

SUMMARY
The interleukin-6 (IL-6) membrane receptor and its circulating soluble form, sIL-6R, can be targeted by antibody therapy to reduce deleterious immune signaling caused by chronic overexpression of the proinflammatory cytokine IL-6. This strategy may also hold promise for treating acute hyperinflammation, such as observed in coronavirus disease 2019 (COVID-19), highlighting a need to define regulators of IL-6 homeostasis. We found that conventional dendritic cells (cDCs), defined in mice via expression of the transcription factor Zbtb46, were a major source of circulating sIL-6R and, thus, systemically regulated IL-6 signaling. This was uncovered through identification of a cDC-dependent but T cell-independent modality that naturally adjuvants plasma cell differentiation and antibody responses to protein antigens. This pathway was then revealed as part of a broader biological buffer system in which cDC-derived sIL-6R set the in-solution persistence of IL-6. This control axis may further inform the development of therapeutic agents to modulate pro-inflammatory immune reactions.
In Brief
Hyper-elevated IL-6 underscores cytokine storming and chronic inflammatory disorders. Yousif et al. demonstrate that conventional dendritic cells are a major source of sIL-6R, which forms a buffer system that sets the in-solution persistence of IL-6, regulating inflammatory immune reactions.
Graphical Abstract
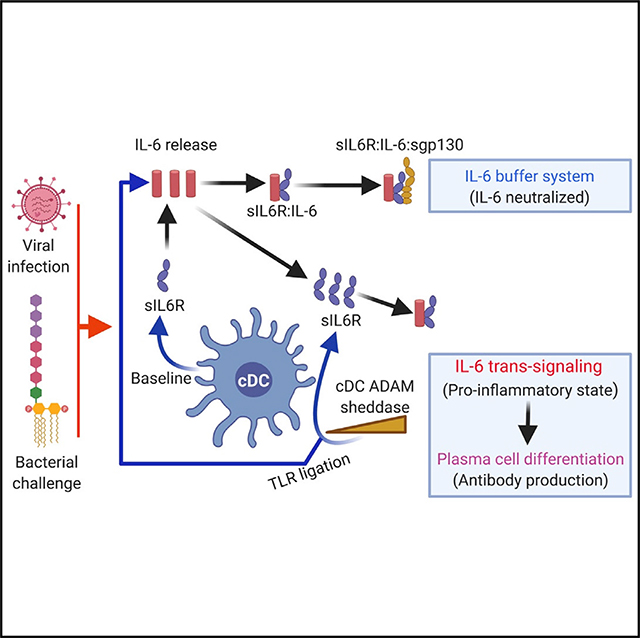
INTRODUCTION
Interleukin-6 (IL-6) is a potent pro-inflammatory cytokine released following immune challenge or tissue injury, where it broadly stimulates innate and adaptive immune reactions (Kishimoto, 2010; Tanaka et al., 2014). Overexpression of IL-6 can lead to chronic inflammatory disorders (Spencer et al., 2019; Tanaka et al., 2016) and potentially fatal hyperinflammation, such as seen during advanced coronavirus disease 2019 (COVID-19) (McElvaney et al., 2020; Mehta et al., 2020). Hyperinflammatory states can be treated by antibody blockade therapies targeting the IL-6 receptor (IL-6R) and its soluble circulating form (sIL-6R) (Tanaka et al., 2016, 2018), and some early clinical studies have reported successful application to treat COVID-19 (Biran et al., 2020; Somers et al., 2020; Xu et al., 2020), underscoring the importance of defining the homeostatic factors that regulate immune signaling though IL-6.
IL-6 classic signaling occurs when this cytokine engages IL-6R on a target cell co-displaying gp130, the receptor’s signaling subunit (Rose-John, 2012; Wolf et al., 2014). IL-6R expression, and thus IL-6 classic signaling, is largely restricted to immune cells and hepatocytes (Rose-John, 2012; Tanaka et al., 2014). In contrast, surface gp130 is more widely expressed, enabling IL-6 trans signaling via the complex formed between IL-6 and circulating sIL-6R (Rose-John, 2012; Wolf et al., 2014). Operationally, these IL-6 signaling modes can be distinguished by the circulating form of gp130 (sgp130), which inhibits the latter but not former (Jostock et al., 2001; Tenhumberg et al., 2008). IL-6 classic signaling is implicated in acute-phase responses, whereas IL-6 trans signaling typically initiates pro-inflammatory pathways (Rose-John, 2012; Scheller et al., 2006). IL-6 classic signaling is also important for production of hepatocyte growth factor during liver regeneration but can be substituted by the trans signaling mode (Fazel Modares et al., 2019).
sIL-6R and sgp130 are also thought to comprise a biological buffer system that is predicted to regulate IL-6 half-life in blood (Rose-John, 2012, 2017; Scheller and Rose-John, 2012). In healthy humans, serum IL-6 is nearly undetectable (2–6 pg/mL), whereas the concentrations of sIL-6R and sgp130 are in molar excess (Rose-John, 2012, 2017; Scheller and Rose-John, 2012), stoichiometries that are also conserved in mice (Doganci et al., 2005; Schuett et al., 2012). IL-6 binds sIL-6R with nanomolar affinity, and the IL-6:sIL-6R complex engages inhibitory sgp130 with picomolar affinity, suggesting that free IL-6 is captured and neutralized rapidly at steady state (Rose-John, 2012, 2017; Scheller and Rose-John, 2012). In pro-inflammatory states, sIL-6R is elevated, whereas sgp130 stays relatively constant, enabling IL-6 trans signaling through molar excess of sIL-6R (Rose-John, 2012, 2017; Scheller and Rose-John, 2012; Tanaka et al., 2014).
The capacity of sIL-6R to set the half-life of free IL-6 and restrict its systemic signaling has not been validated experimentally in vivo (Baran et al., 2018). One issue is that sIL-6R is generated post-translationally and is difficult to manipulate. Cell culture systems have implicated A disintegrin and metalloenzymes (ADAMs) as sheddases that catalyze sIL-6R release from surface IL-6R (Chalaris et al., 2007; Garbers et al., 2014; Matthews et al., 2003; Müllberg et al., 1994, 1995; Riethmueller et al., 2016; Vollmer et al., 1996), but their genetic depletion is lethal in mice, and cell-specific depletion of ADAM sheddases has failed to alter sIL-6R set point in vivo (Garbers et al., 2011; Schumacher et al., 2015, 2016). The cellular sources of sIL-6R are also only broadly defined; cell-specific depletion of IL-6R using Alb cre and lyz2 cre shows 40% and 60% reduction in circulating sIL-6R (McFarland-Mancini et al., 2010). Alb is selective for murine hepatocytes (Weisend et al., 2009), but Lyz2 is expressed in numerous cell lineages, including monocytes and macrophages, CD11c+ dendritic cells, granulocytes, and type II lung alveolar cells (Shi et al., 2018).
Using a combination of bone marrow chimeras and validated cell-specific approaches to deplete IL-6R, we show that conventional dendritic cells (cDCs), defined in mice by expression of the transcription factor Zbtb46 (Meredith et al., 2012; Satpathy et al., 2012a), play a central role in maintaining systemic sIL-6R. DCs connect the innate and adaptive immune systems by priming T cells (Alvarez et al., 2008; Henrickson et al., 2013), which is necessary for thymus-dependent (TD) B cell responses, underscoring humoral immunity (Stebegg et al., 2020; Tesfaye et al., 2019). We identified cDC control over systemic sIL-6R by uncovering a modality that modulates TD antibody output independent from T cell priming. In this pathway, cDC-derived sIL-6R enabled IL-6 trans signaling at the B cell surface, promoting differentiation to antibody-secreting plasma cells. We further demonstrate that cDC-derived sIL-6R captures the free IL-6 released following bacterial and viral challenges. Here induction of sIL-6R release required Toll-like receptor (TLR) ligation followed by ADAM sheddase activity. Our study establishes cDC-derived sIL-6R as a core component of a biological buffer system regulating in-solution persistence of IL-6 and, consequently, immune signaling through this cytokine.
RESULTS
A cDC-dependent but T cell-independent axis that regulates antibody output
We first evaluated splenic antibody responses following intravenous challenge with influenza viral glycoprotein hemagglutinin (HA), an extensively described protein antigen (Altman et al., 2018; Angeletti et al., 2017). Immunoglobulin G (IgG) and IgM responses were lost following splenectomy, whereas only IgG output was reduced in Tcra−/− mice (Figure 1A). When cDCs were depleted in Zbtb46-diphtheria toxin receptor (DTR) bone marrow chimeras (Meredith et al., 2012; Satpathy et al., 2012a), IgG and IgM output was reduced significantly (Figure 1A). The chimeras did not show a defect in antigen uptake within the spleen (Figure S1B). Reduction in IgG was expected in the absence of DC-primed T cell help (O et al., 2014), but co-reduction in the T cell receptor (TCR)-independent IgM response suggested that cDCs may also regulate humoral immunity independent of T cell priming. We confirmed this for other non-HA antigens, including HIV gp120 and ovalbumin (Figures S1C and S1D). The IgM response could be rescued in mixed chimeras containing 50% Zbtb46 DTR:50% major histocompatibility complex (MHC) class II-deficient (homozygous H2dlAb1-Ea); diphtheria toxin (DT)-treated animals raised no IgG (indicating successful depletion of cDC-bearing MHC class II) but retained the native IgM response in the MHC class II-deficient background (Figure 1A). Accordingly, we proceeded to use IgM elicitation as a readout to identify factors responsible for T cell-independent but cDC-dependent control over humoral immunity (Figure 1B).
Figure 1. cDC-dependent but TCR-independent control over IgM responses to influenza hemagglutinin (HA), a model TD antigen.

(A) Serum IgM versus IgG responses against HA that was injected intravenously along with lipid A adjuvant (n = 5 animals per group). The responses were elicited in the following groups: splenectomized versus sham control, Tcra−/− versus WT, Zbtb46-DTR chimeras receiving versus not receiving DT, and chimeras containing 50% Zbtb46-DTR:50% MHC class II-deficient (homozygous H2dlAb1-Ea) receiving versus not receiving DT. The dilution curves (mean ± SD) were quantified by endpoint dilution; **p < 0.02, ***p < 0.001, Student’s t test.
(B) These data provide functional evidence of a cDC-dependent but T cell-independent mechanism modulating humoral output (red arrow).
(C) Recapitulation of the IgM (and IgG) response deficits to HA via genetic ablation of IL-6 and IRF5 but not IL-12 (n = 5 animals per group). Antibody response deficits could be rescued by addition of recombinant murine IL-6. The dilution curves were quantified by endpoint dilution; **p < 0.005, *p < 0.05, ANOVA with Tukey’s test. See Figure S1 for chimera methodology, orthogonal demonstration using non-HA antigens, and antibody responses in Ifnar1−/− mice.
Genetic ablation of IRF5 and IL-6 (but not IL-12 or IFNAR) functionally recapitulates the IgM response defect
We hypothesized that cDCs may exert effects through secreted molecules and that interferon regulatory factor 5 (IRF5) could be important because it is needed for generating IL-12 and IL-6 (Takaoka et al., 2005), cytokines that support humoral immune reactions (Dubois et al., 1998; Karnowski et al., 2012; Kim et al., 2008; Tanaka et al., 2014). Indeed, we recapitulated the IgM response deficit in the Irf5−/− and Il6−/− genotypes but not in Il12p40−/− or Ifnar1−/− mice (Figures 1C and S1E). Injection of Irf5−/− mice with recombinant IL-6 partially rescued IgM and IgG titers (Figure 1C), consistent with IL-6 dependence and an independence from interferon-α/β receptor (IFNAR) signaling. All immunizations were performed using Sigma adjuvant, which deploys bacterial lipid A, a well-described stimulator of IL-6 release (Matsuura et al., 1999; Wang et al., 1991).
cDCs set the circulating concentration of sIL-6R, which is needed for efficient antibody responses
If the IgM response deficits represented the same IL-6-dependent but IFNAR-independent pathway, we hypothesized that IRF5 signaling was needed to produce IL-6 by cDCs and/or that IL-6R derived from cDCs was needed to support IL-6 signaling. Accordingly, we generated mice in which IL-6R or IRF5 was depleted under Zbtb46. B cell, T cell, macrophage, and DC subsets were phenotypically similar in these genotypes (Figure S2), and cDCs showed selective depletion of IL-6R in cDC-IL6ra−/− (Figure 2A) and reduction in total IRF5 in cDC-Irf5−/− (Figure 2B). Antibody responses were unaffected in cDC-Irf5−/−, but IgM (and IgG) response deficits occurred in cDC-IL6ra−/− (Figure 2C). Serum sIL-6R was also strongly reduced in this genotype (Figure 2D), and injection of recombinant sIL-6R rescued IgM and IgG output (Figure 2E). Thus, cDCs are a major source of sIL-6R, and sIL-6R is one factor in T cell-independent control over B cell priming.
Figure 2. cDCs set the circulating concentration of sIL-6R, and this regulates antibody responses.

(A) Histograms reporting surface IL-6R in immune cells from WT, cDC-Irf5−/−, and cDC-IL6ra−/− mice (B cells = B220+/CD3−, T cells = CD3+/B220−, cDCs = CD3−/B220−/MHCII+/CD11chi, tissue resident macrophages = CD3−/B220−/CD11b+/F4–80+). Quantification of these data is presented as the proportion of cells expressing surface IL-6R (mean ± SD, n = 3 per group, ***p < 0.001, ANOVA with Tukey’s test).
(B) Total IRF5 was measured in cDCs (B220−/CD3−/CD11chi/MHCII+) isolated by fluorescence-activated cell sorting (FACS) on splenocytes of WT and cDC-Irf5−/− animals. The sorted cDCs (200,000 cells) were lysed, separated by SDS-PAGE, and immunoblotted for either IRF5 or β-actin.
(C) Serum IgM versus IgG responses against HA (with lipid A adjuvant) in transgenic mice (mean ± SD, n = 4 animals per group). The dilution curves were quantified by endpoint dilution; **p < 0.01, ANOVA with Tukey’s test.
(D) Serum sIL-6R as measured in WT, cDC-Irf5−/−, and cDC-IL6ra−/− (mean ± SD, n = 4 animals per group); ****p < 0.0001, ANOVA with Tukey’s test.
(E) Co-injection of recombinant murine sIL-6R rescued the IgM and IgG responses to HA (with lipid A adjuvant) in cDC-IL6ra−/− animals (mean ± SD, n = 4 animals per group). The dilution curves were quantified by endpoint dilution; **p < 0.01, ANOVA with Tukey’s test.
See also Figures S2 and S3 for immune cell subsets in cDC-IL6ra−/− and cDC-IL6ra−/−, sIL6R concentration in cDC-DTR, and IL6R expression by cDC1 and cDC2 in cDC-IL6ra−/− and Batf3−/−.
The majority of the serum sIL-6R was also lost in Zbtb46-DTR chimeras and restored in mixed chimeras (50% Zbtb46 DTR:50% MHC class II deficient) (Figure S3A). We also examined Batf3−/− mice, which are depleted for cDC1 (Hildner et al., 2008; Figure S3B). Zbtb46 does not discriminate between cDC1 or cDC2 (Figure SB). We found that cDC1 (a minor cDC component) accounted for a minor fraction of circulating sIL-6R (Figure S3C) and that antibody responses were not affected in Batf3−/− (Figure S3D).
IL-6 trans signaling through cDC-derived sIL-6R naturally adjuvants humoral responses by promoting differentiation to plasma cells in vivo
B cells are limited in surface gp130, which is needed for IL-6 trans signaling (Larousserie et al., 2006). We found that gp130 increased upon B cell differentiation to plasmablasts and verified that these cells are specifically receptive to IL-6 trans signaling, as measured by induction of STAT3 phosphorylation by IL6-sIL-6Rα (hyper-IL6) (Fischer et al., 1997; Xu et al., 2017; Figures 3A–3D). The number of plasmablasts could be increased by pre-immunizing with our lipid A adjuvant (3 days post vaccination; Figures 3B and 3D). To define the significance of IL-6 trans signaling in vivo, we repeated our immunization regimen containing the lipid A adjuvant and measured plasmablast differentiation to plasma cells 1 week post vaccination. We found that differentiation was attenuated in cDC-IL6ra−/− but partially restored by injection of recombinant sIL-6R (Figure 3E). cDC-Irf5−/− mice showed no defect compared with wild-type (WT) animals. Collectively, this reveals a natural B cell-adjuvating activity of cDC-derived sIL-6R (Figure 3F).
Figure 3. cDC-derived sIL-6R gates antibody responses by controlling plasma cell differentiation.

(A) Surface expression of gp130 on B cells (B220+/CD3−, gate I) versus plasmablasts (CD3−/B220lo/CD138+, gate II) isolated from spleen of WT mice. The splenocytes were either left unstimulated or exposed to sIL-6Rα or IL6-sIL-6Rα (hyper-IL-6).
(B) Same as in (A), except that animals were first immunized with lipid A adjuvant and spleen was harvested 3 days later.
(C) Quantification of the data in (A). Signaling induced within gate I and gate II was measured as the percentage of STAT3-activated cells (sIL-6Rα-stimulated minus unstimulated and IL-6-sIL-6Rα-stimulated minus unstimulated; mean ± SD, n = 4 animals per group, ***p < 0.001, Student’s t test).
(D) Quantification of the data in (B) (as in C).
(E) Differentiation from plasmablasts (CD3−/B220lo/CD138+/MHCII+) to plasma cells (CD3-/B220lo/CD138+/MHC class IIlo/CD93+) was monitored within each genotype 1 week post-immunization with lipid A adjuvant, and cDC-IL6ra−/− mice were also co-injected with recombinant sIL-6R (as in Figure 2E). The proportion of plasmablasts differentiating into plasma cells (indicated by a box in flow plots) was then quantified below (mean ± SD, n = 3 animals per group; **p < 0.005, ANOVA with Tukey’s test).
(F) Model for T cell-independent, cDC-dependent control over antibody responses.
cDCs regulate TLR4-dependent induction of sIL-6R release and serum persistence of IL-6 following bacterial immune stimulation
sIL6R and sgp130 are proposed to form a biological buffer system that regulates IL-6 half-life in blood (Rose-John, 2012, 2017; Scheller and Rose-John, 2012), but this has not been demonstrated experimentally in vivo (Baran et al., 2018). To evaluate this, we intravenously injected our transgenic animals with bacterial lipid A to mimic sepsis and then tracked the pharmacokinetics of IL-6 release and capture in the bloodstream (Figure 4A). After injection, sIL-6R concentration became elevated and then declined in WT and cDC-Irf5−/− mice but remained low and did not change in cDC-IL6ra−/− (Figure 4A). Induction of sIL-6R release did not occur in Tlr4−/−, but these animals retained a normal sIL-6R set point. Free IL-6, induced by lipid A exposure, was only detectable in cDC-IL6ra−/−, where it showed delayed clearance (Figure 4A). Similar dysregulation was seen in Zbtb46-DTR chimeras (Figure 4B). Restoration of serum sIL-6R in cDC-IL6ra−/− via gene delivery by adeno-associated virus (AAV) largely rescued IL-6 capture after inoculating with bacterial lipid A (Figure S4A). In contrast to bacterial lipopolysaccharide (LPS), IL-6 treatment alone did not stimulate sIL-6R release, as evaluated in bone marrow-derived DCs (BMDCs) (Figure 4C).
Figure 4. cDCs regulate the systemic sIL-6R set point and induction of release, regulating the pharmacokinetics of free IL-6 generated in response to diverse immune challenges.

(A) Animals were injected intravenously with bacterial lipid A to mimic sepsis and promote IL-6 release. Serum concentrations of sIL-6R and IL-6 were measured in WT, cDC− Irf5−/−, Tlr4−/−, and cDC-IL6ra−/− (mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA with Tukey’s test; n = 3 per group).
(B) sIL-6R and IL-6 serum concentrations during the same time course in Zbtb46-DTR chimeras receiving and not receiving DT (mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA with Tukey’s test; n = 3 per group).
(C) BMDCs were cultured and exposed to recombinant IL-6 or bacterial LPS, and sIL-6R was measured in the supernatant (mean SD; ****p < 0.0001, ANOVA with Tukey’s test).
(D) Animals were infected with a sublethal dose of NC99 influenza virus, and serum sIL-6R and IL-6 concentrations were measured in WT, Tlr4−/−, Tlr7−/−, and cDC-IL6ra−/− (mean ± SD; *p < 0.05, ***p < 0.001, ANOVA with Tukey’s test; n = 3 per group). In bacterial and viral challenge models, the defect in IL-6 capture within cDC-IL6ra−/− was rescued by restoring sIL-6R using AAV gene delivery (Figures S4A and S4B). Restoration of sIL-6R also reduced the morbidity rate after lethal challenge with influenza virus (Figure S4C).
(E and F) cDC-Adam17−/− had no defect in the sIL-6R set point but significantly blunted sIL-6R release following stimulation with bacterial lipid A (E) or infection with influenza virus (F) (mean ± SD; *p < 0.04, **p < 0.01, F test for slope comparison; n = 5 per group).
(G) Regulating free IL-6 and IL-6 trans signaling via cDC-derived sIL-6R.
Please also see Figure S4.
cDCs regulate TLR7-dependent induction of sIL-6R biogenesis and serum persistence of IL-6 following infection with influenza virus
We infected WT, Tlr4−/−, Tlr7−/−, and cDC-IL6ra−/− mice with New Caledonia/20/1999 (NC99), an H1N1 virus that propagates but does not cause disease in mice (Amitai et al., 2020; Glaser et al., 2007; Sangesland et al., 2019). We observed induction of sIL-6R release in all genotypes except Tlr7−/− and cDC-IL6ra−/−, where TLR7 recognizes influenza viral RNA (Lund et al., 2004). Influenza virus is also a strong inducer of IL-6 release (Dutta et al., 2013; Vogel et al., 2014), and we found that downstream capture of the released IL-6 was dysregulated in cDC-IL6ra−/− (Figure 4D). This buffer activity was rescued by AAV-mediated restoration of sIL-6R (Figure S4B).
We next performed the same AAV-mediated rescue of sIL-6R in a severe model of infection. Animals received a lethal dose of A/Puerto Rico/8/34 (PR8) influenza virus in which lung disease is accompanied by cytokine storming (Dutta et al., 2013; Vogel et al., 2014). We found that restoration of sIL-6R significantly slowed the morbidity rate, as measured by daily weight loss (Figure S4C).
Induction of sIL-6R release, but not the set point, is regulated by cDC-ADAM17
Induction of sIL-6R release following immune stimulation is needed to generate molar excess relative to sgp130 to enable IL-6 trans signaling and pro-inflammatory reactions (Rose-John, 2012, 2017; Scheller and Rose-John, 2012; Tanaka et al., 2014). The cell surface sheddase ADAM17 has been implicated in sIL-6R release from BMDC following incubation with LPS in vitro (Garbers et al., 2011). We found that induction of sIL-6R release in vivo was significantly blunted in cDC-Adam17−/− mice following inoculation with bacterial lipid A and after infection with influenza virus (Figures 4E and 4F). Notably, the sIL-6R set point was unaffected in cDC-Adam17−/−, suggesting that the set point and immune-stimulated release rely on different mechanisms (Figure 4G).
DISCUSSION
The textbook model of humoral immunity predicts that cDC removal should only reduce T cell-dependent antibody responses because of lack of DC-primed T cell help (Adler et al., 2017; Andersen et al., 2019; O et al., 2014). However, we found that co-reduction in TCR-independent IgM output was a general property of TD antigens. Reduced IgM responses have also been seen in Itgax-DTR chimeras (Gonzalez et al., 2010) but have not been explained. This defect appears to be reversible because longer-term exposure to DT in Zbtb46-DTR chimeras (18 weeks) fails to sustain cDC depletion, and anti-DT immunoglobulin becomes detectable (Rombouts et al., 2017). We demonstrated that the cDC-dependent IgM response could be functionally rescued within our 2-week DT exposure period when non-depleted cDCs were present in the MHC class II-deficient background. We therefore applied this IgM response as a functional readout to identify cDC-dependent but T cell-independent factors regulating humoral immunity.
DCs have long been demonstrated to directly prime B cells through antigen uptake and display (Balázs et al., 2002; Bergtold et al., 2005; Chappell et al., 2012; MacLennan and Vinuesa, 2002; Qi et al., 2006). However, we found that antigen uptake into the spleen was unaffected following cDC depletion, suggesting that other factors may also be in operation. Notably, genetic depletion of IL-6 and its master regulator IRF5 (Takaoka et al., 2005) recapitulated the IgM response defect. Although independent of IFNAR, the phenotype could be partially rescued by recombinant IL-6, a known promoter of humoral reactions (Dubois et al., 1998; Karnowski et al., 2012; Tanaka et al., 2014). A previous study has shown that loss of IL-6 function in a mouse model of rheumatoid arthritis could only be fully rescued by injection of hyper-IL-6 (Nowell et al., 2003). Because our inoculum contained bacterial lipid A adjuvant, a potent inducer of IL-6 release (Matsuura et al., 1999; Wang et al., 1991), we hypothesized that cDCs could modulate such IL-6-dependent effects through IL-6 production and/or by manipulating access to IL-6R. Our transgenic mice suggested that the latter was true, where cDC-IL6ra−/− recapitulated the IgM response deficit. This was not seen in cDC-Irf5−/−, suggesting that cDC production of IL-6, which is not needed for systemic IL-6 release (Heink et al., 2017), was also not required here. Notably, circulating sIL-6R was strongly reduced in cDC-IL6ra−/−, and injecting sIL-6R partially restored IgM and IgG output. This does not preclude a role of cDC-IL-6R (Heink et al., 2017) but strongly implicates cDC-derived sIL-6R in gating IL-6-dependent and T cell-independent control over B cell priming. B cells are limited in surface gp130 (Larousserie et al., 2006), but we found that gp130 was elevated in plasmablasts, rendering these cells receptive to IL-6 trans signaling. Moreover, addition of recombinant sIL-6R to partially restored defective plasma cell differentiation in cDC-IL6ra−/− mice. These findings unveil a cDC-governed pathway for naturally adjuvating antibody responses through IL-6 trans signaling.
Our results also demonstrate that cDCs regulate the concentration of sIL-6R in blood. Zbtb46 is also expressed by endothelial cells (Satpathy et al., 2012a), but these cells do not express IL-6R (Romano et al., 1997), and our Zbtb46-DTR chimeras confirmed cDC-dependent sIL-6R reduction when the endothelium was unaltered. Our orthogonal analyses of Batf3−/−, which are depleted in cDC1 (minority cDCs) but preserve cDC2 (majority cDCs) (Hildner et al., 2008), suggest an additive contribution to the sIL-6R in circulation.
Induction of sIL-6R release, as stimulated by diverse immune challenges, was also cDC dependent. This additional sIL-6R release underscores the proinflammatory state and is needed to bring sIL-6R to molar excess over sgp130 and enable IL-6 trans signaling (Rose-John, 2012, 2017; Scheller and Rose-John, 2012). We found that immune release of sIL-6R after inoculation with bacterial lipid A or after infl viral infection required ligation of the appropriate innate immune receptor followed by cDC-dependent sIL-6R production. The latter step relied on sheddase activity by ADAM17, consistent with studies on BMDCs in vitro (Garbers et al., 2011; Schumacher et al., 2015). Notably, the sIL-6R set point was not dependent on cDC-ADAM17, suggesting that set point and induction are regulated by distinct pathways, perhaps involving multiple sheddases and/or vesicular release mechanisms (Garbers et al., 2011; Schumacher et al., 2015, 2016). Our results do not preclude production of sIL-6R by non-cDC cell types, and cDC contribution to sIL-6R set point is likely included when assessed with lyz2-cre (McFarland-Mancini et al., 2010; Shi et al., 2018).
Although IL-6 release is an immunodefensive action, inflammatory disorders and cytokine storms are often marked by hyperelevated concentrations of this cytokine (Narazaki et al., 2017; Tanaka et al., 2016, 2018) and can signify terminal states, as in COVID-19 (McElvaney et al., 2020; Mehta et al., 2020). Defects in the human IL-6R gene are also linked to elevated IL-6 (Spencer et al., 2019). We experimentally demonstrated that cDC-derived sIL-6R regulates the concentration of free IL-6 in blood. IL-6 showed delayed clearance when cDC-derived sIL-6R was lacking, and restoration of sIL-6R rescued in-solution capture of IL-6 in our bacterial and virus challenge models. Restoration of sIL-6R also slowed morbidity after lethal influenza viral challenge, where respiratory distress is underscored by cytokine storming (Dutta et al., 2013; Vogel et al., 2014). Regulating the in-solution persistence of IL-6 will also be reliant on sgp130, where our results reinforce the view that sIL-6R is a buffering agent that guards against systemic IL-6 signaling (Rose-John, 2012, 2017; Scheller and Rose-John, 2012; Tanaka et al., 2016). IL-6 buffering may also have general therapeutic properties: a SNP in human IL-6R enhances ADAM17 sheddase activity in vitro (Müllberg et al., 1994), is matched with higher sIL-6R serum concentration in vivo, and is associated with reduced risk of congestive heart disease, atrial fibrillation, abdominal aortic aneurism, and rheumatoid arthritis (Sarwar et al., 2012; Ferreira et al., 2013; Interleukin-6 Receptor Mendelian Randomization Analysis (IL6R MR) Consortium et al., 2012; Scheller and Rose-John, 2012).
Limitations of Study
Our conclusions rely on Zbtb46 expression as a specific marker of cDCs in mice (Diao et al., 2018; Durai and Murphy, 2016; Loschko et al., 2016a, 2016b; Lu et al., 2017; Macdougall et al., 2018; Meredith et al., 2012; Rombouts et al., 2017; Satpathy et al., 2012a, 2012b; Sun et al., 2017; Zhang et al., 2019). There is no comparable marker for human cDCs, and the contribution of this immune cell lineage to serum sIL-6R in humans remains unclear. Nevertheless, our finding that sIL-6R serves to buffer the in-solution persistence of free IL-6 will be independent of the cellular source of sIL-6R.
STAR★METHODS
RESOURCE AVAILABILITY
Further information and requests for reagents should be directed to and will be fulfilled by Daniel Lingwood.
Lead contact
Daniel Lingwood (dlingwood@mgh.harvard.edu).
Materials availability
There are no restrictions to the availability of materials in this study
Data and code availability
N/A
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For all experiments, animals were maintained within Ragon Institute’s HPPF barrier facility and were conducted with institutional IACUC approval (MGH protocols 2014N000252 and #2014N000005).
Transgenic mice
The following mice were purchased from Jackson Laboratories: C57BL/6, Tcra−/−, Zbtb46-DTR+/+, MHCII deficient (homozygous H2dlAb1-Ea), Il6−/−, Il12p40−/−, Zbtb46-cre+/+, Il6raflox/flox, Irf5flox/flox, Adam17flox/flox, Batf3−/−, Tlr4−/−, Tlr7−/− and Ifnar1−/−. The cDC-Il6ra−/−, cDC-Irf5−/− and cDC-Adam17−/− genotypes were generated by crossing the Zbtb46-cre+/+ with Il6raflox/flox, Irf5 flox/flox, or Adam17flox/flox, respectively. The Irf5−/− animals were as described (Zhao et al., 2019). All genotypes were confirmed by PCR, according to the instructions provided by Jackson Laboratories. Animal experiments were performed on mice aged 6–12 weeks.
Bone marrow chimeras
Bone marrow chimeras were generated as described (Meredith et al., 2012). C57BL/6 mice were lethally irradiated at 950 cGrey and intravenously received 1 × 107 BM cells per mouse from one of two donors: either Zbtb46-DTR+/+ (generating chimeras with 100% zDCs) or a mixture of Zbtb46-DTR (50%) + MHCII deficient (homozygous H2dlAb1-Ea) (50%). The bone marrow was reconstituted for 8 weeks and our immune challenges were then conducted over the next two-week period during which time diphtheria toxin (DT) (Sigma D0564) was administered IP, first with an initial shot 20ng/g body weight and then 4ng/g every other day.
Splenectomized mice
Mouse splenectomies were as described (Rubtsov et al., 2005). Prior to the surgery animals were treated buprenorphine HCl (0.1 mg/kg sc) 30 minutes prior to the surgical procedure as pre-emptive analgesia. The left flank skin was then shaved and then swabbed with a sterile sponge moistened with 5% betadine and then 70% ethanol. Following this, the animals were anesthetized with ketamine (0.10 mg/g i.p.) and xylazine (0.012 mg/g i.p.). After confirming the depth of anesthesia using toe pinch, a 1.5–2cm incision was made within left hypochondrium using surgical scissors. Animals were closely observed for normal rhythmic breathing pattern throughout this time. The spleen was then exposed and gently pushed free of adjacent tissue using blunt-ended scissors and forceps. The splenic artery was then tied off at the superior polus by a single knot tie of 3–0 plane CATGUT. An additional knot was then made at the efferent venule, exiting the spleen at the inferior polus. The connective tissue was cut away and the spleen was removed by gently cutting on the splenic side of each knotted blood vessel. The surgical wound was then closed with sterile suturing of the peritoneum. The skin layer was then closed with four to five autoclips, using an autoclip wound-clip applicator. The animals were then with gentamicin IM (40mg/kg). For sham controls the same surgery was performed, except the spleen was not removed and the blood vessels were not ligated. After surgery, the mice were observed daily for two weeks to ensure recovery before beginning immunization experiments. Autoclips were removed 7 to 10 days post-surgery, using autoclip-removing forceps.
METHOD DETAILS
Protein antigens
Trimeric influenza hemagglutinin (HA) or and monomeric gp120 (resurfaced core 3) were purified as described (Weaver et al., 2016; Wu et al., 2010). 293F cells grown in Freestyle media (Life Technologies) were transfected with 500 μg/L of HA or probe plasmid (293fectin™ Reagent, Life Technologies). At day 5, the culture was centrifuged (2,000 × g, 10 min) and the supernatant was filtered (VacuCap 8/0.2 μm filters, Pall Corporation) and loaded on Ni Sepharose FF resin (GE Healthcare) by gravity flow. The resin was washed (6 column volumes of BS containing 20mM imidazole) and then HA or gp120 was eluted in 500mM imidazole. The proteins were then concentrated (Amicon Ultra concentrators, 30kDa, cut off), and were separated by size exclusion FPLC using a Superdex 200 10/300 column (GE Healthcare). Trimeric HA or monomeric gp120 was then collected, concentrated and stored at −80°C. To construct a fluorescent version of HA, trimers bearing Avi tag (a site specific biotinylation sequence) were biotinylated using the commercial BirA biotin ligase (Avidity) and then labeled fluorescently with streptavidin conjugated APC (Life Technologies) as described previously (Weaver et al., 2016). Ovalbumin was purchased commercially (Biosearch Technologies).
Immunization regimens
Mice were intravenously immunized with 15μg of either HA, gp120 or ovalbumin, each supplemented with Sigma adjuvant system (Sigma). Sigma adjuvant (formally known as Ribi (Yassine et al., 2015)) is an oil-in-water emulsion which was initially adjusted with ddH20 so that its immunostimulant, the TLR4 agonist monophosphoryl Lipid A and potent inducer of IL-6 release (Matsuura et al., 1999; Wang et al., 1991), is present in the inoculum at 12.5μg [~100x less than the LD50 for lipid A-induction of septic shock in B6 mice (Galanos et al., 1979)]. To capture the primary antibody response from the spleen, blood was sampled at days 3, 7 and 14 post-immunization [the primary IgM + IgG response against these proteins peaked at day 14, consistent with previous studies of blood-based responses against protein antigens in mice (Rubtsov et al., 2005; Sangesland et al., 2020; Song and Cerny, 2003)]. In some experiments, the HA immunization regimen also included intraperitoneal injection with recombinant murine IL-6 (rIL6) (R&D systems) or recombinant murine sIL6R (R&D systems). These IP injections occurred daily (100ng/mouse), starting one day before the IV immunization of HA antigen, and up to day 14. To analyze plasmablast differentiation into plasma cells during these responses, animals were also harvested for splenocytes at days 3 and 7 days post-immunization.
ELISA
To evaluate antibody responses in the serum, MaxSorp plates (Nunc) were coated overnight with 0.2 μg/well of either HA, gp120, or ovalbumin. The plates were blocked by 3% skimmed milk for (1h) at room temperature. After blocking, the plates were incubated for 1h with serial dilutions of pre- and post-immune sera. The samples were initially diluted at 1:20 in PBS followed by a 1:5 serial dilution in the same buffer. The wells were then washed 3x with PBST, incubated for 1h with a 1:5000 dilution of either: goat anti-murine IgM (IgG-HRP, Southern Biotech) or sheep anti-murine IgG (HRP-IgG, GE Healthcare). The plates were washed (3x PBST) and then developed using TMB substrate (Dako). The developer reaction was quenched by the addition of 1N sulphuric acid and the plates were then read at 450nm using a Spectomax Plus (Molecular Devices). Antibody endpoint dilutions were interpolated using Graphpad Prism version 5.0 (GraphPad Software Inc.), as previously described (Kanekiyo et al., 2013; Yassine et al., 2015). The concentrations of IL-6 and sIL6R in mouse sera were quantified according to the instructions provided in commercial sandwich ELISA kits from R&D Systems (Quantikine-M6000B) and Abcam (ab203360), respectively.
Imaging antigen uptake by immunofluorescence
To visualize antigen uptake from the spleen marginal zone into the follicles, zDCs chimeras (DT treated and untreated) mice were injected intravenously by 30 μg of HA-APC. The spleens were then removed at 1 and 4 h post-injection and then cut into ~5mm thick pieces. These pieces were submersed in O.C.T Compound (Tissue-Tek; Sakura) and then frozen by immersing the sample in isobutene that was housed in a metal container externally exposed to liquid nitrogen. The resultant frozen tissue block was stored at −80°C. For immunostaining, the tissue sections were first sectioned on a cryostat (40 μm thick) and air-dried overnight. The sections were then fixed in ice-cold acetone for 10 min, outlined with a wax pen, and rehydrated in PBS for 5 min. The sections were then blocked with Background sniper (Biocare) for 15 min (room temperature within a dark humid chamber) and then incubated for 1 h in the same chamber with anti-CD169-FITC (BioRad) to stain metalophilic macrophages and delineate the spleen follicles (the antibody was diluted 1:500 in PBS containing 0.1% Tween20). The sections were then washed with PBS in a coplin jar (3x, five min each) and then slide-mounted using ProLong Gold Antifade (Invitrogen). Images of the fluorescent APC and FITC were then acquired using a confocal fluorescent microscope (Zeiss LSM 510). To quantify antigen uptake from the spleen marginal zone, the mean fluorescence intensity of HA-APC present within the CD169-delinated spleen follicles was quantified using Zeiss “Zen” 2009 LSM imaging acquisition software in 10 replicates within each of n = 3 DT-treated or n = 3 DT-untreated zDCs chimeras.
Isolation of splenocytes and flow cytometry
Mouse spleens were first disrupted in R10 (RPMI with 10% FBS) containing 1mg/ml collagenase D (Roche collagenase D). After 30 min, the suspension was passed through a 70 μm cell strainer and then lysed with the ACK lysis buffer system to remove erythrocytes (Lonza). The splenocytes were then washed in PBS and then stained with viability dye (Aqua or Blue Viability at 0.025 mg/ml; Thermofisher) before our staining panels were applied. Staining panels were derived from the following fluorescently conjugated anti-murine antibodies, each used at a final dilution of 1:100 in PBS: CD11c-PE Cy7 (N418, Biolegend); B220-BV605 (RA3–6B2, BD Horizon); B220-FITC (RA3–6B2, BD PharMingen); MHCII-BV510 (M5/114.15.2, Biolegend); CD3-BV785 (17A2, Biolegend); CD19-PerCP/Cy5.5 (6D5, Biolegend); IL6R-APC (D7715A7, Biolegend); CD11b-APC (M1/70, Biolegend); B220-FITC (RA3–6B2, BD PharMingen); CD138-BV421 (281–2, BD Horizon); gp130-APC (KGP130, eBioscience); CD11b-FITC (M1/70, Biolegend); Siglec-H-APC (551, Biolegend); CD93 (AA4.1, Biolegend); CD8α-BV605 (53–6.7, Biolegend); DCIR2-PE (33D1, Biolegend); Ly6C-BV650 (HK1.4, Biolegend); F4/80-Alexa 488 (BM8, Thermofisher). Samples were assayed on a 5 laser LSR Fortessa (BD Biosciences) and data was analyzed using FlowJo software version 9.3.2 (TreeStar).
FACS and immunoblotting
IRF5 is a cytosolic protein, so we isolated cDCs by fluorescence-activated cell sorting (FACS) to confirm reduction in this cell lineage within the cDC-Irf5−/− genotype. Splenocytes from WT and cDC-Irf5−/− mice were processed as above and then stained with Blue viability dye (0.025mg/ml; Thermofisher) and then the following antibody flow panel (wherein each antibody was used at a final dilution of 1:100): CD3-BV785 (17A2, Biolegend); B220-FITC (RA3–6B2, BD PharMingen); CD11c-PE Cy7 (N418, Biolegend); and MHCII-BV510 (M5/114.15.2, Biolegend). We sorted 200.000 sorted cDCs (B220−/CD3−/CD11chi/MHCII+) from each animal genotype. For immunoblotting, the cDCs cells were lysed in 100μl of 1X RIPA buffer (ThermoFisher) and the lysate was then immersed in 1x laemmeli buffer (Sigma). After boiling the sample (5 min at 100°C) the samples were separated by SDS-PAGE. The proteins were then transferred overnight (4° C at 30V) onto nitrocellulose membrane. The membranes were blocked with 5% BSA and then probed for 1.5h at room temperature with a rabbit anti-murine IRF5 antibody (ab21689, Abcam) at a dilution of 1/1000 in TBS containing 1% BSA. The membranes were then washed (3x TBST) and incubated for 1h with secondary antibody (anti Rabbit-HRP) at a dilution of 1/2500 in TBS containing 1% BSA. Following four times washing with TBST the membrane was developed with ECL Plus reagent (Pierce). As a loading control, the samples were also blotted with mouse anti-β-Actin (8H10D10, Cell Signaling Technology) (1/10000 in 1% BSA) and anti-mouse HRP (1/2500).
B cell stimulation in vitro
Splenocytes from both immunized and unimmunized animals were harvested as described above. From each animal, four replicates of five million splenocyte cultures were grown for 20 minutes at 37°C in 5 mL of serum-free (SF) culture medium (RPMI-1640, phenol red free, 1% glutamine and 5% penicillin–streptomycin) or SF containing freshly 400ng/ml IL6-sIL6Rα (= Hyper-IL6; Fischer et al., 1997) (9038-SR-025, R&D Systems). After this culture period, the cells were pelleted, fixed and then permeabilized to enable intracellular staining using pY705 STAT3-PE (BioLegend) along with extracellular markers [B220-FITC, MHCII-BV510, CD138-BV421, CD3-BV786 and gp130-APC], according to the True-Phos™ Protocol provided by Biolegend (Xu et al., 2017). The samples were assayed on a 5 laser LSR Fortessa (BD Biosciences) and data was analyzed using FlowJo software version 9.3.2 (TreeStar) as described above.
Simulation of bone marrow-derived dendritic cells in vitro
Bone marrow-derived dendritic cells (BMDCs) cells were generated as performed previously (Park et al., 2019). In this method, bone marrow cells from femurs and tibia were first flushed with PBS and the red blood cells were then depleted using ACK lysis buffer. The remaining marrow cells were plated in 10 cm non-treated plate (CytoOne) in RPMI-1640 media supplemented with 10% FBS, 1% penicillin/streptomycin, murine GM-CSF (10ng/ml; eBioscience) and murine IL-4 (10ng/ml; MBL). These cells were cultured at 37°C, 5%CO2 for six days, where fresh media was added every 2 days. On day six, the floating and loosely attached cells [representing the BMDCs (Park et al., 2019)] were collected. BMDC were then divided into three groups for immune stimulation (106 cells/well within 48-well flat-bottomed plates containing RPMI-1640 media): 1) PBS control; 2) stimulation with 1 μg/ml of recombinant murine IL-6 (rIL6) (R&D systems); or stimulation with 1 μg/ml of bacterial LPS (Sigma). The stimulation was for 6h in supernatants were then analyzed by ELISA to measure sIL6R as described earlier.
AAV sIL6R production
AAV virus encoding amino acid residues 1–357 of murine sIL6R was cloned into an AAV transfer vector. This vector was previously described (Balazs et al., 2011) but modified to contain the CAG promoter and rabbit beta globin polyadenylation signal. Briefly, recombinant AAV was produced via transient transfection of 293T cells with a mixture of plasmid DNAs consisting of the transfer vector, pAAV2/8-SEED and pHELP (Applied Viromics) and purified via PEG precipitation of collected supernatants, CsCl2 ultracentrifugation and fractionation followed by buffer exchange using a 100kDa MWCO centrifugal filter (Millipore) (Balazs et al., 2011). The virus was titered as using primers specific for the CMV enhancer region to obtain a final titer via SYBR Green-based qPCR against a diluted plasmid standard. Aliquots of tittered vector were stored at −80°C and were thawed slowly on ice and diluted in PBS to achieve the predetermined dose (1.00E+10 genome copies / 20 g mouse) in a 200μl volume.
Bacterial and viral immune challenges
To explore the pharmacokinetics of IL-6 post-immune stimulation in our transgenic animals, Sigma Adjuvant containing 12.5μg of bacterial lipid A was injected IV, as described earlier, and blood was sampled both before and then 3, 7, and 14 days post challenge. In some experiments, this was preceded by AAV-gene delivery of murine sIL6R in cDC-Il6ra−/− mice, using the AAV construct described above and which was administered IV at 1.00E+10 genome copies / 20 g mouse. Three weeks after delivering the AAV construct, the lipid A IV challenge regimen was then performed. Serum concentration of sIL6R and IL-6 were measured by ELISA at each sampling period.
To explore the pharmacokinetics of IL-6 following viral challenge in our transgenic animals, we deployed two murine models of influenza virus infection. In the first model of infection, mice anesthetized by isoflurane inhalation and then intranasally infected using a sublethal dose of New Caledonia/20/1999 (NC99) influenza virus (105.0 TCID50 units/ml). NC99 infects and propagates within mice but does not cause disease (Amitai et al., 2020; Glaser et al., 2007; Sangesland et al., 2019). Blood was sampled both before and then 3, 7, and 14 days post-viral challenge. In some experiments, this was preceded by AAV-gene delivery of murine sIL6R in cDC-Il6ra−/− mice, as described above and serum sIL6R and IL-6 concentrations were measured by ELISA at each sampling period. In the second model of infection, cDC-Il6ra−/− mice in which sIL6R concentration was and was not restored by AAV mediated gene delivery, were anesthetized by isoflurane inhalation and then intranasally infected with a 5xLD50 dose of A/Puerto Rico/8/34 (PR8) influenza virus. Subsequent morbidity (weight loss) was measured daily and the animals were humanly euthanized when they reached < 80% body weight.
QUANTIFICATION AND STATISTICAL ANALYSIS
Multiple comparisons were evaluated by ANOVA with Tukey’s test and single comparisons were assessed by two-sided Students t tests. Morbidity rates were evaluated using F-tests to compare slopes. An alpha value of 0.05 was deployed throughout. All statistical analyses were performed using Graphpad Prism version 5.0 (GraphPad Software Inc.).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-murine CD19-PerCP-Cy5.5 | BioLegend | Cat#115533; RRID: AB_2259869 |
| Rat anti-murine -B220-FITC | BD Pharmingen | Cat#553087; RRID: AB_394617 |
| Rat anti-murine -MHCII-BV510 | BioLegend | Cat#107636; RRID: AB_2734168 |
| Rat anti-murine -CD3-BV785 | BioLegend | Cat#100232; RRID: AB_2562554 |
| Hamster anti-murine -CD11c-PE/Cy7 | BioLegend | Cat#117318; RRID:AB_493568 |
| Rat anti-murine-CD169-FITC | BioRad | Cat#MCA884FT; RRID:AB_1100895 |
| Rat anti-murine CD138-BV421 | BD Horizon | Cat#562610 |
| Rat anti-murine B220-BV605 | BD Horizon | Cat#563708 |
| Rat anti-murine IL6R-APC | BioLegend | Cat#115812; RRID: AB_2296238 |
| Rat anti-murine CD11b-APC | BioLegend | Cat#101212; RRID: AB_312795 |
| Rat anti-murine gp130-APC | eBiosciences | Cat#17–1302-82; RRID: AB_10670874 |
| Rat anti-murine CD11b-FITC | BioLegend | Cat#101206; RRID: AB_312789 |
| Rat anti-murine Siglec H-APC | BioLegend | Cat#129612; RRID: AB_10641134 |
| Rat anti-murine CD93-PE | BioLegend | Cat#136503; RRID: AB_1967094 |
| Rat anti-murine CD8α-BV605 | BioLegend | Cat#100744; RRID: AB_2562609 |
| Rat anti-murine DCIR2-PE | BioLegend | Cat#124905; RRID: AB_1186128 |
| Rat anti-murine Ly6C-BV650 | BioLegend | Cat#128035; RRID: AB_2562352 |
| Rat anti-murine F4/80-Alexa 488 | Thermo Fisher | Cat#53–4801-82; RRID: AB_469915 |
| Rat anti-murine pY705 STAT3-PE | BioLegend | Cat#651004; RRID: AB_2571892 |
| Goat anti-murine IgM-HRP | Southern Biotech | Cat#1021–05; RRID:AB_2794240 |
| Sheep anti-murine IgG-HRP | GE Healthcare | Cat# NA931–1ML |
| Bacterial and Virus Strains | ||
| New Caledonia/20/1999 (NC99) influenza virus | International Reagent Resource | Cat# FR-395 |
| A/Puerto Rico/8/34 (PR8) influenza virus | Balazs et al. 2013 | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| LIVE/DEAD Fixable Blue Dead Cell Stain | Thermo Fisher | Cat#L34961 |
| Sigma Adjuvant System | Sigma-Aldrich | Cat#S6322 |
| Collagenase D (Roche collagenase D) | Roche | Cat#11088858001 |
| ACK lysis buffer | Lonza | Cat#10–548E |
| Tissue-Tek O.C.T Compound | Sakura | Cat#4583 |
| Background Sniper Blocking Reagent | Biocare Medical | Cat#902–966-082317 |
| HA Trimer (NC99) | Produced in house (Weaver et al., 2016) |
N/A |
| gp120 monomer | Produced in house (Wu et al., 2010) |
N/A |
| ovalbumin | Biosearch Technologies | Cat#O-1000–100 |
| 293fectin Reagent | Invitrogen | Cat#12347019 |
| Ni-Sepharose excel Affinity Medium | GE Healthcare | Cat#GE17–3712–02 |
| LPS (Escherichia coli O111:B4) | Sigma | Cat#L2630 |
| Tetramethylbenzidine (TMB) | Sigma | Cat#T0440 |
| Murine GM-CSF | eBiosciences | Cat#14–8331-80 |
| Murine IL-4 | MBL | Cat#JM-4138–10 |
| Murine recombinant IL-6 | R&D systems | Cat#406-ML-005 |
| Murine recombinant IL-6-IL6Rα | R&D systems | Cat#9038-SR-025 |
| Murine recombinant IL6R | R&D systems | Cat#1830-SR-025 |
| Diphtheria toxin (DT) | Sigma | Cat#D0564 |
| Streptavidin conjugated APC | ThermoFisher | Cat# S32362 |
| Critical Commercial Assays | ||
| Mouse IL-6 ELISA Kit | R&D systems | Cat#M6000B |
| Mouse IL6Rα ELISA Kit | Abcam | Cat#ab203360 |
| BirA Biotin-Protein Ligase Bulk Reaction Kit | Avidity | Cat#Bulk BirA |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson laboratory | Stock no. 000664 |
| Mouse: Tcra−/− | Jackson laboratory | Stock no. 002116 |
| Mouse: Zbtb46-DTR | Jackson laboratory | Stock no. 019506 |
| Mouse: MHCII deficient (H2dlAb1-Ea) | Jackson laboratory | Stock no. 003584 |
| Mouse: Irf5−/− | Zhao et al. 2019 | N/A |
| Mouse: Il6−/− | Jackson laboratory | Stock no.002650 |
| Mouse: Il12p40−/− | Jackson laboratory | Stock no.002693 |
| Mouse: Zbtb46 cre | Jackson laboratory | Stock no.028538 |
| Mouse: IL6raflox/flox | Jackson laboratory | Stock no.012944 |
| Mouse: Adam17flox/flox | Jackson laboratory | Stock no.009597 |
| Mouse: Irf5flox/flox | Jackson laboratory | Stock no.017311 |
| Mouse: Batf3−/− | Jackson laboratory | Stock no.013755 |
| Mouse: Tlr4−/− | Jackson laboratory | Stock no.029015 |
| Mouse: Tlr7−/− | Jackson laboratory | Stock no.008380 |
| Mouse: Ifnar1−/− | Jackson laboratory | Stock no.32045-JAX |
| Mouse: cDC-Il6r−/− | This paper | |
| Mouse: cDC-Irf5−/− | This paper | |
| Mouse: cDC-Adam17−/− | This paper | |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| FlowJo v.9.3.2 | TreeStar | https://www.flowjo.com; RRID: SCR_008520 |
| Prism 7 | GraphPad | https://www.graphpad.com; RRID: SCR_002798 |
| Other | ||
| 96 well microtiter plates | Fisher Scientific | Cat#439454 |
| 48 well non-treated plates | CytoOne | Cat#CC7672–7548 |
| 10 cm non-treated plates | CytoOne | Cat#CC7672–3614 |
| Superdex 200 10/300 Column | GE Healthcare | Cat#17517501 |
Highlights.
DC-derived sIL-6R gates plasma cell differentiation to adjuvant antibody responses
DC-derived sIL-6R captures IL-6 released after bacterial or viral challenge
TLR ligation, then cell surface sheddase activity, mediates sIL-6R release from DCs
ACKNOWLEDGMENTS
D.L. was supported by NIH grants (R01AI137057, DP2DA042422, R01AI124378, and R01AI153098), the Harvard University Milton Award, and the Gilead Research Scholars Program. H.C.R. was supported by the NIH (DK068181, AI113333, DK043351, and GM138599). We thank Lingwood lab technicians past and present for generating the recombinant protein antigens used in this study (Grant Weaver, Rina Villar, Celina Keating, Matthew Smoot, Julia Bals, and Vintus Okonkwo). The authors also thank Masaru Kanekiyo, Jose Ordovas-Montanes, Shiv Pillai, Facundo Batista, Ulrich von Andrian, and Lingwood lab members for helpful discussions. We gratefully acknowledge Michal Grzybek for assistance with figure graphics and Thomas Diefenbach for help with microscopy and imaging analyses.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2020.12.001.
REFERENCES
- Adler LN, Jiang W, Bhamidipati K, Millican M, Macaubas C, Hung SC, and Mellins ED (2017). The Other Function: Class II-Restricted Antigen Presentation by B Cells. Front. Immunol. 8, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman MO, Angeletti D, and Yewdell JW (2018). Antibody Immunodominance: The Key to Understanding Influenza Virus Antigenic Drift. Viral Immunol. 31, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez D, Vollmann EH, and von Andrian UH (2008). Mechanisms and consequences of dendritic cell migration. Immunity 29, 325–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai A, Sangesland M, Barnes RM, Rohrer D, Lonberg N, Lingwood D, and Chakraborty AK (2020). Defining and Manipulating B Cell Immunodominance Hierarchies to Elicit Broadly Neutralizing Antibody Responses against Influenza Virus. Cell Syst. Published online May 18, 2020 10.2139/ssrn.3586996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen TK, Huszthy PC, Gopalakrishnan RP, Jacobsen JT, Fauskanger M, Tveita AA, Grødeland G, and Bogen B (2019). Enhanced germinal center reaction by targeting vaccine antigen to major histocompatibility complex class II molecules. NPJ Vaccines 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti D, Gibbs JS, Angel M, Kosik I, Hickman HD, Frank GM, Das SR, Wheatley AK, Prabhakaran M, Leggat DJ, et al. (2017). Defining B cell immunodominance to viruses. Nat. Immunol. 18, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs AB, Bloom JD, Hong CM, Rao DS, and Baltimore D (2013). Broad protection against influenza infection by vectored immunoprophylaxis in mice. Nat. Biotechnol. 31, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs AB, Chen J, Hong CM, Rao DS, Yang L, and Baltimore D (2011). Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 481, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balázs M, Martin F, Zhou T, and Kearney J (2002). Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune re sponses. Immunity 17, 341–352. [DOI] [PubMed] [Google Scholar]
- Baran P, Hansen S, Waetzig GH, Akbarzadeh M, Lamertz L, Huber HJ, Ahmadian MR, Moll JM, and Scheller J (2018). The balance of interleukin (IL)-6, IL-6$soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 293, 6762–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergtold A, Desai DD, Gavhane A, and Clynes R (2005). Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 23, 503–514. [DOI] [PubMed] [Google Scholar]
- Biran N, Ip A, Ahn J, Go RC, Wang S, Mathura S, Sinclaire BA, Bednarz U, Marafelias M, Hansen E, et al. (2020). Tocilizumab among patients with COVID-19 in the intensive care unit: a multicentre observational study. Lancet Rheumatol. 2, e603–e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, and Scheller J (2007). Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 110, 1748–1755. [DOI] [PubMed] [Google Scholar]
- Chappell CP, Draves KE, Giltiay NV, and Clark EA (2012). Extrafollicular B cell activation by marginal zone dendritic cells drives T cell-dependent antibody responses. J. Exp. Med. 209, 1825–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Gu H, Tang M, Zhao J, and Cattral MS (2018). Tumor Dendritic Cells (DCs) Derived from Precursors of Conventional DCs Are Dispensable for Intratumor CTL Responses. J. Immunol. 201, 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad B, Lehr HA, Schmitt E, Bopp T, et al. (2005). The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J. Clin. Invest. 115, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Massacrier C, Vanbervliet B, Fayette J, Brière F, Banchereau J, and Caux C (1998). Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J. Immunol. 161, 2223–2231. [PubMed] [Google Scholar]
- Durai V, and Murphy KM (2016). Functions of Murine Dendritic Cells. Immunity 45, 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Miaw SC, Yu JS, Chen TC, Lin CY, Lin YC, Chang CS, He YC, Chuang SH, Yen MI, and Huang CT (2013). Altered T-bet dominance in IFN-γ-decoupled CD4+ T cells with attenuated cytokine storm and preserved memory in influenza. J. Immunol. 190, 4205–4214. [DOI] [PubMed] [Google Scholar]
- Fazel Modares N, Polz R, Haghighi F, Lamertz L, Behnke K, Zhuang Y, Kordes C, Häussinger D, Sorg UR, Pfeffer K, et al. (2019). IL-6 Transsignaling Controls Liver Regeneration After Partial Hepatectomy. Hepatology 70, 2075–2091. [DOI] [PubMed] [Google Scholar]
- Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, Kaptoge S, Clarke P, Boreham C, Coulson RM, et al. (2013). Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 9, e1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A, Grötzinger J, and Rose-John S (1997). I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 15, 142–145. [DOI] [PubMed] [Google Scholar]
- Galanos C, Freudenberg MA, and Reutter W (1979). Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc. Natl. Acad. Sci. USA 76, 5939–5943.293694 [Google Scholar]
- Garbers C, Jänner N, Chalaris A, Moss ML, Floss DM, Meyer D, Koch-Nolte F, Rose-John S, and Scheller J (2011). Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 286, 14804–14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbers C, Monhasery N, Aparicio-Siegmund S, Lokau J, Baran P, Nowell MA, Jones SA, Rose-John S, and Scheller J (2014). The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim. Biophys. Acta 1842, 1485–1494. [DOI] [PubMed] [Google Scholar]
- Glaser L, Conenello G, Paulson J, and Palese P (2007). Effective replicaion of human influenza viruses in mice lacking a major alpha2,6 sialyltransferase. Virus Res. 126, 9–18. [DOI] [PubMed] [Google Scholar]
- Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim YA, Cloninger MJ, Martinez-Pomares L, Gordon S, Turley SJ, and Carroll MC (2010). Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 11, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, Husterer V, Croxford AL, Möller-Hackbarth K, Bartsch HS, et al. (2017). Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 18, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrickson SE, Perro M, Loughhead SM, Senman B, Stutte S, Quigley M, Alexe G, Iannacone M, Flynn MP, Omid S, et al. (2013). Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 39, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, Guo Y, Chung C, Peasey A, et al. (2012). The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379, 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, and Rose-John S (2001). Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167. [DOI] [PubMed] [Google Scholar]
- Kanekiyo M, Wei CJ, Yassine HM, McTamney PM, Boyington JC, Whittle JR, Rao SS, Kong WP, Wang L, and Nabel GJ (2013). Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 499, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D’Costa K, Tarlinton DM, Kallies A, and Corcoran LM (2012). B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J. Exp. Med. 209, 2049–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Caton M, Wang C, Khalil M, Zhou ZJ, Hardin J, and Diamond B (2008). Increased IL-12 inhibits B cells’ differentiation to germinal center cells and promotes differentiation to short-lived plasmablasts. J. Exp. Med. 205, 2437–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T (2010). IL-6: from its discovery to clinical applications. Int. Immunol. 22, 347–352. [DOI] [PubMed] [Google Scholar]
- Larousserie F, Charlot P, Bardel E, Froger J, Kastelein RA, and Devergne O (2006). Differential effects of IL-27 on human B cell subsets. J. Immunol. 176, 5890–5897. [DOI] [PubMed] [Google Scholar]
- Loschko J, Rieke GJ, Schreiber HA, Meredith MM, Yao KH, Guermonprez P, and Nussenzweig MC (2016a). Inducible targeting of cDCs and their subsets in vivo. J. Immunol. Methods 434, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschko J, Schreiber HA, Rieke GJ, Esterházy D, Meredith MM, Pedicord VA, Yao KH, Caballero S, Pamer EG, Mucida D, and Nussenzweig MC (2016b). Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J. Exp. Med. 213, 517–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E, Dang EV, McDonald JG, and Cyster JG (2017). Distinct oxysterol requirements for positioning naïve and activated dendritic cells in the spleen. Sci. Immunol. 2, eaal5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, and Flavell RA (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 101, 5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall CE, Wood EG, Loschko J, Scagliotti V, Cassidy FC, Robinson ME, Feldhahn N, Castellano L, Voisin MB, Marelli-Berg F, et al. (2018). Visceral Adipose Tissue Immune Homeostasis Is Regulated by the Crosstalk between Adipocytes and Dendritic Cell Subsets. Cell Metab. 27, 588–601.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan I, and Vinuesa C (2002). Dendritic cells, BAFF, and APRIL: innate players in adaptive antibody responses. Immunity 17, 235–238. [DOI] [PubMed] [Google Scholar]
- Matsuura M, Kiso M, and Hasegawa A (1999). Activity of monosaccharide lipid A analogues in human monocytic cells as agonists or antagonists of bacterial lipopolysaccharide. Infect. Immun. 67, 6286–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews V, Schuster B, Schütze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, and Rose-John S (2003). Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 278, 38829–38839. [DOI] [PubMed] [Google Scholar]
- McElvaney OJ, Hobbs BD, Qiao D, McElvaney OF, Moll M, McEvoy NL, Clarke J, O’Connor E, Walsh S, Cho MH, et al. (2020). A linear prognostic score based on the ratio of interleukin-6 to interleukin-10 predicts outcomes in COVID-19. EBioMedicine 61, 103026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland-Mancini MM, Funk HM, Paluch AM, Zhou M, Giridhar PV, Mercer CA, Kozma SC, and Drew AF (2010). Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J. Immunol. 184, 7219–7228. [DOI] [PubMed] [Google Scholar]
- Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, and Manson JJ; HLH Across Speciality Collaboration, UK (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, and Nussenzweig MC (2012). Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 209, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müllberg J, Durie FH, Otten-Evans C, Alderson MR, Rose-John S, Cosman D, Black RA, and Mohler KM (1995). A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J. Immunol. 155, 5198–5205. [PubMed] [Google Scholar]
- Müllberg J, Oberthür W, Lottspeich F, Mehl E, Dittrich E, Graeve L, Heinrich PC, and Rose-John S (1994). The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J. Immunol. 152, 4958–4968. [PubMed] [Google Scholar]
- Narazaki M, Tanaka T, and Kishimoto T (2017). The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev. Clin. Immunol. 13, 535–551. [DOI] [PubMed] [Google Scholar]
- Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N, Williams AS, and Jones SA (2003). Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J. Immunol. 171, 3202–3209. [DOI] [PubMed] [Google Scholar]
- O E, Lee YT, Ko EJ, Kim KH, Lee YN, Song JM, Kwon YM, Kim MC, Perez DR, and Kang SM (2014). Roles of major histocompatibility complex class II in inducing protective immune responses to influenza vaccination. J. Virol. 88, 7764–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Omatsu T, Zhao Y, Yoshida N, Shah P, Zagani R, and Reinecker HC (2019). T cell fate following Salmonella infection is determined by a STING-IRF1 signaling axis in mice. Commun. Biol. 2, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Egen JG, Huang AY, and Germain RN (2006). Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science 312, 1672–1676. [DOI] [PubMed] [Google Scholar]
- Riethmueller S, Ehlers JC, Lokau J, Düsterhöft S, Knittler K, Dombrowsky G, Grötzinger J, Rabe B, Rose-John S, and Garbers C (2016). Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteolysis by ADAM10 and ADAM17. Sci. Rep. 6, 25550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, et al. (1997). Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 6, 315–325. [DOI] [PubMed] [Google Scholar]
- Rombouts M, Cools N, Grootaert MO, de Bakker F, Van Brussel I, Wouters A, De Meyer GR, De Winter BY, and Schrijvers DM (2017). Long-Term Depletion of Conventional Dendritic Cells Cannot Be Maintained in an Atherosclerotic Zbtb46-DTR Mouse Model. PLoS ONE 12, e0169608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-John S (2012). IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-John S (2017). The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 102, 591–598. [DOI] [PubMed] [Google Scholar]
- Rubtsov A, Strauch P, Digiacomo A, Hu J, Pelanda R, and Torres RM (2005). Lsc regulates marginal-zone B cell migration and adhesion and is required for the IgM T-dependent antibody response. Immunity 23, 527–538. [DOI] [PubMed] [Google Scholar]
- Sangesland M, Ronsard L, Kazer SW, Bals J, Boyoglu-Barnum S, Yousif AS, Barnes R, Feldman J, Quirindongo-Crespo M, McTamney PM, et al. (2019). Germline-Encoded Affinity for Cognate Antigen Enables Vaccine Amplification of a Human Broadly Neutralizing Response against Influenza Virus. Immunity 51, 735–749.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangesland M, Yousif AS, Ronsard L, Kazer SW, Zhu AL, Gatter GJ, Hayward MR, Barnes RM, Quirindongo-Crespo M, Rohrer D, et al. (2020). A Single Human VH-gene Allows for a Broad-Spectrum Antibody Response Targeting Bacterial Lipopolysaccharides in the Blood. Cell Rep. 32, 108065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, Nelson CP, et al. ; IL6R Genetics Consortium Emerging Risk Factors Collaboration (2012). Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 379, 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM (2012a). Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 209, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Wu X, Albring JC, and Murphy KM (2012b). Re(de)fining the dendritic cell lineage. Nat. Immunol. 13, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller J, Ohnesorge N, and Rose-John S (2006). Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand. J. Immunol. 63, 321–329. [DOI] [PubMed] [Google Scholar]
- Scheller J, and Rose-John S (2012). The interleukin 6 pathway and atherosclerosis. Lancet 380, 338. [DOI] [PubMed] [Google Scholar]
- Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A, Bavendiek U, von Felden J, Divchev D, Kempf T, et al. (2012). Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 32, 281–290. [DOI] [PubMed] [Google Scholar]
- Schumacher N, Meyer D, Mauermann A, von der Heyde J, Wolf J, Schwarz J, Knittler K, Murphy G, Michalek M, Garbers C, et al. (2015). Shedding of Endogenous Interleukin-6 Receptor (IL-6R) Is Governed by A Disintegrin and Metalloproteinase (ADAM) Proteases while a Full-length IL-6R Isoform Localizes to Circulating Microvesicles. J. Biol. Chem. 290, 26059–26071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher N, Schmidt S, Schwarz J, Dohr D, Lokau J, Scheller J, Garbers C, Chalaris A, Rose-John S, and Rabe B (2016). Circulating Soluble IL-6R but Not ADAM17 Activation Drives Mononuclear Cell Migration in Tissue Inflammation. J. Immunol. 197, 3705–3715. [DOI] [PubMed] [Google Scholar]
- Shi J, Hua L, Harmer D, Li P, and Ren G (2018). Cre Driver Mice Targeting Macrophages. Methods Mol. Biol. 1784, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers EC, Eschenauer GA, Troost JP, Golob JL, Gandhi TN, Wang L, Zhou N, Petty LA, Baang JH, Dillman NO, et al. (2020). Tocilizumab for treatment of mechanically ventilated patients with COVID-19. Clin. Infect. Dis. Published online July 11, 2020. 10.1093/cid/ciaa954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, and Cerny J (2003). Functional heterogeneity of marginal zone B cells revealed by their ability to generate both early antibody-forming cells and germinal centers with hypermutation and memory in response to a T-dependent antigen. J. Exp. Med. 198, 1923–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer S, Köstel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, Gürel M, Zhang Y, Sun G, Sabroe RA, et al. (2019). Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J. Exp. Med. 216, 1986–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebegg M, Bignon A, Hill DL, Silva-Cayetano A, Krueger C, Vanderleyden I, Innocentin S, Boon L, Wang J, Zand MS, et al. (2020). Rejuvenating conventional dendritic cells and T follicular helper cell formation after vaccination. eLife 9, e52473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Rojas OL, Li C, Ward LA, Philpott DJ, and Gommerman JL (2017). Intestinal Batf3-dependent dendritic cells are required for optimal antiviral T-cell responses in adult and neonatal mice. Mucosal Immunol. 10, 775–788. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, and Taniguchi T (2005). Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, and Kishimoto T (2014). IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 6, a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, and Kishimoto T (2016). Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 8, 959–970. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, and Kishimoto T (2018). Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 10, a028456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenhumberg S, Waetzig GH, Chalaris A, Rabe B, Seegert D, Scheller J, Rose-John S, and Grötzinger J (2008). Structure-guided optimization of the interleukin-6 trans-signaling antagonist sgp130. J. Biol. Chem. 283, 27200–27207. [DOI] [PubMed] [Google Scholar]
- Tesfaye DY, Gudjonsson A, Bogen B, and Fossum E (2019). Targeting Conventional Dendritic Cells to Fine-Tune Antibody Responses. Front. Immunol. 10, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel AJ, Harris S, Marsteller N, Condon SA, and Brown DM (2014). Early cytokine dysregulation and viral replication are associated with mortality during lethal influenza infection. Viral Immunol. 27, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer P, Walev I, Rose-John S, and Bhakdi S (1996). Novel pathogenic mechanism of microbial metalloproteinases: liberation of membrane-anchored molecules in biologically active form exemplified by studies with the human interleukin-6 receptor. Infect. Immun. 64, 3646–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MH, Flad HD, Feist W, Brade H, Kusumoto S, Rietschel ET, and Ulmer AJ (1991). Inhibition of endotoxin-induced interleukin-6 production by synthetic lipid A partial structures in human peripheral blood mononuclear cells. Infect. Immun. 59, 4655–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver GC, Villar RF, Kanekiyo M, Nabel GJ, Mascola JR, and Lingwood D (2016). In vitro reconstitution of B cell receptor-antigen interactions to evaluate potential vaccine candidates. Nat. Protoc. 11, 193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisend CM, Kundert JA, Suvorova ES, Prigge JR, and Schmidt EE (2009). Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis 47, 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J, Rose-John S, and Garbers C (2014). Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine 70, 11–20. [DOI] [PubMed] [Google Scholar]
- Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al. (2010). Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu E, Pereira MMA, Karakasilioti I, Theurich S, Al-Maarri M, Rappl G, Waisman A, Wunderlich FT, and Brüning JC (2017). Temporal and tissue-specific requirements for T-lymphocyte IL-6 signalling in obesity-associated inflammation and insulin resistance. Nat. Commun. 8, 14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Han M, Li T, Sun W, Wang D, Fu B, Zhou Y, Zheng X, Yang Y, Li X, et al. (2020). Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 117, 10970–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, Gallagher JR, Wang L, Zhang Y, Joyce MG, et al. (2015). Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 21, 1065–1070. [DOI] [PubMed] [Google Scholar]
- Zhang J, Supakorndej T, Krambs JR, Rao M, Abou-Ezzi G, Ye RY, Li S, Trinkaus K, and Link DC (2019). Bone marrow dendritic cells regulate hematopoietic stem/progenitor cell trafficking. J. Clin. Invest. 129, 2920–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zagani R, Park SM, Yoshida N, Shah P, and Reinecker HC (2019). Microbial recognition by GEF-H1 controls IKKε mediated activation of IRF5. Nat. Commun. 10, 1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
N/A