

Abstract
Objective
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer with limited treatment options. Cabozantinib, an orally bioavailable multikinase inhibitor is now approved by Food and Drug Administration (FDA) for HCC patients. We evaluated the therapeutic efficacy of cabozantinib, either alone or in combination, in vitro and in vivo.
Design
Human HCC cell lines and HCC mouse models were used to assess the therapeutic efficacy and targeted molecular pathways of cabozantinib, either alone or in combination with the pan-mTOR inhibitor MLN0128 or the checkpoint inhibitor anti-PD-L1 antibody.
Results
Cabozantinib treatment led to stable disease in c-Met/β-catenin and Akt/c-Met mouse HCC while possessing limited efficacy on Akt/Ras and c-Myc liver tumours. Importantly, cabozantinib effectively inhibited c-MET and ERK activity, leading to decreased PKM2 and increased p21 expression in HCC cells and in c-Met/β-catenin and Akt/c-Met HCC. However, cabozantinib was ineffective in inhibiting the Akt/mTOR cascade. Intriguingly, a strong inhibition of angiogenesis by cabozantinib occurred regardless of the oncogenic drivers. However, cabozantinib had limited impact on other tumour microenvironment parameters, including tumour infiltrating T cells, and did not induce programmed death-ligand 1 (PD-L1) expression. Combining cabozantinib with MLN0128 led to tumour regression in c-Met/β-catenin mice. In contrast, combined treatment with cabozantinib and the checkpoint inhibitor anti-PD-L1 antibody did not provide any additional therapeutic benefit in the four mouse HCC models tested.
Conclusion
c-MET/ERK/p21/PKM2 cascade and VEGFR2-induced angiogenesis are the primary targets of cabozantinib in HCC treatment. Combination therapies with cabozantinib and mTOR inhibitors may be effective against human HCC.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fourth leading cause of cancer-related deaths world-wide.1 The treatment options for advanced HCCs are instead very limited.2 Sorafenib has been the standard first-line therapy against HCC but shows limited efficacy. During the past years, the treatment landscape for advanced HCC has been significantly expanded with the approval of multiple drugs, including cabozantinib, for HCC treatment.3
Cabozantinib is an orally bioavailable multikinase inhibitor that targets receptor tyrosine kinases including c-MET, VEGFR2, AXL and c-KIT.4 c-MET plays a critical role in HCC development and progression.5 Overexpression of c-MET is found in 20%–48% human HCC samples,6 where it promotes HCC proliferation, survival, invasiveness and angiogenesis as well as resistance to chemotherapy and radiotherapy.5,7 The oncogenic potential of c-MET in hepatocarcinogenesis has been proven in mice, as c-Met transgenic mice develop HCC.8 Further studies suggest that overexpression of c-Met alone via hydrodynamic transfection is unable to induce HCC formation in vivo,9 but it cooperates with other genes, such as activated Wnt/β-catenin or Akt/mTOR cascades, to trigger hepatocarcinogenesis in mice.10,11
In a phase III clinical trial for advanced HCC patients who have previously received sorafenib treatment, it was found that cabozantinib significantly improved the overall survival rate as well as progression-free survival when compared with placebo.12 This led to the recent approval of cabozantinib as a second-line treatment for progressed HCC.3 In a preclinical study, it was found that cabozantinib suppresses HCC cell growth and metastasis by the combined blockade of VEGFR and c-MET proteins.13 However, the study was conducted using only HCC cell lines, which may have relatively limited value in recapitulating drug responsiveness in vivo.
In the present manuscript, we investigated the therapeutic efficacy of cabozantinib, either alone or in combination with the pan-mTOR inhibitor MLN0128 or the checkpoint inhibitor anti-PD-L1 antibody in in vitro and in vivo models of HCC.
MATERIALS AND METHODS
Plasmids and reagents
The plasmids used in this study, including pT3-EF1α-c-Met (human c-MET), pT3-EF1α-ΔN90-β-catenin (with N terminal Myc tag), pT3EF1α-c-Myc, pT3-EF1α-HA-myr-Akt (with N terminal HA tag), pT2CAGGS-NRasV12 and pCMV/sleeping beauty transposase (SB), have been described previously in detail.14,15 Cabozantinib and MLN0128 were purchased from LC Laboratories (Woburn, Massachusetts, USA). Anti-mouse PD-L1 Ab was obtained from Genentech (South San Francisco, California, USA).
Hydrodynamic injection and mouse treatment
Female wild-type FVB/N mice were obtained from Charles River Laboratories (Wilmington, Massachusetts, USA). SB-mediated hydrodynamic injection was performed as previously described.16 Cabozantinib, MLN0128, cabozantinib+MLN0128 or vehicle was orally administered via gavage. Anti-mouse PD-L1 Ab (or isotype IgG) was intraperitoneally injected into mice as illustrated in the figures.
Statistical analysis
GraphPad Prism V.6.0 (GraphPad Software Inc) was used to analyse the data. Statistical analyses were conducted using Student’s t-test, Tukey-Kramer test and linear regression analyses. Data are presented as mean±SD. Values of p<0.05 were considered statistically significant.
Additional information is available in supporting information.
RESULTS
Cabozantinib inhibits HCC cell growth in vitro
First, we treated a panel of 14 HCC cell lines with cabozantinib (online supplemental figure 1). The IC50 value of cabozantinib for each cell line was calculated (online supplemental figure 2A), and the potential targets of cabozantinib in all HCC cell lines were evaluated using western blot analysis (online supplemental figure 2B). We found that phosphorylated/activated (p)-MET, total (t)-AXL and p-ERK1/2 levels demonstrated a statistically significant negative correlation with cabozantinib IC50, whereas other proteins, such as total c-MET, did not show any significant correlation (online supplemental figure 2C).
To elucidate the pathways regulated by cabozantinib, two HCC cell lines with robust activation of c-MET, namely MHCC97-H and HLE cells, were treated with cabozantinib at ~IC50 values for 24 hours. BrdU labelling studies revealed that cabozantinib strongly suppressed HCC cell cycle progression (online supplemental figure 3). This was the consequence of reduced proliferation as well as induction of apoptosis in both cell lines (online supplemental figure 4). Cabozantinib treatment suppressed p-MET, p-AXL and p-ERK1/2 expression (online supplemental figure 5A), whereas it did not inhibit the AKT/mTOR pathway and its downstream targets, including p-S6 and lipogenic proteins (FASN and ACAC) (online supplemental figure 5A,D). Cabozantinib decreased the expression of PCNA, Cyclin D1 and Cyclin E proteins, while it increased p21 levels, in HCC cells (online supplemental figure 5B). Interestingly, cabozantinib treatment led to decreased PKM2 protein levels (online supplemental figure 5D), suggesting that it might modulate tumour glycolysis while not affecting lipogenesis. Consistently, we found that cabozantinib reduced glucose uptake (a sign of glycolysis), but not triglycerides and cholesterol levels, in the two cell lines (online supplemental figure 6). Moreover, we found downregulation of the antiapoptotic protein survivin, which was paralleled by increase of the marker of apoptosis cleaved caspase 3, in accordance with a promoting role of programmed cell death by cabozantinib (online supplemental figure 5C).
In summary, our data suggest that HCC cells have variable sensitivity to cabozantinib by suppressing c-MET/p-ERK cascade.
Cabozantinib treatment leads to stable disease in c-Met/β-catenin and Akt/c-Met mouse lesions but has no efficacy on Akt/Ras or c-Myc mouse tumours
As HCC is a highly heterogeneous disease, we attempted to model its heterogeneity by using various combinations of oncogenes. We chose four different murine HCC models, which overexpress the following gene combinations: c-Met/β-catenin,10 Akt/c-Met,11 Akt/Ras17 and c-Myc.15 All these models have been extensively validated by our group as well as by independent investigations to be relevant to subsets of human HCC.10,11,14,15 First, we analysed the expression levels of major signalling molecules in the late stage HCC tissues from these mice together with normal liver from uninjected mice. We found that c-Met protein could be readily detected in all four liver tumour samples (online supplemental figure 7). However, p-Met could only be observed in c-Met/β-catenin and Akt/c-Met mouse HCCs. p-Akt was found to be expressed at higher levels in all HCC samples compared with normal livers, whereas p-Erk1/2 expression was found to be elevated in c-Met/β-catenin, Akt/c-Met and Akt/Ras HCCs, but not c-Myc HCC (online supplemental figure 7).
Subsequently, we used these mouse models to investigate the therapeutic potential of cabozantinib in vivo. Based on the published studies,18,19 the dose of cabozantinib used was 60 mg/kg/day. Our preliminary studies using wild-type mice showed that cabozantinib treatment at this dosage was well tolerated in mice over the course of 13 days of treatment (online supplemental figure 8A), with similar serum TP, ALB, ALKP and AST levels, and moderately higher ALT levels (online supplemental figure 8B), when compared with control mice. For the efficacy studies, mice were hydrodynamically injected with plasmids encoding the aforementioned oncogene(s) and aged until they had moderate liver tumour burden (online supplemental figure 9). Due to the diffused nature of our murine HCC models, it was difficult to accurately measure tumour nodule number. Thus, we chose to use total liver weight and percentage of microscopic tumour area as the measurement of burden. Mice were randomly separated into three groups. Specifically, mice in group 1 were harvested as pretreatment cohort and displayed a liver weight between 3 and 5 g, presenting moderate liver tumour burden. Group 2 mice were instead treated with the vehicle, whereas group 3 mice were treated with cabozantinib at 60 mg/kg/day, as previously described.20 Mice were harvested if they developed large abdominal masses and were required to be euthanised based on our institutional animal care and use committee (IACUC) protocol or after 3 weeks of treatment (online supplemental figure 9).
We treated c-Met/β-catenin, Akt/c-Met, Akt/Ras and c-Myc tumour-bearing mice with vehicle or cabozantinib (figures 1A–4A). In c-Met/β-catenin and Akt/c-Met mouse models, all vehicle treated mice had to be euthanised during the treatment course, whereas cabozantinib treated mice appeared to be healthy (figures 1B and 2B). Based on the liver weight measurement, vehicle treated mice had significant higher tumour burden than the pretreatment mice, consistent with tumour progression in the vehicle cohort. In contrast, the cabozantinib cohort demonstrated significantly lower tumour burden than that of vehicle-treated mice and a similar tumour burden to the pretreatment mice (figures 1C and 2C). Similar results were obtained if we used percentage tumour area as the measurement of tumour burden (online supplemental figure 10A,B). The data suggest that cabozantinib treatment leads to stable disease in c-Met/β-catenin and Akt/c-Met mice. In contrast, in Akt/Ras and c-Myc liver tumour models, cabozantinib treatment did not affect Akt/Ras or c-Myc mouse survival rate, and tumour progression occurred in both vehicle and cabozantinib treated mice (figures 3B and 4B), as also indicated by the liver tumour burden, analysed using total liver weight (figures 3C and 4C) and percentage tumour area (online supplemental figure 10C,D). Thus, cabozantinib treatment led to progressive disease in Akt/Ras and c-Myc liver tumour models.
Figure 1.
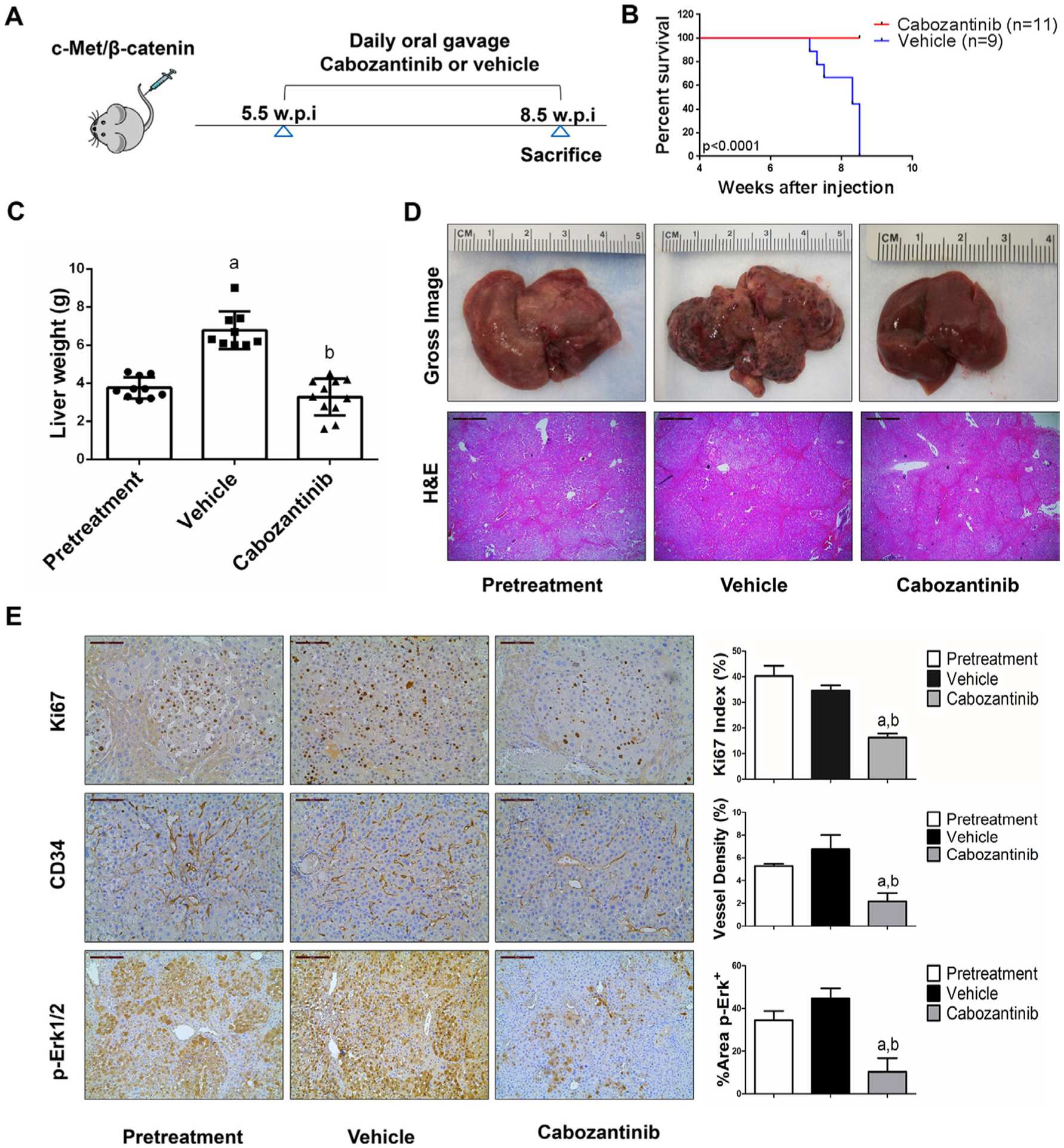
Cabozantinib treatment induces stable disease in c-Met/β-catenin mice. (A) Scheme of the experiment. (B) Survival curve of vehicle-treated and cabozantinib-treated c-Met/β-catenin mice. (C) Liver weight of pretreatment, vehicle-treated and cabozantinib-treated c-Met/β-catenin mice. (D) Gross images and H&E staining of livers from pretreatment, vehicle-treated and cabozantinib-treated c-Met/β-catenin mice. Magnification ×40; scale bar=500 μm. (E) Ki-67 (magnification ×200; scale bar=100 μm), CD34 (magnification ×200; scale bar=100 μm) and p-Erk (magnification,×100; scale bar=200 μm) staining in livers from c-Met/β-catenin mice. Ki-67 positive cells were counted and quantified as proliferation index. CD34 and p-Erk staining were quantified and represented as the percentage of the positive staining area of the whole section area. Tukey-Kramer test: at least p<0.05. a, versus pretreatment; b, versus vehicle.
Figure 4.
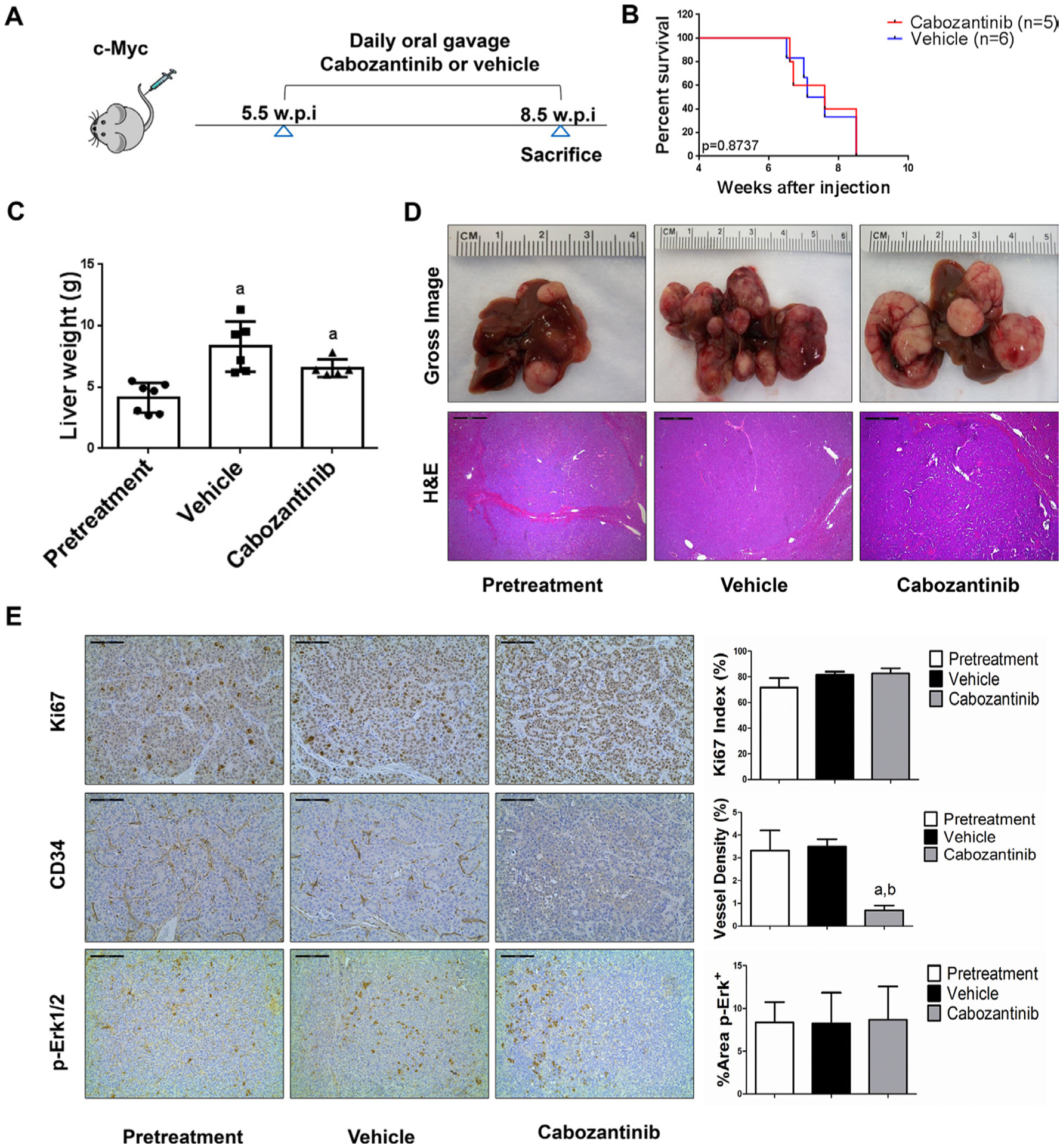
Limited efficacy of cabozantinib on c-Myc mouse liver tumours. (A) Study design. (B) Kaplan-Meier survival of pretreatment, vehicle-treated and cabozantinib-treated c-Myc mice. (C) Liver weight of pretreatment, vehicle-treated and cabozantinib-treated c-Myc mice. (D) Macroscopic and microscopic appearance of livers from pretreatment, vehicle-treated and cabozantinib-treated c-Myc mice. Magnification ×40; scale bar=500 μm. (E) Ki-67 (magnification ×200; scale bar=100 μm), CD34 (magnification ×200; scale bar=100 μm) and p-Erk (magnification ×100; scale bar=200 μm) staining in livers from c-Myc mice. Ki-67–positive cells were counted and quantified as proliferation index. CD34 and p-Erk staining were quantified and represented as the percentage of the positive staining area of the whole section area. Tukey-Kramer test: at least p<0.05. a, versus pretreatment; b, versus vehicle.
Figure 2.
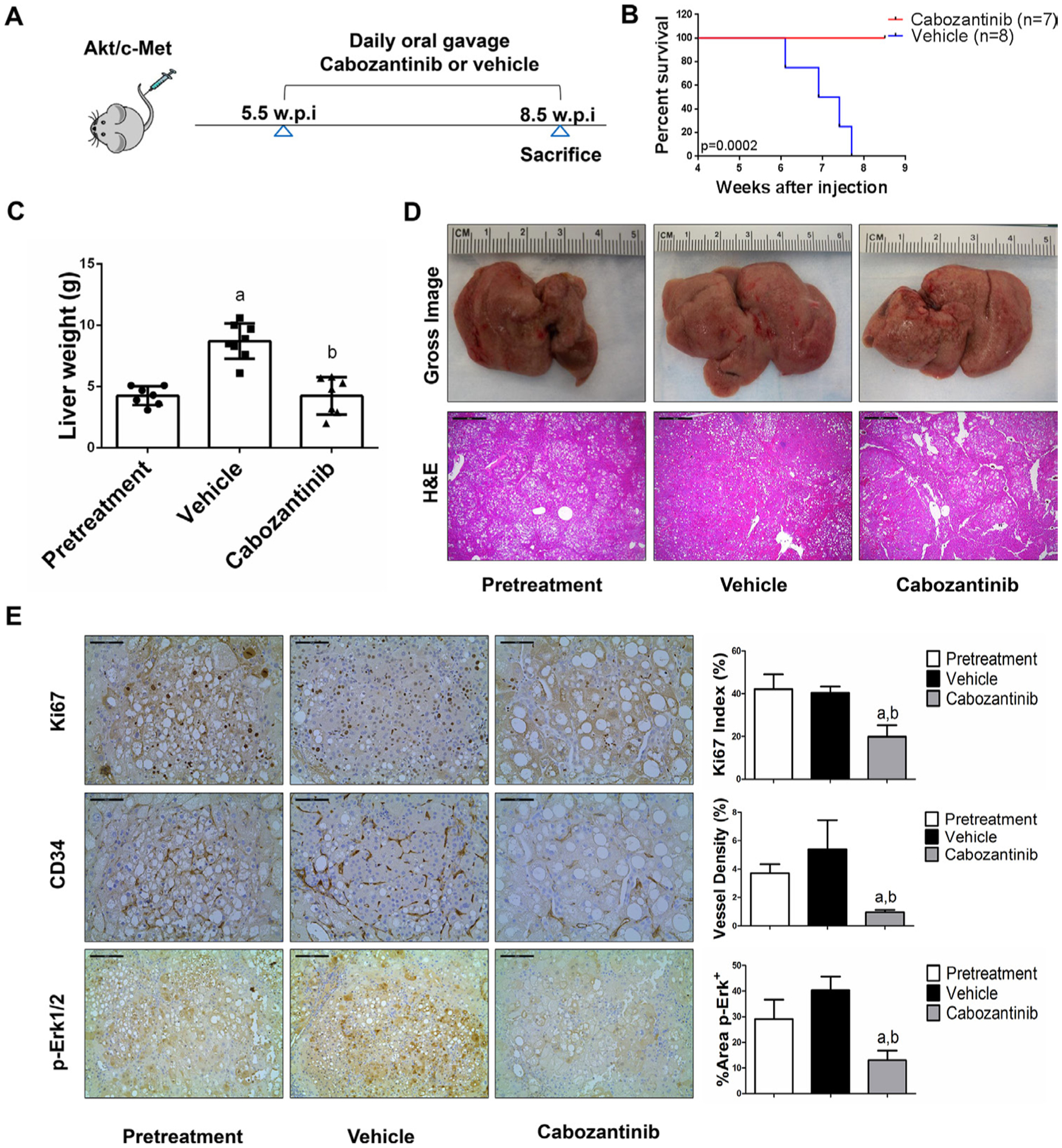
Cabozantinib treatment determines stable disease in Akt/c-Met mice. (A) Scheme of the experiment. (B) Survival curve of Akt/c-Met pretreatment, vehicle-treated and cabozantinib-treated cohort. (C) Liver weight of pretreatment, vehicle-treated and cabozantinib-treated Akt/c-Met mice. (D) Gross images and H&E staining of livers from pretreatment, vehicle-treated and cabozantinib-treated Akt/c-Met mice. Magnification ×40; scale bar=500 μm. (E) Ki-67 (magnification ×200; scale bar=100 μm), CD34 (magnification ×200; scale bar=100 μm) and p-Erk (magnification ×100; scale bar=200 μm) staining in livers from Akt/c-Met mice. Ki-67–positive cells were counted and quantified as proliferation index. CD34 and p-Erk staining were quantified and represented as the percentage of the positive staining area of the whole section area. Tukey-Kramer test: at least p<0.05. a, versus pretreatment; b, versus vehicle.
Figure 3.
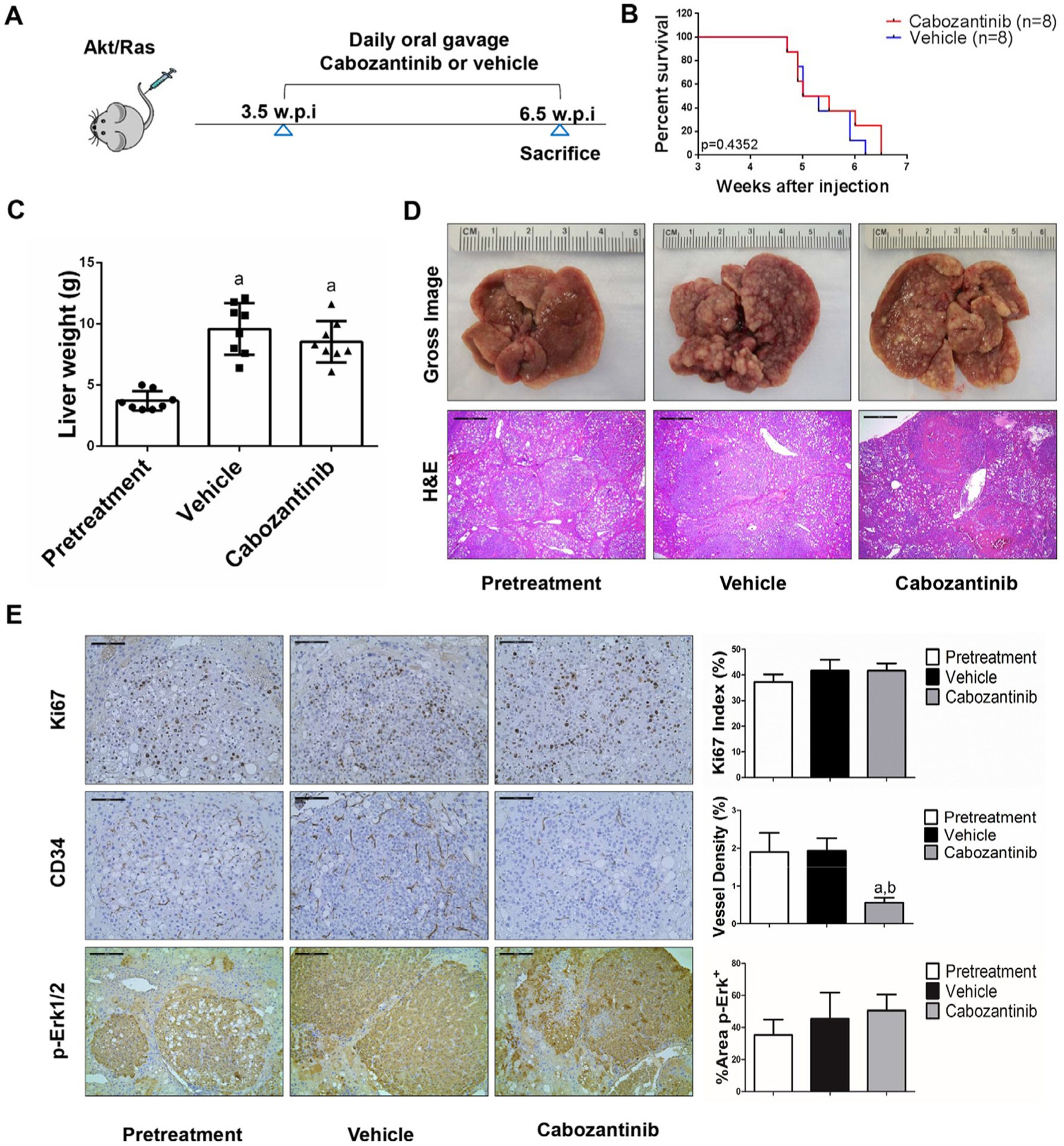
Effects of cabozantinib on Akt/Ras driven hepatocarcinogenesis. (A) Study design. (B) Kaplan-Meier survival of pretreatment, vehicle-treated and cabozantinib-treated Akt/Ras mice. (C) Liver weight of pretreatment, vehicle-treated and cabozantinib-treated Akt/Ras mice. (D) Macroscopic and microscopic appearance of livers from pretreatment, vehicle-treated and cabozantinib-treated Akt/Ras mice. Magnification ×40; scale bar=500 μm. (E) Ki-67 (magnification ×200; scale bar=100 μm), CD34 (magnification ×200; scale bar=100 μm) and p-Erk (magnification ×100; scale bar=200 μm) staining in livers from Akt/Ras mice. Ki-67–positive cells were counted and quantified as proliferation index. CD34 and p-Erk staining were quantified and represented as the percentage of the positive staining area of the whole section area. Tukey-Kramer test: at least p<0.05. a, versus pretreatment; b, versus vehicle.
In summary, we demonstrated that cabozantinib treatment induces stable disease in c-Met/β-catenin and Akt/c-Met mouse HCC models, but it has no efficacy in Akt/Ras and c-Myc mouse HCC.
Cabozantinib treatment suppresses HCC progression via the p-Met/p-Erk cascade, but it does not affect Akt/mTOR signaling in vivo
Subsequently, we investigated the molecular mechanisms underlying the effects of cabozantinib in mouse models. Histological evaluation revealed that cabozantinib treatment did not affect the overall histopathological features of the tumours (figures 1D–4D). Using Ki67 as a proliferation marker, we discovered that in both c-Met/β-catenin and Akt/c-Met mouse HCC, cabozantinib treatment led to significantly decreased cell proliferation (figures 1E and 2E). In contrast, cabozantinib did not affect HCC growth proliferation in Akt/Ras and c-Myc mice (figures 3E and 4E). As concerns apoptosis, a significant induction of programmed cell death was detected in all models, with the highest levels being observed in AKT/Ras and c-Myc mouse neoplastic lesions, following cabozantinib treatment. In these two models, extensive areas of necrosis were also frequently observed (online supplemental figure 11).
At the molecular level, in c-Met/β-catenin and Akt/c-Met HCC, cabozantinib strongly inhibited p-Met expression, leading to decreased p-Erk1/2 levels (figure 5A,B). In contrast, p-Erk1/2 expression was not affected by cabozantinib administration in Akt/Ras and c-Myc mouse HCC (figure 5C,D). Intriguingly, p-Akt, p-mTOR and p-Rps6 levels did not change significantly following cabozantinib treatment in all models investigated, suggesting that cabozantinib does not affect the AKT/mTOR signalling in human HCC cell lines and in mouse models. As concerns markers of proliferation, Pcna levels were downregulated and p21 expression was upregulated by cabozantinib in c-Met/β-catenin and Akt/c-Met HCC models (figure 5A,B). In addition, consistent with the in vitro studies, cabozantinib treatment resulted in the drastic decreased expression of Pkm2 in c-Met/β-catenin and Akt/c-Met HCC (online supplemental figure 12).
Figure 5.
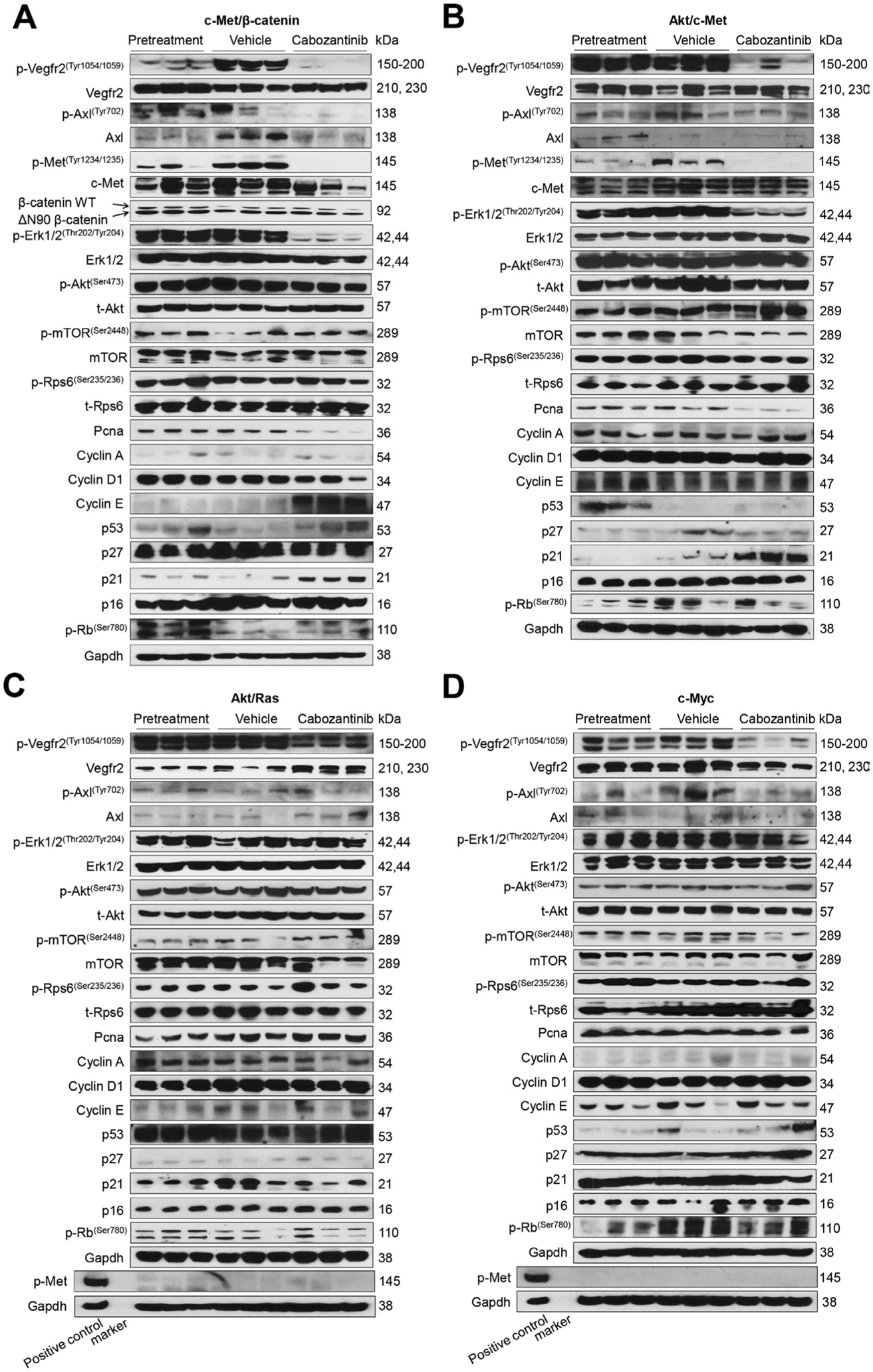
Effects of cabozantinib treatment on the levels of putative target proteins in livers from c-Met/β-catenin, Akt/c-Met, Akt/Ras and c-Myc mice. Western blot analysis was performed to determine the levels of phosphorylated Vegfr2, phosphorylated Axl, phosphorylated c-Met, downstream components in the c-Met cascade as well as proliferation markers in pretreatment, vehicle-treated and cabozantinib-treated c-Met/β-catenin mice (A), Akt/c-Met mice (B), Akt/Ras mice (C) and c-Myc mice (D). p, phosphorylated; t, total.
Our data indicate that cabozantinib functions to inhibit of activated c-Met dependent p-Erk activation. To further investigate this phenotype, we overexpressed c-MET in two human HCC cell lines, Huh7 and Hep40 cells, which have low c-MET expression (online supplemental figure 13A,B). Overexpression of c-MET resulted in increased p-MET and p-ERK1/2 expression (online supplemental figure 13A,B). c-MET-overexpressing cell lines demonstrated significant augmented sensitivity to cabozantinib (online supplemental figure 13C,D). These cell lines exhibited also enhanced sensitivity to the MEK inhibitor PD901, further implying the important role of the MEK/ERK pathway downstream of c-MET in HCC (online supplemental figure 14).
Cabozantinib modulates tumour microenvironment in vivo
The use of murine HCC models enabled us to investigate whether cabozantinib modulates the HCC microenvironment. As VEGFR2 and its mediated angiogenesis have been considered a major mechanism by which cabozantinib suppresses tumour growth, we analysed VEGFR2 levels and tumour vasculature in the four mouse HCC models treated with cabozantinib. In all models, cabozantinib effectively inhibited p-VEGFR2 levels (figure 5A–D). Consistent with this observation, using immunostaining with the anti-CD34 antibody, we found that cabozantinib reduced microvascular density in all HCC models (figures 1E–4E). Similar results were observed with another endothelial cell marker, CD31 (online supplemental figure 15). Intriguingly, despite the strong antiangiogenesis effects by cabozantinib in all mouse models, it did not lead to increased hypoxia, as indicated by the lack of increased HIF-1α expression (online supplemental figure 12).
Studies have suggested the key role of cancer-associated fibroblasts (CAFs) and fibrosis in tumour progression, including HCC.21 Using Sirius Red staining as well as immunostaining with anti-Vimentin antibody, we found that cabozantinib treatment did not affect CAFs and fibrosis in the mouse lesions (online supplemental figures 16 and 17).
It has been suggested that cabozantinib may modulate the HCC immune microenvironment.22 We found that in both c-Met/β-catenin and Akt/c-Met mouse lesions, the tumour-infiltrating macrophages decreased along tumour progression. Cabozantinib treatment was able to normalise the presence of macrophages within the HCC nodules in c-Met/β-catenin and Akt/c-Met mouse HCCs (online supplemental figure 18). However, no consistent changes of genes associated with polarisation of macrophage in the cabozantinib-treated tumour tissues were detected (online supplemental figures 19 and 20). Cabozantinib had no effect on macrophages in Akt/Ras and c-Myc mouse HCCs. As concerns CD3(+) T lymphocytes, only few of these cells were detected in Akt/c-Met and Akt/Ras mouse HCCs by immunostaining. Scattered CD3(+) T cells were found in c-Met/β-catenin HCC, whereas a high number of CD3(+) T lymphocytes were present in c-Myc HCC (online supplemental figure 21). Cabozantinib treatment did not affect the number or distribution of CD3(+) T lymphocytes in the lesions from these mouse models.
In summary, our study demonstrates that cabozantinib is able to modulate tumour microenvironment as it inhibits tumour angiogenesis regardless of the oncogenic drivers. However, it has limited impact on CAFs, macrophages and tumour infiltrating T cells.
Cabozantinib synergises with MLN0128 to inhibit HCC cell growth in vitro
In the TCGA LIHC protein dataset, ~44% of HCC samples showed coactivation of AKT/mTOR and c-MET (online supplemental figure 22A). Levels of phosphorylated/activated p-AKT(S473) and p-MET(Y1234/1235) were subsequently determined in our collection of human HCC specimens (n = 64) by immunohistochemistry (online supplemental figure 22B). We found that 33% HCC specimens exhibited concomitantly elevated p-AKT and p-MET immunoreactivity (online supplemental figure 22B). These data suggest that ~40% of human HCCs have coactivation of AKT/mTOR and c-MET signalling and suggest that inhibition of mTOR signalling may synergise with cabozantinib to suppress the growth of this subset of HCC.
To test this hypothesis, we chose to use the pan-mTOR inhibitor MLN0128.23 Two HCC cell lines, MHCC97-H and HLE, were treated with cabozantinib and MLN0128, either alone or in combination. We found that concomitant treatment of HCC cells with cabozantinib and MLN0128 resulted in stronger growth inhibitory potency (figure 6A and C) and cell cycle arrest (online supplemental figure 23). We calculated the combination index (CI),24 and all CI values were lower than one in Cabozantinib and MLN0128 treatment groups (figure 6B and D), suggesting a synergistic antitumour activity by the combination therapy. At the molecular level, cabozantinib was effective in inhibiting p-MET and p-ERK levels, whereas MLN0128 strongly suppressed p-AKT, p-mTOR, p-4EBP1 and p-RPS6 expression (figure 6E). The combination treatment resulted in a strong inhibition of these proteins, leading to a stronger inhibition of PCNA expression and HCC cell proliferation (figure 6F).
Figure 6.
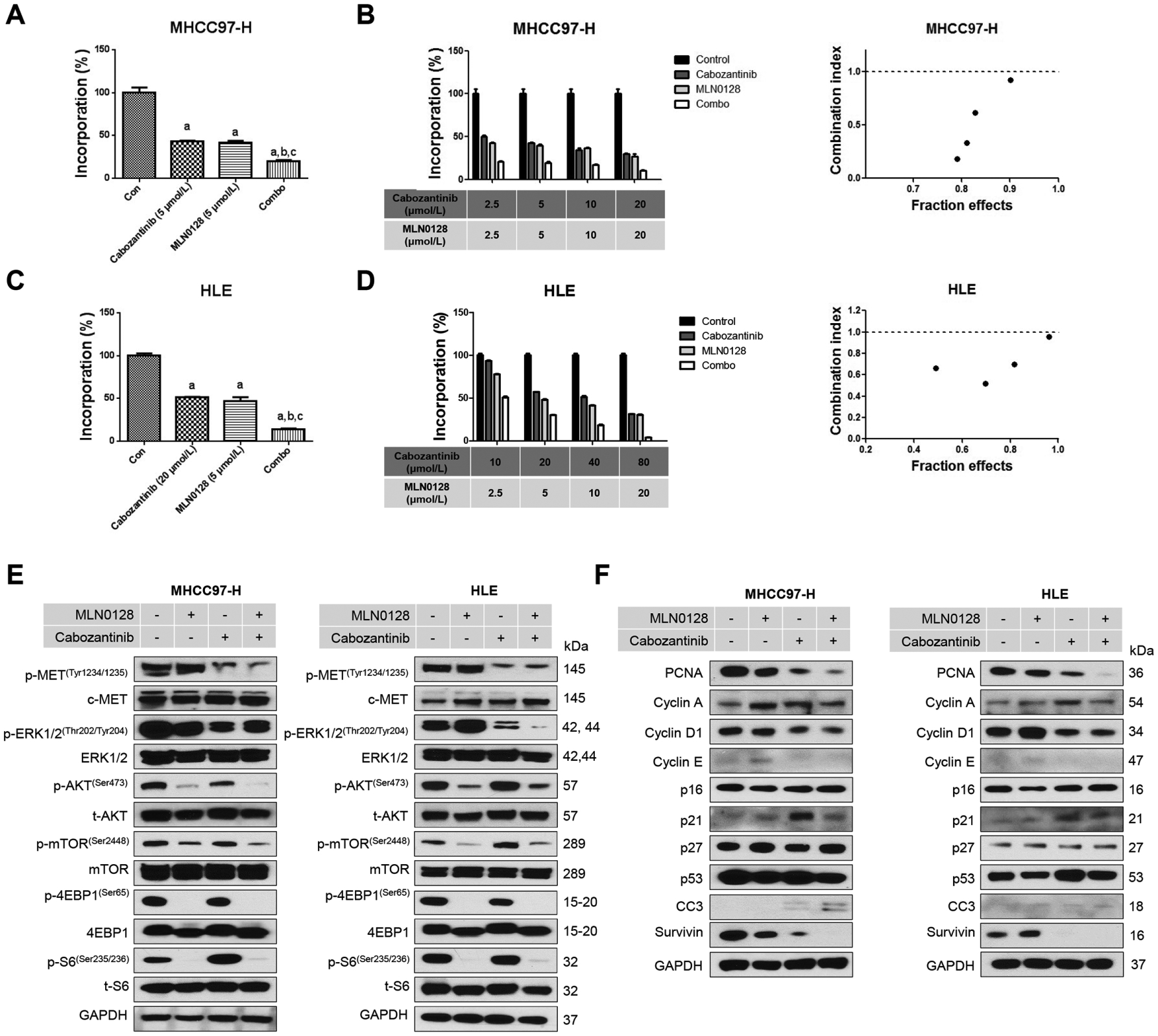
Effects of combined cabozantinib/MLN0128 treatment on HCC cell proliferation and cell cycle. (A and C) Combined cabozantinib/MLN0128 treatment reduced cell proliferation in MHCC97-H and HLE cells. (B and D) The enhanced inhibitory effect of cabozantinib/MLN0128 combination on HCC in vitro growth is a synergistic action. (E and F) Representative western blot analysis phosphorylated MET, AKT/mTOR pathway, MAPK pathway and proliferation pathway in MHCC97-H and HLE cell lines. CC3, cleaved caspase 3; combo, combined cabozantinib/MLN0128 treatment; p, phosphorylated; t, total.
In summary, our data suggest that cabozantinib synergises with MLN0128 to suppress HCC cell growth in vitro.
Combined cabozantinib and MLN0128 treatment induces tumour regression in c-Met/β-catenin mice
To further substantiate the in vitro findings, we tested the hypothesis in vivo using the c-Met/β-catenin mouse model. First, we tested the maximum doses of cabozantinib and MLN0128 that mice could tolerate. Using total body weight as the measurement of overall mouse health, we discovered that treatment of cabozantinib at 60 mg/kg/day and MLN0128 at 0.5 mg/kg/day were well tolerated in mice. Mice were injected with c-Met/β-catenin and randomly separated into five cohorts (figure 7A). The first group of mice was harvested 5.5 weeks after injection as ‘Pretreatment’ cohort. The remaining mice were treated with vehicle, cabozantinib, MLN0128 or cabozantinib plus MLN0128 (combination group) for up to 3 weeks. Vehicle treated mice developed large tumours and were required to be euthanised between 6.7 and 8.3 weeks (figure 7B). MLN0128 treatment did not demonstrate any therapeutic efficacy (figure 7B,C). Consistent with our previous studies, cabozantinib, used as single agent, led to stable disease (figure 7B,C). In striking contrast, combination treatment resulted in tumour regression (figure 7B,C). Specifically, the total liver weight in the combination group was lower than that of ‘pretreatment’ mice (figure 7C). Similar results were obtained when using the percentage of tumour area as the measurement of tumour burden (figure 7E). Histologically, HCC lesions were identified in all cohorts. While vehicle and MLN0128 treated mice had tumour lesions throughout the liver, pretreatment and cabozantinib mice had relatively smaller lesions. Tumour lesions were very small in combination treated mice (figure 7D).
Figure 7.
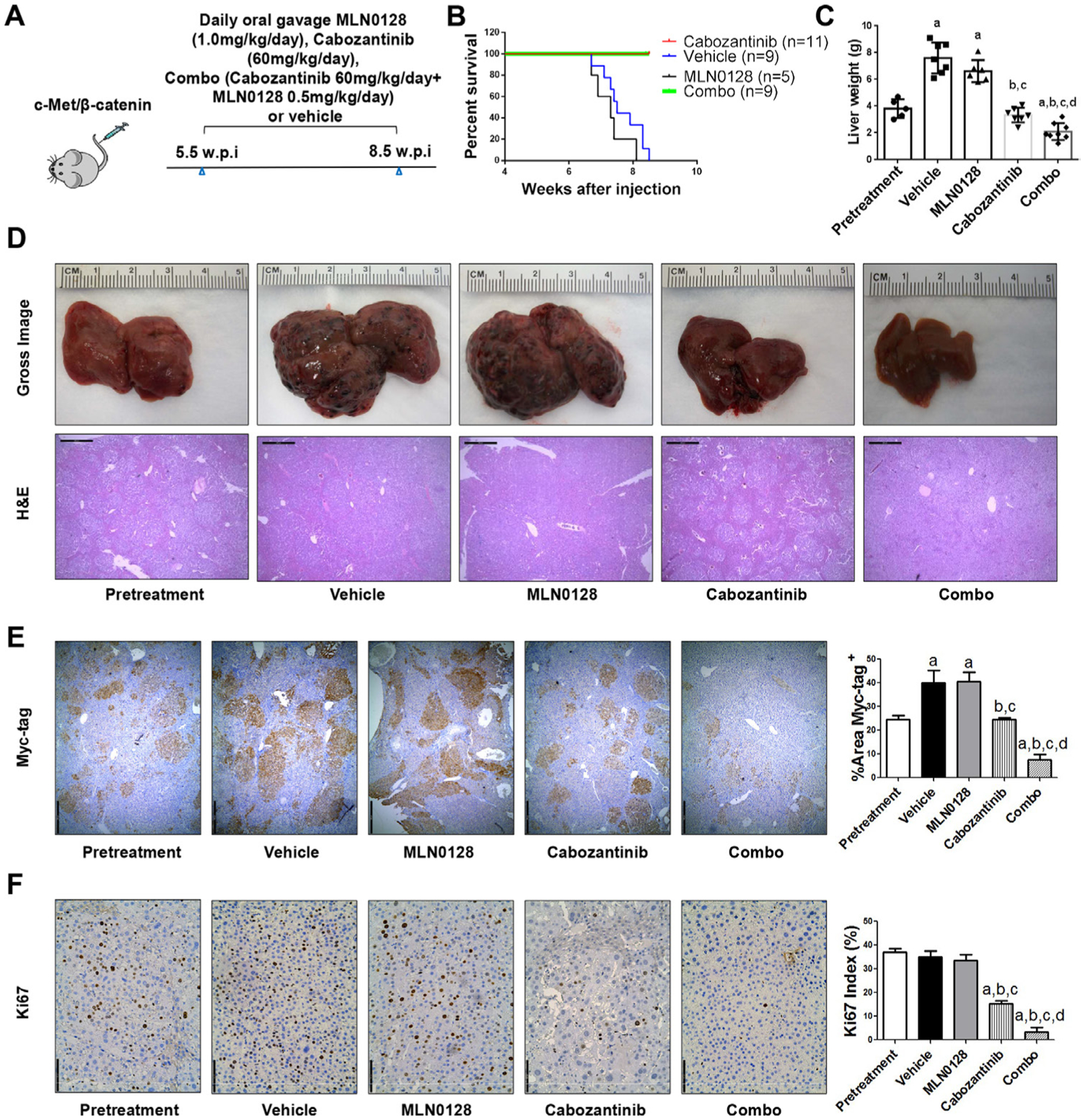
Combined cabozantinib and MLN0128 treatment has a potent antineoplastic effect in HCC lesions from c-Met/β-catenin mice. (A) Scheme of the experiment. (B) Survival curve of c-Met/β-catenin mice pretreated, treated with vehicle, cabozantinib, MLN0128 and cabozantinib/MLN0128. (C) Liver weight of pretreatment, vehicle-treated, MLN0128-treated, cabozantinib-treated and cabozantinib/MLN0128-treated c-Met/β-catenin mice. (D) Gross images and H&E staining of livers from pretreatment, vehicle-treated, MLN0128-treated, cabozantinib-treated and cabozantinib/MLN0128-treated c-Met/β-catenin mice. Magnification ×40; scale bar=500 μm. (E) Myc-tag (magnification ×40; scale bar=500 μm) and (F) Ki-67 (magnification ×100; scale bar=200 μm) staining in livers from c-Met/β-catenin mice. Myc-tag staining was quantified and represented as the percentage of the positive staining area of the whole section area. Ki-67–positive cells were counted and quantified as proliferation index. Tukey-Kramer test: at least p<0.05. a, versus pretreatment; b, versus vehicle; c, versus MLN0128; d, versus cabozantinib. combo, combined cabozantinib/MLN0128 treatment.
At the cellular level, MLN0128 did not affect tumour cell proliferation, whereas cabozantinib treatment significantly decreased Ki67(+) cells in the liver. Furthermore, combination treatment resulted in an even stronger inhibition of tumour cell proliferation (figure 7F). In terms of apoptosis, both cabozantinib and MLN0128 administration induced higher cell death than that observed in the pretreatment and vehicle groups, whereas the combination treatment resulted in large areas of necrosis and elevated apoptosis in the tumour lesions (online supplemental figure 24). As concerns tumour angiogenesis, cabozantinib, but not MLN0128, led to decreased tumour vasculature. Combination treatment had a stronger effect in inhibiting tumour angiogenesis (figure 8A). Mechanistically, cabozantinib was effective in inhibiting p-Vegfr2, p-Met and p-Erk; MLN0128 strongly inhibited p-Akt and p-Rps6; and combination treatment suppressed both MAPK and AKT/mTOR cascades, leading to decreased Pcna, Survivin and Cyclin D1 levels (figure 8B, C and D).
Figure 8.
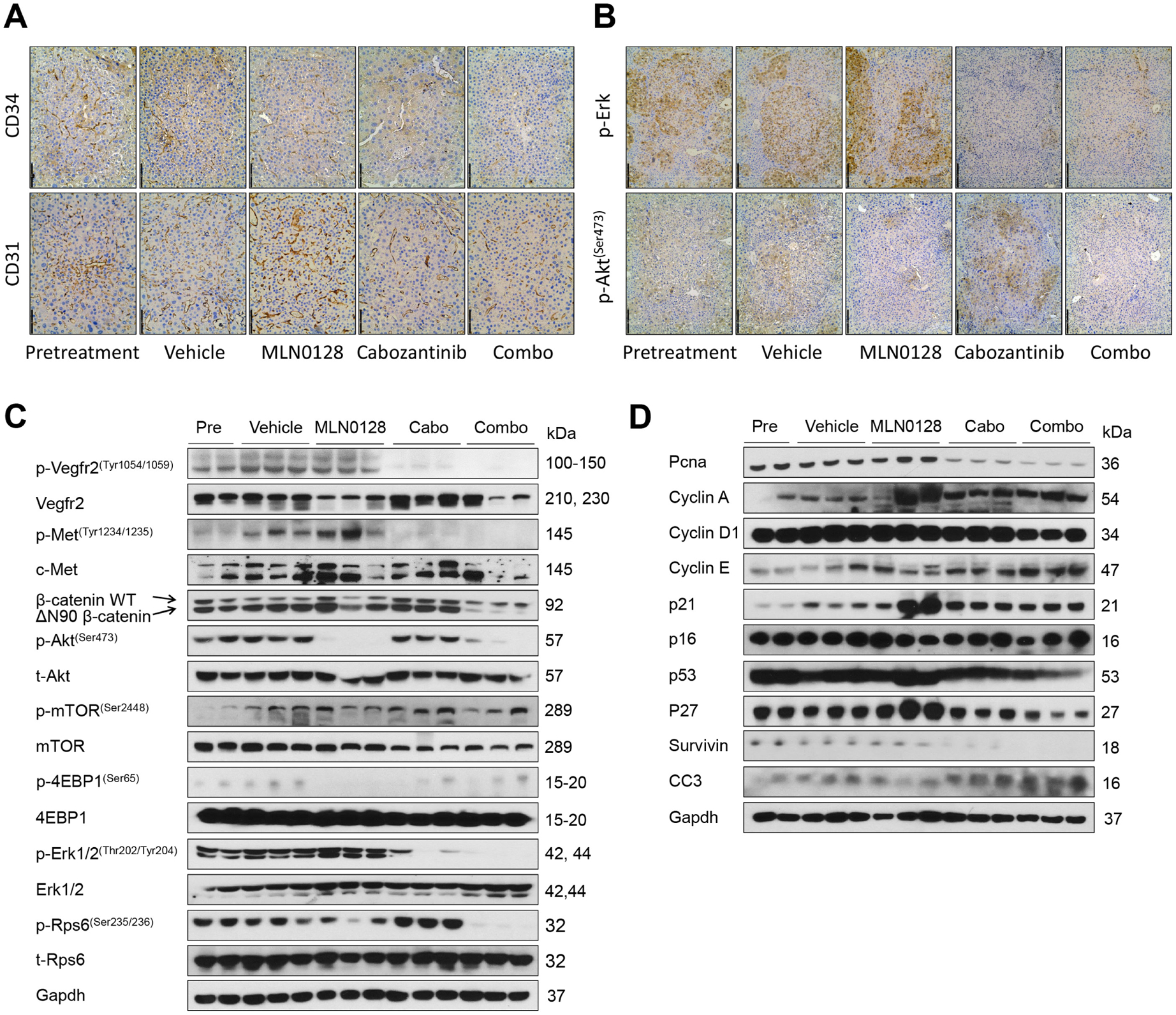
Effect of combined cabozantinib and MLN0128 treatment on tumour microvascular density and putative target proteins in livers from c-Met/β-catenin mice. (A) CD34 (magnification ×200; scale bar=100 μm) and CD31 (magnification,×200; scale bar=100 μm) staining in livers from c-Met/β-catenin mice. (B) p-Akt (Ser473) (magnification ×100; scale bar=200 μm) and p-Erk (magnification, ×100; scale bar=200 μm) staining in livers from c-Met/β-catenin mice. Western blot analysis was performed to determine the levels of phosphorylated Vegfr2, phosphorylated c-Met, Akt/mTOR and MAPK (C), as well as proliferation and apoptosis markers (D) in c-Met/β-catenin mice. Cabo, Cabozantinib; CC3, cleaved caspase 3; combo, combined cabozantinib/MLN0128 treatment.
Overall, our study demonstrates that combining cabozantinib and MLN0128 induces HCC regression in c-Met/β-catenin mice by strongly inhibiting tumour cell proliferation and angiogenesis and inducing elevated cell death.
Combined cabozantinib and anti-PD-L1 treatment does not lead to increased therapeutic efficacy in the four mice HCC models
Since it was reported that inhibition of c-MET resulted in increased PD-L1 expression,25 we examined PD-L1 expression in cabozantinib-treated human HCC cells and mouse HCC samples. Overall, no consistent results were obtained, and cabozantinib did not affect PD-L1 expression in the four mouse models (online supplemental figure 25).
A clinical trial testing the therapeutic efficacy of combined cabozantinib and anti-PD-L1 for advanced HCC is ongoing (Trial ID: NCT03755791). We investigated whether combined cabozantinib and anti-PD-L1 treatment can increase growth inhibition in the four preclinical models. Mice were treated with cabozantinib and anti-PD-L1, either alone or in combination (figure 9A and online supplemental figure 26A). Treatment with anti-PD-L1 alone did not demonstrate any therapeutic efficacy in the models tested (figure 9B–E, online supplemental figure 26B–E and online supplemental figures 27–30). Furthermore, combined cabozantinib and anti-PD-L1 treatment neither led to increased efficacy when compared with cabozantinib alone in c-Met/β-catenin and Akt/c-Met HCC models (figure 9B–E, online supplemental figures 27 and 28), nor showed any efficacy in Akt/Ras and c-Myc HCC models (online supplemental figure 26B–E, online supplemental figures 29 and 30).
Figure 9.
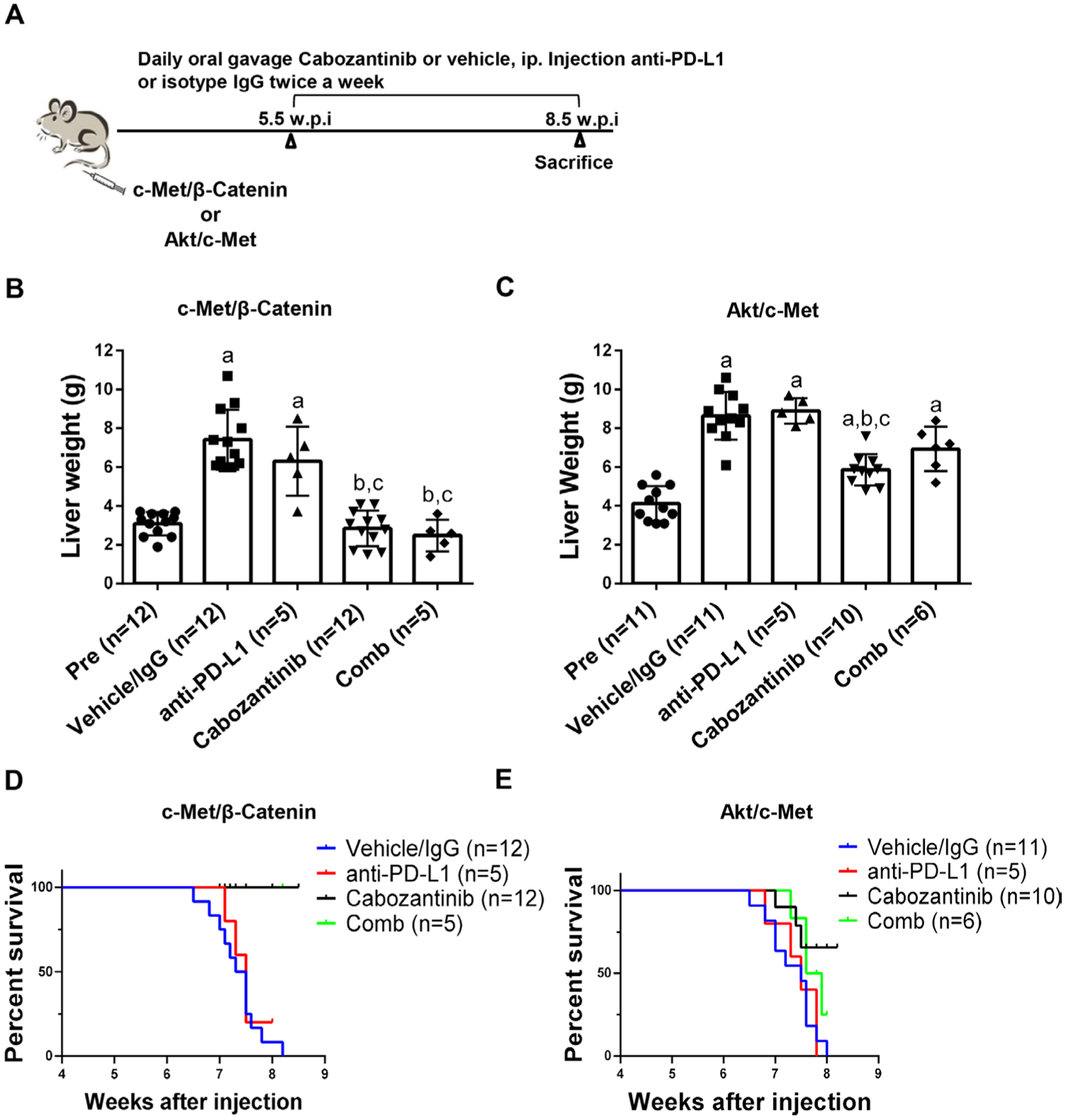
Effect of combined cabozantinib and anti-PD-L1 treatment in c-Met/β-catenin mice and Akt/c-Met mice. (A) Study design. (B) Liver weight of pretreatment-treated, vehicle/isotype IgG-treated, cabozantinib-treated, PD-L1-treated and cabozantinib/anti-PD-L1-treated c-Met/β-catenin mice. (C) Liver weight of pretreatment-treated, vehicle/isotype IgG-treated, cabozantinib-treated, PD-L1-treated and cabozantinib/anti-PD-L1-treated Akt/c-Met mice. (D-E) Survival curve of c-Met/β-Catenin and Akt/c-Met mice treated with vehicle/isotype IgG, Cabozantinib, PD-L1 and Cabozantinib/anti-PD-L1. Tukey-Kramer test: at least p<0.05. a, versus pretreatment; b, versus vehicle; c, versus anti-PD-L1. Cabo, cabozantinib; Comb, combined cabozantinib and anti-PD-L1 treatment; Pre, pretreatment.
Next, we performed detailed analysis of cabozantinib and anti-PD-L1 combination therapies in c-Met/β-catenin and Akt/c-Met HCC models. Histological evaluation of the lesions revealed that all treatment strategies did not affect the overall histological features of the tumours in the two models (online supplemental figures 27 and 28). Subsequently, we investigated immune cell populations, including CD3+ CD8+, CD3+ CD4+, CD3+ CD8+ PD-1+, CD3+ CD4+ PD-1+, CD3+ CD8+ CTLA4+ and CD3+CD4 +CTLA4+ in liver tissues as well as in spleen of c-Met/β-catenin (online supplemental figure 31) and Akt/c-Met mice (online supplemental figure 32). When compared with vehicle/IgG treated control mice, cabozantinib treatment did not result in significant changes of the immune cell populations tested in both models. In Akt/c-Met mouse HCC, combining cabozantinib and anti-PD-L1 led to the increased spleen CD3+ CD8+ and CD3+ CD4+ PD-1+ cells (online supplemental figure 32), as well as CD3 + CD8+ PD-1+ cells in liver tumour tissues. However, these changes were not observed in c-Met/β-catenin mouse lesions.
Altogether, our study suggests that cabozantinib does not induce PD-L1 expression in HCC cells. Combined cabozantinib and anti-PD-L1 treatment does not lead to an increased therapeutic efficacy in the murine models tested.
DISCUSSION
In the present study, we investigated the therapeutic potential of cabozantinib using a large panel of human HCC cell lines and four distinct oncogene-driven HCC murine models. The combined in vitro and in vivo studies suggest that p-MET levels, but not total c-MET expression, could be a reliable biomarker for cabozantinib efficacy. Clearly, our mouse studies were limited by the fact that we had to overexpress c-Met in mice to activate the c-Met pathway in vivo. Additional studies, using mouse models in which c-Met is activated by other oncogenes or mutations would be required to further validate this conclusion.
Mechanistically, we discovered that cabozantinib inhibits activated c-MET dependent MAPK activation, leading to decreased expression of PKM2 and increased expression of p21. Since p21 overexpression has been linked with senescence-associated secretory phenotype (SASP), we investigated whether an increased senescence occurred in cabozantinib treated c-Met/β-catenin mouse liver tumours. We found that cabozantinib treatment did not lead to increased senescence (online supplemental figure 33), suggesting that cabozantinib-driven increased p21 expression likely contributes to the decreased cell proliferation, while does not increases SASP in HCC.
We show that cabozantinib leads to stable disease in c-Met/β-catenin HCC models when mice were harvested at 3 weeks post-treatment. It is worth to note that an increase in serum AST and ALT levels occurred during tumour progression. The elevated AST and ALT levels were not normalised by cabozantinib despite the decreased tumour burden (online supplemental figure 34A). The precise mechanisms underlying this observation remain to be defined. As cabozantinib induces HCC cell apoptosis, it is possible that cabozantinib treatment triggers the release of liver enzymes from HCC cells into the serum.
Long-term treatment with targeted therapies has been shown to induce drug resistance. To test whether a similar event follows cabozantinib treatment, additional c-Met/β-catenin tumour bearing mice were treated with cabozantinib and harvested 3 weeks and 6 weeks post-treatment (online supplemental figure 34B). While mice treated for 6 weeks with cabozantinib demonstrated decreased tumour burden than that of vehicle treated mice, they showed higher tumour burden than that of pretreatment mice and mice treated for 3 weeks with cabozantinib, consistent with tumour progression. These results suggest the emergence of cabozantinib resistance clones within the tumour lesions over long-term Cabozantinib treatment. Additional studies are required to elucidate the molecular mechanisms underlying cabozantinib resistance in HCC.
A major conclusion of our study is that cabozantinib and pan-mTOR inhibitors possess synergistic effects in inhibiting HCC growth in vitro and driving tumour regression in mice with concomitant activation of c-MET and AKT/mTOR pathways. Since our subsequent data indicate that ~40% human HCCs exhibit coactivation of c-MET and AKT/mTOR cascades, it can be envisaged that combining cabozantinib and mTOR inhibitors might be highly useful for the treatment of this HCC subset.
During the past years, immunotherapy using checkpoint inhibitors (anti-PD-1 and anti-PD-L1) has shown promising results in patients with advanced HCC.26 Importantly, in the IMBRAVE150 trial for patients with unresectable HCC, anti-PD-L1 antibody combined with anti-VEGF antibody results in better overall survival than that from sorafenib,27 supporting the usefulness of combined immunotherapy and targeted therapy for HCC treatment. Currently, two clinical trials are testing the therapeutic efficacy of combining cabozantinib and immune checkpoint inhibitors (anti-PD1 or anti-PD-L1) in advanced HCC (trial ID: NCT03299946 and NCT03755791). However, preclinical studies supporting the application of such combination therapies for HCC treatment are limited. In the present investigation, we show that cabozantinib does not consistently increase PD-L1 expression in HCC cell lines, and the combined anti-PD-L1 and cabozantinib treatment does not induce an augmented antineoplastic potency in the examined mouse models. It should be underlined that HCC is a highly heterogeneous disease, and the combined anti-PD-L1 and cabozantinib therapy was only tested in a limited number of murine HCC models, not fully recapitulating the heterogeneity of the human disease. Additional models harbouring other common genetic events, such as TP53 and ARID1A/2 mutations, should be employed to further investigate the therapeutic efficacy of cabozantinib, either alone or in combination with checkpoint inhibitors, for HCC treatment.
Supplementary Material
Significance of this study.
What is already known on this subject?
Hepatocellular carcinoma (HCC) is one of the most frequent malignancies with limited treatment options.
Cabozantinib is an orally bioavailable multikinase inhibitor targeting c-MET, VEGFR2 and AXL proteins, now approved by FDA for HCC patients.
Combination therapies provide a synergistic effect and reduce drug resistance for the treatment of cancer.
What are the new findings?
Cabozantinib possesses antineoplastic activity in mouse models of HCC by restraining the p-MET/p-ERK cascade but not the AKT/mTOR signalling.
Concomitant activation of c-MET and AKT/mTOR pathways occurs in ~40% human HCCs. Combined cabozantinib and mTOR inhibitor treatment results in tumour regression in c-Met/β-catenin mouse HCC.
Administration of cabozantinib neither induces PD-L1 expression in HCC cells nor increases tumour-infiltrating T cells in mice. Combining cabozantinib and anti-PD-L1 treatment does not lead to increased therapeutic efficacy in the four mouse HCC models examined.
How might it impact on clinical practice in the foreseeable future?
Our preclinical findings suggest that combining cabozantinib and mTOR inhibitors may represent a novel therapeutic approach against HCC with activated c-MET and AKT/mTOR cascades.
Funding
This study was supported by grants from the National Institutes of Health: R01CA239251 and R03CA249236 to XC and P30DK026743 to UCSF Liver Center.
Footnotes
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. The data used to support the findings of this study are available from the corresponding author on request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
REFERENCES
- 1.Villanueva A Hepatocellular carcinoma. N Engl J Med 2019;380:1450–62. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118–27. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Montal R, Sia D, et al. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599–616. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz JN, Fancher KM. Cabozantinib: a multitargeted oral tyrosine kinase inhibitor 2018;38:357–69. [DOI] [PubMed] [Google Scholar]
- 5.Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting Met in cancer: rationale and progress. Nat Rev Cancer 2012;12:89–103. [DOI] [PubMed] [Google Scholar]
- 6.Venepalli NK, Goff L. Targeting the HGF-cMET axis in hepatocellular carcinoma. Int J Hepatol 2013;2013:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouattour M, Raymond E, Qin S, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018;67:1132–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tward AD, Jones KD, Yant S, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A 2007;104:14771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SA, Ladu S, Evert M, et al. Synergistic role of Sprouty2 inactivation and c-Met up-regulation in mouse and human hepatocarcinogenesis. Hepatology 2010;52:506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao J, Xu E, Zhao Y, et al. Modeling a human hepatocellular carcinoma subset in mice through coexpression of Met and point-mutant β-catenin. Hepatology 2016;64:1587–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, Che L, Li L, et al. Co-Activation of Akt and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016;6:20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abou-Alfa GK, Meyer T, Cheng A-L, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med 2018;379:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang Q, Chen W, Ren M, et al. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and Met. Clin Cancer Res 2014;20:2959–70. [DOI] [PubMed] [Google Scholar]
- 14.Wang C, Cigliano A, Jiang L, et al. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by Akt and N-ras proto-oncogenes in mice. Hepatology 2015;61:200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu P, Ge M, Hu J, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017;66:167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho C, Wang C, Mattu S, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012;55:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu T, Chen C, Wang A, et al. Repurposing cabozantinib to GISTs: overcoming multiple imatinib-resistant cKIT mutations including gatekeeper and activation loop mutants in GISTs preclinical models. Cancer Lett 2019;447:105–14. [DOI] [PubMed] [Google Scholar]
- 19.Daudigeos-Dubus E, Le Dret L, Bawa O, et al. Dual inhibition using cabozantinib overcomes HGF/Met signaling mediated resistance to pan-VEGFR inhibition in orthotopic and metastatic neuroblastoma tumors. Int J Oncol 2017;50:203–11. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen HM, Ruppender N, Zhang X, et al. Cabozantinib inhibits growth of androgen-sensitive and castration-resistant prostate cancer and affects bone remodeling. PLoS One 2013;8:e78881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Affo S, Yu L-X, Schwabe RF. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol 2017;12:153–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Y-Y, Tan C-T, Chen C-W, et al. Immunomodulatory effects of current targeted therapies on hepatocellular carcinoma: implication for the future of immunotherapy. Semin Liver Dis 2018;38:379–88. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S, Song X, Cao D, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol 2017;67:1194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006;58:621–81. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Li C-W, Li X, et al. Met inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology 2019;156:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahipal A, Tella SH, Kommalapati A, et al. Immunotherapy in hepatocellular carcinoma: is there a light at the end of the tunnel? Cancers 2019;11. doi: 10.3390/cancers11081078. [Epub ahead of print: 30 Jul 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894–905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.