrow progenitor cells, as compared to mice transplanted with PE wild-type progenitor cells (Supplementary Fig. S10). Consistently, while mice transplanted with ΔE-NOTCH1-expressing bone marrow progenitor cells from PE wild-type mice developed T-ALL in 60–80 days (Fig. 5B), mice transplanted with ΔE-NOTCH1-expressing bone marrow progenitor cells from mice lacking one copy of PE developed T-ALL with significantly faster kinetics, which was further accelerated by bi-allelic deletion of PE (strain #6, 20% lower median survival; strain #15, 13% lower median survival; strain #18, 19% lower median survival), underscoring a dose-dependent tumor suppressor role for PE in T-cell leukemia generation (Fig. 5B).
Figure 5.
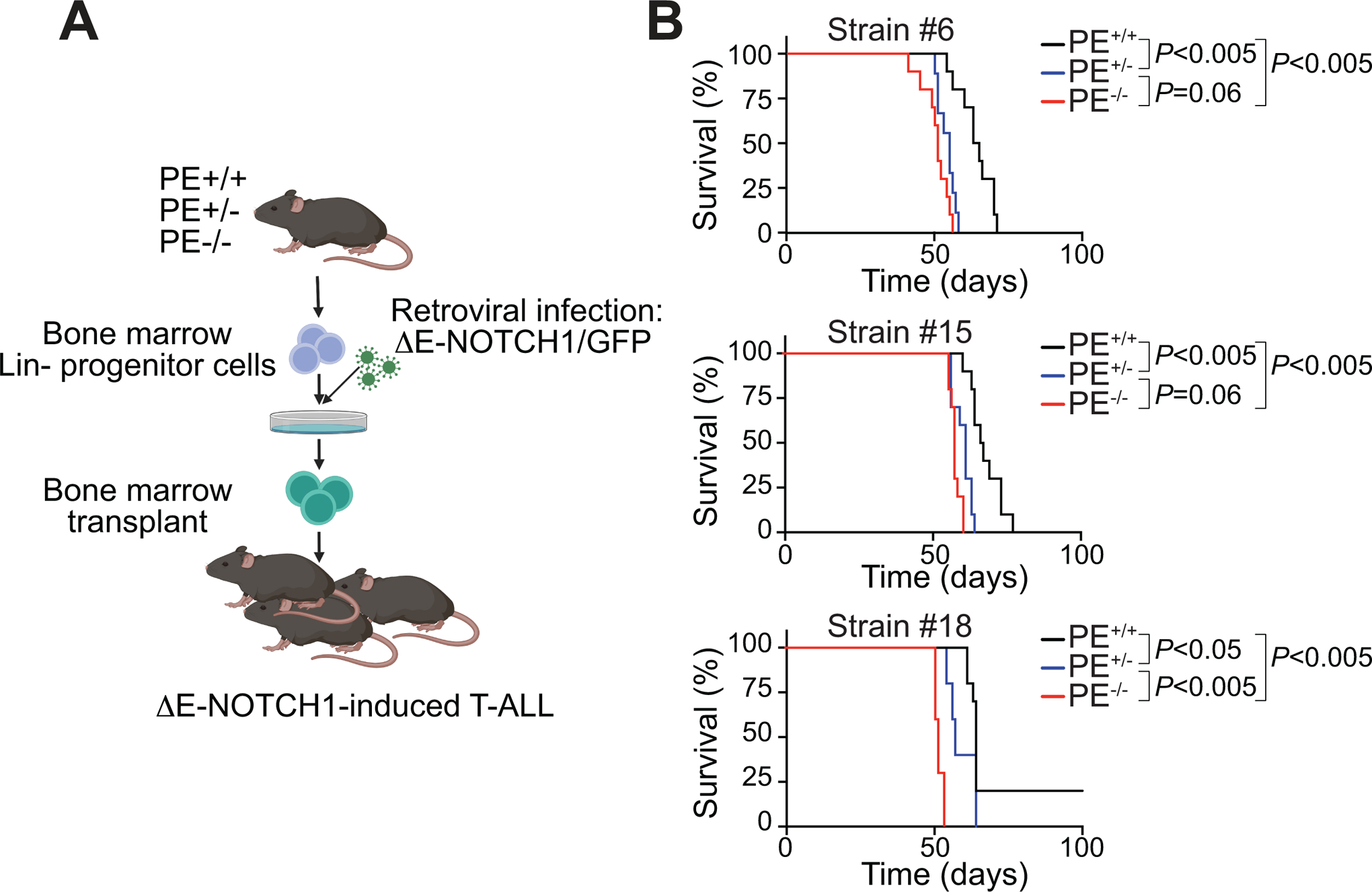
PE loss leads to accelerated NOTCH1-induced T-ALL development. A, Schematic of retroviral-transduction protocol for the generation of NOTCH1-induced T-ALLs from PE+/+, PE+/− and PE−/− mice. B, Kaplan-Meier curves of mice transplanted with ΔE-NOTCH1 infected PE+/+, PE+/− and PE−/− hematopoietic progenitors (n=10 per genotype). P values were calculated using the log-rank test.
Next, in order to test the effects of PE loss in leukemia progression and maintenance, we generated PE conditional knockout mice (Supplementary Fig. S11A–B). These mice were subsequently bred with a mouse strain harboring a tamoxifen-inducible Cre from the Rosa26 locus (6). In this context, we generated PE conditional homozygous knockout leukemias following a similar strategy (Fig. 6A). Transplant of these already generated leukemias in a secondary cohort of mice led to rapid disease progression, however, tamoxifen-induced isogenic deletion of PE still resulted in a significant acceleration (32% median lower survival) of T-ALL progression (Fig. 6B). Of note, this acceleration is in line with what we previously observed and reported upon secondary complete loss of Pten in already established leukemias, which led to a 35% lower median survival (Pten WT=20 days; Pten KO=13 days) (11). Analyses of these leukemic cells after tamoxifen-induced isogenic deletion of PE revealed markedly reduced Pten mRNA and protein levels (Fig. 6C and D). Furthermore, global gene expression profiling analyses upon inactivation of PE in these leukemias unveiled Pten as one of the most significantly downregulated genes (Fig. 6E and Supplementary Fig. S12A), whereas Rnls levels were undetectable, and other genes located in the TADs neighboring the Pten TAD did not show significant differences either (Supplementary Fig. S12B). In addition, gene set enrichment analyses (36) against our previously published gene expression signature obtained upon isogenic deletion of Pten in T-ALL using Pten conditional knockout leukemias (11) revealed that the transcriptional program elicited by deletion of PE is driven by Pten loss (Fig. 6F), and further GSEA analyses against the C2 database revealed 342 gene sets significantly downregulated (nominal P < 0.01) upon loss of PE (Supplementary Table S2). Altogether, our results demonstrate that loss of the PE enhancer in vivo results in an acceleration of T-ALL generation and progression, characterized by reduced expression levels of Pten.
Figure 6.
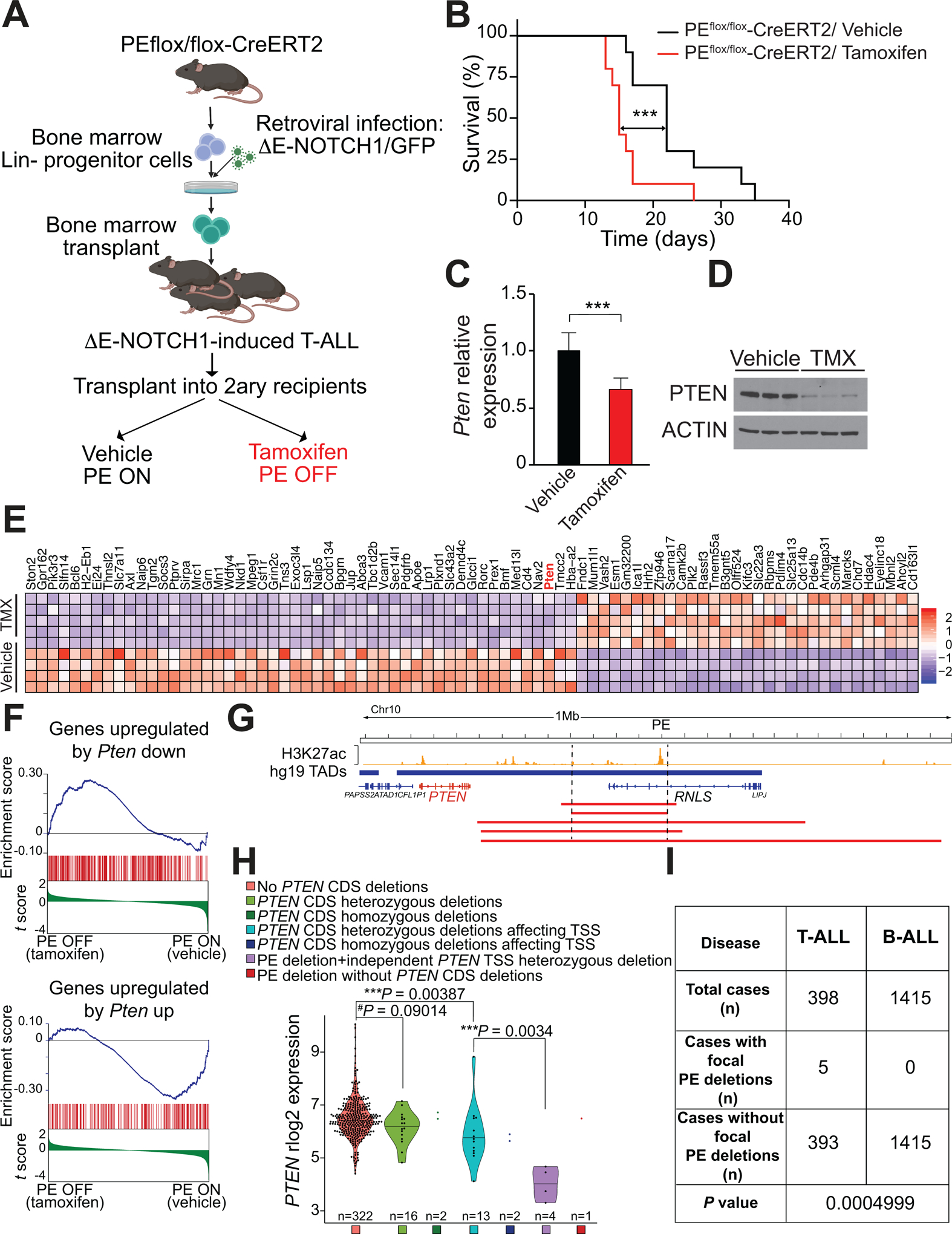
Secondary loss of PE leads to accelerated NOTCH1-induced T-ALL progression and reduced levels of PTEN in mouse and human T-ALL. A, Schematic of retroviral-transduction protocol or the generation and analysis of PE conditional knockout NOTCH1-induced T-ALL. B, Kaplan-Meier curves of mice transplanted with PE conditional knockout ΔE-NOTCH1-induced T-ALL and treated in vivo with vehicle (control) or tamoxifen, to induce isogenic deletion of PE. ***P ≤ 0.005 values calculated using the log-rank test. C, Quantitative RT-PCR analysis of Pten expression in tumor cells isolated from PE conditional knockout leukemia–bearing mice treated with vehicle only (n = 7) or tamoxifen (n = 8) in vivo. Graph show the mean values, and the error bars represent the s.d. ***P ≤ 0.005 was calculated using two-tailed Student’s t test. D, Western blot analysis of PTEN expression in tumor cells isolated from PE conditional knockout leukemia–bearing mice treated with vehicle only (n =3) or tamoxifen (n = 3) in vivo. E, Heat map representation of the top 81 differentially expressed genes between control and tamoxifen-treated PE conditional knockout NOTCH1-induced leukemias. Cutoffs used: Wald statistic < −5 or > 5; P-adjusted value < 1E-04; sorted based on mean expression levels in tamoxifen-treated samples (for full list of significantly downregulated genes upon tamoxifen treatment, see Supplementary Fig. S12A). The scale bar shows color-coded differential expression with red indicating higher levels of expression and blue indicating lower levels of expression. F, Gene Set Enrichment Analysis (GSEA) of genes regulated by PTEN in vehicle only-treated compared to tamoxifen-treated PE conditional knockout NOTCH1-induced leukemia cells in vivo. G, H3K27ac ChIP-seq mark in DND41 T-ALL cells along the PTEN-containing TAD and schematic representation of chromosome 10q23 focal deletions (red bars) found in human T-ALL. H, PTEN mRNA expression levels in human primary T-ALL samples (n=360). Samples are subdivided according to the presence/absence of PTEN coding sequence (CDS) deletions, the presence/absence of PTEN CDS deletions affecting it transcriptional start site (TSS) and the presence/absence of PE focal deletions. P value was calculated using t-test. I, PE focal deletions found specifically in T-ALL but not B-ALL. P value calculated using Fisher’s exact test.
Recurrent deletions encompassing PE in human T-ALL
Finally, in order to test whether genetic lesions in PE might contribute to T-ALL pathogenesis in human patients, we analyzed a panel of 398 T-ALL cases via SNP arrays (n=398), whole-genome sequencing (n=73) and whole transcriptome sequencing (n=360). These analyses uncovered 5 cases (1.25%) with focal deletions encompassing PE without affecting the PTEN coding sequence (CDS) (Fig. 6G). Interestingly, 2 of these samples show homozygous deletions of the PE enhancer (Supplementary Fig. S13A). However, 4/5 of these cases show additional simultaneous deletions targeting the coding sequence of PTEN independently (Supplementary Fig. S13A). In addition, 3/5 of these cases show mutations in NOTCH1 or FBXW7 (Supplementary Fig. S13A). As expected, cases with heterozygous deletions in the PTEN CDS show lower expression levels of PTEN as compared to cases without deletions (Fig. 6H). In addition, further reduced levels are seen when these deletions affect specifically the transcriptional start site (TSS) of PTEN (Fig. 6H). Among cases with loss of PE, there is only 1 sample that does not concomitantly present with deletions in the PTEN CDS, therefore precluding any relevant comparison in this group. The other 4 cases with PE loss present with heterozygous deletions affecting the PTEN TSS. However, these 4 cases show markedly and significantly reduced PTEN levels when compared to other samples similarly having heterozygous deletions in the PTEN TSS but without concomitant loss of PE (Fig. 6H). Furthermore, we performed comparative analyses in a cohort of 1415 B-ALL cases. Interestingly, unlike in T-ALL, there was no case with focal deletion of PE among these 1415 B-ALLs (Fig. 6I), underscoring the relevance of PE in T-ALL. Lastly, we also analyzed the methylation status of the PE DNA sequence in T-ALL. We found 4 CpG sites in the PE DNA sequence, 2 of which are hypomethylated and 2 of which are hypermethylated in normal T-cells (Supplementary Fig. 13B). However, both T-ALL cell lines and primary T-ALL cells maintain the hypermethylation in one of these CpG sites, whereas the other three are hypomethylated (Supplementary Fig. 13C–D). Overall, these results support that the loss of the PE enhancer does contribute to reduce the expression levels of PTEN specifically in T-ALL, and suggests that DNA hypermethylation is likely not involved in the dysregulation of the PE enhancer in T-ALL.
Discussion
PTEN, a key negative regulator of the PI3K/AKT pathway, is the second most frequently mutated TSG (37) and is deregulated in a wide range of cancer types (38). Specifically, loss of PTEN expression is seen in 15–20% of T-ALL patients (9,10), where it leads to secondary resistance to NOTCH1 inhibitory therapies in vivo (11). In addition, several studies suggest that genetic lesions driving loss of PTEN expression are associated with poor prognosis and outcome both in adult and pediatric T-ALL patients (39), highlighting its relevance in this hematological malignancy. However, and most notably, ~5% of T-ALL patients show loss of PTEN expression that cannot be currently explained by the typical mutations or deletions of its coding sequence (10), underscoring that the mechanisms leading to loss of PTEN expression in cancer remain incompletely understood. Here, we identify a previously unknown long-range enhancer region of PTEN located in a highly conserved intronic region of the neighboring RNLS gene, ~550Kb downstream of its transcriptional start site. We further prove that this enhancer region, named PE for PTEN enhancer, acts as a hub for a plethora of major T-cell transcription factors, shows bona-fide enhancer marks and open chromatin in human T-ALL, physically interacts with the PTEN promoter in normal and transformed T-cells and drives increased PTEN transactivation in reporter assays. In addition, we demonstrate that CRISPR-Cas9-induced deletion of PE in human T-ALL cells leads to reduced PTEN levels and a proliferative advantage. Still, even if PE seems to play a prominent role in the control of PTEN expression, the PTEN promoter interacts with additional regions along the PTEN TAD that also show several enhancer features, suggesting that some of these interacting regions might also play relevant roles in the regulation of PTEN.
In order to analyze PE effects in T-cell development and transformation in vivo, we generated mice with germinal or conditional deletion of PE. These novel mouse models unveiled that loss of PE is associated with reduced expression of Pten in developing thymocytes, whereas a wide variety of other solid tissues obtained from PE-knockout mice failed to show any difference in Pten levels, suggesting PE might behave as an hematopoietic-specific enhancer prominently active in the lymphoid compartment. In addition, thymocyte scRNA-seq analyses demonstrated that Pten levels in PE knockout mice are reduced in CD4−CD8−-double negative, CD8+-immature single positive and CD4+CD8+-double positive cells; however, this reduction does not lead to increased cellularity nor altered T-cell development in the thymus. Moreover, in line with the majority of cells in the thymus being CD4+CD8+-double positive cells, and consistent with the striking difference in PTEN protein levels observed by immunohistochemistry, scRNA-seq analyses revealed that CD4+CD8+-double positive thymocytes express the highest levels of Pten across all thymocyte populations, and it is in this specific population where the most significant decrease in Pten is observed in PE knockout thymocytes. Still, even if PE clearly plays a prominent role in T-cells and T-ALL, epigenetic and chromosome conformation analyses via ATAC-seq, ChIP-seq and Hi-C in earlier and/or non-T hematopoietic populations in both mice and humans highlight that PE might play a relevant role in the control of PTEN expression beyond the T-cell lineage, as PE shows H3K27ac mark and open chromatin in multiple hematological populations, and this is also accompanied by physical interaction with the PTEN promoter in non-T populations. Overall, these data suggest that the activity of the PE enhancer might be very prominently controlled by transcription factors that might be specifically present/absent in different stages of development, in addition to (or rather than) by how open its chromatin is. Subsequent follow-up studies are therefore warranted to thoroughly dissect its effects in each specific population and lineage. In addition, it is also worth mentioning that multiple reports have shown abnormalities in different hematopoietic lineages upon loss of PTEN either using an Mx-Cre allele (40–42), an Scl-CreERT allele (39) or a CD4-Cre allele (43,44), whereas we failed to detect noticeable abnormalities in T-cell development in PE knockout mice. However, all of these studies investigated the effects of complete loss of PTEN (i.e., using a Ptenflox/flox background, not a Ptenflox/+ background). In our case, even after complete loss of PE, we still detect PTEN expression even if at lower levels, which is what would be expected from the loss of an enhancer region that modulates the expression of its target gene. Thus, the different levels of reduction in PTEN expression might help explain the discordant phenotypes observed. In addition, specific loss of PTEN in T-cells using an Lck-Cre allele showed no differences in T-cell development at 6-weeks of age (29), similar to what we observed in PE knockout mice. However, even if PE-null mice do not show complete loss of PTEN, we can’t rule out at this point that PE loss might result in T-cell abnormalities at later time points beyond 6-weeks of age or might also lead to some developmental defects in earlier stages of hematopoietic development and/or other hematological lineages beyond T-cells.
Regarding its effects in cancer development, it is well known that PTEN acts as a haploinsufficient TSG, and even minor changes in its expression can lead to measurable effects in cancer development and progression (38). Consistently, germinal loss of one copy of the PE enhancer led to a significant acceleration in the development of NOTCH1-induced T-ALL in vivo, which was further accelerated by deletion of both allelic copies of PE. Furthermore, secondary deletion of PE in mice harboring already established PE conditional knockout T-ALL led to accelerated disease progression in vivo, reduced levels of Pten expression and a gene expression signature driven by Pten loss. Overall, our results underscore a strong dose-dependent effect of the PE enhancer in T-cell leukemia generation and progression.
We have previously demonstrated the importance of long-range regulatory sequences in T-ALL pathogenesis in vivo, as MYC expression is exquisitely dependent on the NOTCH1-driven N-Me MYC enhancer in T-cells (1,6). Indeed, enhancer deregulation is now considered to be a common mechanism contributing to cancer development (5). However, most of the cases described to date are related to oncogenic enhancers (5–7,45–47). Recent studies have suggested that a regulatory region of the tumor suppressor PAX5 might be affected in B-cell leukemia (48). Similarly, deleterious mutations in an enhancer region of ACTRT1 have been described in inherited and sporadic basal cell carcinoma (49), and a non-coding regulatory variant in an enhancer region of PER2 has recently been shown to affect the growth of AML cells (50). Finally, and more specifically related to PTEN, a different study suggested that p53 might regulate PTEN expression through a 5’ proximal regulatory region in epithelial cells. However, this region falls in the promoter of the neighboring CFL1P1 gene and seems to also affect its expression, as well as the expression of other neighboring genes (51). Moreover, whether this region physically interacts with the PTEN promoter or if it has any relevance in vivo remains unknown (51). In this context, to the best of our knowledge, our study is the first demonstration of the existence of an enhancer region of a major tumor suppressor gene and its relevance in cancer development in vivo. A fascinating question still to be addressed is how the PE enhancer might be developmentally regulated, including what pioneering factor/s control its chromatin opening, and what transcription factors control its effects on PTEN transactivation, both positively or negatively. PTEN regulation is notoriously complex, involving both transcriptional and post-transcriptional mechanisms (38). Key recent findings in the regulation of PTEN include the discovery of alternative translation start sites leading to the expression of PTEN-β which negatively regulates rDNA transcription and ribosomal biogenesis in the nucleus (52) and PTEN-Long, which is secreted and can exert tumor suppressive effects in a paracrine manner (53). However, the transcriptional regulation of PTEN is still poorly understood, with only a handful of transcription factors that have been suggested to transcriptionally activate or repress PTEN in certain contexts (54). Follow-up studies to dissect the mechanisms controlling the activity of PE will shed light on the dynamic processes and factors involved in the activation of this enhancer region.
Finally, analyses of a panel of 398 T-ALL cases uncovered 5 cases with deletions encompassing PE. Of note, T-ALL cases with loss of PE show reduced PTEN levels, underscoring its relevance in the pathophysiology of human T-ALL. While this is a relatively small number of cases (1.25%), it is in a similar range to what has been previously described for CNVs affecting other enhancer regions such as the MYC N-Me enhancer (5% of cases), the MYC BENC enhancer (3% of cases), the MYC-LASE enhancer (2% of cases) or the MYC-ECSE enhancer (4% of cases) (7). In addition, no case with loss of PE was detected among a cohort of 1415 B-ALL cases, highlighting the specific relevance of PE in T-ALL. Loss of PTEN expression is associated with resistance to NOTCH1 inhibition (9,11) and to glucocorticoids (55), together with increased risk of relapse and long-term adverse outcomes in T-ALL patients (56,57). Thus, PE status could be clinically used in the short-term as an additional biomarker to refine risk stratification and to select risk-adapted therapeutic strategies in patients. In line with this, it is also worth noting that Mcm2-deficient mice have been previously shown to develop T-ALL harboring genetic lesions affecting Pten and whole-genome sequencing of eight Mcm2-deficient T-ALLs uncovered one case (12.5%) harboring a deletion encompassing PE (58), which might have facilitated leukemia development. Still, additional mechanisms beyond genetic deletions may also lead to impaired activity of the PE enhancer. While we did not find evidence that DNA hypermethylation might play a role in the dysregulation of this enhancer, mutations in the PE sequence or single-nucleotide polymorphisms affecting the binding of important transcription factors controlling the activity of PE might conceivably result in decreased levels of PTEN. Similarly, epigenetic silencing of the PE enhancer resulting in closed chromatin along this region might also occur. In addition, chromosome conformation changes leading to alterations in interactions within TADs, as well as their boundaries, have also been recently shown to affect enhancer activity (19); thus, another potential mechanism driving impaired PE activity might involve the establishment of a TAD boundary gain separating the PTEN promoter from the PE enhancer. It is also tempting to speculate that additional regions yet to be identified might control PTEN expression in other types of cancer. Further studies to elucidate how widespread PE activity might be impaired in T-ALL, and whether PTEN is regulated by additional enhancer regions in other tumor types, are therefore warranted. Overall, our study clearly demonstrates that impaired activity of the PE PTEN enhancer is a novel mechanism that cooperates in leukemia development and, more broadly, paves the way for the discovery and dissection of enhancers of other TSGs in additional malignancies.
Methods
Cell culture
We performed cell culture of cell lines in standard conditions in a humidified atmosphere at 37°C under 5% CO2. DND41 (#ACC-525; RRID:CVCL_2022), JURKAT (#ACC-282; RRID:CVCL_0065), HPB-ALL (#ACC-483; RRID:CVCL_1820) cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). We purchased HEK293T cells from American Type Culture Collection (ATCC #CRL-3216; RRID:CVCL_0063). No cell authentication was performed. T-ALL cells were cultured in HyClone RPMI 1640 Media (SH3002701, Fisher Scientific) supplemented with 10% FBS (900–108, Gemini Bio-Products) and 1% penicillin-streptomycin (45000–652, VWR).
Generation of DND41 cells with CRISPR/Cas9-induced loss of PE
DND41 PE+/− cells were generated following a previously described protocol (59). Briefly, custom primers (Sigma-Aldrich) (Supplementary Table S3) were used to generate sgRNA DNA templates. Evaluation for potential off-targets was performed through CRISPRscan (https://www.crisprscan.org). In vitro transcription of sgRNAs from templates was performed following the protocol guidelines. 500ng of each purified sgRNA was independently incubated with 500ng of Cas9 (1074181, IDT) for 20 minutes at RT. Subsequently, one million DND41 cells were electroporated with 1μg of each complex per transfection using the Neon Transfection System (MPK5000, Thermo Fisher Scientific) and 100μL electroporating tips (MPK10025, Thermo Fisher Scientific). Electroporation conditions: pulse voltage 1350v, pulse width 10ms, pulse number 3, cell density 107/ml. Single cell clones were sorted using the Influx High Speed Sorter (BD Biosciences) and were subsequently grown in tissue culture and screened for PE loss by PCR using REDTaq ReadyMix (R2523–100RXN, Sigma Aldrich) following standard conditions and using custom primers (Supplementary Table S3).
Genomic analysis of primary T-ALL and B-ALL samples
Genomic profiling of patient samples was performed by SNP array (Affymetrix SNP 6.0, T-ALL; n = 398, B-ALL; n = 1415), whole genome sequencing (WGS, n = 73) and whole transcriptome sequencing (WTS, n = 360). Sequencing libraries for WGS were prepared from genomic DNA sheared on LE220 ultrasonicator (Covaris) with HyperPrep Library Preparation Kits (Kapa Biosystems). WTS libraries were generated by using TruSeq Stranded Total RNA Library Prep Kit (Illumina). Libraries were analyzed for insert size distribution with a 2100 BioAnalyzer High Sensitivity kit (Agilent Technologies) or Caliper LabChip GX DNA High Sensitivity Reagent kit (PerkinElmer), and were quantified by using the Quant-iT PicoGreen dsDNA assay (Life Technologies). Paired-end sequencing was performed with HiSeq2000/2500 or NovaSeq 6000 (Illumina).
The detection of copy number variations on PTEN CDS and PE regions was performed by using SNP array data as previously described (60). For WGS, reads were mapped to the GRCh37 reference human genome assembly using BWA, sorted and de-duplicated using and Samtools (https://www.htslib.org/). Multiple softwares on Genomon Project (https://github.com/Genomon-Project) were applied to call SNVs and indels (GenomonFisher, GenomonMutationFilter and EBFilter) or structural variants (GenomonSV). Expression of PTEN was calculated from gene-level read counts, generated by HTseq-count (61). After normalization with DESeq2 Bioconductor R package (62), a regularized log-transformed (rlog) value was calculated by variance-stabilizing transformation (VST) of DESeq2. All statistical analyses were performed using R (v3.6.0) software (www.r-project.org).
Statistical significance on PTEN expression was assessed by t-test (ggpubr R package; https://rpkgs.datanovia.com/ggpubr/), and values were considered statistically significant at P < 0.05. Associations between categorical values were examined using Fisher’s exact test. Data are available at the St. Jude PeCan Data Portal (63) (https://pecan.stjude.cloud/), the Therapeutically Applicable Research to Generate Effective Treatment (TARGET) data portal (https://ocg.cancer.gov/programs/target), dbGaP and the European Genome Phenome Archive, as previously described (64,65).
DNA methylation analysis
DNA methylation status of PE was determined using the Illumina MethylationEPIC BeadChip 850K microarray (66). Briefly, 600 ng of DNA of the studied samples (T-cell normal donor samples, T-ALL cell lines and primary malignancies) was used to hybridize to the BeadChip and scanned using HiScan SQ system (Illumina). Raw signal intensity data were initially QC’d and pre-processed from resulting idat files in R statistical environment (v3.6.2) using minfi Bioconductor package (v1.32.0). A number of quality control steps were applied to minimize errors and remove erratic probe signals, such as failed probes (detection p value >0.01), cross-reacting probes and probes that overlapped single nucleotide variants within +/− 1 bp of CpG sites followed by background correction and dye-based normalization using ssNoob algorithm (single-sample normal-exponential out-of-band). DNA methylation scores for each CpG were represented as a β-values ranging between standard 0 and 1 where 1 represents fully methylated CpGs and 0, fully unmethylated. All downstream analyses were performed under R statistical environment (v3.6.1). Unsupervised hierarchical clustering analysis using euclidean distance with complete-linkage clustering and heatmaps of methylation CpG Beta-values in the studied samples were used to represent the DNA methylation data. Raw DNA methylation data is available upon request.
ChIP-seq and Gro-Seq analysis
We analyzed PE occupancy of chromatin marks, epigenetic and transcription factors using the following T-ALL publicly available ChIP-seq datasets from GEO: GSE29611, GSE54379, GSE54380, GSE29600, GSE72300, GSE59657, GSE58406, GSE83777, GSE17954, GSE85524, GSE83777, GSE109653, GSE94391, GSE62264, GSE29180, GSE50622, GSE76783, GSE115893, GSE60482 and GSE104992. We analyzed nascent RNA around the PE locus using the following T-ALL publicly available GRO-seq data set from GEO: GSE115894.
ChIPmentation analysis
50 to 100,000 CD45+ sorted T-ALL cells were fixed using 1% formaldehyde (Thermo Fisher Scientific 28906) and quenched by glycin (125 mM). Next, cell pellets were lysed in 100 μL of short-term complete lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 0.25 % SDS, 20 mM NaBu histone deacetylase inhibitor, 1X complete protease inhibitors cocktail EDTA-free (Roche, 5056489001)). Chromatin was sheared on the Bioruptor Pico (Diagenode) using a 15 seconds on / 30 seconds off, 7 cycle regimen. Sheared chromatin was magnetically immunoprecipitated using a polyclonal anti-H3K27ac antibody (Supplementary Table S4) and subsequently tagmentated using the Auto ChIPmentation Kit for Histones (Diagenode, C01011010) on the IP-Star Compact Automated System (Diagenode, B03000002), according to the manufacturer’s instructions. Input DNA was de-crosslinked, purified (MinElute, Qiagen), and tagmentated (Nextera DNA Library Prep, Illumina). Stripping, end repair and library amplification were performed according to the ChIPmentation Kit guidelines. Libraries were sequenced using the NextSeq500 (SR75, High Output). Reads were trimmed by Trimmomatic and aligned to hg38 with Bowtie2 using the parameters -N 1 -k 1. Peaks were called with MACS2 with the respective input control for each patient sample.
We analyzed H3K27ac mark around the PE sequence in normal human thymocytes using previously published ChIPmentation results (GSE151078).
ATAC-seq analysis
Analyses of open chromatin regions around the PE sequence in a mouse NOTCH1-induced T- ALL cell line (previously described (1,6)) was done by Active Motif, following well-established protocols (67). We analyzed open chromatin regions around the PE sequence in normal human thymocytes and human T-ALL primary samples using publicly available ATAC-seq data sets from GEO (GSE151075 and GSE124223).
Conservation analysis
The conservation track in Supplementary Figure S2 shows a subset of the vertebrates’ genomic sequence alignment as produced by the UCSC Genome Browser based on the MULTIZ algorithm (68).
4C-seq Analyses
We performed 4C-seq analysis in JURKAT, HPB-ALL and DND41 human T-ALL cells and HDΔP-NOTCH1-induced or ΔE-NOTCH1-induced mouse primary T-ALL lymphoblasts (previously described(11)) following published protocols (69) and using the restriction enzymes HindIII (R3104M, NEB) and DpnII (R0543M, NEB). We constructed sequencing libraries from 4C DNA including barcoded Illumina adapters to the 5′ end of each PCR primer (Supplementary Table S3). We sequenced pooled libraries using a HiSeq 2500 sequencer (Illumina). Processing of 4C-seq data was done as previously described (12,69). For visualization purposes, signal was averaged across three replicates using 25Kb bins with 90% overlap for smoothening.
Hi-C, Hi-ChIP and Capture-HiC analyses
We analyzed Hi-C of human normal CD4 cells or T-ALLs, as well as Hi-ChIP data in CUTTL1 T-ALL cells using available data sets from GEO (GSE115896). We analyzed tagHi-C data of mouse hematopoietic populations using an available data set from GEO (GSE152918). Hi-C data was represented using Juicebox (70). Capture Hi-C data was represented using Capture Hi-C plotter (71).
Luciferase reporter assays
We performed reporter assays using a pGL3-Promoter Vector (E1761, Promega) luciferase construct alone and coupled with human and mouse PE enhancer sequences (hPE: hg19; 90,144,758–90,149,109; mPE: mm10; chr19:33,220,867–33,224,929; both sequences were synthesized by Genewiz, RRID:SCR_003177), cloned in the forward and reverse orientations. In these assays, we electroporated JURKAT cells with a Neon Transfection System Device (MPK5000, Thermo Fisher Scientific) using 100μL tips (MPK10025, Thermo Fisher Scientific). Electroporation conditions: pulse voltage 1350v, pulse width 10ms, pulse number 3, cell density 107/ml. PE constructs were transfected together with a plasmid driving the expression of the Renilla luciferase gene (pCMV-Renilla) used as an internal control. We measured luciferase activity 42h after electroporation with the Dual-Luciferase Reporter Assay kit (E1980, Promega).
Mice and animal procedures
All animals were maintained in specific pathogen-free facilities at New Brunswick RBHS Rutgers Campus. All animal procedures were approved by Rutgers Institutional Animal Care and Use Committee (IACUC). PE knockout and PE conditional knockout mice were generated by the Rutgers Cancer Institute of New Jersey Genome Editing Shared Resource.
To generate PE germinal knockout mice, CRISPR sgRNAs flanking the PE region (Supplementary Table S3) and HiFiCas9 (1081060, IDT) were used. sgRNAs were complexed with Cas9 protein and electroporated into C57BL/6J embryos (JAX:000664; RRID:IMSR_JAX:000664). Three correctly targeted founders were identified and bred to C57BL/6NTac (Taconic) to confirm germline transmission.
To generate PE conditional knockout mice, we introduced LoxP sites at mouse genomic positions GRCm38/mm10 chr19: 33,220,867–33,224,930, flanking the mouse PE enhancer. CRISPR sgRNAs flanking the PE region (Supplementary Table S3) and HiFiCas9 (1081060, IDT) were used to insert LoxP sites at the specified locations. sgRNAs were complexed with Cas9 protein, mixed with a donor oligo containing LoxP sites and electroporated into C57BL/6J embryos. LoxP sites were inserted sequentially with the 5’ LoxP site being done first. A male mouse with a 5’ LoxP site correctly inserted was used to perform IVF with wild-type oocytes and the 3’ LoxP was inserted in these embryos. Both LoxP sites were confirmed in founders and bred to C57BL/6NTac mice to confirm the presence of both loxP sites on the same allele.
To generate conditional inducible PE knockout mice, we bred PE conditional knockout mice with animals harboring a tamoxifen-inducible form of the Cre recombinase from the ubiquitous Rosa26 locus (72). Phenotypic characterization of PE knockout mice included male and female mice and isogenic age- and gender-matched controls without blinding. For secondary deletion of PE using tamoxifen-induced activation of Cre recombinase in vivo, animals were randomly assigned to the vehicle (corn oil, C8267, Sigma-Aldrich) or tamoxifen (T5648, Sigma-Aldrich; 3mg per mouse in corn oil) groups and disease progression was followed without blinding.
Immunohistochemistry
Freshly isolated thymi from 6-week-old PE wild-type, heterozygous or homozygous knockout littermates were fixed overnight in 3.7% zinc formalin solution (175, Anatech). We performed immunohistochemical stainings following standard protocols using hematoxylin and eosin stain or a monoclonal antibody against PTEN (Supplementary Table S4).
Generation of NOTCH1-induced leukemias
We generated NOTCH1-induced T-ALL tumors in mice as previously described (6,11). Briefly, we performed retroviral transduction of bone marrow cells enriched in lineage (Lin)-negative cells isolated using magnetic beads (130-090-858, Miltenyi Biotec) with an activated form of the NOTCH1 oncogene (ΔE-NOTCH1). Primary mouse lineage negative bone marrow cells were cultured in Opti-MEM media (51985091, Life Technologies) supplemented with 10% FBS, penicillin G (100UmL-1), streptomycin (100μgmL-1), β-mercaptoethanol (55 μM), IL3 (10ngmL- 1; 213–13, PeproTech), IL6 (10ngmL-1; 216–16, PeproTech), IL7 (25ngmL-1; 217–17, PeproTech), SCF (50ngmL-1; 250–03, PeproTech), and Flt3L (50ngmL-1; 250–31L, PeproTech). NOTCH1-infected bone marrow progenitor cells were then transplanted by intravenous injection into lethally irradiated female isogenic recipients (C57BL/6NTac). No blinding was performed.
Flow cytometry analysis of hematopoietic populations
All flow cytometry data were collected on an Attune NxT Flow Cytometer (Thermo Fisher Scientific), and was later analyzed using FlowJo Software (BD; RRID:SCR_008520).
Single-cell suspensions of total thymocytes or splenocytes from 6-week-old PE wild-type, heterozygous and homozygous knockout littermates were prepared by pressing tissues through a 70μm filter. Red cells in spleen samples were removed by incubation with red blood cell lysis buffer (155mM NH4Cl, 12mM KHCO3 and 0.1mM EDTA) for 5min at room temperature.
For flow cytometry–based analysis of thymocyte and splenocyte populations and discrete stages of T cell development, cells were stained with anti-mouse fluorochrome-conjugated antibodies (Supplementary Table S4). T cell subsets in the thymus: DN1 (CD4−CD8−CD44+CD25−), DN2 (CD4−CD8−CD44+CD25+), DN3 (CD4−CD8−CD44−CD25+) and DN4 (CD4−CD8−CD44−CD25−), DP (CD4+CD8+), SP CD4+ (CD4+CD8−) and SP CD8+ (CD4−CD8+).
For PTEN intracellular staining, DND41 control cells or with heterozygous loss of PE were fixed and permeabilized with BD Cytofix/Cytoperm solution (554722, BD), washed with BD Perm/Wash buffer (554723, BD) and stained with a mouse anti-PTEN or a mouse isotype control antibody (Supplementary Table S4), following datasheet guidelines.
RNA-seq gene expression profiling
PE conditional knockout NOTCH1-induced T-ALL–bearing mice were treated with vehicle only (corn oil, C8267, Sigma-Aldrich) or tamoxifen (T5648, Sigma-Aldrich; 3mg per mouse in corn oil), to induce isogenic loss of PE, via intraperitoneal injection. Single-cell suspensions of total leukemic splenocytes were prepared by pressing leukemic spleens through a 70μm filter. We removed red cells in spleen samples by incubation with red blood cell lysis buffer (155mM NH4Cl, 12mM KHCO3 and 0.1mM EDTA) for 5min at room temperature. RNA was extracted using QIAshredder (#79656) and Rneasy Mini (#74106) QIAGEN kits. RNA library preparations and next-generation sequencing was performed at Genewiz (RRID:SCR_003177) using an Illumina Next-Seq platform (Illumina). We calculated gene counts for each sample using kallisto (73), and normalized the counts and evaluated differential expression between the samples in R using DESeq (74) and DESeq2 (62), respectively. We used GSEA to assess enrichment of Pten signature genes as previously described in (11), based on 10,000 permutations of the gene list (36).
Single-cell RNA-seq
Single-cell suspensions of total thymocytes from 6-week-old wild-type and PE homozygous knockout littermates were prepared by pressing thymic lobes through a 70μm filter. This single- cell suspension was enriched in CD4−CD3−-immature thymocyte progenitor cells by staining with biotinylated antibodies against CD3 and CD4 (Supplementary Table S4), followed by magnetic depletion of antibody-labeled cells using streptavidin microbeads (Miltenyi Biotec 130-048-101). CD4−CD3−- enriched thymocytes were then mixed 1:1 with single-cell suspensions from total unenriched thymocytes. We then loaded thymocyte suspensions (10,000 cells per sample) isolated from 6-week-old mice on a 10x Chromium instrument (10x Genomics) and prepared single-cell RNA-seq libraries using the Chromium Single Cell 3′ v3 Reagent Kit (10x Genomics PN-1000092) and sequenced them at the Princeton University Genomics Core Facility using NovaSeq instruments (Illumina). We demultiplexed the reads and aligned them to the mm10 reference using 10x Cell Ranger version 3.0.2. We loaded single-cell data into count matrices and merged the PE wild-type and PE knockout thymocytes using Seurat v3.0 (75), and calculated each cell’s hybrid doublet score using scds (76). We retained cells that expressed 200 to 4,000 genes, contained less than 12% of transcripts mapping to mitochondrial genes, and had a hybrid doublet score ≤ 1. We then scaled and log-normalized gene expression measurements for each cell and performed principal component analysis using the top 2,000 genes selected by variance stabilizing transformation approach as implemented in Seurat. We used Harmony to integrate PE wild-type and knockout thymocyte populations and to remove dataset-specific characteristics (77). We used UMAP to visualize the distribution of cells in the projection of the significant principal components. We then extracted markers for the different thymocyte populations from data produced by the IMMGEN project (GSE127267) (78), and annotated cells accordingly, using singleR (79). The percentiles for each population were compared with population percentages determined by flow cytometry analysis in each genotype as ground truth. Cells with multiple or non-T cell population calls were excluded from the final analysis. We statistically evaluated differential expression between the PE wild-type and PE knockout populations within each cell type using the Wilcoxon rank-sum test, and adjusted for the multiple hypotheses using the Bonferroni correction.
Quantitative real-time PCR
We performed reverse transcription reactions with the High-Capacity cDNA Reverse Transcription Kit (4368814, Applied Biosystems) and analyzed the resulting cDNA products by quantitative real-time PCR using PowerUp SYBR Green Master Mix (A25778, Applied Biosystems) using a QuantStudio 3 Real-Time PCR System (Applied Biosystems). Relative expression levels were normalized using Gapdh or Actin as a reference control (Supplementary Table S3).
Western blotting
Western blot analysis was performed using standard procedures. Antibodies used are listed in Supplementary Table S4.
Statistical analyses
Unless otherwise noted, we evaluated statistical significance using two-tailed Student’s t test, assuming normality and equal distribution of variance between the different groups analyzed. Survival in mouse experiments was represented with Kaplan-Meier curves, and significance was estimated with the log-rank test. We performed all statistical analyses using Prism GraphPad (RRID:SCR_002798).
Supplementary Material
Statement of significance.
Here we identify a PTEN enhancer which is recurrently deleted in T-ALL patients. Deletion of this enhancer leads to reduced PTEN levels in T-cells together with accelerated generation and progression of NOTCH1-induced leukemia in vivo. These results uncover long-range regulation of tumor suppressor genes as a relevant mechanism in cancer.
Acknowledgments
We are grateful to Adolfo A. Ferrando (Columbia University Medical Center) and Antonio Maraver (IRCM, Montpellier) for their constructive criticism and support. We thank Peter Romanienko and Ghassan Yehia (Rutgers University) for outstanding technical assistance in the generation of PE germinal and conditional knockout mice. We thank Jessica Wiggins, Wei Wang and the Princeton University Genomics Core Facility for the single-cell library preparation and sequencing. We thank Iannis Aifantis and Aristotelis Tsirigos (New York University School of Medicine) for sharing already processed HiC files with us. We also thank Lorenzo Brunetti (University of Perugia) for his advice with the CRISPR/Cas9 electroporation protocol. We also thank everyone involved with JuanLord for their support. Figures 5A and 6A were created using BioRender.com.
Financial support: Work in the laboratory of DH is supported by the US National Institutes of Health (R00CA197869 and R01CA236936), the American Cancer Society (RSG-19-161-01-TBE), the Alex’s Lemonade Stand Foundation, the Leukemia Research Foundation, the Children’s Leukemia Research Association, the Gabrielle’s Angel Foundation for Cancer Research and the Rutgers Cancer Institute of New Jersey. Work in the laboratory of H.K. is supported by the National Institutes of Health (R01CA233662) and the V Foundation (T2019-012), as well as by Rutgers Office of Advanced Research Computing (NIH 1S10OD012346-01A1). In addition, Rutgers Cancer Institute of New Jersey shared resources supported in part by the National Cancer Institute Cancer Center Support Grant P30CA072720 were instrumental for this project, including Biomedical Informatics Shared Resource (P30CA072720-5917), Flow Cytometry and Cell Sorting Shared Resource (P30CA072720-5921) and Genome Editing Shared Resource (P30CA072720-5922). Work in the lab of P.R. is supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Development (NICHD). We also thank the NICHD molecular genomics core and computational resources of the NIH HPC Biowulf cluster (hpc.nih.gov). Work in the labs of P.V.V and T.T. is supported by the European Research Council (grant no. StG-639784), the Fund for Scientific Research Flanders (FWO) and the Cancer Research Institute Ghent (CRIG). Fellowships from the New Jersey Commission on Cancer Research support the work of L.T. (DCHS20PPC010), JW.L. (DFS18PPC017) and V.dD. (DCHS19PPC008).
Footnotes
Conflict of interest disclosure:
The authors declare no competing financial interests.
Data availability
All generated data was deposited in NCBI Gene Expression Omnibus (GEO; RRID:SCR_005012) under the following accession numbers: 4C-seq data, GSE160427; mouse PE conditional knockout T-ALL RNA-seq data GSE150894; mouse thymus scRNA-seq data, GSE156070; human H3K27ac T-ALL ChIPmentation data, GSE155332; mouse T-ALL ATAC-seq data, GSE160620.
References
- 1.Belver L, Yang AY, Albero R, Herranz D, Brundu FG, Quinn SA, et al. GATA3-Controlled Nucleosome Eviction Drives MYC Enhancer Activity in T-cell Development and Leukemia. Cancer Discov 2019;9(12):1774–91 doi 10.1158/2159-8290.CD-19-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell 2014;157(1):13–25 doi 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 2011;12(4):283–93 doi 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 2014;15(4):272–86 doi 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 5.Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer 2016;16(8):483–93 doi 10.1038/nrc.2016.62. [DOI] [PubMed] [Google Scholar]
- 6.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014;20(10):1130–7 doi 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lancho O, Herranz D. The MYC Enhancer-ome: Long-Range Transcriptional Regulation of MYC in Cancer. Trends Cancer 2018;4(12):810–22 doi 10.1016/j.trecan.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306(5694):269–71 doi 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 9.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 2007;13(10):1203–10 doi 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuurbier L, Petricoin EF 3rd, Vuerhard MJ, Calvert V, Kooi C, Buijs-Gladdines JG, et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 2012;97(9):1405–13 doi 10.3324/haematol.2011.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 2015;21(10):1182–9 doi 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raviram R, Rocha PP, Muller CL, Miraldi ER, Badri S, Fu Y, et al. 4C-ker: A Method to Reproducibly Identify Genome-Wide Interactions Captured by 4C-Seq Experiments. PLoS Comput Biol 2016;12(3):e1004780 doi 10.1371/journal.pcbi.1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonis M, Klous P, Homminga I, Galjaard RJ, Rijkers EJ, Grosveld F, et al. High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nat Methods 2009;6(11):837–42 doi 10.1038/nmeth.1391. [DOI] [PubMed] [Google Scholar]
- 14.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485(7398):376–80 doi 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016;164(6):1110–21 doi 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015;162(4):900–10 doi 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159(7):1665–80 doi 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, et al. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep 2015;10(8):1297–309 doi 10.1016/j.celrep.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet 2020;52(4):388–400 doi 10.1038/s41588-020-0602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature 2014;507(7493):455–61 doi 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melgar MF, Collins FS, Sethupathy P. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol 2011;12(11):R113 doi 10.1186/gb-2011-12-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74 doi 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510(7504):278–82 doi 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao X, Dang L, Zheng X, Lu Y, Lu Y, Ji R, et al. Targeting Super-Enhancer-Driven Oncogenic Transcription by CDK7 Inhibition in Anaplastic Thyroid Carcinoma. Thyroid 2019;29(6):809–23 doi 10.1089/thy.2018.0550. [DOI] [PubMed] [Google Scholar]
- 25.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet 1998;19(4):348–55 doi 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 26.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol 2003;1(3):E59 doi 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003;4(3):209–21 doi 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 28.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012;13(5):283–96 doi 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 29.Xue L, Nolla H, Suzuki A, Mak TW, Winoto A. Normal development is an integral part of tumorigenesis in T cell-specific PTEN-deficient mice. Proc Natl Acad Sci U S A 2008;105(6):2022–7 doi 10.1073/pnas.0712059105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, et al. The cis-Regulatory Atlas of the Mouse Immune System. Cell 2019;176(4):897–912 e20 doi 10.1016/j.cell.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C, Xu Z, Yang S, Sun G, Jia L, Zheng Z, et al. tagHi-C Reveals 3D Chromatin Architecture Dynamics during Mouse Hematopoiesis. Cell Rep 2020;32(13):108206 doi 10.1016/j.celrep.2020.108206. [DOI] [PubMed] [Google Scholar]
- 32.Roels J, Kuchmiy A, De Decker M, Strubbe S, Lavaert M, Liang KL, et al. Distinct and temporary-restricted epigenetic mechanisms regulate human alphabeta and gammadelta T cell development. Nat Immunol 2020;21(10):1280–92 doi 10.1038/s41590-020-0747-9. [DOI] [PubMed] [Google Scholar]
- 33.Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016;167(5):1369–84 e19 doi 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016;16(8):494–507 doi 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- 35.Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med 1996;183(5):2283–91 doi 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene 2008;27(41):5443–53 doi 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 38.Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol 2018;19(9):547–62 doi 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 39.Tesio M, Trinquand A, Macintyre E, Asnafi V. Oncogenic PTEN functions and models in T-cell malignancies. Oncogene 2016;35(30):3887–96 doi 10.1038/onc.2015.462. [DOI] [PubMed] [Google Scholar]
- 40.Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008;453(7194):529–33 doi 10.1038/nature06933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006;441(7092):475–82 doi 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006;441(7092):518–22 doi 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Karnell JL, Yin B, Zhang R, Zhang J, Li P, et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J Clin Invest 2010;120(7):2497–507 doi 10.1172/JCI42382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newton RH, Lu Y, Papa A, Whitcher GH, Kang YJ, Yan C, et al. Suppression of T-cell lymphomagenesis in mice requires PTEN phosphatase activity. Blood 2015;125(5):852–5 doi 10.1182/blood-2014-04-571372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018;553(7689):515–20 doi 10.1038/nature25193. [DOI] [PubMed] [Google Scholar]
- 46.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346(6215):1373–7 doi 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sur IK, Hallikas O, Vaharautio A, Yan J, Turunen M, Enge M, et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 2012;338(6112):1360–3 doi 10.1126/science.1228606. [DOI] [PubMed] [Google Scholar]
- 48.Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015;526(7574):519–24 doi 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 49.Bal E, Park HS, Belaid-Choucair Z, Kayserili H, Naville M, Madrange M, et al. Mutations in ACTRT1 and its enhancer RNA elements lead to aberrant activation of Hedgehog signaling in inherited and sporadic basal cell carcinomas. Nat Med 2017;23(10):1226–33 doi 10.1038/nm.4368. [DOI] [PubMed] [Google Scholar]
- 50.Li K, Zhang Y, Liu X, Liu Y, Gu Z, Cao H, et al. Noncoding Variants Connect Enhancer Dysregulation with Nuclear Receptor Signaling in Hematopoietic Malignancies. Cancer Discov 2020;10(5):724–45 doi 10.1158/2159-8290.CD-19-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pappas K, Xu J, Zairis S, Resnick-Silverman L, Abate F, Steinbach N, et al. p53 Maintains Baseline Expression of Multiple Tumor Suppressor Genes. Mol Cancer Res 2017;15(8):1051–62 doi 10.1158/1541-7786.MCR-17-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang H, Chen X, Yin Q, Ruan D, Zhao X, Zhang C, et al. PTENbeta is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat Commun 2017;8:14771 doi 10.1038/ncomms14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013;341(6144):399–402 doi 10.1126/science.1234907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Worby CA, Dixon JE. Pten. Annu Rev Biochem 2014;83:641–69 doi 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 55.Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 2013;24(6):766–76 doi 10.1016/j.ccr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paganin M, Grillo MF, Silvestri D, Scapinello G, Buldini B, Cazzaniga G, et al. The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br J Haematol 2018;182(5):705–11 doi 10.1111/bjh.15449. [DOI] [PubMed] [Google Scholar]
- 57.Szarzynska-Zawadzka B, Kunz JB, Sedek L, Kosmalska M, Zdon K, Biecek P, et al. PTEN abnormalities predict poor outcome in children with T-cell acute lymphoblastic leukemia treated according to ALL IC-BFM protocols. Am J Hematol 2019;94(4):E93–E6 doi 10.1002/ajh.25396. [DOI] [PubMed] [Google Scholar]
- 58.Rusiniak ME, Kunnev D, Freeland A, Cady GK, Pruitt SC. Mcm2 deficiency results in short deletions allowing high resolution identification of genes contributing to lymphoblastic lymphoma. Oncogene 2012;31(36):4034–44 doi 10.1038/onc.2011.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brunetti L, Gundry MC, Kitano A, Nakada D, Goodell MA. Highly Efficient Gene Disruption of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. J Vis Exp 2018(134) doi 10.3791/57278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008;453(7191):110–4 doi 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 61.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 2016;48(1):4–6 doi 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 2017;49(8):1211–8 doi 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481(7380):157–63 doi 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016;8(3):389–99 doi 10.2217/epi.15.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10(12):1213–8 doi 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 2004;14(4):708–15 doi 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rocha PP, Raviram R, Fu Y, Kim J, Luo VM, Aljoufi A, et al. A Damage-Independent Role for 53BP1 that Impacts Break Order and Igh Architecture during Class Switch Recombination. Cell Rep 2016;16(1):48–55 doi 10.1016/j.celrep.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson JT, Turner D, Durand NC, Thorvaldsdottir H, Mesirov JP, Aiden EL. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst 2018;6(2):256–8 e1 doi 10.1016/j.cels.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schofield EC, Carver T, Achuthan P, Freire-Pritchett P, Spivakov M, Todd JA, et al. CHiCP: a web-based tool for the integrative and interactive visualization of promoter capture Hi-C datasets. Bioinformatics 2016;32(16):2511–3 doi 10.1093/bioinformatics/btw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo K, McMinn JE, Ludwig T, Yu YH, Yang G, Chen L, et al. Disruption of peripheral leptin signaling in mice results in hyperleptinemia without associated metabolic abnormalities. Endocrinology 2007;148(8):3987–97 doi 10.1210/en.2007-0261. [DOI] [PubMed] [Google Scholar]
- 73.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34(5):525–7 doi 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 74.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11(10):R106 doi 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell 2019;177(7):1888–902 e21 doi 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bais AS, Kostka D. scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics 2020;36(4):1150–8 doi 10.1093/bioinformatics/btz698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 2019;16(12):1289–96 doi 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gal-Oz ST, Maier B, Yoshida H, Seddu K, Elbaz N, Czysz C, et al. ImmGen report: sexual dimorphism in the immune system transcriptome. Nat Commun 2019;10(1):4295 doi 10.1038/s41467-019-12348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019;20(2):163–72 doi 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.