

Abstract
Unlike mammals, teleost fish can regenerate an injured retina and restore lost visual function. Although retina regeneration has been studied for decades little is known of the molecular events that govern it. We previously showed that in the damaged zebrafish retina Müller glia re-enter the cell cycle, increase α1tubulin (α1T) promoter activity and generate new neurons and glia for retinal repair. Here we report the identification of an E-box in the α1T promoter that is necessary for its induction during retina regeneration. We show that the proneural basic helix-loop-helix transcription factor achaete-scute complex-like 1a (ascl1a) transactivates the α1T promoter via this particular E-box. More importantly, we show that ascl1a is essential for retina regeneration. Within 4 hrs following retinal injury ascl1a is induced in Müller glia. Knockdown of ascl1a expression in the injured retina blocks the induction of the regeneration markers α1T and Pax6, as well as Müller glial proliferation, consequently preventing the generation of retinal progenitors and their differentiated progeny. These data suggest ascl1a is required to convert quiescent Müller glia into actively dividing retinal progenitors, and that ascl1a is a key regulator in initiating retina regeneration.
Keywords: ascl1a, basic helix-loop-helix, Müller glia, stem cells, regeneration, retina, zebrafish, tubulin, E-box
Introduction
Recent studies suggest that following retinal injury Müller glia can dedifferentiate and function as retinal stem cells in mammals, birds and fish1-3. However, unlike mammals and birds where the capacity of Müller glia to regenerate new neurons is extremely limited2,3, teleost fish mount a robust regenerative response that not only regenerates all damaged retinal neurons1,4,5 but also results in restoration of visual function6. Therefore teleost fish, such as zebrafish, provide an ideal model system for identifying the molecular mechanisms underlying a robust regenerative response following retinal injury and may suggest new strategies for repairing the damaged mammalian retina.
Numerous genes that are either expressed in retinal progenitors or are thought to be involved in maintenance of stem cells are induced in the zebrafish retina in response to injury including: pax61,7, notch and cadherin8,9, delta, vsx and rx8, olig25, and stat310. Low levels of pax6 protein have been detected in Müller glia11, but surprisingly little is known about what role pax6 and other genes play in regulating the response of Müller glia to retinal injury and whether they are necessary for regeneration. Investigations of the signaling pathways that regulate Muller glia proliferation have shown that FGF and insulin signaling in the chick12 and wnt/β-catenin signaling in the rodent13 can influence Müller glia proliferation. In addition retinal disease or injury stimulates endothelin signaling between photoreceptors and Müller glia14. Whether any of these gene or signaling pathways participate in transforming Müller glia into retinal progenitors is not currently known.
Our lab developed a transgenic zebrafish model to study central nervous system (CNS) regeneration15. Using this model we found α1T promoter activity is dramatically increased in Müller glia following retinal injury1,16. We also showed that Müller glia are a source of multipotent progenitors which contribute newborn cells toward regeneration1. In order to identify genes that regulate this process, we focused on DNA elements that mediate α1T transgene expression in proliferating Müller glia. Here we report the identification of an E-box that is required for α1T transgene expression in vivo and provide evidence that the basic helix-loop-helix transcription factor, ascl1a, regulates the α1T promoter via this E-box. To investigate the role ascl1a plays in the process of retina regeneration, we developed a technique to inhibit gene expression in the injured retina using antisense morpholino oligonucleotides. As suggested by our in vitro studies, we found that ascl1a induction is necessary for α1T promoter activation in Müller glia following retinal injury. Most importantly, we found that ascl1a is induced within 4 hrs following retinal injury and knockdown of ascl1a prevents induction of α1T and pax6, in addition to inhibiting Müller glia from proliferating in response to injury, thereby preventing retina regeneration. To our knowledge, this is the first gene to be identified that is required for retina regeneration.
Results
An E-box is required for α1T promoter activation in proliferating Müller glia following retinal injury
The −1696α1T:GFP transgene is induced in proliferating Müller glia following retinal injury1. To begin to identify the mechanism by which Müller glia respond to injury-induced signals and produce a dividing population of retinal stem cells, we wanted to identify α1T promoter elements mediating its induction following retinal injury. We hypothesized that these elements would lead us to transcription factors which regulate retina regeneration. Transgenic fish harboring different α1T promoter deletions driving GFP expression were screened for transgene expression in proliferating Müller glia following retinal injury. This screen identified Δ-1046-846α1T:GFP and −907α1T:GFP as transgenes that no longer responded to retinal injury by inducing GFP expression in proliferating Müller glia (Fig. 1). This lack of expression is not due to promoter inactivation since these promoters can drive transgene expression during development17 (and unpublished observations). These results suggest that a DNA element located between nucleotides −1016 and −907 of the α1T promoter is required for transgene expression in proliferating Müller glia.
Figure 1. A 109 bp region of the α1T promoter is required for transgene expression in dedifferentiating Müller glia.

(a) Schematic representation of α1T promoter constructs. The bars represent promoter sequence and the numbers indicate relative position from the start codon. −1696 is the wild-type promoter described previously15,16. Δ-1046-846 has been described17. The −1016 promoter directs transgene expression in Müller glia1. The −907 promoter lacks 789 bp of upstream sequence. (b) Transgenic fish received retinal injuries on day 0 and were given a 4 hr pulse of BrdU at 4 days post injury. Transgenic fish which carry the required DNA element express GFP in BrdU labeled Müller glia (−1016 panel, arrows), while transgenic fish lacking the element do not (Δ-1046-846 and −907 panels). Two independent lines of Δ-1046-846 and three independent lines of −907 transgenic fish all display a lack of GFP expression in BrdU labeled cells (arrowheads in Δ-1046-846 panels). The images for −1016 and Δ-1046-846 are from the same sections. Because the −907 transgenic fish display very weak GFP expression in general, we used serial sections to obtain the −907 images. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
To further narrow-in on this element we searched for potential transcription factor binding sites within this 109 bp region by performing gel electrophoretic mobility shift assays. Radiolabeled oligonucleotide probes spanning this region combined with nuclear extracts prepared from zebrafish or rat brain (Fig. 2a) or zebrafish retina (Fig. 2d) revealed specific binding to probe 4 (Fig. 2a, arrow) which contains a single E-box sequence CATGTG (Fig. 2b). Probes harboring 2 bp mutations within the E-box (Fig. 2b, probes 4-3 and 4-4) disrupted protein binding (Fig. 2c, lanes 3 and 4) while mutations in surrounding nucleotides (Fig. 2b, probes 4-1, 4-2, and 4-5) had no effect on binding (Fig. 2c, lanes 1, 2 and 5). In addition, E-box mutations prevented unlabeled probes from competing for binding with wild-type radiolabeled probes even at 50-fold molar excess (Fig. 2c, lanes 8 and 9). Although nuclear extracts prepared from brain or retina bound probe 4 (Fig. 2), those prepared from rat liver, fish gill, fish muscle and fish kidney did not (data not shown). Interestingly, probe Eb (Fig. 2e) which contains an E-box (CAGATG) that is not required for transgene expression following retinal injury but is required for transgene induction in RGCs following optic nerve crush16 does not bind nuclear extracts from zebrafish brain (Fig. 2e, lanes 4-6). These results suggest that the binding we observe is specific to the E-box at position −954.
Figure 2. An E-box within the 109 bp region that is required for transgene expression in Müller glia binds nuclear extracts from zebrafish and rat brain and zebrafish retina.

(a) Electrophoretic mobility shift assay (EMSA) using a probe from the 109 bp region binds nuclear extracts from zebrafish and rat brain. The arrow indicates specific binding. Cold indicates where 50-fold molar excess unlabeled probe was added as competition. Extract indicates whether zebrafish (zf) or rat brain extracts were added. (b) Nucleotide sequence of the probes used for EMSA. The E-box is outlined in probe 4 with a box. Mutations are indicated by italicized and underlined text. (c) Mutations to the E-box (lanes 3 & 4) render the probe unable to bind nuclear extracts from zebrafish brain, while mutations to non-E-box nucleotides do not affect binding (lanes 1, 2 and 5). Probes correspond to those shown in (b). Unlabeled mutant probes compete with wild-type probe binding when the E-box is intact (lanes 6, 7 and 10), but not when the E-box is mutated (lanes 8 and 9), even at 50-fold molar excess. (d) Nuclear extracts from zebrafish retina bind specifically to the E-box. (e) An E-box probe from a different region of the promoter (Eb) is unable to compete with the E-box from probe 4 (lane 3), and does not bind to zebrafish brain nuclear extracts (lane 4).
Because the E-box resides within the 109 bp region required for transgene expression in proliferating Müller glia following retinal injury, we hypothesized that this E-box is necessary for transgene expression in these cells. To test this hypothesis, we placed the 2 bp mutation from probe 4-4 (Fig. 2b) in the full length −1696α1T promoter to create TG-954CAα1T:GFP transgenic zebrafish (Fig. 3a). Three independent lines of transgenic fish were identified and examined for transgene expression during development. The TG-954CA promoter directs transgene expression to the brain, spinal cord and retina during development, which is similar to the wild-type promoter (Supplementary Fig. 1). We previously reported that mature fish harboring the −1696α1T:GFP or −1016α1T:GFP transgene express GFP in retinal stem cells located at the circumferential germinal zone (CGZ)1,15 where new retinal cells are continually added as the eye grows. Interestingly, TG-954CAα1T:GFP fish do not express the transgene in the CGZ (Supplementary Fig. 2), suggesting the E-box is required for transgene expression in retinal stem cells. Consistent with the idea that this particular E-box is essential for α1T expression in adult retinal stem cells is our observation that following retinal lesion TG-954CAα1T:GFP fish do not express GFP in proliferating Müller glia (Fig. 3b). Occasionally we noticed GFP+ cells in the ganglion cell layer following retinal lesion (Fig. 3b, arrows). These appeared to be axotomized retinal ganglion cells which are known to induce expression from the −1696α1T:GFP transgene16,17. We confirmed this in TG-954CAα1T:GFP fish by observing robust GFP expression in retinal ganglion cells following optic nerve crush (Supplementary Fig. 3). Therefore, the identified E-box is required for transgene expression in adult retinal stem cells residing in either the retinal periphery (CGZ) or in the central retina following injury (proliferating Müller glia).
Figure 3. Mutation of −954 E-box in the wild-type −1696 α1T promoter prevents transgene induction in proliferating Müller glia following retina injury.

(a) Schematic representation of α1T promoter:GFP constructs. The wild-type −1696α1T:GFP transgene (−1696) and the 2 bp mutation created in the E-box at position −954 in the −1696 construct (TG-954CA) are indicated. (b) Three independent lines of transgenic fish received retinal injuries on day 0 and were given a 4 hr pulse of BrdU at 4 days post injury. Note that none of the lines harboring the 2 bp mutation induced transgene GFP expression in BrdU-labelled Müller glia (arrowheads). See Fig. 1 for comparison to the wild-type −1696α1T:GFP transgene response. The arrows in the lowest right GFP panel show transgene induction in retinal ganglion cells whose axons were presumably lesioned when we injured the retina in this fish.
Ascl1a is rapidly induced in Müller glia following retinal injury
The results described above suggest basic helix-loop-helix transcription factors may be crucial for retina regeneration. One such protein, ascl1a, attracted our attention because: 1) the chick homolog of ascl1a is expressed in the injured chick retina2; 2) ascl1a is induced during regeneration of the zebrafish retina; although its spatial and early temporal expression pattern was not characterized10,18; and 3) the mouse homolog of ascl1a is expressed in retinal19 and neural progenitors20,21. To determine whether ascl1a was induced in proliferating Müller glia, we examined injured retinas from −1016α1T:GFP fish for ascl1a expression. We observed that ascl1a expressing cells correspond precisely to GFP+ Müller glia at 4 dpi (Fig. 4a-c) which we previously identified as proliferating Müller glia-derived retinal progenitors1.
Figure 4. Ascl1a is induced in proliferating Müller glia following retinal injury.

(a-c) Ascl1a expression is detected by in situ hybridization (a) in GFP+ Müller glia (b) at 4 days post injury (arrowheads). Panel (c) shows the merge image of panels (a) and (b). (d-g) In situ hybridization for ascl1a from 6-24 hours post injury (hpi). Ascl1a is not expressed in the uninjured retina (d). Ascl1a is induced in cells of the INL at 6 hpi (arrowheads in e). Ascl1a expression gradually increases and is easily detected at 24 hpi (arrowheads in g). (h-m) Ascl1a in situ hybridization (red, panels h and k) and glutamine synthetase immunostaining (green, panels i and l) show ascl1a is induced in Müller glia at 6 hpi (arrows in merge panel j) and 24hpi (arrows in merge panel m). DAPI nuclear staining is shown in the merged panels. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
To determine if ascl1a expression precedes Müller glia proliferation and α1T induction which begin around 24 hrs post-injury (hpi)1, we examined retinas at 6, 12, 18 and 24 hpi for ascl1a expression by in situ hybridization (ISH). We detected a low level of expression at 6, 12 and 18 hpi and stronger expression at 24 hpi (6, 12 and 24 hpi shown in Fig 4d-g). Co-localization of ascl1a and glutamine synthetase at these early time points confirmed ascl1a-expressing cells are Müller glia (Fig 4h-m). These results indicate that ascl1a is induced in Müller glia at least 18 hours before they enter the cell cycle and begin expressing the α1T transgene.
To put ascl1a and α1T gene induction in the context of other genes induced in the injured retina which are known to be involved in retina development and induced during retina regeneration, we compared ascl1a and α1T expression with pax6, delta, notch and olig2 at various times after retinal injury using ISH. The results of this analysis are summarized in Supplementary Table 1 and demonstrate ascl1a is the earliest induced gene in the injured retina. Ascl1a was first detectable at 4 hpi. Its expression increased at 24 hpi and reached peak levels by 48 hpi. Surprisingly, delta, notch and olig2 are induced between 48 and 96 hpi, well after ascl1a and α1T induction. Because pax6 is expressed by amacrine cells7 it was difficult to detect its induction until ectopic expression appeared in the inner and outer nuclear layers at 96 hpi. This result is consistent with the observation that not all GFP+ Müller glia induce pax6 at 4dpi1, suggesting pax6 induction in Müller glia occurs after GFP induction. Although low levels of pax6 protein have been reported in Müller glia11, we did not detect pax6 induction in transgene expressing Müller glia at 6 or 24 hpi (Supplementary Fig. 4).
Ascl1a regulates α1T expression
Because injury-induced expression of the −1696α1T:GFP transgene requires an E-box at position −954 and E-boxes bind basic helix-loop-helix transcription factors like ascl1a, we suspected ascl1a would regulate α1T promoter activity via its −954 E-box. Consistent with this idea, we found that ascl1a transactivated a minimal β-globin promoter harboring 3 copies of the −954 E-box (CATGTG) but did not transactivate the β-globin promoter with 3 copies of a mutant E-box (CATGCA) (Fig. 5a). In addition, mutation of this E-box in the context of the −1696α1T promoter also blocked transactivation by ascl1a (Fig. 5b), although transactivation was less dramatic, possibly due to high basal expression because of endogenous transcriptional activators which act at sites other than the E-box. We were unsuccessful in demonstrating a direct interaction between ascl1a and the −954 E-box using gel electrophoretic mobility shift assasys (unpublished observations) which may suggest that ascl1a activation of the α1T promoter is indirect.
Figure 5. Ascl1a regulates the α1T promoter through the −954 E-box.

(a) Luciferase reporter vectors with either a minimal β-globin promoter alone (light grey bars), or three copies of the −954 E-box (dark grey bars) or the mutant E-box from probe 4-4 (see Fig. 2b) (black bars) were transfected in combination with ascl1a into HEK293T cells. Ascl1a transactivates the reporter when a functional E-box is present, but not when the E-box is mutated. (b) Full-length α1T constructs harboring either the wild-type (−1696) or mutant E-box promoters were transfected into HEK293T cells with or without ascl1a. Ascl1a induces reporter expression when the E-box is intact, but not when the E-box is mutated. Error bars indicate standard error of the mean for three replicates.
Despite the lack of evidence that ascl1a can bind the −954 E-box, the very early induction of ascl1a following retinal injury and its ability to regulate α1T promoter activity suggest it may regulate α1T transcription in vivo. In order to test this hypothesis we suppressed ascl1a expression in Müller glia following retinal injury. To do this we electroporated injured adult retinas with lissamine-labeled antisense morpholino oligonucleotides (MOs) that have been previously demonstrated to knockdown ascl1a expression when injected into single cell zebrafish embryos22. We compared the effects of control or ascl1a MOs on transgene expression in our −1016α1T:GFP transgenic fish. Normally, GFP+ Müller glia are visible at 2 dpi, and by 4 dpi, there are numerous GFP+ Müller glia (Fig. 1, −1016 panel). It was readily apparent that ascl1a knockdown suppressed transgene expression in Müller glia following retinal injury at both 2 and 4 dpi (4 dpi shown in Fig. 6). Specificity was confirmed using a second MO (ascl1a5′UTR) which gave similar results. We counted the number of lissamine+/GFP+ cells in the inner nuclear layer (INL) and outer nuclear layer (ONL) by analyzing confocal images and found that 13% of the control MO-treated cells expressed GFP at 2 dpi while only 4% the ascl1a MO treated cells expressed GFP, (three retinas were counted for each group, p=0.02). At 4 dpi, 21.6% of the control MO treated cells expressed GFP while only 2.3% and 2.4% of the ascl1a and ascl1a5′UTR MO treated cells expressed GFP respectively (three retinas were counted for each group, p=0.03 for the ascl1a MO and p=0.04 for the ascl1a5′UTR MO). These quantitative data confirm our qualitative observations and suggest ascl1a is required for α1T:GFP transgene expression in vivo.
Figure 6. Ascl1a is required for transgene expression in vivo.
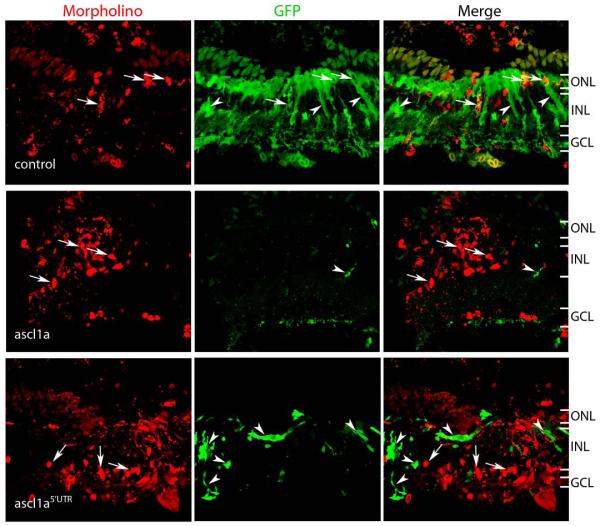
−1016 transgenic fish received retinal injuries and morpholino treatment on day 0. Eyes were harvested on day 4. (a) Control morpholino treatment has no effect on transgene expression in Müller glia (arrows) and there are many GFP+ Müller glia (arrowheads). Morpholinos targeting ascl1a (ascl1a and ascl1a5′UTR panels) prevent transgene expression in treated cells (arrows), and cause reduced transgene expression in general, although Müller glia that did not receive ascl1a morpholinos are able to express GFP (arrowheads). ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Since ascl1a knockdown prevented the α1T transgene from being induced in response to injury, we wondered whether endogenous gene induction was also blocked. For these experiments we used our −1016α1T:GFP transgenic fish so we could monitor transgene GFP expression by fluorescence microscopy and endogenous α1T expression by in situ hybridization in control and ascl1a MO treated retinas at 4 dpi. While the control MO had no effect on transgene expression or endogenous α1T induction (Fig. 7a-c), ascl1a knockdown almost completely abolished transgene and endogenous α1T expression in the morpholino treated portion of the injured retina (Fig. 7d-f).
Figure 7. Ascl1a knockdown prevents induction of endogenous α1T and pax6.

Retinas from −1016 transgenic fish were injured and electroporated with lissamine-labeled morpholinos (MO) on day 0 and harvested on day 4. (a-f) α1T expression detected by in situ hybridization (ISH) is shown as a dark brown deposit (a, d), native GFP expression is shown in green (b, e) and lissamine labeled MO is shown in red (b, e). The injury site is marked by an asterisk. (a-c) Control MO treatment does not affect endogenous or transgene α1T induction (arrows). (d-f) Ascl1a knockdown prevents endogenous and transgene α1T induction. Note the lack of α1T and GFP expression between the asterisk and the arrows, where the retina is treated with MO. Where the retina did not receive MO, α1T and GFP are expressed (arrows). (g-l) Pax6 expression detected by ISH is shown as a blue/purple deposit, native GFP expression is shown in green and MO is shown in red. (g-i) Control MO treatment does not affect pax6 induction at 4dpi. Arrows indicate pax6+/MO+/GFP+ Müller glia. (j-l) Ascl1a MO treatment prevents pax6 induction. Arrowheads indicate MO+/pax6− cells. Rare GFP+ cells are sometimes present but were not treated with MO (arrow).
Pax6 expression in injury-induced proliferating Müller glia is used as an indication that these cells have become multipotent progenitors. However pax6 is also expressed by amacrine cells located in the INL (Fig. 7j). Therefore, to determine whether ascl1a knockdown prevented pax6+ progenitors from being generated, we compared ectopic pax6 expression in the INL and ONL of control and ascl1a MO treated/injured retinas by ISH (Fig. 7g-i). Interestingly, ascl1a MO treated retinas exhibited a dramatic reduction in ectopic pax6 expression (Fig 7j-l) compared to control MO treated retinas (Fig. 7g-i). The observation that pax6 expression persisted in amacrine cells of the ascl1a MO treated/injured retina (Fig. 7i) suggests that these cells control pax6 expression in an ascl1a-independent manner and demonstrate the ascl1a MOs are not causing a general decrease in gene expression.
Ascl1a is required to convert Müller glia into multipotent progenitors
The early induction of ascl1a following retinal injury and its requirement for α1T expression suggests ascl1a induction may be necessary for retina regeneration to occur. One of the hallmarks of retina regeneration is the production of a cycling population of retinal progenitors derived from Müller glia. To determine if ascl1a induction is necessary for the generation of this cycling progenitor pool, we delivered ascl1a-targeted or control MOs to Müller glia in the injured retina. Since the vast majority of BrdU-labeled cells in the injured retina are derived from Müller glia1, MO+/BrdU+ cells represent injury-induced retinal progenitors. We therefore injured and electroporated −1016:GFP transgenic retinas with control or ascl1a MOs and labeled dividing cells by housing the fish in BrdU-treated water from 36-60 hours, when Müller glia begin dividing. We harvested the retinas at 10 dpi and found that in control MO treated retinas, many MO+/BrdU+ cells could be identified (Fig. 8); however, we did not detect MO+/BrdU+ cells in ascl1a MO treated retinas (Fig. 8). In fact, we observed an almost complete absence of BrdU+ cells in the INL, suggesting ascl1a MO treatment prevents proliferation or prevents proliferating cells from surviving. To determine whether ascl1a knockdown blocks proliferation, we examined whether ascl1a MOs block the appearance of proliferating cells at 2dpi. Here also the control MO had no effect on proliferation, while ascl1a MOs prevented Müller glia from entering the cell cycle (Supplementary Fig. 5). Although the presence of ascl1a MO treated cells at 10 dpi suggests these MOs do not cause cell death, we examined whether any MO treated cells were caspase+ at 2 or 4 dpi and did not detect MO+/caspase+ cells, further suggesting ascl1a MOs do not cause cell death (data not shown). Taken together, these results suggest ascl1a knockdown prevents Müller glia from entering the cell cycle following injury.
Figure 8. Ascl1a is required for proliferation of Müller glia.

Morpholinos were electroporated into injured −1016 retinas on day 0 and fish were housed in BrdU treated water from 36-60 hours post injury to label dividing cells. The fish recovered until day 10 when eyes were harvested. The control morpholino does not prevent treated cells from labeling with BrdU (arrowheads, top 3 control panels). Morpholinos targeting ascl1a (ascl1a and ascl1a5′UTR) prevent cells from labeling with BrdU (arrows in panels ascl1a and ascl1a5′UTR). Some cells are able to proliferate, but were not treated with morpholino (arrowheads panels ascl1a and ascl1a5′UTR). ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Discussion
Zebrafish provide an ideal system for exploring the mechanisms underlying nervous system regeneration because of their robust regenerative response to injury and their amenability to experimental manipulation. We have generated transgenic zebrafish models for studying regeneration in the zebrafish nervous system and these models have further facilitated our ability to study the regenerative process 1,15-17. In particular we have used the α1T promoter driving GFP expression as a reporter of the regenerative response. Transgenic fish harboring wild-type or mutant α1T promoters led us to a critical E-box that is necessary for transgene induction in retinal progenitors following injury. Investigation of the proteins that may activate the α1T promoter via this E-box led us to ascl1a which proved to be a critical regulator of not only α1T promoter activity but also the regenerative response itself. Ascl1a is one of the earliest gene inductions in the injured retina, preceding α1T, olig2, pax6, notch and delta by at least 18 hrs. Based on their temporal expression pattern most of these latter genes may regulate cell type specification. Delta-notch signaling has long been known to regulate cell fate decisions in drosophila23, and in zebrafish notch signaling regulates neuronal cell type specification in the spinal cord24. Pax6 has also been shown to play a role in specifying certain retinal cell types since its removal at optic cup stages reduces the formation of most retinal neurons25.
Ascl1a regulates α1T transgene expression in proliferating Müller glia
We identified a critical E-box (CATGTG) at position −954 within the α1T promoter that is necessary for transgene expression in proliferating Müller glia following retinal injury. Although we demonstrated that this E-box mediates ascl1a-dependent promoter induction we were unable to show a direct interaction between ascl1a and this E-box in gel electrophoretic mobility shift assays. This may suggest the conditions we used are not appropriate for binding, or that ascl1a has an indirect effect on α1T promoter activity. Although we cannot distinguish between these possibilities we note that the critical E-box in the α1T promoter deviates from the consensus mouse achaete-scute complex-like 1 (Ascl1) binding site (CAGCTG)26. In addition, α1T activation in the injured retina lags behind ascl1a induction by at least 14 hrs suggesting ascl1a may induce expression of other basic helix-loop-helix proteins or must cooperate with later induced proteins to bind the α1T promoter's −954 E-box. Nonetheless, knockdown of ascl1a in the injured retina dramatically suppressed α1T:GFP transgene and endogenous α1T expression. By using two different MOs targeting different ascl1a sequences we ensure specificity of these results. Therefore, we conclude that ascl1a regulates α1T expression in proliferating Müller glia.
Ascl1a activates quiescent Müller glia to become multipotent progenitors
Müller glia appear to be quiescent stem cells which respond to injury by generating multipotent retinal progenitors. α1T transgene expression, cell cycle re-entry and expression of other injury-induced genes are all indications that multipotent progenitors have been formed. What causes these slowly cycling stem cells to become actively dividing retinal progenitors? Although the signaling mechanisms are still unknown, the results presented here suggest ascl1a expression is necessary for converting quiescent Müller glia into actively proliferating progenitors. In mice, Ascl1 specifies neural precursors in the brain and olfactory epithelium27,28 and seems to play a role in directing stem cells to differentiate into neural progenitors20. We suspect ascl1a expression in Müller glia initiates a similar transition from a stem cell state into an actively dividing progenitor state since inhibition of ascl1a expression in the injured retina blocks Müller glia proliferation. Ascl1a may commit Müller glia to become injury-induced progenitors by initiating expression of target genes such as α1T. There are likely other genes which are regulated by ascl1a which may include regulators of the cell cycle including cyclin family members10. Interestingly, there is a CATGTG E-box in the zebrafish cyclinD1 promoter at roughly the same position as the α1T E-box (data not shown).
The rapid induction of ascl1a in Müller glia after retinal injury suggests it may be regulated by transcription factors already present in Müller glia. One such candidate is Stat3 which is expressed in quiescent Müller glia and may be activated by injury-induced extracellular cues10. Interestingly, FGF can cause Müller glia to re-enter the cell cycle in the absence of injury12 and ascl1a appears to act downstream of FGF signaling in developing zebrafish pituitary gland29,30. Another potential regulator is the wnt signaling pathway. In rats, adding wnt3a to retinal explant cultures increases Müller glia proliferation13. Wnt signaling is mediated by nuclear localization of β-catenin which activates genes harboring TCF binding sites. Whether wnt3a/β-catenin mediates its effect via ascl1a is not known. Future studies will begin to address the role these signal transduction cascades play in regeneration of the zebrafish retina.
Experimental Procedures
Animals
The animals used in this study were treated in accordance with the guidelines of the University Committee on Use and Care of Animals at the University of Michigan. Fish were obtained from our breeding colony and raised with a 14:10 light/dark cycle at a temperature of 28°C.
Transgenic zebrafish
1696α1TIpEGFP, del1046-846α1TIpEGFP, and 1016α1TIpEGFP transgenic fish have been described previously1,15,17. −907α1TIpEGFP and TG-954CAα1TIpEGFP constructs were resuspended in injection buffer, and single-cell zebrafish embryos were injected, raised to adulthood and screened for transgenic progeny as previously described15,17.
Optic nerve lesions
Fish were anesthetized in 0.02% tricaine methane sulfonate (Sigma, St. Louis, MO) before surgery. Optic nerve crushes were performed as described previously1,16,31.
Eye lesions and Morpholino mediated gene knockdown
Eye lesions were performed as described previously1,16. To deliver morpholinos to the injured retina, a 30-gauge needle was attached to a Hamilton syringe (Hamilton, Reno, NV) containing 1mM morpholino (Gene Tools, Philomath, OR). Approximately 0.5 μl was injected into the vitreous after inserting the needle to the length of the bevel. We used the following lissamine labeled morpholinos: Control MO 5′CCTCTTACCTCAGTTACAATTTATA-3′; ascl1a MO, 5′ATCTTGGCGGTGATGTCCATTTCGC-3′; ascl1a5′UTR MO, 5′AAGGAGTGAGTCAAAGCACTAAAGT-3′ (The latter two MOs have been described previously as ash1a MOs22). Custom electrodes were then placed across the head of the fish with the cathode on the left eye and the anode on the right eye. An ECM 830 Electro Square Porator (BTX, San Diego, CA) was used to deliver 5 consecutive 50 ms pulses at 70 V with a 950 ms interval between pulses. The uninjected eye served as a negative control. One observer assigned letters to control and ascl1a MOs. A second observer then electroporated these MOs into fish and assigned the fish with numbers. This way, both observers could score MO treated cells for GFP expression without any bias. For cell counts, morpholino treated cells were scored for either GFP or BrdU labeling. The total number of morpholino labeled cells that were labeled with GFP or BrdU was tallied for each retina to determine the percentage of MO treated cells which also labeled with GFP or BrdU. The percentage of double labeled cells for each group was calculated by taking the average from three retinas. A student's T-test, assuming equal variance, was used to calculate p values.
Bromodeoxyuridine labeling
To identify dividing cells, fish were either given a single injection of BrdU as described1 or housed in 10mM BrdU for 24 hours (from 24-48 or 36-60 hpi). Fish were transferred to tanks with fresh water and killed at various times after BrdU administration to harvest the retinas.
Tissue preparation
Fish were given an overdose of tricaine methane sulfonate and eyes from adult fish were dissected, enucleated, fixed and sectioned as previously described1,16.
Immunohistochemistry
Immmunohistochemistry was performed as previously described1,16. The following primary antibodies were used: rat anti-BrdU (dividing cell marker; 1:250; Harlan; Sera-Lab); rabbit anti-GFP (1:1000; Molecular Probes, Eugene, OR); and mouse anti-glutamine synthetase (GS; glial marker;) (1:500; Chemicon).
In situ hybridization
In situ hybridizations were performed with digoxigenin labeled cRNA probes as described33. Ascl1a was a gift from Eric Weinberg, University of Pennsylvania; Pax6 cRNA was prepared from a full length Pax6a cDNA clone (Open Biosystems, Huntsville, AL); α1T probe was described previously31; notch1b and notch3 were a gift from Michael Lardelli, University of Adelaide; deltaA, deltaD and olig2 were a gift from Bruce Appel, Vanderbilt University; and deltaB was a gift from Julian Lewis, University College London. For timeline expression analysis (Supplemental Table 1), notch3 hybridization was done individually and in combination with notch1b; similar results were obtained in each case. Delta in situ hybridization was done with deltaA individually and in combination with deltaB and deltaD; similar results were obtained in each case. Negative results were repeated twice for the 24 and 48 hpi time points except for olig2.
Imaging
Slides were examined using a Zeiss (Oberkochen, Germany) Axiophot or Olympus Fluoview FV1000 laser scanning confocal microscope. Images were captured using a digital camera adapted onto the Axiophot microscope or Olympus confocal microscope. Images were processed and annotated with Adobe Photoshop CS.
Vectors for creating transgenic zebrafish
−907α1TIpEGFP expression vector contains 907 bp of 5′ flanking α1-tubulin DNA, exon 1, and the first intron fused in frame to the GFP sequence. This promoter fragment is similar to the full-length −1696α1TIpEGFP expression vector from previous work15-17 except that it is lacking 789 bp from the 5′ end. A 2 bp mutation (TG-CA) was introduced into the full length α1T promoter by amplifying a PCR product containing the 2bp E-box mutation and cloning it into the α1TIpEGFP vector. The TG-954CAα1TIpEGFP construct is identical to the −1696 expression vector except for the TG:CA mutation at position −954.
Vectors for in vitro assays
Luciferase reporter vectors are based on the pXP2 construct32. The α1TpXP2 reporter (wild-type) contains 1696 bp of 5′ flanking α1T sequence and its first exon cloned in frame and upstream of the luciferase coding sequence. The TG-954CAα1T-pXP2 vector is identical to the wild-type vector except for a 2 bp substitution at position 954 (TG-CA). The E-box-pXP2 vector contains 3 copies of the α1T E-box at position 954 (5′CATGTG 3′) upstream of a minimal β-globin promoter in pXP2. The mE-box-pXP2 vector is identical to the E-box-pXP2 vector except each E-box has a 2 bp mutation (CATGTG to CATGCA). The coding sequence of ascl1a was cloned into pCS2+ to create pCS2+ascl1a. pCS2+β-globin was a gift from Audrey Seasholtz, (University of Michigan, Ann Arbor, Michigan).
Transactivation Assays
HEK293T cells were plated in 24 well plates 24 hr before transfection. Cells were transfected via calcium phosphate DNA precipitation. Luciferase assays were performed in duplicate for each sample and values were normalized to β-globin expression.
Nuclear Extracts
Nuclear extracts were isolated from zebrafish brain, retina, and HEK293T transfected cells using standard protocols.
Electophoretic Mobility Shift Assays
Oligonucleotide probes labeled with 32P-dCTP (ICN, San Diego, CA) were incubated with 5-10μg of nuclear extracts prepared from: zebrafish brain or retina, rat brain, or with in vitro synthesized proteins using TNT Coupled Transcription/Translation system (Promega, Madison, WI). Cold oligonucleotides were incubated at >50 fold excess to test binding specificity. The protein:DNA mix was then run on a non-denaturing polyacrylamide gel and DNA migration visualized by exposing the gel to Kodak imaging film.
Supplementary Material
(a) The wild-type promoter directs GFP expression in the eye, brain and spinal cord of two day old zebrafish larvae. (b-d) Transgenic fish harboring a mutation to the E-box also express GFP in the eye, brain and spinal cord, although the expression is not as intense and uniform. (e) Immunostaining indicates GFP is expressed in the developing retina of wild-type transgenic fish at 3 days. (f-h) GFP expression in TG-945CA transgenic retinas is similar to wild-type GFP expression.
Retinas from TG-954CA transgenic fish received a single injection of BrdU 4hr prior to harvesting the retina. Sections were stained for GFP (green), BrdU (red) and DAPI (blue). The BrdU labeled cells are proliferating at the CGZ. Note the lack of GFP expression in BrdU labeled cells. The dotted line indicates the edge of the retina.
GFP is induced in ganglion cells six days after optic nerve crush in all three lines of transgenic fish harboring the −954 E-box mutation (arrows in GFP panel). DAPI staining is shown in blue. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Injured retinas were examined at 6 and 24 hpi for induction of pax6 by in situ hybridization (red). Müller glia are shown by glutamine synthetase immunostaining (green). At these early time points, pax6 expression does not correspond to glutamine synthetase+ Müller glia.
Retinas from 1016α1T:GFP transgenic fish were injured and electroporated with control or ascl1a MOs on day 0 and housed in BrdU treated water from 24-48 hpi at which time the retinas were harvested. Cells treated with control MO also labeled with BrdU (arrowheads, top 3 panels) while cells treated with ascl1a MO did not (lower 3 panels).
Acknowledgements
We thank members of the Goldman lab along with Professors Ben Novitch and Dave Turner for comments and suggestions on this work. We would also like to thank Eric Weinberg, Matthias Hammerschmidt, Bruce Appel, Julian Lewis, Michael Lardelli, and Audrey Seasholtz for providing cDNA clones. This work was supported by a research grant from the Michigan Life Sciences Corridor, MEDC-38, a grant from NEI and a grant from the University of Michigan EBS awarded to DG. BVF was partially supported by a NIH Vision Research Training Grant.
References
- 1.Fausett BV, Goldman D. A role for alpha1 tubulin-expressing Müller glia in regeneration of the injured zebrafish retina. J. Neurosci. 2006;26:6303–6313. doi: 10.1523/JNEUROSCI.0332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer AJ, Reh TA. Müller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat. Neurosci. 2001;4:247–252. doi: 10.1038/85090. [DOI] [PubMed] [Google Scholar]
- 3.Ooto S, et al. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. PNAS. 2004;101:13654–13659. doi: 10.1073/pnas.0402129101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vihtelic TS, Hyde DR. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J. Neurobiol. 2000;44:289–307. doi: 10.1002/1097-4695(20000905)44:3<289::aid-neu1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 5.Fimbel SM, Montgomery JE, Burket CT, Hyde DR. Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J. Neurosci. 2007;27:1712–1724. doi: 10.1523/JNEUROSCI.5317-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mensinger AF, Powers MK. Visual function in regenerating teleost retina following surgical lesion. Vis. Neurosci. 2007;24:1–9. doi: 10.1017/S0952523807070265. [DOI] [PubMed] [Google Scholar]
- 7.Hitchcock PF, Macdonald RE, VanDeRyt JT, Wilson SW. Antibodies against Pax6 immunostain amacrine and ganglion cells and neuronal progenitors, but not rod precursors, in the normal and regenerating retina of the goldfish. J. Neurobiol. 1996;29:399–413. doi: 10.1002/(SICI)1097-4695(199603)29:3<399::AID-NEU10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Raymond PA, Barthel LK, Bernardos RL, Perkowski JJ. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev. Biol. 2006;6:36. doi: 10.1186/1471-213X-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu DM, et al. Cones regenerate from retinal stem cells sequestered in the inner nuclear layer of adult goldfish retina. Invest. Ophthalmol. Vis. Sci. 2001;42:2115–2124. [PubMed] [Google Scholar]
- 10.Kassen SC, et al. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Developmental Neurobiology. 2007;67:1009–1031. doi: 10.1002/dneu.20362. [DOI] [PubMed] [Google Scholar]
- 11.Bernardos RL, Barthel LK, Meyers JR, Raymond PA. Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J. Neurosci. 2007;27:7028–7040. doi: 10.1523/JNEUROSCI.1624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer AJ, McGuire CR, Dierks BD, Reh TA. Insulin and fibroblast growth factor 2 activate a neurogenic program in Müller glia of the chicken retina. J. Neurosci. 2002;22:9387–9398. doi: 10.1523/JNEUROSCI.22-21-09387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osakada F, et al. Wnt signaling promotes regeneration in the retina of adult mammals. J. Neurosci. 2007;27:4210–4219. doi: 10.1523/JNEUROSCI.4193-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rattner A, Nathans J. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J. Neurosci. 2005;25:4540–4549. doi: 10.1523/JNEUROSCI.0492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldman D, Hankin M, Li Z, Dai X, Ding J. Transgenic zebrafish for studying nervous system development and regeneration. Transgenic Res. 2001;10:21–33. doi: 10.1023/a:1008998832552. [DOI] [PubMed] [Google Scholar]
- 16.Senut M, Gulati-Leekha A, Goldman D. An element in the α1-tubulin promoter is necessary for retinal expression during optic nerve regeneration but not after eye injury in the adult zebrafish. J. Neurosci. 2004;24:7663–7673. doi: 10.1523/JNEUROSCI.2281-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman D, Ding J. Different regulatory elements are necessary for alpha1 tubulin induction during CNS development and regeneration. Neuroreport. 2000;11:3859–3863. doi: 10.1097/00001756-200011270-00051. [DOI] [PubMed] [Google Scholar]
- 18.Cameron DA, Gentile KL, Middleton FA, Yurco P. Gene expression profiles of intact and regenerating zebrafish retina. Mol. Vis. 2005;11:775–791. [PubMed] [Google Scholar]
- 20.Torii M, et al. Transcription factors Mash-1 and Prox-1 delineate early steps in differentiation of neural stem cells in the developing central nervous system. Development. 1999;126:443–456. doi: 10.1242/dev.126.3.443. [DOI] [PubMed] [Google Scholar]
- 19.Jasoni CL, Reh TA. Temporal and spatial pattern of MASH 1 expression in the developing rat retina demonstrates progenitor cell heterogeneity. J. Comp. Neurol. 1996;369:319–327. doi: 10.1002/(SICI)1096-9861(19960527)369:2<319::AID-CNE11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 21.Yun K, et al. Modulation of the notch signaling by Mash1 and Dlx1/2 regulates sequential specification and differentiation of progenitor cell types in the subcortical telencephalon. Development. 2002;129:5029–5040. doi: 10.1242/dev.129.21.5029. [DOI] [PubMed] [Google Scholar]
- 22.Cau E, Wilson SW. Ash1a and neurogenin1 function downstream of floating head to regulate epiphysial neurogenesis. Development. 2003;130:2455–2466. doi: 10.1242/dev.00452. [DOI] [PubMed] [Google Scholar]
- 23.Bray S. Notch signalling in Drosophila: three ways to use a pathway. Seminars in Cell and Developmental Biology. 1998;9:591–597. doi: 10.1006/scdb.1998.0262. [DOI] [PubMed] [Google Scholar]
- 24.Shin J, Poling J, Park H, Appel B. Notch signaling regulates neural precursor allocation and binary neuronal fate decisions in zebrafish. Development. 2007 doi: 10.1242/dev.001602. [DOI] [PubMed] [Google Scholar]
- 25.Marquaqrdt T, et al. Pax6 is required for multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 26.Hu Y, Wang T, Stormo GD, Gordon JI. RNA interference of the achaete-scute homolog 1 in mouse prostate neuroendocrine cells reveals its gene targets and DNA binding sites. Proc. Natl. Acad. Sci. USA. 2004;101:5559–5564. doi: 10.1073/pnas.0306988101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. doi: 10.1242/dev.126.3.525. [DOI] [PubMed] [Google Scholar]
- 28.Cau E, Gradwohl G, Fode C, Guillemot F. Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development. 1997;124:1611–1621. doi: 10.1242/dev.124.8.1611. [DOI] [PubMed] [Google Scholar]
- 29.Herzog W, et al. Genetic analysis of adenohypophysis formation in zebrafish. Mol. Endocrinol. 2004;18:1185–1195. doi: 10.1210/me.2003-0376. [DOI] [PubMed] [Google Scholar]
- 30.Pogoda HM, et al. The proneural gene ascl1a is required for endocrine differentiation and cell survival in the zebrafish adenohypophysis. Development. 2006;133:1079. doi: 10.1242/dev.02296. [DOI] [PubMed] [Google Scholar]
- 31.Hieber V, Dai X, Foreman M, Goldman D. Induction of alpha1-tubulin gene expression during development and regeneration of the fish central nervous system. J. Neurobiol. 1998;37:429–440. doi: 10.1002/(sici)1097-4695(19981115)37:3<429::aid-neu8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 32.Nordeen SK. Luciferase reporter gene vectors for analysis of promoters and enhancers. BioTechniques. 1988;6:454–458. [PubMed] [Google Scholar]
- 33.Barthel LK, Raymond PA. In situ hybridization studies of retinal neurons. Methods Enzymol. 2000;316:579–590. doi: 10.1016/s0076-6879(00)16751-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) The wild-type promoter directs GFP expression in the eye, brain and spinal cord of two day old zebrafish larvae. (b-d) Transgenic fish harboring a mutation to the E-box also express GFP in the eye, brain and spinal cord, although the expression is not as intense and uniform. (e) Immunostaining indicates GFP is expressed in the developing retina of wild-type transgenic fish at 3 days. (f-h) GFP expression in TG-945CA transgenic retinas is similar to wild-type GFP expression.
Retinas from TG-954CA transgenic fish received a single injection of BrdU 4hr prior to harvesting the retina. Sections were stained for GFP (green), BrdU (red) and DAPI (blue). The BrdU labeled cells are proliferating at the CGZ. Note the lack of GFP expression in BrdU labeled cells. The dotted line indicates the edge of the retina.
GFP is induced in ganglion cells six days after optic nerve crush in all three lines of transgenic fish harboring the −954 E-box mutation (arrows in GFP panel). DAPI staining is shown in blue. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Injured retinas were examined at 6 and 24 hpi for induction of pax6 by in situ hybridization (red). Müller glia are shown by glutamine synthetase immunostaining (green). At these early time points, pax6 expression does not correspond to glutamine synthetase+ Müller glia.
Retinas from 1016α1T:GFP transgenic fish were injured and electroporated with control or ascl1a MOs on day 0 and housed in BrdU treated water from 24-48 hpi at which time the retinas were harvested. Cells treated with control MO also labeled with BrdU (arrowheads, top 3 panels) while cells treated with ascl1a MO did not (lower 3 panels).