

Abstract
DNA shuffling and directed evolution were employed to develop a novel adeno-associated virus (AAV) vector capable of crossing the seizure-compromised blood–brain barrier (BBB) and transducing cells in the brain. Capsid DNA from AAV serotypes 1–6, 8, and 9 were shuffled and recombined to create a library of chimeric AAVs. One day after kainic acid–induced limbic seizure activity in rats, the virus library was infused intravenously (i.v.), and 3 days later, neuron-rich cells were mechanically dissociated from seizure-sensitive brain sites, collected and viral DNA extracted. After three cycles of selection, green fluorescent protein (GFP)–packaged clones were administered directly into brain or i.v. 1 day after kainic acid–induced seizures. Several clones that were effective after intracranial administration did not transduce brain cells after the i.v. administration. However, two clones (32 and 83) transduced the cells after direct brain infusion and after i.v. administration transduced the cells that were localized to the piriform cortex and ventral hippocampus, areas exhibiting a seizure-compromised BBB. No transduction occurred in areas devoid of BBB compromise. Only one parental serotype (AAV8) exhibited a similar expression profile, but the biodistribution of 32 and 83 diverged dramatically from this parental serotype. Thus, novel AAV vectors have been created that can selectively cross the seizure-compromised BBB and transduce cells.
Introduction
Recent studies have established the potential of viral vector gene therapy as a viable treatment option for intractable epilepsy. Adeno-associated virus (AAV)–mediated expression and constitutive secretion of the neuroactive peptide, galanin, attenuates focal seizure sensitivity, prevents seizure-induced neuronal cell death, and blocks kainic acid–induced limbic seizure activity.1,2 Similarly, studies by Richichi et al.3 have shown that AAV-mediated expression of prepro-neuropeptide Y exerts a range of antiseizure effects that include increasing the electrical seizure threshold in the hippocampus and significantly retarding the rate of kindling epileptogenesis. Although a number of additional viral vector–based approaches have exhibited varying degrees of antiseizure success (for review see ref. 4), for any of these approaches the clinical application would entail microinfusion of the viral vectors directly into brain areas that exhibit identified seizure activity.
Recent advances in AAV engineering techniques have vastly expanded the ability to develop novel AAV serotypes. When combined with directed evolution where specific characteristics can be selected without a priori knowledge of the physical determinants, novel vectors can be identified that exhibit the desired, specific tropisms. As an initial proof of principle, Maheshri et al.5 introduced random changes in the AAV2 capsid by error-prone PCR and then selected novel AAV vectors that lacked heparin-binding properties or evaded the in vivo immune response. More recently, Li et al.6 utilized DNA shuffling of AAV capsid genes from a number of serotypes and directed evolution to identify a novel chimeric serotype that exhibited increased efficacy for melanoma cell transduction. Thus, by using a combination of AAV capsid DNA shuffling and directed evolution one can select mutants that exhibit specific, desirable properties.
Given these recent advances in AAV vector engineering, development of an alternative, peripheral therapeutic approach to epilepsy might be attained by taking advantage of the fact that seizures compromise the blood–brain barrier (BBB). Although the BBB tightly controls the movement of molecules both into and out of the brain,7 evidence has accumulated in both experimental animals and epileptic patients that seizures cause a local compromise in BBB function.8–13 This compromise in the BBB leads to the extravasation of large molecules such as albumin into the parenchyma of the brain.13 If viral vectors could be developed that selectively cross this seizure-compromised BBB and transduce cells in the area of compromise, one potentially would have a therapeutic conduit to those areas of brain involved in seizure generation.
The following studies utilized AAV capsid DNA shuffling and subsequent directed evolution in order to identify novel clones that exhibited the ability to cross the seizure-compromised BBB after intravenous (i.v.) administration and subsequently transduce cells in the central nervous system (CNS). The generation of such mutant AAV vectors would establish the possibility to deliver a therapeutic gene to areas of seizure activity using peripheral administration instead of direct microinfusion into the brain parenchyma.
Results
Kainic acid seizure–induced compromise of the BBB
In order to validate that indeed the kainic acid–induced seizures caused a compromise in the BBB at the time of library administration, rats were treated with kainic acid (10 mg/kg, intraperitoneally) where class IV limbic seizure activity developed over a period of 70–100 minutes post-treatment. Twenty-four hours after the appearance of the first class IV seizure, intracardiac infusion of fluorescein isothiocyanate–conjugated bovine serum albumin revealed specific areas of BBB compromise similar to previous findings with pilocarpine-induced limbic seizures10 (Figure 1). More importantly, the areas of BBB compromise were primarily localized to areas known to modulate the kainic acid–induced seizure activity, such as the piriform cortex and hippocampus.
Figure 1. Blood–brain barrier integrity is disrupted 24 hours after kainic acid administration and class IV seizures.
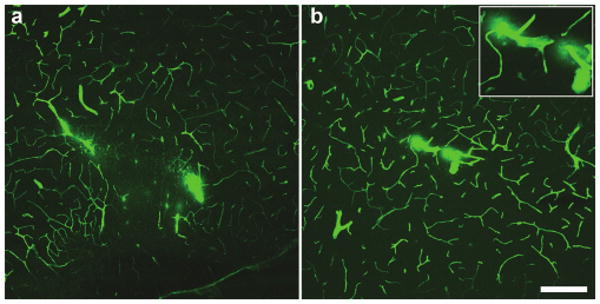
Confocal micrographs of cerebral blood vessels perfused with fluorescein isothiocyanate–albumin illustrating irregular, cloud-like patches of albumin leakage in the (a) piriform cortex and (b) ventral hippocampus. Inset in b shows magnified image of microvascular compromise in the ventral hippocampus. Bar = 200 μm for a and b at equal magnification.
Directed evolution and recovery of chimeric clones
As previously described,6 the initial chimeric library was composed of shuffled capsids from AAV serotypes 1–6, 8, 9, and an AAV8 with E531K mutation. This infectious and replication-competent library was injected (0.5 ml; 1.6 × 1013 viral particles/ kg) into the tail vein of rats 24 hours after kainic acid induction of class IV limbic seizure activity. Three days later, cells were mechanically dissociated from the piriform cortex and the hippocampus as previously described14 (Supplementary Figure S1), and viral capsid DNA was extracted from these neuron-rich samples. The pooled PCR products were cloned back into a wild-type (WT) AAV backbone (pSSV9) and pooled clones were used to generate the next round's starting library. This selection process was repeated two times and then individual clones were made into recombinant virus that contained a green fluorescent protein (GFP) reporter gene. A total of three rounds of selection were performed, and a sampling of clones was sequenced after each round of selection (Figure 2, Supplementary Table S1). We observed several instances where multiple clones had identical overall serotype composition but contained silent, missense, and nonsense point mutations throughout the capsid gene. In an assay for the ability of the recovered clones to make virus equivalent to AAV2, only 14–27% of recovered clones were viable (Supplementary Table S1). The presence of multiple point mutations and the limited viability of recovered clones suggest that the PCR recovery step was mutagenic. Thus, not only did these mutations likely influence the ability to package recombinant virus, but PCR point mutants also provided additional capsid variability beyond the original input library. In the VP3 region of the capsid where the surface would be exposed for receptor interaction,15 the capsids were primarily composed of AAV1, 8, and 9 (Figure 2).
Figure 2. Directed evolution and recovery of chimeric clones.
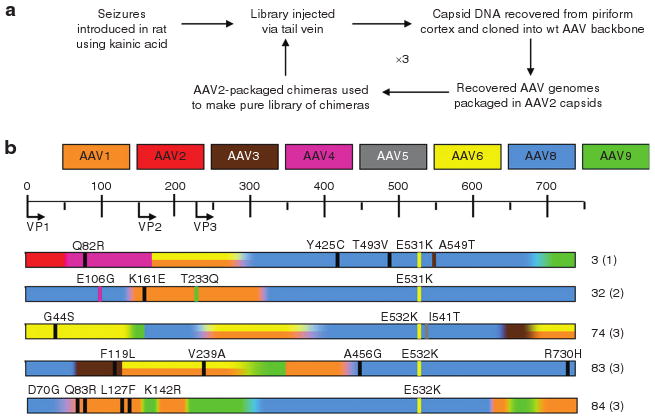
(a) Using a shuffled library based on adeno-associated virus (AAV) 1–6, 8, and 9, a directed evolution strategy was employed to select for an AAV capsid that can cross the seizure-compromised blood–brain barrier (BBB) and transduce cells in the areas of seizure influence. (b) Viable recovered clones that were tested in these studies. The scale at the top corresponds to the amino acid residues, with start sites for VP1, VP2, and VP3 indicated. A color-coding scheme is used to indicate the overall serotype composition at each location, and graded colors within each clone indicate regions of shared identity between neighboring serotypes. Where AAV1 and 6 are indistinguishable by amino acid sequence, a double stripe is drawn. Missense point mutations are shown as thin vertical lines, with the color corresponding to a serotype with that amino acid or a black line to indicate an amino acid not present in any parent. To the right of each amino acid map is the number assigned to that clone and the round from which it was recovered (in parentheses). wt, wild type.
In vivo assessment of transduction efficacy after direct infusion into the brain
A number of recombinant clones (packaging self-complementary chicken β actin-GFP genomes), were infused into the rat piriform cortex (1 μl) to test the ability to transduce cells in the brain. This initial test was conducted, because a clone incapable of transducing CNS cells after direct administration certainly would not be expected to transduce cells after peripheral administration. Clone 83 (2.1 × 1012 viral genomes/ml) exhibited substantial transduction in the piriform cortex, whereas clone 32 (2.8 × 1012 viral genomes/ ml) exhibited more moderate levels of transduction (Figure 3a,b). Clones 74 (3.3 × 1011 viral genomes/ml) and 84 (1.1 × 1011 viral genomes/ml) also transduced cells in the piriform cortex, although not to the same degree as clones 32 and 83 (Figure 3c,d). In all cases, the majority of the transduced cells exhibited a morphology resembling neurons. The titer attained for clone 3 was 1.2 × 1011 viral genomes/ml, and after direct piriform cortex infusion, no transduction was found. Because this infusion of clone 3 was not sufficient to transduce cells upon direct infusion into the CNS, further studies were not conducted with this clone.
Figure 3. Immunohistochemical analysis of green fluorescent protein transgene expression, 2 weeks following injections of infectious clones into piriform cortex.
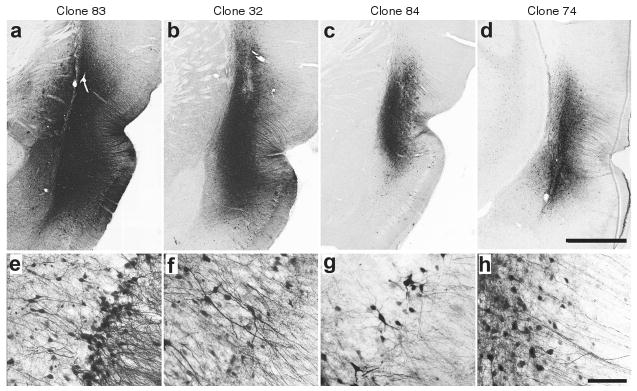
(a–d) Low-power photomicrographs of representative coronal sections showing the maximal dorsoventral extent of transduction around the site of injection. Bar = 1 mm for a–d at equal magnification. (e–h) Higher power images from sites depicted in a–d, demonstrating predominant neuronal tropism. Bar = 50 μm for e–h at equal magnification. (a,e) Clone 83; (b,f) clone 32; (c,g) clone 84; (d,h) clone 74.
In vivo assessment of transduction patterns after postseizure i.v. administration
When the various AAV clones were infused i.v. (0.5 ml) 24 hours after kainic acid–induced seizures in rats, a unique pattern emerged. I.v. administration of clones 74 (7.8 × 1011 viral genomes/kg) and 84 (3.8 × 1012 viral genomes/kg) did not result in any transduced cells in the brain 2 weeks after vector administration (data not shown). In marked contrast, post-seizure, i.v. administration of clone 32 (5.4 × 1012 viral genomes/kg) resulted in a number of GFP-positive cells that were confined to the piriform cortex (Figure 4a). More impressively, postseizure i.v. administration of clone 83 (3.5 × 1012 viral genomes/kg) caused substantial transduction of cells throughout the anterior–posterior extent of the piriform cortex, as well a number of cells in the ventral hippocampus (Figure 4b–f). For both clones 32 and 83, no transduced cells occurred in brain areas that are normally not associated with seizures or seizure-induced BBB compromise, such as the striatum and hypothalamus.8,10
Figure 4. Transduction patterns of clones 32 and 83 in rat brain two weeks after class IV seizures and intravenous vector administration.
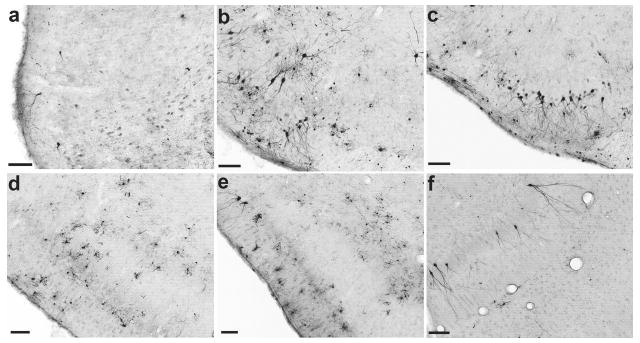
(a) Representative coronal section illustrating green fluorescent protein (GFP)–positive cells transduced by clone 32. Although not shown here, systemic injection of clone 32 results in GFP-positive cells throughout the rostro-caudal extent of the piriform cortex. (b–f) After intravenous injection of clone 83, GFP-positive cells were found in greater numbers compared to clone 32 throughout the full rostral–caudal extent of the piriform cortex as well as in the ventral hippocampus. (b) rostral piriform, 1.0 mm and (c) 0.2 mm anterior to bregma; (d) middle piriform, 2.2 mm posterior to bregma; (e) caudal piriform, 3.6 mm posterior to bregma; (f) and ventral hippocampus, 5.8 mm posterior to bregma. Bar = 100 μm for all panels.
Because the predominant composition of clones 32 and 83 included portions from AAV1, 8, and 9, these parental serotypes were evaluated. Certainly recent studies have demonstrated that AAV9 can cross the intact BBB.16,17 As would be expected, all three serotypes effectively transduced cells in the piriform cortex after direct infusion (Supplementary Figure S2). I.v. administration of AAV1 (2.1 × 1012 viral genomes/kg) 1 day after kainic acid–induced seizures resulted in sporadic GFP-positive neurons and glia in the paraventricular nucleus, hippocampus (Figure 5a,b), and piriform cortex. However, widely dispersed, cerebrovascular endothelial cells comprised the vast majority of GFP-positive cells (Figure 5c). In contrast, the same i.v. protocol with AAV8 (7.8 × 1012 viral genomes/kg) produced a pattern of transduction that was very similar to the pattern seen after i.v. administration of clones 32 and 83. GFP-positive cells were found in the piriform cortex and the hippocampus, whereas only a few scattered cells were found in brain areas with an intact BBB (Figure 5d–f). Finally, after kainic acid–induced seizures i.v. administration of AAV9 (4.3 × 1012 viral genomes/kg) resulted in sparse, widely scattered GFP-positive cells across a wide range of brain structures (Figure 5g–i). Although the ability of AAV9 to cross the intact BBB agrees with previous studies,16,17 the lower dose employed in this study (4.3 × 1012 viral genomes/kg) transduced far fewer cells than previously reported for a higher dose of AAV9 (2 × 1014 viral genomes/kg) (ref. 16). Moreover, in contrast to AAV8, AAV9 transduced only a few widely scattered cells in the entire piriform cortex.
Figure 5. Transduction patterns of adeno-associated virus (AAV) 1, 8, and 9 in rat brain 2 weeks after class IV seizures and intravenous vector administration.
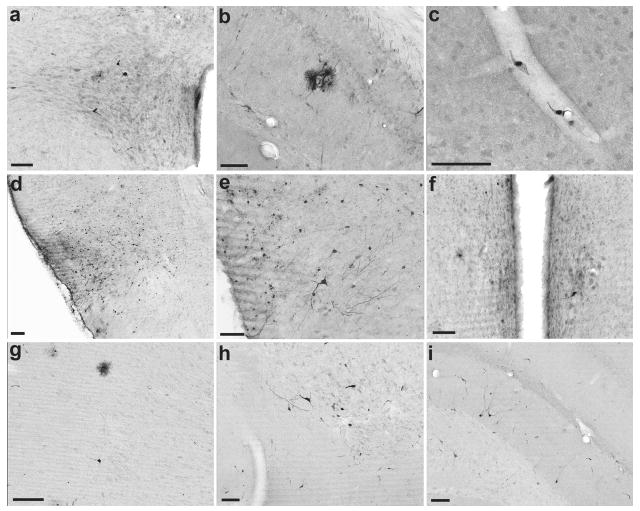
(a–c) Systemic injection of AAV1 resulted in transduction of endothelial cells predominantly, with sporadic labeling of neurons and glia throughout the brain. (a) Neurons in the paraventricular nucleus of the hypothalamus; (b) green fluorescent protein (GFP)-labeled cells (presumably astrocytes) surrounding a blood vessel in the hippocampus; (c) endothelial cells lining a capillary in the motor cortex. (d–f) Intravenous injection of AAV8 resulted in a pattern of GFP labeling that was similar to, but more widespread than clones 32 and 83. (d) Middle piriform cortex, 2.2 mm posterior to bregma; (e) caudal piriform cortex, 3.2 mm posterior to bregma; (f) neuron-like cells in the medial preoptic nucleus of the hypothalamus. (g–i) Systemic injection of AAV9 resulted in scattered GFP-labeled cells throughout the brain. (g) Cells with glial and neuronal morphology in motor cortex; (h) a cluster of neurons in the nucleus of the horizontal limb of the diagonal band; (i) neurons in the dorsal hippocampus. Bar = 100 μm for all panels.
Double-label immunofluorescence shows that clone 83 transduced both oligodendrocytes and neurons (Figure 6a,b). However, no GFP colocalization was found with astrocytes or microglia (data not shown). Similarly, double-label immunofluorescence shows that after i.v. AAV8 administration GFP expression colocalized predominantly with neurons and oligodendrocytes (Figure 6c,d). For AAV1, the vast majority of GFP-positive cells colocalized with RECA-1, a marker for endothelial cells (Figure 6e).
Figure 6. Clone 83 and adeno-associated virus (AAV)8 transduce neurons and oligodendrocytes, whereas AAV1 transduces primarily endothelial cells after intravenous administration and kainic acid injection.
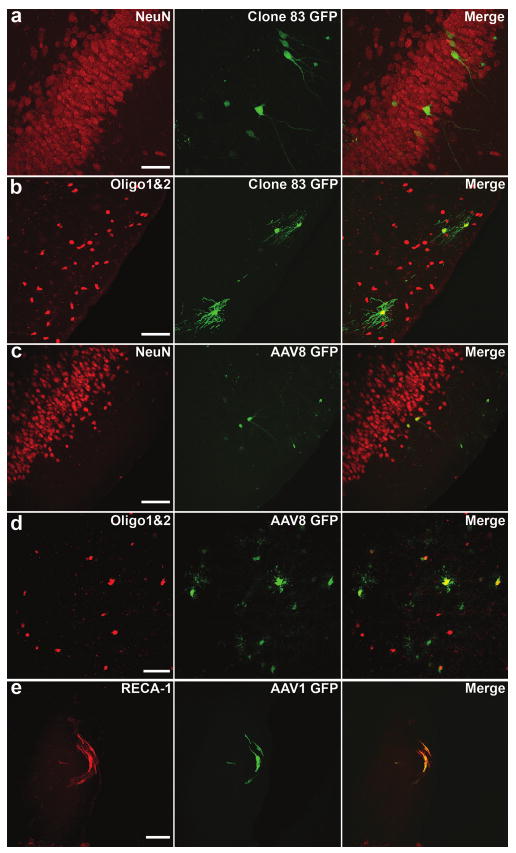
Fluorescence confocal micrographs illustrating clone 83-transduced cells [green fluorescent protein (GFP), green] colabel (a) with the neuronal marker NeuN in ventral hippocampus, and (b) with the oligodendrocyte markers Olig1 and Olig2 in the piriform cortex. Colabeling of GFP transduced by (c) AAV8 with NeuN, and (d) with Olig1 and Olig2, both in the piriform cortex. (e) The endothelial marker RECA-1 colocalizes with GFP-positive cells transduced by AAV1 in a large blood vessel near the cortical surface. Bar = 50 μm for all panels.
Biodistribution of clones 32 and 83
To assess the biodistribution of clones 32 and 83 compared to their parent serotypes, 5 × 1010 viral genomes of AAV1, 8, 9, clone 32, or clone 83 (packaging self-complementary chicken β actin-GFP genomes) was injected via the tail vein of adult mice (N = 3/group). This 2.5 × 1012 viral genome/kg body-weight dose was somewhat lower than the dose used in Figure 4 rat studies, but was required in order to deliver equivalent doses across all vectors. Biodistribution of GFP relative to mouse genomic DNA in the liver, heart, lung, kidney, spleen, skeletal muscle (gastrocnemius), and brain was performed using quantitative PCR (qPCR). Compared to AAV1, 8, and 9, clones 32 and 83 had very limited tropism for all organs tested except the spleen and kidney (Figure 7; Supplementary Tables S2 and S3). For example, the liver concentration of clone 83 was 30-fold lower than AAV1, 250-fold lower than AAV9, and 1,100-fold lower than AAV8 (Supplementary Table S2). These results suggest that clones 32 and 83 are detargeted from most systemic organs beyond that of the parent serotypes. Furthermore, the relative lack of clones 32 and 83 in the brain indicate that these clones do not cross the intact BBB.
Figure 7. Biodistribution of clones 32 and 83.
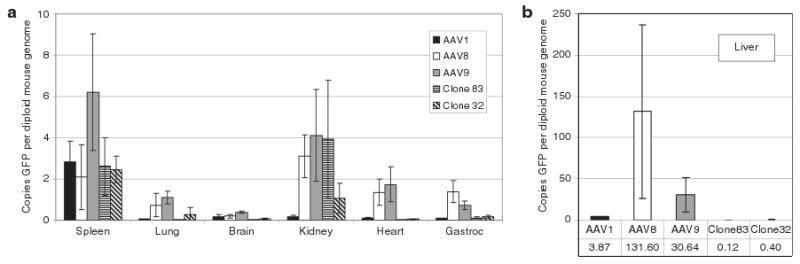
Mice were injected intravenously with 5 × 1010 vector genomes of each serotype, then after 10 days the mice were killed and DNA was isolated from the brain, heart, liver, spleen, lung, kidney, and gastrocnemius muscle. Values were calculated as copies of GFP per diploid copy of mouse genomic DNA (laminB2 locus). (a) Spleen, lung, brain, kidney, heart, and gastrocnemius muscle. (b) Biodistribution in liver (note difference in scale). Error bars indicate standard deviation. Numerical values for all groups and P values are provided in Supplementary Tables S2 and S3, respectively.
Discussion
The results from these studies clearly illustrate the vast potential of combining AAV capsid DNA shuffling with directed evolution in the CNS. The in vivo selection process was designed to be very stringent where only those capsid clones that crossed the seizure-compromised BBB would be recovered. Indeed clones 32 and 83 exhibited transduction patterns that proved localized to brain areas that experience a seizure-compromised BBB, though clone 83 appeared to transduce substantially more cells than clone 32. As importantly, these clones did not transduce cells in areas of the brain that do not exhibit a seizure-induced compromise in the BBB, such as the striatum or hypothalamus. Another unintentional but beneficial outcome involved the biodistribution patterns. Clones 32 and 83 lost the parental tropism for non-CNS organs, such as the heart, lung, and liver. By testing the parental serotypes that compose clones 32 and 83, it appears that the shuffling and selection process combined the ability of AAV8 to gain CNS access through the seizure-compromised BBB with the peripheral organ detargeting properties of AAV1.
The success of this CNS selection process depended on a unique combination of two crucial elements. In a number of previous studies where the selection occurred in vitro, the library clones were rescued and amplified using WT adenovirus.6,18 Even when clones were rescued in vivo using a “biopanning” technique, animals were infected with WT adenovirus in order to rescue the AAV clones.18 This use of WT adenovirus was not practical in the CNS for two reasons. First, WT adenovirus would need to be injected directly into the CNS, but soon after direct injection adenovirus toxicity would kill many if not most of the infected cells (T.J. McCown and R.J. Samulski unpublished results). Second, even if some cells survived the WT adenovirus infection, the selection process would be extremely biased by the specific cellular tropism of adenovirus in the CNS. Thus, mutant AAV rescue from the CNS required PCR techniques. Another key element involved the tissue selection. Even though several previous peripheral shuffling/directed evolution studies have used PCR rescue techniques, these investigations extracted DNA from whole organs19 or intact tumors.20 Whole-tissue extraction in the CNS, however, would include not only all CNS cells, but also endothelial cells of the cerebral vasculature. Thus, it would be possible to select a mutant AAV that targets the cerebral vasculature. In order to circumvent this problem, cells were mechanically dissociated from seizure-sensitive areas of the CNS, and the viral DNA was extracted. This approach allowed the extraction and amplification of AAV DNA from neuron-rich, site-specific cell populations. Given that the virus DNA was amplified by PCR, however, it must be emphasized that the vectors were not selected for their ability to transduce neurons per se, but only on their ability to penetrate the compromised BBB and bind to cells in regions modulating seizure activity. Thus, we likely recovered some clones that can bind to neurons and other contaminating cells but have defects in cellular entry, trafficking, or uncoating. For this reason, multiple clones needed to be screened rather than continuing the selection until clonal homogeneity was attained. Clearly, the present findings validate the utility of this approach.
Four previously published studies have described some level of AAV delivery to the adult rodent brain after i.v. injection of naturally occurring AAV vectors. Foust et al.16 and Duque et al.17 delivered high doses (2 × 1014 vector genomes/kg and 1 × 1014 vector genomes/kg, respectively) of recombinant AAV9 (rAAV9) to achieve widespread transduction of cells throughout the brain. Towne et al.21 injected 1 × 1013 vector genomes/kg of rAAV6 to achieve a lower level of widespread brain transduction, whereas more recently McCarty et al.22 found widespread brain transduction by optimizing the time interval between mannitol BBB disruption and subsequent i.v. rAAV2 administration (2 × 1013 vector genomes/kg). Although in all of these cases substantial transduction was achieved, the patterns of transduction lacked any regional specificity. In contrast, clones 32 and 83 only transduced cells in areas known to experience seizure-induced BBB compromise. This specific pattern would restrict gene expression to those areas of seizure-induced pathology, a property that should minimize unwanted side effects. Also, in the case of clone 83, this selective transduction was achieved using 3.5 × 1012 vector genomes/kg, a dose that contained 3- to 60-fold fewer viral particles, when compared to these previous studies.
Another beneficial property of clones 32 and 83 involves the dramatic divergence in biodistribution from that of the parental serotypes. Unlike AAV8 or 9, clones 32 and 83 exhibited virtually no tropism for the liver, heart, muscle, or lung. Also, although AAV1 exhibits a biodistribution that is similar to clones 32 and 83, the liver concentration of clone 83 was 30-fold less than the AAV1 concentration (0.12 genomes/cell versus 3.87 genomes/cell; Supplementary Table S2). This substantial reduction in peripheral biodistribution proves to be quite important in light of studies on AAV-mediated liver transduction. In recent clinical trials for hemophilia B, when rAAV2/factor IX vector was delivered to the liver at doses of 4 × 1011 vector genomes/kg or 2 × 1012 vector genomes/kg, an asymptomatic immune response to the capsid cleared the vector and the accompanying gene expression.23,24 Furthermore, in nonhuman primates intraportal administration of rAAV7 at doses ranging from 0.7 to 3.5 × 1012 vector genomes/ kg provoked a cytotoxic T-cell response, which led to a loss of transgene expression.25 In a separate clinical trial for hemophilia B, this immune response was not observed in the muscle at doses up to 1.8 × 1012 vector genomes/kg, where the rAAV2 vector was well tolerated and transgene expression persisted for over 3 years (ref. 26). These findings suggest that liver transduction likely poses a major barrier to systemic delivery in humans. The apparent inability of clones 32 and 83 to accumulate in the liver should minimize immune responses to these mutant AAV vectors following peripheral administration.
Tests of the parental serotypes provided important insight into the structural features of the capsid that allowed the mutant AAV vectors to cross a seizure-compromised BBB, as well as detarget peripheral organs. I.v. administration of AAV8 after kainic acid–induced seizures produced a pattern of CNS transduction that was highly similar to the pattern produced by clones 32 and 83. Thus, it appears that the AAV8 component of clones 32 and 83 conveys the ability to cross the seizure-compromised BBB. This conclusion is reinforced by the observation that clones 32 and 83 were the only clones where the AAV8 component extended to the C-terminus of VP3. Although cerebrovascular endothelial cells comprised the dominant cell type transduced by AAV1 after i.v. administration, AAV1 exhibited a biodistribution pattern that was far more similar to clones 32 and 83 than to AAV8 and 9. Thus, the AAV1 portion of clones 32 and 83 likely contributed to this peripheral detargeting. However, the concentration of clone 83 was significantly lower than that of AAV1 in all organs tested except the spleen and gastrocnemius muscle (Supplementary Table S3). Therefore, other factors, such as PCR point mutations, also must contribute to the peripheral detargeting of clone 83.
In conclusion, the application of AAV capsid DNA shuffling and directed evolution allowed the identification of two mutant AAV clones that upon i.v. administration, selectively cross the seizure-compromised BBB and transduce cells. Coincident with this property was the unexpected but advantageous finding that these clones also exhibit a unique, favorable biodistribution. Future studies will focus on both the therapeutic efficacy of peripheral administration, as well as further mutagenesis focused on increasing the transduction efficacy. Thus, these studies not only illustrate the power of combining capsid DNA shuffling and directed evolution, but also provide a potentially novel avenue of seizure gene therapy.
Materials and Methods
DNA shuffling and directed evolution
The original library used consisted of shuffled capsids from AAV serotypes AAV serotypes 1–6, 8, 9, and an AAV8 with E531K mutation. It was produced exactly as previously described.6 Rats received an injection of kainic acid (10 mg/kg, intraperitoneally), and within 70–100 minutes after kainic acid treatment, the rats exhibited class IV limbic seizure activity as described by Racine27 (raising up with forelimb clonus). Approximately 24 hours after kainic acid treatment, the shuffled library was injected into the tail vein (0.5 ml). Three days later, the rats were killed and cells were mechanically dissociated from the piriform cortex and dorsal hippocampus of 400-μm thick brain slices as previously described.14 Briefly, the slices were transferred to a recording chamber containing HEPES-buffered artificial cerebrospinal fluid (145 mmol/l NaCl, 5 mmol/l KCl, 10 mmol/l HEPES, 2 mmol/l CaCl2, and 10 mmol/l glucose, pH 7.4: 340 mOsM/l). Then, a 0.3-mm glass probe was lowered to just touch the surface of the submerged tissue over either the pirirform cortex or the dorsal hippocampus. The probe was vibrated using a piezoelectric transducer (∼0.5 mm amplitude at 10–50 Hz) for 2 minutes. The dissociated cells were collected from the artificial cerebrospinal fluid. DNA was recovered from neuron-rich samples using a DNeasy blood and tissue kit (cat. no. 69506; Qiagen, Valencia, CA) and subsequently concentrated by ethanol precipitation. Error-prone PCR was employed to further diversify the library between rounds, using the Expand Long Template PCR System (cat. no. 11681834001, low starting template, 50 cycles; Roche, Indianapolis, IN) with primers previously described.6 The pooled PCR products were cloned back into a WT AAV backbone (pSSV9) and pooled clones were used to generate the next round's starting library. Pooled clones were transfected into HEK293 cells with an adenovirus helper plasmid (pXX680) and a 10-fold excess of pXR2 containing AAV2 Rep and Cap. By this method, chimeric capsid genomes were packaged into mostly AAV2 capsids. The infectious titer of the AAV2-encapsidated chimeric library was determined using an infectious center assay.28 Then, the AAV2-encapsidated chimeras were added to HEK293 cells at a multiplicity of infection of 0.5 with WT adenovirus at a multiplicity of infection of 5 to predominantly package each chimeric AAV genome in its own capsid. After 72 hours, the cells were harvested and the virus purified as described28 and titered by qPCR. A total of three rounds of selection were performed. A sampling of recovered capsids after each round were subcloned into rAAV pXR2 backbones and SSV9 replication-competent backbones and sequenced.
Viability of recovered clones
Due to the mutagenesis introduced during PCR capsid recovery, many of the recovered capsids were defective for viral production. The recovered capsid sequences were screened for those able to produce virus. Approximately 60% confluent HEK293 cells in 24-well plates were transfected with the adenovirus helper plasmid pXX680 and pSSV9-based plasmids containing either AAV2 capsid or individual recovered chimeric capsids. pXX680 was co-transfected with pUC18 as a negative control. After 72 hours, cells were harvested and pelleted, then resuspended in 100 μl DNase I digestion buffer (10 mmol/l Tris–HCl, pH 8; 10 mmol/l MgCl2, 2 mmol/l CaCl2). After three freeze/ thaw cycles (−80 °C for 15 minutes, 50 °C for 30 minutes), the lysate was centrifuged at 13,000 rpm for 10 minutes and the supernatant retained. Lysates were incubated with 2 μl DNase I at 37 °C for 2 hours to remove unincorporated genomes, and then the reactions were stopped by adding 6 μl 0.5 mol/l EDTA and boiling for 10 minutes. A volume of 92 μl of Proteinase K solution (1 mol/l NaCl, 1% sarkosyl, 0.1 mg/ml Proteinase K) was added to each tube, and incubated at 50 °C for at least 2 hours to liberate the viral genomes. The reaction mixtures were diluted 1:1,000 and 1:10,000 in water and used directly as template for qPCR reactions. Those clones showing titers equivalent to pSSV9 (AAV2) were considered viable for production of virus.
Production of rAAV
rAAV vectors lack the Rep and Cap genes necessary for competent, independent replication, and the rAAV vectors were produced by the University of North Carolina Vector Core using a triple-transfection method in HEK293 cells as described.28 All recombinant vectors packaged self-complementary genomes with enhanced GFP under the control of a miniature chicken β actin promoter and cytomegalovirus enhancer. Highly pure recombinant virus containing self-complementary genomes were recovered using two sequential CsCl gradients; then the peak fractions were dialyzed in phosphate-buffered saline (PBS) containing 5% D-sorbitol. Viral titers were determined by both dot blot28 and qPCR.
CNS site microinfusions
All care and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals [DHHS publication no. (NIH)85-23], and all procedures received prior approval by the University of North Carolina Institutional Animal Care and Usage Committee. The animals were pathogen-free male Sprague-Dawley rats (250–350 g) obtained from Charles River Laboratories (Wilmington, MA). The animals were maintained in a 12-hour light–dark cycle and had free access to food and water. For virus vector infusions, the rats (N = 2 per vector) were first anesthetized with 50 mg/kg pentobarbital and placed into a stereotaxic frame. Using a 32-gauge stainless steel injector and a Sage infusion pump, animals received 1 μl of each vector (clones 3, 32, 74, 83, 84, AAV1, 8, or 9) over 10 minutes into the piriform cortex (interaural line 6.7 mm, lateral 6.0 mm, vertical 7.4 mm, according to the atlas of Paxinos and Watson29). The injector was left in place for 5 minutes after infusion to allow diffusion from the injectors. The incision was sutured, and the animals were allowed 2 weeks to recover.
Immunohistochemistry, microscopy, and image processing
To assess BBB integrity, rats were deeply anesthetized with 100 mg/kg pentobarbital intraperitoneally 24 hours after kainic acid seizure induction. Subsequently, the rats (N = 3) were infused intracardially with 10 ml/kg fluorescein isothiocyanate–albumin (5 mg/ml; Sigma, St Louis, MO) as previously described.10 The brains were removed and immediately immersed into a solution of 4% paraformaldehyde in 100 mmol/l phosphate buffer (pH 7.4). After immersion fixation for 48 hours at 4 °C, 40-μm thick coronal sections were cut using a vibrating blade microtome and mounted onto glass slides. Fluorescein isothiocyanate fluorescence was visualized by confocal microscopy as described below.
Two weeks after intracerebral or intravascular injection of shuffled clones, animals received an overdose of pentobarbital (100 mg/kg intraperitoneally) and were perfused transcardially with ice-cold 100 mmol/l sodium PBS (pH 7.4), followed by 4% paraformaldehyde in phosphate buffer (pH 7.4). After brains were postfixed 12–48 hours at 4 °C in the paraformaldehyde–phosphate buffer, 40-μm thick coronal sections were collected as described above and subjected to immunohistochemical processing for GFP and neural cell markers.
Colorimetric GFP detection was performed by incubating free-floating sections with a rabbit polyclonal antibody to GFP (1:500; Millipore, Billerica, MA). Briefly, sections were blocked in 10% normal goat serum and 0.1% Triton X-100 in PBS for 1 hour. The sections were then incubated with anti-GFP primary antibody (prepared in 3% normal serum, 0.2% Triton X-100) for 48–72 hours at 4 °C with gentle agitation. Immunoperoxidase staining was then performed using the anti-rabbit Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) as described in the manufacturer's instructions, with 3,3′-diaminobenzidine substrate and nickel–cobalt intensification of the reaction product. Brain sections were mounted on slides, coverslipped, and digitized using a Scan-Scope slide scanner (Aperio Technologies, Vista, CA). Virtual slides were viewed using ImageScope software package (v. 10.0; Aperio Technologies), which allowed sections to be viewed at equivalent magnifications. Images were digitally adjusted for contrast and brightness and assembled into figures in Adobe Photoshop CS, v8.0.
For immunofluorescence colocalization, sections were incubated simultaneously with anti-GFP (prepared as described above) and one of the following monoclonal antibodies to cellular markers: the neuronal marker NeuN (1:500; Chemicon, Temecula, CA); the microglial marker OX-42 (1:500; AbD Serotec, Oxford, UK); the astrocyte marker glial fibrillary acidic protein (1:4,000; Dako, Glostrup, Denmark), a combination of markers for oligodendrocytes consisting of Olig1 (1:500; Chemicon) and Olig2 (1:500, Millipore), or the endothelial cell marker RECA-1 (MCA970R, 1:500; AbD Serotec). Following incubation for 48–72 hours in primary antibodies, the sections were rinsed with PBS and blocked again for 30 minutes at room temperature in PBS containing 10% goat serum. Sections were then co-incubated for 45 minutes at 4 °C with Alexafluor 488-conjugated goat-anti-rabbit IgG (1:500; Invitrogen, Carlsbad, CA) and Alexafluor 594-conjugated goat anti-mouse IgG (1:500; Invitrogen) in PBS containing 3% goat serum. For the astrocyte marker, the polyclonal glial fibrillary acidic protein antibody was detected using Alexafluor 594-conjugated goat anti-rabbit IgG (1:500; Invitrogen) while native GFP was visualized. Rinsed sections were mounted and fluorescence was visualized by confocal microscopy.
Confocal imaging was performed at the Michael Hooker Microscopy Center at UNC-Chapel Hill using a Zeiss Axiovert LSM510 laser confocal microscope (Carl Zeiss Jena, Jena, Germany). Images were processed using Zeiss LSM Image Browser (Carl Zeiss Jena) and Adobe Photoshop software. Fluorescein isothiocyanate–albumin and dual-label immunofluorescence images were reconstructed from a minimum of five consecutive steps in a Z-series taken at 1-μm intervals through the section of interest using a ×40 objective.
Biodistribution
All care and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals [DHHS publication no. (NIH)85-23], and all procedures received prior approval by the University of North Carolina Institutional Animal Care and Usage Committee. Six-month-old female BALB/C mice purchased from Jackson Laboratory (Bar Harbor, ME) each were injected in the tail vein with 5 × 1010 vg of vector (∼2.5 × 1012 vg/kg body weight) in 200 μl of PBS with 5% d-sorbitol. Each serotype was injected into three mice. After 10 days, mice were killed and the brain, lungs, heart, liver, kidney, spleen, and gastrocnemius muscle were harvested for biodistribution analysis of the GFP vectors. Total DNA from each organ was extracted with a DNeasy blood and tissue kit (cat. no. 69506; Qiagen) and total copies of GFP and mouse genomic DNA were determined for each by qPCR. Biodistribution of clones 32 and 83 were compared to their parent AAV serotypes 1, 8, and 9 using Student's t-test (unpaired, one-tailed), and P values are presented in Supplementary Table S3.
qPCR
qPCR was used to determine viral titer and for biodistribution studies. All reactions were done using the SyBR Green-based Lightcycler fast start DNA master mix (cat. no. 03003230001; Roche) on a Roche 480 Lightcycler instrument, following the manufacturer's instructions. To prepare virus samples for titer, they were treated with DNase I for 1 hour, then the DNase I was inactivated with the addition of EDTA and heating at 70 °C for 10 minutes. To liberate the encapsidated genomes for qPCR analysis, the reaction mixtures were digested with Proteinase K for at least 2 hours at 50 °C, then boiled for 10 minutes to inactivate the Proteinase K. Samples were diluted in PCR-grade water and used as template for qPCR reactions. For GFP or virus quantitation, plasmid DNA was used as the standard. For quantification of mouse genomic DNA, purified and quantified mouse genomic DNA was used as a standard. All successful reactions gave a single product by melting curve analysis, used a standard curve with an R2 value of 1, and had a reaction run in parallel containing no template that gave no product. GFP primers were as follows: (forward) 5′-AGCAGCACGACTTCTTCAACTCC-3′, (reverse) 5′-TGTAGTTGTACTCCAGCTTGTGCC-3′. Rep primers for quantitation of library virus were as follows: (forward) 5′-TCCATGGTTTT GGGACGTTTCC-3′, (reverse) 5′-TTAGTCCACGCCCACTGGAG-3′. LaminB2 primers for quantitation of mouse genomic DNA were as follows: (forward) 5′-GTTAACACTCAGGCGCATGGGCC-3′, (reverse) 5′-CCATCAGGGTCACCTCTGGTTCC-3′.
Supplementary Material
Figure S1. Mechanical dissociation of AAV2-GFP positive neurons from the piriform cortex.
Figure S2. Immunohistochemical analysis of GFP transgene expression, 2 weeks following injections of AAV1, AAV8 or AAV9 into piriform cortex.
Table S1. Recovered clones.
Table S2. Biodistribution of clones 32 and 83 relative to their parent serotypes: quantitation.
Table S3. Biodistribution of clones 32 and 83 relative to their parent serotypes: statistical significance.
Acknowledgments
We thank Huijing Sun and Swati Yadav for expert technical assistance and the UNC Gene Therapy Vector Core for recombinant vector production. We also thank James Wilson at the University of Pennsylvania for providing AAV2/8 and AAV2/9 helper plasmids. These studies were supported by NINDS grant NS059518 (T.J.M.) and the International Rett Syndrome Foundation (S.J.G.).
References
- 1.Haberman RP, Samulski RJ, McCown TJ. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nat Med. 2003;9:1076–1080. doi: 10.1038/nm901. [DOI] [PubMed] [Google Scholar]
- 2.McCown TJ. Adeno-associated virus-mediated expression and constitutive secretion of galanin suppresses limbic seizure activity in vivo. Mol Ther. 2006;14:63–68. doi: 10.1016/j.ymthe.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Richichi R, Lin ED, Stefanin D, Colella D, Ravizza T, Grignaschi G, et al. Anticonvulsant and antiepileptic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24:3051–3059. doi: 10.1523/JNEUROSCI.4056-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foti SB, Russek ST, Brooks-Kayal AR, McCown TJ. Viral vector gene therapy for epilepsy. In: Baraban SC, editor. Animal Models of Epilepsy: Methods and Innovations. Humana Press; New York: 2009. pp. 235–250. [Google Scholar]
- 5.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 6.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 7.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 8.Nitsch C, Klatzo I. Regional patterns of blood-brain barrier breakdown during epileptiform seizures induced by various convulsive agents. J Neurol Sci. 1983;59:305–322. doi: 10.1016/0022-510x(83)90016-3. [DOI] [PubMed] [Google Scholar]
- 9.Ruth RE. Increased cerebrovascular permeability to protein during systemic kainic acid seizures. Epilepsia. 1984;25:259–268. doi: 10.1111/j.1528-1157.1984.tb04185.x. [DOI] [PubMed] [Google Scholar]
- 10.Marchi N, Oby E, Batra A, Uva L, De Curtis M, Hernandez N, et al. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48:1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liwnicz BH, Leach JL, Yeh HS, Privitera M. Pericyte degeneration and thickening of basement membranes of cerebral microvessels in complex partial seizures: electron microscopic study of surgically removed tissue. Neurosurgery. 1990;26:409–420. doi: 10.1097/00006123-199003000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Hufnagel A, Weber J, Marks S, Ludwig T, De Greiff A, Leonhardt G, et al. Brain diffusion after single seizures. Epilepsia. 2003;44:54–63. doi: 10.1046/j.1528-1157.2003.07802.x. [DOI] [PubMed] [Google Scholar]
- 13.van Viliet EA, da Costa Arujo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- 14.Criswell HE, Ming Z, Kelm MK, Breese GR. Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J Pharmacol Exp Ther. 2008;326:596–603. doi: 10.1124/jpet.107.135418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Z, Asokan A, Grieger JC, Govindasamy L, Agbandje-McKenna M, Samulski RJ. Single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. J Virol. 2006;80:11393–11397. doi: 10.1128/JVI.01288-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci USA. 2009;106:3946–3951. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michelfelder S, Kohlschütter J, Skorupa A, Pfennings S, Müller O, Kleinschmidt JA, et al. Successful expansion but not complete restriction of tropism of adeno-associated virus by in vivo biopanning of random virus display peptide libraries. PLoS ONE. 2009;4:e5122. doi: 10.1371/journal.pone.0005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Towne C, Raoul C, Schneider BL, Aebischer P. Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol Ther. 2008;16:1018–1025. doi: 10.1038/mt.2008.73. [DOI] [PubMed] [Google Scholar]
- 22.McCarty DM, DiRosario J, Gulaid K, Muenzer J, Fu H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009;16:1340–1352. doi: 10.1038/gt.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 24.Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2007;7:316–324. doi: 10.2174/156652307782151425. [DOI] [PubMed] [Google Scholar]
- 25.Gao G, Wang Q, Calcedo R, Mays L, Bell P, Wang L, et al. Adeno-associated virus-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum Gene Ther. 2009;20:930–942. doi: 10.1089/hum.2009.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 27.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 28.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 29.Paxinos G, Watson G. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mechanical dissociation of AAV2-GFP positive neurons from the piriform cortex.
Figure S2. Immunohistochemical analysis of GFP transgene expression, 2 weeks following injections of AAV1, AAV8 or AAV9 into piriform cortex.
Table S1. Recovered clones.
Table S2. Biodistribution of clones 32 and 83 relative to their parent serotypes: quantitation.
Table S3. Biodistribution of clones 32 and 83 relative to their parent serotypes: statistical significance.