

Abstract
Genotype imputation across populations of mixed ancestry is critical for optimal discovery in large-scale genome-wide association studies (GWAS). Methods for direct imputation of GWAS summary-statistics were previously shown to be practically as accurate as summary statistics produced after raw genotype imputation, while incurring orders of magnitude lower computational burden. Given that direct imputation needs a precise estimation of linkage-disequilibrium (LD) and that most of the methods using a small reference panel for example, ~2,500-subject coming from the 1000 Genome-Project, there is a great need for much larger and more diverse reference panels. To accurately estimate the LD needed for an exhaustive analysis of any cosmopolitan cohort, we developed DISTMIX2. DISTMIX2: (a) uses a much larger and more diverse reference panel compared to traditional reference panels, and (b) can estimate weights of ethnic-mixture based solely on Z-scores, when allele frequencies are not available. We applied DISTMIX2 to GWAS summary-statistics from the psychiatric genetic consortium (PGC). DISTMIX2 uncovered signals in numerous new regions, with most of these findings coming from the rarer variants. Rarer variants provide much sharper location for the signals compared with common variants, as the LD for rare variants extends over a lower distance than for common ones. For example, while the original PGC post-traumatic stress disorder GWAS found only 3 marginal signals for common variants, we now uncover a very strong signal for a rare variant in PKN2, a gene associated with neuronal and hippocampal development. Thus, DISTMIX2 provides a robust and fast (re)imputation approach for most psychiatric GWAS-studies.
Keywords: direct imputation, GWAS, genetics, summary statistcis
1 |. INTRODUCTION
Genotype imputation (B. L. Browning & Browning, 2009; Howie, Donnelly, & Marchini, 2009; Nicolae, 2006; Servin & Stephens, 2007) methods are commonly used to increase the genomic resolution for large-scale multi-ethnic genome-wide association studies (GWAS) meta-analyses (Consortium, 2015; Ripke et al., 2013; Ripke et al., 2011; Sklar et al., 2011; Sladek et al., 2007) by predicting genotypes at unmeasured single nucleotide polymorphisms (SNPs) markers based on cosmopolitan reference panels, for example, 1000 Genomes (1 KG) Project (Genomes Project et al., 2012). However, genotypic imputation is computationally burdensome and requires access to subject-level genetic data, which is harder and slower to obtain than summary statistics.
To overcome these limitations, researchers proposed summary statistics-based imputation methods, for example, DIST (Lee, Bigdeli, Riley, Fanous, & Bacanu, 2013) and ImpG (Pasaniuc et al., 2013). These methods can directly impute summary statistics (two-tailed Z-scores) for unmeasured SNPs from summary statistics of GWAS or called variants from sequencing studies. The methods were shown to i) substantially reduce the computational burden and ii) be practically as accurate as commonly used genotype imputation methods. These methods were successfully applied in gene-level joint testing of functional variants and functional enrichment analyses. However, these first wave of direct imputation methods were only amenable for imputation in ethnically homogeneous cohorts.
To accommodate cosmopolitan cohorts, DIST method was extended (Lee et al., 2015) to directly imputing summary statistics for unmeasured SNPs from Mixed ethnicity cohorts (DISTMIX). It: (a) predicted a study’s proportions (weights) of ethnicities from a multi-ethnic reference panel based only on cohort allele frequencies (AFs) for (common) SNPs from the studied cohort or taking prespecified ethnic weights, (b) computed an ethnicity-weighted correlation matrix based on the estimated/prespecified weights and genotypes of ethnicities from the reference panel and then, (c) used the weighted correlation matrix for accurate imputation. Currently, due to privacy concerns (Homer et al., 2008), cohort AFs are lately only rarely provided. To circumvent the lack of AFs, the ARDISS method (Togninalli, Roqueiro, Investigators, & Borgwardt, 2018) extended the inference to cosmopolitan cohorts using a gaussian field approach based only on Z-scores. However, ARDISS software provides only a 1 KG-derived reference panel. Unlike DISTMIX that is implemented as a standalone C++ software, ARDISS is implemented in Python that is more temperamental in diverse installing/working environments with various versions of libraries.
The direct imputation was already used in practice to impute GWAS data with success (Endo et al., 2018; Khor et al., 2018; Revez et al., 2020). Moreover, direct imputation is also performed in the background of other applications. For instance, unlike many alternatives, our transcriptome wide association study (TWAS) tools (Chatzinakos et al., 2020; Chatzinakos et al., 2018; Lee et al., 2015; Lee et al., 2016), internally impute statistics for expression Quantitative Trait Loci (eQTL) SNPs that are not reported in GWASAn additional need for direct imputation methods is to be able analyze studies (a) not reporting AFs and (b) also, containing a non-trivial fractions of non-European subjects (Cai et al., 2017; Ripke et al., 2013). To increase the resolution and relevance to such cosmopolitan cohorts, DIST/DISTMIX enlarged the reference panel by including genotypes from large sequencing studies, such as Haplotype Reference Consortium (HRC) (McCarthy et al., 2016) and CONVERGE (Consortium, 2015). The inclusion of >11 K Han Chinese cohort from the CONVERGE consortium complement nicely the (largely European) HRC panel and provides accurate linkage-disequilibrium (LD) information for the second most studied continental population after Europeans.
In this paper we propose DISTMIX2 method/software, which addresses the above shortcomings by including two critical components. First, we provide a novel method to accurately estimate ethnic weights of the cohort which uses only summary statistics, for example, Z-scores. Second, we build a larger, more diverse reference panel with 33 K subjects, which combines the subjects from the publicly available part of HRC and CONVERGE. While implementing the above 2 features, DISTMIX2: (a) adequately controls the false positive rate, and (b) provides much improved resolution when compared to methods based on older reference panels. Based on simulated data sets we provide guidance on the significance thresholds for rare and/or low information variants. Furthermore, in a practical application to reported summary statistics from studies of psychiatric disorders, we uncover numerous regions harboring signals. Most of these novel signals are associated with rarer variants that could not be robustly interrogating using the smaller panels from previous methods. Some of the new findings provide strong signals in new regions for traits that reported only marginal signals. In the quest to provide enhanced resolution for ethnically mixed GWAS studies, DISTMIX2 provides a robust and substantially faster alternative to the laborious genotype imputation.
2 |. METHOD
2.1 |. Larger and more diverse reference panel
To facilitate imputation of rarer variants, the current version uses the 33,000 subjects (33 K) as reference panel. It consists of 20,281 - Europeans, 10,800 East Asians, 522 South Asians, 817 Africans and 533 Native Americans (Text S1, Table S1 in SI). The reference panel includes the publicly available 22,691 subjects from Haplotype Reference Consortium (HRC) and 10,262 CONVERGE. For CONVERGE subjects, we used province of origin to assign them to four populations (China North East - CNE, China Central East - CCE, China South East - CSE and China Central South - CCS). HRC subjects coming from the small Orkney (ORK) island provided the basis for an extra European population, that is, ORK. Subjects from 1 KG in HRC sample, CONVERGE and ORK along with their (a) population label, (b) first 20 ancestry principal components were used to train a quadratic discriminant model for predicting population label from principal components. Subsequently, to have more homogeneous populations in the panel, all available subjects were assigned(reassigned) population labels based on model prediction. Consequently, a subject might be re-assigned to a different (but related) population.
To have reasonably accurate SNP LD estimators, we eliminate the rarest SNPs which did not have at least: (a) 20 alleles in European or East Asian superpopulations or (b) 5 in African, South Asian and America native superpopulations. Our final cosmopolitan reference panel contains 26 million SNPs.
2.2 |. Converge haplotypes
2.2.1 |. DNA sequencing
DNA was extracted from saliva samples using the Oragene protocol. A barcoded library was constructed for each sample. Sequencing reads obtained from Illumina Hiseq machines were aligned to Genome Reference Consortium Human Build 37 patch release 5 (GRCh37.p5) with Stampy (v1.0.17) (Lunter & Goodson, 2011) using default parameters, after filtering out reads containing adaptor sequencing or consisting of more than 50% poor quality (base quality <= 5) bases. Samtools (v0.1.18) (Li et al., 2009) was used to index the alignments in BAM format (Li et al., 2009) and Picardtools (v1.62) was used to mark PCR duplicates for downstream filtering. The Genome Analysis Toolkit’s (GATK, version 2.6). Base quality score recalibration (BQSR) was then applied to the mapped sequencing reads using Base-Recalibrator in Genome Analysis Toolkit (GATK, basic version 2.6) (DePristo et al., 2011) with the known insertion and deletion (INDEL) variations in 1000 Genomes Projects Phase 1 (Genomes Project et al., 2010) and known single nucleotide polymorphisms (SNPs) from dbSNP (v137, excluding all sites added after v129) excluded from the empirical error rate calculation. GATKlite (v2.2.15) was then used to output sequencing reads with the recalibrated base quality scores while removing reads without the “proper pair” flag bit set by Stampy (1–5% of reads per sample) using the –read_filter ProperPair option (if the “proper pair” flag bit is set for a pair of reads, it means both reads in the mate-pair are correctly oriented, and their separation is within 5 standard deviations from the mean insert size between mate-pairs).
2.2.2 |. Variant calling, imputation, and phasing
Variant discovery and genotyping (for both SNPs and INDELs) at all polymorphic SNPs in 1000G Phase1 East Asian (ASN) reference panel(Genomes Project et al., 2012) was performed simultaneously using post-BQSR sequencing reads from all samples using the GATK’s UnifiedGenotyper (version 2.7–2-g6bda569). Variant quality score recalibration (VQSR) was then performed with GATK’s VariantRecalibrator (v2.7–4-g6f46d11) in SNP variant calls using the SNPs in 1000 Genomes Phase 1 ASN Panel (Genomes Project et al., 2010) as the known, truth and training sets. A sensitivity threshold of 90% to SNPs in the 1000G Phase1 ASN panel was applied for SNP selection for imputation after optimizing for Transition to Transversion (TiTv) ratios in SNPs called. Genotype likelihoods (GLs) were calculated at selected sites using a sample-specific binomial mixture model implemented in SNPtools (version 1.0), and imputation was performed at those sites without a reference panel using BEAGLE (version 3.3.2) (S. R. Browning & Browning, 2007). The second round of imputation was performed with BEAGLE on the same GLs, but only at biallelic SNPs polymorphic in the 1000G Phase 1 ASN panel using the 1000G Phase 1 ASN haplotypes as a reference panel. The genotypes derived from Beagle imputation were phased using Shapeit (version 2, revision 790) (Delaneau, Howie, Cox, Zagury, & Marchini, 2013). Genetic maps were obtained from the Impute2 (Howie et al., 2009) website. Chromosomes 13–22 and X were phased using 12 threads and default parameters. Chromosomes 1–12 were phased using 12 threads in four chunks that overlap by 1 MB. The phased chunks were ligated together using ligateHAPLOTYPES, available from the Shapeit website. A final set of allele dosages and genotype probabilities was generated from these two datasets by replacing the results in the former with those in the latter at all sites imputed in the latter. We then applied a conservative set of inclusion threshold for SNPs for genome-wide association study (GWAS): a) p-value for violation HWE > 10−6, b) Info score > 0.9, c) Minor-AF (MAF) in CONVERGE >0.5% to arrive at the final set of 6,242,619 SNPs. Details can be found in (Cai et al., 2017).
2.3 |. Automatic detection of cohort composition
Our group has previously described, in DISTMIX paper (Lee et al., 2015), a method to estimate the ethnic composition when the cohort AF are available. However, lately some consortia do not provide such measure; they often provide only the AF for Caucasian / European cohorts. Consequently, there is a great need to estimate the ethnic composition of the cohort even when no AFs are provided.
Below is the theoretical outline of such method. Suppose that the cohort genotype is a mixture of genotypes belonging k ethnic groups from the reference panel. The Gij denotes the genotype for the i-th subject at the j-th SNP which belongs to the l-th group, let be the frequency of the reference allele frequency for this SNP in the l-th group. Let be the normalized genotype, that is, the transformation to a variable with zero mean and unit variance. Near H0, SNP Z-score statics Zj′ s have the approximately the same correlation matrix as the genotypes used to construct it, G*j′s (Lee et al., 2014); given that G′*j′s are linear combination (with positive slope) of G*j′s, it follows that Z-scores have the same correlation structure G′*j′s. However, given that both G′*j′ s and Zj′ s have unit variance, it follows that the two have the same covariance (i.e. not only the same correlation) structure. Therefore, for any s ≥ 1.
E(ZjZj + s) = E(G′*jG′*j + s), which, due independence of genotypes in different ethnic groups becomes:
| (1) |
where w(l) is the expected fraction of subjects from the entire cohort that belong to the l-th group.
While is unknown, it can be easily approximated using their reference panel counterparts. Thus, the weights, w(l), can be estimated by simply regressing the product of reasonably close SNP Z-scores, Z′jZ′j+s, on correlations between normalized genotypes at the same SNP pairs for all subpopulations in the reference panel. To increase bias power, we chose the parameter s, such as to maximize the variance of the within-panel ethnic group correlations while keeping j + s-th SNP no more than 50Kb away from j−th SNP. Because some GWAS might have numerous large signals, for example, latest height meta-analysis (Ripke et al., 2013; Wood et al., 2014), a more accurate estimation of the weights is very likely to be obtained by substituting expected gaussian quantiles for (see Nonparametric robust estimation of weights subsection).
Due to the strong LD among SNPs, the estimation of the correlation using all SNPs in a genome might lead to a poor regression estimate in (1). To avoid this, we sequentially split GWAS SNPs into 1000 non-overlapping SNP sets, for example, first set consists of the 1st, 1001st, 2001st, etc. map ordered SNPs in the study. The large distances between SNPs in the same set make them quasi-independent which, thus, improves the accuracy of the estimated correlation. W = (w(l)) is subsequently estimated as the average of the weights obtained from the 1000 SNP sets. Finally, we set to zero the negatives weights and normalize the remaining weights to sum to 1 (Chatzinakos et al., 2018). This method should be even more useful when we already know the approximate continental (EUR, ASN, SAS, AFR and AMR) weights (as estimated from study information) but it is not always clear how these proportions should be allocated among continental subpopulations. This further apportioning of continental weights is likely to be extremely important when the GWAS cohorts contain many admixed populations, for example, African Americans and American native populations. Consequently, when continental proportions are provided by the users, we use our automatic detection to distribute these weights to the most likely subpopulations in the reference panel. To eliminate unforeseen artifacts, we strongly recommend to the users to provide continental proportions when AFs are not available.
2.4 |. Nonparametric robust estimation of weights
To estimate robust weights and to avoid false positives, we apply a two-step, robust algorithm to the Z-scores of the SNPs. First, let , where σ indicates the permutation of indices for sorting in increasing order Z-scores, Z, for the m SNPs. Second, , where Φ−1 is the inverse normal cumulative distribution function. Subsequently, these transformed risk scores are used for estimating ethnic weights.
2.5 |. Simulation
To estimate the accuracy and false positive rates of DISTMIX2, for five different cosmopolitan studies scenarios, we simulated (under H0: no association between trait and variants) 100 cosmopolitan cohorts of 10,000 subjects for autosomal SNPs in Ilumina 1 M panel (Lee et al., 2015) using 1 KG haplotype patterns (Text S1, Table S2 in SI). The subject phenotypes were simulated independent of genotypes as a random Gaussian sample. SNP phenotype–genotype association summary statistics were computed from a correlation test.
The accuracy of the procedure was assessed by masking 5% of the SNPs (Experiment 1, Table 1 – three type of parameter settings). Subsequently, the true values and the imputed values at these masked SNPs were used to compute: (a) their correlation and (b) the mean squared error of the imputation. We assess these measures at four different levels of MAF. To compare the Type I error rate of our proposed method, DISTMIX2, we estimated the relative Type I error (the empirical divided by the nominal Type I error rate) as a function of the nominal Type I error rate, for the same four MAF levels for all the cohorts. Finally, for all the combinations between MAFs and Info we performed DISTMIX2 analyses with three different parameters for the length of the predicted window (the length of the predicted window also depends on the minimum number of measured SNPs it encompasses).
TABLE 1.
Experiment 1 parameter settings
| MAF levels | Panel | Window length |
|---|---|---|
| MAF<5% | 1 K | 250Kb |
| 5 % <MAF<10% | 33 K | 500Kb |
| 10%<MAF<20% | 1000Kb | |
| 20%<MAF<50% | ||
Abbreviation: MAF, minor-AF.
To assess the reliability of DISTMIX2 results for rare and very rare variants, for the above cohorts, we also estimate DISTMIX2 size of the test for very low MAFs (rare variants), (Experiment 2, Table 2 – two type of parameter settings). The size of the test is assessing for 5 imputation Info intervals and 6 MAF intervals.
TABLE 2.
Experiment 2 variable parameter settings. Fixed parameters for this experiment: 33 K panel and 500Kb length of predicting window
| MAF levels | Info levels |
|---|---|
| 0.05% <MAF<0.5% | Info<20% |
| 0.5% <MAF<1% | 20%<Info<40% |
| 1 % <MAF<2% | 40%<Info<60% |
| 2% <MAF<5% | 60%<Info<80% |
| 5% <MAF<10% | Info>80% |
| 10% <MAF<50% | |
Abbreviation: MAF, minor-AF.
However, given that: 1) the simulated cohorts might not reflect real data and 2) these data sets do not have the sample sizes needed to detect very rare SNPs (e.g. MAFs <0.05%), which is important for DISTMIX2 inference in practical applications, we used real data sets to create so-called nullified data sets (Experiment 3, Table 3 – two types of parameter settings). These nullified data are based on 20-real and mostly Caucasian GWAS schizophrenia (SCZ), attention deficit hyperactive disorder (ADHD), autism (AUT), major depressive disorder (MDD) and 16 GWAS meta-analyses that are not yet publicly available. This approximation for null data is obtained by substituting the expected quantile of the Gaussian distribution for the (ordered) Z-score. We note that, while the quantile estimation adjusts the noncentrality parameter (enrichment) of the statistics to zero, it does not change the order of the statistics. One effect of this fact is that imputing statistics within/near the peak signals in original GWASs might result in increased false positive rates and, thus, the genome-wide false positive rates might appear to be moderately inflated.
TABLE 3.
Experiment 3 variable parameter settings. Fixed parameters for this experiment: 33 K panel and 500Kb window length
| MAF levels | Info levels |
|---|---|
| MAF<0.05% | Info<20% |
| 0.05% <MAF<0.5% | 20 % <Info<40% |
| 0.5% <MAF<1% | 40 % <Info<60% |
| 1% <MAF<2% | 60 % <Info<80% |
| 2 % <MAF<5% | Info>80% |
| 5 % <MAF<10% | |
| 10% <MAF<50% |
Abbreviation: MAF, minor-AF.
2.6 |. Applications in psychiatric GWAS
We applied DISTMIX2 to a subset of the psychiatric summary datasets available for download from Psychiatric Genetics Consortium (PGC- http://www.med.unc.edu/pgc/), that is, SCZ, ADHD, AUT, eating disorder (ED), bipolar (BIP) disorder, MDD and post-traumatic stress disorder (PTSD) (see Table 4 for references). For PTSD, we also analyzed an admixed African (PTSD-AA) GWAS by combing 20 PTSD African cohorts, which were part of the recent PTSD study (Nievergelt et al., 2019), by using METAL (Willer, Li, & Abecasis, 2010). Based on the results from simulations under the null hypothesis (Experiment 1), for all these applications we used: a) the larger 33 K size panel and b) a length of the predicted window (500Kb). To improve the imputation of the unmeasured SNPs for SCZ, we denote as “measured SNPs” only those with very high information (Info>0.997). For the ADHD, AUT, BIP, MDD, PTSD and PTSD-AA data sets, because the imputation information is not available, we accept as measured SNPs the set consisting of the intersection between SNPs in each GWAS and the above SCZ’s “measured” SNPs. Where available (e.g. MDD), we also filtered out SNPs with effective sample sizes below the maximum.
TABLE 4.
Description of data sets used for practical application
2.7 |. Increasing power of TWAS tools
TWAS methods (Barbeira et al., 2018; Chatzinakos et al., 2020; Chatzinakos et al., 2018; Mancuso et al., 2018), are based on genetically regulated gene expression (GReX) models (Gamazon et al., 2015) in order to estimate gene-associations with the trait. These GReX models are practically a tissue-specific linear combination of eQTL SNPs for each gene. Often the GWAS (i.e. TWAS input) does not include all those eQTL SNPs from GReX models. To assess the decrease in power of TWAS tools not imputing SNP internally, we applied TWAS JEPEGMIX2 (Chatzinakos et al., 2018) using GTEx version 6 release GReX models (Barbeira et al., 2018; Gamazon et al., 2015) to PGC data sets above and to Re-Experiencing Symptoms GWAS (PTSD-REX) (Gelernter et al., 2019) [having summary statistics for only 4,374,623 SNPs] with and without re-imputing GWAS first.
3 |. RESULTS
For Illumina 1 M SNPs (Marenne et al., 2011) that were masked, and then imputed, DISTMIX2 with our novel automatic ethnic weight detection, controls the false positive rates at or below nominal threshold, even at very low type I error, for example, 10−6 (Text S2, Figure S1 in SI). R2 between true values and estimated ones is above 0.92 for our five simulated mixed-cohort scenarios (Text S2, Figures S2–S6 in SI). Also, DISTMIX2 imputed statistics had very good mean squared error (RMS) (Text S2, Figures S7–S11 in SI). For the above three measurements (size of the test, R2 and RMS) the setting of 250Kb for the length of the predicted window was the least precise, while 500Kb and 1000Kb had practically identical precision.
For rare and very rare variants, the size of the test was up to 300–1,000X higher than the nominal one and even up to 5,000–10,000X for cohorts that have large fractions of subpopulations that are underrepresented in the reference panel (e.g. Americans, Africans etc.), especially for the setting Minor Allele Frequency (MAF), 0.05%<MAF<0.5% and Information (Info), Info<0.2 (Text S2, Figures S12–S47 in SI).
For the “nullified” data sets, for example, those obtained from real data sets by substituting the study Z-scores by their expected quantile under the null hypothesis (H0) (Method evaluation section and Text S2, Figures S42–S48 in SI), DISTMIX2 controlled reasonably well the size of the test - up to 20X higher than the nominal rate (even for SNPs with low MAFs and low Info). The minimum GWAS p-values for the nullified data sets that were imputed ranged between 8.13 * 10−7 and 1.11 * 10−11. By fitting a normal distribution to −log10(minimum p-values), we estimated the mean to be 8.655 and the standard deviation to be 1.172. Using as criterion the conservative three standard deviations above the mean, we obtain from these realistic data a 12.17 as the upper bound for the −log10 (p-value). That is in DISTMIX2 applications in PGC GWAS, a conservative threshold for significance is 10−12, regardless of imputation Info and SNP MAF. Consequently, in all applied analyses in this paper we added this very stringent threshold for DISTMIX2 imputed summary statistics. Using as criterion the even more conservative five standard deviations above the mean (the very conservative Chebyshev inequality for the upper bound of the p-value of exceeding this threshold =), we obtained a 14.515 upper bound for the −log10 (p-values), that is a super-conservative significance threshold of 3 * 10−15.
For the applications to PGC GWAS (Table 4), we constructed Manhattan plots for all autosome chromosomes (1–22) and, individually, for chromosomes harboring novel signals (defined as imputed SNPs with statistically significant p-values that are at least 250Kb away from the reported GWAS signal) (Figure 1 and Figure 2, Text S3, Figure S49–S61 in SI). Furthermore, in order to investigate the potential risk of genomic inflation, we constructed Q-Q plots for the following three scenarios (i) all the SNPs, (ii) rare SNPs and (iii) common SNPS, for all the traits (Figure 3, Text S3, Figures S62–S84 in SI). Finally, we compared DISTMIX2 with ARDISS (Text S3, Figures S85–S91 in SI). Since ARDISS software did not provide minor allele frequency and Info estimation, we subset the imputed signals according to DISTMIX2 for MAF > 0.05. For all Manhattan plots we drew two dash lines denoting threshold for statistically significant signals. The red line is the default genome-wide threshold of p = 5 * 10−8, which is applicable to signals from measured SNPs and common imputed SNPs with high Info values. The purple line at p = 10−12 is the threshold to be used for rare/very rare variants and/or variants with low information; it corresponds to the above mentioned upper bound for nullified data. As an illustration, we present PTSD Manhattan plot for all chromosomes, only for chromosome 1 and Q-Q plots for all signals (Figure 1, Figure 2 and Figure 3 respectively).
FIGURE 1.
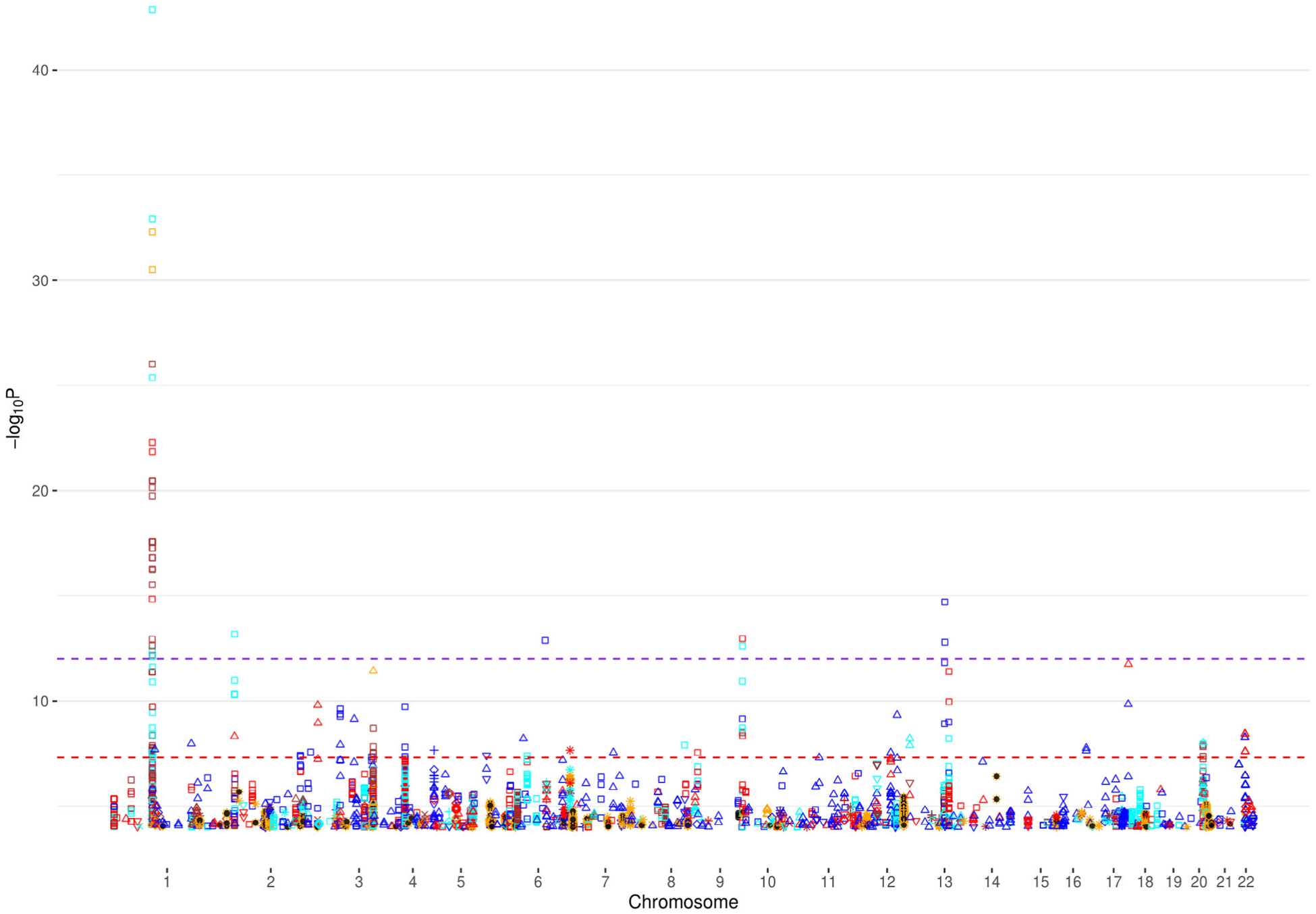
Manhattan plot for chromosomes 1–22 for PTSD. ● denotes reported signals from the original GWAS and the remain symbols and colors denote DISTMIX2 imputed signals. Among imputed signals blue denotes info<0.2, red denotes 0.2 < info<0.4, cyan denotes 0.4 < info<0.6, brown denotes 0.6 < info<0.8, orange denotes info>0.8, □ denotes MAF <0.05%, △ denotes 0.05% < MAF < 0.5%, ▽denotes 0.5% < MAF < 1%, + denotes 1% < MAF < 2%, ◊ denotes 2% < MAF < 5%, x denotes 5% < MAF < 10% and * denotes 10% < MAF < 50%. The red line is the default genome-wide threshold of p = 5*10e-8, which is applicable common SNPs with moderate to large Info values. The purple line at p = 10e-12 is the threshold to be used for rare and/or low Info variants. GWAS, genome-wide association studies; MAF, minor-AF; PTSD, post-traumatic stress disorder; SNPs, single nucleotide polymorphisms
FIGURE 2.
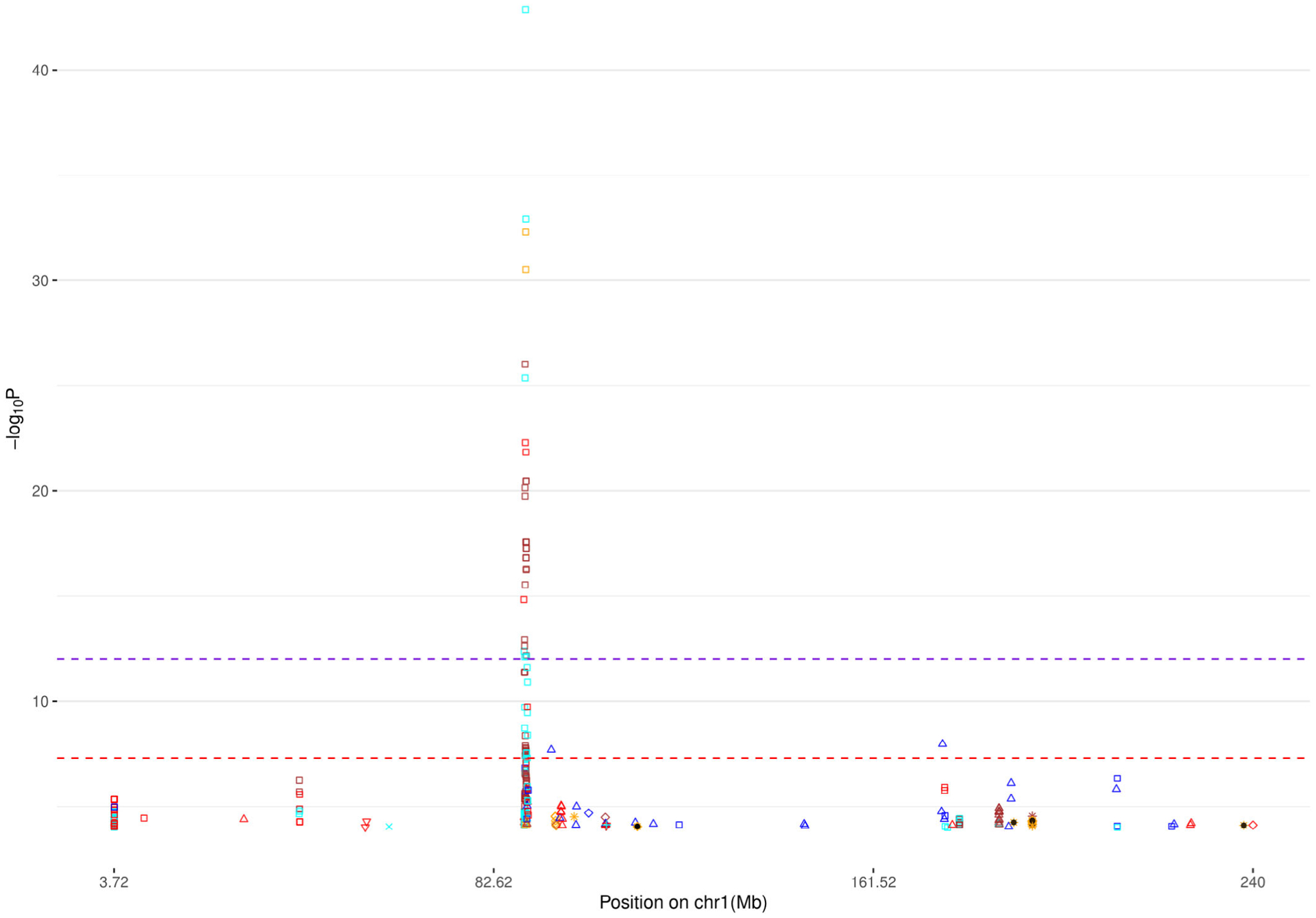
Manhattan plot for chromosome 1 for PTSD (see Figure 1 for background). PTSD, post-traumatic stress disorder
FIGURE 3.
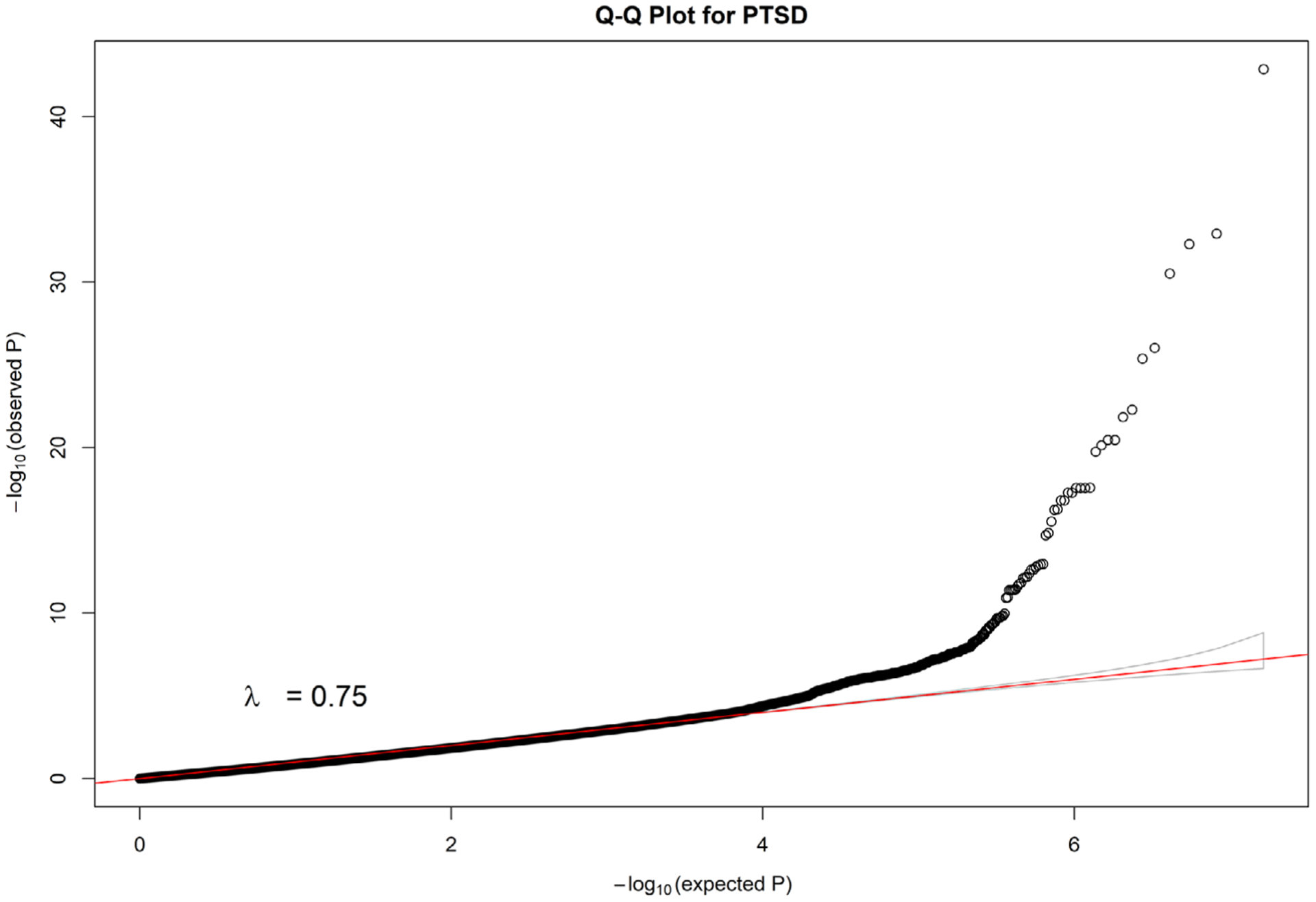
Q-Q plot for all SNPs of imputed PTSD GWAS. GWAS, genome-wide association studies; PTSD, post-traumatic stress disorder; SNPs, single nucleotide polymorphisms
These applications of DISTMIX2 to PGC data sets suggested the existence of numerous new signals, most associated with rare SNPs (see Table 5). For instance, in chromosome 12 for schizophrenia (rs143374), with MAF=0.0007, Info=0.245 and p-value=9.26 * 10−46 the magnitude of the p-value along with the lambda of the correspond Q-Q plot of the SCZ trait suggested that this signal is likely not to be an artifact (above the most stringent threshold), in chromosome 11 for ADHD (rs5681132) where the MAF=0.0004, the Info= 0.018 and p-value=7.40 * 10−16, in chromosome 22 for AUT (rs1380986), with MAF=0.0006, Info=0.498 and p-value=8.01 * 10−15, in chromosome 7 for BIP (rs76350051), with MAF=0.0004, Info=0.04 and p-value=2.47 * 10−37, in chromosome 12 for MDD (rs567868887), with MAF=0.0009, Info=0.28 and p-value=1.57 * 10−55, in chromosome 1 for PTSD (rs150642422), with MAF = 0.0002, Info = 0.5512 and p-value=1.3 * 10−43, and in chromosome 1 for PTSD-AA (rs111819353), with MAF = 0.001, Info = 0.3121 and p-value=1.16 * 10−17.
TABLE 5.
Best three signals for each PGC dataset
| Trait | rs_id | chr | bp | P pval | Info | MAF | Genes/Distance (Kbp) |
|---|---|---|---|---|---|---|---|
| ADHD | rs568113293 | 11 | 54,899,533 | 7.40 * 10 −16 | 0.0189 | 0.00049 |
TRIM48,130.125
RP11–72 M10.2,135.351 |
| rs544637819 | 3 | 15,310,737 | 1.78 * 10−14 | 0.1543 | 0.00171 |
SH3BP5, 0
SH3BP5-AS1,4.737 |
|
| chr6:30450452 | 6 | 30,450,452 | 6.44 * 10−13 | 0.0698 | 0.00151 |
RANP1, 3.265
HLA-E, 6.792 |
|
| AUT | rs138098629 | 22 | 36,584,165 | 8.01 * 10−15 | 0.4980 | 0.00063 |
APOL4, 1.007 MTND1P10, 8.732 |
| BIP | rs76350051 | 7 | 64,164,245 | 2.42 * 10 −37 | 0.0417 | 0.00046 |
ZNF107, 0
RP11–561 N12.7, 16.474 |
| rs138549126 | 3 | 52,592,843 | 6.65 * 10 −16 | 0.073 | 0.00052 |
SMIM4, 0
PBRM1, 0 |
|
| rs149257260 | 15 | 71,600,045 | 1.40 * 10−15 | 0.4246 | 0.00017 |
THSD4, 0
RP11–592 N21.1, 33.421 |
|
| ED | rs78958069 | 8 | 43,539,021 | 4.17 * 10−10 | 0.005 | 0.0002 |
RP11–643 N23.2, 10.803 AC134698.1, 123.105 |
| rs144485994 | 20 | 4,963,320 | 5.18 * 10−9 | 0.15 | 0.0001 |
SLC23A2, 0 RP5–1116H23.1, 30.846 |
|
| MDD | rs567868887 | 12 | 31,931,432 | 1.57 * 10 −55 | 0.2800 | 0.00098 |
H3F3C, 12.691
RP11–467 L13.5, 23.377 |
| rs112241719 | 11 | 111,514,969 | 8.14 * 10 −45 | 0.4900 | 0.00025 |
SIK2, 0
AP000925.2, 26.711 |
|
| rs182264017 | 1 | 188,992,506 | 5.05 * 10 −44 | 0.2775 | 0.00035 |
LINC01035, 0
CLPTM1LP1, 12.586 |
|
| PTSD | rs150642422 | 1 | 89,223,553 | 1.3 * 10 −43 | 0.5512 | 0.0002 |
PKN2, 0
RNU6–125P, 58.909 |
| rs7521099 | 1 | 88,875,371 | 1.4 * 10−15 | 0.4521 | 0.0002 | GBP3, 248.796 | |
| rs111229512 | 2 | 31,800,662 | 6.7 * 10−14 | 0.6325 | 0.0002 |
BP7, 373.881
GBP4, 423.278 |
|
| PTSD-AA | rs111819353 | 7 | 127,907,569 | 1.1 * 10 −17 | 0.312 | 0.001 |
LEP, 9.8
RBM28, |
| rs370549636 | 20 | 58,131,312 | 7.2 * 10−15 | 0.37 | 0.001 |
PHACTR3, 21.252
PIEZO1P1,95.42 |
|
| rs554371692 | 13 | 20,632,168 | 3.2 * 10−12 | 0.21 | 0.001 |
ZMYM2, 0 KRR1P1, 21.222 |
|
| SCZ | rs559199817 | 3 | 17,267,731 | 1.30 * 10 −87 | 0.0213 | 0.00073 |
TBC1D5, 0
AC090644.1, 90.517 |
| rs143337489 | 12 | 11,2089,686 | 9.26 * 10 −46 | 0.2464 | 0.00019 |
BRAP, 0
PCNPP1, 17.97 |
|
| rs193224736 | 16 | 8,593,132 | 3.79 * 10 −21 | 0.28476 | 0.00018 |
RP11-483 K5.3, 11.117
TMEM114, 26.37 |
Abbreviations: ADHD, attention deficit hyperactive disorder; AUT, autism; BIP, bipolar; ED, eating disorder; MAF, minor-AF; MDD, major depressive disorder; PTSD, post-traumatic stress disorder; PTSD-AA, Africanpost-traumatic stress disorder; SCZ, schizophrenia.
Note: Bolded and underlined entries correspond to the most stringent threshold of p < 3 * 10−15, not bolded but underlined to the second most conservative threshold 3 * 10−15 < p < 10−12 and not bolded not underlined 10−12 < p < 5 * 10−8.
When imputing in parallel SNPs regions of 40 Mbp, the analysis of each data set had a running time of less than 5 days on a cluster node with 4x Intel Xeon 6 core 2.67 GHz.
By imputing GWAS before a generic TWAS analysis (Table 6), we could impute TWAS signals for a larger number of genes, especially for the traits with the smallest number of variants (i.e. PTSD-REX). Additionally, the Q-Q plots (Text S3, Figures S92–S96 in SI) showed that all the analysis gained statistical power when we applied the imputation step.
TABLE 6.
Number genes with successful TWAS prediction under no imputation and imputation scenarios
| Trait | Number of original variants | Number of Imputed variants | Number of gene-tissue pairs with TWAS statistics under no imputation scenario | Number of gene-tissue pairs with TWAS statistics under imputation scenario |
|---|---|---|---|---|
| BIP | 13,413,244 | 13,800 | 26,772 | 26,894 |
| MDD | 13,554,550 | 14,805 | 26,734 | 26,889 |
| PTSD | 9,766,174 | 27,439 | 26,513 | 26,892 |
| PTSD-REX | 4,374,623 | 189,823 | 21,634 | 26,894 |
| SCZ | 14,225,895 | 6,444 | 26,846 | 26,894 |
Abbreviation: TWAS, transcriptome wide association study.
4 |. DISCUSSION
DISTMIX2, is a software/method for “off-the-shelf” direct imputation of the unmeasured SNP statistics in cosmopolitan cohorts. The main features of the updated version are: (a) a much larger (33 K subjects) and more diverse (includes ~11 K Han Chinese) reference panel and (b) a procedure for estimating the ethnic composition of the cohort without the need for AF information. Using simulated and the very novel nullified (real) data sets we propose conservative and very conservative significance thresholds for low info and low MAF signal. Application of DISTMIX2 to PGC data sets provides numerous new signal regions, most harboring rarer variants.
It is noteworthy that we uncovered a potentially very strong signal in PGC PTSD (p < 10−42) in a rare variant of PKN2 gene, when the initial publication reported only 3 marginal signals on common variants. While PKN2 has not been extensively characterized, it would be a potentially interesting target in PTSD given that it has been associated with Rho/Rock and mTOR cell pathways previously associated with fear learning and processing (Lachmann et al., 2011; Schmidt, Durgan, Magalhaes, & Hall, 2007; Wallace, Magalhaes, & Hall, 2011). It has also been associated with hippocampal functioning and development (Buchser, Slepak, Gutierrez-Arenas, Bixby, & Lemmon, 2010; Schmidt et al., 2007). All these cellular and neurobiological processes have been established as important in PTSD development and recovery (Maddox, Hartmann, Ross, & Ressler, 2019; Parsons & Ressler, 2013).
Due to our reassignment of subjects to subpopulations when constructing the 33 K reference panel, the naive assignment of the pre-estimated weights to only specific subpopulations from the reference panel that are considered the closest ones to the perceived cohort composition, can greatly increase the type I error (false positives). For that reason, when AF is not available, we recommend that users provide continental cohort weights (i.e. European [EUR], East Asian [ASN], South Asian [SAS], African [AFR] and America native [AMR]) and our software automatically will allocate these meta-weights to the most likely within-continent subpopulations. However, when AF is available there is no need to provide this additional information.
DISTMIX2 maintains the type I error reasonably accurately, even for low MAFs and low Info variants, especially for mostly European and East Asian cohorts that are overrepresented in our reference panel. When MAF > 5% (common variants), DISTMIX2 appears to maintain the false positive rates up to an order of magnitude higher than the nominal ones for all levels of information. Simulation results suggest that, when a larger part of study cohort consist of subpopulations underrepresented in our reference panel, it is reasonable to lower (by a factor of ~10,000) the genome-wide Bonferroni threshold of significance for p-values of imputed rarer variants. For imputed variants (especially rarer or with lower Info) in study of Europeans, we also use novel nullified data sets to propose a conservative threshold for significance of p = 10−12 and a very conservative threshold of p = 3 * 10−15. The yield of just one strong signal close to the LEP gene for PTSD-AA sample also suggests that the guidance also holds for the continental cohorts less represented in the reference panel. These rules-of-thumb are likely to be useful for similar methods when users enlarge their reference panels.
The length of the prediction window (250Kb, 500Kb, 1000Kb) is an important design parameter due to its implications for speed and precision of analyses. Simulations results suggest that, while the accuracies for 500Kb and 1000Kb estimates are very close, the computational burden increases ~2.5 times for the 1000 kb window. For that reason, we recommend that researchers use a 500Kb prediction window.
While mentioned only briefly in this manuscript, for application we used as “measured” SNP in the input summary statistic file only the GWAS SNPs reported to have close to perfect information and/or effective sample size. Our approach is rooted in preserving the cardinal assumption, of our and all but one other imputation methods (Rueger, McDaid, & Kutalik, 2018), that the LD between SNP Z-scores is very well approximated by the LD of the same SNPs in the reference panels. It is well known that when there are non-negligible missing rates for the variant pair this assumption is not met (Rueger et al., 2018). While the LD of Z-scores can be estimated by making reasonably realistic assumptions about co-missingness patterns of such SNP pairs, to avoid even the rarer circumstances in which these assumptions might not be met, we decided to avoid such an approach. Consequently, we employed (and recommend) the conservative approach of deeming as measured only SNPs with close to perfect imputation information and/or effective sample sizes in the original GWAS.
In the applications to PGC GWAS, the very low MAF and Info for some SNPs were associated with up to four orders of magnitude inflation in false positive rates, especially when the cohorts contain many subjects belonging to populations that are underrepresented in the reference panel. While signals for rarer SNPs from PGC data sets reported in this paper can be viewed as “softer” signals than the ones associated with common and high Info variants, the very low p-values for some of them (e.g. p < 10−42 in PKN2 gene for PTSD) suggest that most of these signals are likely to be real. This suggestion is enhanced by the fact that, to avoid the pitfalls of estimating covariances from just very few minor alleles, we did not include in the imputation panel SNPs that do not have at least: (a) 20 minor alleles in the Europeans or East Asians or (b) 5 minor alleles in all other continental groups. Nonetheless, we recognize that signals for these SNPs should be treated with more skepticism than the more common/higher Info variants and subjected to more stringent wet-lab validations.
Finally, we recommend users to re-impute summary statistics using the latest reference panels before employing most omics-based prediction tools, for example, TWAS (Chatzinakos, Georgiadis, & Daskalakis, 2021). The re-imputation of GWAS summary statistics is likely to increase the number of gene signals, especially for studies that employed older and smaller imputation panels. For instance, besides increasing the number of genes with TWAS predictions, the PTSD-REX imputation increased the significance of the MAPT (TWAS p = 1.4 * 10−5 to p = 1.12 * 10−9) and PLEKHM1 (TWAS p = 1.82 * 10−8 to p = 2.72 * 10−9) genes for the Cortex tissue analyses. There is no such need when using TWAS methods from our group (Chatzinakos et al., 2020; Chatzinakos et al., 2018; Lee et al., 2015; Lee et al., 2016) because they impute all missing eQTL SNPs by default].
Supplementary Material
ACKNOWLEDGMENTS
The authors want to thank Adam Maihofer and Caroline M Nievergelt for providing summary statistics files from the PGC PTSD cohorts. This study was supported by the 2019 Seed Grant from Silvio O. Conte Center for Stress Peptide Advanced Research, Education, & Dissemination (NIMH P50MH115874) to Chris Chatzinakos, NIMH (R01MH106595) to Kerry J. Ressler, an appointed KL2 award from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences KL2TR002542, UL1TR002541) to Nikolaos P. Daskalakis, and NIMH (R21MH121909) to Nikolaos P. Daskalakis.
Footnotes
Corrections added after online publication, 15 February, 2021: A new affiliation has been added for Dr. Chatzinakos & Dr. Daskalakis, and other affiliations were reordered to match journal style.]
CONFLICT OF INTEREST
Nikolaos P. Daskalakis has held a part-time paid position at Cohen Veteran Biosciences, has served as a paid consultant for Sunovion Pharmaceuticals and is on the scientific advisory board for Sentio Solutions, Inc. for unrelated work. The remaining authors have nothing to disclose.
DATA AVAILABILITY STATEMENT
The data (GWAS) that support the findings of this paper, except PTSD-AA and PTSD-REX, are openly available in https://www.med.unc.edu/pgc/
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- Anney RJL Ripke S, et al. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Molecular Autism 8, 21, 10.1186/s13229-017-0137-9 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, … Im HK (2018). Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature Communications, 9(1), 1825. doi: 10.1038/s41467-018-03621-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, & Browning SR (2009). A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. American Journal of Human Genetics, 84(2), 210–223.doi: 10.1016/j.ajhg.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, & Browning BL (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. American Journal of Human Genetics, 81(5), 1084–1097.doi: 10.1086/521987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchser WJ, Slepak TI, Gutierrez-Arenas O, Bixby JL, & Lemmon VP (2010). Kinase/phosphatase overexpression reveals pathways regulating hippocampal neuron morphology. Molecular Systems Biology, 6, 391. doi: 10.1038/msb.2010.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai N, Bigdeli TB, Kretzschmar WW, Li Y, Liang J, Hu J, … Flint J (2017). 11,670 whole-genome sequences representative of the Han Chinese population from the CONVERGE project. Scientific Data, 4, 170011. doi: 10.1038/sdata.2017.11 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chatzinakos C, Georgiadis F, & Daskalakis NP (2021). GWAS meets transcriptomics: from genetic letters to transcriptomic words of neuropsychiatric risk. Neuropsychopharmacology, 46(1), 255–256.doi: 10.1038/s41386-020-00835-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzinakos C, Georgiadis F, Lee D, Cai N, Vladimirov VI, Docherty A, … Bacanu SA (2020). TWAS pathway method greatly enhances the number of leads for uncovering the molecular underpinnings of psychiatric disorders. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics, 183(8), 454–463.doi: 10.1002/ajmg.b.32823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzinakos C, Lee D, Webb BT, Vladimirov VI, Kendler KS, & Bacanu SA (2018). JEPEGMIX2: improved gene-level joint analysis of eQTLs in cosmopolitan cohorts. Bioinformatics, 34(2), 286–288.doi: 10.1093/bioinformatics/btx509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium C (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature, 523(7562), 588–591, 10.1038/nature14659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Howie B, Cox AJ, Zagury JF, & Marchini J (2013). Haplotype estimation using sequencing reads. American Journal of Human Genetics, 93(4), 687–696.doi: 10.1016/j.ajhg.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, … Neale BM (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature Genetics, 51(1), 63–75.doi: 10.1038/s41588-018-0269-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, … Daly MJ (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43(5), 491–498.doi: 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan L, Yilmaz Z, Gaspar H, Walters R, Goldstein J, Anttila V, … Bulik CM (2017). Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. The American Journal of Psychiatry, 174(9), 850–858.doi: 10.1176/appi.ajp.2017.16121402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo C, Johnson TA, Morino R, Nakazono K, Kamitsuji S, Akita M, … Kawashima M (2018). Genome-wide association study in Japanese females identifies fifteen novel skin-related trait associations. Scientific Reports, 8(1), 8974. doi: 10.1038/s41598-018-27145-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, … Im HK (2015). A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics, 47(9), 1091–1098.doi: 10.1038/ng.3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Sun N, Polimanti R, Pietrzak R, Levey DF, Bryois J, … Million Veteran P (2019). Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in >165,000 US veterans. Nature Neuroscience, 22(9), 1394–1401.doi: 10.1038/s41593-019-0447-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, … McVean GA (2010). A map of human genome variation from population-scale sequencing. Nature, 467(7319), 1061–1073.doi: 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, … McVean GA (2012). An integrated map of genetic variation from 1,092 human genomes. Nature, 491 (7422), 56–65.doi: 10.1038/nature11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N, Szelinger S, Redman M, Duggan D, Tembe W, Muehling J, … Craig DW (2008). Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS. Genetics, 4(8), e1000167. doi: 10.1371/journal.pgen.1000167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, & Marchini J (2009). A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics, 5(6), e1000529. doi: 10.1371/journal.pgen.1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor SS, Morino R, Nakazono K, Kamitsuji S, Akita M, Kawajiri M, … Johnson TA (2018). Genome-wide association study of self-reported food reactions in Japanese identifies shrimp and peach specific loci in the HLA-DR/DQ gene region. Scientific Reports, 8(1), 1069. doi: 10.1038/s41598-017-18241-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann S, Jevons A, De Rycker M, Casamassima A, Radtke S, Collazos A, & Parker PJ (2011). Regulatory domain selectivity in the cell-type specific PKN-dependence of cell migration. PloS one, 6(7), e21732. doi: 10.1371/journal.pone.0021732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Bigdeli TB, Riley BP, Fanous AH, & Bacanu SA (2013). DIST: direct imputation of summary statistics for unmeasured SNPs. Bioinformatics, 29(22), 2925–2927.doi: 10.1093/bioinformatics/btt500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Bigdeli TB, Williamson VS, Vladimirov VI, Riley BP, Fanous AH, & Bacanu SA (2015). DISTMIX: direct imputation of summary statistics for unmeasured SNPs from mixed ethnicity cohorts. Bioinformatics, 31(19), 3099–3104.doi: 10.1093/bioinformatics/btv348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Williamson VS, Bigdeli TB, Riley BP, Webb BT, Fanous AH, … Bacanu SA (2016). JEPEGMIX: gene-level joint analysis of functional SNPs in cosmopolitan cohorts. Bioinformatics, 32(2), 295–297.doi: 10.1093/bioinformatics/btv567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, … Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079.doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, & Goodson M (2011). Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Research, 21(6), 936–939.doi: 10.1101/gr.111120.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Hartmann J, Ross RA, & Ressler KJ (2019). Deconstructing the Gestalt: Mechanisms of Fear, Threat, and Trauma Memory Encoding. Neuron, 102(1), 60–74.doi: 10.1016/j.neuron.2019.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, … consortium P (2018). Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nature Communication, 9(1), 4079. doi: 10.1038/s41467-018-06302-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenne G, Rodriguez-Santiago B, Closas MG, Perez-Jurado L, Rothman N, Rico D, … Malats N (2011). Assessment of copy number variation using the Illumina Infinium 1M SNP-array: a comparison of methodological approaches in the Spanish Bladder Cancer/EPICURO study. Human Mutation, 32(2), 240–248.doi: 10.1002/humu.21398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, … Haplotype Reference, C. (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nature genetics, 48(10), 1279–1283.doi: 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL (2006). Testing untyped alleles (TUNA)-applications to genome-wide association studies. Genetic epidemiology, 30(8), 718–727.doi: 10.1002/gepi.20182 [DOI] [PubMed] [Google Scholar]
- Nievergelt CM, Maihofer AX, Klengel T, Atkinson EG, Chen CY, Choi KW, … Koenen KC (2019). International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nature communications, 10(1), 4558. doi: 10.1038/s41467-019-12576-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, & Ressler KJ (2013). Implications of memory modulation for post-traumatic stress and fear disorders. Nature Neuroscience, 16(2), 146–153.doi: 10.1038/nn.3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasaniuc B, Zaitlen N, Shi H, Bhatia G, Gusev A, Pickrell J, … Price AL (2013). Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Quantity Biology,Cornell University Library. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revez JA, Lin T, Qiao Z, Xue A, Holtz Y, Zhu Z, … McGrath JJ (2020). Genome-wide association study identifies 143 loci associated with 25 hydroxyvitamin D concentration. Nature Communications, 11(1), 1647. doi: 10.1038/s41467-020-15421-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, … Sullivan PF (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature Genetics, 45(10), 1150–1159.doi: 10.1038/ng.2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, … Gejman PV (2011). Genome-wide association study identifies five new schizophrenia loci. Nature Genetics, 43(10), 969–976 10.1038/ng.940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueger S, McDaid A, & Kutalik Z (2018). Evaluation and application of summary statistic imputation to discover new height-associated loci. PLoS Genetics, 14(5), e1007371. doi: 10.1371/journal.pgen.1007371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511(7510), 421–427.doi: 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Durgan J, Magalhaes A, & Hall A (2007). Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. The EMBO journal, 26(6), 1624–1636.doi: 10.1038/sj.emboj.7601637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servin B, & Stephens M (2007). Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS genetics, 3(7), e114. doi: 10.1371/journal.pgen.0030114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, … Purcell SM (2011). Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature Genetics, 43(10), 977–983 10.1038/ng.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, … Froguel P (2007). A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature, 445(7130), 881–885.doi: 10.1038/nature05616 [DOI] [PubMed] [Google Scholar]
- Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, … Bipolar Disorder Working Group of the Psychiatric Genomics, C. (2019). Genome-wide association study identifies 30 loci associated with bipolar disorder. Nature Genetics, 51(5), 793–803.doi: 10.1038/s41588-019-0397-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togninalli M, Roqueiro D, Investigators CO, & Borgwardt KM (2018). Accurate and adaptive imputation of summary statistics in mixed-ethnicity cohorts. Bioinformatics, 34(17), i687–i696.doi: 10.1093/bioinformatics/bty596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SW, Magalhaes A, & Hall A (2011). The Rho target PRK2 regulates apical junction formation in human bronchial epithelial cells. Molecular and cellular biology, 31(1), 81–91.doi: 10.1128/MCB.01001-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, & Abecasis GR (2010). METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26(17), 2190–2191.doi: 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, … Frayling TM (2014). Defining the role of common variation in the genomic and biological architecture of adult human height. Nature Genetics, 46(11), 1173–1186.doi: 10.1038/ng.3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, … Major Depressive Disorder Working Group of the Psychiatric Genomics, C. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics, 50(5), 668–681.doi: 10.1038/s41588-018-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.