

Abstract
The stimulator of interferon genes (STING) is an endoplasmic-reticulum transmembrane protein targeted by therapeutics for infectious disease and cancer. However, early-phase clinical trials of small-molecule STING agonists have shown limited antitumor efficacy and dose-limiting toxicity. Here, we show that a polyvalent STING agonist, a pH-sensitive polymer bearing a seven-membered ring with a tertiary amine (PC7A), activates innate-immunity pathways through the polymer-induced formation of STING–PC7A condensates. Unlike the natural STING ligand cGAMP (cyclic guanosine monophosphate–adenosine monophosphate), PC7A stimulates the prolonged production of proinflammatory cytokines by binding to a non-competitive STING surface site that is distinct from the cGAMP binding pocket. PC7A induces antitumor responses dependent on STING expression and on CD8+ T-cell activity, and the combination of PC7A and cGAMP led to synergistic therapeutic outcomes (including the activation of cGAMP-resistant STING variants) in mice bearing subcutaneous tumours and in resected human tumours and lymph nodes. The activation of the STING pathway via polymer-induced STING condensation may offer new therapeutic opportunities.
The stimulator of interferon genes (STING) plays a central role in innate immunity during infection and cancer1–4. STING is endogenously activated by 2’,3’-cyclic-GMP-AMP (cGAMP), a cyclic dinucleotide synthesized by cGAMP synthase (cGAS) in response to cytosolic DNA as a danger signal5,6. Activation of STING mediates a multifaceted type I interferon (IFN-I) response that promotes the maturation and migration of dendritic cells (DCs), and primes cytotoxic T lymphocytes and nature killer (NK) cells for spontaneous immune responses7–11. In recent years, STING has emerged as an important target that activates antitumor immune pathways for cancer immunotherapy12–17. Previous studies have observed punctate structures upon the addition of cGAMP to STING, indicating that oligomerization or even higher order architecture may be critical for activation18–21. Therapeutic attempts to deliver cGAMP into the cytosol of target cells, where STING is located, have been limited by its inherent properties as a small, dual negatively charged molecule22. Moreover, the rapid enzymatic degradation and clearance as well as off-target toxicity of cGAMP have hindered its further clinical application23,24. Therefore, the pharmaceutical industry has devoted great efforts to the chemical modification of natural cyclic dinucleotides (CDNs) as well as new STING agonists to improve their bioavailability and pharmacological activity25,26. Despite therapeutic promise, several small molecule agonists of STING have shown limited antitumor efficacy and dose-limiting toxicity in early phase clinical trials27,28.
Polyvalent phase condensation has been shown to regulate diverse biological processes, including ribosome assembly, gene expression, and signal transduction29,30. Phase separation involves the assembly of macromolecular complexes through multivalent interactions31. A previous study has shown that DNA-induced liquid phase separation of cGAS triggers innate immunity32. By forming such biomolecular condensates, proteins involved in signaling cascades can be easily enriched in membrane-less assemblies and amplify responses to small changes in the microenvironment. These biomolecular condensates are typically hundreds of nanometers to micrometers in size and are transient and dynamic in response to specific stimuli or stress33,34.
Previously we synthesized a library of pH sensitive polymers with linear or cyclic tertiary amine structures, among which a polymer with a cyclic 7-membered ring (PC7A) has shown strong vaccine adjuvant effect through the STING-dependent pathway17. In this study, we report PC7A is a polyvalent STING agonist. It acts through polymer-induced phase separation of STING for innate immune activation with prolonged cytokine expressions than cGAMP. Level of STING activation depends on the length of the polymer thereby valency of the interaction. We further demonstrate that PC7A nanoparticles loaded with cGAMP lead to robust tumor growth inhibition and enhanced survival in two animal tumor models and synergistic STING activation in resected human tumors and lymph nodes. Our study provides a proof-of-principle for new cancer immunotherapy strategies targeting the STING pathway.
Results
PC7A polymer activates STING with a spatiotemporal profile distinct from cGAMP.
To understand how PC7A-induced STING activation differs from cGAMP20,21, we first investigated the intracellular distribution of GFP-labeled STING and the downstream signals in live cells in response to treatment. Remarkably, the temporal profile of PC7A-induced STING puncta formation and maturation is distinct from those induced by cGAMP. When primed by cGAMP, STING puncta formation occurs rapidly, producing a strong immune response which peaks around 6 h after stimulation, followed by rapid degradation and subsequent immune silence (Fig. 1a–c). In contrast, PC7A generates a durable STING activation profile, with sustained expression of interferon-stimulated genes (ifn-β and cxcl10) over 48 h. STING degradation is delayed after PC7A stimulation, as indicated by the limited fusion of STING puncta with lysosomes even at 48 h (Fig. 1d and Supplementary Fig. 1c). We observed a similar effect of delayed STING degradation in cGAMP-treated cells pre-incubated with bafilomycin A1 (Baf A1), a vacuolar H+ ATPase inhibitor which blocks lysosome acidification, and in cells treated with combined cGAMP and PC7A (Supplementary Fig. 1d, e). Overall, these data suggest the endo-lysosomal pH buffering capability of PC7A may be responsible for slow STING degradation35.
Fig. 1 |. PC7A polymer activates STING with a spatiotemporal profile distinct from cGAMP.
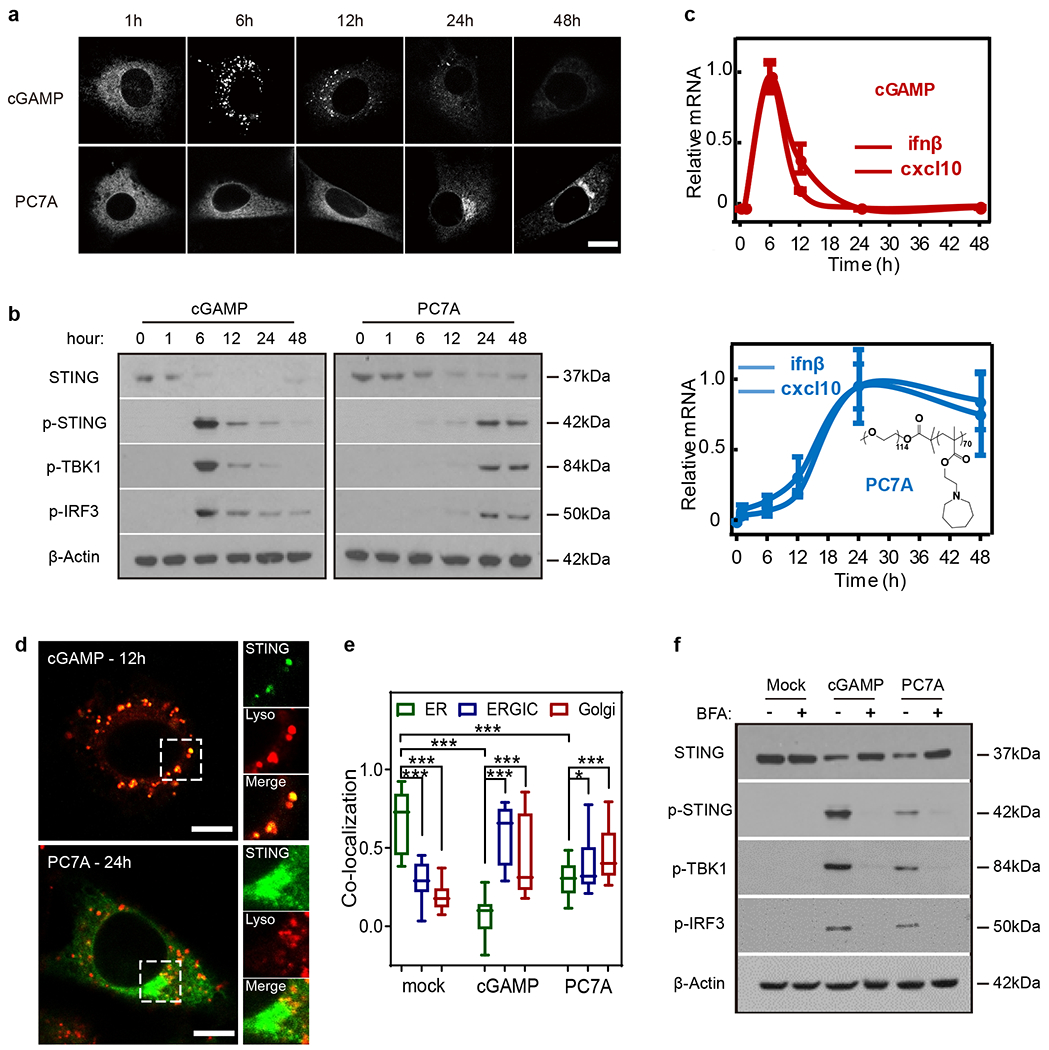
a, MEF cells primed by cGAMP or PC7A exhibit different geometric and temporal patterns of GFP-STING punctate formation and depletion. Cells were first incubated with cGAMP (10 μM, PEI was used for cytosolic delivery, Supplementary Fig. 1a, b) or PC7A micelles (10 μM) for 1 h, then media was exchanged, and cells were incubated for indicated periods prior to imaging. Scale bar, 10 μm. b, THP1 cells treated with cGAMP display a burst effect of TBK1/IRF3 phosphorylation followed by rapid STING degradation, while treatment by PC7A leads to sustained TBK1/IRF3 phosphorylation and slower STING degradation. c, Relative ifn-β and cxcl10 mRNA levels show slower but prolonged STING activation in THP1 cells by PC7A compared to cGAMP. Values are mean ± SD, n=3 biologically independent experiments. d, STING-GFP colocalizes with lysosomes in MEFs 12 h after cGAMP treatment, supporting rapid degradation. In contrast, PC7A inhibits lysosomal degradation of GFP-STING, as indicated by lack of colocalization and persistent GFP fluorescence. Scale bar, 5 μm. e, cGAMP and PC7A induce similar STING translocation from ER to ERGIC and Golgi apparatus. Colocalization was quantified by Pearson’s correlation coefficient. Box and whisker, mean ± min/max, lower quartile: 25th percentile, upper quartile: 75th percentile, n=20 cells examined over 3 independent experiments. Two-tailed Student’s t-test (in PC7A treatment group: ER vs. ERGIC P=0.029, ER vs. Golgi P=0.0005; in all other comparisons: P<0.0001). f, STING translocation is necessary for downstream signaling as BFA, an inhibitor of protein transport from ER to Golgi, prevents phosphorylation of TBK1/IRF3 by cGAMP or PC7A. Confocal images in a and d are representative of at least three biologically independent experiments.
Despite the differences in size and kinetics of puncta formation, intracellular STING foci resulting from cGAMP or PC7A treatment follow a similar course of translocation from the endoplasmic reticulum (ER) to the ER-Golgi intermediate compartments (ERGIC) and the Golgi apparatus (Fig. 1e and Supplementary Fig. 2a). During transportation, STING forms clusters and phosphorylates TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3, Fig. 1f), which initiates the downstream production of type I IFNs. In the presence of brefeldin A (BFA), which blocks protein trafficking between ER and Golgi36, both cGAMP and PC7A fail to trigger p-TBK1/p-IRF3 production and proinflammatory cytokine expression (Fig. 1f and Supplementary Fig. 2b–d).
PC7A binds to STING and forms biomolecular condensates.
To investigate the biophysical mechanism of PC7A-mediated STING clustering and activation, we first determined the binding affinity between PC7A and STING (human AA137-379, C-terminal domain) by isothermal calorimetry (ITC). STING binds strongly to PC7A (apparent Kd=72 nM), but weakly to other polymers with the same backbone, such as PEPA (apparent Kd=671 nM, Supplementary Fig. 3a–c). Notably, polymers with cyclic side chains exhibit higher affinity to STING than linear analogs, and the seven-membered-ring of PC7A elicits the strongest binding. To investigate whether PC7A was sufficient to induce clustering of STING in vitro, we incubated cyanine-5 (Cy5)-labeled STING CTD dimer with PC7A or PEPA at pH 6.5 (both P7CA and PEPA have apparent pKa’s at 6.9, and stay as cationic unimers at pH 6.5). PEPA was used as a negative control due to its poor binding affinity to STING. Upon mixing of Cy5-STING and PC7A, liquid droplets were observed within minutes and grew over time, with approximately 85% of STING proteins present in the condensates after 4 h (Fig. 2a). Incubation of Cy5-STING with PC7A labeled with aminomethylcoumarin acetate (AMCA) confirms co-localization of PC7A with STING puncta (Fig. 2b). Similar condensates were also observed in GFP-STING-expressing cell lysates after PC7A incubation (Supplementary Fig. 3d). The biomolecular condensates are hydrophobic as indicated by the increased fluorescence intensity and red-shifted maximum emission wavelength in a Nile-Red assay37 (Supplementary Fig. 3e). Fluorescence resonance energy transfer (FRET) from GFP-STING to tetramethylrhodamine (TMR)-PC7A further confirms the formation of a biomolecular condensate consisting of PC7A and STING in human-STING-overexpressing mouse embryonic fibroblasts (MEFs) (Fig. 2c). The downstream protein product, p-TBK1, was also found in this macromolecular cluster (Fig. 2d). In contrast, no obvious STING condensation or activation was observed when PEPA was used in these studies (Fig. 2 and Supplementary Fig. 3d). At pH 7.4, few PC7A-STING condensates were formed (Supplementary Fig. 3f) due to micellization of PC7A polymers above its pKa (6.9) and PEG shielding38,39.
Fig. 2 |. PC7A polymer induces STING condensation and immune activation.
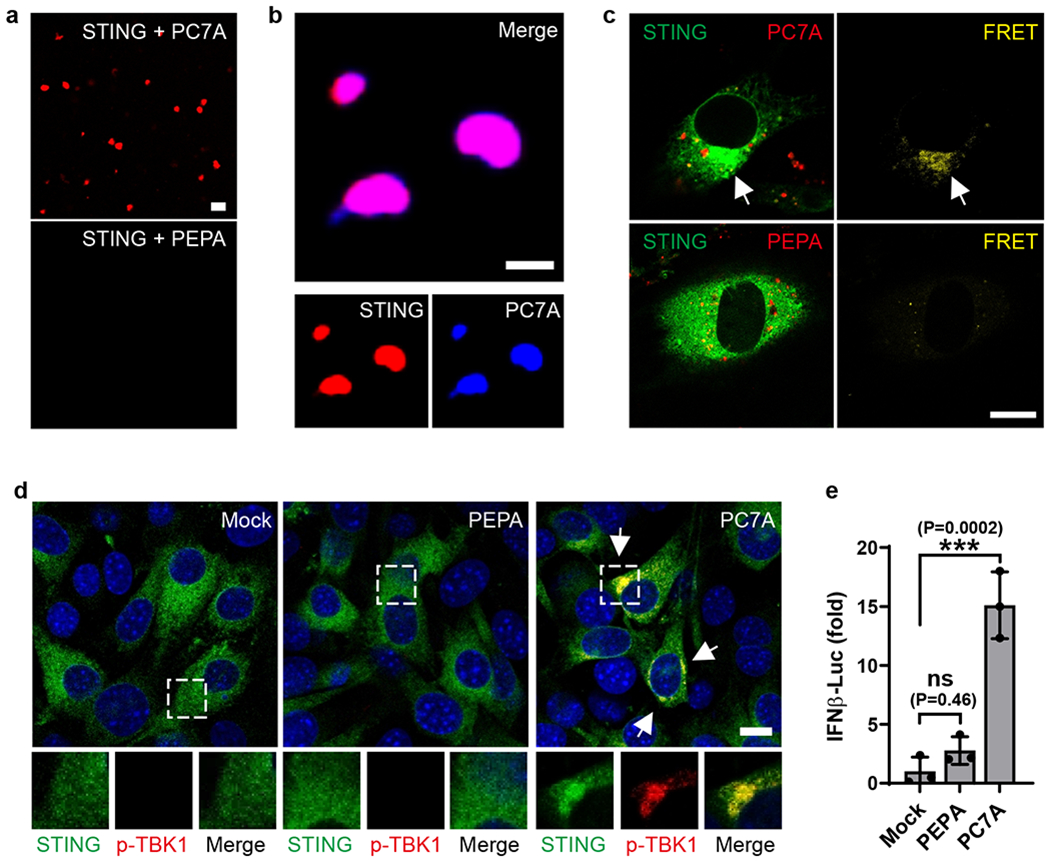
a, PC7A, but not PEPA, induces STING (Cy5-labeled) phase condensation after 4 h incubation. Scale bar, 10 μm. b, STING (4 μM, Cy5-labeled) and PC7A polymer (2 μM, AMCA-labeled) are colocalized within the condensates. Scale bar, 5 μm. c, Hetero-FRET from GFP-STING to TMR-PC7A illustrates colocalization of STING and PC7A in MEF cells. Energy transfer was not observed from GFP-STING to TMR-PEPA. Cell culture conditions identical to Fig. 1. GFP (lex/lem=488/515 nm) and TMR (555/580 nm) signals are shown in the left panels as green and red, respectively. FRET signals (488/580 nm) are shown as yellow in the right panels. Scale bar, 10 μm. d, p-TBK1 is recruited in the STING/PC7A condensates. Scale bar, 10 μm. e, PC7A, not PEPA, induces expression of IFNβ–luciferase in ISG-THP1 cells. Values are mean ± SD, n=3 biologically independent experiments. One-way ANOVA. Confocal images in a-d are representative of at least three biologically independent experiments.
PC7A induces STING activation through polyvalent interactions.
Recent studies revealed STING oligomerization upon cGAMP binding is responsible for the recruitment and activation of downstream TBK1 and IRF3 proteins18–21. We hypothesize that PC7A polymer can serve as a supramolecular scaffold and directly engage polyvalent interactions to multimerize STING molecules for activation (Fig. 3a). To test this idea, we first labeled STING proteins using a FRET pair (TMR and Cy5) and mixed the two differentially labeled proteins in a 1:1 ratio. Upon addition of PC7A, we observed strong energy transfer from TMR to Cy5 (Supplementary Fig. 4a), indicating close proximity of STING dimers after polyvalent binding to PC7A. Fluorescence recovery after photobleaching (FRAP) experiments40,41 on STING-PC7A condensates revealed that while both PC7A polymer and STING protein are exchangeable with surrounding molecules, PC7A exhibited a slower recovery rate than STING (Fig. 3b and Supplementary Fig. 4b, c).
Fig. 3 |. PC7A polymer induces STING condensation and immune activation through polyvalent interactions.
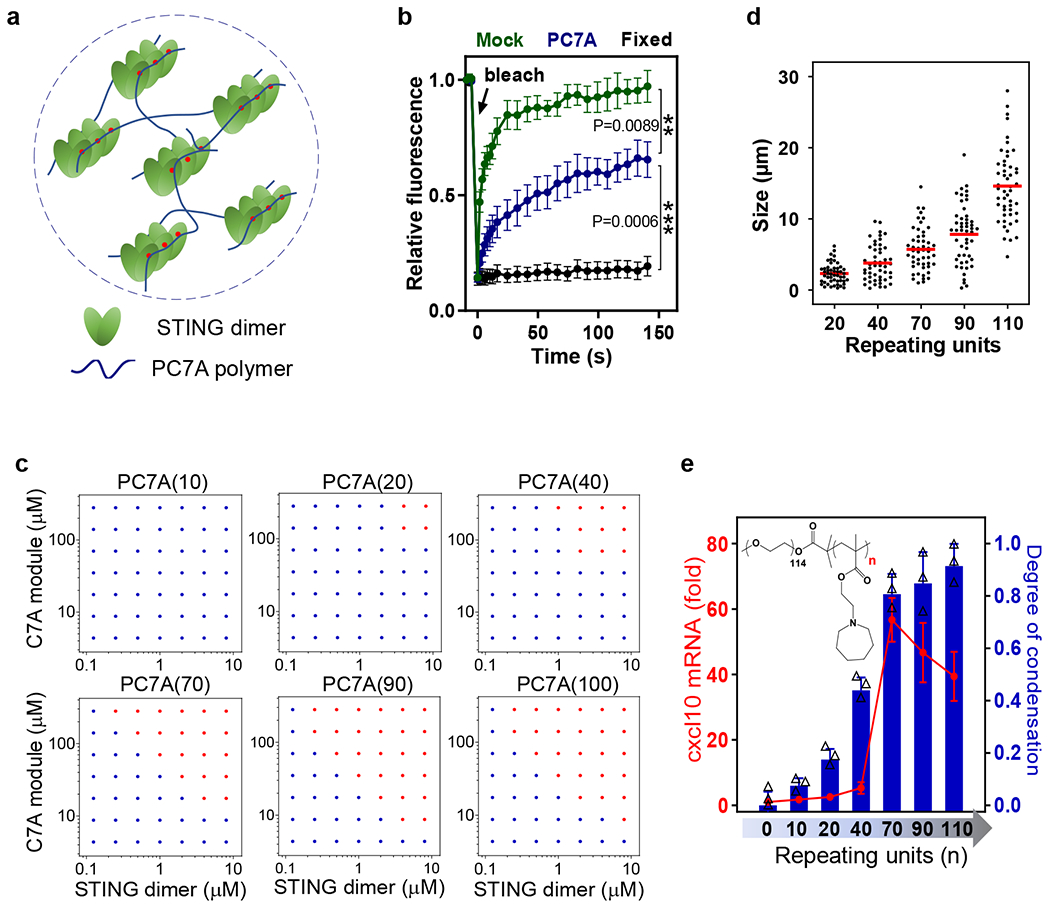
a, Schematic of STING oligomerization and condensation driven by PC7A through polyvalent interactions. b, PC7A decreases the molecular mobility of GFP-STING in the condensates compared to free GFP-STING in MEF cells. Bleaching was performed 24 h after PC7A treatment, and recovery was monitored over 150 s. Untreated (mock) and fixed cells were used as mobile and stationary STING controls, respectively. Values are mean ± SD, n=5 cells examined over 2 independent experiments. One-way ANOVA. c, Biomolecular condensation of STING and PC7A is dependent on PC7A valency. Red dots indicate phase separation while blue dots indicate no phase separation. d, Size distributions of STING condensates induced by PC7A increase with higher PC7A valency. Condensate size was calculated as the average of longest and shortest axis, n=50 condensates examined over 2 independent experiments. e, STING activation in THP1 cells correlates with the PC7A valency, with optimal cxcl10 expression induced by PC7A(70). Values are mean ± SD, n=3 biologically independent experiments. In experiments c-e, polymers with different repeating units were used at the same C7A modular concentrations.
To examine the effects of binding valence, we synthesized a series of PC7A polymers with an increasing number of repeating units. PC7A(n) refers to a polymer with n repeating units of C7A methacrylate monomer. We incubated the PC7A of increasing lengths with STING dimer under a matrix of concentrations in vitro to generate a phase diagram, which shows a minimum requirement of 20 repeating units for condensation (Fig. 3c). No phase separation was observed for PC7A(10). Higher degree of PC7A polymerization resulted in larger condensates (Fig. 3d and Supplementary Fig. 5a–c). For PC7A(110), over 90% of STING proteins were found in the condensates, compared to 17% when PC7A(20) was used (Supplementary Fig. 5d). PC7A with higher degree of polymerization exhibited lower phase reversibility and slower recovery rate of STING after photobleaching (Supplementary Fig. 5e, f). To investigate the relationship between condensate formation and STING activation in live cells, we treated THP1 cells with PC7A of varying lengths, and compared mRNA expression levels of cxcl10. Longer polymers induced higher cxcl10 expression, with peak levels observed at 70 repeating units of PC7A (Fig. 3e). Further elongation of chain length (e.g., 110) led to reduced cxcl10 expression, probably because of the weaker signaling capacity of oversized condensates with excessive crosslinking and poor molecular dynamics41,42.
PC7A binds to a distinct surface site from the cGAMP-binding pocket.
The STING-PC7A condensates are sensitive to high concentrations of salt or the presence of other proteins. While STING-PC7A condensates were formed at a physiological concentration of NaCl (150 mM), no phase separation was observed when salt concentration was raised to 600 mM (Supplementary Fig. 6a, b). When bovine serum albumin (BSA) was added, the condensates decreased in number and size (Supplementary Fig. 6c). To further investigate the specificity of PC7A induced condensate, we labeled STING with Cy5 and BSA with boron-dipyrromethene (BODIPY) dyes. In the presence of PC7A, only Cy5-STING was present in the condensates, whereas the majority of BODIPY-BSA was excluded (Supplementary Fig. 6d). As controls, mixtures of BSA/PC7A or STING/BSA did not form condensates.
Based on the pH (Supplementary Fig. 3f) and salt effects (Supplementary Fig. 6a, b) on the PC7A-STING interactions and computational modeling (data not shown), we hypothesized that negatively charged surface sites on STING may be responsible for PC7A binding. To test this hypothesis, we constructed STING mutants with several negatively charged amino acids in the α5-β5-α6 region replaced by alanine and investigated their PC7A binding affinity, phase condensation, and STING activation both in vitro and in live cells. Notably, the mutation of two acidic residues (E296A/D297A) on the α5 helix was sufficient to abolish polymer binding and biomolecular condensation, whereas two other mutants (D319A/D320A and E336A/E337A/E339A/E340A) exhibited marginal effects (Fig. 4a, b and Supplementary Table 1). We then transfected HEK293T cells with mutant STING plasmids and measured downstream activation. Consistent with the abrogation of PC7A binding and condensation, the E296A/D297A mutant was deficient in forming condensate structures and inducing TBK1 phosphorylation and ifn-β/cxcl10 expression in cells (Fig. 4c and Supplementary Fig. 7a, d). In contrast, these STING mutants did not impact cGAMP-mediated STING activation (Supplementary Fig. 7b, c). Together, these data suggest that the E296D297 site on the α5 helix of STING, which is distinct from the cGAMP binding site, is responsible for PC7A binding and induced activation.
Fig. 4 |. STING condensation and activation by PC7A polymer occurs through a distinct binding site from cGAMP.
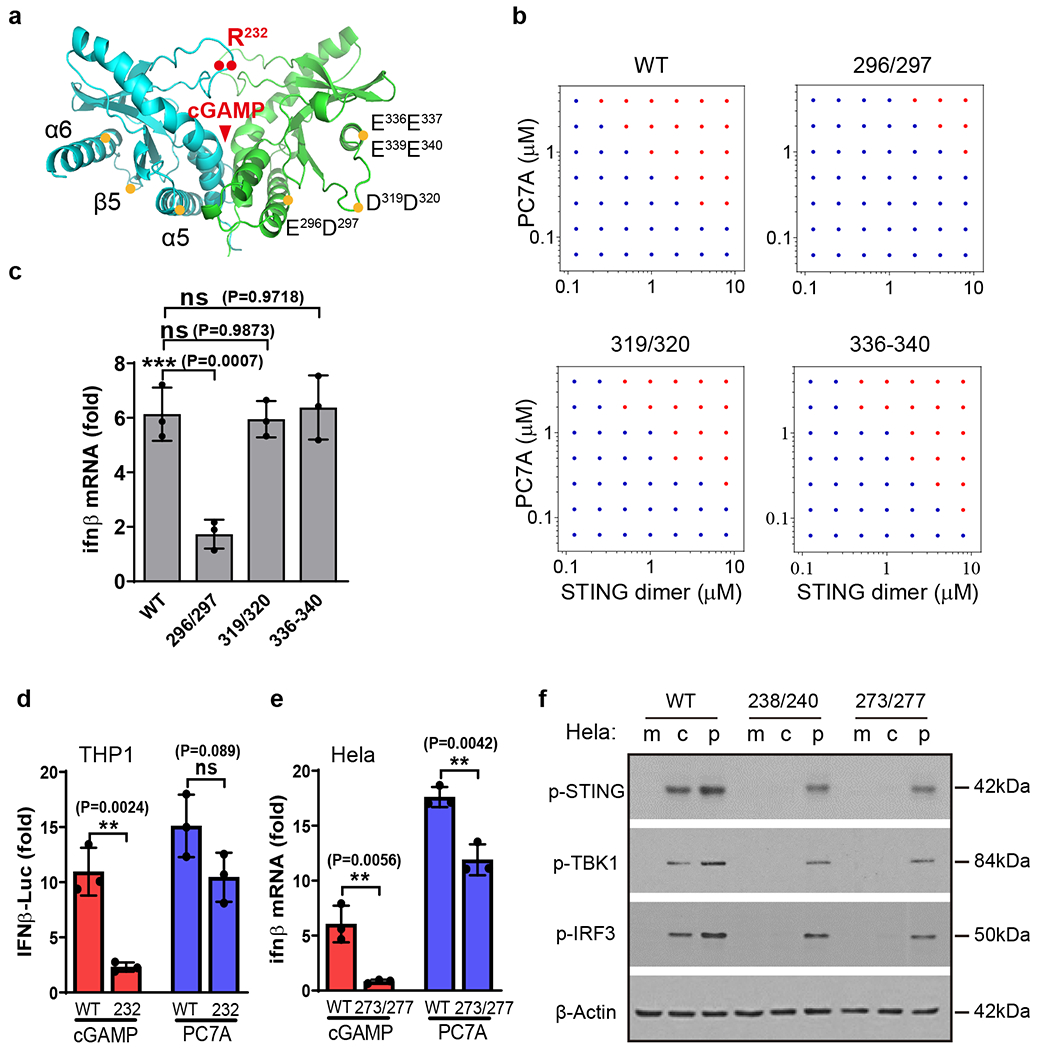
a, Schematic of site-directed mutagenesis on the STING structure. Mutation sites are distinct from the cGAMP binding pocket. Mutation of E296A-D297A abolishes STING condensation (b) and immune activation (c) in response to PC7A. Other mutations of STING do not affect PC7A-induced STING activation. Values are mean ± SD, n=3 biologically independent experiments. One-way ANOVA. d-f, PC7A retains immune activity in several cGAMP-resistant STING variants. R232H in THP1 cells or R238A/Y240A in Hela cells abrogate cGAMP binding. Q273A/A277Q, which disrupts the tetramer interface and cGAMP mediated STING oligomerization, abolishes STING activation by cGAMP but not by PC7A. Values are mean ± SD, n=3 biologically independent experiments. Two-tailed Student’s t-test. m: mock; c: cGAMP; p: PC7A polymer.
Endogenous STING agonists (cGAMP or other CDNs) bind to the STING dimer interface covered by a lip of four-stranded antiparallel β sheet (human AA 219-249)43,44. A natural STING variant (R232H) occurring in ~14% of the human population exhibits a reduced response to small molecule STING agonists45. Since PC7A binds to a STING site different from the cGAMP binding pocket, we tested the biological activity of PC7A in THP1 cells harboring the STING R232H variant. Whereas the cGAMP response was expectedly abrogated in these cells, PC7A was still able to elevate IFN-β-Luc expression (Fig. 4d). Additional studies in mutant Hela cells (R238A/Y240A or Q273A/A277Q mutations that abolish cGAMP binding or prevent STING oligomerization upon cGAMP binding, respectively)20,21 show persistent PC7A-induced STING activation, whereas cGAMP-mediated effects were abolished (Fig. 4e, f and Supplementary Fig. 7e–h). Collectively, these results demonstrate PC7A stimulates STING through cGAMP-independent mechanisms.
PC7A prolongs innate activation in vivo and synergizes with cGAMP in antitumor immunity.
In vitro cell culture studies show PC7A NP generated durable STING activation over free cGAMP (Fig. 1a–c). To test whether PC7A NP prolongs STING activation in vivo, we intratumorally injected cGAMP (50 μg), PC7A NP (50 μg), and cGAMP-loaded PC7A NP (2.5/50 μg) in MC38 tumors (~100 mm3) and measured the expression of interferon-stimulated genes in both tumors and draining lymph nodes over time. Because of PC7A’s ability for STING activation and cytosolic delivery of cGAMP, we chose a lower dose of cGAMP (~5wt% loading) in cGAMP-PC7A NP for the majority of in vivo studies. cGAMP-PC7A NP was prepared by a base titration method, resulting in spherical micelles of 29.9±2.5 nm in diameter and over 90% loading efficiency (Supplementary Fig. 8). Consistent with our in vitro studies, mice treated with free cGAMP showed rapid ifn-β/cxcl10 expression 6 h after intratumoral injection while the activity dramatically decreased over 48 h in both tumor and nodal tissues (Supplementary Fig. 9). In contrast, PC7A-induced STING activity is minimal at 6 h but reaches the maximum level at 24 h. cGAMP-PC7A NP yields the most optimal STING activity profile, which exhibits a rapid rise of ifn-β/cxcl10 expression over PC7A (50 μg) at 6 h, and unlike free cGAMP, this response is also sustained over 48 h.
Next we investigated the antitumor efficacy in MC38 and TC-1 tumor models (Fig. 5). In MC38 tumors, we performed three intratumoral injections of free cGAMP (2.5 or 50 μg, see Supplementary Fig. 10a for high dose data), PC7A NP (50 μg), or cGAMP-PC7A NP (2.5/50 μg) when tumors reached ~50 mm3 in size. As a negative control, we injected mice with a 5% glucose solution (all treatment groups were prepared in 5% glucose solutions). Results show all mice in the control group died within 50 days after MC38 inoculation. cGAMP (2.5 μg) or PC7A alone groups significantly extended the survival over the control group while the difference between the two treatment groups is not statistically significant. cGAMP-PC7A NP treatment achieved the most efficacious outcome with 4 out of 7 mice remaining tumor free over 100 days after tumor inoculation. In the TC-1 model, all mice in the control group died within 26 days. cGAMP or PC7A alone conferred a minor degree of immune protection, extending median survival by 4 or 8 days, respectively. The cGAMP-PC7A NP treatment showed significantly improved tumor growth inhibition and long-term survival over either treatment alone.
Fig. 5 |. PC7A and cGAMP show synergistic antitumor efficacy in tumor-bearing mice.
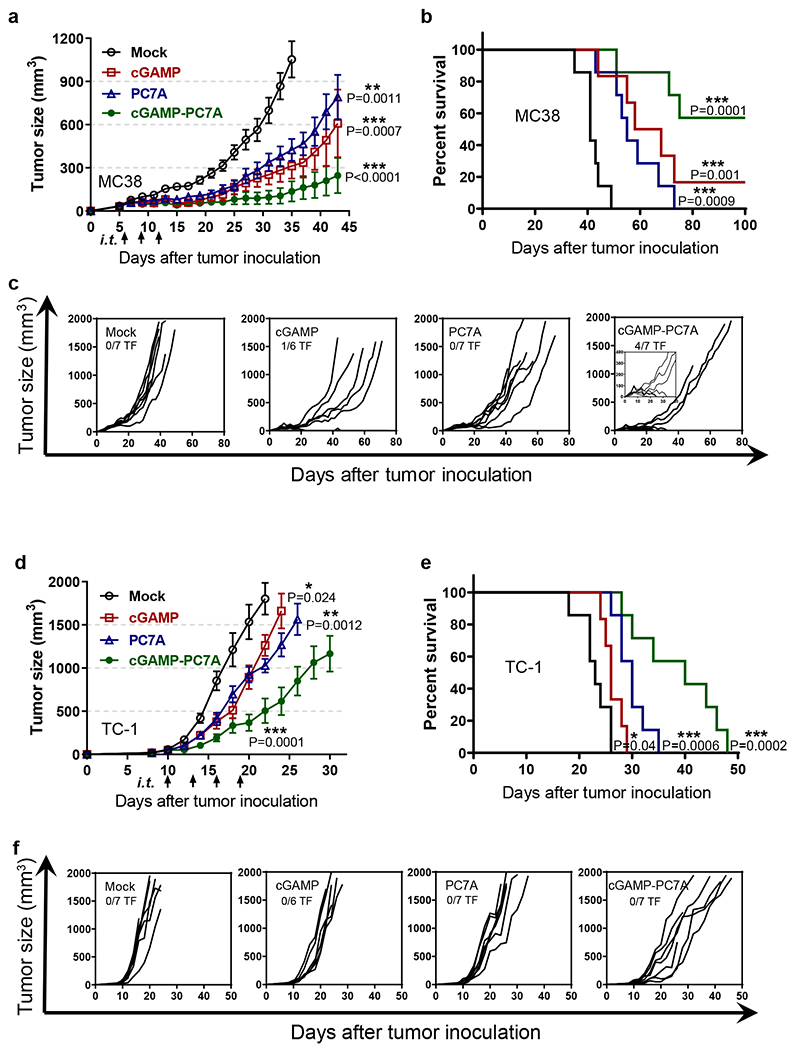
(a-c) MC38 and (d-f) TC-1 tumor-bearing mice were injected intratumorally with 5% glucose (mock), cGAMP (2.5 μg), PC7A (50 μg), or cGAMP-loaded PC7A nanoparticles at indicated time points. Mean tumor volume (a, d), Kaplan–Meier survival curves (b, e), and spider plots of individual tumor growth curves (c, f) are shown. PC7A NP or cGAMP alone offers some degree of immune protection. cGAMP-loaded PC7A NP confers a synergistic anti-tumor immune response, with significantly improved survival and 4 of 7 mice in the MC38 model tumor-free. In tumor growth studies, values represent mean ± SEM, n=7 (mock), n=6 (cGAMP), n=7 (PC7A), n=7 (cGAMP-PC7A) of biologically independent mice in each tumor model, two-tailed Student’s t-test (versus mock). In survival studies, Mantel–Cox test.
In MC38 tumors, high dose (50 μg) of free cGAMP treatment did not lead to significantly improved tumor growth inhibition over the low dose group (2.5 μg, Supplementary Fig. 10a). In contrast, systemic side effects were observed at the higher cGAMP dose, as evidenced by the elevated levels of alanine transaminase and aspartate transaminase (liver), urea (kidney) and systemic cytokine (e.g., IL10) (Supplementary Fig. 10b–e). cGAMP-PC7A NP treatment did not show significant increase in toxic side effects over the control group.
Previous studies have shown an association between elevated type I IFN production and increased tumor infiltration of PD-1+ cytotoxic T lymphocytes7,46–48. We hypothesized that STING activation by cGAMP-PC7A NP may synergize with PD-1 blockade. We found the combination provided significantly improved efficacy, with 100% of mice remaining tumor-free after 100 days in the murine MC38 colorectal tumor model (Supplementary Fig. 11a–c). The therapeutic efficacy is also improved in the more aggressive TC-1 tumor model, with over 50% of mice bearing TC-1 tumors surviving over 45 days (Supplementary Fig. 11d–f).
STING status and immune cell type on PC7A-induced antitumor immunity.
Using an in vivo cell killing assay, our previous study showed that the generation of antigen-specific T cells by the PC7A nanoparticle vaccine was dependent on the STING-type I interferon pathway17. To confirm the importance of the STING pathway and to determine whether host or cancer cell STING status plays a more dominant role on PC7A-induced antitumor immunity, we performed tumor growth inhibition assays in host Tmem173−/− (Tmem173 encodes STING) mice/wildtype MC38 tumors and wildtype mice/Tmem173−/− MC38 tumors (Supplementary Fig. 12a–c). Without treatment, wildtype MC38 cancer cells grew faster in Tmem173−/− mice than in wildtype mice, indicating the role of the STING pathway in immune protection by the host alone. The antitumor efficacy improvement of PC7A and cGAMP-PC7A NPs was abolished in Tmem173−/− animals compared to wildtype mice. In contrast, comparable antitumor efficacy by PC7A and cGAMP-PC7A NPs was observed when treating Tmem173 knockout vs. wildtype MC38 tumors in wildtype mice.
To further investigate the immune cell-dependent contribution to antitumor immunity, we evaluated tumor growth inhibition by antibody blockade of CD8 T cells, NK cells and in CD11c-DTR transgenic mice with depletion of dendritic cells49. Blockade of CD8 T cells abolished the antitumor efficacy by PC7A treatment whereas blockade of NK cells showed minimal effect (Supplementary Fig. 12d, e). Results with CD11c-DTR mice showed that DC depletion reduced the therapeutic efficacy after treatments, albeit to a lesser extent when compared to CD8 T cell blockade (Supplementary Fig. 12f).
STING activation in human tissues.
To explore the translational potential, we investigated the feasibility of STING activation in human tissues. We acquired freshly resected squamous cell carcinoma from the base/lateral of tongue, cervical tumor tissues, and a sentinel lymph node. We locally injected these tissues with cGAMP, PC7A NP, or cGAMP-PC7A NP, incubated them in cell culture medium for 24 h at 37 °C, and detected IFN related gene expression. Free cGAMP had a marginal effect on ifn-β and cxcl10 mRNA expressions over the control due to limited bioavailability. In contrast, PC7A NP elevated downstream signals by 5-20 folds. A remarkable increase of cytokine expression (100-200 folds, Fig. 6a–d and Supplementary Fig. 13) was observed with cGAMP-PC7A NP in all tissue types. Notably, CD45+ myeloid cell populations in the tumor showed higher level of STING activation by PC7A NP and cGAMP-PC7A NP treatment over CD45− cells (Fig. 6e, f), indicating that leukocytes, instead of cancer cells, are the primary targets for STING-mediated immunomodulation by nanoparticles.
Fig. 6 |. PC7A and cGAMP show synergistic STING activation in fresh human tissues.
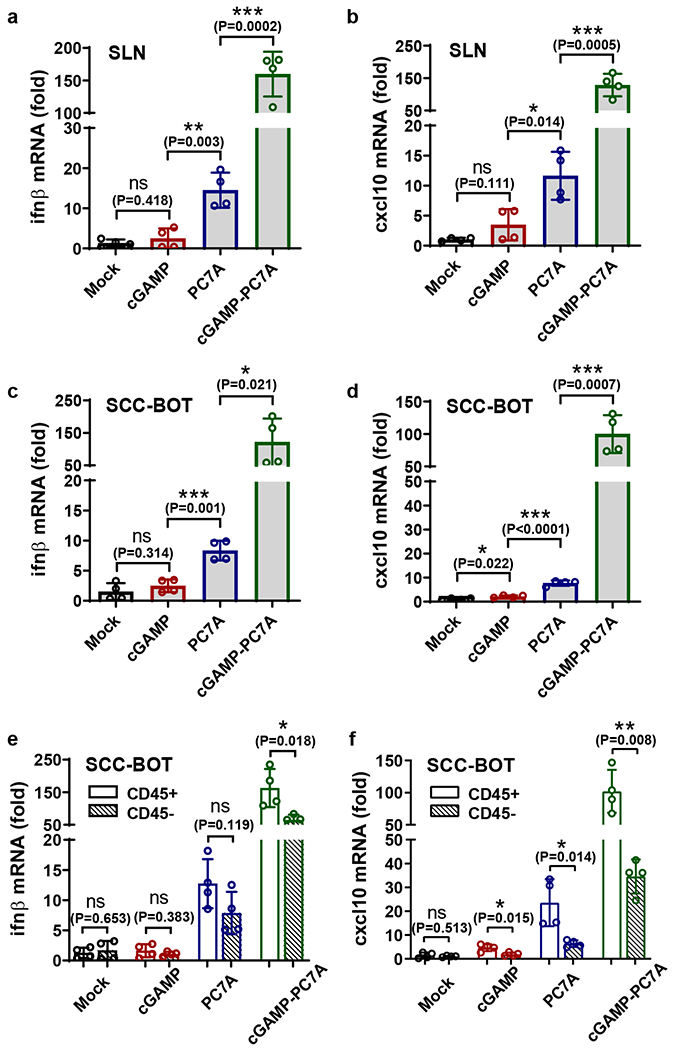
Free cGAMP alone is unable to activate STING, while PC7A NP and cGAMP-loaded PC7A NP demonstrate effective STING activation. Fresh surgically resected sentinel lymph node (SLN) (a, b) or squamous cell carcinoma from the base of tongue (SCC-BOT) (c-f) were divided into multiple sections (1-5 mm3) and injected with 5% glucose, free cGAMP, PC7A NP, or cGAMP-loaded PC7A NP in 5% glucose solutions. Ifn-β and cxcl10 gene expressions were measured after 24 h incubation. (e, f) The CD45+ cell population exhibits enhanced level of STING activation compared with CD45− cells. Values are mean ± SD, n=4 SLN sections from the same patient in a and b, and n=4 SCC-BOT tumor sections from the same patient in c-f. Two-tailed Student’s t-test.
Discussion
The mechanistic insights and therapeutic strategies described in this study exploit noncanonical STING activation with cell intrinsic pathways for cancer immunotherapy. First, we determined a distinctive surface binding site on the STING protein by the PC7A polymer that is different from cGAMP or other cyclic dinucleotides. Although previous report showed PC7A nanoparticle vaccine worked through STING for T cell activation17, it is not clear whether PC7A competes with cGAMP for the same binding pocket at the STING dimer interface. Discovery of non-competitive binding sites formulates a basis to combine PC7A with cGAMP for synergistic STING activation while allowing PC7A to activate cGAMP-resistant STING variants (Fig. 4d–f). In humans, STING consists of several haplotypes (e.g., 14% human population have R232H phenotype) that exhibit reduced innate activity in response to CDN agonists50,51. PC7A presents an alternative STING activation strategy in these STING variant patient populations. Second, we uncovered a PC7A-induced protein condensation mechanism for STING activation. We used a synthetic polymer to induce polyvalent phase condensation for biological activation. Phase condensates are shown to impact a broad range of biological processes and are under intensive investigations in biophysics and cell biology29,30. The current study provides the proof of concept to install polymer-induced protein condensation as an emerging bioengineering principle for biological interrogation and pharmaceutic development.
STING remains as a promising target for cancer immunotherapy, but several small-molecule STING agonists showed limited efficacy and dose-limiting toxicity in early stage clinical trials27,28. In the current study, intratumoral injection of high dose cGAMP (50 μg) did not lead to significant tumor growth inhibition over low dose (2.5 μg) but resulted in increased systemic toxicity (Supplementary Fig. 10), corroborating clinical observations. We attribute the limited therapeutic window to the poor pharmacokinetics and mechanism of action in STING activation. Because of its small size (674 Da) and water solubility, blood perfusion can quickly remove cGAMP from tumor site to systemic circulation, limiting STING activation to a few hours inside tumors (Supplementary Fig. 9).
Compared to cGAMP, the PC7A polymer induces a slower but more sustained STING activity in vitro and in vivo. We attribute this kinetic difference to several factors. First, endosomal escape followed by cytosolic transport to reach ER-bound STING target is likely faster for cGAMP than the PC7A polymer (molecular weight = 21 kDa). Second, cGAMP-induced conformational change of STING and subsequent oligomerization20,21 may also occur faster than PC7A-induced STING condensate formation for immune activation. Lastly, buffering of endosomal pH and disruption of endosomal membranes by PC7A deter STING degradation through the endosome-lysosome pathway. With PC7A’s ability to activate STING and cytosolic delivery of cGAMP, we demonstrate cGAMP-PC7A NP achieved rapid and sustained STING activation across 6-48 h in both MC38 tumors and draining lymph nodes (Supplementary Fig. 9), which allow for an optimal time window for DC maturation and T cell priming (normally requires 1-2 days)52,53. This is supported by the synergistic therapeutic outcomes of cGAMP-PC7A NP in MC38 and TC-1 tumor treatment over single therapy alone.
A growing number of studies report the importance of STING pathway in cancer immunotherapy12–17. However, it is unclear whether STING activity in the cancer cells, immune cells or stromal cells are playing a more critical role in antitumor immunity. Our studies revealed the importance of host STING activity in the cGAMP-PC7A NP therapy (Supplementary Fig. 12). Data also show tumor growth inhibition is abolished by antibody blockade of CD8 T cells but not NK cells, indicating that CD8 T cells are the ultimate effector cells against tumor. We also show partial reduction of antitumor efficacy in DC-depleted mice, suggesting that additional immune cells (e.g., macrophage, B cells) or stromal cells (e.g., fibroblasts) may also contribute to the T cell-mediated antitumor immunity. Further investigations are warranted to elucidate the contributions from other immune cell types or subset of immune cells (e.g., tumor-resident CD103+ DCs)11,54 which may help identify key biomarkers for clinical translation.
In summary, this study highlights the use of a synthetic polymer to induce STING condensation for activation of an important innate immune pathway with spatiotemporal dynamics distinct from a natural STING ligand. Combination of polyvalent STING activation by PC7A with cell-intrinsic cGAMP stimulation further offers a synergistic and robust strategy to mount antitumor immunity for cancer immunotherapy.
Methods
Syntheses of polymers.
Monomers including 2-hexamethyleneiminoethyl methacrylate (C7A-MA), 2-(4-methylpiperidineleneimino)ethyl methacrylate (C6S1A-MA), 2-heptamethyleneiminoethyl methacrylate (C8A-MA), 2-diisopropylaminoethyl methacrylate (DPA-MA), and 2-ethylpropylaminoethyl methacrylate (EPA-MA) were synthesized following previous publications39,55. PEG-b-PR copolymers were synthesized using an atom transfer radical polymerization (ATRP) method. Poly(ethylene glycol)-b-poly(2-hexamethyleneiminoethyl methacrylate) with 70 repeating units, PC7A(70), is used as an example to illustrate the procedure. First, C7A-MA (1.5 g, 7 mmol), MeO-PEG114-Br (0.5 g, 0.1 mmol, Sigma Aldrich), and N,N,N′,N″,N″-Pentamethyldiethylenetriamine (PMDETA, 21 μL, 0.1 mmol, Sigma Aldrich) were dissolved in a mixture of 2-propanol (2 mL) and dimethylformamide (2 mL) in a Schlenk flask. Oxygen was removed by three cycles of freeze-pump-thaw, then CuBr (14 mg, 0.1 mmol, Alfa Aesar) was added under nitrogen protection. Polymerization was carried out in vacuo at 40 °C overnight. After polymerization, the reaction mixture was diluted in tetrahydrofuran (10 mL), then passed through a neutral Al2O3 column to remove the catalyst. The organic solvent was removed by rotary evaporation. The residue was dialyzed in distilled water and lyophilized to obtain a white powder. After syntheses, the product was characterized by 1H NMR and gel permeation chromatography. The four other polymers, including PC6S1A, PC8A, PDPA, and PEPA, were all synthesized with 70 repeating units. PC7A polymers with different repeating units were synthesized by adjusting the initial ratio of C7A-MA monomer over the MeO-PEG114-Br initiator.
Syntheses of dye-conjugated copolymers followed a similar procedure39,55. Primary amino groups (aminoethyl methacrylate or AMA-MA, Polysciences) were introduced into each polymer chain by controlling the feeding ratio of AMA-MA monomer to the initiator (3:1). After synthesis, PEG-b-(PR-r-AMA) was dissolved in dimethylformamide, and dye-N-hydroxylsuccinimidal ester was added (3 molar equivalences to the primary amino group, Lumiprobe). After overnight reaction, the copolymer was purified by ultracentrifugation (MW = 10 kDa cutoff) three times to remove free dye molecules. The product was lyophilized and stored at −80 °C.
Preparation of micelle nanoparticles.
Micelle nanoparticles for cellular studies were prepared following a solvent evaporation method as previously reported39,55. Briefly, polymer (4 mg) was first dissolved in methanol (0.4 mL) and then added dropwise into distilled water (3.6 mL) under sonication. Methanol was removed by ultrafiltration (MW = 100 kDa cutoff) three times with fresh distilled water. Sterile PBS was added to adjust the concentration to 200 μM as a stock solution.
cGAMP-loaded nanoparticles were prepared by mixing 2′3′-cGAMP in PC7A polymer solution containing 5% D-glucose at pH 4, followed by adjusting to pH 7.4 using NaOH. After micelle formation, the nanoparticles were analyzed by dynamic light scattering to measure size and zeta potential, and transmission electron microscopy for particle morphology. The cGAMP loading efficiency (> 90%) was quantified by high performance liquid chromatography.
Expression, purification and labeling of recombinant STING proteins.
Human STING C-terminal domain (CTD, amino acid sequence between 139–379) plasmid containing His6 tag encoded in pET-SUMO vector (provided by Dr. Z. J. Chen, UT Southwestern) was used as a template to generate E296A/D297A, D319A/D320A, and E336A/E337A/E339A/E340A mutants using a Q5 site directed mutagenesis kit (NEB). Overexpression of WT or mutant protein was induced in Escherichia coli. (E.coli) strain BL21/pLys with 0.8 mM isopropyl-β-D-thiogalactoside (IPTG) at 16 °C for 18 h. Bacterial cells were collected, suspended (50 mM Tris-Cl, 300 mM NaCl, 20mM imidazole, pH 8.0), and disrupted by sonication on ice. Cellular debris was removed by centrifugation at 20,000 g at 4 °C for 1 h. The supernatant was loaded onto a Ni2+-nitrilotriacetate affinity resin (Ni-NTA, QIAGEN). After 4 h incubation at 4 °C, the resin was rinsed three times with washing buffer (50 mM Tris-Cl, 1 M NaCl, 20 mM imidazole, pH 8.0). The SUMO tag was then removed by digesting the proteins using ULP1 SUMO protease at 4 °C overnight. Proteins were eluted with elution buffer (20 mM Tris-Cl, 50 mM NaCl, 20 mM imidazole, pH 7.5). Subsequently, the eluted proteins were subjected to size-exclusion chromatography using a Superdex 200 column (GE Healthcare), and the fractions were collected, concentrated, and dialyzed against a buffer containing 25 mM HEPES and 150 mM NaCl (pH 7.5)43.
For dye-conjugation, protein solution was mixed with Cy5-NHS in NaHCO3 (pH 8.4) at 4 °C overnight. Free dye molecules were removed by using a desalting column (7K, Thermo Scientific). Dye-labeled proteins were collected, concentrated, and used in phase separation studies.
Isothermal titration calorimetry (ITC).
A MicroCal VP-ITC was used to measure the binding affinity between protein and polymer. STING dimer concentration was held at 12.5 μM and PC7A(70) at 10 μM. The titrations were performed at 20 °C in a buffer containing 25 mM HEPES and 150 mM NaCl (pH 6.5). Twenty-nine injections were performed in 3 min spacing time. The titration traces were integrated by NITPIC 1.2.7, the curves were fitted by SEDFHAT 15.2b, and the figures were prepared using GUSSI 1.4.2 software.
Nile Red assay.
Nile Red assay is used for studying protein-protein interaction and interruption in protein structure37. Briefly, Nile Red (final concentration 5 μM, Thermo Scientific), STING dimer (2.1 μM), and PC7A (0, 0.6, 1.2, 3, 6, or 12 μM) were mixed for 4 h. Their max excitation wavelengths and fluorescence intensities were recorded on a fluorescence spectrophotometer (Hitachi F-7000 model).
Phase condensation assay.
Wild type (WT) or mutant human STING CTD (Cy5-labeled) was mixed with PC7A polymers of varying repeating units in a 96-well glass plate (coated with mPEG-silane) at 25 °C. After 4 h, the mixture was centrifuged at 13,000 g for 5 minutes, and the supernatant was transferred to another plate. Fluorescent intensity of the supernatant was measured by a plate reader (CLARIOstar). Data are representative of at least three independent measurements. The degree of condensation (D) was calculated by the following equation:
where Fi is the fluorescent intensity of the supernatant for a specific group i and F0 is the Cy5-STING intensity at the same concentration without PC7A addition.
For phase reversibility assay, STING CTD (Cy5-labeled) and PC7A polymer were first mixed. After condensate formation, the mixture was diluted ten times in pH = 6.5 HEPES buffer, and shaken on a plate shaker for 24 h. The fluorescent intensity of supernatant was measured, and reversibility (R) was calculated by following equation:
where DRi was the new DPS value after 24 h recovery.
For microscopy examination, STING protein (Cy5-labeled) was mixed with PC7A polymer in a 4-well glass chamber (Thermo Scientific, coated with mPEG-silane) at 25 °C, and images were acquired over a 140-s time course in 4-s intervals with the built-in software (ZEN 2.6) of Zeiss 700 confocal laser scanning microscope. Size was calculated as the average of longest and shortest axis of each condensate. The size distribution was plotted using GraphPad Prism 7.
Animals and cells.
All animals were maintained at the animal facilities under specific pathogen-free conditions and all animal procedures were performed with ethical compliance and approval by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. Female C57BL/6 mice (6–8 weeks old) were obtained from the UT Southwestern breeding core. Host Tmem173−/− C57BL/6 mice56 were provided by Y-X. Fu and CD11c-DTR transgenic C57BL/6 mice were purchased from the Jackson Laboratory. Mice were housed in a barrier facility with a 12h light/dark cycle and maintained on standard chow (2916 Teklad Global). The temperature range for the housing room is 68-79 °F (average is around 72 °F) and the humidity range is 30-50% (average is around 50%).
STING-GFP MEFs (provided by Dr. Nan Yan, UT Southwestern), HEK293T (ATCC), B16F10 (ATCC), MC38 (ATCC), Tmem173-KO MC38 (provided by Y-X. Fu)56, TC-1 (provided by T. C. Wu, John Hopkins University) cells were cultured in complete DMEM media supplemented with 10% fetal bovine serum (FBS). THP-1 cells (ATCC) were cultured in RPMI media supplemented with 10% FBS and 0.05 mM β-mercaptoethanol (β-ME). All cells were grown at 37 °C in 5% CO2. THP-1 monocytes were differentiated into macrophages by phorbol 12-myristate 13-acetate (PMA, 150 nM, InvivoGen) before use.
In cell mutagenesis assay, GFP tagged full-length WT STING plasmid (provided by N. Yan) was used as a template to generate E296A/D297A, D319A/D320A, and E336A/E337A/E339A/E340A mutants. HEK293T cells were transfected with lipofectamine 2000 (Invitrogen) carrying full-length WT or mutant STING-GFP plasmid for 24 h and allowed to recover for 12 h before use. WT or R232H THP-1 reporter cells were purchased from Invitrogen. R238A/Y240A and single or dual Q273A/A277Q Hela mutants (provided by Z. J. Chen)20,21 was used as cGAMP-resistant STING mutant cells.
Microscopy.
Cells were grown in a 4-well glass chamber and treated with cGAMP or PC7A polymer for indicated time. In STING degradation assay, LysoTracker Red DND-99 (Thermo Scientific) was used to stain lysosomes in live cells. In STING trafficking assay, cells were fixed in 4% paraformaldehyde, then permeabilized and stained for ER (Calnexin, 1:200, Abcam), ERGIC (p58, 1:1000, Sigma Aldrich), Golgi (GM130, 1:50, BD Biosciences), or p-TBK1 (Ser 172, 1:50, Cell Signaling) using an immunofluorescence kit (Cell Signaling). Samples were mounted in prolong gold antifade with Dapi stain (Thermo Scientific) and imaged with the built-in software (ZEN 2.6) of Zeiss 700 confocal laser scanning microscope with a 63× oil objective. ImageJ 1.52d was used to quantify co-localization by Pearson’s correlation coefficient. Data are representative of at least twenty cells. In inhibitor assay, cells were pre-treated with Brefeldin A (BFA, 10 μM, Selleckchem) for 1 h before cGAMP/PC7A addition.
Fluorescence recovery after photobleaching (FRAP) experiments.
FRAP method is a versatile tool for determining diffusion and exchange properties of biomacromolecules57. Both in vitro and cellular FRAP experiments were performed on a Zeiss 700 confocal laser scanning microscope at 25 °C. In a typical procedure, a 2 μm diameter spot in the condensation was photobleached with 100% laser power for 5 seconds using a 633 nm laser. Images were acquired over a 150-s time course with 4-s intervals. Fluorescent intensity of the region of interest (ROI) was corrected by an unbleached control region and then normalized to pre-bleached intensity of the ROI. At least five biologically independent samples were measured. The mean intensity of the bleached spot was fit to a single exponential model32 by Graph Pad Prism 7 software.
Western blot analysis.
All solutions were purchased from Bio-Rad and antibodies against STING (1:1000), p-STING (S366, 1:1000), p-TBK1 (Ser 172, 1:1000) and p-IRF3 (Ser 369, 1:1000) were obtained from Cell Signaling. Briefly, cells were lysed in SDS sample buffer (with protease and phosphatase inhibitor cocktail) and heated for denaturation. Supernatant was loaded onto a 4-15% Mini-PROTEAN gel (Bio-Rad), and run at 50 V for 20 min followed by 100 V for 60 min. Electrotransfer was performed using 100 V for 60 min on ice. After transfer, the membrane was blocked either in 5% non-fat milk or BSA (phosphorylated protein) for 1 h at room temperature, and incubated with primary antibody overnight at 4 °C. Goat anti-mouse or goat anti-rabbit IgG HRP-linked secondary antibody (1:3000, Bio-Rad) was used for 1 h at room temperature before detection on X-ray film (GE Healthcare). Membrane was stripped in stripping buffer for 30 min and reused for β-actin (Sigma Aldrich) detection.
RT-qPCR.
Total RNAs were extracted from cells or human tissues by using RNeasy mini kit (QIAGEN). RNA quantity and quality were confirmed using NanoDrop (DeNovix DS-11). Genomic DNA was removed and cDNA was synthesized using an iScript™ gDNA clear cDNA synthesis kit (Bio-Rad). Bio-Rad SsoAdvanced™ universal SYBR green supermix and CFX connect real-time system were used for PCR analysis. Results were corrected by β-actin or GAPDH in Excel Office 365 and plotted in Graph Pad Prism 7 software. DNA primers are listed as follows.
Mouse ifn-β: ATGAGTGGTGGTTGCAGGC, TGACCTTTCAAATGCAGTAGATTCA.
Mouse cxcl10: GGAGTGAAGCCACGCACAC, ATGGAGAGAGGCTCTCTGCTGT.
Mouse β-actin: ACACCCGCCACCAGTTCGC, ATGGGGTACTTCAGGGTCAGGATA.
Human ifn-β: GTCTCCTCCAAATTGCTCTC, ACAGGAGCTTCTGACACTGA.
Human cxcl10: TGGCATTCAAGGAGTACCTC, TTGTAGCAATGATCTCAACACG.
Human β-actin: GGACTTCGAGCAAGAGATGG, AGGAAGGAAGGCTGGAAGAG.
Human gapdh: ATGACATCAAGAAGGTGGTG, CATACCAGGAAATGAGCTTG.
Evaluation of STING activation in tumor-bearing mouse.
Mice were subcutaneously inoculated with MC38 cells (1 × 106) into the right flank. One intratumoral injection of different agents (50 μl of 5% glucose, 50 μg PC7A polymer, 2.5 or 50 μg cGAMP, or a formulation with 2.5 μg cGAMP in 50 μg PC7A NP) was performed when tumor size reached 100 ± 20 mm3. Mice were euthanized at different time points post-injection, and tumors and draining lymph nodes were collected. Total RNAs were extracted by TRIzol (Invitrogen), and the expression of interferon-stimulated genes (ifn-β and cxcl10) were measured via RT-qPCR.
Safety studies.
Mice were subcutaneously inoculated with MC38 cells (1 × 106) into the right flank. Intratumoral injections of different agents (50 μl of 5% glucose, 50 μg PC7A polymer, 2.5 or 50 μg cGAMP, or a formulation with 2.5 μg cGAMP in 50 μg PC7A NP) was performed when tumor size reached ~50 mm3 (around day 6). Two additional injections were performed on day 9 and 12. One day after the last administration, 1 mL of blood sample was collected from each mouse without heparinization and then centrifuged at 4,000 rpm for 5 mins to obtain serum. The activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and urea were measured using specific kits (Abcam, #105134, 105135, 83362). The systemic concentration of interleukin-10 was measured using an Elisa assay (Invitrogen #88-7105-22). Statistical analysis was performed using GraphPad Prism 7.
Tumor therapy experiments.
Mice were subcutaneously inoculated with MC38 cells (1 × 106) or TC-1 cells (1 × 105) into the right flank. Tumor size was measured every 2 or 3 days via a digital caliper, and tumor volume was calculated as 0.5 × length × width2. On reaching sizes of ~50 mm3, tumors were injected with different STING agonists (50 μl of 5% glucose, 50 μg PC7A polymer, 2.5 or 50 μg cGAMP, or 2.5 μg cGAMP in 50 μg PC7A nanoparticles), and some groups were intraperitoneally injected with 200 μg depletion antibodies (anti-mCD8α, BioXcell, BP0117 or anti-mNK1.1, BioXcell, BP0036) or 200 μg checkpoint inhibitors (anti-mPD-1, BioXcell, BE0146) every 3 days for comparison or synergy evaluation. For systemic DC depletion, CD11c-DTR transgenic mice were injected intraperitoneally with 100 ng diphtheria toxin (DT, Sigma-Aldrich) every 3 days after tumor inoculation. Mice were injected 3× in MC38 model and 4× in TC-1 model with STING agonist treatments spaced 3 d apart. Mice were euthanized at a tumor burden endpoint of 2,000 mm3. Statistical analysis was performed using GraphPad Prism 7.
Evaluation of STING activation in resected human tissues.
Patients were consented to the use of biospecimens for research as approved by the UT Southwestern Institutional Review Board. Freshly resected human tissues (squamous cell carcinoma from the base/lateral of tongue, cervical tumor tissues, and a sentinel lymph node) were rinsed and divided into several sections (1-5 mm3) using a scalpel, followed by injection at multiple sites using 5% glucose control, free cGAMP (80 ng), PC7A polymer (50 μg), or cGAMP-PC7A nanoparticles (80 ng cGAMP in 50 μg PC7A nanoparticles) in 5% glucose solution within 30 min of resection. Each section was cultured in 0.5 mL RPMI 1640 medium (supplemented with 10% heat-inactivated human serum, 1% insulin-transferrin-selenium, 1% glutamax, and 1% penicillin-streptomycin) in a 24-well plate for 24 h. RNA was isolated and RT-qPCR was performed as previously described. For CD45 selection, tumor tissues were first digested by 1 mg/mL collagenase IV and 0.2 mg/mL DNase I (Sigma Aldrich) for 45 minutes at 37°C, then passed through a 70 μm nylon cell strainer to obtain single cells. CD45+ leukocytes and CD45− cell populations were collected via magnetic separation using CD45 TIL microbeads and MS columns (Miltenyi Biotec) according to the manufacturer’s instructions before RT-qPCR analysis.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank Z. J. Chen, M. Du, C. Zhang, C. Han, and N. Yan for sharing plasmids, STING mutant cell lines, and STING knockout animals. M. Rosen for discussion on biomolecular condensation, S. Y. Wu and H. T. Lai for the construction of STING mutant plasmids, Y. Li and L. You on polymer synthesis and characterization, C. Chen on computational modeling, S. Tso for ITC experiments and J. Lea for procurement of cervical tumor tissues. We also thank G. Huang, T. Huang, Z. Sun, and Z. Liu for helpful discussions. This work was supported by a grant from the National Institutes of Health (U54 CA244719) and Mendelson-Young Endowment in Cancer Therapeutics to J.G.
Footnotes
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. All data generated in this study, including source data and the data used to generate the figures, are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.13356464.
Competing interests
B.D.S. and J.G. are scientific co-founders and advisors of OncoNano Medicine, Inc.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-01X-XXXX-X.
References
- 1.Barber GN STING: infection, inflammation and cancer. Nat. Rev. Immunol 15, 760–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Yum S, Li M, Frankel AE & Chen ZJ Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu. Rev. Cancer Biol 3, 323–344 (2019). [Google Scholar]
- 4.Ishikawa H, Ma Z, & Barber GN STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun L, Wu J, Du F, Chen X & Chen ZJ Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu J, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, & Kroemer G Type I interferons in anticancer immunity. Nat. Rev. Immunol 15, 405–414 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Dunn GP, Koebel CM, & Schreiber RD Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol 6, 836–848 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Woo SR, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su T, et al. STING activation in cancer immunotherapy. Theranostics 9, 7759–7771 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vatner RE & Janssen EM STING, DCs and the link between innate and adaptive tumor immunity. Mol. Immunol 110, 13–23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramanjulu JM, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Haag SM, et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Yang H, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest 129, 4350–4364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shae D, et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol 14, 269–278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miao L, et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol 37, 1174–1185 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Luo M, et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ergun SL, Fernandez D, Weiss TM & Li L STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 178, 290–301 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Zhao B, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569, 718–722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shang G, Zhang C, Chen ZJ, Bai XC & Zhang X Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 567, 389–393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koshy ST, Cheung AS, Gu L, Graveline AR & Mooney DJ Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy. Adv. Biosys 1, 1600013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol 10, 1043–1048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato K, et al. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun 9, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu JJ, et al. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rev 1–25 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Lian Y, Duffy KJ, & Yang J STING Activation and its Application in Immuno-Oncology. Curr. Top. Med. Chem 19, 2205–2227 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Meric-Bernstam F, et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol 37, 2507 (2019). [Google Scholar]
- 28.Harrington KJ, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol 29, mdy424–015 (2018). [Google Scholar]
- 29.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyman AA, Weber CA & Jülicher F Liquid-liquid phase separation in biology. Annu. Rev. Cell. Dev. Biol 30, 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Hyman AA, & Simons K Beyond oil and water—phase transitions in cells. Science 337, 1047–1049 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Du M & Chen ZJ DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin Y & Brangwynne CP Liquid phase condensation in cell physiology and disease. Science 357, 4382 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Alberti S, Gladfelter A & Mittag T Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, et al. A nanobuffer reporter library for fine-scale imaging and perturbation of endocytic organelles. Nat. Commun 6, 8524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobbs N, et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host & Microbe 18, 157–168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sackett DL & Wolff J Nile red as a polarity-sensitive fluorescent probe of hydrophobic protein surfaces. Anal. Biochem 167, 228–234 (1987). [DOI] [PubMed] [Google Scholar]
- 38.Feng Q, Wilhelm J & Gao J Transistor-like Ultra-pH-Sensitive Polymeric Nanoparticles. Acc. Chem. Res 52, 1485–1495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou K, et al. Multicolored pH-tunable and activatable fluorescence nanoplatform responsive to physiologic pH stimuli. J. Am. Chem. Soc 134, 7803–7811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li P, et al. Phase transitions in the assembly of multivalent signaling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin Y, Protter DS, Rosen MK & Parker R Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molliex A, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ouyang S, et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36, 1073–1086 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi G, et al. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PloS one 8, e77846 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacquelot N, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 29, 846–861 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore E, et al. Established T cell–inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol. Res 4, 1061–1071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foote JB, et al. A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunol. Res 5, 468–479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jung S, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17, 211–220 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin L, et al. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun. 12, 263–269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel S, & Jin L TMEM173 variants and potential importance to human biology and disease. Genes Immun. 20, 82–89 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mempel TR, Henrickson SE & Von Andrian UH T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427, 154–159 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Obst R The timing of T cell priming and cycling. Front. Immunol 6, 563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spranger S, Dai D, Horton B & Gajewski TF Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou K, et al. Tunable, ultrasensitive pH-responsive nanoparticles targeting specific endocytic organelles in living cells. Angew. Chem. Int. Ed 50, 6109–6114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mender I, et al. Telomere stress potentiates STING-dependent anti-tumor immunity. Cancer Cell 38, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Axelrod D, Koppel DE, Schlessinger J, Elson E & Webb WW Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 16,1055–1069 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.