

Abstract
The incidence of autism spectrum disorder (ASD) has been rising, however ASD-risk biomarkers remain lacking. We previously identified the presence of maternal autoantibodies to fetal brain proteins specific to ASD, now termed maternal autoantibody-related (MAR) ASD. The current study aimed to create and validate a serological assay to identify ASD-specific maternal autoantibody patterns of reactivity against 8 previously identified proteins (CRMP1, CRMP2, GDA, NSE, LDHA, LDHB,STIP1 and YBOX) that are highly expressed in developing brain, and determine the relationship of these reactivity patterns with ASD outcome severity. We used plasma from mothers of children diagnosed with ASD (n=450) and from typically developing children (TD, n=342) to develop an ELISA test for each of the protein antigens. We then determined patterns of reactivity a highly significant association with ASD, and discovered several patterns that were ASD-specific (18% in the training set and 10% in the validation set vs 0% TD). The three main patterns associated with MAR ASD are CRMP1 + GDA (ASD%= 4.2 vs TD%=0, OR 31.04, p= <0.0001), CRMP1+ CRMP2 (ASD%= 3.6 vs TD%=0, OR 26.08, p= 0.0005) and NSE + STIP1 (ASD%= 3.1 vs TD%=0, OR 22.82, p= 0.0001). Additionally, we found that maternal autoantibody reactivity to CRMP1 significantly increases the odds of a child having a higher Autism Diagnostic Observation Schedule (ADOS) severity score (OR 2.3; 95% CI: 1.358– 3.987, p= 0.0021). This is the first report that uses machine learning sub-group discovery to identify with 100% accuracy MAR ASD-specific patterns as potential biomarkers of risk for a subset of up to 18% of ASD cases in this study population.
INTRODUCTION
Autism spectrum disorder (ASD) is characterized by social and behavioral impairments, along with restricted interests and repetitive behaviors. In 2018, the CDC estimated that 1 in 59 children are affected in the USA 1, making ASD an important health concern and a substantial socioeconomic burden for affected families and the healthcare system 2, 3.
We previously described specific maternal autoantibody reactivity against seven proteins highly expressed in the developing brain including collapsin response mediator proteins 1 and 2 (CRMP1, CRMP2), guanine deaminase (GDA), lactate dehydrogenase A and B (LDHA, LDHB), stress-induced phosphoprotein-1 (STIP1) and Y-box binding protein 1 (YBOX). These earlier studies identified autoantibody reactivity against these antigens by Western blot in plasma from mothers whose children were diagnosed with ASD (23%) with only 1% in the typically developing group (TD) 4. More recently, we discovered an additional target autoantigen, neuron-specific enolase (NSE) 5. Further, we performed autoantibody epitope mapping for each of the eight antigens and found peptide sequences recognized only by maternal samples from the ASD group5, 6. We have termed this subtype of ASD as Maternal Autoantibody Related (MAR) autism.
In the current study, our primary goal was to improve upon our previous findings through the development of a highly accurate and specific ELISA test for the assessment of maternal autoantibody reactivity against the eight antigens, thus enabling the ability to predict the risk of having a child with ASD. To achieve this goal, we used machine learning (ML) techniques to identify and evaluate the precision for the patterns of reactivity to the eight autoantigens. Success in the current study will allow the future development of this technology and exploration of these autoantibody patterns as predictors of an ASD diagnosis.
MATERIALS AND METHODS:
Study subjects
Biologic samples for this study were from mothers enrolled in the Childhood Autism Risks from Genetics and Environment (CHARGE) study 7. This project included mothers of children diagnosed with ASD (n=450) and of children selected from the general population and evaluated as neurotypical (typically developing, TD; n=342). The participants provided written consent and fulfilled the recruitment and eligibility criteria. All the children underwent diagnostic evaluation including medical, social and cognitive assessment as previously described 7, 8. The demographic information related to these samples is shown in Supplementary Table 1.
Sample collection and preparation
Maternal blood was collected in citrate dextrose (BD Diagnostic) and plasma was separated, labeled, aliquoted, and stored at −80˚C. Prior to use, samples were thawed at room temperature (RT), vortexed and centrifuged at 13,000 RPM for 10 minutes.
Experimental groups
The samples were randomly divided into two experimental groups: 1) The training set (n=375; ASD=206, TD=169), to determine the reactivity patterns and evaluate the association between reactivity and diagnosis (ASD), and 2) the validation set (n=418; ASD=244, TD=174) which served to corroborate the patterns discovered using the training set and the association of those patterns with an ASD diagnosis.
Proteins
An important aspect of the recombinant proteins used to build this assay was the removal of the His-Tag from the antigens due to the non-specific binding by human plasma samples to His-Tag used in prokaryotic expression systems. The tag-less proteins CRMP1 #MBS7074427 and LDHA #MBS949692 were from MyBioSource (San Diego CA), GDA # NBP2–49692, LDHB #NBP2–49694, NSE #NBC1–18342 and STIP1 #NBP2–49685 were from Novus Biologicals (Centennial, CO), while CRMP2 and YBOX were custom made by Expression Systems using a baculovirus system (Davis, CA).
Enzyme Linked Immunosorbent Assay (ELISA)
Autoantibody reactivity of plasma samples against protein antigens was determined by Enzyme-Linked Immunosorbent Assay (ELISA) and corroborated by Western Blot (WB) using commercially available proteins as previously described 5. The protein concentration and plasma sample dilutions were optimized for each antigen for both assays. In summary, microplates were coated with 100μl of antigen (1.5–3µg/µl) in carbonate coating buffer pH 9.6, incubated overnight at 4°C, washed four times with Phosphate Buffered Saline Tween-20 (PBST) 0.05%, and blocked with 2% Super Block (Thermo Scientific, Rockford, lL) for 1 hour at RT. The plasma samples were diluted 1:250–1:1000, and run in duplicate. Following dilution, 100 µl of the diluted sample was added to each well, incubated for 1.5 hours, washed 4 times in PBST 0.05%, and then washed four times with (PBST) 0.05%, and incubated with goat anti-human IgG-HRP IgG (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MA) diluted 1:10,000 for 1 hour. The plates were then washed four times with (PBST) 0.05%, and detection was performed by adding 100 µl of BD optEIA liquid substrate for ELISA (BD Biosciences, San Jose, CA). After 4 minutes, the reaction was stopped with 50µl of 2N HCl. The absorbance was measured at 490–450 nm using an iMark Microplate Absorbance Reader (Biorad, Hercules, CA, USA). Of note, we only examine IgG reactivity since it is the only isotype able to cross the placenta, which is a key component of the MAR ASD mechanism.
Statistical Analysis
Receiver Operating Characteristic (ROC) curve
For the ELISA assay, a positive cutoff value for reactivity to each antigen was determined using an ROC curve as previously described 5. Youden’s index was used to calculate the specific numerical threshold cutoff for each protein, for each set after plate-plate normalization. The cut-off was optimized for each set and antigen 9, 10. The seven control-positive samples that were used to create the ROC curves were not included in the pattern discovery analysis.
Pattern identification:
We identified as positive samples that were reactive for a combination of 2 or more of the 8 antigens and that were perfectly specific for ASD diagnosis and compared these patterns of reactivity between TD and ASD groups by Fisher Exact test (P<0.05). To determine if the association of specific patterns of reactivity with ASD outcome was greater than would be obtained by chance, we conducted a permutation analysis 11, 12. All permutation analysis was done using SAS® software version 9.4 (Cary, NC).
To detect high-precision ASD indicator patterns, we employed methods from subgroup analysis and exceptional model mining. In particular, we applied a depth-first search algorithm to identify patterns based on a family of interestingness measures that judges the quality of pattern by weighing its precision against its support 13, 14. We focused on patterns with high precision by setting the weighting parameter a=0.01. For the current study, we limited the length of the patterns to three. The results of this analysis are shown in Supplementary Tables 2 and 3. To visualize the relations between patterns, we built a dependency network, where each pattern is represented by a binary vector based on the instances it covers. Based on this, the patterns are then embedded into a two-dimensional space via PCA. The patterns are connected based on the relationship between their descriptions and covered instances. This enables deeper insights into which patterns may be redundant with regard to their descriptions or related with regard to the samples they cover. Finally, we evaluated several predictive models, one based directly on the patterns we have found as well as several multi-variate models that are based on state-of-the-art machine learning approaches. These models were trained and tested on the previously defined training and validation set. Parameter optimization was solely performed on the training set. For the pattern-based algorithm, we extracted top-k patterns from the training set and predicted ASD for a new subject if any of these patterns matched this subject’s reactivity profile.
ADOS severity score correlation
To investigate if autoantibody reactivity was relevant to the ADOS severity score, we analyzed 254 ASD samples that had autoantibody reactivity to at least one antigen. We used a least absolute shrinkage and selection operator (LASSO) ordinal logistic regression model to determine important autoantibody-antigen combinations that describe the ADOS severity score. LASSO helped increase model interpretability by eliminating irrelevant autoantibody-antigens that were not associated with ADOS severity score and reduced model overfitting. We assessed the model predictive performance using the Schwarz Bayesian criterion (SBC) and cross validation. Once relevant interactions were selected, we used a generalized ordinal logistic regression model for ordered outcome (1–10). Computing was done in SAS® software version 9.4 (Cary, NC).
RESULTS:
Autoantibody reactivity against fetal brain antigens:
As mentioned previously, we divided the samples into two experimental sets 1) the training set to determine autoantibody reactivity combinations associated with an ASD outcome, and 2) the blinded validation set to test the accuracy of patterns identified in the training set for prediction of ASD. Supplementary Table 4 shows a summary of maternal antibody reactivity to the eight target autoantigens.
For the training set, we found that of 375 samples, 229 had autoantibody reactivity to at least one antigen (ASD=134, 65% and TD= 96 57%; p= 0.1108), demonstrating that autoantibodies against brain-antigens are not individually correlated with diagnosis, as previously observed 4. Additionally, we did not observe any differences in the reactivity level (low, medium, high) to the individual protein-antigens between the ASD and TD groups. However, we found several ASD-specific autoantibody reactivity patterns for combinations of two or more antigens (Supplementary Table 2). To identify and validate these patterns of reactivity, we performed ML subgroup discovery analysis, evaluating all possible combinations containing at least of three of the eight antigens (CRMP1, CRMP2, GDA, LDHA, LDHB, NSE, STIP1 and YBOX). We then evaluated the association of a particular pattern with ASD by Fisher’s Exact Test of Independence. Supplementary Table 5 shows the performance of the ASD specific patterns found in the training set when tested on the validation set.
Pattern discovery:
The patterns of reactivity discovered via subgroup analysis are tightly interconnected in their description as well as in what samples they cover, shown in Figure 1. These patterns of reactivity were used to build a high precision model to predict ASD outcome (quality analysis; supplementary Figure 1). Each node represents a pattern; nodes that are close together cover similar sample sub-populations and the node size is directly proportional to the number of samples covered.
Figure 1: Dependency network of maternal autoantibody reactivity patterns.
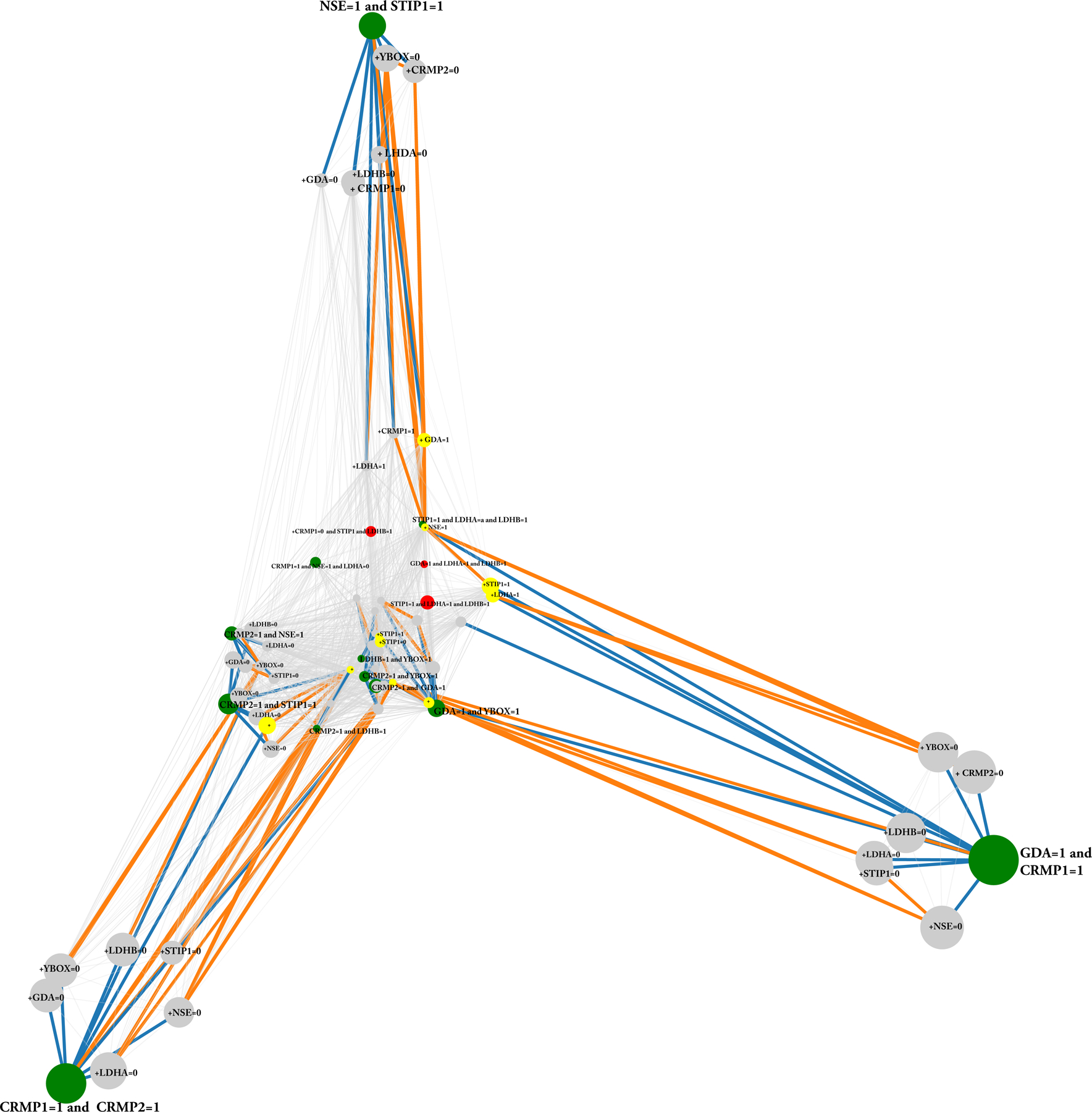
This network shows the top 70 patterns of autoantibody reactivity predictive of autism spectrum disorder. Each node represents a pattern. The closer two nodes are, the more similar are the sets of samples they cover. The bigger a node and the label, the more samples are covered by the corresponding pattern. Patterns identified by green nodes and bold fonts have 100% precision in the training as well as in the validation set; Grey patterns are patterns that also have 100% precision in the training and the validation set, but are sub-patterns of at least one a green pattern, i.e., they cover a subset of samples of the green parent pattern (given this relation, they may be considered redundant); Red patterns are patterns that are perfect with regard to precision in the training set but some samples fail in the validation set. Yellow patterns are also 100% precise in the training set but are absent in the validation set. Orange connections mean that the set of samples the bigger of one of the connected patterns covers is a subset of the set of samples the smaller connected pattern covers, while blue connections mean that the description of one pattern is a subset of another pattern (note that blue implies orange). Abbreviations: CRMP1 and CRMP2, collapsin response mediator 1 and 2; GDA, guanine deaminase; NSE, neuron specific enolase; LDHA-B, lactate dehydrogenase A and B; STIP1, stress induced phosphoprotein 1 and YBOX, Y-box binding protein 1. We see that ASD is identified by three major patterns namely “CRMP1=1 AND CRMP2=1”, “STP=1 AND NSE=1” as well as “CRMP=1 AND GDA=1”. Most of the other patterns are sub-patterns or represent only a very small set of covered samples.
Green nodes represent patterns that have 100% precision in the training and in the validation set, and are the most abundant (CRMP1+CRMP2, CRMP1+GDA, and NSE+STIP1). Grey nodes are patterns that also have 100% precision in the training and the validation sets and are sub-patterns of the green patterns. Red and yellow nodes are perfect patterns (patterns that are only found in the ASD population) in the training set, but some fail in the validation set (red) or are absent in validation set (yellow). These patterns are highly interconnected, and orange connections mean that the set of samples the smaller of the connected pattern covers is a subset of the set of samples the bigger pattern covers. Blue connections mean that the connected pattern is a subset of another pattern. The latter (description relation) implies the former (instance-relation). We see that three prominent pattern groups emerge at the outer areas of the network, with non-overlapping sample populations, while the other patterns are mostly specializations (sub-patterns) of these main antigen combinations. Table 1 shows the most representative patterns detected by subgroup discovery, with GDA + CRMP1, CRMP1 + CRMP2 and STIP1 + NSE (and sub-patterns containing reactivity to these antigens) as the main combinations associated with ASD outcome. These patterns cover up to 18% of the ASD cases with 100% precision in the training set, and up to 10% of the ASD cases in the validation data.
Table 1.
Summary of most relevant autoantibody-antigen reactivity combinations that are 100% specific with an ASD diagnosis in training and validation sets.
| Patterns | ASD + Training set n (%) | TD + Training set n (%) | Fisher exact p-value | ASD + Validation set n (%) | TD + Validation set n (%) | Training/ Validation precision (%) | Fisher exact p-value |
|---|---|---|---|---|---|---|---|
| CRMP1+GDA | 15 (7) | 0 (0) | 0.0002 | 4 (2) | 0 (0) | 100 | 0.1443 |
| CRMP1+CRMP2 | 12 (6) | 0 (0) | 0.0007 | 4 (2) | 0 (0) | 100 | 0.1443 |
| NSE+STIP1 | 8 (4) | 0 (0) | 0.0093 | 6 (2) | 0 (0) | 100 | 0.0435 |
| CRMP2+STIP1 | 6 (3) | 0 (0) | 0.0346 | 3 (1) | 0 (0) | 100 | 0.2693 |
| GDA+YBOX | 5 (2) | 0 (0) | 0.0669 | 1 (0) | 0 (0) | 100 | 1 |
| CRMP2+GDA | 4 (2) | 0 (0) | 0.1303 | 1 (0) | 0 (0) | 100 | 1 |
| CRMP2+NSE | 4 (2) | 0 (0) | 0.1303 | 3 (1) | 0 (0) | 100 | 0.2693 |
| LDHA+NSE+STIP1 | 3 (1) | 0 (0) | 0.2553 | 1 (0) | 0 (0) | 100 | 1 |
| CRMP1+GDA+LDHB | 3 (1) | 0 (0) | 0.2553 | 1 (0) | 0 (0) | 100 | 1 |
| CRMP2+YBOX | 3 (1) | 0 (0) | 0.2553 | 2 (1) | 0 (0) | 100 | 0.5129 |
| CRMP1+NSE+STIP1 | 3 (1) | 0 (0) | 0.2553 | 2 (1) | 0 (0) | 100 | 0.5129 |
| CRMP1+CRMP2+NSE | 3 (1) | 0 (0) | 0.2553 | 1 (0) | 0 (0) | 100 | 1 |
Abbreviations: ASD, Autism Spectrum Disorders; TD, Typically Developing. CRMP1 and CRMP2, collapsin response mediator proteins 1 and 2; GDA, guanine deaminase; NSE, neuron specific enolase; LDHA-B, lactate dehydrogenase A and B; STIP1, stress induced phosphoprotein 1 and YBOX, Y-box binding protein 1. The training set was composed of 375 samples (ASD=206, TD=169) and the validation set was composed of 418 samples (ASD= 244, TD= 174).
Fisher’s exact test (two-sided) was used to evaluate the association of the patterns with ASD diagnosis and p-values > 0.05 were bolded and considered significant.
Using the training set, we identified 12 autoantibody pattern combinations that were considered ASD-relevant based on having 3 or more positive ASD and no TD subjects with a particular pattern of reactivity and were also present in the validation set (Table 1). CRMP1 + GDA (n= 15, p=0.0002), CRMP1 + CRMP2 (n= 12, p=0.0007), NSE + STIP1(n= 8, p=0.0093), CRMP2 + STIP1(n= 6, p=0.0346) were the most highly represented combinations with the greatest statistical significance. We then used the validation sample set to evaluate pattern accuracy for the prediction of an ASD outcome.
In the validation set, the most abundant pattern was NSE + STIP1 (n= 6, p=0.0435), followed by CRMP1 + GDA and CRMP1 + CRMP2 (both n=4, p=0.1443), CRMP2+STIP1 and CRMP2+NSE (both n=3, p=0.2693) that were ASD-specific patterns in training and validation sets but did not reach statistical significance. Of interest, we identified STIP1 + YBOX as an ASD-specific pattern in the validation set (n= 7, p=0.0448), however, that pattern was not ASD specific in the training set (4 ASD vs 1 TD), resulting in 92% of samples having an ASD diagnosis when considering both sample sets (Table 2 and Supplementary Tables 5 and 6).
Table 2.
Summary of clinically relevant statistics of autoantibody-antigen reactivity combinations that are from 100 to 90% specific with ASD diagnosis in the training and validation set (combined data).
| Pattern | Training ASD + /subgroup | Validation ASD + /subgroup | ASD/ Subgroup | Precision | Precision drop | Total ASD % (n=450) | Fisher’s exact test p-value | OR (95% CI)a | OR p-value |
|---|---|---|---|---|---|---|---|---|---|
| CRMP1+GDA | 15/15 | 4/4 | 19/19 | 1.00 | 0.00 | 4.2 | <0.0001 | 31.04 (1.8678 to 516.0620) | 0.0166 |
| CRMP1+CRMP2 | 12/12 | 4/4 | 16/16 | 1.00 | 0.00 | 3.6 | 0.0001 | 26.08 (1.5596 to 436.4170) | 0.0233 |
| NSE+STIP1 | 8/8 | 6/6 | 14/14 | 1.00 | 0.00 | 3.1 | 0.0005 | 22.82 (1.3565 to 383.9379) | 0.0299 |
| CRMP2+STIP1 | 6/6 | 3/3 | 9/9 | 1.00 | 0.00 | 2 | 0.0064 | 14.78 (0.8573 to 254.8841) | 0.0637 |
| LDHA+YBOX | 1/1 | 5/5 | 6/6 | 1.00 | 0.00 | 1.3 | 0.0393 | 10.04 (0.5640 to 178.9532) | 0.1164 |
| LDHB+YBOX | 2/2 | 4/4 | 6/6 | 1.00 | 0.00 | 1.3 | 0.0393 | 10.04 (0.5640 to 178.9532) | 0.1164 |
| GDA+YBOX | 5/5 | 1/1 | 6/6 | 1.00 | 0.00 | 1.3 | 0.0393 | 10.04 (0.5640 to 178.9532) | 0.1164 |
| STIP1+YBOX | 4/5 | 7/7 | 11/12 | 0.92 | 0.08 | 2.4 | 0.0161 | 8.56 (1.1010 to 66.7020) | 0.0402 |
| CRMP1+STIP1 | 15/17 | 3/3 | 18/20 | 0.90 | 0.10 | 4 | 0.0022 | 7.10 (1.6371 to 30.8292) | 0.0088 |
Abbreviations: ASD, Autism Spectrum Disorders; OR: odds ratio; CI, confidence interval. CRMP1 and CRMP2, collapsin response mediator proteins 1 and 2; GDA, guanine deaminase; NSE, neuron specific enolase; LDHA-B, lactate dehydrogenase A and B; STIP1, stress induced phosphoprotein 1 and YBOX, Y-box binding protein 1.
Fisher’s exact (two-sided) was used to evaluate the association of the patterns with ASD diagnosis and p-values > 0.05 were bolded and considered significant.
A 0.5 continuity correction was applied to OR calculations for observations with zero cell counts, since all the patterns in this table are ASD specific, all the TD cells were equal to zero, the correction was applied to all OR calculations in this table.
Table 2 presents a summary of clinically-relevant statistics of autoantibody-antigen reactivity combinations that are 90–100% specific with ASD diagnosis in the training and validation sets. In order to evaluate the association of a given pattern with ASD, we used the Fisher Exact Test and calculated the odds ratios (ORs) with 95% confidence intervals (95% CIs) for each primary pattern (including the sub-patterns) from the entire sample set (ASD=450, TD=343). The ASD-specific combinations that had odd ratios ≥ 10 and were statistically significant (p=0.05) included CRMP1 + GDA (OR 31.04, 95% CI: 1.8678–516.0620, p<0.0001), CRMP1 + CRMP2 (OR 26.08, 95% CI: 1.5596–436.4170, p= 0.0005), NSE + STIP1 (OR 22.82, 95% CI: 1.3565–383.9379, p= 0.0001) and CRMP2 + STIP1 (OR 14.78, C95% CI: 0.8573– 254.8841, p= 0.0064). We found two patterns that were not 100% specific for ASD: STIP1 + YBOX (ASD=11 vs TD=1, p=0.0161) and CRMP1 +STIP1(ASD=18, TD=2, p=0.0022), with an ASD prediction accuracy of 92 and 90% respectively. The association of these two patterns with ASD was statistically significant in both cases.
ADOS Correlation:
To study the relationship between autoantibody reactivity against the 8 antigens with the ADOS severity score, we evaluated the 254 ASD samples that were positive for any given antigen using stepwise and LASSO selection models and calculated the odds ratio and 95% confidence interval. Out of the eight antigens, CRMP1 had the strongest correlation with ADOS severity, with an odds ratio of 2.3 (95% CI: 1. 358– 3.987, p= 0.0021), meaning that having autoantibodies against CRMP1 increases the risk of having a more severe overall ADOS score by 2.3 (Table 3).
Table 3.
Ordinal Logistic Regression for ADOS Severity
| Variable | Point Estimate | 95% Confidence Interval | p value |
|---|---|---|---|
| CRMP1 | 2.327 | 1.358 to 3.987 | 0.0021 |
| CRMP2 | 0.589 | 0.284 to 1.22 | 0.1541 |
| GDA | 0.702 | 0.421 to 1.172 | 0.1764 |
| LDHA | 1.011 | 0.601 to 1.702 | 0.9676 |
| LDHB | 0.947 | 0.544 to 1.649 | 0.8484 |
| NSE | 1.106 | 0.598 to 2.046 | 0.7479 |
| STP1 | 0.927 | 0.575 to 1.493 | 0.7544 |
| YBOX | 0.858 | 0.451 to 1.63 | 0.6391 |
Abbreviations: ADOS, Autism Diagnostic Observation, Schedule; CRMP1 and CRMP2, collapsin response mediator proteins 1 and 2; GDA, guanine deaminase; NSE, neuron specific enolase; LDHA-B, lactate dehydrogenase A and B; STIP1, stress induced phosphoprotein 1 and YBOX, Y-box binding protein 1; LASSO, Least Absolute Shrinkage and Selection Operator.
Stepwise and LASSO selection models were used to calculate the odds ratio and 95% confidence interval and p-values > 0.05 were bolded and considered significant
Discussion
Several groups have shown that the presence of deleterious maternal autoantibodies against fetal brain proteins can result in permanent neurodevelopmental and behavioral alterations in the progeny 15–23. The mechanisms and dynamics of how the maternal antibodies are able to cross the fetal blood brain barrier, transfer to the fetal brain parenchyma where are taken up by the neural progenitor cells to bind the intracellular targets is still unknown. Further, it has been proposed that autoantibodies against brain antigens can act as agonistic, antagonist or co-agonist antibodies on surface receptors, altering receptor signaling, fix complement and/or activating Fc surface receptors (cell death) 24.
To address the potential pathogenicity of the MAR autoantibodies, we previously created several animal models, both passive transfer models using human IgG reactive to the antigens25–28, as well as the creation of an endogenous mouse model in which we generated clinically-relevant autoantibodies in the dam prior to breeding29. In our MAR rodent models, we have not observed tissue damage histologically, but we have found that maternal autoantibodies affect progenitor cell maturation resulting in altered dendritic maturation. For example, in Martínez-Cerdeño et al., when biotin-labeled human ASD-specific IgG antibodies to LDHA, LDHB, STIP1, CRMP1 were injected into the mouse cerebral ventricles at embryonic day 14.5, we noted specific intracellular autoantibody deposition in radial glial stem cells, and further noted abnormal radial glial cell proliferation, maturation, and alteration of mature dendritic structure 30,31. These findings demonstrate that the maternal IgG antibodies can bind to their intracellular targets in vivo. The mechanism of this uptake by the proliferating radial glial cells is currently under investigation.
In the endogenous mouse model, the developing pups were exposed throughout gestation to pathogenic antibodies against LDHA, LDHB, STIP1, and CRMP1. Exposed pups showed ASD-like behavioral alterations, including reduced vocalizations, increased repetitive self-grooming, and aberrant social interactions 29, demonstrating for the first time the true pathological significance of these autoantibodies.
We have reported in each of our studies that reactivity to an individual autoantigen is present to some degree in both groups (ASD and TD) and does not correlate with an ASD diagnosis 4. Instead, reactivity to a combination of two or more autoantigens is necessary to determine an association of risk for ASD. This phenomenon, where detection of more than one autoantibody its necessary to accurately predict disease risk, has been reported for other autoimmune diseases, such as Type 1 diabetes 32.
Other groups have searched for individual IgG-targeted autoantigens that could serve as a biomarker for ASD. Lee et al. demonstrated the neurotoxic effects of gestational exposure to monoclonal anti-NMDAR (N-methyl-D-aspartate) that resulted in morphological alterations in the developing brain causing long-term cognitive effects in the exposed pups. However, these offspring did not exhibit the specific behavioral changes related to ASD 23. Maternal antibodies to contactin-associated protein-like 2 (CASPR2) have been reported to be associated with neurodevelopmental alterations and behavioral aberrations related to ASD 22. However, such an association was not observed in a recent Danish study concluding that maternal autoantibodies to CASPR2 were highly associated with child diagnosis of intellectual disability and/or psychological development disorders, but not with ASD 33. Therefore, antibodies that interfere with NMDAR and CASPR2 function appear to have profound effects in neurodevelopment, causing brain abnormalities (observed in murine models) and permanent behavioral aberrations, yet additional studies are necessary to evaluate their utility as ASD biomarkers 21, 23, 34.
The primary goal of the current study was to build upon our previous findings to develop an optimized, quantitative ELISA assay able to detect the presence of maternal autoantibodies specific to ASD risk using ML tools. We found that the most common MAR-ASD patterns were CRMP1+CRMP2, CRMP1+GDA, NSE+STIP1, and GDA + YBOX. These patterns were 100% accurate for the prediction of ASD in both the training and validation sets, suggesting that autoantibodies to these antigen combinations are highly related to an ASD diagnosis and have the potential to be used as biomarkers of MAR-ASD risk. As illustrated in Figure 2, the autoantigens are highly interconnected by their tissue expression, biological function, and/or structural similarities as represented in the STRING network 35. The target proteins are highly expressed in the developing brain, and play important roles in neurogenesis, metabolism, and homeostasis 15, 16, 18. Therefore, it is possible that antibody binding during this vulnerable period could impact proper protein functionality affecting significant neurodevelopmental pathways with a lasting effect on the developing brain.
Figure 2. STRING protein-protein interaction network of the known MAR-ASD antigens.
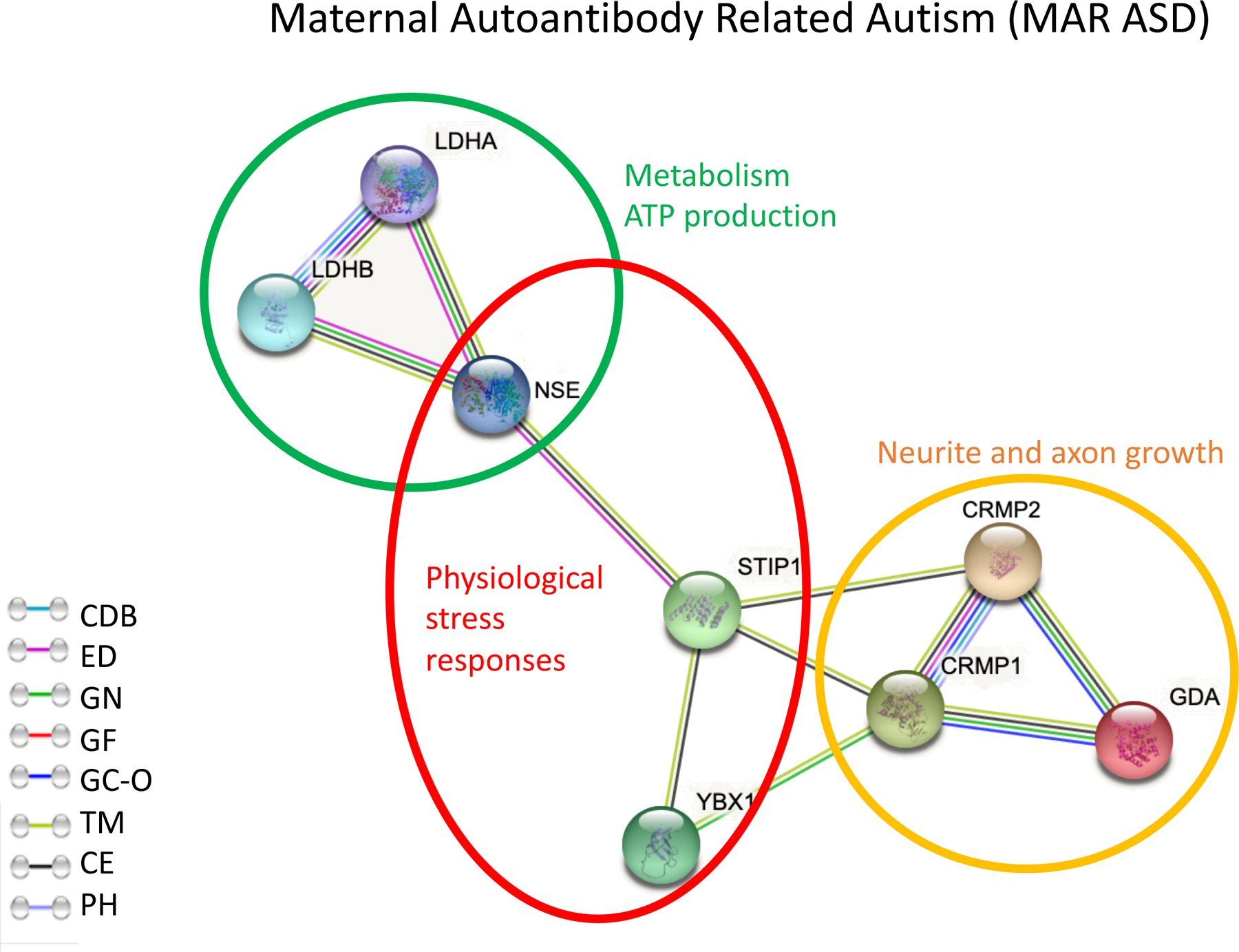
Each antigen is presented as a network node, and the edges represent meaningful protein-protein associations. Abbreviations. CDB; curated data bases, ED; experimentally determined, GN; gene neighborhood, GF; gene fusions, GC-O; gene co-occurrence, TM; text mining, CE; co-expression, PH; protein homology.
One of the primary MAR-ASD-specific patterns, CRMP1 and CRMP2 are involved in axon and neurite guidance in the nervous system 36,37,38. Knockout mice (CRMP1−/− and CRMP2 −/−) present behavioral deficits including hyperactivity, increased locomotion, and deficits in social behavior and memory 39. CRMP1/2 expression and phosphorylation has been proposed as a biomarker for several neuropsychiatric pathologies including Alzheimer disease, schizophrenia, and epilepsy 36. GDA (Cypin) has important catabolic and structural functions in neurons, thus regulating dendrite patterning and synaptic development and plasticity 40. Therefore, autoantibodies to GDA + CRMP1 -the most abundant pattern found in the current study- could interfere with two independent neurite and axon development pathways, which could have serious implications during neurodevelopment.
STIP1 modulates several biological process including physiological stress responses, signal transduction, transcription, and cell cycle regulation 41. These observations are complemented by animal studies where STIP1+/− mice (lower expression of STIP1) showed ASD-like behaviors including attention deficits and hyperactivity 42. These findings suggest that reduced expression or functionality of STIP1 could be used as a biomarker for ASD. NSE is an enolase enzyme with glycolytic activity involved in ATP, and has been shown to mediate the PI3K activation pathway, having neuroprotective or neurogenerative effects depending on the strength of the signal 43. As noted, NSE + STIP1 is the third most abundant ASD-specific pattern, and both proteins have been described to play important roles in neurodevelopment, brain homeostasis, and especially neuroprotection under physiologic stress conditions. Thus, interference in the function of both STIP1 and NSE could result in neurodevelopmental abnormalities and an insufficient response to cellular stress.
It was previously reported that maternal autoantibody reactivity to fetal brain antigens correlated with distinct ASD manifestations in the affected children8 including increased irritability and language deficits. One intriguing finding in the current study was the association between autoantibodies against CRMP1 and worse ASD manifestations based on the ADOS severity score. While additional studies are underway to better understand the endophenotypes within MAR-ASD as they relate to the various MAR patterns, our current findings serve as a strong foundation to further examine these interactions using a larger data set as well as looking at other ASD metrics and subcategories.
Although this is the largest study to date in terms of sample size for MAR-ASD, we are still limited in our ability to significantly detect ASD patterns with lower frequencies. Additionally, this is a retrospective study, as the samples were collected approximately 2–3 years after delivery and at the time of the child’s diagnosis. Recognizing the importance of a prospective analysis, we are currently evaluating the MAR-ASD patterns described herein in additional prospective studies as well as in geographically distinct study populations. Likewise, new animal models are underway to evaluate the patterns of reactivity described in the current clinical population allowing assessment of the pathogenic effect of antibodies against individual or specific combinations of antigens.
In conclusion, this is the first report that uses ML to identify a set of biomarkers that demonstrate an association with MAR-ASD with 100% accuracy. This is a novel serological risk assessment test for women at high risk of having a child with ASD; for example, those mothers that have previously had a child diagnosed on the spectrum or that have other ASD-associated maternal co-morbidities such as metabolic syndrome during pregnancy44. While the use of this technology in the clinical population will require substantial clinical validation and testing, this study provides a strong foundation for such studies in the future and provides a framework for understanding the biologic implications of MAR-autoantibodies in future animal models.
Supplementary Material
Acknowledgements
We would like to thank the families that participated in the CHARGE study, and the staff at the UC Davis MIND institute. We especially want to acknowledge Dr. Daniel Tancredi for providing statistical consultation with respect to implementing permutation analysis. Special acknowledgement to the Van de Water Lab: Lori Happen, Matthew Bruce and Victor Walters for experimental and technical support.
Funding: This study was funded by the NIEHS Center for Children’s Environmental Health and Environmental Protection Agency (EPA) grants (2P01ES011269–11, 83543201 respectively), the NIEHS-funded CHARGE study (R01ES015359), the NICHD funded IDDRC P50 (P50HD103526), Consejo Nacional de Ciencia y Tecnologia (CONACYT- UC MEXUS) Doctoral Fellowships, and NIH grant R35 GM138353.
Footnotes
Conflict of interest: Dr. Van de Water has patents issued for this technology and has founded a UC Davis startup company to develop this technology for commercial use. Dr. Aghaeepour and Dr. Becker served as consultants for MARA Bio. The other authors have no conflicts of interest.
Supplementary information is available at MP’s website.
Literature Cited:
- 1.Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. Morbidity and mortality weekly report Surveillance summaries (Washington, DC : 2002) 2018; 67(6): 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cidav Z, Marcus SC, Mandell DS. Implications of childhood autism for parental employment and earnings. Pediatrics 2012; 129(4): 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavelle TA, Weinstein MC, Newhouse JP, Munir K, Kuhlthau KA, Prosser LA. Economic Burden of Childhood Autism Spectrum Disorders. Pediatrics 2014; 133(3): e520–e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P et al. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Translational psychiatry 2013; 3: e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramirez A, Edmiston E, Schauer J, Vu T, Van de Water J. Peptides of neuron specific enolase as potential ASD biomarkers: from discovery to epitope mapping. Brain, Behavior, and Immunity 2019. [DOI] [PMC free article] [PubMed]
- 6.Edmiston E, Jones KL, Vu T, Ashwood P, Van de Water J. Identification of the antigenic epitopes of maternal autoantibodies in autism spectrum disorders. Brain, behavior, and immunity 2017. [DOI] [PMC free article] [PubMed]
- 7.Hertz-Picciotto I, Croen LA, Hansen R, Jones CR, van de Water J, Pessah IN. The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environ Health Perspect 2006; 114(7): 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braunschweig D, Duncanson P, Boyce R, Hansen R, Ashwood P, Pessah IN et al. Behavioral correlates of maternal antibody status among children with autism. Journal of autism and developmental disorders 2012; 42(7): 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fluss R, Faraggi D, Reiser B. Estimation of the Youden Index and its Associated Cutoff Point. Biometrical Journal 2005; 47(4): 458–472. [DOI] [PubMed] [Google Scholar]
- 10.Hajian-Tilaki K Receiver Operating Characteristic (ROC) Curve Analysis for Medical Diagnostic Test Evaluation. Caspian J Intern Med 2013; 4(2): 627–635. [PMC free article] [PubMed] [Google Scholar]
- 11.Blackford JU, Salomon RM, Waller NG. Detecting change in biological rhythms: a multivariate permutation test approach to Fourier-transformed data. Chronobiol Int 2009; 26(2): 258–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.León-Novelo L, Kemppainen KM, Ardissone A, Davis-Richardson A, Fagen J, Gano K et al. TWO APPLICATIONS OF PERMUTATION TESTS IN BIOSTASTICS. Bol Soc Mat Mex 2013; 19(2): 255–266. [PMC free article] [PubMed] [Google Scholar]
- 13.Lemmerich F Novel Techniques for Efficient and Effective Subgroup Discovery. 2014.
- 14.Klösgen W Explora: A multipattern and multistrategy discovery assistant. 1996: 249–271.
- 15.Gata-Garcia A, Diamond B. Maternal Antibody and ASD: Clinical Data and Animal Models. Front Immunol 2019; 10: 1129–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones KL, Van de Water J. Maternal autoantibody related autism: mechanisms and pathways. Molecular psychiatry 2018. [DOI] [PMC free article] [PubMed]
- 17.Matelski L, Van de Water J. Risk factors in autism: Thinking outside the brain. J Autoimmun 2016; 67: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meltzer A, Van de Water J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2017; 42(1): 284–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braunschweig D, Van de Water J. Maternal autoantibodies in autism. Arch Neurol 2012; 69(6): 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox-Edmiston E, Van de Water J. Maternal Anti-Fetal Brain IgG Autoantibodies and Autism Spectrum Disorder: Current Knowledge and its Implications for Potential Therapeutics. CNS Drugs 2015; 29(9): 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coutinho E, Menassa D, Jacobson L, West S, Domingos J, Moloney T et al. Maternal CASPR2 antibodies and neurodevelopmental disorders in the offspring: epidemiological findings and an animal model. The Lancet 2017; 389: S18. [Google Scholar]
- 22.Brimberg L, Mader S, Jeganathan V, Berlin R, Coleman TR, Gregersen PK et al. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Molecular psychiatry 2016; 21(12): 1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JY, Huerta PT, Zhang J, Kowal C, Bertini E, Volpe BT et al. Neurotoxic autoantibodies mediate congenital cortical impairment of offspring in maternal lupus. Nat Med 2009; 15(1): 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT. Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol 2009; 9(6): 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braunschweig D, Golub MS, Koenig CM, Qi L, Pessah IN, Van de Water J et al. Maternal autism-associated IgG antibodies delay development and produce anxiety in a mouse gestational transfer model. Journal of neuroimmunology 2012; 252(1–2): 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin LA, Ashwood P, Braunschweig D, Cabanlit M, Van de Water J, Amaral DG. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav Immun 2008; 22: 806–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM et al. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Translational psychiatry 2013; 3: e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camacho J, Jones K, Miller E, Ariza J, Noctor S, Van de Water J et al. Embryonic intraventricular exposure to autism-specific maternal autoantibodies produces alterations in autistic-like stereotypical behaviors in offspring mice. Behav Brain Res 2014; 266: 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones KL, Pride MC, Edmiston E, Yang M, Silverman JL, Crawley JN et al. Autism-specific maternal autoantibodies produce behavioral abnormalities in an endogenous antigen-driven mouse model of autism. Mol Psychiatry 2018. [DOI] [PMC free article] [PubMed]
- 30.Martínez-Cerdeño V, Camacho J, Fox E, Miller E, Ariza J, Kienzle D et al. Prenatal Exposure to Autism-Specific Maternal Autoantibodies Alters Proliferation of Cortical Neural Precursor Cells, Enlarges Brain, and Increases Neuronal Size in Adult Animals. Cerebral Cortex 2014; 26(1): 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ariza J, Hurtado J, Rogers H, Ikeda R, Dill M, Steward C et al. Maternal autoimmune antibodies alter the dendritic arbor and spine numbers in the infragranular layers of the cortex. PLoS One 2017; 12(8): e0183443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leslie D, Lipsky P, Notkins AL. Autoantibodies as predictors of disease. The Journal of Clinical Investigation 2001; 108(10): 1417–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coutinho E, Jacobson L, Pedersen MG, Benros ME, Nørgaard-Pedersen B, Mortensen PB et al. CASPR2 autoantibodies are raised during pregnancy in mothers of children with mental retardation and disorders of psychological development but not autism. Journal of Neurology, Neurosurgery & Psychiatry 2017; 88(9): 718–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coutinho E, Menassa DA, Jacobson L, West SJ, Domingos J, Moloney TC et al. Persistent microglial activation and synaptic loss with behavioral abnormalities in mouse offspring exposed to CASPR2-antibodies in utero. Acta Neuropathologica 2017; 134(4): 567–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 2018; 47(D1): D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM. CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases. Molecular psychiatry 2015; 20(9): 1037–1045. [DOI] [PubMed] [Google Scholar]
- 37.Makihara H, Nakai S, Ohkubo W, Yamashita N, Nakamura F, Kiyonari H et al. CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes Cells 2016; 21(9): 994–1005. [DOI] [PubMed] [Google Scholar]
- 38.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem 2009; 284(45): 31375–31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q et al. Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nature Communications 2016; 7(1): 11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, Firestein BL. Cypin regulates dendrite patterning in hippocampal neurons by promoting microtubule assembly. Nature Neuroscience 2004; 7(2): 145–152. [DOI] [PubMed] [Google Scholar]
- 41.Linden R The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front Mol Neurosci 2017; 10: 77–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beraldo FH, Thomas A, Kolisnyk B, Hirata PH, De Jaeger X, Martyn AC et al. Hyperactivity and attention deficits in mice with decreased levels of stress-inducible phosphoprotein 1 (STIP1). Dis Model Mech 2015; 8(11): 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haque A, Polcyn R, Matzelle D, Banik NL. New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain sciences 2018; 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL et al. Maternal Metabolic Conditions and Risk for Autism and Other Neurodevelopmental Disorders. Pediatrics 2012; 129(5): e1121–e1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.