

Abstract
Recognition of psychostimulants such as cocaine and the amphetamines by the dopamine transporter (DAT) protein is principally responsible for the euphoria and addiction associated with these drugs. Using as a template the crystal structure of a distantly related bacterial leucine transporter, 3-D DAT computer molecular models have been generated. Ligand docking to such models has revealed potential substrate and inhibitor binding pockets, subsequently confirmed by in vitro pharmacology. An inhibitor pocket defined by the DAT model to be within the “extracellular vestibule”, just to the extracellular side of the external gate of the primary substrate pocket, was used for virtual screening of a structural library of compounds. High-throughput docking and application of pharmacophore constraints within this vestibular inhibitor pocket identified a compound structurally dissimilar to the classic monoamine (dopamine, norepinephrine and serotonin) transporter (MAT) inhibitors. The compound displaced binding of radiolabeled cocaine analogs at all three MATs, usually with nanomolar Ki values and within two fold of cocaine's affinity at the norepinephrine transporter. Although a very weak dopamine uptake inhibitor itself, this compound reduced by three fold the potency of cocaine in inhibiting DAT-mediated cellular uptake of dopamine. To our knowledge, the present findings are the first to successfully employ “receptor-based” computer modeling to identify moderate-to-high affinity MAT ligands. In silico ligand screening using MAT models provides a rapid, low cost discovery process that should accelerate identification of novel ligand scaffolds and provide lead compounds in combating psychostimulant addiction and in treating other monoamine-related CNS diseases.
Keywords: addiction, cocaine, docking, pharmacophore, virtual screening, neurotransmitter transporter
Introduction
Addiction to cocaine is a worldwide scourge for which there are few answers. Cocaine use is associated with alertness, increased energy and motor activity, enhancement of sensation, and euphoria (1). The reinforcing effects of cocaine and its analogs generally correspond with the ability of the drugs to inhibit the dopamine transporter (DAT). Blockade of this protein increases synaptic levels of the neurotransmitter dopamine in the nucleus accumbens and other brain regions critical to euphoria and addiction (2). Cocaine also increases synaptic concentrations of serotonin and norepinephrine by blockade of the cognate transporters, resulting in stimulant and mood-altering effects. Despite extensive research, no therapeutic is available at this time to manage cocaine abuse and addiction; there is great interest in finding such a medication. The computational approach described herein employs an in silico screening system toward identifying novel DAT ligands and possibly anti-cocaine therapeutic lead compounds.
A DAT 3-D molecular computer model was previously constructed using comparative modeling methods (3, 4) with the bacterial leucine transporter protein LeuT as a template (5). Substrates and inhibitors appear to have a primary DAT binding pocket, recently labeled “S1” (6), midway through the lipid bilayer, between internal and external ligand gating residues (3, 4). Ligand docking studies and subsequent mutagenesis/pharmacology revealed a secondary substrate pocket for the DAT, located several angstroms to the extracellular side of the primary substrate binding pocket (4, 7). The secondary pocket, in the DAT extracellular vestibule just above the external gate of the primary pocket, approximately colocalizes with that for imipramine and related tricyclic antidepressant (TCA) drugs (8, 9) and serves as a substrate staging area, or “waiting room”, prior to passage through the external gate (4, 7). Our docking of classic DAT inhibitors including cocaine (Figure 1), methylphenidate (Ritalin™) and benztropine (Cogentin™) resulted in the ligands binding in the extracellular vestibule (S2). A detergent molecule that inhibits transport has recently been confirmed to bind in this secondary substrate pocket (labeled “S2”) for LeuT (6).
Figure 1.
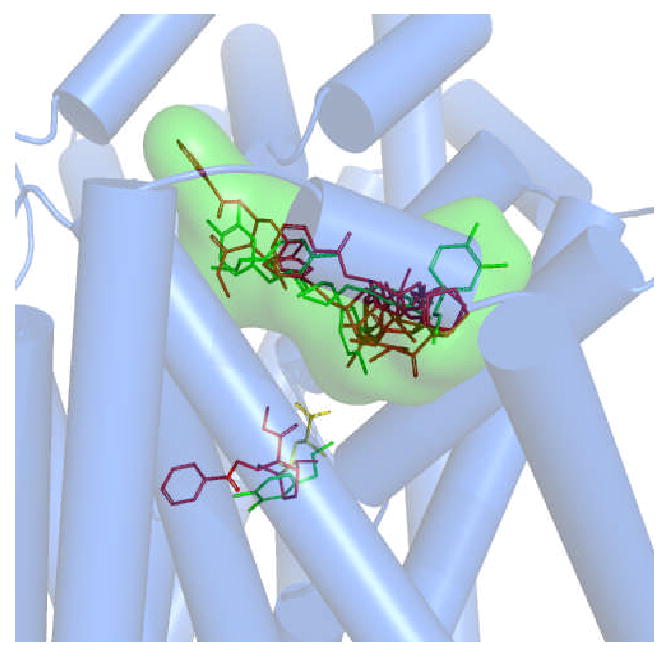
Confirmed and postulated substrate and inhibitor binding pockets within a DAT molecular model. A superposition of DAT-ligand docking poses is shown. Leucine (yellow sticks) is positioned in the pocket analogous to that found in the LeuT crystal structure (5). The primary binding pocket (S1) for dopamine (green sticks) colocalized with that of the analogous leucine pocket (4). A secondary dopamine pocket (S2) was found in the extracellular vestibule (4, 7). The primary inhibitor site is represented by cocaine (red sticks), overlapping the primary substrate binding site (3). The secondary (vestibular) inhibitor pocket used for in silico screening is delineated by a Connolly surface (green cloud); various poses of docked cocaine and dopamine are shown. MOE 2007 was used to generate the models and docking and the Pymol (DeLano Scientific) educational version (2007) was used to render the complex.
It is debatable if a therapeutic drug can be obtained that blocks DAT binding of cocaine without also blocking dopamine uptake; a compound that blocks both would be predicted to be another abused psychostimulant. The cramped primary substrate/inhibitor pocket (S1) probably does not allow for such a therapeutic (3). The vestibular secondary pocket (coinciding with S2), on the other hand, provides more opportunities for preferred inhibition of a nonsubstrate ligand. In the present work, the DAT vestibular cocaine pocket, has been defined by docking iterations and pharmacophore filtering. The refined pocket was next used in virtual screening (VS) of a structural library of compounds. A handful of VS “hits” was then screened in vitro for DAT affinity and inhibition of dopamine uptake.
Results and Discussion
DAT Model Vestibular Binding Pocket Ligand Docking Validation Using Classic DAT Inhibitors
The ultimate destination of cocaine and its analogs appears to be the more interior, primary substrate binding site of the DAT (3); however, no inhibitor has been reported to reach a monoamine transporter (MAT) primary substrate binding site via docking iterations. This may be due to interference from the external gate MAT residues, or due to a focus on the wrong transporter conformation. As a caveat, the field is working under the assumption that the outward-facing LeuT model derived from the 2A65 X-ray structure (5) is capable of binding nonsubstrate inhibitors. The recent publication of the nucleobase-cation-symporter-1 (10) and Na+/betaine symporter (11) structures offer other possible conformations for 12 TM domain transporters. Conceivably, MAT proteins could assume these conformations to provide high affinity inhibitor sites. These possibilities were not explored here. Given that DAT inhibitors only docked in the secondary substrate pocket (S2) found in the external vestibule, this pocket was chosen as the focus for the in silico ligand screen. This relatively roomy vestibular pocket presumably provides a greater chance for a high affinity compound to block cocaine binding and still allow some substrate access to the permeation pore.
X-ray structures of LeuT complexed with different TCA drugs (PDBs 2QEI, 2Q72, 2Q6H and 2QB4) (8, 9) were utilized to assess the best docking/scoring protocol for high throughput VS (HTVS) at the vestibular ligand pocket of our DAT molecular model (4). The X-ray structure of LeuT complexed with clomipramine (PDB 2QEI) was first compared to the previously documented vestibular DAT pocket (4). Superposition and binding pocket analysis of the DAT comparative model and the LeuT X-ray structure revealed several DAT residues positioned similarly to their LeuT analogs (identified in Figure 2). In contrast, the DAT residues Trp84 (Leu29 in LeuT), Pro386 (Phe320 in LeuT), and Tyr88 (Val33 in LeuT) diverged in orientation from their LeuT counterparts. A pairwise percentage residue identity calculation for the vestibular binding pocket residues yielded 43% identity between the DAT model and LeuT X-ray based model, twice that of the 21% overall sequence identity between the proteins. The two models displayed considerable overlap of vestibular binding site residues, with a root mean square deviation (RMSD) correlation matrix of approximately 1 Ǻ (Figure 2).
Figure 2.
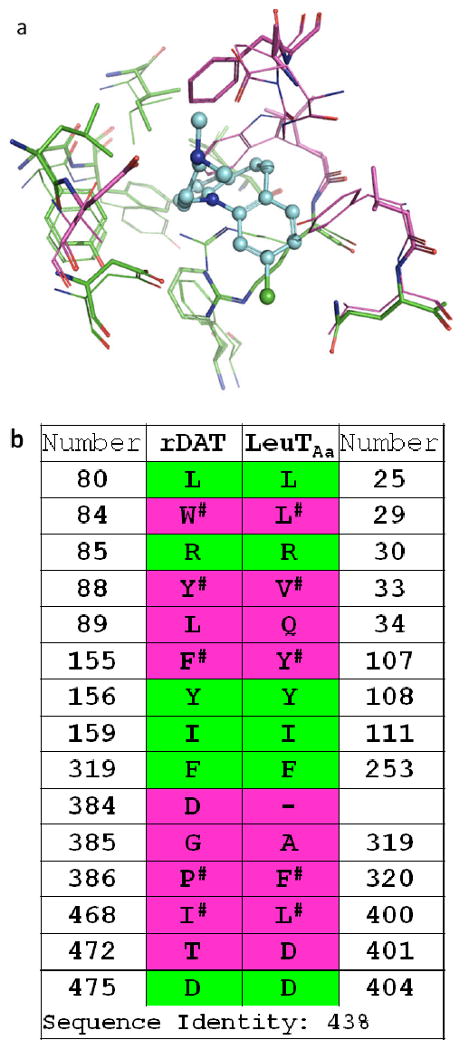
DAT – LeuT conservation of putative vestibular binding pocket residues. Panel a: The DAT comparative model (lines) and an X-ray structure-based LeuT model (sticks) are superposed with docked clomipramine (cyan/blue, ball-and-stick). Residues within 5 Ǻ of the ligand are pictured; a RMSD correlation matrix of approximately 1 Ǻ was observed. Panel b: Pairwise sequence alignment and consensus calculation of putative binding pocket residues. For residues in analogous positions, sequence identity (green) and divergence (pink) is indicated in both panels. Analogous residues that are nonidentical but similar with respect to hydrophobic, aromatic or polar features are indicated (#).
Because the DAT and LeuT vestibular ligand pockets correlated well, the RMSDs of LeuT-TCA crystals versus the predicted TCA-bound LeuT conformations were calculated for the different methods. The triangle matcher with GRID or Forcefield pose refinement in combination with the Affinity dG scoring function demonstrated a robust correlation between RMSD and scores (the more negative the score, the lower the RMSD of predicted poses vs. X-ray bound ligands). This docking/scoring method was employed to predict binding modes of DAT inhibitors within the protein. Once the method with the best predictive ability was found, docking studies employed the DAT inhibitors cocaine, WIN 35,428, oxa-norcocaine, 4-ARA-127, benztropine, GBR 12,909, methylphenidate and mazindol. The ultimate goal of this validation was to assess the DAT homology model reliability based on pKi calculations, the only parameter that offered experimental values (12). The scoring function (Affinity dG) was employed to set a score threshold for the selection of possible inhibitors, and was based on well-documented DAT inhibitors. Intuitively, compounds with better score profiles than the established inhibitors might act as DAT ligands. The pKi calculation and score of each candidate pose was aimed to enhance the chances of finding active compounds. Compounds with low scores (below “-5”, approximately) and/or higher predicted pKi values (above “+5”, approximately) were retained for further inspection and eventual pharmacological testing. The DAT model performed optimally with respect to pKi calculations for cocaine and analogs, and was slightly less accurate for other DAT ligands (Table 1).
Table 1.
Experimental and theoretical calculations using known DAT ligands.
| Drug | E pKi[a] | T pKi[b] | Score |
|---|---|---|---|
| Cocaine | 6.89 | 7.10 ± 0.19 | -6.20 ± 0.67 |
| 8-Oxa-norcocaine | 5.81 | 5.77 ± 0.09 | -6.21 ± 0.10 |
| WIN 35,428 | 7.70 | 7.00 ± 0.37 | -6.12 ± 0.27 |
| 4-ARA-127 | 5.48 | 6.67 ± 0.28 | -5.18 ± 0.18 |
| Methylphenidate | 7.24 | 7.21 ± 0.27 | -5.57 ± 0.21 |
| Mazindol | 7.89 | 8.82 ± 0.26 | -6.04 ± 0.27 |
| Benztropine | 6.90 | 7.66 ± 0.20 | -6.62 ± 0.16 |
| GBR 12,909 | 7.51 | 9.91 ± 0.33 | -8.15 ± 0.11 |
(Experimental pKi reported by Ukairo et al. (12)
(Theoretical pKi)
HTVS Docking with Pharmacophore Filtering
The difficulty in discovering bioactive compounds increases when virtual screening employs comparative protein models with low sequence identity to the template and disallows protein flexibility during the docking iterations. In order to surmount this limitation, the introduction of a pharmacophore model with spherical features of approximately 1.5 Ǻ as a pre-filter for docking was created within the vestibular pocket of the DAT model. This was meant to recapture some of the structural flexibility sacrificed due to rigid protein docking.
A broad chemical database of approximately 140,000 compounds was culled from the Sigma-Aldrich catalog using in silico filters that invoked Lipinski's Rule of Five and excluded toxic functional groups (13). The resultant filtered database, now consisting of “drug-like” compounds, was utilized in high throughput docking with the pharmacophore filter. The docking site (DAT vestibular binding pocket) harbored the pharmacophore query with five features (Figure 3): F1, F2 and F3, donor/cation interaction; F4, donor/acceptor interaction; F5, excluded volume. These features can be linked to amino acids with ligand binding potential and that are highly conserved between the LeuT X-ray structures and DAT model (Figure 2). The transmembrane domain (TM) 1 and TM 10 residues Arg85, Asp475 and Thr472 showed potential for cation-π stacking (Arg85), hydrogen bonding (Arg85, Asp475 and Thr472) or hydrophobic interactions (Arg85 and Thr472). A final visual inspection of the top-scoring compounds (Affinity dG) retrieved ∼100 compounds; re-docking and a final visual inspection retained ∼50 compounds. Ten of these compounds (coded MI-1 through MI-10) were selected and purchased based on their optimal scores, predicted pKi values, visual binding site fitting, and to some extent price and availability (Figure 4).
Figure 3.
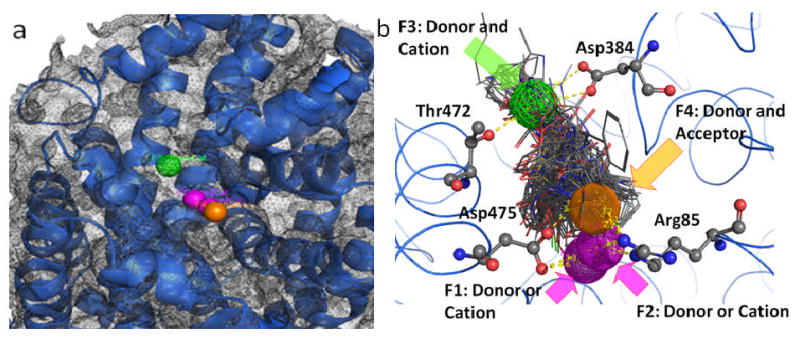
Pharmacophore query for the vestibular DAT binding pocket obtained by inhibitor and substrate docking. Panel a: Five pharmacophoric features were created: F1 and F2, donor/cation (pink spheres); F3, donor/cation (green wire mesh sphere); F4, donor/acceptor (orange sphere); F5, excluded volume (grey wire mesh spheres). Panel b: The amino acids with potentiality for ligand binding (Arg85, Asp384, Thr472 and Asp475) and their spatial relationship with the pharmacophoric features are depicted as atom colored sticks. Possible H-bond interactions with pharmacophore features are depicted as yellow dashed lines.
Figure 4.
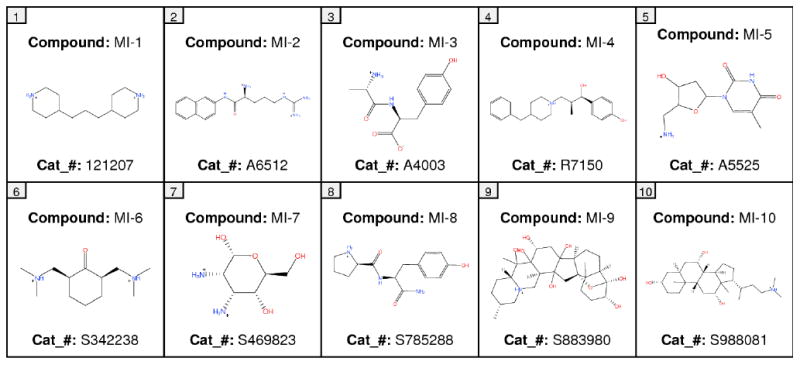
Two-dimensional representations and Sigma-Aldrich catalog numbers (cat_#) for the VS hit compounds MI-1 to MI-10.
Pharmacology
DAT binding affinity and dopamine uptake inhibition potency (DUIP) for the ten VS “hit” compounds purchased were initially assessed via [3H]-WIN 35,428 (cocaine analog) displacement and [3H]-dopamine uptake inhibition assays, respectively, in N2A neuroblastoma cells stably expressing wild type hDAT. The pre- and co-incubation of compounds with [3H]-WIN 35,428 in initial binding displacement assays demonstrated that only MI-4 was able to block [3H]-WIN 35,428 binding in a statistically significant fashion; the method of incubation was irrelevant. In this binding screen, MI-4 inhibition did not approach the potency of the positive control inhibitor mazindol, which possesses low nM DAT affinity. The [3H]-dopamine uptake inhibition screening assay indicated that MI-4 did not appreciably inhibit dopamine uptake (Figure 5). With hDAT N2A cells, MI-4 displayed a DAT affinity of 6 μM as measured by WIN 35,428 displacement, and a DUIP of 28 μM (Figure 6).
Figure 5.
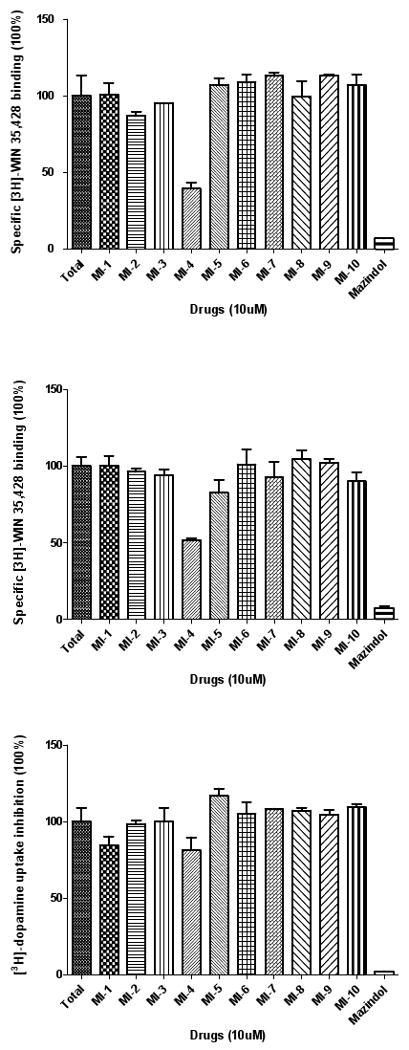
Initial DAT binding affinity and dopamine uptake inhibition screening for MI-1 to MI-10 at N2A neuroblastoma cells stably expressing WT hDAT. Representative relative DAT binding affinities assessed via [3H]-WIN 35,428 displacement when MI-1 to MI-10 (10 μM) were coincubated (upper panel) or preincubated for 10 minutes (middle panel) with the radioligand. Assessment of [3H]-dopamine uptake inhibition by MI-1 to MI-10 (10 μM) (lower panel).
Figure 6.
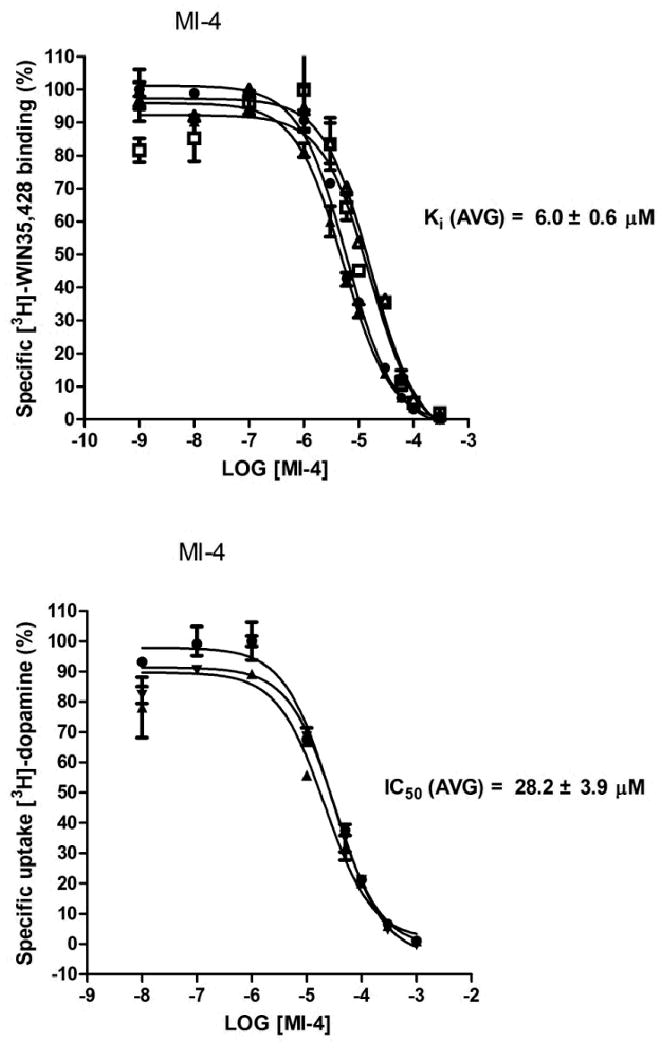
MI-4 DAT binding affinity and dopamine uptake inhibition potency in N2A cells. MI-4 inhibition of [3H]-WIN 35,428 binding (upper panel) and inhibition of [3H]-dopamine uptake (lower panel) in N2A neuroblastoma cells stably expressing WT hDAT (n = 3 or more experiments for each assay). Results were analyzed with one-way ANOVA (P < 0.05) with a post-hoc Dunnett's Multiple Comparison Test.
Given that the DUIP of MI-4 was so weak as to be negligible, the possibility that MI-4 could reduce cocaine DUIP and thus antagonize the actions of cocaine was addressed. Using the hDAT N2A cell line, cocaine inhibition of [3H]-dopamine uptake was measured in the absence and presence of 10 μM MI-4. The DUIP IC50 value of 291 ± 56 nM for cocaine alone was shifted approximately 3-fold (845 ± 118 nM) in the presence of MI-4 (Figure 7). Thus, MI-4 lacks appreciable inhibition of dopamine uptake at a concentration (10 μM) that can partially displace a classic cocaine analog from the DAT.
Figure 7.
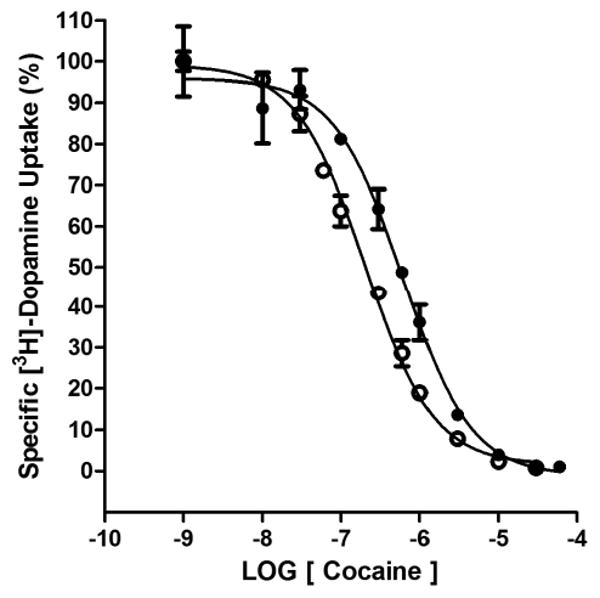
MI-4 decreases the DUIP of cocaine. The inhibition of [3H]-dopamine uptake by cocaine in the absence (open circles) or presence (closed circles) of 10 μM MI-4 at hDAT/N2A neuroblastoma cells (n = 3 experiments).
To investigate MI-4 binding and substrate uptake inhibition at the human norepinephrine transporter (hNET) and human serotonin transporter (hSERT), human embryonic kidney (HEK) cells stably transfected with either NET or SERT cDNAs were employed. In order to directly compare DAT results with those of NET and SERT, a stably-transfected hDAT HEK cell line was tested in parallel. The cocaine analog [125I] RTI-55 served as the binding radioligand in all cases, a compound similar to WIN 35,428 in DAT affinity and identical to WIN 35,428 except for a para- iodine-for-fluorine substitution at the C-3 phenyl ring. Substrate uptake was monitored with [3H]-dopamine at hDAT, [3H]-norepinephrine at hNET, and [3H]-serotonin at hSERT. As an internal reference, cocaine was assessed in parallel with MI-4. DAT affinity for MI-4 increased almost 2-fold with the switch to HEK cells but was still 20-fold lower than that for cocaine; DUIP increased 4-fold (Table 2). Interestingly, MI-4 affinity at hNET was 10-fold higher than at hDAT, and thus only two fold lower than that for cocaine. Substrate uptake inhibition potency of MI-4 at hNET was also greater than that at hDAT, but only by 3 – 4 fold. The cognate substrates for these transporter proteins differ only by norepinephrine's additional hydroxyl group, and each protein efficiently transports the other's substrate (14-16). The hDAT and hNET are very similar in transmembrane amino acid sequence, so isolating the residues responsible for the 10-fold MI-4 affinity difference between the two should be possible. As a caveat, the cocaine-like affinity of MI-4 at the hNET may be due to MI-4 ultimately docking in the primary substrate/inhibitor pocket. MI-4 affinity at hSERT was 5-fold higher than at hDAT, and substrate uptake inhibition potency was greatest at SERT among the three transporters. In all cases, cocaine affinity and substrate inhibition potency were within expected values (Table 2).
Table 2.
MI-4 binding and substrate uptake inhibition at HEK-hDAT, HEK-hSERT and HEK-hNET cellsa
| HEK-hDAT cells | MI-4 | Cocaine |
|---|---|---|
| [125l]-RTI-55 binding (Ki, nM) | 3460 ± 260 | 167 ± 25 |
| Hill coefficient | -0.80 ± 0.10 | -0.81 ± 0.03 |
| [3H]-Dopamine uptake (IC50, nM) | 6800* | 304 ± 55 |
| HEK-hSERT cells | ||
| [125l]-RTI-55 binding (Ki, nM) | 670 ±100 | 294 ± 83 |
| Hill coefficient | -1.09 ± 0.04 | -1.12 ± 0.03 |
| [3H]-Serotonin uptake (IC50, nM) | 790 ±170 | 390 ±140 |
| HEK-hNET cells | ||
| [125l]-RTI-55 binding (Ki, nM) | 365 ± 68 | 188 ± 96 |
| Hill coefficient | -1.04 ± 0.07 | -0.71 ± 0.06 |
| [3H]-NE uptake (IC50, nM) | 2090 ± 280 | 286 ± 50 |
Values represent the mean ± SEM from at least three independent experiments, each conducted in duplicate (for binding assays) or triplicate (for uptake assays) determinations. For MI-4 inhibition of dopamine uptake, some experiments yielded Ki values greater than 10 μM; these were assigned a value of 10 μM to allow calculation of an average. The actual value is greater than the average, and no standard error is reported.
Computationally-Derived Binding Mode of MI-4, an Ifenprodil Analog
The best-scoring binding mode of MI-4 (ΔG = -8.4 kcal/mol) obtained from the DAT comparative model indicates an array of interactions within the vestibular binding pocket (Figure 8). The benzylic hydroxyl group establishes H-bonds with Arg85 (TM 1) and Asp475 (TM 10). This ring is within reach of Tyr88, creating the potential for π-π aromatic stacking. In the current pose, the charged amine from the benzylpiperidine is able to H-bond with Thr472 (TM 10). A hydroxyl group of MI-4 may also H-bond with Asp384 in the fourth extracellular loop (ECL4a-4b). The TM 10 residues Ile468 and Tyr469 (not shown) contribute to a pocket surrounding the aromatic portion of the benzylpiperidine moiety. A similar MI-4 binding mode is observed in our hSERT model (Figure 9). A π-cation interaction is observed between Arg103 in TM 1 (the DAT Arg85 analog) and the aromatic region of the MI-4 benzylic hydroxyl moiety. An additional long-range ionic interaction between Glu494 of hSERT and the benzylpiperidine functional group of MI-4 is also suggested. This interaction and other TM 10 – MI-4 contacts pictured in Figure 9 could partially explain the higher SERT affinity for MI-4.
Figure 8.
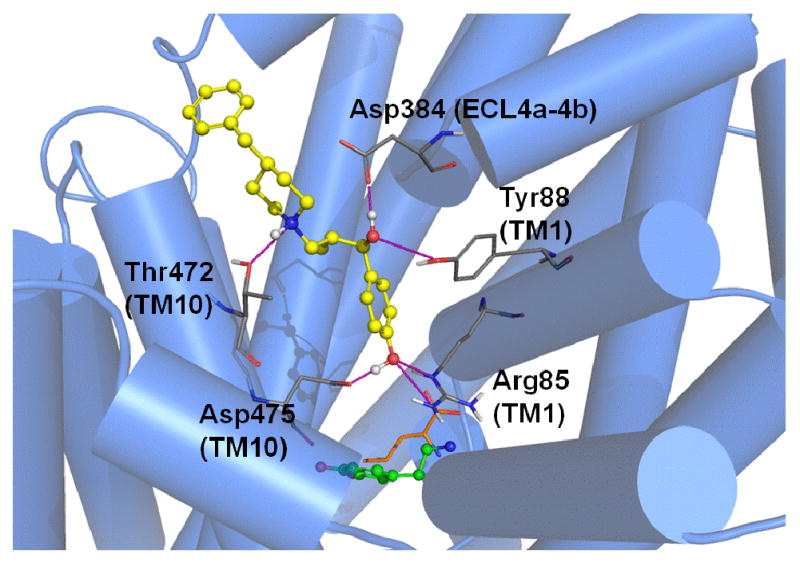
MI-4 docking in the DAT model. Optimal positioning of MI-4 in the DAT vestibular pocket is depicted by the top-ranked pose generated by MOE-Dock 2007. For reference, the primary substrate/inhibitor pocket (below the vestibular pocket) is indicated by superpositions of docked dopamine (green) and the analogous position of leucine (orange, sticks) in LeuT. DAT vestibular pocket side chains (atom colored) within 5 Å of the docked MI-4 molecule (yellow / atom colored) are shown. Intermolecular H-bond interactions are indicated (pink dashed lines).
Figure 9.
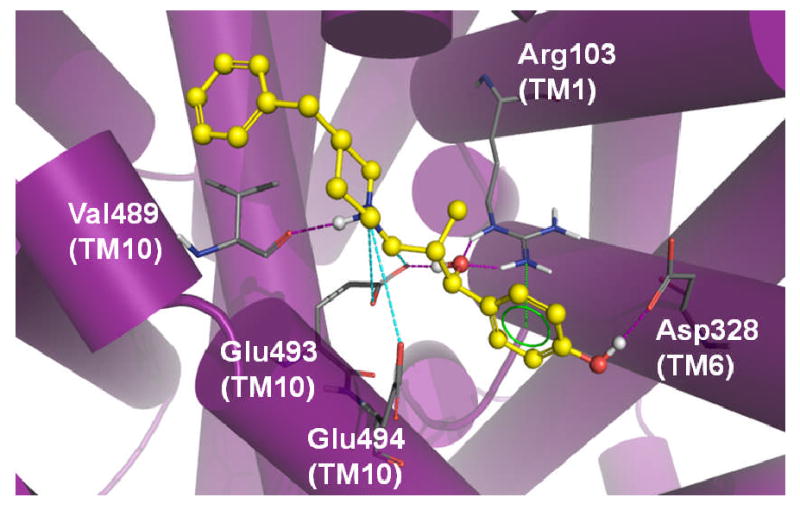
MI-4 docking in the SERT model. Optimal positioning of MI-4 in the SERT vestibular pocket is depicted by the top-ranked pose generated by MOE-Dock 2007. Ligand pocket side chains (atom colored) within 5 Å of the docked MI-4 molecule (yellow / atom colored) are shown. Intermolecular H-bond (pink dashed lines), π-cation (green dashed lines) and ionic (cyan dashed lines) interactions are indicated.
The MI-4 nomenclature codes for Ro-25-6981 ([R-(R*, S*)]-α-(4-Hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol hydrochloride), a second generation ifenprodil analog and a known NMDA (glutamate) receptor antagonist that crosses the blood-brain barrier (17). Blockade of NMDA receptors can delay or even prevent ischemic damage to the brain via attenuation of glutamate excitotoxicity. NMDA antagonists potentiate the antiparkinsonian effects of L-DOPA in a non-DAT related fashion, and generate analgesic effects by diminution of glutamatergic cell firing (17). Intriguingly, NMDA antagonists attenuate cocaine-induced behavioral toxicity and cocaine overdose (18, 19). The ability of Ro-25-6981 to cross the blood-brain barrier and antagonize NMDA receptors to prevent brain ischemia, coupled with its ability to block MAT binding of cocaine analogs with notably less effect on substrate uptake, suggests that the drug may be a therapeutic “lead compound” candidate.
Implications for Rational Discovery and Design of Therapeutic MAT Inhibitors
Despite the advances in VS and HTVS, the percentage of “hit” compounds is typically less than 1% of all compounds screened (20, 21). Moreover, when homology models and not X-ray structures of targets are employed, only low micromolar activities are typically obtained (22-29). Finding VS hits with low nM MAT affinity was not expected. It was hoped that the VS experiment would yield an affordable commercial compound that could serve as a lead compound for development of a medication that modulated dopamine levels; it is generally too much to expect the VS hit compound itself to be therapeutically useful. Still, MI-4 interfered with cocaine binding at the DAT to the extent that cocaine potency was reduced 3-fold. Alternatively, the differential MAT affinities observed with MI-4 might actually be desirable. Modafinil, a drug used to treat narcolepsy, possesses a low μM DAT binding affinity (30) yet is studied as a possible cocaine anti-addiction medication (31-34). The low DAT affinity and somewhat indiscriminate receptor binding of modafinil suggests that nondopaminergic mechanisms contribute to its pharmacology. Modafinil nevertheless interacts with DAT sites in the rat brain, a property shared with medications under investigation for treating cocaine dependence, and does not exhibit higher affinity at any other known target (30, 35-37). GBR-12909, benztropine and their analogs are under development as anti-cocaine medications (38), and modafinil shares the signature diphenylmethyl moiety with these two compounds. It is conceivable that similar DAT binding modes and sites are employed by the three (30). An example of another unorthodox therapeutic candidate is the dual dopamine/serotonin releasing agent PAL287, which demonstrated low abuse potential while maintaining the ability to suppress drug-seeking behavior (39). The serotonin-releasing property of this compound may prevent the activation of mesolimbic dopamine neurons linked to abuse liability (39-41).
A MAT molecular model has been successfully employed herein to discover a high affinity ligand with a structural scaffold distinct from those of classic MAT inhibitors. Only with such a methodology could MI-4 have been identified as a MAT ligand. High throughput virtual screening of small molecule structural libraries has yielded a variety of therapeutically promising lead compounds (42-45). Once reliable molecular models for all CNS-relevant transporter and receptor proteins are obtained, simultaneous in silico screening of structural libraries each containing millions of structural compounds should greatly accelerate therapeutic drug discovery.
Conclusion
The present work reflects a structure-based ligand discovery effort based on DAT inhibitor docking studies and driven by the hypothesis that the external vestibule (S2) substrate/inhibitor binding pocket of MAT proteins can be used in virtual screening experiments to find novel inhibitors of one or more members of this transporter family. The successful identification of the ifenprodil analog MI-4 provides a new structural scaffold for creation of structure-activity series of MAT ligands. The ability to screen millions of small molecule MAT ligand candidates in silico instead of in vitro should drastically reduce the time and expense associated with drug discovery. Reliable ligand-MAT docking poses identify binding pocket residues to be tested via site-directed mutagenesis and subsequent pharmacology, the results of which will further fine-tune the MAT models. The lead compounds discovered using the refined models may yield novel anti-addiction medications as well as therapeutics to combat depression, anxiety, Parkinson's disease, narcolepsy, chronic pain, and other CNS-related maladies.
Experimental Section
Structural Small Molecule Library Database Preparation
Compounds found in the Sigma-Aldrich catalogue were obtained as a structure data file (sdf). The classic DAT inhibitors cocaine (methyl (1R,2R,3S,5S)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate), WIN 35,428 (methyl (1R,2S,3S,5S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate), benztropine ((1R,5R)-3-benzhydryloxy-8-methyl-8-azabicyclo[3.2.1]octane), methylphenidate (methyl 2-phenyl-2-piperidin-2-ylacetate) and mazindol (5-(4-chlorophenyl)-2,3-dihydroimidazo[2,1-a]isoindol-5-ol) were retrieved as SMILES notations from PubChem (http://www.ncbi.nlm.nih.gov/pccompound). The remaining classical inhibitors oxa-norcocaine (methyl (1R,2R,3S,5S)-3-benzoyloxy-8-oxabicyclo[3.2.1]octane-2-carboxylate), 4-ARA-127 ((1S, 3S, 4R)-4-(chlorophenyl)-3-[hydroxy(methyloxo)methyl]-1-methylpiperidin-1iium) and GBR 12,909 (1-(30)-4-(3-phenylpropyl)piperazine-1,4-diium) were obtained from the literature (12). Both the sdf, SMILES files or 2D images were transformed into 3D structures via MOE; tautomer creation, partial charge calculation and energy minimization were conducted using the Merck Molecular Force Field 94X (MMFF94X). Commercial compounds without toxic chemical features (13) and adhering to Lipinski's Rule of Five were retained for the VS process.
Pharmacophore Query Creation
The DAT inhibitors cocaine, WIN 35,428, oxa-norcocaine, 4-ARA-127, benztropine, GBR 12,909, methylphenidate and mazindol were docked in the vestibular pocket of the DAT model, and the ligand functional groups that established the more prominent interactions were used as pharmacophore centers. The comparative model utilized in the VS process was constructed as described previously (4). The residues Arg85, Asp475 and Thr472 showed potential for cation-π stacking (Arg85), hydrogen bonding (Arg85, Asp475 and Thr472) and hydrophobic interactions (Arg85 and Thr472). The inhibitor functional groups that created the maximum number of hydrogen bonds with these amino acids were used to manually create pharmacophoric features with a radius of 2 Å (except for F5). Five pharmacophore features were created: F1, F2 and F3, donor/cation interaction; F4, donor/acceptor interaction; F5, excluded volume.
LeuT / DAT Vestibular Binding Site Analysis and Docking of Potential Ligands
The X-ray structure of LeuT complexed with clomipramine (PDB 2Q6H) was utilized to compare the previously found secondary pocket (4). Superimposition and binding site residue analysis of the DAT comparative model and the LeuT X-ray structure was performed with Molecular Operating Environment (MOE) software. A general superposition was performed with the Pro-Superpose module of MOE. Residues within 5 Ǻ of clomipramine were selected for the superposition. A pairwise percentage residue identity was calculated after the final vestibular binding pocket superposition. A RMSD correlation matrix was obtained and plotted using Pro-Superpose. Poses found within the figures were rendered using Pymol Educational Version 2007 (DeLano Scientific).
TCA-LeuT docking poses were obtained using MOE-Dock 2007.0902 and compared to the original crystal structure. The value of the predictions was assessed based on the RMSDs of TCAs bound in the crystal versus the predicted bound TCAs for the different methods. A maximum of 10,000 poses were generated for each ligand with the Triangle Matcher feature, and a default selection of the best 1000 poses was based on the GB/VI score implemented in MOE for further relaxation and final scoring. After this step, one of three pose relaxation options was employed: no relaxation, GRID and Forcefield refinement. All final poses were retained and scored. Several scoring functions were evaluated (London dG, ASEScore, Alpha HB and Affinity). High throughput docking of compounds from the final database that passed the pharmacophore query filtering step were docked within the extracellular vestibule of the DAT model utilizing the same protocol used in the LeuT docking validation step (Triangle Matcher method with a GRID minimization of poses and a final Affinity dG scoring) in a receptor-rigid environment.
In Vitro Substrate Uptake Inhibition Assays
For assays involving hDAT N2A neuroblastoma cells, monolayers were prepared in 6-well plates. The VS-identified compounds were dissolved in 50% DMSO without any signs of insolubility. In the initial screening, a final concentration of 10 μM of the VS compound was added to the cells 10 minutes before addition of [3H]-dopamine. Nonspecific binding was assessed by addition of 10 μM mazindol in control wells. The monolayer was washed 2 × 2 ml with “KRH buffer” (25 mM HEPES, pH 7.3, 125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM Mg2SO4, 1.2 mM KH2PO4, 5.6 mM glucose), and uptake was initiated by addition of 1 ml of [3H]-dopamine (10 nM final concentration) and 50 mM ascorbic acid (AA) in KRH to duplicate or triplicate cell monolayers. Uptake was quenched after 5 minutes at 22°C by washing the monolayer with 2 × 2 ml KRH + AA. Cell monolayers were solubilized in 1 ml of 1% SDS and incubated at room temperature for 1 hour with gentle shaking. The lysate was transferred to scintillation vials containing 5 ml of ScintSafe and radioactivity was counted using a liquid scintillation counter. VS compounds demonstrating the ability to decrease net uptake of [3H]-dopamine in this assay were characterized further using a range of concentrations, typically 0.1 – 60,000 nM, to determine IC50 values (GraphPad Prism 5, La Jolla, CA).
Assays involving stably transfected HEK cells were conducted by the NIDA Addiction Treatment Discovery Program. Methods employed with HEK cells were adapted from (14). HEK293 cells expressing hDAT, hSERT or hNET inserts were grown to 80% confluence on 150 mm culture dishes. Monolayers were washed with 10 ml of calcium- and magnesium-free phosphate-buffered saline. After a 10 min incubation with lysis buffer (10 ml; 2 mM HEPES, 1 mM EDTA), cells were scraped from plates and centrifuged 30,000 × g for 20 min. Supernatant was decanted and pellet resuspended in 12-32 ml of 0.32 M sucrose (Polytron at setting 7 for 10 sec). The resuspension volume depended on the density of binding sites within a cell line and was chosen to reflect binding of 10% or less of the total radioactivity. Krebs-HEPES (350 μl: 122 mM NaCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 10 μM pargyline, 100 μM tropolone, 0.2% glucose and 0.02% ascorbic acid, buffered with 25 mM HEPES, pH 7.4) and the compound of interest or buffer alone (50 μl) were added to 1 ml vials and incubated at 25°C. Specific uptake was defined as the difference in uptake observed in the presence and absence of 5 μM mazindol (HEK-hDAT and HEK-hNET) or 5 μM imipramine (HEK-hSERT). Cells (50 μl) were added and preincubated with the compound for 10 min. The assay was initiated by the addition of 50 μl [3H]-dopamine, [3H]-serotonin, or [3H]-norepinephrine (20 nM final concentration). Filtration through Whatman GF/C filters presoaked in 0.05% polyethylenimine was used to terminate uptake after 10 min. IC50 values were calculated via GraphPad Prism for triplicate curves.
In Vitro Inhibitor Binding Assays
For hDAT N2A cells, this assay only differed from the dopamine uptake inhibition assay in that [3H]-dopamine was replaced with 1 nM of the cocaine analog [3H]-WIN 35,428, and the VS compound and radioligand were added simultaneously to the cells and incubated for 15 min. VS compound concentrations were as indicated above for dopamine uptake inhibition. Nonspecific binding was assessed by addition of 10 μM mazindol. Screening results were analyzed with one-way ANOVA (P < 0.05) with a post-hoc Dunnett's Multiple Comparison Test. For saturation binding assays, data were analyzed with GraphPad Prism 5.0 software to obtain Ki values.
For stably transfected HEK cells, each assay tube contained 50 μl of membrane preparation (about 10-15 μg of protein), 25 μl of the compound of interest, the drug used to define non-specific binding, or Krebs-HEPES buffer alone, 25 μl of the cocaine analog [125I]-RTI-55 (40-80 pM final concentration) and additional buffer sufficient to bring up the final volume to 250 μl. Membranes were preincubated with VS compounds for 10 min prior to the addition of the [125I]-RTI-55; the assay tubes were further incubated at 25°C for 90 min. Binding was terminated by filtration over GF/C filters using a Tomtec 96-well cell harvester. Filters were washed for 6 sec with ice-cold saline. Scintillation fluid was added to each square and radioactivity remaining on the filter was determined using a Wallace alpha- or beta-plate reader. Specific binding was defined as the difference in binding observed in the presence and absence of 5 μM mazindol (HEK-hDAT and HEK-hNET) or 5 μM imipramine (HEK-hSERT). Three independent competition experiments were conducted with duplicate determinations. GraphPad Prism generated IC50 values, converted to Ki values using the Cheng-Prusoff equation (Ki=IC50/ (1+ ([RTI-55]/Kd RTI-55)).
Acknowledgments
The authors thank Dr. Jane Acri of NIDA and the NIDA Addiction Treatment Discovery Program for data on HEK cell DAT, NET and SERT ligand binding and inhibition of substrate uptake, generated through Contract #N01-DA-7-8877 with Dr. Aaron Janowsky at the PVAMC. NIDA Drug Supply graciously supplied selected nonradioactive MAT ligands. We thank Dr. Ling Chan of Chemical Computing Group (Montreal) for providing the beta version of MOE docking software and for helpful discussions. We thank Judy Froehlich (Sigma-Aldrich) for providing academic pricing to compounds from the Rare Chemical Library.
Funding Sources: This work was supported by NIDA grants DA016604 and DA026530 (to C.K.S.) and DA027806 (to J.D.M.) and equipment grants P116Z050331 and P116Z080180 from the Department of Education.
References
- 1.Preti A. New developments in the pharmacotherapy of cocaine abuse. Addict Biol. 2007;12:133–51. doi: 10.1111/j.1369-1600.2007.00061.x. [DOI] [PubMed] [Google Scholar]
- 2.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 3.Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–9. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Indarte M, Madura JD, Surratt CK. Dopamine transporter comparative molecular modeling and binding site prediction using the LeuT(Aa) leucine transporter as a template. Proteins. 2008;70:1033–46. doi: 10.1002/prot.21598. [DOI] [PubMed] [Google Scholar]
- 5.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–23. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 6.Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci U S A. 2009;106:5563–8. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–77. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–6. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–3. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, Carpenter EP, Rutherford NG, Hadden JM, O'Reilly J, Ma P, Saidijam M, Patching SG, Hope RJ, Norbertczak HT, Roach PC, Iwata S, Henderson PJ, Cameron AD. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008;322:709–13. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature. 2009;458:47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 12.Ukairo OT, Bondi CD, Newman AH, Kulkarni SS, Kozikowski AP, Pan S, Surratt CK. Recognition of benztropine by the dopamine transporter (DAT) differs from that of the classical dopamine uptake inhibitors cocaine, methylphenidate, and mazindol as a function of a DAT transmembrane 1 aspartic acid residue. J Pharmacol Exp Ther. 2005;314:575–83. doi: 10.1124/jpet.105.085829. [DOI] [PubMed] [Google Scholar]
- 13.Kazius J, McGuire R, Bursi R. Derivation and validation of toxicophores for mutagenicity prediction. J Med Chem. 2005;48:312–20. doi: 10.1021/jm040835a. [DOI] [PubMed] [Google Scholar]
- 14.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–85. [PubMed] [Google Scholar]
- 15.Giros B, Wang YM, Suter S, McLeskey SB, Pifl C, Caron MG. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine-norepinephrine transporters. J Biol Chem. 1994;269:15985–8. [PubMed] [Google Scholar]
- 16.Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. 2002;22:389–95. doi: 10.1523/JNEUROSCI.22-02-00389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006;6:68–74. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Cai SX. Glycine/NMDA receptor antagonists as potential CNS therapeutic agents: ACEA-1021 and related compounds. Curr Top Med Chem. 2006;6:651–62. doi: 10.2174/156802606776894465. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto RR, Brackett RL, Kanthasamy AG. Novel NMDA/glycine site antagonists attenuate cocaine-induced behavioral toxicity. Eur J Pharmacol. 1997;338:233–42. doi: 10.1016/s0014-2999(97)81926-7. [DOI] [PubMed] [Google Scholar]
- 20.Bailey D, Brown D. High-throughput chemistry and structure-based design: survival of the smartest. Drug Discov Today. 2001;6:57–59. doi: 10.1016/s1359-6446(00)01596-8. [DOI] [PubMed] [Google Scholar]
- 21.Schneider G, Bohm HJ. Virtual screening and fast automated docking methods. Drug Discov Today. 2002;7:64–70. doi: 10.1016/s1359-6446(01)02091-8. [DOI] [PubMed] [Google Scholar]
- 22.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–7. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 23.Enyedy IJ, Lee SL, Kuo AH, Dickson RB, Lin CY, Wang S. Structure-based approach for the discovery of bis-benzamidines as novel inhibitors of matriptase. J Med Chem. 2001;44:1349–55. doi: 10.1021/jm000395x. [DOI] [PubMed] [Google Scholar]
- 24.Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D, Wang S. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J Med Chem. 2001;44:4313–24. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 25.Li R, Chen X, Gong B, Selzer PM, Li Z, Davidson E, Kurzban G, Miller RE, Nuzum EO, McKerrow JH, Fletterick RJ, Gillmor SA, Craik CS, Kuntz ID, Cohen FE, Kenyon GL. Structure-based design of parasitic protease inhibitors. Bioorg Med Chem. 1996;4:1421–7. doi: 10.1016/0968-0896(96)00136-8. [DOI] [PubMed] [Google Scholar]
- 26.Que X, Brinen LS, Perkins P, Herdman S, Hirata K, Torian BE, Rubin H, McKerrow JH, Reed SL. Cysteine proteinases from distinct cellular compartments are recruited to phagocytic vesicles by Entamoeba histolytica. Mol Biochem Parasitol. 2002;119:23–32. doi: 10.1016/s0166-6851(01)00387-5. [DOI] [PubMed] [Google Scholar]
- 27.Rajnarayanan RV, Dakshanamurthy S, Pattabiraman N. “Teaching old drugs to kill new bugs”: structure-based discovery of anti-SARS drugs. Biochem Biophys Res Commun. 2004;321:370–8. doi: 10.1016/j.bbrc.2004.06.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selzer PM, Chen X, Chan VJ, Cheng M, Kenyon GL, Kuntz ID, Sakanari JA, Cohen FE, McKerrow JH. Leishmania major: molecular modeling of cysteine proteases and prediction of new nonpeptide inhibitors. Exp Parasitol. 1997;87:212–21. doi: 10.1006/expr.1997.4220. [DOI] [PubMed] [Google Scholar]
- 29.Zuccotto F, Zvelebil M, Brun R, Chowdhury SF, Di Lucrezia R, Leal I, Maes L, Ruiz-Perez LM, Gonzalez Pacanowska D, Gilbert IH. Novel inhibitors of Trypanosoma cruzi dihydrofolate reductase. Eur J Med Chem. 2001;36:395–405. doi: 10.1016/s0223-5234(01)01235-1. [DOI] [PubMed] [Google Scholar]
- 30.Zolkowska D, Jain R, Rothman RB, Partilla JS, Roth BL, Setola V, Prisinzano TE, Baumann MH. Evidence for the involvement of dopamine transporters in behavioral stimulant effects of modafinil. J Pharmacol Exp Ther. 2009;329:738–46. doi: 10.1124/jpet.108.146142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson AL, Reid MS, Li SH, Holmes T, Shemanski L, Slee A, Smith EV, Kahn R, Chiang N, Vocci F, Ciraulo D, Dackis C, Roache JD, Salloum IM, Somoza E, Urschel HC, 3rd, Elkashef AM. Modafinil for the treatment of cocaine dependence. Drug Alcohol Depend. 2009;104:133–9. doi: 10.1016/j.drugalcdep.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart CL, Haney M, Vosburg SK, Rubin E, Foltin RW. Smoked cocaine self-administration is decreased by modafinil. Neuropsychopharmacology. 2008;33:761–8. doi: 10.1038/sj.npp.1301472. [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Raga J, Knecht C, Cepeda S. Modafinil: a useful medication for cocaine addiction? Review of the evidence from neuropharmacological, experimental and clinical studies. Curr Drug Abuse Rev. 2008;1:213–21. doi: 10.2174/1874473710801020213. [DOI] [PubMed] [Google Scholar]
- 34.Umanoff DF. Trial of modafinil for cocaine dependence. Neuropsychopharmacology. 2005;30:2298. doi: 10.1038/sj.npp.1300866. author reply 2299-300. [DOI] [PubMed] [Google Scholar]
- 35.de Saint Hilaire Z, Orosco M, Rouch C, Blanc G, Nicolaidis S. Variations in extracellular monoamines in the prefrontal cortex and medial hypothalamus after modafinil administration: a microdialysis study in rats. Neuroreport. 2001;12:3533–7. doi: 10.1097/00001756-200111160-00032. [DOI] [PubMed] [Google Scholar]
- 36.Madras BK, Xie Z, Lin Z, Jassen A, Panas H, Lynch L, Johnson R, Livni E, Spencer TJ, Bonab AA, Miller GM, Fischman AJ. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther. 2006;319:561–9. doi: 10.1124/jpet.106.106583. [DOI] [PubMed] [Google Scholar]
- 37.Murillo-Rodriguez E, Haro R, Palomero-Rivero M, Millan-Aldaco D, Drucker-Colin R. Modafinil enhances extracellular levels of dopamine in the nucleus accumbens and increases wakefulness in rats. Behav Brain Res. 2007;176:353–7. doi: 10.1016/j.bbr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 38.Rothman RB, Baumann MH, Prisinzano TE, Newman AH. Dopamine transport inhibitors based on GBR12909 and benztropine as potential medications to treat cocaine addiction. Biochem Pharmacol. 2008;75:2–16. doi: 10.1016/j.bcp.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothman RB, Blough BE, Baumann MH. Dual dopamine/serotonin releasers as potential medications for stimulant and alcohol addictions. Aaps J. 2007;9:E1–10. doi: 10.1208/aapsj0901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negus SS, Mello NK, Blough BE, Baumann MH, Rothman RB. Monoamine releasers with varying selectivity for dopamine/norepinephrine versus serotonin release as candidate “agonist” medications for cocaine dependence: studies in assays of cocaine discrimination and cocaine self-administration in rhesus monkeys. J Pharmacol Exp Ther. 2007;320:627–36. doi: 10.1124/jpet.106.107383. [DOI] [PubMed] [Google Scholar]
- 41.Rothman RB, Blough BE, Baumann MH. Dual dopamine-5-HT releasers: potential treatment agents for cocaine addiction. Trends Pharmacol Sci. 2006;27:612–8. doi: 10.1016/j.tips.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 42.Betzi S, Restouin A, Opi S, Arold ST, Parrot I, Guerlesquin F, Morelli X, Collette Y. Protein protein interaction inhibition (2P2I) combining high throughput and virtual screening: Application to the HIV-1 Nef protein. Proc Natl Acad Sci U S A. 2007;104:19256–61. doi: 10.1073/pnas.0707130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jorgensen WL. Efficient drug lead discovery and optimization. Acc Chem Res. 2009;42:724–33. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segers K, Sperandio O, Sack M, Fischer R, Miteva MA, Rosing J, Nicolaes GA, Villoutreix BO. Design of protein membrane interaction inhibitors by virtual ligand screening, proof of concept with the C2 domain of factor V. Proc Natl Acad Sci U S A. 2007;104:12697–702. doi: 10.1073/pnas.0701051104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]