

Abstract
Objective:
Receptor interacting proteins kinase 1 and 3 (RIPK1 and RIPK3) have been shown to play essential roles in the pathogenesis of abdominal aortic aneurysms (AAAs) by mediating necroptosis and inflammation. We previously discovered a small molecular inhibitor GSK2593074A (GSK’074) that binds to both RIPK1 and RIPK3 with high affinity and prevents AAA formation in mice. In this study, we evaluated whether GSK’074 can attenuate progression of existing AAA in the calcium phosphate model.
Methods:
C57BL6/J mice were subjected to the calcium phosphate model of aortic aneurysm generation. Mice were treated with either GSK’074 (4.65 mg/kg/day) or dimethylsulfoxide (DMSO) controls starting 7 days after aneurysm induction. Aneurysm growth was monitored via ultrasound imaging every 7 days until harvest on day 28. Harvested aortas were examined via immunohistochemistry. The impact of GSK’074 on vascular smooth muscle cells and macrophages were evaluated via flow cytometry and transwell migration assay.
Results:
At the onset of treatment, mice in both the control (DMSO) and GSK’074 groups showed similar degree of aneurysmal expansion. The weekly ultrasound imaging showed a steady aneurysm growth in DMSO-treated mice. The aneurysm growth was attenuated by GSK’074 treatment. At humane killing, GSK’074-treated mice had significantly reduced progression in aortic diameter from baseline as compared with the DMSO-treated mice (83.2% ± 13.1% [standard error of the mean] vs 157.2% ± 32.0% [standard error of the mean]; P < .01). In addition, the GSK’074-treated group demonstrated reduced macrophages (F4/80, CD206, MHCII), less gelatinase activity, a higher level of smooth muscle cell-specific myosin heavy chain, and better organized elastin fibers within the aortic walls compared with DMSO controls. In vitro, GSK’074 inhibited necroptosis in mouse aortic smooth muscle cells; whereas, it was able to prevent macrophage migration without affecting Il1b and Tnf expression.
Conclusions:
GSK’074 is able to attenuate aneurysm progression in the calcium phosphate model. The ability to inhibit both vascular smooth muscle cell necroptosis and macrophage migration makes GSK’074 an attractive drug candidate for pharmaceutical treatment of aortic aneurysms.
Clinical Relevance:
Previous clinical trials evaluating pharmaceutical treatments in blocking aneurysm progression have failed. However, most agents used in those trials focused on inhibiting only one mechanism that contributes to aneurysm pathogenesis. In this study, we found GSK’074 is able to attenuate aneurysm progression in the calcium phosphate model by inhibiting both vascular smooth muscle cell necroptosis and macrophage migration, which are both key processes in the pathogenesis of aneurysm progression. The ability of GSK’0474 to inhibit multiple key pathologic mechanisms makes it an attractive therapeutic candidate for aneurysm progression.
Keywords: Abdominal aortic aneurysm, Macrophages, Vascular smooth muscle cells, Necroptosis, Vascular inflammation
Abdominal aortic aneurysms (AAA) are a common aortic disorder and 10th leading cause of death among men over 65 years of age.1 Current guidelines recommend a one-time screening via ultrasound examination for men aged 65 to 75 years who have ever smoked, selective screening for men aged 65 to 75 who have never smoked, or women aged 65 to 75 who have ever smoked or have a family history of AAA.2 The feared complication of AAAs is rupture, which has an associated mortality rate of more than 80%.3 As such, an AAA that is greater than 5.5 cm in diameter is recommended for elective repair via either open or endovascular surgical repair. Pharmaceutical strategies for patients with AAAs are limited to treating associated risk factors, such as hypertension and hyperlipidemia. Although the majority of newly diagnosed AAA cases are small, the natural history of aneurysms is progressive growth. Currently, no medical therapies have been clinically proven to halt or attenuate degeneration of the aortic wall in hopes of preventing the need for surgical repair.4
The depletion of vascular smooth muscle cell (VSMC) death is a major pathologic characteristic of AAA.3,5,6 Death of VSMCs contributes to aneurysm pathophysiology by inducing vascular inflammation. Necroptosis, or programmed necrosis, has been recognized as a major cell death mechanism alternative to apoptosis.7 Being a form of necrosis, necroptosis evokes immune responses by releasing danger signals. Receptor interacting protein kinase 3 (RIPK3) is a key mediator of the necroptotic pathway that, through its interaction with RIPK1, phosphorylates mixed lineage kinase domain-like pseudokinase (MLKL), which subsequently leads to disruption of the cell membrane.8 Previously, our group demonstrated that levels of RIPK1 and RIPK3 are increased in human AAA tissues. In addition, gene deletion of Ripk3 prevents AAA formation by inhibiting VSMC necroptosis and vascular wall inflammation.9 Further, we showed that inhibition of RIPK1 in mice with small aneurysms attenuated disease progression,10 which proved, in concept, that targeting the necroptosis pathway may block aneurysm growth.
Although the complex formation between RIPK1 and RIPK3 is essential for necroptosis and triggered by death receptors, necroptosis initiated by Toll-like receptors and other mechanisms may only require RIPK3.11 Additionally, RIPK1 and RIPK3 can participate directly in various inflammatory responses. Therefore, we sought to develop compounds with dual inhibitory effects against both kinases. By screening three libraries of kinase inhibitors, we identified GSK’074 as a novel dual RIPK1/RIPK3 inhibitor that blocks necroptosis in both human and murine cells with excellent potency and limited toxicity.12 Further, we demonstrated that daily intraperitoneal injections of GSK’074 starting at the time of AAA induction prevented aneurysm formation in two AAA models (angiotensin II and calcium phosphate).12
The purpose of the current study was to translate the RIPK1/3 inhibitor to a clinically applicable pharmaceutical treatment by determining whether GSK’074 can halt or attenuate aneurysm progression in mice with existing aortic dilation. We found that GSK’074 significantly decreased aneurysm growth in mice after daily intraperitoneal treatments starting 7 days after aneurysm induction. In addition, GSK’074-treated mice had significantly improved aortic remodeling and decreased vascular inflammation. This finding suggests that GSK’074 may be a clinically applicable small molecule inhibitor to attenuate aneurysm progression and improve aortic remodeling in patients with aortopathy.
METHODS
Mice.
C57BL/6J mice were purchased from The Jackson Laboratory (Stock #000664, Bar Harbor, Maine). The 8- to 12-week-old male mice were used for experiments because AAAs predominantly affect men.13 All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison (Protocol # M005792). The procedures were carried out in accordance with the approved guidelines.
Calcium phosphate-induced murine AAA.
Mice were subjected to calcium phosphate treatment as previously described.14 Briefly, the abdominal aorta was isolated and wrapped in 0.5 mol/L calcium chloride-soaked gauze perivascularly for 10 minutes followed by application of phosphate-buffered saline (PBS)-soaked gauze for 5 minutes. Mice in the sham group received 0.5 mol/L sodium chloride-soaked gauze for 10 minutes followed by PBS-soaked gauze for 5 minutes. The external diameter of the largest portion of the abdominal aorta was measured with a digital caliper (VWR Scientific, West Chester, Pa) before treatment and at the time of tissue harvest. Aneurysm incidence was defined as an increase of 50% or more in the external width of the abdominal aorta at the time of harvest compared with the time of aneurysm induction. GSK’074 was dissolved in 90:5:5 H2O:dimethylsulfoxide (DMSO):Cremophore EL and intraperitoneally injected into mice at various doses (0.31, 0. 93, 2.33, and 4.65mg/kg/day). Control mice were injected with equal volume of 90:5:5 H2O:DMSO:Cremophore EL.
Ultrasound imaging of the abdominal aorta.
Ultrasound images of the abdominal aorta were obtained in mice anesthetized with isoflurane using a VisualSonics Vevo2100 LAZR imaging system equipped with an MS400 linear array transducer. The abdominal aortic diameter measurements were electrocardiogram-gated and measured in B-mode. External aortic diameters were measured during systole over various time points after aneurysm induction (baseline, day 7, day 14, day 21, and day 28) in both the axial and sagittal planes. The maximum aortic lumen diameter (aortic systolic diameter corresponding with cardiac systole) and the minimum aortic lumen diameter (aortic diastolic diameter corresponding with cardiac diastole) monitored by simultaneous electrocardiogram recordings were measured and used to calculate the aortic expansion index: ([systolic aortic diameter – diastolic aortic diameter]/systolic aortic diameter).15
Histologic and morphometric analysis and immunofluorescent staining.
Mice were perfused with PBS under physiologic pressure and aortas were harvested. Tissues were embedded in O.C.T. compound (Sakura Tissue Tek, Alphen aan den Rijn, the Netherlands) and sectioned in 6 mm thickness using Leica CM3050S cryostat. Hematoxylin and eosin, Verhoeff-Van-Gieson, and Masson trichrome stainings were conducted. Immunostaining was also performed according to standard protocols with the following antibodies: anti-CD206 (AF2535, R&D Systems, Minneapolis, Minn), anti-F4/80 (123120, Biolegend, San Diego, Calif), anti-MHCII (107616, Biolegend), anti-MYH11 (ab53219, Abcam, Cambridge, UK), and anti-IL-1β (AF-401, R&D Systems). A total of three sections, approximately 60 μm apart, were stained and analyzed for each mouse aorta. The integrated density (the sum of the value of the pixels in the image or selection), area of immunofluorescence (square pixels), and number of signal positive cells were measured using ImageJ Software (National Institutes of Health, Bethesda, Md), as previously described.16
In situ gelatin zymography.
In situ gelatin zymography was conducted as described previously.17 Briefly, 1 mg/mL Fluorescein Conjugated DQ Gelatin From Pig Skin (D12054, Invitrogen, Carlsbad, Calif) was diluted 20 times with 70°C 1% UltraPure Low Melting Point Agarose (16520100, Invitrogen) in PBS and immediately applied to the fresh frozen sections followed by coverslip mounting. The samples were then incubated at 37°C for 2 hours and immediately subjected to confocal microscopy. During the capturing, unimaged specimens were stored on ice to slow down the enzyme reaction. A total of three sections, approximately 60 μm apart, were stained and analyzed for each mouse aorta. The integrated density (the sum of the value of the pixels in the image or selection), area of immunofluorescence (square pixels), and number of signal positive cells were measured using ImageJ Software (National Institutes of Health) as previously described.16 The autofluorescence of elastin was obtained by imaging negative controls in which only agarose gel was applied to sections without fluorescent conjugated substrate, and images were captured using the same camera settings. We then subtract the signal of negative control from the overall signal.
Cell culture.
Primary mouse aortic SMCs were isolated from the abdominal aorta as described previously.12 Smooth muscle cells were validated by expression of smooth muscle protein 22-α and smooth muscle-α actin, while lacking endothelial cell marker CD31 or fibroblast marker ER-TR7. Cells between three and seven passages were used. Bone marrow-derived macrophages were isolated and cultured as described before.18 Briefly, bone marrow was flushed from long bones, washed with PBS, and suspended in DMEM supplemented with 10% L-cell conditioned media. L-cell conditioned media was collected from L929 cells cultured in T-75 cm2 filter cap flasks in DMEM for 10 days and subjected to 0.2 μm filters. Seven days after harvest, all nonadherent cells were removed and the remaining cells were split in six-well plates.
Flow cytometry.
Cell death was evaluated using 7-AAD staining (51-68981E, BD Biosciences, San Jose, Calif). Cultures were rinsed with ice-cold PBS and incubated with accutase (Life Technologies, Carlsbad, Calif) at 37°C for 2 minutes. The detached cells were collected via centrifugation (2000 rpm for 5 minutes). Cell pellets were further washed twice with PBS and resuspended in 100 μL 1× binding buffer (51-66121E, BD Biosciences). We added 5 μL of 7-AAD to the cell suspension and incubated it at room temperature for 15 minutes. After incubation, a 400-μL binding buffer was added to each sample. Cells were analyzed using a Becton Dickinson Biosciences FACSCalibur (BD Biosciences).
Transwell migration assay.
Bone marrow-derived macrophages were starved in 0.5% fetal bovine serum in DMEM for 24 hours, then pretreated with 100 nmol/L GSK’074 or DMSO for 2 hours. Then, 2 × 104 cells were seeded into the upper chamber of Costar 24-well transwell plates with 5-μm pore filters (Corning Inc, Corning, NY). Medium containing 100 ng/mL MCP1 and 100 nmol/L GSK’074 or DMSO was added into the lower wells. After 16 hours, cells that migrated to the bottom of the membrane were fixed, stained with 4’,6-diamidino-2-phenylindole, and counted under a fluorescence microscope (Nikon Eclipse Ti inverted microscope system).
RNA isolation and real-time polymerase chain reaction.
Total RNA was extracted from cultured cells using Trizol reagent (15596018, ThermoFisher Scientific, Waltham, Mass) according to manufacturer’s protocols. We used 1 μg total RNA for the first-strand cDNA synthesis followed by real-time polymerase chain reaction. Primer sequences used were Il1b: forward 5′-AAATGCCACCTTTTGACAGTGATG-3′, reverse 5′-AGATTTGAAGCTGGATGCTCTCAT-3′; Tnf: forward 5′- AGGCACTCCCCCAAAAGATG -3′, reverse 5′- CCACTTGGTGGTTTGTGAGTG -3′; and Actb: forward 5′- AGCCTTCCTTCTTGGGTATGG-3′, reverse 5′-AAGGGTGTAAAACGCAGCTCA-3′.
Coimmunoprecipitation.
Mouse aortic smooth muscle cells were lysed in Pierce IP Lysis Buffer (87787, Pierce, Waltham, Mass). Floating cells were collected via centrifugation (2000 rpm for 5 minutes) and combined with attached cells then co-immunoprecipitation experiments were performed using SureBeads magnetic beads (1614013, Biorad, Hercules, Calif) according to the manufacturer’s protocol. In brief, magnetic beads were washed in PBS saline/Tween (PBST) then incubated with anti-RIPK3 antibody (2283, ProSci, Poway, Calif) or its isotype control for 30 minutes at room temperature. Beads were magnetized and washed 3 times with PBST, then incubated with cell lysate for 1 hour at room temperature. After incubation, beads were washed three times with PBST, and immunoprecipitated proteins were eluted in a 1× Laemmli buffer and subjected to Western blotting.
Western blotting.
Samples were loaded and separated by SDS-PAGE and then transferred to polyvinylidene fluoride membranes. The membranes were blocked for 60 minutes with 5% skim milk in Tris-buffered saline plus 0.05% Tween 20 (TBST), and then incubated with the following primary antibodies: anti-RIPK1 (1:1000, 610459, BD Biosciences)oranti-RIPK3 (1:1000, 2283, ProSci)followed by horseradish peroxidase-labeled secondary antibody (1:5000, Jackson ImmunoResearch, West Grove, Pa).
Statistical analysis.
Categorical variables were represented as numbers with proportions. Continuous variables were represented as means with standard error. Pearson’s χ2 and Fisher’s exact tests were used for categorical variables, where appropriate. A nonpaired Student t-test was used to compare means between two groups. One-way analysis of variance and two-way analysis of variance were used to compare three or more groups. All statistical analysis was performed using SPSS v25 (Armonk, NY). A P value of .05 or less was considered statistically significant.
RESULTS
GSK’074 in vivo dose response.
To determine the optimal dosage of GSK’074, we treated mice with five different doses (0, 0.31, 0.93, 2.33, and 4.65 mg/kg/day) for 14 days starting at the time of AAA induction via the calcium phosphate model (Fig 1, A). There were a total of five mice in each group. All dosages were tolerated by mice without causing observable negative effects on weight or behavior. Compared with controls, dosages of 0.93, 2.33, and 4.65 mg/kg/day GSK’074 significantly inhibited aneurysm formation, reflected by diminished expansion in aortic diameter (Fig 1, B and C). The highest dosage of 4.65 mg/kg/day seemed to be most effective in inhibiting aneurysm formation; however, differences among the top three dosages failed to reach statistical significance.
Fig 1.
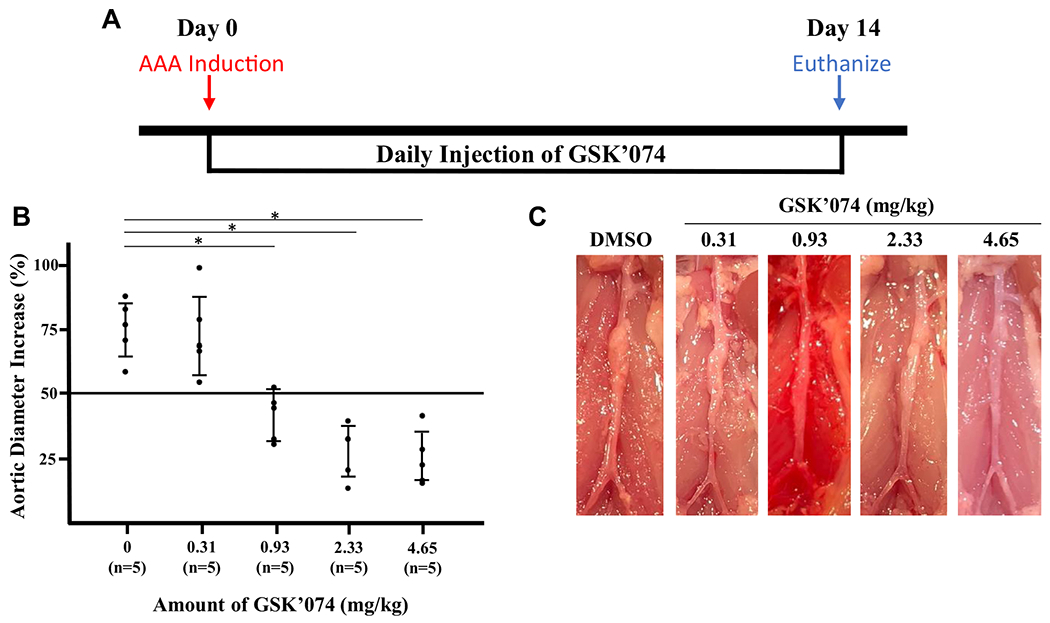
Dose response of GSK2593074A (GSK’074) on aneurysm formation. A, Experimental design. C57BL/6J mice underwent daily intraperitoneal injections with dimethylsulfoxide (DMSO)-control or GSK’074 (0.31, 0.93, 2.33, or 4.65 mg/kg), starting at the time of aneurysm induction by calcium phosphate model. B, Mice were humanely killed 14 days after aneurysm induction and percentage increase in maximal external aortic diameter measured by digital caliper was calculated. C, Representative images of aortas. n = 5 for each group. One-way analysis of variance with a post hoc Tukey’s test was performed. *P ≤ .05. The bars indicate 95% confidence intervals. AAA, Abdominal aortic aneurysm.
GSK’074 attenuates aneurysm progression.
Clinically, medical treatment would be initiated once an aneurysm is diagnosed. However, few studies have focused on whether aneurysms can be halted once aneurysms have already been induced. Therefore, we initiated GSK’074 treatment at a dose of 4.65 mg/kg/day 7 days after AAA was induced (Fig 2, A). Once aortic dilation was confirmed via ultrasound examination, we randomly divided mice into two groups: DMSO or GSK’074 treatment. There were no differences in aortic diameters at baseline (before aneurysm induction) and at day 7 (after aneurysm induction but before treatment initiation) between DMSO controls and GSK’074-treated mice (Fig 2, B and C; Supplementary Fig 1). Seven days after treatment (day 14), the GSK’074-treated mice demonstrated less aortic stiffness, indicated by improvements in aortic expansion compared with DMSO controls (Fig 2, D). However, the inhibitory effect of GSK’074 on external aortic diameters did not become significant until 21 days after aneurysm induction (ie, 14 days after the initiation of treatment) (Fig 2, C). Manual measurement of aortas at the time of euthanizing confirmed that mice treated with GSK’074 had significantly smaller aneurysm (Fig 2, E and F). There were no differences in internal diameter between the two groups (Supplementary Fig 2).
Fig 2.
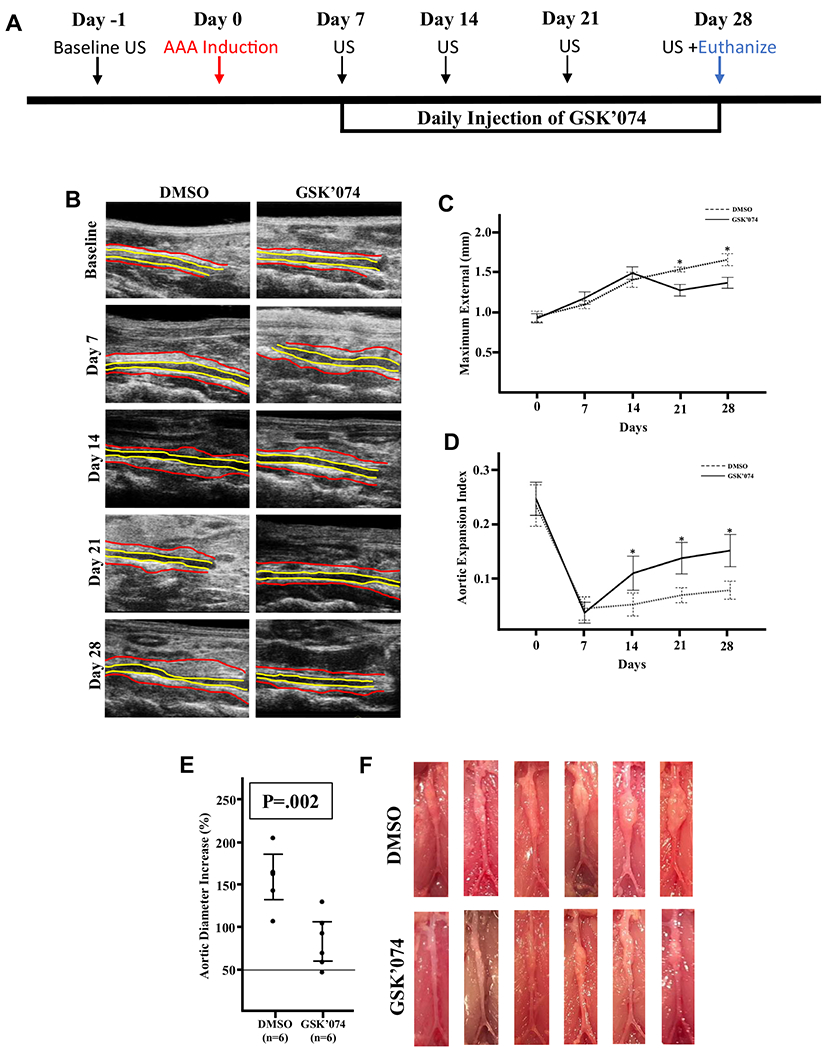
GSK2593074A (GSK’074) attenuates progression of existing aneurysmal dilation. A, Experimental design. C57BL/6J mice underwent ultrasound examination of the infrarenal abdominal aorta at baseline and weekly after aneurysm induction for 28 days. Daily intraperitoneal injections of GSK’074 (4.65 mg/kg) or dimethylsulfoxide (DMSO)-control started 7 days after aneurysm induction. B, Representative ultrasound images of the infrarenal aorta. Red outlines the external diameter and yellow outlines the luminal diameter. C, Maximum external diameter measured from ultrasound images over the course of the study period. Data are presented as mean ± standard error. Two-way analysis of variance with a post-hoc Tukey’s test was performed. D, Aortic expansion index over the course of the study period. Data are presented as mean ± standard error. Two-way analysis of variance with a post hoc Tukey’s test was performed. E, Mice were euthanized 28 days after aneurysm induction and the percentage increase in maximal external aortic diameter measured by digital caliper was calculated. The bars indicate 95% confidence intervals. A two-tailed Student t-test was performed. F, Images of aortas of both groups. n = 6 for each group. *P ≤ .05. AAA, abdominal aortic aneurysm.
GSK’074 treatment improves aortic remodeling.
Aortic expansion index has beenthoughtto bea markerofaortic stiffness and is associated with inflammation and dysregulated aortic remodeling.15,19 Therefore, we hypothesized that GSK’074 treatment improves inflammatory resolution and aortic remodeling. To test this hypothesis, we analyzed the tissue sections from different groups using hematoxylin and eosin, Verhoeff-Van Gieson (elastin), and Masson trichrome (collagen) staining. Compared with sham aortas, the aortic sections of calcium phosphate mice treated with DMSO showed typical aneurysmal pathology characterized by severally thinned and disorganized elastin fibers (Fig 3, A). In contrast, elastin organization was preserved in the GSK’074-treated aortic samples (Fig 3, A). Similarly, collagen deposition in the DMSO control groups seemed to be dispersed throughout all layers of the aortic wall, whereas collagen deposition was confined to the adventitial layer in the GSK’074-treated group (Fig 3, A). Correspondingly, levels of smooth muscle cell-specific myosin heavy chain 11 (MYH11) was significantly increased in the GSK’074-treated group in comparison with DMSO controls (Fig 3, B). These changes were accompanied by a decrease in gelatinase activity in the GSK’074-treated group (Fig 3, C).
Fig 3.
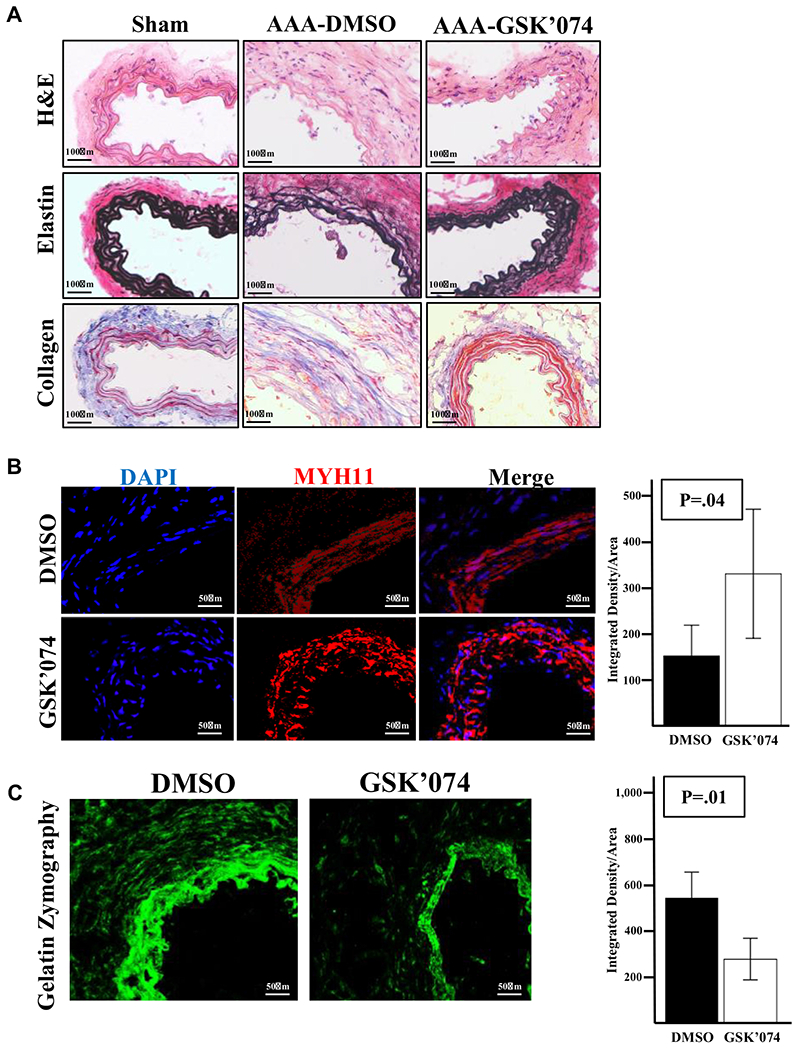
Therapeutic administration of GSK2593074A (GSK’074) improves aortic integrity. A, Representative images of hematoxylin and eosin (H&E), elastin (Van-Gieson), and collagen (trichrome) staining of aortic cross-sections from sham-treated aortas 7 days after surgery and dimethylsulfoxide (DMSO) vs GSK’074 treated aortas 28 days after abdominal aortic aneurysm (AAA) induction. B, Immunostaining and quantification of smooth muscle cell-specific myosin heavy chain 11 (MYH11) on aortic cross-sections from DMSO and GSK’074-treated aortas 28 days after AAA induction. C, In situ gelatin zymography and quantification of aortic cross-sections from DMSO and GSK’074-treated aortas 28 days after AAA induction. n = 6 for each group. Data are presented as mean ± standard error. A two-tailed Student t-test was performed. *P ≤ .05. DAPI, 4’,6-diamidino-2-phenylindole.
GSK’074 decreases inflammatory cell populations within the aortic wall.
Negative aortic remodeling in aneurysms is closely linked with inflammation.20 Previously, we demonstrated that GSK’074 treatment starting at the time of AAA induction decreased the CD68+ cell population within aortas at day 4.12 In this study, we administrated GSK’074 starting 7 days after AAA induction and examined aortas at day 28. We found that there was a significant decrease in inflammatory markers IL-1β and MHCII, as well as macrophage marker F4/80+ (Fig 4, A–C). We also found significant reductions in M2-like macrophage marker CD206 in GSK’074-treated mice (Fig 4, D). No differences were detected in CD3+, CD4+, or FOXP3+ cells between the two groups (data not shown).
Fig 4.
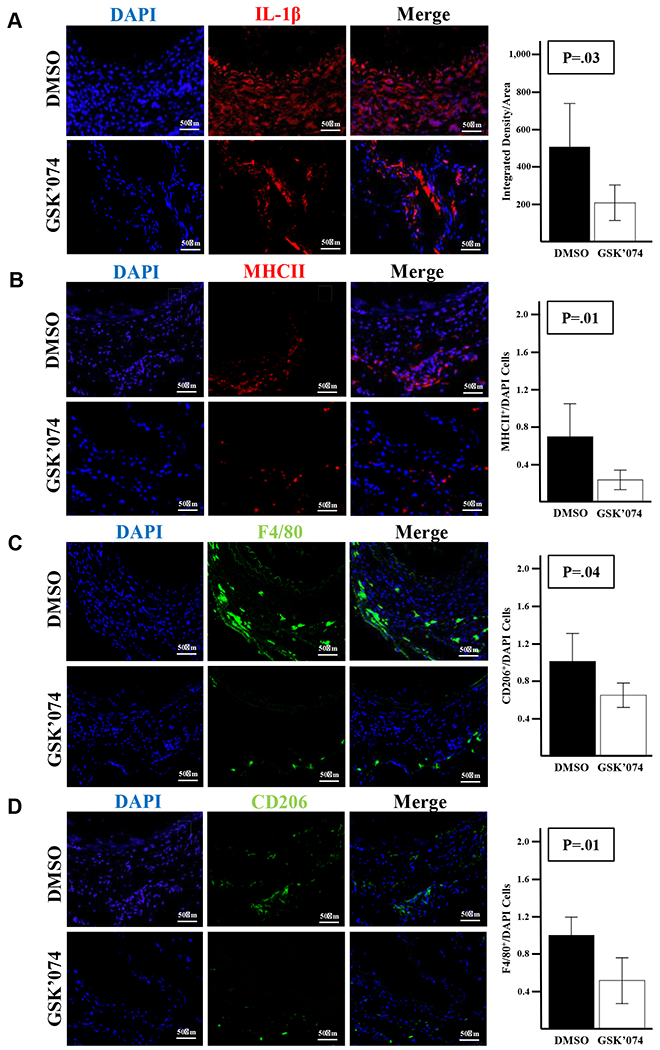
Therapeutic administration of GSK2593074A (GSK’074) decreases macrophage population within the aortic wall. Immunostaining and quantification of F4/80 (A), CD206 (B), major histocompatibility complex II (MHCII) (C), and IL-1β (D) of aortic cross-sections from dimethylsulfoxide (DMSO) and GSK’074-treated aortas 28 days after AAA induction, n = 6 for each group. Data are presented as mean ± standard error. A two-tailed Student t-test was performed. *P ≤ .05. DAPI, 4’,6-diamidino-2-phenylindole.
GSK’074 inhibits VSCM necroptosis and macrophage migration.
The preserved medial expression of smooth muscle cell marker MYH11 suggests that GSK’074-treatment prevents progressive VSMC loss in aneurysms. Using mouse VSMCs, we validated that GSK’074 inhibited necroptosis induced by tumor necrosis factor-α plus zVAD by blocking the essential interaction between RIPK1 and RIPK3 (Fig 5, A and B). Because VSMC death is associated with the release of cytokines and damage-associated molecular patterns,21,22 GSK’074 can affect inflammation by decreasing chemoattractive signals in the aortic wall. However, it is currently unknown whether GSK’074 has an effect on macrophages directly. To this end, we derived macrophages from mouse bone marrow and found that GSK’074 significantly decreased macrophage migration toward MCP-1 (Fig 5, C). Of note, GSK’074 had no effect on macrophage Il1b or Tnf cytokine expression in response to lipopolysaccharide (Fig 5, D). These results suggest that GSK’074 may affect aortic remodeling and aneurysm progression via inhibition of VSMC necroptosis and macrophage recruitment/migration into the aortic wall.
Fig 5.
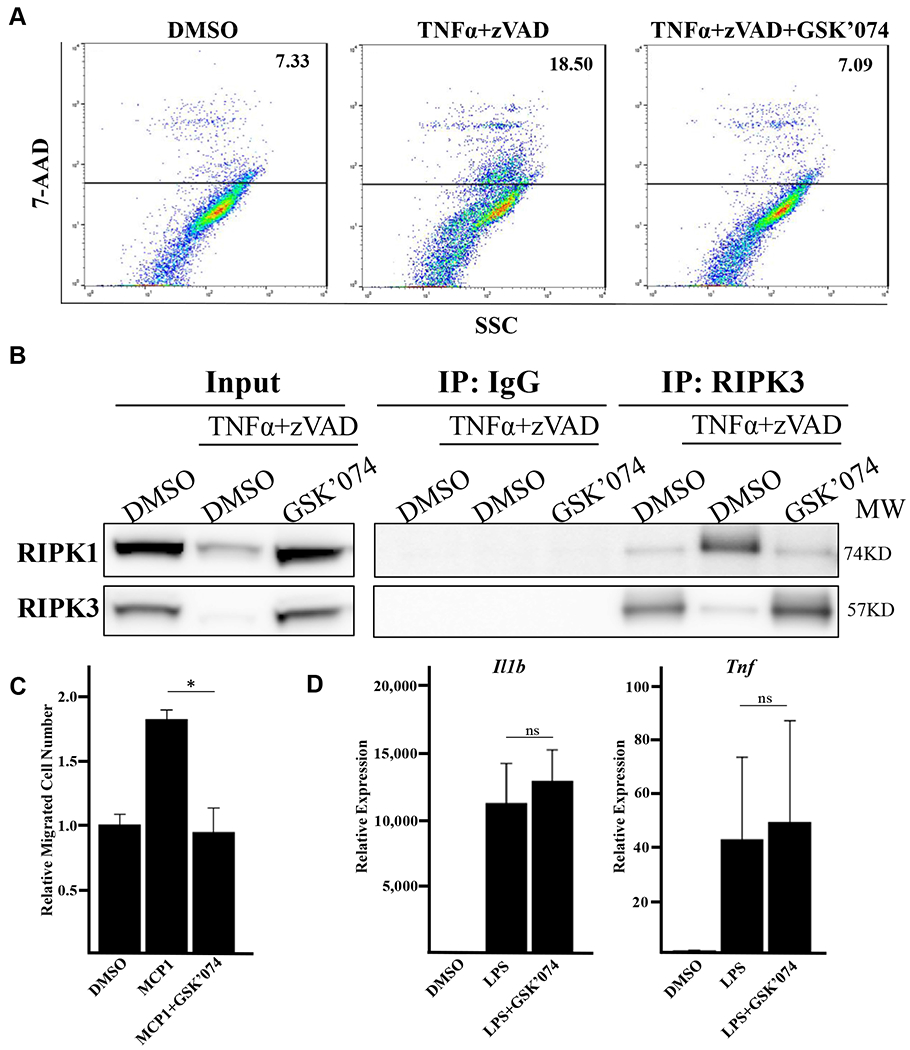
GSK2593074A (GSK’074) directly inhibits vascular smooth muscle cell (VSCM) necroptosis and macrophage migration. A, Mouse primary smooth muscle cells were treated with 100 ng/mL tumor necrosis factor-α (TNF-α) plus 60 μmol/L zVAD and 10 nmol/L GSK’074 for 24 hours. Cells were then stained with 7-AAD and analyzed via flow cytometry. B, Mouse aortic smooth muscle cells were treated with 30 ng/ml TNF-α plus 60 μmol/L zVAD overnight in the presence or absence of 10 nmol/L GSK’074. Receptor interacting proteins kinase (RIPK)1 and RIPK3 complex formation were detected by coimmunoprecipitation with anti-IgG or anti-RIPK3 antibodies followed by Western blotting analysis with the indicated antibodies. C, Mouse bone marrow-derived macrophages (BMDMs) were pretreated with 100nM GSK’074 or dimethylsulfoxide (DMSO) for 2 hours, cell migration toward MCP1 (100ng/ml) was determined via transwell migration assay. One-way analysis of variance with a post hoc Tukey’s test was performed. D, Mouse BMDMs were pretreated with 100 nmol/L GSK’074 or DMSO for 2 hours followed by 1 ng/mL lipopolysaccharide (LPS) stimulation in the presence or absence of GSK’074 for 6 hours. Messenger RNA levels of Il1b and Tnf were determined by real-time polymerase chain reaction. One-way analysis of variance with a post hoc Tukey’s test was performed. Data are presented as mean ± standard error of three independent experiments. *P ≤ .05. ns, Nonsignificant.
DISCUSSION
The current management of AAAs is limited exclusively to surgical repair. Although smaller clinical trials have reported successful stabilization of aortic aneurysms with various pharmaceutical treatment, these experimental drugs failed in larger randomized controlled studies.4 Consequently, no medical therapy exists for the stabilization of AAAs. Previously, we identified GSK’074 as a novel RIPK1/RIPK3 inhibitor that prevented AAA formation when administered at the time of aneurysm induction. The effects of GSK’074 on aortic expansion and aortic deterioration are consistent with the aneurysm-resistant phenotype of Ripk3 gene deficient mice. However, realistically, treatment for AAA would occur after diagnosis. In this study, we found that GSK’074 treatment attenuated AAA growth in mice with existing aortic dilation. Further, we found that GSK’074 treatment improved aortic remodeling as evidenced by a decreased loss of VSMCs, normalized elastin and collagen organization, improvement in aortic expansion index, and decreased macrophage populations within the aortic wall. Mechanistically, GSK’074 directly inhibited macrophage migration. This novel effect, along with its anticipated effect on VSMC necroptosis, suggest that GSK’074 attenuates aneurysm progression via multiple molecular pathways (Fig 6).
Fig 6.
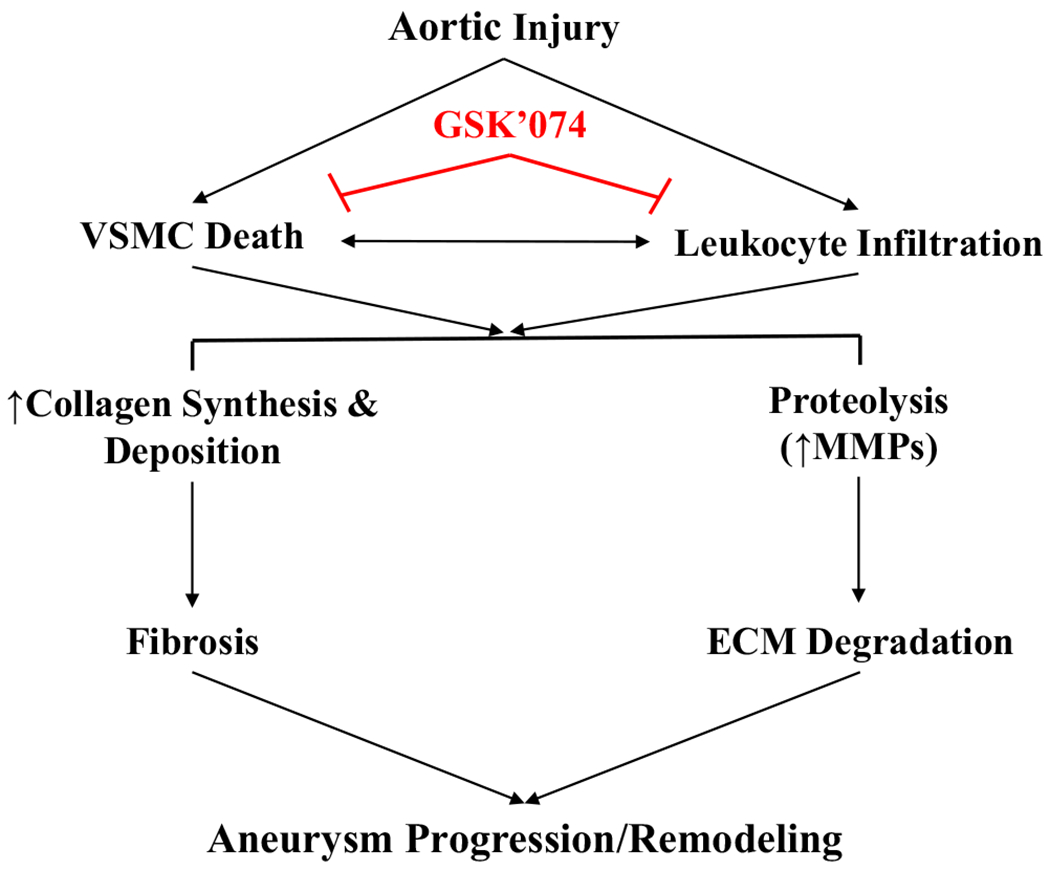
Schematic outlining GSK2593074A (GSK’074) mechanisms of action on blocking existing aneurysmal dilation. Progressive loss of vascular smooth muscle cell (VSMC) and infiltration of leukocytes, especially macrophages, are characteristic features of aneurysm pathology. GSK’074 directly blocks VSMC death and macrophage migration, which result in attenuated fibrosis and extracellular matrix (ECM) degradation, which ultimately inhibits negative aortic remodeling and dilatation of existing aneurysms. MMP, Matrix metalloproteinase.
Clinical data suggest that the majority of small aneurysms continue to expand when left untreated, albeit the rate of growth varies from patient to patient.19,23 This progressive nature is replicated in the murine calcium phosphate model used in this study, evidence by our ultrasound imaging data of the DMSO-treated mice. At the time of initiation of treatment with GSK’074, mouse aortas were dilated with significant VSMC loss and inflammation.12 This finding suggests that, at the time of treatment with GSK’074, some pathologic events, including VSMC depletion, have yet reached an irreversible point. We speculate that VSMCs in the aortic wall 1 week after aneurysm might be analogous to the ischemic penumbra after neurologic ischemic strokes where there are cells at risk for death, but are potentially salvageable.24 The at risk but potentially salvageable VSMCs may be an important target to prevent further aneurysmal degeneration/progression. In the absence of noninvasive high-resolution imaging technology, we are unable to determine the molecular and cellular changes in the aortic wall over the course of GSK’074 treatment. However, ultrasound data demonstrated that small AAAs responded to GSK’074 with halted aortic expansion 14 days after treatment. Histologic analyses of aortic tissues harvested at the end of the study (28 days after aneurysm induction) showed reserved smooth muscle marker MYH11 compared with the DMSO controls, suggesting that GSK’074 successfully prevented progressive loss of VSMCs.
Literature suggests that multiple types of cell death may take place in aneurysmal aortic tissues.5,22,25 We have previously demonstrated the presence of apoptotic, necrotic, and necroptotic cells in experimental aneurysm tissues.9,12 Because RIPK1 is also involved in regulation of apoptosis, the selective RIPK1 inhibitor necrostatin-1 attenuated both necroptosis and apoptosis in mouse AAA models.10 Being able to bind and inhibit both RIPK1 and RIPK3, GSK’074 can prevent medial cell depletion by inhibiting both necroptosis and apoptosis.12 Although it is difficult to distinguish GSK’074’s antine-croptosis effect from its potential antiapoptosis effect, we have previously shown that the pan-caspase inhibitor quinoline-Val-Asp-difluorophenoxymethylketone is unable to block aneurysm progression, despite effectively preventing aneurysm formation.26 Many recent advances have been made in the understanding of molecular mechanisms of apoptosis, necroptosis, and other forms of cell death. However, a knowledge of human small aneurysm is necessary to evaluate the relationships between apoptosis and necroptosis during the progression of aneurysmal tissue deterioration.
Inflammation is another widely investigated process believed to contribute to AAA growth. Various molecular pathways causing inflammation have been implicated in the pathogenesis of aortic aneurysms.27 Necroptosis induces sterile inflammation upon release of its intracellular contents into the extracellular space. Specifically, VSMCs undergoing necroptosis increase expression of various cytokines that lead to leukocyte infiltration.9 We have previously reported that GSK’074 inhibited cytokine expression by VSMCs through inhibition of RIPK3.12 In this study, we unexpectedly found that GSK’074 had direct effects on macrophage migration. However, using macrophages derived from mouse bone marrow, we found GSK’074 had no detectable effect on the expression of Il1b or Tnf upon stimulation with lipopolysaccharide. It is possible that GSK’074 inhibits macrophages through a mechanism unrelated to necroptosis and its mediators. For example, one of the known off-targets of GSK’074 identified by molecular binding studies is dual specificity mitogen-activated protein kinase 5,12 which has been shown to have an active role in supporting cell migration of various cell types.28,29 However, we cannot rule out the possibility that RIPK1 and/or RIPK3 are involved in regulation of macrophage migration because both kinases have been found to regulate inflammatory processes via cell death-independent mechanisms.30 Nevertheless, it is possible that GSK’074 decreases aortic wall inflammation through both indirect and direct mechanisms. Targeting multiple relevant pathways simultaneously may prove to be beneficial in pharmaceutical treatment of aneurysms; previous clinical studies directed at one mechanism have failed.4
Our histologic analyses and ultrasound imaging indicated that GSK’074 was able to improve aortic remodeling as evident by the organization in collagen deposition and improvements in aortic expansion index. Aortic expansion index is a measure of aortic compliance and stiffness. Prior clinical studies have highlighted the association between aortic stiffness and rupture risk.31,32 Our observation that GSK’074 increased aortic expansion index raises a possibility that this inhibitor may reduce rupture risk by reducing aortic stiffness. Further, aortic remodeling is a result of chronic inflammatory responses, extracellular matrix degradation, and matrix metalloproteinase upregulation.33 Various macrophage subsets have been known to have different functions in extracellular matrix remodeling and aneurysm progression. Although macrophages exist in a large spectrum of different phenotypes, M1-like macrophages are classically thought to be proinflammatory and M2-like macrophages as anti-inflammatory. M2-like macrophages are typically characterized via their expression of CD206 and secretion of factors such as IL-10 and transforming growth factor-β (TGF-β). TGF-β signaling has a well-established role in tissue fibrosis.34–36 Further, it has recently been shown that M2-like macrophages contribute to aortic stiffness and fibrosis primarily mediated by TGF-β.37 Thus, we deduce that GSK’074 primarily improves aortic remodeling by decreasing the overall macrophage population, including CD206+ cells, and subsequently TGF-β signaling.
This study has several limitations to consider. First and foremost is the use of a murine model to induce and mimic human aneurysm development. Although the calcium phosphate model is a well-validated murine model in the study of aortic aneurysms,14 the translation of such findings to humans should always proceed with caution. GSK’074 was administered starting from day 7 of this model, a time point in which aortic dilation is present, but has not yet reached the threshold of aortic aneurysm. The rationale for starting GSK’074 early is that the majority of cell death, the primary function of RIPK1/RIPK3, occurs between induction and day 7 in this mouse aortic aneurysm model.9,12,14 The molecular and cellular events taking place in small human aneurysms remains unknown; however, we detected high levels of RIPK1 and RIPK3 in late stage aneurysmal tissues removed during surgical repair.9,10 Although our findings in the CaCl2 model is encouraging, assessing GSK’074 treatment in more chronic models is necessary before proceeding to clinical trials. In addition, we are unable to determine the tissue distribution of GSK’074 over the course of treatment or its real time effects on the various cellular events during aneurysm progression. We have previously demonstrated in vitro that GSK’074 inhibits the kinase activities of RIPK1 as well as RIPK3. At early stage in mouse aneurysm model induced by CaCl2, we showed that GSK’074 treatment dramatically decreased MLKL-phosphorylation, apoptosis, and necroptosis in the aortic wall using immunostaining, terminal uridine nick-end labeling, and propidium iodide staining as surrogates.12 Because cell death activities subside as aneurysm progresses in mouse aneurysm models, we were unable to detect MLKL-phosphorylation, apoptosis, or necroptosis in the aortic samples harvested 28 days after aneurysm induction in either control or treated group. As such, data presented here do not provide direct proof that GSK’074 inhibited RIPK1 and RIPK3 activities at late stage of aneurysm development. However, the observation that GSK’074 inhibited macrophage migration in itself suggests that this small molecule inhibitor can decrease aortic wall inflammation both directly and indirectly.
CONCLUSIONS
GSK’074 attenuates aneurysm progression in the calcium phosphate model. The ability to inhibit both VSCM necroptosis and macrophage migration makes GSK’074 an attractive drug candidate for pharmaceutical treatment of aortic aneurysms.
Supplementary Material
ARTICLE HIGHLIGHTS.
Type of Research: Single-center experimental study
Key Findings: Administration of GSK2593074A after aneurysm induction blocked aneurysm progression and improved aortic remodeling in mice subjected to the calcium phosphate model.
Take Home Message: GSK2593074A is an attractive drug candidate for pharmaceutical treatment of aortic aneurysms.
Acknowledgments
The authors thank Ms Ashley M. Weichmann for performing ultrasound scanning and Ms Sierra Raglin for performing histologic staining.
Supported by the National Institutes of Health R01HL088447 (BL), R01HL122562 (BL), T32HL110853 (MK), and American Heart Association 17POST33680095 (TZ) and 17PRE33670082 (KG). The funders had no role in the decision to publish or preparation of the manuscript. Ultrasound imaging and analysis was conducted at UW Small Animal Imaging & Radiotherapy Facility (S10 OD018505, P30 CA014520).
Footnotes
Author conflict of interest: none.
The editors and reviewers of this article have no relevant financial relationships to disclose per the JVS-Vascular Science policy that requires reviewers to decline review of any manuscript for which they may have a conflict of interest.
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics - 2018 update: a report from the American Heart Association. Circulation 2018;137:e67–492. [DOI] [PubMed] [Google Scholar]
- 2.Owens DK, Davidson KW, Krist AH, Barry MJ, Cabana M, Caughey AB, et al. Screening for abdominal aortic aneurysm: US Preventive Services Task Force recommendation statement. JAMA 2019;322:2211–8. [DOI] [PubMed] [Google Scholar]
- 3.Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet 2005;365:1577–89. [DOI] [PubMed] [Google Scholar]
- 4.Lindeman JH, Matsumura JS. Pharmacologic management of aneurysms. Circ Res 2019;124:631–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta K, Phan N, Wang Q, Liu B. Necroptosis in cardiovascular disease - a new therapeutic target. J Mol Cell Cardiol 2018;118:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the pathophysiology of disease. Am J Pathol 2020;190:272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135–47. [DOI] [PubMed] [Google Scholar]
- 8.Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem 2016;85:743–63. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q, Liu Z, Ren J, Morgan S, Assa C, Liu B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ Res 2015;116:600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Zhou T, Liu Z, Ren J, Phan N, Gupta K, et al. Inhibition of receptor-interacting protein kinase 1 with necrostatin-1s ameliorates disease progression in elastase-induced mouse abdominal aortic aneurysm model. Sci Rep 2017;7:42159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 2013;288:31268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou T, Wang Q, Phan N, Ren J, Yang H, Feldman CC, et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis 2019;10:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kent KC, Zwolak RM, Egorova NN, Riles TS, Manganaro A, Moskowitz AJ, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg 2010;52:539–48. [DOI] [PubMed] [Google Scholar]
- 14.Yamanouchi D, Morgan S, Stair C, Seedial S, Lengfeld J, Kent KC, et al. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vasc Surg 2012;56:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basu R, Fans D, Kandalams V, Lee J, Das SK, Wang X, et al. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J Biol Chem 2012;287:44083–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morioka N, Miyauchi K, Miyashita K, Kochi T, Zhang FF, Nakamura Y, et al. Spinal high-mobility group box-1 induces long-lasting mechanical hypersensitivity through the toll-like receptor 4 and upregulation of interleukin-1β in activated astrocytes. J Neurochem 2019;150:738–58. [DOI] [PubMed] [Google Scholar]
- 17.Hadler-Olsen E, Kanapathippillai P, Berg E, Svineng G, Winberg JO, Uhlin-Hansen L. Gelatin in situ zymography on fixed, paraffin-embedded tissue: zinc and ethanol fixation preserve enzyme activity. J Histochem Cytochem 2010;58: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Morgan S, Ren J, Wang Q, Annis DS, Mosher DF, et al. Thrombospondin-1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circ Res 2015;117:129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brady AR, Thompson SG, Fowkes FGR, Greenhalgh RM, Powell JT. Abdominal aortic aneurysm expansion: risk factors and time intervals for surveillance. Circulation 2004;110:16–21. [DOI] [PubMed] [Google Scholar]
- 20.Jana S, Hu M, Shen M, Kassiri Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp Mol Med 2019;51:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wortmann M, Skorubskaya E, Peters AS, Hakimi M, Böckler D, Dihlmann S. Necrotic cell debris induces a NF-κB-driven inflammasome response in vascular smooth muscle cells derived from abdominal aortic aneurysms (AAA-SMC). Biochem Biophys Res Commun 2019;511:343–9. [DOI] [PubMed] [Google Scholar]
- 22.Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation 1999;99:96–104. [DOI] [PubMed] [Google Scholar]
- 23.Bhak RH, Wininger M, Johnson GR, Lederle FA, Messina LM, Ballard DJ, et al. Factors associated with small abdominal aortic aneurysm expansion rate. JAMA Surg 2015;150:44–50. [DOI] [PubMed] [Google Scholar]
- 24.Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009;40:e331–9. [DOI] [PubMed] [Google Scholar]
- 25.López-Candales A, Holmes DR, Liao S, Scott MJ, Wickline S a, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 26.Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu B. Effects of caspase inhibitor on angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2010;30:702–7. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2006;26:987–94. [DOI] [PubMed] [Google Scholar]
- 28.Stecca B, Rovida E. Impact of ERK5 on the hallmarks of cancer. Int J Mol Sci 2019;20:1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira DM, Gomes SE, Borralho PM, Rodrigues CMP. MEK5/ERK5 activation regulates colon cancer stem-like cell properties. Cell Death Discov 2019;5:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton K RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol 2015;25:347–53. [DOI] [PubMed] [Google Scholar]
- 31.Kolipaka A, Illapani VSP, Kenyhercz W, Dowell JD, Go MR, Starr JE, et al. Quantification of abdominal aortic aneurysm stiffness using magnetic resonance elastography and its comparison to aneurysmdiameter. J Vasc Surg 2016;64:966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vorp DA, Vande Geest JP. Biomechanical determinants of abdominal aortic aneurysm rupture. Arterioscler Thromb Vasc Biol 2005;25:1558–66. [DOI] [PubMed] [Google Scholar]
- 33.Cheng Z, Zhou YZ, Wu Y, Wu QY, Liao XB, Fu XM, et al. Diverse roles of macrophage polarization in aortic aneurysm: destruction and repair. J Transl Med 2018;16:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 2017;127:3770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol 2011;43:154–62. [DOI] [PubMed] [Google Scholar]
- 36.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 2016;12:325–38. [DOI] [PubMed] [Google Scholar]
- 37.Sharma N, Dev R, Belenchia AM, Aroor AR, Whaley-Connell A, Pulakat L, et al. Deficiency of IL12p40 (interleukin 12 p40) promotes Ang II (angiotensin II)-induced abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 2019;39:212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.