

Abstract
TET2 is frequently mutated in myeloid neoplasms. Genetic TET2 deficiency leads to skewed myeloid differentiation and clonal expansion, but minimal residual TET activity is critical for survival of neoplastic progenitor and stem cells. Consistent with mutual exclusivity of TET2 and neomorphic IDH1/2 mutations, here we report that IDH1/2 mutant-derived 2-hydroxyglutarate is synthetically lethal to TET-dioxygenase deficient cells. In addition, a TET-selective small molecule inhibitor decreased cytosine hydroxymethylation and restricted clonal outgrowth of TET2 mutant, but not normal hematopoietic precursor cells in vitro and in vivo. While TET-inhibitor phenocopied somatic TET2 mutations, its pharmacologic effects on normal stem cells were, unlike mutations, reversible. Treatment with TET inhibitor suppressed the clonal evolution of TET2 mutant cells in murine models and TET2-mutated human leukemia xenografts. These results suggest that TET inhibitors may constitute a new class of targeted agents in TET2 mutant neoplasia.
Keywords: TET2, MDS, IDH, α-ketoglutarate, 2-hydroxygluterate, 5hmC
Introduction
Somatic mutations are essential pathogenic lesions in myeloid leukemias. A subset of these mutations may serve as targets for drug development either directly or through modulation of up/down-stream pathways and regulatory signaling networks critical for survival and proliferation of malignant cells(1). DNA methylation-demethylation is central to epigenetic gene regulation. In humans, 60–80% of CpGs are methylated in somatic cells as a default state(2). Demethylation of enhancer and promoter CpG islands establishes transcription programs that determine cell lineage, survival and proliferation(3, 4). TET-dioxygenases are indirect erasers of methylation from mCpG containing DNA(5) with TET2 accounting for more than 50% of activity in hematopoietic stem and progenitor cells (HSPCs). Somatic mutations in the TET2 gene (TET2MT) are among the most common genetic defects in myeloid neoplasia(6, 7). In particular, TET2MT in myelodysplastic syndromes (MDS) increase with age, with >70% of MDS patients 80 years or older having TET2MT(8). Approximately 50% of TET2MT are founder lesions(9). In addition, TET2MT are also detected in blood leukocytes of otherwise healthy older adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP) associated with a risk for subsequent myeloid neoplasia and cardiovascular disorders(8, 10). The TET2 gene, like TET1/3, is an iron(II) and α-ketoglutarate (α-KG) dependent DNA dioxygenase. TET2MT cause partial loss of dioxygenase catalyzed oxidation of 5-methyl cytosine (5mC)→ 5hydroxy methyl cytosine (5hmC)→ 5-formyl cytosine (5fC)→5-carboxyl cytosine(5caC). TET catalyzed reactions require a radical equivalent to abstract a hydrogen from 5mC by cleaving the O-O bond of O2. For this purpose, it uses 2e− gained by decarboxylation of αKG via a Fe2+/Fe3+ redox reaction in two single-electron transfers. Ultimately, 5hmC generated by TET2 passively prevents maintenance methylation due to DNA methyltransferase’s inability to recognize 5hmC or causes demethylation as a result of base excision repair of 5fC and 5caC. TET2MT leads to HSC expansion due to perturbation in differentiation programs resulting in a skewed differentiation toward monocytic predominance(11). In addition, hypermethylation in TET2 mutant cells resulting from 5mC accumulation may increase background C>T mutation rates via mC deamination(12).
The high incidence of TET2MT in MDS and related myeloid neoplasia with a strong age-dependence suggests that TET2 is a key pathogenetic factor. Targeting founder TET2MT could disrupt clonal proliferation at its origin and therefore can be a rational target, both for therapeutics in MDS and for strategies preventing CHIP evolution. To date, the only TET2MT targeted therapeutic compound aimed at restoration of TET2 activity is vitamin C, a co-factor in TET2 catalysis(13, 14). However, several recent reports demonstrated that the effects of ascorbic acid in myeloid neoplasms are complex and context dependent and often fail to restore TET function in the presence of certain mutations and post translational modifications(15–17). Basal TET function is essential for the expression of several 5mC-sensitive transcription factors including Myc(18), and Runx1(19). TET2-deficient leukemic cells rely on the remaining TET-activity largely from TET3 and weakly expressed TET1. They must compensate for TET2 loss as evidenced by the persistence of hydroxymethylation in cells with biallelic inactivation of TET2 in human leukemias and in Tet2−/− mice(20). Hence, we hypothesized that TET2MT cells might be more vulnerable to TET-inhibition compared to normal HSPCs. The murine model suggests that TET1/3 may play an important compensatory role; a knockout of all 3 Tet genes in mouse models is embryonically lethal(21), and in a zebrafish model system(22), results in loss of hematopoietic stem cells. The series of experiments presented in this report indicate that inhibition of the essential residual DNA dioxygenases activity in TET2MT cells may lead to selective synthetic lethality that can be experimentally exploited to study the role of TET enzymes in HSPCs biology. Most importantly, our results indicate that TET-specific inhibitors (TETi) may constitute a new class of therapeutics effective in TET2MT associated myeloid neoplasia.
Results
Lessons from natural α-KG antagonist, 2HG
A comprehensive analysis of the configurations of TET2MT in myeloid neoplasia including MDS (n=1809) and acute myeloid leukemia (AML) (n=808), showed mutual exclusivity between TET2MT and 2HG producing neomorphic IDH1/2MT (Fig. 1A), consistent with earlier reports with much smaller cohorts(23). In a separate MDS cohort (495/1809 TET2MT cases) only 9 double mutant exceptions were found; they included small TET2MT (or IDH1/2MT) 5/9 subclones, 2/9 were missense N-terminal TET2MT and one case involved non-2-hydroxyglutarate (2HG) producing missense IDH1MT (Table S1). Similarly, in an AML cohort, there were 166/808 IDH1/2MT cases, but only 8 had co-occurrence of TET2MT. Among these overlapping TET2MT and IDH1/2MT, 4/8 had non-deleterious missense TET2 variants, 2/8 had low VAF implicating small subclones, and one had a non-neomorphic IDH1 variant (Fig.1A and Table S1). Analysis of TET2 expression in 97 healthy and 909 MDS/MPN or AML patients from two independent studies (Table S2) showed that IDH1/2MT cases have significantly higher TET2 expression (Fig.1B) and were absent in the lower 25th percentile of TET2 expressing patients (Fig.1C). These observations suggest that the product of neomorphic IDH1/2MT, 2HG may further inhibit the residual TET-activity (TET1/3) and cause synthetic lethality to cells affected by the loss-of-function TET2MT and decreased expression. As a result, cells with functional TET2 and adequate TET-activity may survive and proliferate even after transient and partial TET-inhibition, while the cells with defective TET2 are eliminated or growth restricted. However, the mutual exclusivity of TET2MT and IDH1/2MT may also arise due to the functional redundancy of these two mutations. To clarify which of these mechanisms is operative, we tested the hypothesis that 2HG, a natural TET inhibitor produced by neomorphic IDH1/2MT may have selective toxicity to TET2MT clones. When we transduced a natural TET2MT cell line SIGM5 (TET2 p.I1181fs, p.F1041fs) with IDH1WT or IDH1R132C under doxycycline inducible tet-on promoter (Fig.1D), a significant increase in 2HG (~3,000-fold over baseline) with a concurrent decrease in 5hmC was observed following IDH1R132C induction with doxycycline (Figs.1D–F). SIGM5-tet-on-IDH1R132C cells displayed a profound growth inhibition upon doxycycline induction compared to SIGM5-tet-on-IDH1 cells (Fig.1G). To further verify the in vivo effects of the neomorphic IDH1R132C in eliminating TET2MT cells, we implanted SIGM5-tet-on-IDH1R132C cells in the flanks of NSG mice and induced expression of IDH1R132C by treating with doxycycline after the tumor was established. Indeed, doxycycline-induced 2HG production led to a significant inhibition of tumor growth (Figs.1H–I).
Figure 1. Effect of 2HG producing IDH1/2 mutations on TET2MT cells.
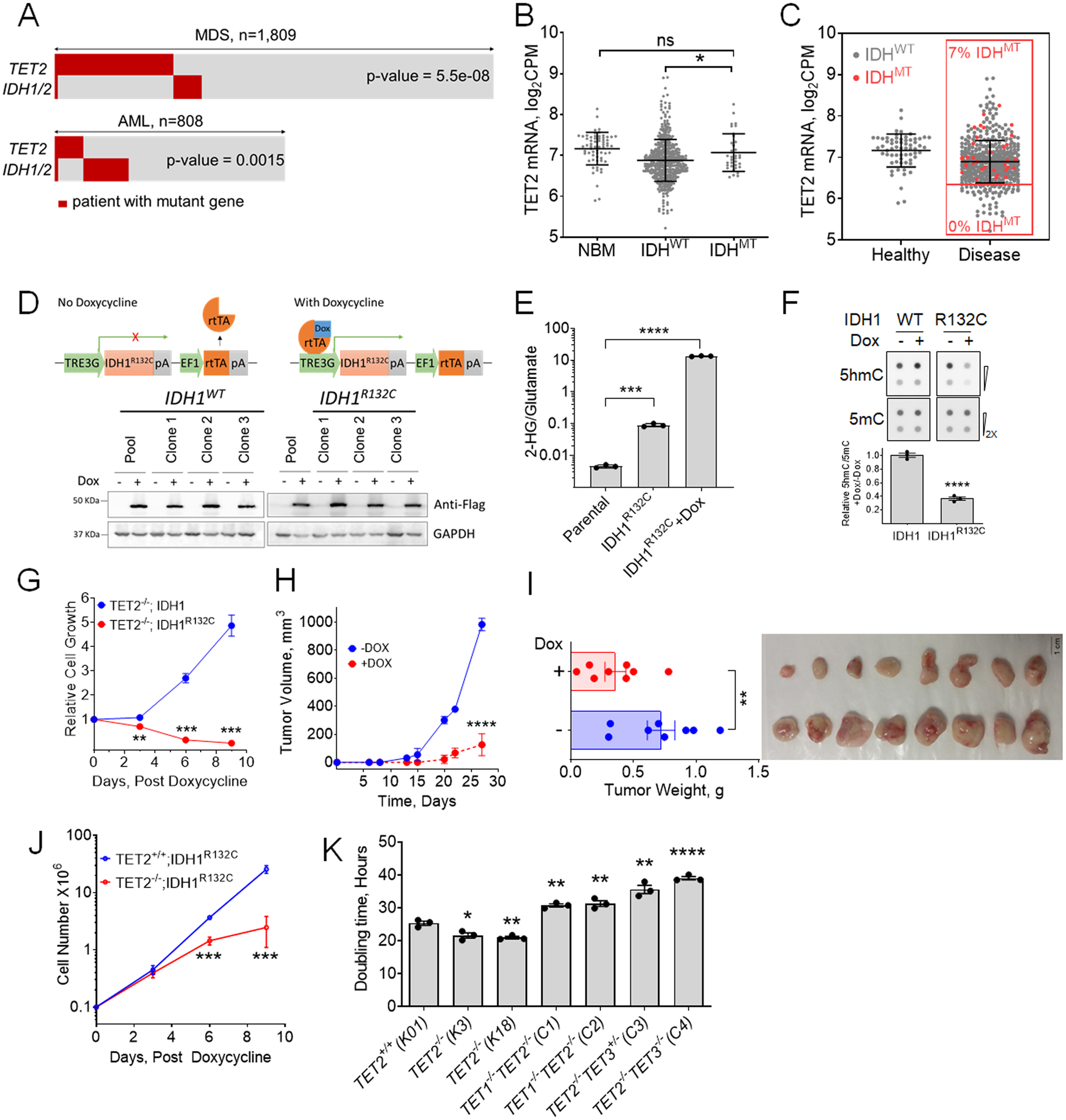
A, Analysis of TET2 and IDH1/2 mutations in MN patients. IDH1/2MT in patients with TET2MT within a CCF cohort of 1,809 MDS patients and TCGA & Beat AML cohorts of 808 AML patients were analyzed for the co-occurrence of TET2 and IDH1/2 mutations (p values are from Fisher’s exact test). B, Comparison of TET2 expression among normal healthy donor and myeloid neoplasia patients carrying wild type or mutant IDH1/2 from different cohorts including Beat AML (www.Vizome.org). C, Distribution of IDH1/2MT and TET2 expression in myeloid neoplasia patients. Red dots are mutant IDH1/2, no mutations were observed in low TET2 expressing population defined as half of the mean expression of controls. D, Inducible expression of 3XFlag-IDH1 or -IDH1R132C in SIGM5 cells. Cells were treated with 1 μg/ml doxycycline (Dox) for 3 days. Anti-Flag antibody was used in western blot for the detection of induced IDH1, and IDH1R132C. Three independent clones from each cell lines along with their pool were analyzed for the expression analysis. E, Production of 2HG measured by LC-MS/MS. Cells were treated with or without 1 μg/ml Dox for 3 days followed by 2HG extraction and analysis. F, Dot blot analysis and quantification of 5hmC and 5mC in IDH1 inducible SIGM5 cell line. Cells were treated with or without 1 μg/ml Dox for 3 days. Sodium ascorbate at final concentration of 100 μM was added 12 hours before harvesting the cells for DNA extraction. G, Effect of 2HG producing IDH1R132C on the growth of SIGM5 cells. Cells (105/ml) were treated with 1 μg/ml Dox and cell proliferation was monitored. The total cell output was plotted as a function of time. H-I, Tumor growth of SIGM5-IDH1R132C cells in NSG mice (n=8/group) were monitored upon Doxycycline treatment. J, Cell growth curve of K562 TET2+/+ and TET2−/− cells after inducing IDH1R123C expression. K, doubling time of K562 isogenic TET mutant cells. Indicated TET dioxygenase genes were knocked out using CRISPR-Cas9 and the genotypes were confirmed by western blot analysis and Sanger sequencing. The doubling times were determined by exponential growth curve fitting in GraphPad prism. Three independent clones of each cell line were used in E-G and J. Three biological replicates were used in K. Experiments were performed at least twice for E-G and J-K. Data are shown as mean ±SEM; statistical significance (p values) from two tailed t-test (except for A) are indicated; * p<0.05; ** p<0.01; *** p<0.001; **** p<0.001; ns: not significant.
To further understand if the synthetic lethality of neomorphic IDH1/2MT is due to the loss of TET2, we generated isogenic TET2 knockout K562 cells (Figs. S1A-B and Table S3) using CRISPR-Cas9 and transduced with IDH1R132C under a doxycycline-induced promoter (Fig. S1C). We did not observe any change in the growth rate of these cells in the absence of doxycycline, most likely caused by some increase of basal 2HG levels due to leakiness of IDHR132CTET-on expression construct(24) that may have slowed the growth of TET2−/−;IDH1R132C cells while increasing the rate of TET2+/+;IDH1R132C cells (Fig. S1D). However, doxycycline induction of neomorphic IDH1R132C led to a significant growth inhibition of K562 TET2−/− vs. control K562 TET2+/+ cells (Fig. 1J).
To further support the hypotheses that a threshold level of DNA dioxygenases activity is essential for the growth and survival of leukemia cells, we used genetic approaches to sequentially inactivate TET1 and TET3 in TET2 knockout cells (Figs. S1A-B and Tables S3–4). As expected, deletion of TET2 gave proliferative advantage to K562 over the control cells however, further deletion of either TET1 or TET3 led significant reduction of global 5hmC correlating with severe growth impairment (Fig. 1K) due to G2/M cell cycle arrest and increased (2-fold for TET2−/−TET1−/− and 3-fold for TET2−/−TET3MT) basal levels of apoptotic cells (Figs. S1E-F).
Intrinsically, SIGM5 cells express significant levels of TET3 and very low levels of TET1 (Fig. S1G) and therefore, they rely heavily on TET3 for their DNA dioxygenase activity. Every attempt to completely inactivate TET3 in SIGM5 cells failed (Table S5). Instead, we were only able to obtain TET3 heterozygous deletion resulting in nearly 2-fold decrease in 5hmC and a significant growth impairment (Figs. S1B). The reliance on residual TET3 and TET1 activity was further confirmed by either double TET knockouts or inducible knockdown of TET3 in TET2−/− leukemic cell line SIGM5 transduced with shTET3 tet-on vector. Doxycycline-induced knockdown of TET3 in SIGM5 resulted in a significant growth retardation (Figs. S1H-I).
TET inhibitors (TETi)
TET-dioxygeases not only determine the transcriptional program that guides cell lineage determination, they are also important for efficient transcription of target genes essential for proliferation and survival of malignant cells(4, 18). While the loss-of-function TET2MT leads to skewing towards myeloid precursors their survival and proliferation are critically dependent on residual TET-activity derived mostly from TET3 and to some extent on TET1. We hypothesized that transient suppression of this residual DNA dioxygenase activity with a small molecule inhibitor (e.g., designed based on a 2HG scaffold in the α-KG binding site of TET2 catalytic domain) may preferentially suppress and eventually eliminate TET-deficient/TET2MT leukemia initiating clones. 2HG, N-oxalylglycine (NOG) and dimethyl fumarate (DMF) are known to inhibit a variety of α-KG dependent dioxygenases and hence lack specificity, pharmacologic properties and potency(23, 25). Therefore, we utilized the co-crystal structure of the TET2 catalytic domain (TET2CD) in complex with pseudo substrate NOG and performed in silico docking to design and synthesize several compounds where the C4-position was substituted with either -keto, -olefin, -methyl, or -cyclopropyl functional groups and the C2-position was single or double substituted with –chloro, -fluoro, -hydroxy, -methyl, or –trifluoromethyl groups (Figs. 2A–B, S2A–B). These compounds were tested at 25 μM in SIGM-5 (a TET2−/− leukemia cell line) cells in vitro and their ability to induce cell death followed by their ability to inhibit TET-dioxygenase activity were used to select the top three compounds (Figs. 2C–D). For further selection of most potent and pharmacologically active compounds we calculated the therapeutic index using ratio of LD50 of normal bone marrow derived mononuclear cells (NBM) and TET2 deficient leukemia cells (SIGM5 and MOLM13) (Figs. 2E and S2C). Interestingly, in this series of compounds, 2-methelene and 4-hydroxy (TETi76) are critical to maintain TET-inhibitory and cell killing activity of these compounds (Figs. 2D, 2F and S2B). The substitution of the C4 proton in TETi76 either with -CH3 (TETi187) or -CF3 (TETi220) leads to a decrease of the therapeutic index from 5.4 to 2.5, suggesting some off target effect of these compounds (Fig. 2E). The 2-methelene and 4-hydroxy derivative of α-KG binds to TET2CD similar to pseudo substrate NOG that putatively involved H1801, H1381 and S1898 (Figs. 2A andS2D). These residues are conserved among TET1, TET2 and TET3 (Fig. S2E), therefore we tested the TETi76 against these three dioxygenases in cell free assay using recombinant proteins and found that it inhibits all three TETs with 1.5, 9.4 and 8.8 μM IC50 respectively (Figs. 2G and S2F). The effect of TETi76 on TET2CD can be reversed by increasing α-KG but not by Fe2+ (Fig. 2H). We performed the direct binding interaction of TET2CD and TETi76 using highly sensitive and label free MicroScale Thermophoresis (MST) technique. Analysis of thermophoresis binding curves for the association and dissociation of TETi76 with TET2CD using MO.Affinity software supplied with the instrument demonstrated that TETi76 specifically binds to TET2CD with a dissociation constant of 0.3 μM (Fig. 2I) evaluated with in-built Kd model that utilized the normalized signal with increasing concentration of ligands.
Figure 2. Lead optimization and efficacy enhancement of TETi.
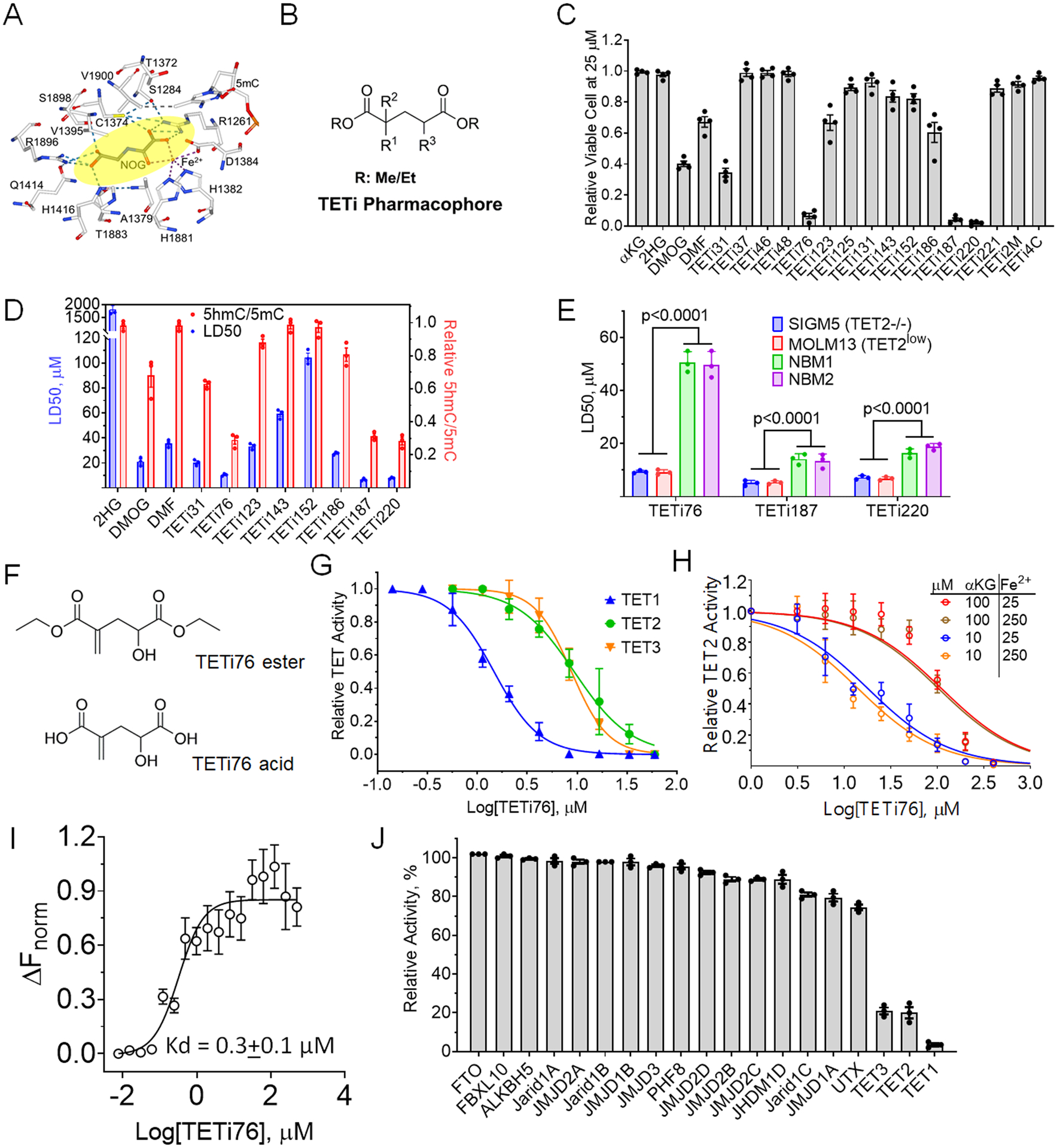
A, Consensus binding site for known pseudo substrate NOG. B, 2D structure TETi pharmacophore. C, TET2−/− leukemia cell killing effect of 20 compounds. TET2 deficient SIGM5 AML cells at 105/ml were treated with 25 μM compounds for 3 days and viable cells were determined by trypan blue exclusion method. D, LD50 and TET inhibitory effect of 11 selected compounds. SIGM5 cells at 105/ml were treated with increasing concentrations of TET inhibitors for 3 days and surviving cells were counted by trypan blue exclusion on automated Vi-Cell counter and LD50 was calculated from viable cell number in GraphPad Prism. TET-dioxygenase activity was performed by measuring 5hmC in a dot blot assay. SIGM5 cells at 3×105/ml were treated with 25 μM compounds for 12 hours in the presence of 100 μM sodium ascorbate and the relative 5hmC/5mC quantification using dot blot were performed on genomic DNA. E, LD50 of TET inhibitor against normal bone marrow derived mononuclear cells (NBM) and TET2 mutant and TET2-deficient leukemia cells (SIGM5 and MOLM13). Cells were treated with increasing doses of indicated TETi for 3 days and LD50 was calculated from viable cell number in GraphPad Prism. F, Structure of ester and acid forms of TETi76. Acid forms were used for cell free assays while diethyl esters were used in cell culture. G-H, Dose dependent inhibition of TET activity by TETi76 in cell free condition. Catalytic domains of recombinant TET1, TET2 and TET3 were incubated with TETi76 separately in the presence of different concentrations of co-factors (αKG and Fe2+) and the 5hmC was monitored by ELISA using anti 5hmC antibodies. I, Measurement of direct interaction of TETi76 with TET2CD by Microscale Thermophoresis (MST). Binding interaction of TETi76 with TET2CD was monitored using label-free Microscale Thermophoresis on Monolith NT.LableFree instrument. The normalized change in MST signal ΔFnorm= (Fbound-Ffree)/Response amplitude at different TETi76 concentration is plotted. Data were analyzed using MO.Affinity analysis V2.3 software using built-in Kd model for data fitting and calculation of dissociation constant. J, Testing inhibitory effect of TETi76 on α-KG and Fe2+ dependent demethylases. Purified enzymes were used with their substrate in the presence or absence of 15μM TETi76 in alpha screen (https://bpsbioscience.com) and the relative activity was determined for a vehicle control. Each assay was accompanied by a positive control (see Supplementary Table S6). Data are representative for experiments performed in triplicates at least twice (C-E and G-I). Data are shown as mean ±SEM.
To test if TETi76 can inhibit other αKG/Fe2+ dependent enzymes, we performed in vitro enzyme assays against 16 other known family members including most closely related RNA dioxygenase FTO. Interestingly, TETi76 do not inhibit any of the enzymes at a concentration of 15 μM, well above the IC50 for TET-dioxygenases. Therefore, TETi76 is a TET-specific inhibitor as determined by in-vitro enzyme activity assays (Fig. 2J and Table S6). We synthesized and purified R- and S- enantiomers of TETi76, performed in vitro TET inhibition assay, and found that there were no significant differences in the IC50 of the two enantiomers under cell free in vitro conditions (Fig. S2G).
Efficacy and selectivity of TETi76 in cell culture model
TETi76 preferentially inhibits the catalytic function of TET-dioxygenases in a cell free system, therefore, we further analyze its specificity in cell culture models and examined if it mimics loss of TET function. For this purpose, we generated cell permeable diethyl ester of TETi76 and treated different human leukemia cell lines (K562, MEG01, SIGM5, OCI-AML5, MOLM13) and measured 5hmC using dot blot assay. Consistent with cell free data, we observed a dose dependent decrease in the global 5hmC content in a variety of TET2 proficient and TET2 deficient human leukemia cells (Figs. 3A, S3A and S2C). With 50% inhibition of 5hmC ranging from 20 to 37 μM (Fig. 3A). To characterize target specificity of TETi76, we performed global gene expression analyses of K562 TET2+/+ and TET2−/− control cells; TETi76 mimicked expression signatures generated by the loss of TET2 in K562. The addition of Ascorbic Acid (AA), known to enhance TET-activity, counteracted the changes induced by TETi76 (Figs. 3B–C).
Figure 3. TETi76 mimics loss of TET activity and preferentially restricts the growth of TET-dioxygnease deficient neoplastic cells.
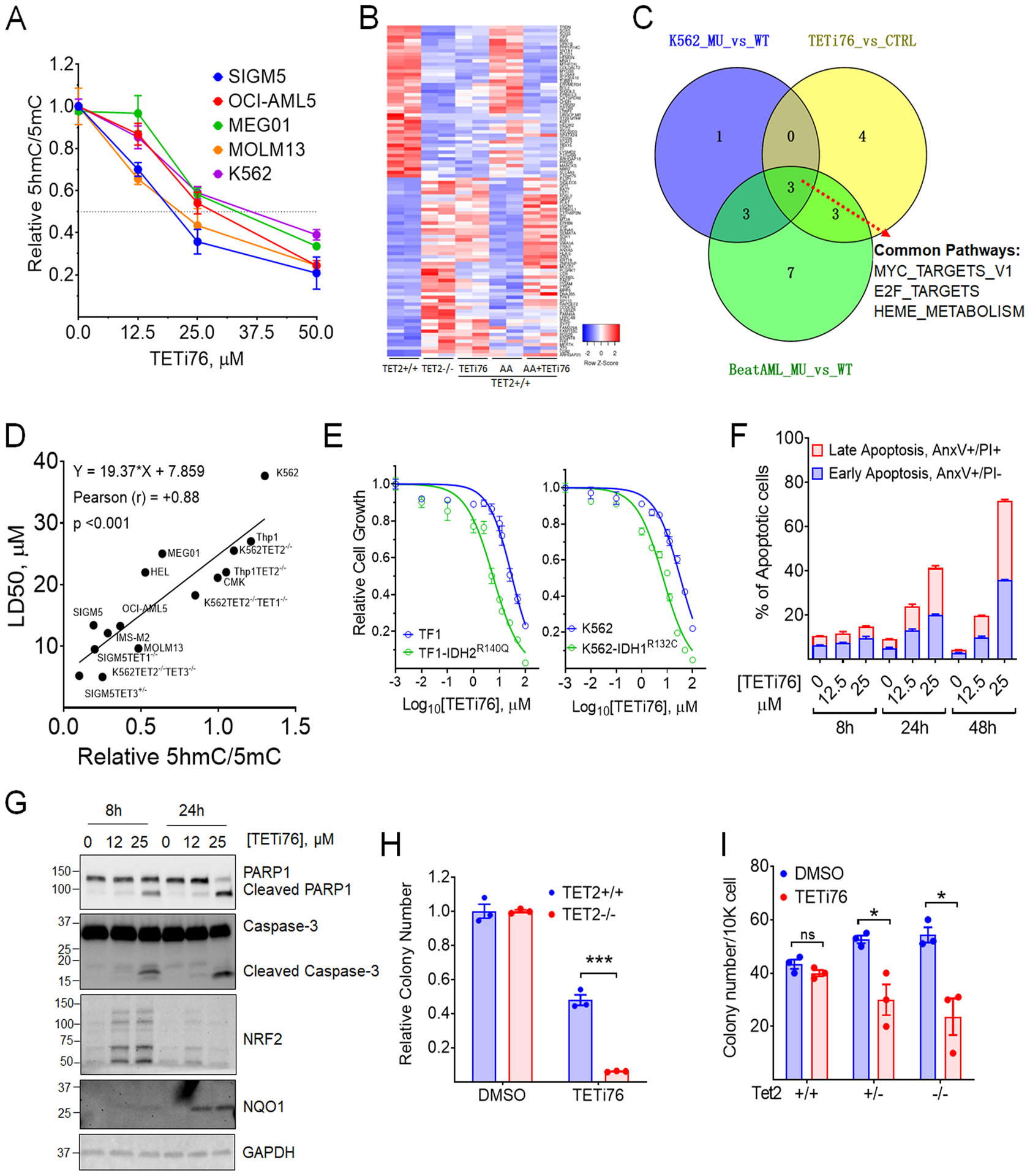
A, Dose dependent inhibition of 5hmC by TETi76 in different leukemia cells. Cells were treated with increasing concentrations of TETi76 in the presence of 100 μM sodium ascorbate for 12 hours. Genomic content of 5hmC and 5mC were detected by dot blot analysis using specific antibodies and the ratio of 5hmC/5mC are plotted. B, Heatmap of significantly up and down regulated genes in K562 TET2+/+ (K01) or K562 TET2−/− (K18) cells treated with 25 μM TETi76 or 100 μM ascorbic acid (AA) for 24 hours followed by RNAseq analysis. C, Venn diagram of pathway analysis of K562 TET2−/− (K18, MU) vs K562 TET2+/+ (K01, WT), K562 TET2+/+ (K01) treated with 25 μM TETi76 for 24 hours vs vehicle (DMSO) control (CTRL) or BeatAML RNAseq data of TET2 mutant (MU) vs TET2 wild type (WT) (vizome.org/aml/) performed by hallmark gene set enrichment analysis. D, Correlation Analysis of LD50 of TETi76 against different leukemia cells with TET-activity. The TET activities in different cells were measured by relative ratio of 5hmC/5mC in 15 different leukemia cell lines including 8 isogenic TET1/2/3 knockout cell lines. Each cell lines were treated with increasing concentrations of TETi76 for 72 hours and the LD50 was calculated from viable cell monitored by methylene blue exclusion on vi-cell counter. Pearson correlation coefficient (r) and significance were calculated in GraphPad Prism. E, Effect of TETi76 on cells expressing neomorphic IDH1/2 mutants. Commercially available TF1-IDH2R140Q, house-made K562-IDH1R132C (Fig. 1J) and their parental cells (TF1 and K562) were treated with different concentrations of TETi76 for 3 days. Both K562 and K562-IDH1R132C cells were supplied with 1 μg/ml doxycycline during TETi76 treatment. Relative cell growth was measured by CellTiter-Glo assay. F, TETi76 induces programmed cell death in TET2 deficient cells. SIGM5 cells were treated with TETi76 and the apoptotic cell population was determined by annexin V and propidium iodide staining using flow cytometer. TETi76 treatment demonstrate a dose and time dependent increase in early and late apoptotic cells. G, Western blot analysis of PARP1, Caspase-3, NRF2 and NQO1 after TETi76 treatment in SIGM5 cells. H, Colony forming abilities of K562 TET2+/+ and TET2−/− cells in the presence and absence of TETi76. I, Bone marrow from Tet2+/+, Tet2+/− and Tet2−/− mice were harvested and cultured in MethoCult in the presence or absence of TETi76. Data is representative of 3 independent experiments performed separately. Data are shown as mean or mean±SEM; statistical significance (p values) from two tailed t-test are indicated; * p<0.05; *** p<0.001; ns: not significant.
Suspension cultures followed by determination of 5hmC indicated that TET2 deficiency was associated with decreased levels of 5hmC and increased proliferation. However, any further suppression of TET-dioxygenases by inactivating either TET1 or TET3 induced profound growth suppression in a variety of leukemia cell lines (Fig. 1K). Cell lines with lower TET-dioxygenase activity (SIGM5, MOLM13 and TET deficient isogenic engineered cells lines) were more sensitive to TETi76, compared to TET-dioxygenase proficient cells like K562 and CMK cells with higher activity which showed less sensitivity to TETi76-mediated growth inhibition (Fig. 3D). The isogenic TET1/2/3 single or double knockout leukemia cells also showed dioxygenase activity-dependent sensitivity to TETi76 (Figs. 3D, and S1A–B). For example, the parental control K562 cells has an IC50 of 40 μM, while TET2 and TET3 double knockout K562 cells exhibited nearly 4-fold lower IC50. A consistent positive correlation between TET-activity and the IC50 was observed in 15 different cell lines including 8 isogenic TET knockout human leukemia cell lines (Fig. 3D).
It has been shown that neomorphic IDH1/2 mutations phenocopy loss of TET2 in acute myeloid leukemia and other malignancies(23, 26, 27). Consistent with previous observations, ectopic inducible expression of IDH1R132C in K562 or stably expressing of IDH2R140Q in TF1 demonstrated loss of TET-dioxygenase activity as observed by significant reduction in global 5hmC that can further be suppressed by TETi76 (Fig. S3B). To test if TET-inhibition in IDH1/2 mutant expressing cells by TETi76 restrict their growth, we treated IDH1R132C (K562) and IDH2R140Q (TF1) cells with increasing concentration of TETi76 and observed a 5-fold decrease in the IC50 of IDH1/2 mutant expressing cells (Fig. 3E).
TETi76 treatment induces apoptotic cell death in TET-dioxygenase deficient leukemia cells in dose and time dependent manner. The treatment of SIGM5 cells with TETi76 induced early and late stages of apoptotic cell death as probed by the fractions of Annexin V and propidium iodide positive cells (Figs. 3F and S3C), a finding further confirmed by PARP1 and caspase-3 cleavage, hallmark of programmed cell death (Figs. 3G and S3D).
Global gene expression analyses (RNASeq) of SIGM5 cells after treatment with TETi76 demonstrated a significant upregulation of TNF-α signaling and the down regulation of Interferon-α signaling (Figs. S3E–F). Interestingly, we also observed significant up-modulation of oxidative stress response pathway genes consistent with the inhibition of dioxygenases. In particular, TETi76 treatment induces 8-fold increase of oxidative stress sensor NQO1 (Figs. 3G and S3D) a NRF2 target gene that has been shown earlier to induce pro-apoptotic cell death in cancer cells(28).
Consistent with TET-dioxygenase deficiency and sensitivity to TETi76 in suspension cells we also observed that TETi76 preferentially restricted the growth of colony forming units of TET-dioxygenase deficient cells. The effect of TETi76 on TET2−/− K562 was more profound than TET2 proficient isogenic cells (Fig. 3H). This observation was striking in murine bone marrow derived from Tet2+/+, Tet2+/− and Tet2−/− C57BL6 mice. Interestingly we did not observe any significant growth inhibitory or anti-clonogenic effects of TETi76 on bone marrow derived mononuclear cells from TET-dioxygenase proficient wild type mice; on the contrary, it significantly restricted the growth of Tet2−/− and Tet2+/− mice bone marrow mononuclear cells in a similar assay (Fig. 3I).
Consistent with in vitro specificity of the inhibitory effect we observed that treatment of cells with TETi76 did not affect the function of α-KG-dependent histone dioxygenases that demethylate histone K4, K27 and K36 lysine residues. For example, we did not see any change in H3K4 methylation upon treatment with TETi76, though a cell permeable α-KG pseudo substrate methyl 2-[(2-methoxy-2-oxoethyl)amino]-2-oxoacetate (DMOG) significantly affected the levels of H3K4 methylation in a dose dependent manner (Figs. S3G-H). Interestingly, analysis of TCA cycle metabolite in TETi76 treated SIGM5 cells showed significant increase in the levels malate, isocitrate, citrate, fumarate and α-KG while a decrease in 2HG and succinate (Fig. S3I).
TETi76 restricts the growth of Tet2mt cells in preventative CHIP model systems
To probe the effects of TETi76 on TET2 deficient cells, Tet2mt/Tet2+/+ BM mononuclear cells were co-cultured at fixed ratios to mimic evolving Tet2mt clones and the differences in surface marker CD45 isoform fractions served as a read out. Marrow from C57BL6 CD45.2 Tet2mt (Tet2+/− or Tet2−/−) vs. C57BL6 CD45.1 Pep Boy (Tet2+/+) mice were co-cultured (3 mice/group) in a 1:2 ratio with/without TETi76. In culture, TETi76 effectively targeted otherwise dominating Tet2mt cells (Figs. 4A–C). As expected Tet2mt cells grew at a faster rate in the controls as reflected in the increased percentage of Tet2mt cells. However, TETi76 treatment selectively restricted the proliferation of Tet2mt cells as determined using CD45.1/CD45.2 surface markers on Tet2+/+ and Tet2mt bone marrow, respectively (Figs. 4B–C).
Figure 4. TETi selectively restricts the growth of TET2 mutant cells.
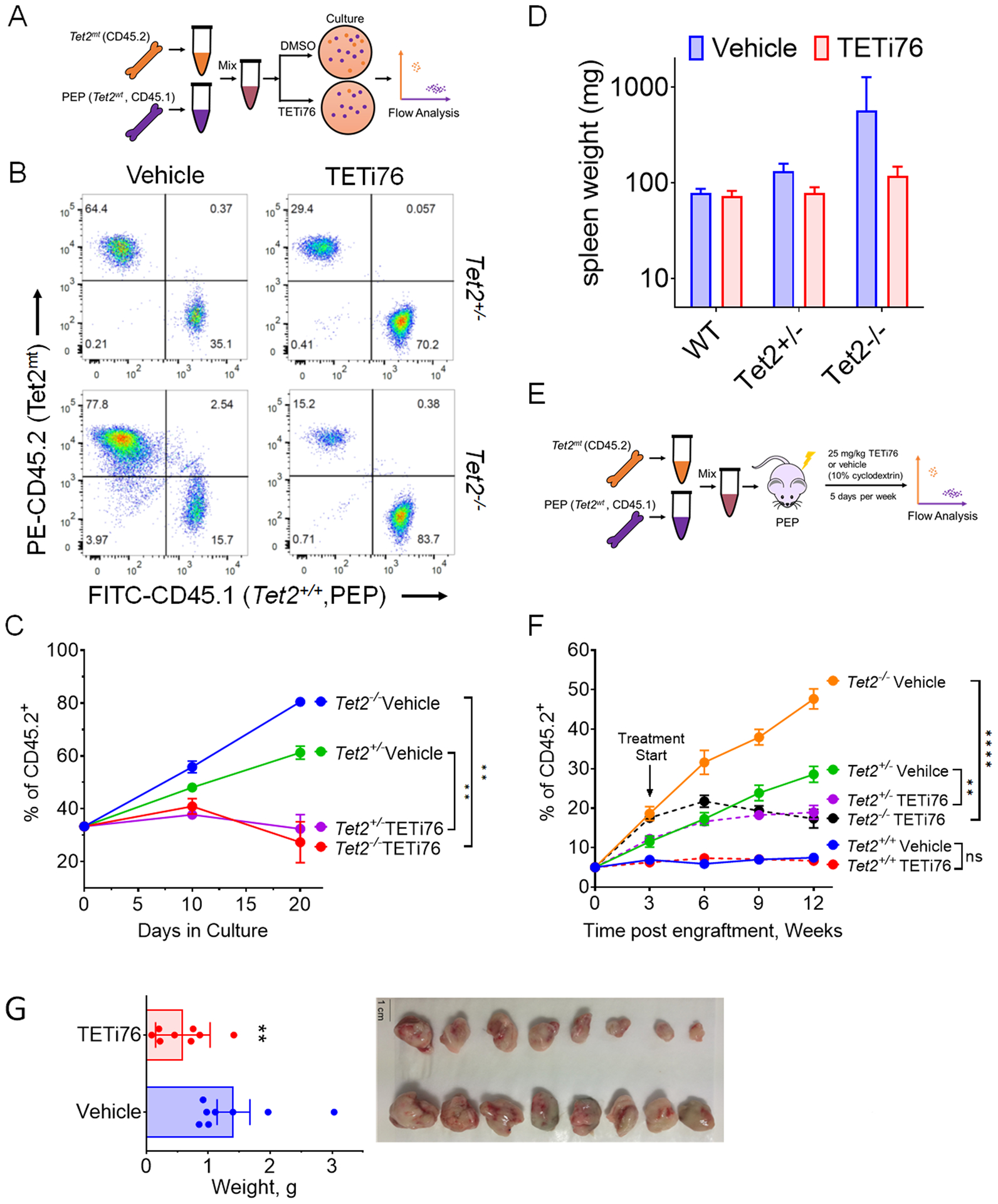
A, Schematic representation of the mixing experiment of Tet2wt and Tet2mt murine bone marrow in colony forming assay. B, Tet2mt bone marrow (CD45.2) cells were mixed in the ratio of 1:2 with Tet2+/+ (CD45.1) and grown in MethoCult for colony formation in the presence or absence of TETi76 (20 μM). On day 10 cells were harvested and the ratio of Tet2mt/Tet2+/+ was measured by flow cytometry using isoform specific antibodies. C, The ratio was plotted for two consecutive platings. D, Tet2mt or TET2wt mice were treated with TETi (50 mg/Kg, p.o., 5 days/week) for 8 weeks. The spleens were harvested and weights were plotted compared to vehicle control. E, Schematics of experimental design for in-vivo transplant experiment. F, C57BL6 PepBoy mice expressing CD45.1 surface marker on mononuclear hematopoietic cells were lethally irradiated and transplanted with a mixture donor mice bone marrows (5% Tet2−/−, CD45.2; and 95% Tet2+/+, CD45.1). Once mice fully recovered after transplant, the engraftement was accessed by isotype specific antibodies and TETi treatment (25mg/kg, s.c.) 5 days a week at 4 weeks were started. The engraftment of Tet2mt cells in the PBMC was monitored and plotted. TETi76 prevented the clonal expansion of Tet2mt cells in vivo. G, Tumor growth of SIGM5 cells in NSG mice (n=8/group) were monitored upon TETi76 treatment. Once controlled reached the maximum allowed limit of tumor burden, tumors were harvested and tumor weight is plotted. TETi76 significantly reduced the tumor size. Data are shown as mean±SEM; statistical significance (p values) from two tailed t-test are indicated; ** p<0.01; **** p<0.0001; ns: not significant.
To further evaluate the in vivo effects of TETi76 treatment in mice we used Tet2+/+, Tet2+/− and Tet2−/− mice and treated them with TETi76 (50mg/Kg, n=3/group, 5-days/week) for 3 months and observed bi-weekly body weight and once a month blood count. We did not observe any impact on the overall body weight or any significant change in the overall blood counts of TETi76 treated mice compared to placebo (Figs. S4A–C). However, TETi76 treatment did decrease spleen sizes in Tet2 deficient mice in a gene dose dependent manner (Fig. 4D).
To determine the in vivo effects of TETi76 in preventing the clonal evolution of Tet2 deficient cells, e.g., on restricting the growth of Tet2+/− or Tet2−/− (Tet2mt) clones, we performed bone marrow competitive reconstitution assays in C57BL6 CD45.1 PepBoy mice. For this purpose, C57BL6 CD45.1 PepBoy mice (n=6 per group) were lethally irradiated followed by injection of 2×106 total bone marrow cells that included 5% of cells from C57BL6 CD45.2 Tet2+/+, Tet2+/− or Tet2−/− and 95% of CD45.1 Pep Boy Tet2+/+ mice. Proliferation of each genotype was monitored by flow cytometry using surface markers. The CD45.2 vs. CD45.1 ratios were plotted as a function of time with treatment and compared with vehicle alone (Fig. 4E). Consistent with several previous reports(20, 29) Tet2mt cells becomes the dominant fractions in vehicle treated mice in a gene dose dependent manner (Fig. 4F). However, treatment with TETi76 preferentially restricted the proliferative advantage of Tet2mt cells compared to vehicle control (Fig. 4F). The ratio of mice receiving Tet2+/+ CD45.2 cells did not change, suggesting that TETi76 restricts the growth of Tet2mt cells only.
TETi76 restricts the growth of TET-deficient leukemia in vivo
To test if TET inhibition would inhibit TET2-deficient leukemic cells under in-vivo conditions we used subcutaneous tumor development in immune compromised mice. Since Tet2mt mice develop a protracted myeloproliferative syndrome rather than overt leukemia, we used a human TET2−/− leukemia cell line xenograft model to test the efficacy of TETi76 for established tumors. SIGM5 a TET2-deficient cells subcutaneously implanted in NSG mice grew very aggressive tumors. Once tumors were established, TETi76 was orally (50 mg/Kg) administered to the implanted mice. Treatment with TETi76 significantly reduced tumor burden compared to vehicle treated mice indicating that TET2MT leukemia may be sensitive to TETi76 in vivo (Fig. 4G).
Discussion
DNA dioxygenases are the key enzymes that catalyze cytosine hydroxymethylation, an essential step for passive DNA demethylation. The high prevalence of somatic loss of function mutations in TET2 underscores the important role of this gene in myeloid neoplasia. In hematopoietic cells TET2 is the most abundantly expressed gene among the TET family of DNA dioxygenases and accounts for nearly 60% of DNA dioxygenase activity explaining why TET1 and TET3 are rarely mutated in leukemia. However, even in cases with biallelic TET2MT, the residual activity of TET3 and TET1 preserves detectable levels of 5hmC in the genome, suggesting a compensatory role for TET1/TET3 in the survival of TET2MT cells. TET2MT are mutually exclusive with IDH1/2MT, which produce 2HG and inhibit TET-dioxygenases(23). It has been proposed previously that the functional redundancy may be responsible for the mutual exclusivity, however, here we demonstrate the elimination of TET2 deficient cells by ectopic expression of neomorphic IDH1R132C mutation.
The results presented here underscore the cellular requirement for preservation of DNA dioxygenase activity and therefore the essential compensatory activity of TET1/TET3 in TET2MT cells. Furthermore, this work suggests a mechanism whereby 2HG is synthetic lethal to TET2 deficient cells and explains the observed mutual exclusivity of TET2 and IDH1/2 mutations.
These observations indicate that chemical dioxygenase inhibitors may be exploited as novel class of agents for TET2MTassociated disorders. The natural Tet2 inhibitor, 2HG, itself affects a broad array of α-KG-utilizing enzymes and hence is not a suitable drug candidate. In addition, its LD50 for TET2MT (in millimolar ranges) cannot be achieved therapeutically. Consequently, using 2HG computer-aided rational drug design and iterative modification, we generated several competitive inhibitors of TET activity. One of these inhibitors, TETi76, proved highly specific and potent and was selected for further studies. We demonstrated using an inducible IDH1/2MT model that produces 2HG, that treatment with TETi76 further decrease the levels of 5hmC resulting in selective growth inhibition of TET2MT cells, while normal bone marrow cells and leukemic cell lines with robust TET-function remained unaffected. In vitro, TETi76 results in selective toxicity (>100-fold that of 2HG) to primary human TET2MT lines, engineered TET2−/− cells as well as Tet2−/− murine hematopoietic cells. The difference in TET activity in TET2WT and TET2MT cells offers a therapeutic window to selectively eliminate TET2MT clones, either preventatively early on in the pathway towards oncogenesis (e.g., in CHIP)(8, 10), or therapeutically in evolved neoplasms.
There have two reports of the co-occurrence of IDH2R172 and TET2MT in cases of angioimmunoblastic T-cell lymphoma (AITL)(30, 31). However, the cellular and extracellular levels of 2-HG were within the normal range(30). In other case, majority of the TET2MT reported have relatively low VAFs and the exact VAF for IDH2 R172 is not clear, therefore it is hard to access the clonal architecture and clonal/sub-clonal mosaicism of the AITL patients. Based on our analysis in MDS and AML patients, most of the co-occurrence were non-damaging or small clone for one or the other variants. In AITL, IDH2 mutations are exclusively restricted to R172 which are commonly mutated to serine or lysine residues(31). It also has been shown that median 2HG level in IDH2R172 AITL patients is significantly lower than the level IDH1- or IDH2- mutated AML(32). Thus, the co-existence of TET2 with weak neomorphic mutations that produces low levels of 2HG may exist under such conditions.
The effects of TET-inhibition observed in TET-deficient malignant HSPCs are consistent with observations in zebrafish models, where loss of all three TET result in loss of HSCs(33). TETi showed potent efficacy in in vivo murine model systems, where the proliferative capacity of Tet2mt bone marrow cells was abrogated. In murine transplant model systems that mimic CHIP, TETi was highly effective in selectively preventing clonal expansion of Tet2−/− hematopoietic cells. Unlike acute leukemia described in genetic mouse models of simultaneous TET inactivation, we did not see evidence in vitro that the pharmacologic manipulation would phenocopy the engineered TET2-deficient leukemia. Moreover, therapeutic intervention with TET inhibitors would be applied transiently and discontinued upon elimination of vulnerable TET2-deficient cells (Fig. 5). In general, the effects of TETi in TET2WT cells were minimal with respect to a stable transcriptional profile and preserved cell viability at levels corresponding to a 10-fold therapeutic index. The effect of TETi is relatively specific to DNA dioxygenases since other α-KG consuming enzymes in humans(34) remained unaffected in vitro. In contrast, 2HG or the aKG pseudo substrate N-oxalylglycine, exhibit ‘off target’ activity, for example against histone lysine demethylases.
Figure 5.
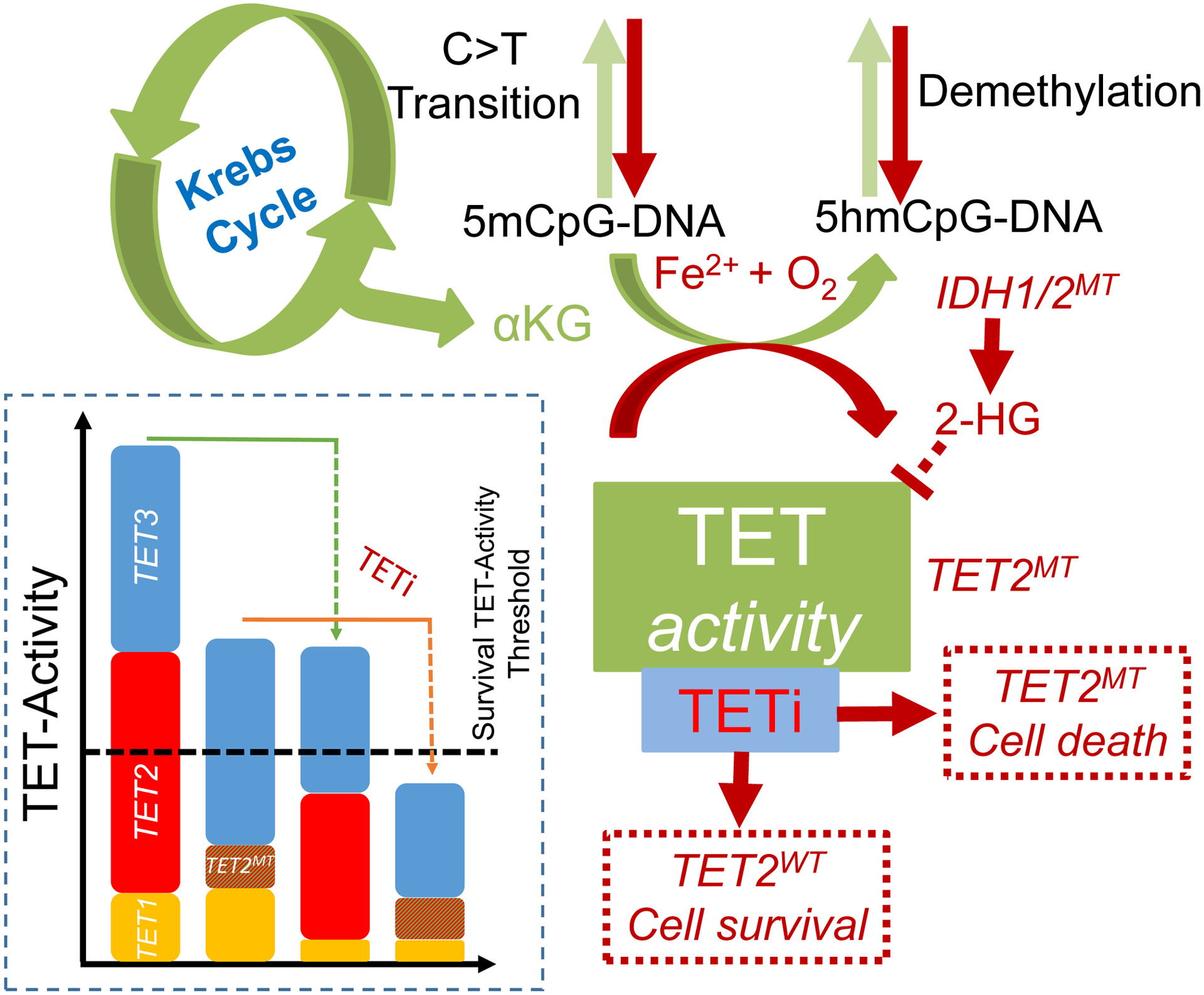
Schematic representation of TETi mechanism of action. TET2 mutant and TET-dixoygenase deficient cells are susceptible to further TET-inhibition. Loss to TET2 increases C→T transition and mutator phenotype leading to neoplastic evolution. Residual TET-dioxygenase activity from TET1 and TET3 in TET2 mutant cells are important for efficient transcription of survival and proliferative genes. Inhibition the residual TET-dioxygenase activity leads to preferential growth restriction and finally elimination of TET2 mutant and TET-dioxygenase deficient clones.
TETi creates transcriptional changes similar to that of the genetically engineered loss of TET2 activity in leukemic cells. The effect of TETi was partially reversed by ascorbic acid. TETi76 induced apoptosis in TET2MT cells, while no detectable apoptosis was seen in either normal TET2WT bone marrow or untreated TET2MT cells. The analysis of RNAseq data and metabolic profile further suggested that TET inhibition leads to a major metabolic shift in TET2MT cells as demonstrated by significantly down modulation of c-MYC target genes, a profile similar to the complete loss of TET2 or the treatment with 2HG(18).
One potential drawback of TETi can be that it may phenocopy TET2MT and drive transformation of normal hematopoietic cells. We did not observe disease acceleration or pro-proliferative effects of TETi administered to Tet2+/+, Tet2+/− or Tet2−/− mice to support accelerated leukemogenesis. One of the key concerns of TET-dioxygenase inhibition stems from a report where combine deletion of Tet2 and Tet3 in early hematopoietic murine cells is suggested to cause an aggressive and transplantable AML(35). To best of our knowledge, TET3 loss of function mutation in human myeloid cancer has not been described, which indicates the importance of TET3 dioxygenase for the survival and the proliferation of hematopoietic cells. Consistent with this observation, Tet2 or Tet3 is required for normal hematopoietic stem cell emergence in zebrafish(33).
The reports of Tet2/Tet3 double deletion develop aggressive myeloid cancer(35) utilized Tet3fl/fl mice crossed with Mx1-Cre or ERT2-Cre recombinase to induce conditional excision of floxed alleles. Polyinosinic–polycytidylic acid (p(rI):p(rC)) or tamoxifen injection respectively are essential to induce conditional Tet3 KO in these models. It has been previously reported that both p(rI):p(rC) as well as tamoxifen can affect the normal hematopoiesis(36, 37), which in part may explain the effect in mice. P(rI):p(rC) in particular mimics RNA virus infection, actives toll-like receptor 3 (TLR3) and leads to the immune response, while Tet2 is required to resolve inflammation to repress IL-6(38). In our study, we showed that both monoallelic and biallelic TET3 knockout K562 TET2−/− leukemia cells are growth impaired. The triple TET knock out of K562 was not feasible. It will be interesting to characterize Tet2−/−Tet3+/− mice compared to Tet2−/− mice and the effect of TET-inhibitor on such cells.
Preferential inhibition of the clonal evolution of TET2MT clone can have huge therapeutic implications in CHIP. In the context of CHIP, eliminating and restricting the growth of TET2MT cells by TET inhibition are highly relevant not only to the myeloid malignancies and associated disorders but also to the increased risk of cardiovascular disorders(10, 39, 40). Transient TET inhibition by small molecule inhibitor such as TETi76 would be devoid of any aberration caused by genetic lesion while lethal against Tet2mt and TET-dioxygenase deficient. Thus the therapeutic benefit of TET inhibitors in the prevention of cardiovascular disease could be obtained.
The utility of TET inhibitor is not limited to CHIP and the myeloid malignancies since a pan TET-dioxygenase small molecule inhibitor (C35) has been reported to achieve somatic cell reprograming with several probable therapeutic utilities(41).
In summary, we demonstrate for the first time that TET inhibition leads to selective synthetic lethality in TET2MT cells. This approach may be developed as a potential targeted therapeutic strategy for TET2MT associated disorders myeloid neoplasia associated with TET-dioxygenase deficiency and may lead to development of a new class of TET-selective DNA-dioxygenase-inhibiting agents.
Materials and Methods
Patient samples
Patient bone marrow samples were obtained from healthy controls or patients with myeloid neoplasia after informed consent in accordance with Cleveland Clinic IRB-approved protocol. Human cord blood was acquired from Cleveland Cord Blood Center, Cleveland, Ohio, and CD34+ cells were isolated by human CD34 MicroBead Kit (Miltenyi Biotec).
Cell Lines
All cell lines were purchased recently (two years or less) and cultured according to the supplier’ guidelines. K562, THP-1, TF1 and TF1-IDH2R140Q cell lines were purchased from ATCC (Manassas, VA), while CMK, MEG-01, MOLM13, HEL, OCI-AML5, and SIG-M5 cell lines were from DMSZ (Braunschweig, Germany). ISM-M2 cells was a gift from Yogen Saunthararajah of Cleveland Clinic and authenticated by short tandem repeat (STR) assay. Normal bone marrow was cultured in IMDM with 10% FBS, 100 U/ml pen-strep, and 10 ng/ml each of SCF, FLT3L, IL3, IL6 and TPO. Mouse bone marrow was cultured in IMDM with 10% FBS, 100 U/ml pen-strep, 50 ng/ml mSCF, 10 ng/ml mIL3, and 10 ng/ml mIL6. SIGM5 was cultured in IMDM while all other leukemic cells were grown in RPMI with 10% FBS and 100 U/ml pen-strep. Medium was supplied with 2 ng/ml recombinant human GM-CSF. Cells were confirmed Mycoplasma negative by using MycoAlert Mycoplasma Detection Kit (Lonza, Catalog #: LT07–118).
Reagents for cell experiments
RPMI-1640 and IMDM cell media was purchased from Invitrogen. FBS was purchased from ATLANTA biologicals (Cat. No.: S11150). Pen/Strep was purchased from Lerner Research Institute Media Core in Cleveland Clinic. Human and murine recombinant growth factors were purchased from PeproTech. MethoCult H4435 and M3434 was purchased from StemCell Technologies (Cambridge, MA). Antibody information is provided as Table S7.
IDH1 and IDH1R132C inducible cell lines generation
Tetracycline-inducible IDH1 and IDH1R132C were generated based on a previously published construct(42),(43). IDH1 was amplified from IDH1 in pDONR221 (Harvard Medical School, Plasmid HsCD00043452) and then R132C mutation was introduced, using In-Fusion HD Cloning Plus CE kit (Takara, 638916). Tet-system approved FBS (Atlanta Biological) was used for doxycycline inducible cell lines.
Measurement of metabolite by LC-MS/MS
5×106 cells were harvested by centrifugation, washed with PBS and re-suspended in 0.5 mL of chilled 80% methanol (20% ddH2O) by dry ice. The cell suspension was freeze-thawed for two cycles on dry ice and centrifuged twice for 15 min at 4 °C at 15,000 g and then the supernatant was collected, dried by N2 and dissolved in 100 uL HPLC grade water. The solution was filtered by 0.22 μm Eppendorf filter and used for LCMS/Ms. Shimadzu LCMS-8050 with a C18 column (Prodigy, 3 μM, 2 × 150mm, Phenomenex) was used for LC-MS/MS following the gradient: 0–2 min 0% B; 2–8 min 0% B to 100% B; 8–16 min 100% B; 16–16.1 min 100% B to 0% B; 16.1–24 min 0% B. Mobile phase A was water + 5 mM AmAc. Mobile phase B was Methanol+ 5 mM ammonium acetate. The flow rate was 0.3mL/min. The MRM transition for 2-Hydroxyglutaric acid was 147>85.
Viable cell count and doubling time measurement
Viable cell count was performed by methylene blue exclusion on vi-cell counter (Beckman Coulter).
CellTiter-Glo based cell proliferation assay for doubling time measurement
Cells were cultured in 96 well plates (Corning, catalog number 3610) at 8000 cells per 100 μl of culture media for each well. Relative Cell number was calculated by using CellTiter-Glo assay (Promega). One to one of 1X CellTiter-Glo: 1X PBS was mixed and then 40uL/ well was added. Plates were shaken for 10 minutes to ensure complete lysis before CellTiter-Glo signal reading. Reading was performed every 24 hrs for 4 days with new plates for each time point. There were 6 replicates per condition. Experiments were repeated for three times. Doubling time was calculated with exponential growth equation in GraphPad Prism 8.
Generation TET dioxygenase knockout cells using CRISPR-Cas9
Zhang lab LentiCRISPR plasmid was used for CRISPR-Cas9 mediated TET2 gene knockout. 5’-caccgGGATAGAACCAACCATGTTG-3’ and 5’-aaacCAACATGGTTGGTTCTATCCc-3’ DNA oligos was ordered from IDT and was used to for cloning. Vector cloning, virus production and virus infection was performed according to the reference paper(44). Virus infected K562 cells were seeded in 96 well plates at the density of 0.5 cell per well and colonies grown from single cells were kept for expansion and analysis. DNA was extracted for sequencing. Sequencing libraries were prepared using Illumina’s Nextera Custom Enrichment panel and sequenced with the HiSeq 2000. Variants were extracted according to GATK Best Practices. The sequencing panel covers all the exons of 170 genes, including TET2, commonly mutated in myeloid malignancies as previously described(7). TET1 and TET3 knockout cells were generated by using IDT genome editing with CRISPR-Cas9 system according to the manufacturer’s protocol. The gRNA details and sequencing results are summarized in supplementary Table S4.
RNAseq and analysis
RNA was purified by using NucleoSpin RNA kit ((Takara Bio USA, Inc Cat #740955) according to the manufacturer’s instruction. RNA quality was validated by RNA integrity number (RIN >9) calculated by Agilent 2100 Bioanalyzer. mRNA was enriched using oligo(dT) beads and then was fragmented randomly by adding fragmentation buffer. The cDNA was synthesized by using mRNA template and random hexamers primer, after which a custom second-strand synthesis buffer (Illumina, dNTPs, RNase H and DNA polymerase I) was added to initiate the second-strand synthesis. After a series of terminal repair, a ligation and sequencing adaptor ligation, the double-stranded cDNA library is completed through size selection and PCR enrichment. The qualified library was fed into Illumina sequencer after pooling and more than 30 million reads was acquired for each sample. The quality of RNA-seq raw reads were checked using FastQC (Galaxy Version 0.72). Raw reads were mapped to human genome, hg19, using RNA STAR. Differential expression and gene set enrichment analysis was assessed using edgeR 3.24.3 and limma 3.38.3 with R 3.5. Computational analysis was performed using Galaxy server (https.usegalaxy.org). The RNAseq data was submitted to the Gene Expression Omnibus (GEO) repository at the National Center for Biotechnology Information (NCBI) archives with assigned GEO accession numbers of GSE162487.
Computational docking and small molecule
The crystal structure of TET2 catalytic domain (TET2CD) in complex with DNA and aKG pseudo substrate NOG and DNA oligo (protein data bank ID 4NM6) was used for all docking simulations and structural activity analysis of TETi by Glide running in Schrodinger Maestro environment. The complex was minimized and the binding site was analyzed in UCSF Chimera 1.8.
TET2CD protein purification
GST-TET2 (1099–1936 Del-insert)(45) expression vector was transformed into Escherichia coli strain BL21(DE3)pLysS. The transformant was grown at 37 °C to an OD600 of 0.6 and switched to 16 °C for another two hours. Ethanol was added to the final concentration of 3% before expression induction by adding isopropyl-b-D-thiogalactopyranoside to the final concentration of 0.05 mM. Cells was cultured for 16 hours at 16 °C. Cells from 2 L of culture were harvested and lysed in 50 ml of lysis buffer (20 mM Tris-HCl pH7.6, 150 mM NaCl, 1X CelLytic B (Sigma C8740), 0.2 mg/ml lysozyme, 50 U/ml Benzonase, 2mM MgCl2, 1 mM DTT, 1X protease inhibitor (Thermo Scientific A32965)) for 30 minutes on ice. Lysate was sonicated by ultrasonic processor (Fisher Scientific FB-505 with ½” probe) with the setting of 70% amplitude and 18 cycles of 20 seconds on and 40 seconds off. Then the lysate was centrifuged twice at 40,000 g for 20 minutes each. Supernatant was filtered through the membrane with the pore size of 0.45 μm. Flowthrough was diluted 4 times with the solution of 20 mM Tris-HCl pH7.6, 150 mM NaCl. GST-TET2 was purified by GE Healthcare AKTA pure by affinity (GSTPrep FF16/10) and gel filtration (Superdex 200 increase 10/300 GL). For gel filtration, buffer of 10 mM phosphate and 140 mM NaCl, pH 7.4, was used. Protein was dialyzed in 50 mM HEPES, pH6.5, contains 10% glycerol, 0.1% CHAPS, 1 mM DTT, and 100 mM NaCl. GST tag was removed by TEV enzyme.
Dot-Blot assay for 5hmC and 5mC detection of genomic DNA
Genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega). DNA samples were diluted in ddH2O, denatured in 0.4 M NaOH/10 mM EDTA for 10 min at 95 °C, neutralized with equal volume of 2 M NH4OAc (pH 7.0), and spotted on a nitrocellulose membrane (pre-wetted in 1 M NH4OAc, pH 7.0) in two-fold serial dilutions using a Bio-Dot Apparatus Assembly (Bio-Rad). The blotted membrane was air-dried and cross-linked by Spectrolinker™ XL-1000 (120 mJ/cm2). Cross-linked membrane was blocked in 5% non-fat milk for 1 hour at room temperature and incubated with anti-5hmC (Active motif, 1:5,000) or anti-5mC (Eurogentec, 1:2,500) antibodies at 4 °C overnight. After 3× 5 min washing with TBST, the membrane was incubated with HRP-conjugated anti-rabbit or anti-mouse IgG secondary antibody (Santa Cruz), treated with ECL substrate and developed using film or captured by ChemiDoc MP Imaging System (Bio-Rad). Membrane was stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) to ensure equal loading of input DNA.
ELISA assay of 5hmC for TET activity detection
The 96-well microtiter plate was coated with 10 pmol avidin (0.66 μg, SIGMA A8706) suspended in 100 μl 0.1 M NaHCO3, pH 9.6, overnight. Biotin labelled DNA substrate (10 pmol) was added for 2 hours. The 60 bp duplex DNA substrate (forward strand: 5’-ATTACAATATATATATAATTAATTATAATTAACGAAATTATAATTTATAATT AATTAAT A-3’ and reverse strand: 5’-Bio-TATTAATTAATTATAAATTATAATTTmCGTTAATTAT AATTAATTATATATATATTGTAAT-3’) was synthesized by IDT. TET2CD, TET1CD (Epigentek, Catalog No. E12002–1) or TET3CD (BPS Bioscience, Catalog # 50163) protein (0.1 μM) in 100 μl reaction buffer containing 50 mM HEPES (pH 6.5), 100 mM NaCl, 1 mM DTT, 0.1 mM ascorbate, 25 μM Fe(NH4)2(SO4)2, and 10 μM αKG was added to each well for 2 hours in 37 °C. Concentrations of Fe(NH4)2(SO4)2 and alpha-ketoglutarate are indicated in the relative figure if different than previously stated. Reaction was stopped by incubating with 100 μl of 0.05 M NaOH on a shaking platform for 1.5 hours at room temperature. After washing, the wells were blocked with 2% BSA dissolved in TBST for 30 minutes, and incubated with anti-5hmC antibody (Active motif, 39769, 1: 3,000) at 4 °C overnight. After washing with TBST, the wells were incubated with HRP-conjugated anti-rabbit secondary antibody (Santa Cruz). Signal was developed by adding TMB (SIGMA, T4444). Reaction was stopped by adding 2M H2SO4.
Label free thermophoresis assay to measure the binding of TET2CD and TETi76
Binding interaction of TETi 76 with TET2CD was monitored using label-free Microscale Thermophoresis (MST)(46). Briefly, TET2CD (0.5 μM) recombinant protein was mixed with different concentrations of TETi76 in modified TET2 reaction buffer without αKG containing 50 mM HEPES (pH 6.5), 100 mM NaCl, 1 mM DTT, 0.1 mM ascorbate and 25 μM Fe(NH4)2(SO4)2, then was loaded to capillary (NanoTemper, MO-Z022) and measurements were made using the Monolith NT.115 (NanoTemper). Data was analyzed using MO.Affinity analysis V2.3 software using in built Kd model for data fitting and calculation of dissociation constant. GraphPad Prism 8.0.2 was used for plotting data.
Dioxygenase inhibition screen
The Dioxygenase inhibition screen was performed by using alpha screen of BPS bioscience (https://bpsbioscience.com).
TET3 Dox-inducible knockdown by shRNA
Dox inducible shRNA lentivirus vector EZ-Tet-pLKO-Blast was used for cloning. The vector was a gift from Cindy Miranti (Addgene plasmid # 85973). Primers 5’-CTAGCGAACCTTCTCTTGCGCTATTTTACTAGTAAATAGCGCAAGAG AAGGTTCTTTTTG-3’ and 5’-AATTCAAAAAGAACCTTCTCTTGCGCTATTTACTAGTAAAAT AGCGCAAGAGAAGGTTCGA-3’ were purchased from IDT. The procedures in a reference paper(47) was followed for cloning, lentivirus production, and stable cell line selection.
Quantitative real-time PCR
Total RNA was extracted from cells using NucleoSpin RNA kit (Takara Bio USA, Inc Cat #740955). The purity of RNA was confirmed by the 260/280 absorption ratio on Nanodrop. cDNA synthesis was performed by using iScript cDNA Synthesis Kit (Bio-Rad, 1708890). 500 ng of total RNA was used as template. qRT-PCR was performed by using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Cat. 1725270) on a CFX96 real-time PCR detection system (Bio-Rad). Primers 5’-CGATTGCGTCGAACAAATAG-3’ and 5’-CTCCTT CCCCGTGTAGATGA-3’ was used for TET3 detection. Primers 5’-CTCCTCCTGTTCGACAGT CAGC-3’ and 5’-CCATGGAATTTGCCATGGGTGG-3’ were used for GAPDH detection. The values obtained for the target gene expression were normalized to GAPDH.
Chemical synthesis of TETi
All novel TET-inhibitors (TETi) were synthesized in-house and purified by using by solid phase extraction on Silica flash column (All di-esters) and C18 sep pak column using Water/Acetonitrile as mobile phase for all corresponding acid or di-lithium salt. The derivatives were synthesized with modified methodology from the previously published literature(48, 49). We used barbier reaction method with acetic acid instead of ammonium chloride, which gave better yield. We used the Dess-Martin-Periodinane reagent for the oxidation and Pd/C hydrogenation for the reduction of the respective hydroxyl and methylene group. The details of the chemistry are attached as a supporting evidence. Briefly, the compounds were characterized by 1HNMR (500 MHz Bruker Asend Avance III HD at room temperature) 13CNMR (125MHz for 13C NMR), in the dutrated solvent and high resolution mass spectrometry was performed on an Agilent Q-TOF. The purity of compounds was always >95%, based on the combination of 1HNMR and chromatography. Detail synthesis protocol is provided as Supplementary Methods. TETi di-esters stock solutions were dissolved in dry DMSO and acid or di-lithium salts were dissolved in ultrapure nuclease and proteinase free water for all in vitro cell culture or cell free assays respectively. The final DMSO content in all assays were always maintained ≤ 0.1% v/v.
Annexin V and Propidium Iodide Staining
Cells were incubated with FITC-Annexin V (BD Pharmingen Catalog No.556420) in a binding buffer (BD Pharmingen catalog number 556454) containing propidium iodide (PI, BD Pharmingen catalog number 556463) and analyzed by flow cytometry (BD FACSVerse).
Mouse Studies
Animal care and procedures was conducted in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee (IACUC), Cleveland Clinic. Tet2 mutant mice (Stock# 023359) were procured from Jackson lab. In cell line derived tumor xenograft, 1×106 doxycycline inducible IDH1R132C SIG-M5 cells or parental SIG-M5 cells were subcutaneously injected into each flank of NSG mouse. For inducible IDH1R132C SIG-M5 cells, mice were intraperitoneally injected with 10 mg/kg doxycycline (Millipore, Cat.No.198955). For parental SIGM5 cells, mice were orally administrated with 50mg/kg TETi76. In transplantation mouse model, recipient mice received two doses of 480 rad (4.8 Gy) irradiation delivered 3 hours apart; 2 million of a mixture of mononuclear cells from donor mice bone marrows (5% Tet2−/− cell, CD45.2; and 95% Tet2+/+, PEP, CD45.1) were injected into the tail veins of recipients after the 2nd irradiation. Mice was maintained on antibiotic food for 3 weeks. 50 ul blood was drew from each mouse for hematology profile analysis by using DREW HEMAVET 950FS and for ratio of CD45.1/CD45.2 analyses by flow cytometry using FITC-CD45.1 (Invitrogen, Catalog # 11–0453-82) and PE- CD45.2 (Invitrogen, Catalog # 12–0454-82). Once mice were fully recovered and engraftment of was confirmed, TETi treatment (25 mg/kg) was started by intraperitoneal injection. All the treatment was performed once a day, five days a week.
Supplementary Material
Statement of significance.
Loss of function somatic TET2 mutations are among the most frequent lesions in myeloid neoplasms and associated disorders. Here we report a novel strategy for selective targeting of residual TET-dioxygenase activity in TET-deficient clones that result in restriction of clonal evolution in vitro and in vivo.
Acknowledgements
We thank our colleagues at the Cleveland Clinic: Amy Graham at Flow Core for help with flow cytometer, Bartlomiej Przychodzen for help with the RNAseq, Dr. Renliang Zhang at Small Molecule Mass Spectroscopy Core for mass spectrometry experiments, Dr. Smarajit Bandyopadhyay at Molecular Biotechnology Core for help with thermophoresis experiment and Phillip Maciejewski for proof reading. This work was supported in parts, by grants from the NIH/NHLBI (R35HL135795-01 and R01132071-01), Leukemia and Lymphoma Society (LLS) TRP, and LLS SCOR and Taub Foundation to JPM and BKJ.
Conflict of Interest: JPM, BKJ, JGP, TR, YG and ADT are inventors on the patent application filed for small molecule TET modulators.
Data and material availability
Request of any specific raw data or material used in this study but not commercially available can be made to the corresponding authors.
References
- 1.Andre F, Mardis E, Salm M, Soria JC, Siu LL, Swanton C. Prioritizing targets for precision cancer medicine. Annals of oncology : official journal of the European Society for Medical Oncology. 2014;25(12):2295–303. [DOI] [PubMed] [Google Scholar]
- 2.Strichman-Almashanu LZ, Lee RS, Onyango PO, Perlman E, Flam F, Frieman MB, et al. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome research. 2002;12(4):543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Molecular cell. 2014;56(2):286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, Ozark PA, Smith ER, Zhao Z, Marshall SA, Rendleman EJ, et al. TET2 coactivates gene expression through demethylation of enhancers. Science advances. 2018;4(11):eaau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schubeler D Function and information content of DNA methylation. Nature. 2015;517(7534):321–6. [DOI] [PubMed] [Google Scholar]
- 6.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirsch CM, Nazha A, Kneen K, Abazeed ME, Meggendorfer M, Przychodzen BP, et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia. 2018;32(8):1751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nature genetics. 2017;49(2):204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Molecular cell. 2014;56(2):286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan F, Wingo TS, Zhao Z, Gao R, Makishima H, Qu G, et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nature communications. 2017;8:15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. 2017;170(6):1079–95 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 2017;549(7673):476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun J, He X, Zhu Y, Ding Z, Dong H, Feng Y, et al. SIRT1 Activation Disrupts Maintenance of Myelodysplastic Syndrome Stem and Progenitor Cells by Restoring TET2 Function. Cell stem cell. 2018;23(3):355–69 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang YW, Wang Z, Xie W, Cai Y, Xia L, Easwaran H, et al. Acetylation Enhances TET2 Function in Protecting against Abnormal DNA Methylation during Oxidative Stress. Molecular cell. 2017;65(2):323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan Y, Greenberg EF, Hasipek M, Chen S, Liu X, Kerr CM, et al. Context dependent effects of ascorbic acid treatment in TET2 mutant myeloid neoplasia. Commun Biol. 2020;3(1):493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen LL, Lin HP, Zhou WJ, He CX, Zhang ZY, Cheng ZL, et al. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell reports. 2018;25(6):1485–500 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu Y, Zhao Z, Sant DW, Zhu G, Greenblatt SM, Liu L, et al. Tet2 Regulates Osteoclast Differentiation by Interacting with Runx1 and Maintaining Genomic 5-Hydroxymethylcytosine (5hmC). Genomics, proteomics & bioinformatics. 2018;16(3):172–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai HQ, Wang BA, Yang L, Chen JJ, Zhu GC, Sun ML, et al. TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature. 2016;538(7626):528–32. [DOI] [PubMed] [Google Scholar]
- 22.Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Developmental cell. 2014;29(1):102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18(6):553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das AT, Zhou X, Metz SW, Vink MA, Berkhout B. Selecting the optimal Tet-On system for doxycycline-inducible gene expression in transiently transfected and stably transduced mammalian cells. Biotechnol J. 2016;11(1):71–9. [DOI] [PubMed] [Google Scholar]
- 25.Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell. 2018;172(1–2):90–105 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waitkus MS, Diplas BH, Yan H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer cell. 2018;34(2):186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Annals of oncology : official journal of the European Society for Medical Oncology. 2016;27(4):599–608. [DOI] [PubMed] [Google Scholar]
- 28.Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer cell. 2016;30(6):940–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell. 2011;20(1):11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Churchill H, Naina H, Boriack R, Rakheja D, Chen W. Discordant intracellular and plasma D-2-hydroxyglutarate levels in a patient with IDH2 mutated angioimmunoblastic T-cell lymphoma. International journal of clinical and experimental pathology. 2015;8(9):11753–9. [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015;126(15):1741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemonnier F, Cairns RA, Inoue S, Li WY, Dupuy A, Broutin S, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci U S A. 2016;113(52):15084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Lan Y, Schwartz-Orbach L, Korol E, Tahiliani M, Evans T, et al. Overlapping Requirements for Tet2 and Tet3 in Normal Development and Hematopoietic Stem Cell Emergence. Cell Rep. 2015;12(7):1133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ. 2-Oxoglutarate-Dependent Oxygenases. Annual review of biochemistry. 2018;87:585–620. [DOI] [PubMed] [Google Scholar]
- 35.An J, Gonzalez-Avalos E, Chawla A, Jeong M, Lopez-Moyado IF, Li W, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun. 2015;6:10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velasco-Hernandez T, Sawen P, Bryder D, Cammenga J. Potential Pitfalls of the Mx1-Cre System: Implications for Experimental Modeling of Normal and Malignant Hematopoiesis. Stem Cell Reports. 2016;7(1):11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez-Aguilera A, Arranz L, Martin-Perez D, Garcia-Garcia A, Stavropoulou V, Kubovcakova L, et al. Estrogen signaling selectively induces apoptosis of hematopoietic progenitors and myeloid neoplasms without harming steady-state hematopoiesis. Cell Stem Cell. 2014;15(6):791–804. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525(7569):389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaasinen E, Kuismin O, Rajamaki K, Ristolainen H, Aavikko M, Kondelin J, et al. Impact of constitutional TET2 haploinsufficiency on molecular and clinical phenotype in humans. Nature communications. 2019;10(1):1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh AK, Zhao B, Liu X, Wang X, Li H, Qin H, et al. Selective targeting of TET catalytic domain promotes somatic cell reprogramming. Proceedings of the National Academy of Sciences of the United States of America. 2020;117(7):3621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagata Y, Narumi S, Guan Y, Przychodzen BP, Hirsch CM, Makishima H, et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood. 2018;132(21):2309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shima H, Koehler K, Nomura Y, Sugimoto K, Satoh A, Ogata T, et al. Two patients with MIRAGE syndrome lacking haematological features: role of somatic second-site reversion SAMD9 mutations. J Med Genet. 2018;55(2):81–5. [DOI] [PubMed] [Google Scholar]
- 44.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155(7):1545–55. [DOI] [PubMed] [Google Scholar]
- 46.Sparks RP, Fratti R. Use of Microscale Thermophoresis (MST) to Measure Binding Affinities of Components of the Fusion Machinery. Methods in molecular biology. 2019;1860:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank SB, Schulz VV, Miranti CK. A streamlined method for the design and cloning of shRNAs into an optimized Dox-inducible lentiviral vector. BMC Biotechnol. 2017;17(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Chen H, Laurini E, Wang Y, Dal Col V, Posocco P, et al. 2-Difluoromethylene-4-methylenepentanoic Acid, A Paradoxical Probe Able To Mimic the Signaling Role of 2-Oxoglutaric Acid in Cyanobacteria. Organic Letters. 2011;13(11):2924–7. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Wang Y, Laurini E, Posocco P, Chen H, Ziarelli F, et al. Structural Requirements of 2-Oxoglutaric Acid Analogues To Mimic Its Signaling Function. Organic Letters. 2013;15(18):4662–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.