

Abstract
Contemporary studies of systemic sclerosis (SSc) consistently demonstrate that interstitial lung (ILD) is a leading cause of disease-related death. This review summarizes morbidity and mortality outcomes in SSc-ILD patients from high-quality observational and interventional studies over the last 50 years. The data presented suggest a trend for improved morbidity and mortality outcomes among present day SSc-ILD patients. Specifically, SSc-ILD patients appear to be living longer from the time of the initial diagnosis. Despite improved survival, the number one cause of death for most SSc-ILD patients remains respiratory failure from ILD. This review describes the most important demographic, clinical, and biological factors, which affect mortality in SSc-ILD, and could be used to help stratify patients for closer monitoring and more aggressive initial treatment. The review concludes with an overview of future research needed to (1) understand how to personalize the care of SSc-ILD patients to improve morbidity and mortality outcomes; and (2) investigate whether novel therapeutic interventions (e.g., anti-fibrotics, hematopoetic stem-cell transplantation) offer any meaningful long-term survival advantage over the current standard of care.
Keywords: systemic sclerosis, interstitial lung disease, mortality, scleroderma, immunosuppression
Introduction
One of the most common – and potentially most devastating – manifestations of systemic sclerosis (SSc) is interstitial lung disease (ILD), a group of chronic lung conditions characterized by fibrosis and/or inflammation of the lung parenchyma. ILD, also called pulmonary fibrosis, is now the leading cause of SSc-related death1 and SSc patients with lung fibrosis have a mortality risk nearly three times greater than SSc patients without lung fibrosis.2
Although estimates of the prevalence of ILD in SSc vary by mode of detection, between 30% and 90% of patients with SSc are believed to develop ILD over the course of the disease.3 In early autopsy series, up to 100% of patients were found to have some degree of parenchymal involvement.4,5 As many as 90% of patients will have interstitial abnormalities on high-resolution computed tomography (HRCT)6 (Figure 1) and 40 to 75% will have pulmonary function test (PFT) changes7,8 Parenchymal lung involvement often appears early after the diagnosis of SSc, with 25% of patients developing clinically significant ILD within 3 years as defined by physiologic, radiographic or bronchoalveolar lavage abnormalities.9
Figure 1. High resolution computed tomography (HRCT) scan of a patient with SSc-ILD.
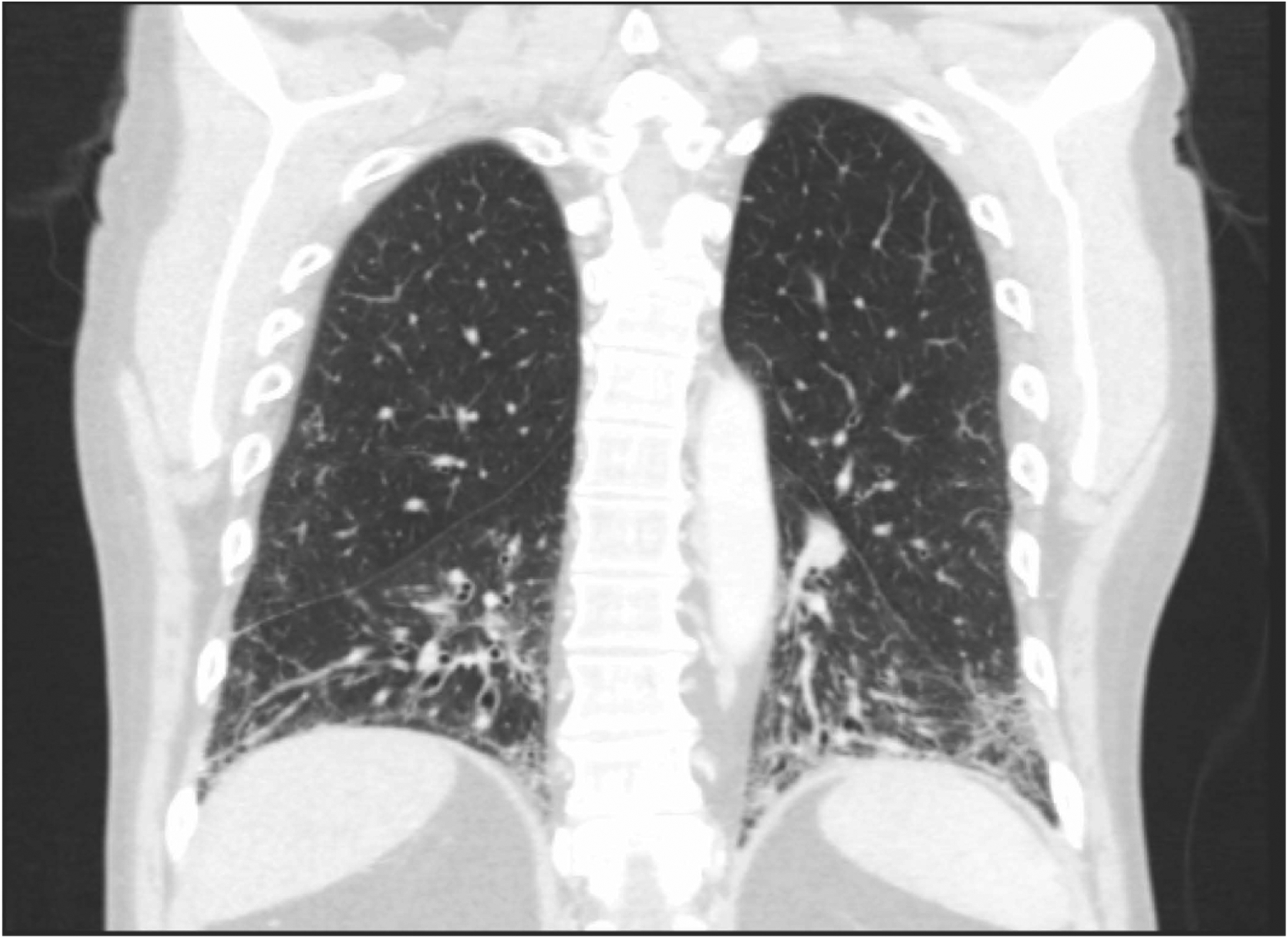
This image demonstrates the radiographic features characteristic of a non-specific interstitial pneumonia pattern commonly observed in patients with SSc-ILD. With permission from AF.
Risk factors for the development of ILD include African American ethnicity, degree of skin involvement, serum creatinine and creatine phosphokinase levels, hypothyroidism and cardiac involvement.9,10 Genetic factors, specific serologic findings (e.g., anti-topoisomerase I antibodies) predict the presence of lung involvement and the pattern of skin disease. Moreover, patients with diffuse skin thickening have a higher incidence of ILD.11,12,13,14 Predictors of severe restrictive lung disease (defined by a forced vital capacity [FVC] ≤ 50%) include African American ethnicity, male gender, the degree of physiologic abnormalities at diagnosis (FVC and carbon monoxide diffusing capacity [DLco]) and younger age.7,15
In this update, we review the historical perspective and changing landscape of mortality associated with ILD in SSc, describe what pivotal SSc-ILD trials have taught us about prognosis, discuss the current understanding of predictors of SSc-ILD progression and conclude with a discussion of future research ideas to address unmet needs in this arena.
Historical perspective on morbidity and mortality in SSc-ILD
A common theme from historical SSc cohorts is that major organ involvement – including lung fibrosis – is associated with worse survival, and prior to the advent of angiotensin converting enzyme inhibitors, renal disease was the most dreaded manifestations (Table 1). In one early study, Bennett and colleagues described a cohort of 67 SSc patients cared for at Hammersmith Hospital in London between 1947–1970.16 The cumulative 5-year survival of the cohort was 73% and the 10-year survival rate was 50%.16 Slightly more than a quarter of the patients had evidence of ILD based on plain chest radiograph.16 The following features at disease onset were associated with a worse prognosis: diffuse skin involvement, age > 40, renal dysfunction, abnormal electrocardiogram and presence of lung fibrosis.16
Table 1.
Cumulative survival rates in select historical SSc observational studies
| Study Author, Year | N | Number of Centers, Location | Study Duration | Survival Rates |
|---|---|---|---|---|
| Bennet et al., 197116 | 67 | Single center (Hammersmith Hospital, London, UK) | 1947–1970 | 5 years = 73% 10 years = 50% |
| Medsger and Masi, 197317 | 358 | VA hospitals, USA | 1963–1968 | 5 years = 44% 7 years = 35% |
| Altman et al., 199118 | 264 | 29 centers, USA | 1973–1977 | 2 years = 80% 8.5 years = 50% 12 years = 30% |
| Steen and Medsger, 200720 | 221–655 | Single center (University of Pittsburgh, USA) | 1972–1996 | 10 years = 54–66% (varied by decade) |
| Ferri et al., 200221 | 1012 | 3 centers (Italy) | 1955–1999 | 10 year = 69% 20 year = 46% |
In another early study, Medsger and Masi analyzed the survival of 358 men with SSc from the Veterans Administration hospital registry between 1963–1968.17 They showed that SSc is associated with poor prognosis, reporting a 5-year survival rate of 44% and a 7-year survival rate of 35%.17 Renal failure was identified in 5% and was rapidly progressive and uniformly fatal – thus constituting the worst prognostic sign in the disease.17 The relative effects on survival of renal, cardiac or pulmonary involvement found at entry were analyzed. All patients with kidney involvement at entry died within 10 months of recognition of this complication and only one lived beyond 3 months.17 Pulmonary involvement also affected survival, but not to the same devastating degree as with renal involvement. Lung disease was described as evidence of fibrotic changes on plain radiograph and was noted in a total of 146 patients (41%) at study entry.17 Of the 112 patients with lung disease without either cardiac or renal involvement, survival was significantly reduced compared to the 178 patients with none of these specified organs involved (p < 0.01).17
Altman and colleagues evaluated SSc-related deaths in 264 patients enrolled in a multicenter SSc registry between 1973–77 with a mean follow up of 5.2 years.18 They reported survival rates of 80% at 2 years, 50% at 8.5 years, and 30% at 12 years from study entry.18 The presence of severe cardiac, renal, gastrointestinal, or pulmonary involvement (defined as either ILD or pulmonary hypertension) predicted worse survival. Renal disease accounted for 39% of deaths compared with 20% due to pulmonary disease.18 Patients with lower FVC had a higher risk of death with a relative risk of 0.98 indicating that a patient with a 1% higher percent predicted FVC had a 2% reduction in hazard of death.18
Steen and Medsger similarly demonstrated that severity of organ involvement predicted early death in SSc.19 Patients with severe skin involvement, FVC <55% (a surrogate for severe ILD), malabsorption, pseudo-obstruction, arrhythmia, congestive heart failure or renal crisis had a 9-year survival rate of only 38%.19 In contrast, those with mild organ involvement had a 9-year survival rate of 78%.19
Changing landscape of morbidity and mortality in SSc-ILD
The current era has witnessed a changing landscape in the etiology of SSc-related deaths. In a landmark study from Pittsburgh, Steen et al.20 analyzed mortality in their SSc cohort between 1972–1996. While the proportion of deaths due to renal crisis declined from 42% to 6%, the proportion of deaths due to lung fibrosis increased from 6% of SSc-related deaths in the 1970s to > 31% of SSc-related deaths in the 1990s.20 Cumulative survival rate at 9 years with severe ILD (FVC < 55%) was ~ 30%, compared with ~ 40% with severe cardiac disease and 68% with severe renal disease after the advent of angiotensin converting enzyme inhibitors.20
Similar to the Pittsburgh experience, other series consisting of cohorts studied after the introduction of angiotensin converting enzyme inhibitors for renal crisis, consistently found ILD as a leading cause of SSc-related deaths. In a very large Italian multicenter study, Ferri and colleagues evaluated 1012 SSc patients recruited between 1955–1999 at three rheumatology specialty centers.21 The observed SSc survival rates were significantly lower than those expected in the Italian general population (p < .00001).21 Of note, patients recruited after 1985 showed a significantly better 10-year survival rate compared with subjects referred before 1985 (76.8% vs 60.6%, p < .0001).21 Significantly worse prognosis was observed in the following subgroups: diffuse cutaneous disease (p < 0.00001), male sex (p < 0.00001), presence of lung involvement (ILD or pulmonary hypertension) (p < 0.00001), presence of cardiac involvement (p < 0.00001), and presence of renal involvement (p < 0.00001).21 Multivariate analysis using a Cox proportional hazard regression model demonstrated that the mortality risk was significantly increased in male patients, in patients with diffuse cutaneous SSc, in patients with lung, heart, and kidney involvement, and in patients with high erythrocyte sedimentation rate (>25 mm/hour) evaluated at patient enrollment.21 Thirty percent of patients died during the follow-up period; the most frequent causes of death were cardiac (36%) and lung (24%) involvement, and cancer (15%).21 In contrast to historical cohorts, renal involvement was a relatively rare complication.21
Finally, more recent data from the EULAR Scleroderma Trials and Research (EUSTAR) cohort solidifies that in the current era, ILD is the leading cause of SSc-related mortality.1 Tyndall and colleagues analyzed 5860 SSc patients enrolled since 2004 from 145 centers in 28 European and 6 non-European countries.1 The mean duration of follow-up was 0.9 years per person. Patients that expired had been followed in the cohort for a mean period of 1.3 years after inclusion.1 Fifty-five percent of deaths were attributed directly to SSc and 41% to non-SSc causes (in 4% the cause of death was not assigned).1 Of the SSc-related deaths, 35% were attributed to ILD, 26% to pulmonary arterial hypertension and 26% to cardiac causes (mainly heart failure and arrhythmias).1 Independent risk factors for mortality and their hazard ratios (HR) were: proteinuria (HR 3.34), the presence of PAH based on echocardiography (HR 2.02), ILD as evidenced by pulmonary restriction (FVC < 80%; HR 1.64), dyspnea above New York Heart Association Class II (HR 1.61), DLco (HR 1.20 per 10% decrease), patient age at onset of Raynaud’s phenomenon (HR 1.30 per 10 years) and the modified Rodnan skin score (HR 1.20 per 10 score points).1
SSc-ILD morbidity and mortality outcomes from clinical trials
Follow up data from pivotal SSc clinical trials augment our understanding of long-term outcomes in SSc-ILD. Clinical trial cohorts represent unique SSc-ILD patient populations (Table 2). The nature of a clinical trial facilitates the collection of longitudinal data in a uniform manner. Participants typically have well characterized ILD and are often enrolled in clinical trials at relatively early disease stages. Moreover, all patients receive standardized treatment and equal access to care and follow up. As a result, confounding due to patient-related factors that may affect survival, such as health insurance coverage and socioeconomic status, may be less of an issue in a clinical trial compared with an observational study.
Table 2.
Mortality outcomes in recent SSc RCTs with >2 years of follow-up
| Study Name | Intervention | ILD Presence | One-year mortality % (N) | Long-term mortality % (N) |
|---|---|---|---|---|
| SLS I22 | Oral CYC for vs. Placebo for 12 months | 100% | CYC: 1% (2/79)Placebo: 5% (4/79) | CYC: 48% (38/79) |
| Placebo: 5% (4/79) | Placebo: 35% (28/79) | |||
| Median follow-up: 8 years | ||||
| SLS II26 | Oral CYC for 12 months vs. MMF for 24 months | 100% | CYC: 11% (8/73) | CYC: 22% (16/73) |
| MMF: 3% (2/69) | MMF: 20% (14/69) | |||
| Median follow up: 3.6 years | ||||
| SCOT28 | HSCT vs. IV CYC for 12 months | 97% | HSCT: | HSCT: 17% (6/36) |
| CYC: | CYC: 28% (11/39) At 4.5 years | |||
| ASTIS29 | HSCT vs. IV CYC | 87% | HSCT: 14% (11/79) | HSCT: 24% (19/79) |
| CYC: 9% (7/77) | CYC: 30% (23/77) | |||
| Median follow up of 5.8 years |
Abbreviations: CYC = cyclophosphamide; MMF = mycophenolate; HSCT = hematopoetic stem cell transplantation; IV = intravenous
Scleroderma Lung Study I
Scleroderma Lung Study (SLS) I was the first randomized controlled (RCT) to compare oral cyclophosphamide (CYC) with placebo in SSc-ILD.22 This study enrolled 158 patients (mean age 48.5 years; 59% with diffuse cutaneous SSc) with relatively early SSc (mean disease duration based on the first non-Raynaud’s symptom of SSc 3.2 years) and evidence of active alveolitis either by HRCT criteria or by inflammation in bronchoalveolar lavage (BAL) specimens. At 12 months, patients randomized to CYC experienced a modest improvement in the primary endpoint of FVC%-predicted compared with placebo (2.53%; p<0.03), as well as improvements in several key secondary endpoints: patient-reported dyspnea, total lung capacity (TLC)%-predicted, quality of life, cutaneous sclerosis, and extent of both visually-assessed and computer-assisted quantitative lung fibrosis.22,23
One year after treatment cessation, the FVC%-predicted returned to baseline in the CYC arm, and a responder analysis demonstrated loss of any significant treatment effect for both the FVC%-predicted and the TLC%-predicted.24 Moreover, a recent study demonstrated no significance difference in long-term survival in patients randomized to CYC versus placebo in SLS I.25 In this study, investigators assessed morbidity and mortality outcomes up to 12 years from the date the first patient was randomized in SLS I. After a median follow up time of 8 years for all patients, 42% of SLS I participants had died (CYC 38; Placebo 28).25 The majority of evaluable deaths were due to respiratory failure from SSc-ILD.25
In terms of morbidity outcomes, 29% of SLS I patients were started on supplemental oxygen during or after the study, while 2% underwent lung transplantation.25 Eight percent developed a malignancy during the long-term follow up period. Types/locations of malignancies included colon (N=2), anus (N=1), vulvar (N=1), prostate (N=1), sarcoma (N=1) and breast (N=1) in patients randomized to CYC, and lung (N=3), colon (N=1), esophageal (N=1) and Hodgkin’s lymphoma (N=1) in patients randomized to placebo.25
Scleroderma Lung Study (SLS) II
SLS II was a RCT comparing 24 months of mycophenolate mofetil (MMF) with 12 months of oral CYC followed by 12 months of placebo in 142 patients with SSc-ILD (mean age 52.3 years; 59% diffuse cutaneous disease; mean disease duration 2.6 years).26 The entry criteria and investigating centers were nearly identical in SLS I and II. The results demonstrated that there was no difference in the course of the FVC%-predicted over 24 months in patients assigned to MMF versus CYC. Both treatment arms experienced significant improvements in their FVC%-predicted over the course of the trial (3.0% for CYC, 3.3% for MMF; p<0.05 for within-treatment group comparison with baseline), as well as significant improvements in dyspnea, cutaneous sclerosis, and quantitative extent of radiographic fibrosis and ILD.27 (Goldin 2018). However, there were no significant between-treatment differences in any of the outcome measures with the exception of the DLCO (favored MMF)26.
Up to 8 years after the first patient was randomized in SLS II, 21% of participants had died (CYC: 16; MMF: 14).25 Similar to SLS I, the majority of deaths were attributable to respiratory failure due to SSc (median follow up time for all participants was 3.6 years).25 In terms of morbidity outcomes, 8% of SLS I patients were started on supplemental oxygen during or after the study, while only 1 patient underwent lung transplantation.25 Two SLS II participants developed malignancies during the follow-up period (MMF: N=1 thyroid cancer, N=1 papillary urothelial carcinoma; CYC: None).25
Taken together, the results of SLS I and II demonstrate that even when patients are treated early and aggressively with immunosuppressive therapy for their SSc-ILD, disease progression occurs in a subgroup of patients and ILD remains the leading cause of death.
Hematopoetic Stem Cell Transplantation (HSCT) Trials
Recent clinical trials evaluating the safety and efficacy of HSCT for SSc have also yielded important data on morbidity and mortality outcomes in SSc-ILD,28,29 as the majority of patients in these landmark trials had underlying ILD (Table 2). With subtle differences in eligibility criteria and the transplant protocol between these studies, the results suggest that intervention with HSCT was consistently associated with improved overall and progression-free survival, as well as skin score, lung function and other indicators of disease activity.28,29
For example, in the SCOT study, 36 patients were randomized to HSCT and 39 were randomized to CYC. ILD was present in 100% of the patients in the HSCT arm and 95% of the patients in the CYC arm.28 At 54 months, 9% and 24% of patients in the HSCT and CYC, arms, respectively, had died (due to any cause).28 In the per-protocol analysis, event-free survival at 54 months and 72 months was 79%/50% and 74%/47%, respectively in the HSCT/CYC arms.28 Moreover, respiratory failure was the most common type of organ failure (5 participants in the HSCT arm and 13 in the CYC arm), and the most frequent cause of death in the CYC arm.28
In the ASTIS trial, 79 patients were randomized to HSCT and 77 patients were randomized to CYC.29 Similar to the SCOT trial, the majority of study patients had underlying ILD (HSCT: 86%, CYC: 87%).29 The all-cause mortality rate in this trial was 24% and 30% of patients in the HSCT and CYC arms, respectively, over a median follow-up time of 5.8 years.29 However, over the course of 4 years, patients randomized to HSCT experienced improved long-term event-free survival compared to those randomized to CYC (p = 0.03).29
The aforementioned trials have demonstrated that HSCT is a viable option for patients with SSc-ILD. While there does appear to be a heightened risk of mortality within the first year of HSCT, the long-term mortality rate appears comparable to other interventional studies (e.g., SLS I and II). However, all of these RCTs illustrate that despite aggressive and early treatment for SSc, mortality is still high and more research is needed to develop safe and effective disease-modifying therapies for this condition.
Lung transplantation in SSc-ILD
An increasing number of patients with SSc-ILD are being referred for lung transplantation (LT). Studies have demonstrated comparable survival rates between SSc patients and non-SSc patients following LT for ILD.30 A systematic review reported data from 7 observational studies (Total N from all studies was 96 patients) investigating SSc post-LT outcomes and demonstrated that survival in these studies ranged from 59–93% at 1-year, 49–80% at 2-years, and 46–79% at 3 years.31 The range of survival rates was likely due to the different inclusion/exclusion criteria for LT at the various study centers. The causes of death included graft failure, infection, cardiac events, hemorrhagic stroke, respiratory failure, malignancy, pulmonary hypertension, complications of bronchiolitis obliterans syndrome, scleroderma renal crisis, and anesthetic complication.31
Notably, none of the aforementioned studies reported recurrence of SSc in the lung allograft. Since a relatively small number of patients with SSc-ILD undergo LT, it is unclear how this intervention affects overall survival rates for SSc-ILD. However, for the individual patient, LT can be a life-saving intervention.
Predicting mortality in SSc-ILD
A number of studies have examined predictors of mortality in unselected, retrospective and prospective SSc cohorts with patients followed for relatively short time intervals.1,20,32–36 While these studies have shed light on important prognostic factors for SSc patients in general, many of these studies included patients who developed SSc over 20 years ago. Given the changes in the medical management of SSc-ILD in the past decade,37 it is unclear whether these study findings are relevant in the modern ILD treatment era. Furthermore, these prognostic factors may or may not affect mortality in an SSc patient with clinically significant ILD. Fewer studies have examined predictors of mortality in SSc patients with well-characterized ILD.37 Among studies in SSc-ILD specifically, demographic factors, disease-specific features of SSc, severity and progression of ILD, as well as candidate biomarkers, have been found to predict mortality in SSc-ILD (Table 3; Figure 2).
Table 3.
Clinical and biological factors associated with increased mortality in SSc-ILD*
| Factor | Effect on Mortality |
|---|---|
| Male sex38,39,46 | Increased risk of mortality |
| African American race7,40 | Potential increased risk of mortality |
| Increased age25,38,39,42 | Consistently found to increase risk of mortality |
| Anti-topoisomerase I antibodies43,44 | Increased risk of mortality |
| Diffuse cutaneous sclerosis43,59 | Increased risk of mortality |
| Greater extent of ILD on HRCT imaging39,46 | Consistently found to increase risk of mortality |
| Early progression of ILD based on pulmonary physiology25,42,45 | An early decline in lung function is associated with an increased risk of mortality in both clinical trial25 and observational cohorts42,45 |
| Baseline FVC7,46,60 | Lower FVC is associated with an increased risk of mortality |
| Baseline DLCO46,60 | Lower DLCO is associated with an increased risk of mortality |
| Presence of PH or PAH46,59 | Increased risk of mortality |
| C-reactive protein (CRP)52,60 | High levels associated with an increased mortality in a single-center, prospective cohort (N=266)52 |
| High levels associated with an increased mortality in a single-center, prsospective cohort (N=75)60 | |
| CC chemokine ligand 18 (CCL18)55,56 | Increased mortality in a single-center, prospective cohort (N=96)55 |
| Increased mortality in 14-center, clinical trial cohort among patients randomized to CYC (N=71)56 |
This table only includes mortality predictor variables that have been identified in at least two SSc-ILD studies, which used HRCT to define the presence of ILD.
Figure 2. Predictors of mortality in SSc-ILD.
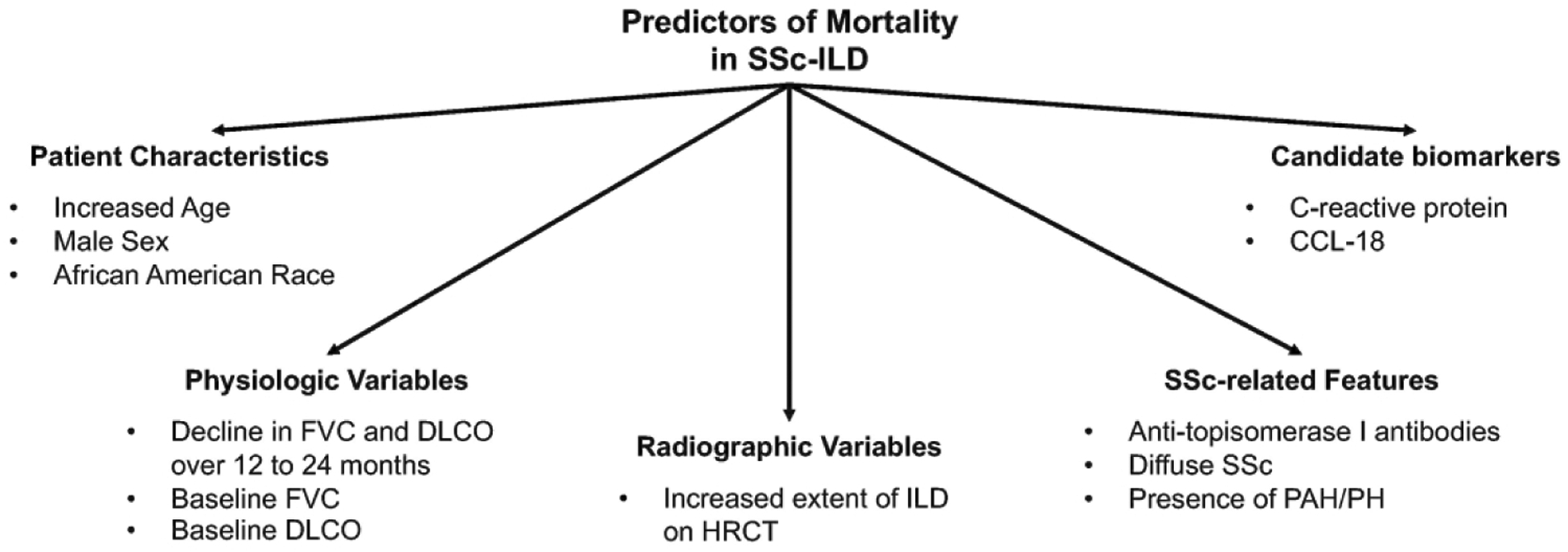
This figure describes the key factors associated with mortality in SSc-ILD. This figure was adapted and modified from the systematic review by Winstone et al.61 to include only those predictor variables, which have been found to predict mortality in two independent SSc-ILD cohorts, which used HRCT to define the presence of ILD.
Demographic factors
Specific demographic characteristics are associated with increased mortality in patients with SSc-ILD. Male sex is associated with increased mortality in observational SSc-ILD studies.38,39 African American race has also been linked with worse survival in patients with SSc-ILD in observational studies.7,40 However, a recent study suggests that socioeconomic disparities may underlie some of the racial differences observed in SSc-ILD observational studies.40 Indeed, an analysis of the SLS I and II cohorts demonstrated no difference in long-term survival between African Americans and non-African American patients who participated in these two trials.41 Finally, older age consistently predicts poor outcomes in SSc-ILD in both observational studies38,39,42 and in clinical trial cohorts.25
Disease-specific factors
Certain disease features of SSc are associated with increased risk of mortality in SSc-ILD, including the presence of the anti-topiosomerase I antibody,43.44 diffuse cutaneous disease,43 and greater extent of ILD on HRCT imaging.39,45,46 A small study in patients with limited cutaneous SSc (N=22) found that those with biopsy-proven nonspecific interstitial pneumonia (NSIP) had improved survival compared to usual interstitial pneumonia (UIP) (15 years versus 3 years).47 However, a subsequent study did not detect an increase in mortality in SSc-ILD with a UIP pattern, but this study used radiographic criteria to define UIP.39
A number of prior observational studies have identified factors associated with SSc-ILD progression, which may in turn diminish survival. These factors include: low FVC,7,43 shorter total disease duration,7 and low DLCO.43,44,48,49 Historical studies identified cigarette smoking as predictor of ILD progression.8,50,51
ILD Progression
Recent data from observational cohorts42,45 and clinical trials25 demonstrate that early progression of ILD independently predicts mortality. Goh and colleagues42 demonstrated that pulmonary function trends at 1- and 2-years predicted both intermediate and long-term mortality in 140 patients followed at a single center over 15 years. Specifically, a FVC decline from baseline of ≥10% or the composite an FVC decline of 5–9% in association with a DLCO decline of ≥15% at 1 year was the most accurate predictor of mortality in patients with extensive ILD.42 At 2 years, a decline in the diffusing capacity coefficient (KCO) of ≥10% was the single best independent predictor of mortality in SSc patients with both limited and extensive ILD based on the criteria established by the same group.46 Since this was an observational study, the investigators could not evaluate treatment effects and their impact on survival.
These findings are consistent with an earlier study by Moore and colleagues45 of 264 SSc patients with HRCT-defined ILD. In this observational cohort, the rates of decline and the percentage change in the FVC, DLCO and KCO predicted 1-year adverse outcomes (defined as death [N=38], initiation of supplemental oxygen [N=10], or lung transplantation [N=1]).45 Specifically, a decline in the FVC of 10% and in the DLCO and KCO of 15% yielded the most optimal sensitivity and specificity for predicting adverse outcomes with a negative predictive value of 92–93%.45
Volkmann and colleagues25 tracked survival in 300 patients who participated in SLS I and II and found that the course of the FVC and DLCO in the first 2 years was a better predictor of survival than the baseline FVC or DLCO.25 Patients who experienced an improvement in their FVC and DLCO in response to immunosuppressive therapy had better survival than those who experienced worsening of these pulmonary function parameters, regardless of baseline disease severity.25 Since this study evaluated patients who participated in a RCT, the investigators were able to control for treatment arm assignment and explore interactions between treatment arm and ILD progression.
Candidate biomarkers
Select peripherally-measured proteins have been associated with mortality in SSc-ILD. For example, in a single-center observational study of 266 SSc patients (44% with underlying ILD), higher baseline C-reactive protein (CRP) levels were associated with increased progression of ILD based on FVC decline, as well as increased mortality.52 Other candidate biomarkers associated with mortality in SSc-ILD include interleukin (IL)-6,53 chemoattractant protein-1 (MCP-1),54 and CCL-18.55,56 The aforementioned studies were all single-center, observational cohort studies with patients on different/no ILD therapies and with varying follow up. Further research to identify reliable, reproducible and affordable serologic biomarkers in SSc-ILD is greatly needed, and clinical trial cohorts may offer unique research opportunities to evaluate whether specific biomarker assessment can improve the prediction of ILD progression or prognostication.
Future research on morbidity and mortality outcomes in SSc-ILD
Using SSc-ILD mortality prediction models for precision medicine
Across disease states, health outcomes improve when evidence-based prediction tools are used to personalize the care of patients. This approach is directly relevant and of paramount importance in SSc-ILD, a disease with varying progression and therapeutic response rates. Personalizing the care of patients with SSc-ILD, may begin with the identification and characterization of specific SSc-ILD subgroups based on disease progression. Specifically, studies are needed to develop valid morbidity and mortality SSc-ILD prediction tools that are reproducible across diverse SSc-ILD populations.
The factors which most consistently predict mortality across various SSc-ILD cohorts include increased age, diffuse cutaneous disease, increased ILD disease severity at baseline, as well as increased progression of ILD early in the disease course. While no valid mortality prediction models exist for SSc-ILD, a recent study examined mortality prediction in an SSc-ILD cohort from a single center (N=156) using mortality risk models for idiopathic pulmonary fibrosis (IPF).57 This study assessed the Composite Physiologic Index (CPI: FVC, FEV1, DLCO), Interstitial Lung Disease- Gender, Age, Physiology Index (ILD-GAP index: Sex, Age, FVC, DLCO), du Bois index (Age, Respiratory hospitalization, FVC, change in FVC), and the modified du Bois index (Age, Respiratory hospitalization, FVC, change in FVC, 6-minute walk distance [6MWD], change in 6-minute walk distance [6MWD]). Among these prediction models, the modified du Bois index yielded the best discrimination and calibration for the prediction of 1-year mortality in SSc.57
Future studies are needed to expand upon this work and develop SSc-specific ILD mortality prediction tools to help identify patients at highest risk for mortality not just at 1-year, but at later time points in the disease course. It is critical to identify those patients who are at greatest risk for disease progression to stratify high risk patients for aggressive ILD therapy and consider referral for HSCT at an early disease stage.
Will novel SSc-ILD therapeutics affect morbidity and mortality outcomes?
Future research efforts in this area should also aim to understand how novel SSc-ILD therapeutics affect mortality. The therapeutic pipeline for SSc-ILD has burgeoned in recent years.58 More clinical trials are being conducted in SSc-ILD than ever before and in addition to evaluating the safety and efficacy of specific agents there is also need to investigate their longer term impact on quality of life and mortality.
Conclusion
Data from observational and clinical trial cohorts have both informed and broadened our understanding of morbidity and mortality outcomes in SSc-ILD. Recent reports suggest that outcomes for SSc-ILD patients have improved in recent years, although it is challenging to compare survival rates from prior studies since the design of historical studies differed considerably from present day studies (i.e., ILD detection and characterization method; study duration; study entry criteria; use of a selected or unselected population; use of prevalent or inception cohorts). Furthermore, unique differences undoubtedly exist between SSc-ILD patients who participate in RCTs and those consecutively enrolled in registry studies.
Data from both historical and modern-day studies have consistently revealed a number of poor prognostic factors in SSc-ILD: increased age, diffuse cutaneous disease, increased ILD disease severity at baseline, as well as increased progression of ILD early in the SSc disease course. Factors which do not consistently predict mortality outcomes across different SSc-ILD populations include male gender, African American race, anti-topoisomerase I antibody status, disease duration, specific ILD pattern (e.g., UIP versus NSIP).
Understanding the factors that predict morbidity and mortality outcomes for SSc-ILD will hopefully allow us to offer a more personalized approach to the care of these patients. The development of a modern day SSc-ILD mortality prediction tool could help clinicians identify patients who may benefit from early, aggressive ILD therapy and possible referral for HSCT. With the potential advent of novel therapeutics in SSc-ILD on the horizon, the assessment of meaningful long-term clinical outcomes, such as quality of life and survival will be important.
Acknowledgement
ERV receives funding from the Rheumatology Research Foundation.
References
- 1.Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69: 1809–15. [DOI] [PubMed] [Google Scholar]
- 2.Rubio-Rivas M, Royo C, Simeón CP, et al. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum 2014; 44: 208–19. [DOI] [PubMed] [Google Scholar]
- 3.Volkmann ER, Tashkin DP. Treatment of systemic sclerosis-related interstitial lung disease: A review of existing and emerging therapies. Ann Am Thorac Soc 2016; 13: 2045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. 1969; 46: 428–40. [DOI] [PubMed] [Google Scholar]
- 5.Weaver AL, Divertie MB, Titus JL. Pulmonary scleroderma. Dis Chest 1968; 54: 490–8. [DOI] [PubMed] [Google Scholar]
- 6.Schurawitzki H1, Stiglbauer R, Graninger W, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology 1990; 176: 755–9. [DOI] [PubMed] [Google Scholar]
- 7.Steen VD, Conte C, Owens GR, Medsger TA Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum 1994; 37: 1283–1289. [DOI] [PubMed] [Google Scholar]
- 8.Steen VD, Owens GR, Fino GJ, et al. Pulmonary involvement in systemic sclerosis (scleroderma). Arthritis Rheum 1985; 28: 759–67. [DOI] [PubMed] [Google Scholar]
- 9.McNearney TA, Reveille JD, Fischbach M, et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum 2007; 57: 318–26. [DOI] [PubMed] [Google Scholar]
- 10.Greidinger EL, Flaherty KT, White B, et al. African-American race and antibodies to topoisomerase I are associated with increased severity of scleroderma lung disease. Chest 1998; 114: 801–7. [DOI] [PubMed] [Google Scholar]
- 11.Briggs DC, Vaughan RW, Welsh KI, et al. Immunogenetic prediction of pulmonary fibrosis in systemic sclerosis. Lancet 1991; 338: 661–2. [DOI] [PubMed] [Google Scholar]
- 12.Steele R, Hudson M, Lo E, Baron M; Canadian Scleroderma Research Group. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res (Hoboken) 2012; 64: 519–24. [DOI] [PubMed] [Google Scholar]
- 13.Morelli S, Barbieri C, Sgreccia A, et al. Relationship between cutaneous and pulmonary involvement in systemic sclerosis. J Rheumatol 1997; 24: 81–5. [PubMed] [Google Scholar]
- 14.Launay D, Remy-Jardin M, Michon-Pasturel U, et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J Rheumatol 2006; 33: 1789–801. [PubMed] [Google Scholar]
- 15.Morgan C, Knight C, Lunt M, et al. Predictors of end stage lung disease in a cohort of patients with scleroderma. Ann Rheum Dis 2003; 62: 146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett R, Bluestone R, Holt PJ, Bywaters EG. Survival in scleroderma. Ann Rheum Dis 1971; 30: 581–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medsger TA Jr, Masi AT. Survival with scleroderma. II. A life-table analysis of clinical and demographic factors in 358 male U.S. veteran patients. J Chronic Dis 1973; 26: 647–60. [DOI] [PubMed] [Google Scholar]
- 18.Altman RD, Medsger TA Jr, Bloch DA, Michel BA. Predictors of survival in systemic sclerosis (scleroderma). Arthritis Rheum 1991; 34: 403–13. [DOI] [PubMed] [Google Scholar]
- 19.Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43: 2437–44. [DOI] [PubMed] [Google Scholar]
- 20.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66: 940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81: 139–53. [DOI] [PubMed] [Google Scholar]
- 22.Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006; 354: 2655–66. [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Brown MS, Elashoff R, et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. Eur Radiol 2011; 21: 2455–2465 [DOI] [PubMed] [Google Scholar]
- 24.Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med 2007; 176: 1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volkmann ER, Tashkin DP, Sim M, et al. Early progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Annals of Rheumatic Diseases 2019; 78: 122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease: Scleroderma lung study II (SLS-II), a double-blind, parallel group, randomised controlled trial. Lancet Respiratory Medicine 2016; 4: 708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldin JG, Kim GH, Volkmann ER, et al. Longitudinal changes in quantitative lung disease on CT after immunosuppression in the Scleroderma Lung Study II. Ann Am Thorac Soc 2018; 15: 1286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. for the SCOT Study Investigators. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med 2018; 378: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Laar JM, Farge D, Sont JK, et al. ; EBMT/EULAR Scleroderma Study Group. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014; 311: 2490–8. [DOI] [PubMed] [Google Scholar]
- 30.Crespo MM, Bermudez CA, Dew MA, et al. Lung Transplant in Patients with Scleroderma Compared with Pulmonary Fibrosis. Short- and Long-Term Outcomes. Ann Am Thorac Soc 2016; 13: 784–92. [DOI] [PubMed] [Google Scholar]
- 31.Khan IY, Singer LG, de Perrot M, et al. Survival after lung transplantation in systemic sclerosis. A systematic review. Respir Med 2013; 107: 2081–7. [DOI] [PubMed] [Google Scholar]
- 32.Mayes MD. Scleroderma epidemiology. Rheum Dis Clin North Am 2003; 29: 239–54. Mayes MD1. [DOI] [PubMed] [Google Scholar]
- 33.Elhai M, Meune C, Boubaya M, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis 2017; 76: 1897–1905. [DOI] [PubMed] [Google Scholar]
- 34.Domsic RT, Nihtyanova SI, Wisniewski SR, et al. Derivation and validation of a prediction rule for two-year mortality in early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol 2014; 66: 1616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beretta L, Santaniello A, Cappiello F, et al. Development of a five-year mortality model in systemic sclerosis patients by different analytical approaches. Clin Exp Rheumatol 2010; 28: S18–27. [PubMed] [Google Scholar]
- 36.Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61: 1403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volkmann ER, Chung A, Tashkin DP. Managing systemic sclerosis-related interstitial lung disease in the modern treatment era. J Scleroderma & Related Disorders 2017; 2: 72–83. [Google Scholar]
- 38.Volkmann ER, Saggar R, Khanna D, et al. Improved transplant-free survival in patients with systemic sclerosis-related pulmonary arterial hypertension plus interstitial lung disease. Arthritis and Rheumatol 2014; 66: 1900–8. [DOI] [PubMed] [Google Scholar]
- 39.Takei R, Arita M, Kumagai S, et al. Radiographic fibrosis score predicts survival in systemic sclerosis-associated interstitial lung disease. Respirology 2018; 23: 385–391. [DOI] [PubMed] [Google Scholar]
- 40.Moore DF, Kramer E, Eltaraboulsi R, Steen VD. Increased Morbidity and Mortality of Scleroderma in African Americans Compared to Non-African Americans. Arthritis Care Res (Hoboken). Epub ahead of print 1 March 2019. DOI: 10.1002/acr.23861. [DOI] [PubMed] [Google Scholar]
- 41.Volkmann ER, Ning L, Khanna D, et al. Long-term outcomes of African American patients with systemic sclerosis-related interstitial lung disease [Abstract]. Ann Rheum Dis 2019; 78: 1808. [Google Scholar]
- 42.Goh NS, Hoyles RK, Denton CP, et al. Short-Term Pulmonary Function Trends Are Predictive of Mortality in Interstitial Lung Disease Associated With Systemic Sclerosis. Arthritis Rheumatol 2017; 69: 1670–1678. [DOI] [PubMed] [Google Scholar]
- 43.Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol 2014; 66: 1625–35. [DOI] [PubMed] [Google Scholar]
- 44.Assassi S, Sharif R, Lasky RE, et al. Predictors of interstitial lung disease in early systemic sclerosis: a prospective longitudinal study of the GENISOS cohort. Arthritis Res Ther 2010; 12: R166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore OA, Proudman SM, Goh N, et al. Quantifying change in pulmonary function as a prognostic marker in systemic sclerosis-related interstitial lung disease. Clin Exp Rheumatol 2015; 33: S111–6 [PubMed] [Google Scholar]
- 46.Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med 2008; 177: 1248–54. [DOI] [PubMed] [Google Scholar]
- 47.Fischer A, Swigris JJ, Groshong SD, et al. Clinically significant interstitial lung disease in limited scleroderma: histopathology, clinical features, and survival. Chest 2008; 134: 601–605. [DOI] [PubMed] [Google Scholar]
- 48.Bryan C, Knight C, Black CM, Silman AJ. Prediction of five-year survival following presentation with scleroderma: development of a simple model using three disease factors at first visit. Arthritis Rheum 1999; 42: 2660–5. [DOI] [PubMed] [Google Scholar]
- 49.Morgan C, Knight C, Lunt M, et al. Predictors of end stage lung disease in a cohort of patients with scleroderma. Ann Rheum Dis 2003; 62: 146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenwald GI, Tashkin DP, Gong H, et al. Longitudinal changes in lung function and respiratory symptoms in progressive systemic sclerosis. Prospective study. Am J Med 1987; 83: 83–92. [DOI] [PubMed] [Google Scholar]
- 51.Peters-Golden M, Wise RA, Schneider P, et al. Clinical and demographic predictors of loss of pulmonary function in systemic sclerosis. Medicine (Baltimore) 1984; 63: 221–31. [DOI] [PubMed] [Google Scholar]
- 52.Liu X, Mayes MD, Pedroza C, et al. Does C-reactive protein predict the long-term progression of interstitial lung disease and survival in patients with early systemic sclerosis? Arthritis Care Res 2013; 65: 1375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol 2013; 40: 435–46. [DOI] [PubMed] [Google Scholar]
- 54.Wu M, Pedroza C, Salazar G, Zhou X, Reveille J, Mayes MD, Assassi S. Plasma MCP-1 and IL-10 levels predict long-term progression of interstitial lung disease in patients with early systemic sclerosis [Abstract]. Arthritis Rheum 2013;65:S742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schupp J, Becker M, Günther J, et al. Serum CCL18 is predictive for lung disease progression and mortality in systemic sclerosis. Eur Respir J 2014; 43: 1530–2. [DOI] [PubMed] [Google Scholar]
- 56.Volkmann ER, Tashkin DP, Masa K, et al. Specific pneumoproteins predict progression of interstitial lung disease in systemic sclerosis patients undergoing treatment with immunosuppression. Accepted to Arthritis and Rheumatology 2019. [Google Scholar]
- 57.Ryerson CJ, O’Connor D, Dunne JV, et al. Predicting Mortality in Systemic Sclerosis-Associated Interstitial Lung Disease Using Risk Prediction Models Derived From Idiopathic Pulmonary Fibrosis. Chest 2015; 148: 1268–1275. [DOI] [PubMed] [Google Scholar]
- 58.Volkmann ER, Varga J. Emerging treatments for systemic sclerosis. Nature Rev Rheumatology 2019; 15: 208–24. [DOI] [PubMed] [Google Scholar]
- 59.De Santis M, Bosello SL, Peluso G, et al. Bronchoalveolar lavage fluid and progression of scleroderma interstitial lung disease. Clin Respir J 2012; 6: 9–17. [DOI] [PubMed] [Google Scholar]
- 60.Le Gouellec N , Faivre JB , Hachulla AL, et al. Prognosis factors for survival and progression-free survival in SSc associated interstitial lung disease [Abstract]. Rheumatology . 2012; 51(suppl 2): ii82. [Google Scholar]
- 61.Winston TA, Assayag D, Wilcox PG, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: A systematic review. Chest 2014; 146: 422–36. [DOI] [PubMed] [Google Scholar]