

SUMMARY
The complete assembly of each human chromosome is essential for understanding human biology and evolution1,2. Using complementary long-read sequencing technologies, we complete the first linear assembly of a human autosome: chromosome 8. Our assembly resolves the sequence of five previously long-standing gaps, including a 2.08 Mbp centromeric α-satellite array, a 644 kbp β-defensin copy number polymorphism important for disease risk, and an 863 kbp variable number tandem repeat at chromosome 8q21.2 that can function as a neocentromere. We show that the centromeric α-satellite array is generally methylated except for a 73 kbp hypomethylated region of diverse higher-order α-satellite enriched with CENP-A nucleosomes, consistent with the location of the kinetochore. Additionally, we confirm the overall organization and methylation pattern of the centromere in a diploid human genome. Using a dual long-read sequencing approach, we complete high-quality draft assemblies of the orthologous chromosome 8 centromere in chimpanzee, orangutan, and macaque for the first time to reconstruct its evolutionary history. Comparative and phylogenetic analyses show that the higher-order α-satellite structure evolved in the great ape ancestor with a layered symmetry, where more ancient higher-order repeats locate peripherally to monomeric α-satellites. We estimate that the mutation rate of centromeric satellite DNA is accelerated >2.2-fold, and this acceleration extends into the flanking sequence.
Since the announcement of the sequencing of the human genome 20 years ago1,2, human chromosomes have remained unfinished due to large regions of highly identical repeats clustered within centromeres, regions of segmental duplication (SD), and the acrocentric short arms of chromosomes. The presence of large swaths (>100 kbp) of highly identical repeats that are themselves copy number polymorphic has meant that such regions have persisted as gaps, limiting our understanding of human genetic variation and evolution3,4. The advent of long-read sequencing technologies and the use of DNA from complete hydatidiform moles (CHMs), however, have now made it possible to assemble these regions from native DNA for the first time5–7. Here, we present the first complete linear assembly of a human autosome: chromosome 8. We chose to assemble chromosome 8 because it carries a modestly sized centromere (approximately 1.5-2.2 Mbp)8,9, where the AT-rich, 171 bp α-satellite repeats are organized into a well-defined higher-order repeat (HOR) array. The chromosome, however, also contains one of the most structurally dynamic regions in the human genome—the β-defensin gene cluster at 8p23.110–12—as well as a recurrent polymorphic neocentromere at 8q21.2, which have been largely unresolved for the last 20 years.
Telomere-to-telomere assembly of chromosome 8.
Unlike the assembly of the human X chromosome13, we took advantage of both ultra-long ONT (Oxford Nanopore Technologies) and PacBio (Pacific Biosciences) high-fidelity (HiFi) data to resolve the gaps in human chromosome 8 (Fig. 1a,b; Methods). We first generated 20-fold sequence coverage of ultra-long ONT data and 32.4-fold coverage of PacBio HiFi data from a CHM (CHM13hTERT; abbr. CHM13; Supplementary Fig. 1). Then, we assembled complex regions in chromosome 8 by creating a library of singly unique nucleotide k-mers (SUNKs)14, or sequences of length k that occur approximately once per haploid genome (here, k = 20), from CHM13 PacBio HiFi data. We validated the SUNKs with Illumina data from the same genome and used them to barcode ultra-long ONT reads (Fig. 1b). Ultra-long ONT reads sharing highly similar barcodes were assembled into an initial sequence scaffold that traverses each chromosome 8 gap (Fig. 1b). We improved the base-pair accuracy of the sequence scaffolds by replacing the raw ONT sequence with concordant PacBio HiFi contigs and integrating them into a linear assembly of human chromosome 8 generated by Nurk and colleagues5 (Fig. 1b; Methods).
Figure 1. Telomere-to-telomere assembly of human chromosome 8.
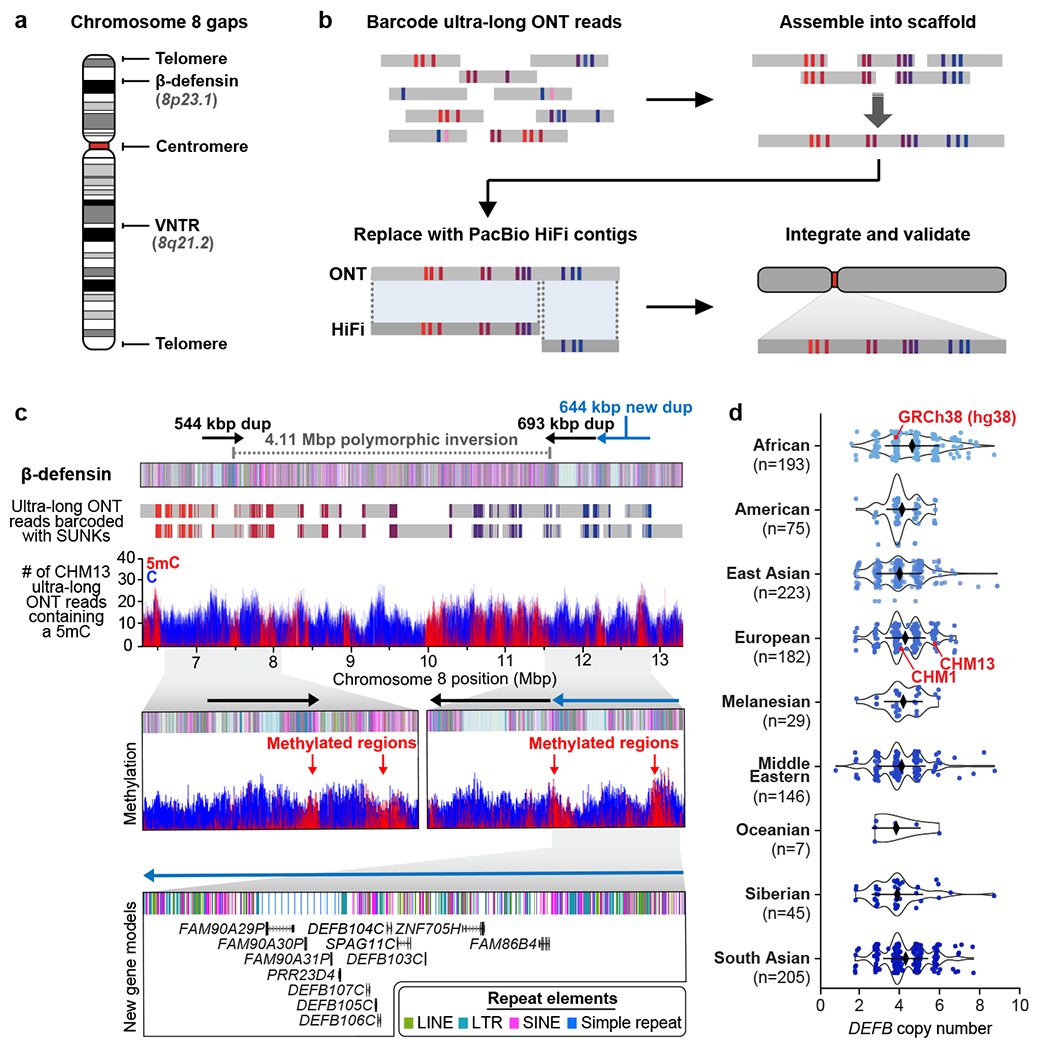
a) Gaps in the GRCh38 chromosome 8 reference. b) Targeted assembly method to resolve complex repeat regions in the human genome. Ultra-long ONT reads (gray) are barcoded with singly unique nucleotide k-mers (SUNKs; colored bars) and assembled into a sequence scaffold. Regions within the scaffold sharing high sequence identity with PacBio HiFi contigs (dark gray) are replaced, improving the base accuracy to >99.99%. The PacBio HiFi assembly is integrated into an assembly of CHM13 chromosome 85 and validated. c) Sequence, structure, methylation status, and genetic composition of the CHM13 β-defensin locus. The CHM13 locus contains three segmental duplications (SDs; dups) at chr8:7098892-7643091, chr8:11528114-12220905, and chr8:12233870-12878079. A 4,110,038 bp inversion (chr8:7500325-11610363) separates the first and second duplications. Iso-Seq data reveal that the third duplication (light blue) contains 12 new protein-coding genes, five of which are DEFB genes (Extended Data Fig. 3g). d) Copy number of the DEFB genes (chr8:7783837–7929198 in GRCh38) throughout the human population, determined from a collection of 1,105 high-coverage genomes (Methods). Median +/− s.d. is shown.
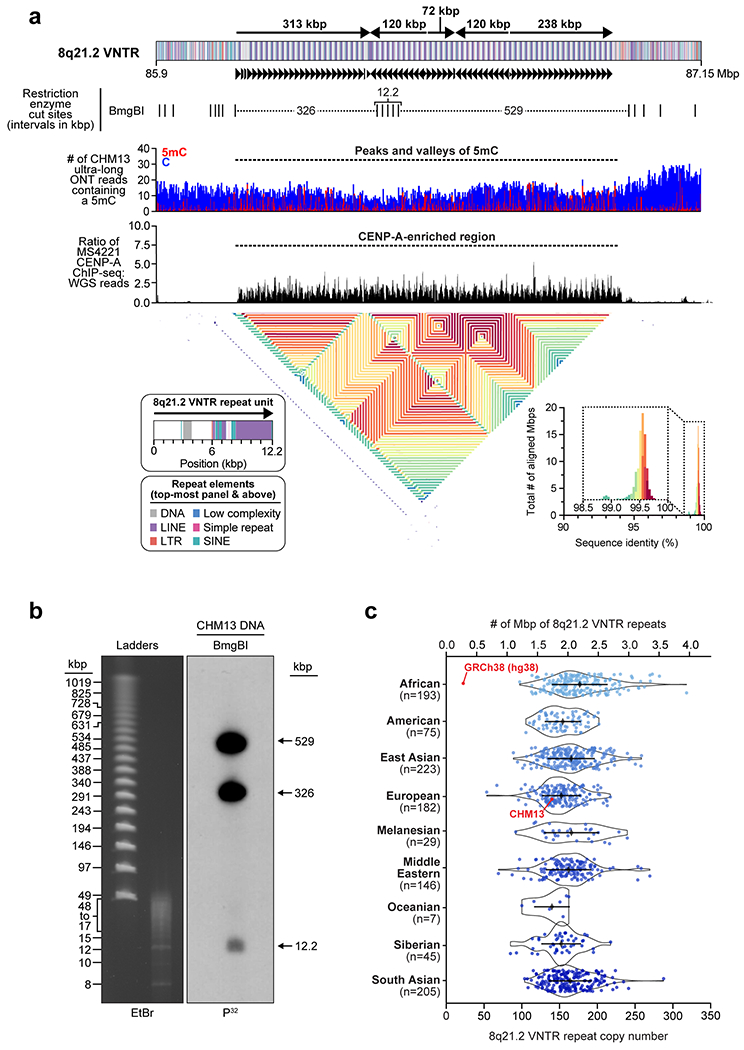
The complete telomere-to-telomere sequence of human chromosome 8 is 146,259,671 bases long and includes 3,334,256 bases missing from the current reference genome (GRCh38). Most of the additions reside within distinct chromosomal regions: a ~644 kbp copy number polymorphic β-defensin gene cluster mapping to chromosome 8p23.1 (Fig. 1c,d); the complete centromere corresponding to 2.08 Mbp of α-satellite HORs (Fig. 2); an 863 kbp 8q21.2 variable number tandem repeat (VNTR) (Extended Data Fig. 1); and both telomeric regions ending with the canonical TTAGGG repeat sequence (Extended Data Fig. 2). We validated the assembly with optical maps (Bionano Genomics), Strand-seq15,16, and comparisons to finished BAC sequence as well as Illumina whole-genome sequencing data derived from the same source genome (Supplementary Fig. 2; Methods). We estimate the overall base accuracy of our chromosome 8 assembly to be between 99.9915% and 99.9999% (quality value [QV] score between 40.70 and 63.19, as determined from sequenced BACs and mapped k-mers17, respectively). An analysis of 24 million human full-length transcripts generated from Iso-Seq data identifies 61 protein-coding and 33 noncoding loci that map better to this finished chromosome 8 sequence than to GRCh38 (Supplementary Table 1; Extended Data Fig. 3a–f), including the discovery of novel genes mapping to copy number polymorphic regions (Fig. 1d, Extended Data Fig. 3g).
Figure 2. Sequence, structure, and epigenetic map of the chromosome 8 centromeric region.
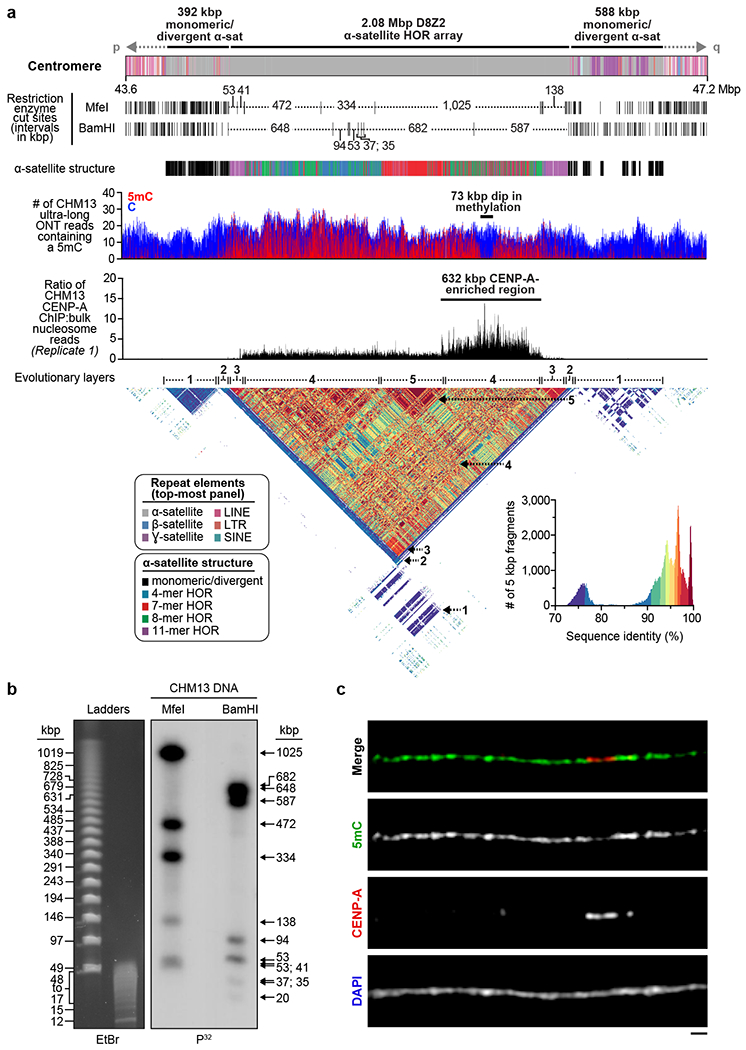
a) Schematic showing the composition of the CHM13 chromosome 8 centromere. The centromeric region is comprised of a 2.08 Mbp D8Z2 α-satellite HOR array flanked by regions of monomeric and/or divergent α-satellite interspersed with retrotransposons, β-satellite, and ɣ-satellite. The predicted restriction digest pattern is shown. The D8Z2 α-satellite HOR array is heavily methylated except for a 73 kbp hypomethylated region, which is encompassed by a 632 kbp CENP-A chromatin domain (Extended Data Fig. 9, Supplementary Fig. 8). A pairwise sequence identity heatmap indicates that the centromere is composed of five distinct evolutionary layers (dashed arrows). b) PFG Southern blot of CHM13 DNA confirms the structure and organization of the chromosome 8 centromeric HOR array. Left: EtBr staining; Right: P32-labeled chromosome 8 α-satellite-specific probe; n = 2. For gel source data, see Supplementary Fig. 9a,b. c) Representative images of a CHM13 chromatin fiber showing CENP-A enrichment in an unmethylated region. n = 3; bar = 1 micron.
Our targeted assembly method successfully resolves the β-defensin gene cluster10 into a single 7.06 Mbp locus, eliminating two 50 kbp gaps in GRCh38 (Fig. 1c, Extended Data Fig. 4). We estimate the base accuracy of this locus to be 99.9911% (QV score 40.48; based on mapped BACs; Extended Data Fig. 5a). Our analysis reveals CHM13 has a more structurally complex haplotype than GRCh38 (Extended Data Fig. 4), consistent with previously published reports10,12. We show that the breakpoints of the largest common human inversion polymorphism (4.11 Mbp) map within large, highly identical duplications that are copy number polymorphic (Fig. 1d; Extended Data Fig. 5b). In contrast to the human reference, which carries two such SDs, there are three SDs in CHM13: a 544 kbp SD on the distal end and two 693 and 644 kbp SDs on the proximal end (Fig. 1c). Each SD cassette carries at least five β-defensin genes and, as a result, we identify five additional β-defensin genes that are virtually identical at the amino acid level to the reference (Fig. 1c, Supplementary Table 2). Because ONT data allow methylation signals to be assessed18, we inferred the methylation status of cytosines across the entire β-defensin locus. All three SDs harbor a 151-163 kbp methylated region residing in the LTR-rich region of the duplication, while the remainder of the SD, including the β-defensin gene cluster, is largely unmethylated (Fig. 1c). Complete sequence resolution of this alternate haplotype is important because the inverted haplotype preferentially predisposes to recurrent microdeletions associated with developmental delay, microcephaly, and congenital heart defects19,20. Copy number polymorphism of the five β-defensin genes has been associated with immune-related phenotypes, such as psoriasis and Crohn’s disease11,21.
Sequence resolution of the chromosome 8 centromere.
Prior studies estimate the length of the chromosome 8 centromere to be between 1.5-2.2 Mbp, based on analysis of the HOR α-satellite array8,9. While HORs of different lengths are thought to comprise the centromere, the predominant species has a unit length of 11 monomers (1881 bp)8,9. During assembly, we spanned the chromosome 8 centromere with 11 ultra-long ONT reads (mean length 389.4 kbp), which were replaced with PacBio HiFi contigs based on SUNK barcoding. Our chromosome 8 centromere assembly consists of a 2.08 Mbp D8Z2 α-satellite HOR array flanked by blocks of monomeric α-satellite on the p- (392 kbp) and q- (588 kbp) arms (Fig. 2a). Both monomeric α-satellite blocks are interspersed with LINEs, SINEs, LTRs, and β-satellite, with tracts of ɣ-satellite specific to the q-arm. Multiple methods were used to validate its organization. First, long-read sequence read-depth analysis from two orthogonal native DNA sequencing platforms shows uniform coverage, suggesting that the assembly is free from large structural errors (Extended Data Fig. 6a). Fluorescent in situ hybridization (FISH) on metaphase chromosomes confirms the long-range organization of the centromere (Extended Data Fig. 6a–c). Droplet digital PCR shows that there are 1344 +/− 142 D8Z2 HORs within the α-satellite array, consistent with our estimates (Extended Data Fig. 6d; Methods). Pulsed-field gel electrophoresis Southern blots on CHM13 DNA digested with two different restriction enzymes supports the banding pattern predicted from the assembly (Fig. 2a,b). Finally, applying our assembly approach to ONT and HiFi data available for a diploid human genome (HG00733; Supplementary Table 3; Methods) generates two additional chromosome 8 centromere haplotypes, replicating the overall organization with only subtle differences in overall length of HOR arrays (Extended Data Fig. 7, Supplementary Table 4).
We find that the chromosome 8 centromeric HOR array is primarily composed of four distinct HOR types represented by 4, 7, 8, or 11 α-satellite monomer cassettes (Fig. 2a, Extended Data Fig. 8). While the 11-mer predominates (36%), the other HORs are also abundant (19-23%) and are all derivatives of the 11-mer (Extended Data Fig. 8b,c). Interestingly, we find that HORs are differentially distributed regionally across the centromere. While most regions show a mixture of different HOR types, we also identify regions of homogeneity, such as clusters of 11-mers mapping to the periphery of the HOR array (92 and 158 kbp in length) and a 177 kbp region in the center composed solely of 7-mer HORs. To investigate the epigenetic organization, we inferred methylated cytosines along the centromeric region and find that most of the α-satellite HOR array is methylated, except for a small, 73 kbp hypomethylated region (Fig. 2a). To determine if this hypomethylated region is the site of the epigenetic centromere (marked by the presence of nucleosomes containing the histone H3 variant CENP-A), we generated CENP-A ChIP-seq data from CHM13 cells and find that CENP-A is primarily located within a 632 kbp stretch encompassing the hypomethylated region (Fig. 2a, Extended Data Fig. 9). Chromatin fiber FISH reveals that CENP-A maps to the hypomethylated region within the α-satellite HOR array (Fig. 2c). Remarkably, the hypomethylated region shows some of the greatest HOR admixture, suggesting a potential optimization of HOR subtypes associated with the active kinetochore (mean entropy over the 73 kbp region = 1.91; Extended Data Fig. 8a; Methods).
To better understand the long-range organization and evolution of the centromere, we generated a heatmap to compare the sequence identity of 5 kbp fragments along the length of the centromere (Fig. 2a, Supplementary Fig. 3). We find that the centromere consists of five major evolutionary layers that show mirror symmetry. The outermost layer resides in the monomeric α-satellite, where sequences are highly divergent from the rest of the centromere but are more similar to each other (Fig. 2a, Arrow 1). The second layer defines the monomeric-to-HOR transition and is a short (57-60 kbp) region. The p and q regions are 87-92% identical with each other but only 78% or less with other centromeric satellites (Arrow 2). The third layer is completely composed of HORs. The p and q regions are 92 and 149 kbp in length, respectively, and share more than 96% sequence identity with each other (Arrow 3) but less than that with the rest of the centromere. This layer is composed largely of homogenous 11-mers and defines the transition from unmethylated to methylated DNA. The fourth layer is the largest and defines the bulk of the HOR α-satellite (1.42 Mbp in total). It shows the greatest variety of different HOR subtypes and, once again, the p and q blocks share identity with each other but are more divergent from the remaining layers (Arrow 4). Finally, the fifth layer encompasses the centermost 416 kbp of the HOR array, a region of near-perfect sequence identity that is divergent from the rest of the centromere (Arrow 5).
Sequence resolution of the chromosome 8q21.2 VNTR.
The layered and mirrored nature of the chromosome 8 centromere is reminiscent of another GRCh38 gap region located at chromosome 8q21.2 (Extended Data Fig. 1). This region is a cytogenetically recognizable euchromatic variant22 that contains one of the largest VNTRs in the human genome22. The 12.192 kbp repeating unit carries the GOR1/REXO1L1 pseudogene and is highly copy number polymorphic among humans22,23. This VNTR is of biological interest because it is the site of a recurrent neocentromere, where a functional centromere devoid of α-satellite has been observed in multiple unrelated individuals24,25. Using our approach, we successfully assembled the VNTR into an 863.5 kbp sequence composed of ~71 repeating units (67 complete and 7 partial units) (Extended Data Fig. 1a). A pulsed-field gel Southern blot confirms the VNTR length and structure (Extended Data Fig. 1a,b), and chromatin fiber FISH estimates 67 +/− 5.2 repeat units, consistent with the assembly (Extended Data Fig. 10; Methods). Among humans, the repeat unit varies from 53 to 326 copies, creating tandem repeat arrays ranging from 652 kbp to 3.97 Mbp (Extended Data Fig. 1c). The higher-order structure of the VNTR consists of five distinct domains that alternate in orientation (Extended Data Fig. 1a), where each domain contains 5 to 23 complete repeat units that are more than 98.5% identical to each other (Extended Data Fig. 1a). Detection of methylated cytosines18 shows that each 12.192 kbp repeat is primarily methylated in the 3 kbp region corresponding to GOR1/REXO1L1, while the rest of the repeat unit is hypomethylated (Extended Data Fig. 1a). Mapping of centromeric chromatin from a cell line harboring an 8q21.2 neocentromere25 shows that approximately 98% of CENP-A nucleosomes map to the hypomethylated region of the repeat unit in the CHM13 assembly (Extended Data Fig. 1a). While this is consistent with the VNTR being the potential site of the functional kinetochore of the neocentromere, sequence and assembly of this and other neocentromere-containing cell lines will be critically important.
Centromere evolutionary reconstruction.
In an effort to fully reconstruct the evolutionary history of the chromosome 8 centromere over the last 25 million years, we applied the same approach to reconstruct the corresponding centromere in chimpanzee, orangutan, and macaque. We first generated 25- to 40-fold HiFi data and 40- to 56-fold ONT data of each nonhuman primate (NHP) genome (Supplementary Table 5). Using this data, we generated two contiguous draft assemblies of the chimpanzee chromosome 8 centromere (one for each haplotype) and one haplotype assembly from the orangutan and macaque chromosome 8 centromeres (Fig. 3). Mapping of long-read data to each assembly shows uniform coverage, indicating a lack of large structural errors (Supplementary Figs. 4, 5). Assessment of base accuracy indicates that the assemblies are 99.9988-100% accurate (QV score > 49.3; Methods). Analysis of each NHP chromosome 8 centromere reveals distinct HOR arrays ranging in size from 1.69 Mbp in chimpanzee to 10.92 Mbp in macaque, consistent with estimates from short-read sequence data and cytogenetic analyses26,27 (Fig. 3). Our data, once again, reveal a mirrored and layered organization, with the chimpanzee organization being most similar to human (Figs. 2a, 3). Each NHP chromosome 8 centromere is composed of four or five distinct layers, with the outermost layer showing the lowest degree of sequence identity (73-78% in chimpanzee and orangutan; 90-92% in macaque) and the innermost layer showing the highest sequence identity (90-100% in chimpanzee and orangutan; 94-100% in macaque). The orangutan structure is striking in that there appears to be very little admixture of HOR units between the layers, in contrast to other apes where the different HOR cassettes are derived from a major HOR structure. The blocks of orangutan HORs (with the exception of layer 3) show reduced sequence identity. This suggests that the orangutan centromere evolved as a mosaic of independent HOR units. In contrast to all apes, the macaque lacks HORs and, instead, harbors a basic dimeric repeat structure26, which is much more homogenous and highly identical (>90%) across the nearly 11 Mbp of assembled centromeric array.
Figure 3. Sequence and structure of the chimpanzee, orangutan, and macaque chromosome 8 centromeres.
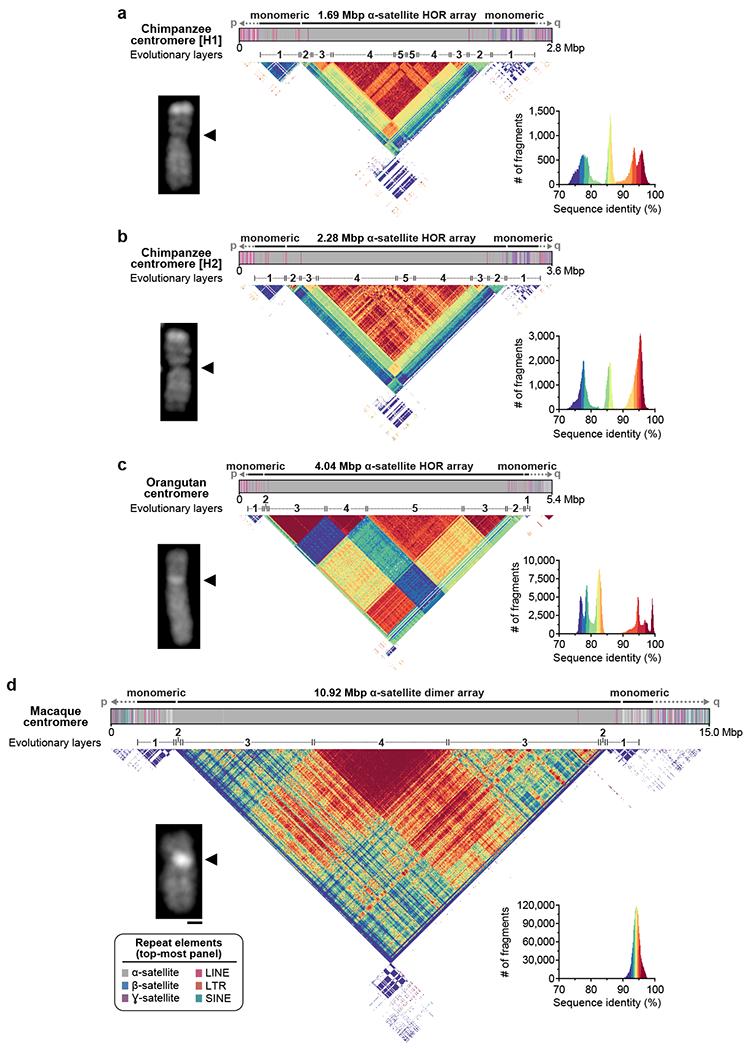
a-d) Structure and sequence identity of the a) chimpanzee H1, b) chimpanzee H2, c) orangutan, and d) macaque chromosome 8 centromeres. Each centromere has a mirrored organization consisting of 4-5 distinct evolutionary layers. The size of each centromeric region is consistent with microscopic analyses, showing increasingly bright DAPI staining with increasing centromere size. See Supplementary Figs. 10 and 11 for sequence identity heatmaps plotted on the same color scale. H1: haplotype 1; H2: haplotype 2; bar = 1 micron.
Phylogenetically (Fig. 4a), we find that all great ape higher-order α-satellite sequences (corresponding to layers 2-5) cluster into a single clade, while the monomeric α-satellite (layer 1) split into two clades separated by tens of millions of years. The proximal clade contains monomeric α-satellite from both the p- and q-arms, while the more divergent clade shares monomeric α-satellite solely from the q-arm, and specifically, the α-satellite nestled between clusters of ɣ-satellite (Supplementary Fig. 6a,b). Unlike great apes, both monomeric and dimeric repeat structures from the macaque group together and are sister clades to the monomeric ape clades, suggesting a common ancient origin restricted to these flanking pericentromeric regions. We used the orthology of flanking primate sequences to understand how rapidly sequences decay over the course of evolution. We assessed divergence based on 10 kbp windows of pairwise alignments in the ~2 Mbp flanking the α-satellite HOR array (Fig. 4b). We find that the mean allelic divergence increases more than threefold as the sequence transitions from unique to monomeric α-satellite. Such increases in divergence are rare in the human genome, where only 1.27-1.99% of nearly 20,000 random loci show comparable levels of divergence (Supplementary Fig. 6c). Using evolutionary models (Methods), we estimate a minimal mutation rate of the chromosome 8 centromeric region of ~4.8 x 10−8 and ~8.4 x 10−8 mutations per base pair per generation on the p- and q-arms, respectively, which is 2.2- to 3.8-fold higher than the basal mean mutation rate (~2.2 x 10−8) (Supplementary Table 6). These analyses provide the first complete comparative sequence analysis of a primate centromere for an orthologous chromosome and a framework for future studies of genetic variation and evolution of these regions across the genome.
Figure 4. Evolution of the chromosome 8 centromere.
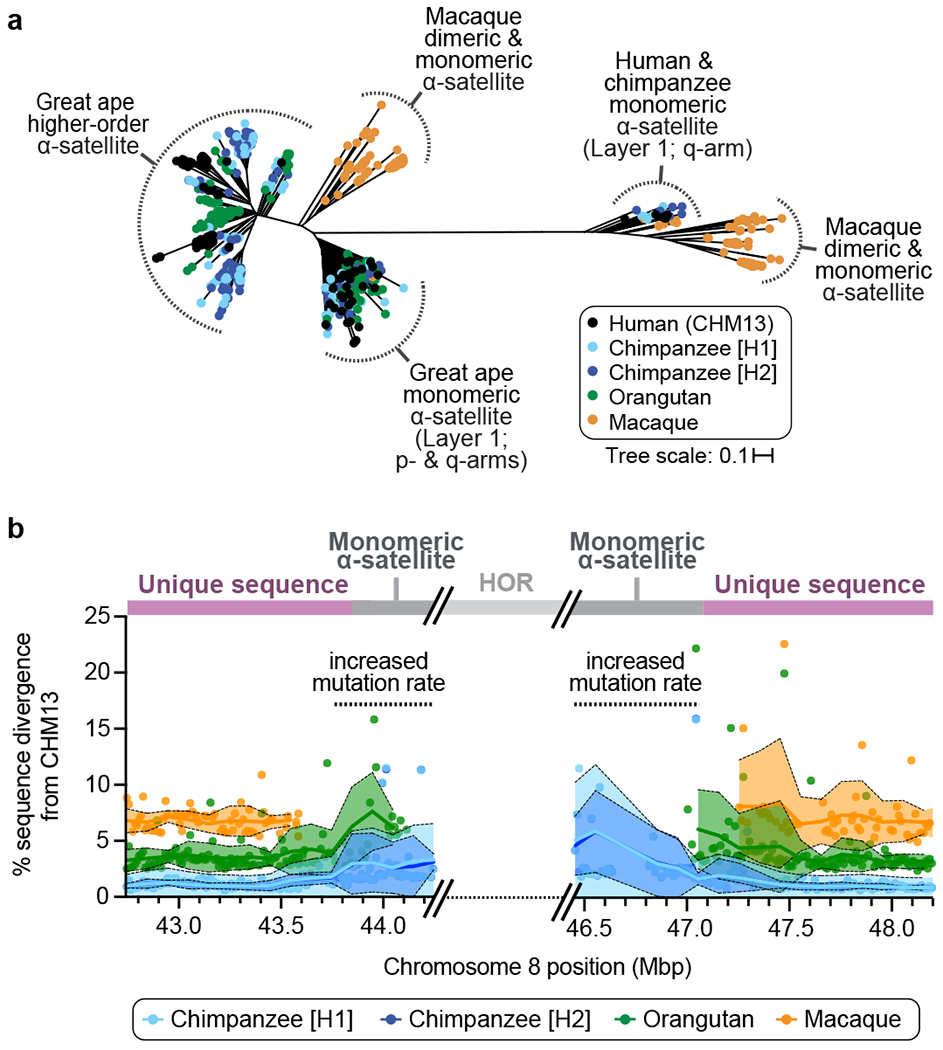
a) Phylogenetic tree of human, chimpanzee, orangutan, and macaque α-satellite from the chromosome 8 centromere (Supplementary Fig. 6a,b). b) Plot showing the sequence divergence between CHM13 and NHPs in the regions flanking the chromosome 8 α-satellite HOR array. See Supplementary Fig. 6d for a model of centromere evolution.
DISCUSSION
Chromosome 8 is the first human autosome to be sequenced and assembled from telomere to telomere and contains only the third completed human centromere to date13,28. Both chromosome X and 8 harbor a pocket of hypomethylation (~61-73 kbp in length), and we show that this region is enriched for the centromeric histone CENP-A, consistent with the functional kinetochore binding site29,30. Interestingly, CENP-A enrichment extends over a broader swath (632 kbp), with its peak centered over the hypomethylated region composed of diverse HORs. The layered and mirrored organization of the chromosome 8 centromere (Supplementary Fig. 7) supports a model of evolution31–33, wherein highly identical repeats expand, pushing older, more divergent repeats to the edges in an assembly-line fashion (Supplementary Fig. 6d). The chromosome 8 centromere reveals five such layers, and this organization is generally identified in other NHP centromeres. We confirm that HOR structures evolved after apes diverged from Old World monkeys (OWMs; <25 million years ago)26,34,35 but also distinguish different classes of monomeric repeats that share an ancient origin with the OWMs. One ape monomeric clade (present only in the q-arm) groups with the macaque’s (Supplementary Fig. 6a,b). We hypothesize that this ~70 kbp segment present in chimpanzee and human, but absent in orangutan, represents the remnants of the ancestral centromere. Sequence comparisons show that mutation rates increase by at least two to fourfold in proximity to the HOR array, likely due to the action of concerted evolution, unequal crossing-over, and saltatory amplification33,39,40. Among three human centromere 8 haplotypes, we identify regions of excess allelic variation and structural divergence (Extended Data Fig. 7), and these locations differ among haplotypes. Nevertheless, the first sequence of a complete human genome is imminent, and the next challenge will be applying the methods to fully phase and assemble diploid genomes36–38.
METHODS
Cell line sources
CHM13hTERT (abbr. CHM13) cells were originally isolated from a hydatidiform mole at Magee-Womens Hospital (Pittsburgh, PA) as part of a research study (IRB MWH-20-054). Cryogenically frozen cells from this culture were grown and transformed using human telomerase reverse transcriptase (TERT) to immortalize the cell line. This cell line has been authenticated via STR analysis, tested negative for mycoplasma contamination, and karyotyped to show a 46,XX karyotype13. Human HG00733 lymphoblastoid cells were originally obtained from a female Puerto Rican child, immortalized with the Epstein-Barr Virus (EBV), and stored at the Coriell Institute for Medical Research (Camden, NJ). Chimpanzee (Pan troglodytes; Clint; S006007) fibroblast cells were originally obtained from a male western chimpanzee named Clint (now deceased) at the Yerkes National Primate Research Center (Atlanta, GA) and immortalized with EBV. Orangutan (Pongo abelii; Susie; PR01109) fibroblast cells were originally obtained from a female Sumatran orangutan named Susie (now deceased) at the Gladys Porter Zoo (Brownsville, TX), immortalized with EBV, and stored at the Coriell Institute for Medical Research (Camden, NJ). Macaque (Macaca mulatta; AG07107) fibroblast cells were originally obtained from a female rhesus macaque of Indian origin and stored at the Coriell Institute for Medical Research (Camden, NJ). The HG00733, chimpanzee, orangutan, and macaque cell lines have not yet been authenticated or assessed for mycoplasma contamination to our knowledge.
Cell culture
CHM13 cell cultured in complete AmnioMax C-100 Basal Medium (Thermo Fisher Scientific, 17001082) supplemented with 15% AmnioMax C-100 Supplement (Thermo Fisher Scientific, 12556015) and 1% penicillin-streptomycin (Thermo Fisher Scientific, 15140122). HG00733 cells were cultured in RPMI 1640 with L-glutamine (Thermo Fisher Scientific, 11875093) supplemented with 15% FBS (Thermo Fisher Scientific, 16000-044) and 1% penicillin-streptomycin (Thermo Fisher Scientific, 15140122). Chimpanzee (Pan troglodytes; Clint; S006007) and macaque (Macaca mulatta; AG07107) cells were cultured in MEM α containing ribonucleosides, deoxyribonucleosides, and L-glutamine (Thermo Fisher Scientific, 12571063) supplemented with 12% FBS (Thermo Fisher Scientific, 16000-044) and 1% penicillin-streptomycin (Thermo Fisher Scientific, 15140122). Orangutan (Pongo abelii; Susie; PR01109) cells were cultured in MEM α containing ribonucleosides, deoxyribonucleosides, and L-glutamine (Thermo Fisher Scientific, 12571063) supplemented with 15% FBS (Thermo Fisher Scientific, 16000-044) and 1% penicillin-streptomycin (Thermo Fisher Scientific, 15140122). All cells were cultured in a humidity-controlled environment at 37°C with 5% CO2.
DNA extraction, library preparation, and sequencing
PacBio HiFi data were generated from the HG00733, chimpanzee, orangutan, and macaque genomes as previously described39 with modifications. Briefly, high-molecular-weight (HMW) DNA was extracted from cells using a modified Qiagen Gentra Puregene Cell Kit protocol40. HMW DNA was used to generate HiFi libraries via the SMRTbell Express Template Prep Kit v2 and SMRTbell Enzyme Clean Up kits (PacBio). Size selection was performed with SageELF (Sage Science), and fractions sized 11, 14, 18, 22, or 25 kbp (as determined by FEMTO Pulse (Agilent)) were chosen for sequencing. Libraries were sequenced on the Sequel II platform (Instrument Control SW v7.1 or v8.0) with three to seven SMRT Cells 8M (PacBio) using either Sequel II Sequencing Chemistry 1.0 and 12-hour pre-extension or Sequel II Sequencing Chemistry 2.0 and 3- or 4-hour pre-extension, both with 30-hour movies, aiming for a minimum estimated coverage of 25X in HiFi reads (assuming a genome size of 3.2 Gbp). Raw data was processed using the CCS algorithm (v3.4.1 or v4.0.0) with the following parameters: –minPasses 3 –minPredictedAccuracy 0.99 –maxLength 21000 or 50000.
Ultra-long ONT data were generated from the CHM13, HG00733, chimpanzee, and orangutan genomes according to a previously published protocol41. Briefly, 5 x 107 cells were lysed in a buffer containing 10 mM Tris-Cl (pH 8.0), 0.1 M EDTA (pH 8.0), 0.5% w/v SDS, and 20 ug/mL RNase A for 1 hour at 37C. Proteinase K (200 ug/mL) was added, and the solution was incubated at 50C for 2 hours. DNA was purified via two rounds of 25:24:1 phenol-chloroform-isoamyl alcohol extraction followed by ethanol precipitation. Precipitated DNA was solubilized in 10 mM Tris (pH 8) containing 0.02% Triton X-100 at 4C for two days. Libraries were constructed using the Rapid Sequencing Kit (SQK-RAD004) from ONT with modifications to the manufacturer’s protocol. Specifically, 2-3 ug of DNA was resuspended in a total volume of 18 ul with 16.6% FRA buffer. FRA enzyme was diluted 2- to 12-fold into FRA buffer, and 1.5 uL of diluted FRA was added to the DNA solution. The DNA solution was incubated at 30C for 1.5 min, followed by 8C for 1 min to inactivate the enzyme. RAP enzyme was diluted 2- to 12-fold into RAP buffer, and 0.5 uL of diluted RAP was added to the DNA solution. The DNA solution was incubated at room temperature (RT) for 2 hours before loading onto a primed FLO-MIN106 R9.4.1 flow cell for sequencing on a GridION using MinKNOW (v2.0 - v19.12).
Additional ONT data was generated from the CHM13, HG00733, chimpanzee, orangutan, and macaque genomes. Briefly, HMW DNA was extracted from cells using a modified Qiagen Gentra Puregene Cell Kit protocol40. HMW DNA was prepared into libraries with the Ligation Sequencing Kit (SQK-LSK109) from ONT and loaded onto primed FLO-MIN106 or FLO-PRO002 R9.4.1 flow cells for sequencing on a GridION or PromethION, respectively, using MinKNOW (v2.0 - v19.12). All ONT data were base called with Guppy 3.6.0 or 4.0.11 with the HAC model.
PacBio HiFi whole-genome assembly
The CHM13 genome was previously assembled from PacBio HiFi data using HiCanu5 and described by Nurk and colleagues5. The HG00733 genome was assembled from PacBio HiFi data (Supplementary Table 3) using hifiasm6 (v0.7). The chimpanzee, orangutan, and macaque genomes were assembled from PacBio HiFi data (Supplementary Table 5) using HiCanu5 (v2.0). Contigs from each assembly were used to replace the ONT-based sequence scaffolds in targeted regions (described below).
Targeted sequence assembly
Gapped regions within human chromosome 8 were targeted for assembly via a SUNK-based method that combines both PacBio HiFi and ONT data. Specifically, CHM13 PacBio HiFi data was used to generate a library of SUNKs (k = 20; total = 2,062,629,432) via Jellyfish (v2.2.4) based on the sequencing coverage of the HiFi dataset. 99.88% (2,060,229,331) of the CHM13 PacBio HiFi SUNKs were validated with CHM13 Illumina data (SRR3189741). A subset of CHM13 ultra-long ONT reads aligning to the CHM1 β-defensin patch (GenBank: KZ208915.1) or select regions within the GRCh38 chromosome 8 reference sequence (chr8:42,881,543-47,029,467 for the centromere and chr8:85,562,829-85,848,463 for the 8q21.2 locus) were barcoded with Illumina-validated SUNKs. Reads sharing at least 50 SUNKs were selected for inspection to determine if their SUNK barcodes overlapped. SUNK barcodes can be composed of “valid” and “invalid” SUNKs. Valid SUNKs are those that occur once in the genome and are located at the exact position on the read. In contrast, invalid SUNKs are those that occur once in the genome but are falsely located at the position on the read, and this may be due to a sequencing or base-calling error, for example. Valid SUNKs were identified within the barcode as those that share pairwise distances with at least ten other SUNKs on the same read. Reads that shared a SUNK barcode containing at least three valid SUNKs and their corresponding pairwise distances (+/−1% of the read length) were assembled into a tile. The process was repeated using the tile and subsetted ultra-long ONT reads several times until a sequence scaffold spanning the gapped region was generated. Validation of the scaffold organization was carried out via three independent methods. First, the sequence scaffold and underlying ONT reads were subjected to RepeatMasker (v3.3.0) to ensure that read overlaps were concordant in repeat structure. Second, the centromeric scaffold and underlying ONT reads were subjected to StringDecomposer42 to validate the HOR organization in overlapping reads. Finally, the sequence scaffold for each target region was incorporated into the CHM13 chromosome 8 assembly generated by Nurk and colleagues5, thereby filling the gaps in the chromosome 8 assembly. CHM13 PacBio HiFi and ONT data were aligned to the entire chromosome 8 assembly via pbmm2 (v1.1.0) (for PacBio data; https://github.com/PacificBiosciences/pbmm2) or Winnowmap43 (v1.0) (for ONT data) to identify large collapses or misassemblies. Although the ONT-based scaffolds are structurally accurate, they are only 87-98% accurate at the base level due to base-calling errors in the raw ONT reads7. Therefore, we sought to improve the base accuracy of the sequence scaffolds by replacing the ONT sequences with PacBio HiFi contigs assembled from the CHM13 genome5, which have a consensus accuracy greater than 99.99%5. Therefore, we aligned CHM13 PacBio HiFi contigs generated via HiCanu5 to the chromosome 8 assembly via minimap244 (v2.17-r941; parameters: minimap2 -t 8 -I 8G -a --eqx -x asm20 -s 5000) to identify contigs that share high sequence identity with the ONT-based sequence scaffolds. A typical scaffold had multiple PacBio HiFi contigs that aligned to regions within it. Therefore, the scaffold was used to order and orient the PacBio HiFi contigs and bridge gaps between them when necessary. PacBio HiFi contigs with high sequence identity replaced almost all regions of the ONT-based scaffolds: ultimately, the chromosome 8 assembly is comprised of 146,254,195 bp of PacBio HiFi contigs and only 5,490 bp of ONT sequence scaffolds (99.9963% PacBio HiFi contigs and 0.0037% ONT scaffold). The chromosome 8 assembly was incorporated into a whole-genome assembly of CHM13 generated by Nurk and colleagues5 for validation via orthogonal methods (detailed below). The HG00733, chimpanzee, orangutan, and macaque chromosome 8 centromeres were assembled via the same SUNK-based method.
Accuracy estimation
The accuracy of the CHM13 chromosome 8 assembly was estimated from mapped k-mers using Merqury17. Briefly, Merqury (v1.1) was run on the chromosome 8 assembly with the following command: eval/qv.sh CHM13.k21.meryl chr8.fasta chr8_v9.
CHM13 Illumina data (SRR1997411, SRR3189741, SRR3189742, SRR3189743) was used to identify k-mers with k = 21. In Merqury, every k-mer in the assembly is evaluated for its presence in the Illumina k-mer database, with any k-mer missing in the Illumina set counted as base-level ‘error’. We detected 1,474 k-mers found only in the assembly out of 146,259,650, resulting in a QV score of 63.19, estimated as follows:
The accuracy percentage for chromosome 8 was estimated from this QV score as:
The accuracy of the CHM13 chromosome 8 assembly and β-defensin locus were also estimated from sequenced BACs. Briefly, 66 BACs from the CHM13 chromosome 8 (BAC library VMRC59) were aligned to the chromosome 8 assembly via minimap244 (v2.17-r941) with the following parameters: -I 8G -2K 1500m --secondary=no -a --eqx -Y -x asm20 -s 200000 -z 10000,1000 -r 50000 -O 5,56 -E 4,1 -B 5. QV was then estimated using the CIGAR string in the resulting BAM, counting alignment differences as errors according to the following formula:
The median QV was 40.6988 for the entire chromosome 8 assembly and 40.4769 for the β-defensin locus (chr8:6300000-13300000; estimated from 47 individual BACs; see Extended Data Fig. 5 for more details), which falls within the 95% confidence interval for the whole chromosome This QV score was used to estimate the base accuracy39 as follows:
The BAC QV estimation should be considered a lower bound, since differences between the BACs and the assembly may originate from errors in the BAC sequences themselves. Vollger and colleagues showed that BACs can occasionally contain sequencing errors that are not supported by the underlying PacBio HiFi reads39. Additionally, the upper bound for the estimated BAC QV is limited to ~53, since BACs are typically ≲200 kbp and, as a result, the maximum calculable QV is 1 error in 200 kbp (QV 53). We also note that the QV of the centromeric region could not be estimated from BACs due to biases in BAC library preparation, which preclude centromeric sequences in BAC clones.
The accuracy of the HG00733, chimpanzee, orangutan, and macaque chromosome 8 centromere assemblies was estimated with Merqury17. Briefly, Merqury (v1.1) was run on the centromere assemblies as described above for the CHM13 chromosome 8 assembly. Ultimately, we detected 248 k-mers found only in the HG00733 maternal assembly out of 3,877,376 bp (estimated QV score of 55.16; base accuracy of 99.9997%); 10,562 k-mers found only in the HG00733 paternal assembly out of 3,597,645 bp (estimated QV score of 38.54; base accuracy of 99.986%); 0 k-mers found only in the chimpanzee H1 assembly out of 2,803,083 bp (estimated QV score of infinity; base accuracy of 100%); 20 k-mers found only in the chimpanzee H2 assembly out of 3,603,864 bp (estimated QV score of 65.7796; base accuracy of 99.9999%); 1302 k-mers found only in the orangutan assembly out of 5,372,621 bp (estimated QV score of 49.3774; accuracy of 99.9988%); and 104 k-mers found only in the macaque assembly out of 14,999,980 bp (estimated QV score of 64.8128; accuracy of 99.9999%). We note that Merqury detects the presence of erroneous k-mers in the assembly that have no support within the raw reads, but it cannot detect the absence of true k-mers (variants) within the assembled repeat copies. Thus, within these highly repetitive arrays, Merqury is useful for comparative analyses but may overestimate the overall accuracy of the consensus.
Strand-seq analysis
We evaluated the directional and structural contiguity of CHM13 chromosome 8 assembly, including the centromere, using Strand-seq data. First, all Strand-seq libraries produced from the CHM13 genome39 were aligned to the CHM13 assembly, including chromosome 8 using BWA-MEM45 (v0.7.17-r1188) with default parameters for paired-end mapping. Next, duplicate reads were marked by sambamba46 (v0.6.8) and removed before subsequent analyses. We used SAMtools47 (v1.9) to sort and index the final BAM file for each Strand-seq library. To detect putative misassembly breakpoints in the chromosome 8 assembly, we ran breakpointR48 on all BAM files to detect strand-state breakpoints. Misassemblies are visible as recurrent changes in strand state across multiple Strand-seq libraries49. To increase our sensitivity of misassembly detection, we created a ‘composite file’ that groups directional reads across all available Strand-seq libraries50,51. Next, we ran breakpointR on the ‘composite reads file’ using the function ‘runBreakpointr’ to detect regions that are homozygous (‘ww’; ‘HOM’ - all reads mapped in minus orientation) or heterozygous inverted (‘wc’, ‘HET’ - approximately equal number of reads mapped in minus and plus orientation). To further detect any putative chimerism in the chromosome 8 assembly, we applied Strand-seq to assign 200 kbp long chunks of the chromosome 8 assembly to unique groups corresponding to individual chromosomal homologues using SaaRclust49,52. For this, we used the SaaRclust function ‘scaffoldDenovoAssembly’ on all BAM files.
Bionano analysis
Bionano Genomics data was generated from the CHM13 genome13. Long DNA molecules labeled with Bionano’s Direct Labeling Enzyme were collected on a Bionano Saphyr Instrument to a coverage of 130X. The molecules were assembled with the Bionano assembly pipeline Solve (v3.4), using the nonhaplotype-aware parameters and GRCh38 as the reference. The resulting data produced 261 genome maps with a total length of 2921.6 Mbp and a genome map N50 of 69.02 Mbp.
The molecule set and the nonhaplotype-aware map were aligned to the CHM13 draft assembly and the GRCh38 assembly, and discrepancies were identified between the Bionano maps and the sequence references using scripts in the Bionano Solve software package -- runCharacterize.py, runSV.py, and align_bnx_to_cmap.py.
A second version of the map was assembled using the haplotype-aware parameters. This map was also aligned to GRCh38 and the final CHM13 assembly to verify heterozygous locations. These regions were then examined further.
Analysis of Bionano alignments revealed three heterozygous sites within chromosome 8 located at approximately chr8:21,025,201, chr8:80,044,843, and chr8:121,388,618 (Supplementary Table 7). The structure with the greatest ONT read support was selected for inclusion in the chromosome 8 assembly (Supplementary Table 7).
TandemMapper and TandemQUAST analysis of the centromeric HOR array
We assessed the structure of the CHM13 and NHP centromeric HOR arrays by applying TandemMapper and TandemQUAST53 (https://github.com/ablab/TandemTools; version from March 20th, 2020), which can detect large structural assembly errors in repeat arrays. For the CHM13 centromere, we first aligned ONT reads longer than 50 kbp to the CHM13 assembly containing the contiguous chromosome 8 with Winnowmap43 (v1.0) and extracted reads aligning to the centromeric HOR array (chr8:44243868-46323885). We then inputted these reads in the following TandemQUAST command: tandemquast.py -t 24 --nano {ont_reads.fa} -o {out_dir} chr8.fa. For the NHP centromeres, we aligned ONT reads to the whole-genome assemblies containing the contiguous chromosome 8 centromeres with Winnowmap43 (v1.0) and extracted reads aligning to the centromeric HOR arrays. We then inputted these reads in the following TandemQUAST command: tandemquast.py -t 24 --nano {ont_reads.fa} -o {out_dir} chr8.fa.
Methylation analysis
Nanopolish18 (v0.12.5) was used to measure CpG methylation from raw ONT reads (>50 kbp in length for CHM13) aligned to whole-genome assemblies via Winnowmap43 (v1.0). Nanopolish distinguishes 5-methylcytosine from unmethylated cytosine via a Hidden Markov Model (HMM) on the raw nanopore current signal. The methylation caller generates a log-likelihood value for the ratio of probability of methylated to unmethylated CpGs at a specific k-mer. We filtered methylation calls using the nanopore_methylation_utilities tool (https://github.com/isaclee/nanopore-methylation-utilities)54, which uses a log-likelihood ratio of 2.5 as a threshold for calling methylation. CpG sites with log-likelihood ratios greater than 2.5 (methylated) or less than −2.5 (unmethylated) are considered high quality and included in the analysis. Reads that do not have any high-quality CpG sites are filtered from the BAM for subsequent methylation analysis. Nanopore_methylation_utilities integrates methylation information into the BAM file for viewing in IGV’s55 bisulfite mode, which was used to visualize CpG methylation.
Iso-Seq data generation and sequence analyses
RNA was purified from approximately 1 x 107 CHM13 cells using an RNeasy kit (Qiagen; 74104) and prepared into Iso-Seq libraries following a standard protocol56. Libraries were loaded on two SMRT Cells 8M and sequenced on the Sequel II. The data were processed via isoseq3 (v8.0), ultimately generating 3,576,198 full-length non-chimeric (FLNC) reads. Poly-A trimmed transcripts were aligned to this CHM13 chr8 assembly and to GRCh38 with minimap244 (v2.17-r941) with the following parameters: -ax splice -f 1000 --sam-hit-only --secondary=no --eqx. Transcripts were assigned to genes using featureCounts57 with GENCODE58 (v34) annotations, supplemented with CHESS v2.259 for any transcripts unannotated in GENCODE. Each transcript was scored for percent identity of its alignment to each assembly, requiring 90% of the length of each transcript to align to the assembly for it to count as aligned. For each gene, non-CHM13 transcripts’ percent identity to GRCh38 was compared to the CHM13 chromosome 8 assembly. Genes with an improved representation in the CHM13 assembly were identified with a cutoff of 20 improved reads per gene, with at least 0.2% average improvement in percent identity. GENCODE (v34) transcripts were lifted over to the CHM13 chr8 assembly using Liftoff60 to compare the GRCh38 annotations to this assembly and Iso-Seq transcripts.
We combined the 3.6 million full-length transcript data (above) with 20,937,742 FLNC publicly available human Iso-Seq data (Supplementary Table 8). In total, we compared the alignment of 24,513,940 FLNC reads from 13 tissue and cell line sources to both the completed CHM13 chromosome 8 assembly and the current human reference genome, GRCh38. Of the 848,048 non-CHM13 cell line transcripts that align to chromosome 8, 93,495 (11.02%) align with at least 0.1% greater percent identity to the CHM13 assembly, and 52,821 (6.23%) to GRCh38. This metric suggests that the chromosome 8 reference improves human gene annotation by ~4.79% even though most of those changes are subtle in nature. Overall, 61 protein-coding and 33 noncoding loci have improved alignments to the CHM13 assembly compared to GRCh38, with >0.2% average percent identity improvement, and at least 20 supporting transcripts (Extended Data Fig. 3a–c, Supplementary Table 1). As an example, WDYHV1 (NTAQ1) has four amino acid replacements, with 13 transcripts sharing the identical open reading frame to CHM13 (Extended Data Fig. 3d).
Pairwise sequence identity heatmaps
To generate pairwise sequence identity heatmaps, we fragmented the centromere assemblies into 5 kbp fragments (e.g., 1-5000, 5001-10000, etc.) and made all possible pairwise alignments between the fragments using the following minimap244 (v2.17-r941) command: minimap2 -f 0.0001 -t 32 -X --eqx -ax ava-ont. The sequence identity was determined from the CIGAR string of the alignments and then visualized using ggplot2 (geom_raster) in R (v1.1.383)61. The color of each segment was determined by sorting the data by identity and then creating 10 equally sized bins, each of which received a distinct color from the spectral pallet. The choice of a 5 kbp window came after testing a variety of window sizes. Ultimately, we found 5 kbp to be a good balance between resolution of the figure (since each 5 kbp fragment is plotted as a pixel) and sensitivity of minimap2 (fragments less than 5 kbp often missed alignments with the ava-ont preset). A schematic illustrating this process is shown in Supplementary Fig. 3.
Miropeats analysis
To compare the organization and orientation of the CHM13 and GRCh38 β-defensin loci, we aligned the two β-defensin regions [CHM13 chr8:6300000-13300000; GRCh38 chr8:6545299-13033398] to each other using the following minimap244 parameters: minimap2 -x asm20 -s 200000 -p 0.01 -N 1000 --cs {GRCh38_defensin.fasta} {CHM13_defensin.fasta}. Then, we applied a version of Miropeats62 that is modified to use minimap244 alignments (https://github.com/mrvollger/minimiro) to produce the figure showing homology between the two sequences.
Analysis of α-satellite organization
To determine the organization of the CHM13 chromosome 8 centromeric region, we employed two independent approaches. First, we subjected the CHM13 centromere assembly to an in silico restriction enzyme digestion wherein a set of restriction enzyme recognition sites were identified within the assembly. In agreement with previous findings that XbaI digestion can generate a pattern of HORs within the chromosome 8 HOR array9, we found that each α-satellite HOR could be extracted via XbaI digestion. The in silico digestion analysis indicates that the chromosome 8 centromeric HOR array is comprised of 1462 HOR units: 283 4-mers, 4 5-mers, 13 6-mers, 356 7-mers, 295 8-mers, and 511 11-mers. As an alternative approach, we subjected the centromere assembly to StringDecomposer42 (https://github.com/ablab/stringdecomposer; version from February 28th, 2020) using a set of 11 α-satellite monomers derived from a chromosome 8 11-mer HOR unit. The sequence of the α-satellite monomers used are as follows: A: AGCATTCTCAGAAACACCTTCGTGATGTTTGCAATCAAGTCACAGAGTTGAACCTTCCGTTTCATAGAGCAGGTTGGAAACACTCTTATTGTAGTATCTGGAAGTGGACATTTGGAGCGCTTTCAGGCCTATGGTGAAAAAGGAAATATCTTCCCATAAAAACGACATAGA; B: AGCTATCTCAGGAACTTGTTTATGATGCATCTAATCAACTAACAGTGTTGAACCTTTGTACTGACAGAGCACTTTGAAACACTCTTTTTTGGAATCTGCAAGTGGATATTTGGATCGCTTTGAGGATTTCGTTGGAAACGGGATGCAATATAAAACGTACACAGC; C: AGCATACTCAGAAAATACTTTGCCATATTTCCATTCAAGTCACAGAGTGGAACATTCCCATTCATAGAGCAGGTTGGAAACACTCTTTTTGGAGTATCTGGAAGTGGACATTTGGAGCGCTTTCTGAACTATGGTGAAAAAGGAAATATCTTCCAATGAAAACAAGACAGA; D: AGCATTCTGAGAAACTTATTTGTGATGTGTGTCCTCAACAAACGGACTTGAACCTTTCGTTTCATGCAGTACTTCTGGAACACTCTTTTTGAAGATTCTGCATGCGGATATTTGGATAGCTTTGAGGATTTCGTTGGAAACGGGCTTACATGTAAAAATTAGACAGC; E: AGCATTCTCAGAAACTTCTTTGTGGTGTCTGCATTCAAGTCACAGAATTGAACTTCTCCTCACATAGAGCAGTTGTGCAGCACTCTATTTGTAGTATCTGGAAGTGGACATTTGGAGGGCTTTGTAGCCTATCTGGAAAAAGGAAATATCTTCCCATGAATGCGAGATAGA; F: AGTAATCTCAGAAACATGTTTATGCTGTATCTACTCAACTAACTGTGCTGAACATTTCTATTGATAGAGCAGTTTTGAGACCCTCTTCTTTTGGAATCTGCAAGTGGATATTTGGATAGATTTGAGGATTTCGTTGGAAACGGGATTATATATAAAAAGTAGACAGC; G: AGCATTCTCAGAAACTTCTTTGTGATGTTTGCATCCAGCTCTCAGAGTTGAACATTCCCTTTCATAGAGTAGGTTTGAAACCCTCTTTTTATAGTGTCTGGAAGCGGGCATTTGGAGCGCTTTCAGGCCTATGCTGAAAAAGGAAATATCTACATATAGAAACTAGACAGA; H: AGCATTCTGAGAATCAAGTTTGTGATGTGGGTACTCAACTAACAGTGTTGATCCATTCTTTTGATACAGCAGTTTTGAACCACACTTTTTGTAGAATCTGCAAGTGGATATTTGGATAGCTGTGAGGATTTCGTTGGAAACGGGAATGTCTTCATAGAAAATTTAGACAGA; I: AGCATTCTCAGAACCTTGATTGTGATGTGTGTTCTCCACTAACAGAGTTGAACCTTTCTTTTGACAGAACTGTTCTGAAACATTCTTTTTATAGAATCTGGAAGTGGATATTTGGAAAGCTTTGAGGATTTCGTTGGAAACGGGAATATCTTCAAATAAAATCTAGCCAGA; J: AGCATTCTAAGAAACATCTTAGGGATGTTTACATTCAAGTCACAGAGTTGAACATTCCCTTTCACAGAGCAGGTTTGAAACAATCTTCTCGTACTATCTGGCAGTGGACATTTTGAGCTCTTTGGGGCCTATGCTGAAAAAGGAAATATCTTCCGACAAAAACTAGTCAGA; K: AGCATTCGCAGAATCCCGTTTGTGATGTGTGCACTCAACTGTCAGAATTGAACCTTGGTTTGGAGAGAGCACTTTTGAAACACACTTTTTGTAGAATCTGCAGGTGGATATTTGGCTAGCTTTGAGGATTTCGTTGGAAACGGTAATGTCTTCAAAGAAAATCTAGACAGA.
This analysis indicated that the CHM13 chromosome 8 centromeric HOR array is comprised of 1515 HOR units: 286 4-mers, 12 6-mers, 366 7-mers, 303 8-mers, 3 10-mers, 539 11-mers, 2 12-mers, 2 13-mers, 1 17-mer, and 1 18-mer, which is concordant with the in silico restriction enzyme digestion results. The predominant HOR types from StringDecomposer42 are presented in Extended Data Fig. 8.
Copy number estimation
To estimate the copy number for the 8q21.2 VNTR and DEFB loci in human lineages, we applied a read-depth based copy number genotyper14 to a collection of 1,105 published high-coverage genomes63–68. Briefly, sequencing reads were divided into multiples of 36-mer, which were then mapped to a repeat-masked human reference genome (GRCh38) using mrsFAST69 (v3.4.1). To increase the mapping sensitivity, we allowed up to two mismatches per 36-mer. The read depth of mappable sequences across the genome was corrected for underlying GC content, and copy number estimate for the locus of interest was computed by summarizing over all mappable bases for each sample.
Entropy calculation
To define regions of increased admixture within the centromeric HOR array, we calculated the entropy using the frequencies of the different HOR units in 10-unit windows (1 unit slide) over the entire array. The formula for entropy is:
where frequency is (# of HORs) / (total # of HORs) in a 10-unit window. The analysis is analogous to that performed by Gymrek and colleagues70.
Droplet digital PCR
Droplet digital PCR was performed on CHM13 genomic DNA to estimate the number of D8Z2 α-satellite HORs, as was previously done for the DXZ1 α-satellite HORs13. Briefly, genomic DNA was isolated from CHM13 cells using the DNeasy Blood & Tissue Kit (Qiagen). DNA was quantified using a Qubit Fluorometer and the Qubit dsDNA HS Assay (Invitrogen). 20 uL reactions were prepared with 0.1 ng of gDNA for the D8Z2 assay or 1 ng of gDNA for the MTUS1 single-copy gene (as a control). EvaGreen droplet digital PCR (Bio-Rad) master mixes were simultaneously prepared for the D8Z2 and MTUS1 reactions, which were then incubated for 15 minutes to allow for restriction digest, according to the manufacturer’s protocol.
Pulsed-field gel electrophoresis and Southern blot
CHM13 genomic DNA was prepared in agarose plugs and digested with either BamHI or MfeI (to characterize the chromosome 8 centromeric region) or BmgBI (to characterize the chromosome 8q21.2 region) in the buffer recommended by the manufacturer. The digested DNA was separated with the CHEF Mapper system (Bio-Rad; autoprogram, 5-850 kbp range, 16 hr run), transferred to a membrane (Amersham Hybond-N+) and blot-hybridized with a 156 bp probe specific to the chromosome 8 centromeric α-satellite or 8q21.2 region. The probe was labeled with P32 by PCR-amplifying a synthetic DNA template #233: 5′-TTTGTGGAAGTGGACATTTCGCTTTGTAGCCTATCTGGAAAAAGGAAATATCTTCCCATGAATGCGAGATAGAAGTAATCTCAGAAACATGTTTATGCTGTATCTACTCAACTAACTGTGCTGAACATTTCTATTGTAAAAATAGACAGAAGCATT-3′ (for the centromere of chromosome 8); #264: 5′-TTTGTGGAAGTGGACATTTCGCCCGAGGGGCCGCGGCAGGGATTCCGGGGGACCGGGAGTGGGGGGTTGGGGTTACTCTTGGCTTTTTGCCCTCTCCTGCCGCCGGCTGCTCCAGTTTCTTTCGCTTTGCGGCGAGGTGGTAAAAATAGACAGAAGCATT-3′ (for the organization of the chromosome 8q21.2 locus) with PCR primers #129: 5′-TTTGTGGAAGTGGACATTTC-3′ and #130: 5′-AATGCTTCTGTCTATTTTTA-3′. The blot was incubated for 2 hr at 65°C for pre-hybridization in Church’s buffer (0.5 M Na-phosphate buffer containing 7% SDS and 100 μg/ml of unlabeled salmon sperm carrier DNA). The labeled probe was heat denatured in a boiling water bath for 5 min and snap-cooled on ice. The probe was added to the hybridization Church’s buffer and allowed to hybridize for 48 hr at 65°C. The blot was washed twice in 2× SSC (300 mM NaCl, 30 mM sodium citrate, pH 7.0), 0.05% SDS for 10 min at room temperature, twice in 2× SSC, 0.05% SDS for 5 min at 60°C, twice in 0.5× SSC, 0.05% SDS for 5 min at 60°C, and twice in 0.25× SSC, 0.05% SDS for 5 min at 60°C. The blot was exposed to X-ray film for 16 hr at −80°C. Uncropped, unprocessed images of all gels and blots are shown in Supplementary Figure 9.
Fluorescence in situ hybridization (FISH) and immunofluorescence (IF)
To validate the organization of the chromosome 8 centromere, we performed FISH on metaphase chromosome spreads according to the Haaf and Ward protocol71 with slight modifications. Briefly, CHM13 cells were treated with colcemid and resuspended in HCM buffer (10 mM HEPES pH7.3, 30 mM glycerol, 1 mM CaCl2, 0.8 mM MgCl2). After 10 minutes, cells were fixed with methanol:acetic acid (3:1), dropped onto previously clean slides, and soaked in 1X PBS. Slides were incubated overnight in cold methanol, hybridized with labelled FISH probes at 68°C for 2 min, and incubated overnight at 37°C. Slides were washed 3× in 0.1X SSC at 65°C for 5 min each before mounting in Vectashield containing 5 μg/ml DAPI. Slides were imaged on a fluorescence microscope (Leica DM RXA2) equipped with a charge-coupled device camera (CoolSNAP HQ2) and a 100x 1.6-0.6 NA objective lens. Images were collected using Leica Application Suite X (v3.7).
The probes used to validate the organization of the chromosome 8 centromere were picked from the human large-insert clone fosmid library ABC10. ABC10 end sequences were mapped using MEGABLAST (similarity=0.99, parameters: -D 2 -v 7 -b 7 -e 1e-40 -p 80 -s 90 -W 12 -t 21 -F F) to a repeat-masked CHM13 genome assembly containing the complete chromosome 8 (parameters: -e wublast -xsmall -no_is -s -species Homo sapiens). Expected insert size for fosmids was set to (min) 32 kbp and (max) 48 kbp. Resulting clone alignments were grouped into the following categories based on uniqueness of the alignment for a given pair of clones, alignment orientation and the inferred insert size from the assembly.
Concordant best: unique alignment for clone pair, insert size within expected fosmid range, expected orientation
Concordant tied: non-unique alignment for clone pair, insert size within expected fosmid range, expected orientation
Discordant best: unique alignment of clone pair, insert size too small, too large or in opposite expected orientation of expected fosmid clone
Discordant tied: non unique alignment for clone pair, insert size too small, too large or in opposite expected orientation of expected fosmid clone
Discordant trans: clone pair has ends mapping to different contigs
Clones aligning to regions within the chromosome 8 centromeric region were selected for FISH validation. The fosmid clones used for validation of the chromosome 8 centromeric region are: 174552_ABC10_2_1_000046302400_C7 for the p-arm monomeric α-satellite region (Cy5; blue), 174222_ABC10_2_1_000044375100_H13 for the p-arm portion of the D8Z2 HOR array (FluorX; green), 171417_ABC10_2_1_000045531400_M19 for the central portion of the D8Z2 HOR array (Cy3; red), 173650_ABC10_2_1_000044508400_J14 for the q-arm portion of the D8Z2 HOR array (FluorX; green), and 173650_ABC10_2_1_000044091500_K11 for the q-arm monomeric α-satellite region (Cy5; blue).
To determine the location of CENP-A relative to methylated DNA (specifically, 5-methylcytosines), we performed IF on stretched CHM13 chromatin fibers as previously described with modifications72,73. Briefly, CHM13 cells were swollen in a hypotonic buffer consisting of a 1:1:1 ratio of 75 mM KCl, 0.8% NaCitrate, and dH2O for 5 min. 3.5 × 104 cells were cytospun onto an ethanol-washed glass slide at 800 rpm for 4 min with high acceleration and allowed to adhere for 1 min before immersing in a salt-detergent-urea lysis buffer (25 mM Tris pH 7.5, 0.5 M NaCl, 1% Triton X-100, and 0.3 M urea) for 15 min at room temperature. The slide was slowly removed from the lysis buffer over a time period of 38 s and subsequently washed in PBS, incubated in 4% formaldehyde in PBS for 10 min, and washed with PBS and 0.1% Triton X-100. The slide was rinsed in PBS and 0.05% Tween-20 (PBST) for 3 min, blocked for 30 min with IF block (2% FBS, 2% BSA, 0.1% Tween-20, and 0.02% NaN2), and then incubated with a mouse monoclonal anti-CENP-A antibody (1:200, Enzo, ADI-KAM-CC006-E) and rabbit monoclonal anti-5-methylcytosine antibody (1:200, RevMAb, RM231) for 3 h at room temperature. Cells were washed 3× for 5 min each in PBST and then incubated with Alexa Fluor 488 goat anti-rabbit (1:200, Thermo Fisher Scientific, A-11034) and Alexa Fluor 594 conjugated to goat anti-mouse (1:200, Thermo Fisher Scientific, A-11005) for 1.5 h. Cells were washed 3× for 5 min each in PBST, fixed for 10 min in 4% formaldehyde, and washed 3× for 1 min each in dH2O before mounting in Vectashield containing 5 μg/ml DAPI. Slides were imaged on an inverted fluorescence microscope (Leica DMI6000) equipped with a charge-coupled device camera (Leica DFC365 FX) and a 40x 1.4 NA objective lens.
To assess the repeat organization of the 8q21 neocentromere, we performed FISH74 on CHM13 chromatin fibers. DNA fibers were obtained following Henry H. Q. Heng’s protocol with minor modifications75. Briefly, chromosomes were fixed with methanol:acetic acid (3:1), dropped onto previously clean slides, and soaked in 1X PBS. Manual elongation was performed by coverslip in NaOH:ethanol (5:2) solution. Slides were mounted in Vectashield containing 5 μg/ml DAPI and imaged on a fluorescence microscope (Leica DM RXA2) equipped with a charge-coupled device camera (CoolSNAP HQ2) and a 100x 1.6-0.6 NA objective lens. The probes used for validation of the 8q21.2 locus were picked from the same ABC10 fosmid library described above and include 174552_ABC10_2_1_000044787700_O7 for Probe 1 (Cy3; red) and 173650_ABC10_2_1_000044086000_F24 for Probe 2 (FluorX; green). Several CHM13 8q21.2 chromatin fibers were imaged. We quantified the number and intensity of the probe signals on a set of CHM13 chromatin fibers using ImageJ’s Gel Analysis tool (v1.51) and found that there were 63 +/− 7.55 green signals and 67 +/− 5.20 red signals (n = 3 independent experiments), consistent with the 67 full and 7 partial repeats in the CHM13 8q21.2 VNTR.
Native CENP-A ChIP-seq and analysis
We performed two independent replicates of native CENP-A ChIP-seq on CHM13 cells as described previously25,73 with some modifications. Briefly, 3-4 x 107 cells were collected and resuspended in 2 mL of ice-cold buffer I (0.32 M sucrose, 15 mM Tris, pH 7.5, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, and 2x Halt Protease Inhibitor Cocktail (Thermo Fisher 78429)). 2 mL of ice-cold buffer II (0.32 M sucrose, 15 mM Tris, pH 7.5, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 0.1% IGEPAL, and 2x Halt Protease Inhibitor Cocktail) was added, and samples were placed on ice for 10 min. The resulting 4 mL of nuclei were gently layered on top of 8 mL of ice-cold buffer III (1.2 M sucrose, 60 mM KCl, 15 mM, Tris pH 7.5, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, and 2x Halt Protease Inhibitor Cocktail (Thermo Fisher 78429)) and centrifuged at 10,000 × g for 20 min at 4°C. Pelleted nuclei were resuspended in buffer A (0.34 M sucrose, 15 mM HEPES, pH 7.4, 15 mM NaCl, 60 mM KCl, 4 mM MgCl2, and 2x Halt Protease Inhibitor Cocktail) to 400 ng/mL. Nuclei were frozen on dry ice and stored at 80°C. MNase digestion reactions were carried out on 200-300 μg chromatin, using 0.2-0.3 U/μg MNase (Thermo Fisher #88216) in buffer A supplemented with 3 mM CaCl2 for 10 min at 37°C. The reaction was quenched with 10 mM EGTA on ice and centrifuged at 500 × g for 7 min at 4°C. The chromatin was resuspended in 10 mM EDTA and rotated at 4°C for 2 h. The mixture was adjusted to 500 mM NaCl, rotated for another 45 min at 4°C and then centrifuged at max speed (21,100 × g) for 5 min at 4°C, yielding digested chromatin in the supernatant. Chromatin was diluted to 100 ng/ml with buffer B (20 mM Tris, pH 8.0, 5 mM EDTA, 500 mM NaCl and 0.2% Tween 20) and precleared with 100 μL 50% protein G Sepharose bead (GE Healthcare) slurry for 20 min at 4°C, rotating. Precleared supernatant (10–20 μg bulk nucleosomes) was saved for further processing. To the remaining supernatant, 20 μg mouse monoclonal anti-CENP-A antibody (Enzo ADI-KAM-CC006-E) was added and rotated overnight at 4°C. Immunocomplexes were recovered by the addition of 200 mL 50% protein G Sepharose bead slurry followed by rotation at 4°C for 3 h. The beads were washed 3x with buffer B and once with buffer B without Tween. For the input fraction, an equal volume of input recovery buffer (0.6 M NaCl, 20 mM EDTA, 20 mM Tris, pH 7.5, and 1% SDS) and 1 mL of RNase A (10 mg/mL) was added, followed by incubation for one hour at 37°C. Proteinase K (100 mg/ml, Roche) was then added, and samples were incubated for another 3 h at 37°C. For the ChIP fraction, 300 μL of ChIP recovery buffer (20 mM Tris, pH 7.5, 20 mM EDTA, 0.5% SDS and 500 mg/mL Proteinase K) was added directly to the beads and incubated for 3–4 h at 56°C. The resulting Proteinase K–treated samples were subjected to a phenol-chloroform extraction followed by purification with a QIAGEN MinElute PCR purification column. Unamplified bulk nucleosomal and ChIP DNA were analyzed using an Agilent Bioanalyzer instrument and a 2100 High Sensitivity Kit.
Sequencing libraries were generated using the TruSeq ChIP Library Preparation Kit - Set A (Illumina IP-202-1012) according to the manufacturer’s instructions, with some modifications. Briefly, 5–10 ng bulk nucleosomal or ChIP DNA was end-repaired and A-tailed. Illumina TruSeq adaptors were ligated, libraries were size-selected to exclude polynucleosomes using an E-Gel SizeSelect II agarose gel, and the libraries were PCR-amplified using the PCR polymerase and primer cocktail provided in the kit. The resulting libraries were submitted for 150 bp, paired-end Illumina sequencing using a NextSeq 500/550 High Output Kit v2.5 (300 cycles). The resulting reads were assessed for quality using FastQC (https://github.com/s-andrews/FastQC), trimmed with Sickle (https://github.com/najoshi/sickle; v1.33) to remove low-quality 5′ and 3′ end bases, and trimmed with Cutadapt76 (v1.18) to remove adapters.
Processed CENP-A ChIP and bulk nucleosomal reads were aligned to the CHM13 whole-genome assembly5 using two different approaches: 1) BWA-MEM77 (v0.7.17) and 2) a k-mer-based mapping approach we developed (described below).
For BWA-MEM mapping, data were aligned with the following parameters: bwa mem -k 50 -c 1000000 {index} {read1.fastq.gz}for single-end data, and bwa mem -k 50 -c 1000000 {index} {read1.fastq.gz} {read2.fastq.gz} for paired-end data. The resulting SAM files were filtered using SAMtools47 with FLAG score 2308 to prevent multi-mapping of reads. With this filter, reads mapping to more than one location are randomly assigned a single mapping location, thereby preventing mapping biases in highly identical regions. Alignments to the chromosome 8 centromere were downsampled to the same coverage and normalized with deepTools78 (v3.4.3) bamCompare with the following parameters: bamCompare -b1 {ChIP.bam} -b2 {Bulk_nucleosomal.bam} --operation ratio --binSize 1000 -o {out.bw}. The resulting bigWig file was visualized on the UCSC Genome Browser using the CHM13 chromosome 8 assembly as an assembly hub.
For the k-mer-based mapping, the initial BWA-MEM alignment was used to identify reads specific to the chromosome 8 centromeric region (chr8:43600000-47200000). K-mers (k = 50) were identified from each chromosome 8 centromere-specific dataset using Jellyfish (v2.3.0) and mapped back onto reads and chromosome 8 centromere assembly allowing for no mismatches. Approximately 93-98% of all k-mers identified in the reads were also found within the D8Z2 HOR array. Each k-mer from the read data was then placed once at random between all sites in the HOR array that had a perfect match to that k-mer. These data were then visualized using a histogram with 1 kbp bins in R (R core team, 2020).
Mappability of short reads within the chromosome 8 centromeric region
To determine the mappability of short reads within the chromosome 8 centromeric HOR array, we performed a simulation where we generated 300,000 random 150 bp fragments from five equally sized (416 kbp) regions across the CHM13 D8Z2 HOR array. We mapped these fragments back to the CHM13 chromosome 8 centromeric region using BWA-MEM (v0.7.17) or the k-mer based approach, as described above. For BWA-MEM mapping, the 150 bp fragments were aligned with the following parameters: bwa mem -k 50 -c 1000000 {index} {fragments.fasta}. The resulting SAM files were filtered using SAMtools47 with FLAG score 2308 to prevent multi-mapping of reads and then converted to a BAM file. BAM files were visualized in IGV55. For the k-mer-based mapping, k-mers (k = 50) were identified from each set of 150 bp fragments using Jellyfish (v2.3.0) and mapped back onto the fragments and the chromosome 8 centromere assembly allowing for no mismatches. K-mers with perfect matches to multiple sites within the centromeric region were assigned to one of the sites at random. These data were visualized using a histogram with 1 kbp bins in R (R core team, 2020).
Phylogenetic analysis
To assess the phylogenetic relationship between α-satellite repeats, we first masked every non-α-satellite repeat in the human and NHP centromere assemblies using RepeatMasker79 (v4.1.0). Then, we subjected the masked assemblies to StringDecomposer42 (version available February 28th, 2020) using a set of 11 α-satellite monomers derived from a chromosome 8 11-mer HOR unit (described in the “Analysis of α-satellite organization subsection” above). This tool identifies the location of α-satellite monomers in the assemblies, and we used this to extract the α-satellite monomers from the HOR/dimeric array and monomeric regions into multi-FASTA files. We ultimately extracted 12,989, 8,132, 12,224, 25,334, and 63,527 α-satellite monomers from the HOR/dimeric array in human, chimpanzee (H1), chimpanzee (H2), orangutan, and macaque, respectively, and 2,879, 3,781, 3,351, 1,573, and 8,127 monomers from the monomeric regions in human, chimpanzee (H1), chimpanzee (H2), orangutan and macaque, respectively. We randomly selected 100 and 50 α-satellite monomers from the HOR/dimeric array and monomeric regions and aligned them with MAFFT80,81 (v7.453). We used IQ-TREE82 to reconstruct the maximum-likelihood phylogeny with model selection and 1000 bootstraps. The resulting tree file was visualized in iTOL83.
To estimate sequence divergence along the pericentromeric regions, we first mapped each NHP centromere assembly to the CHM13 centromere assembly using minimap244 (v2.17-r941) with the following parameters: -ax asm20 --eqx -Y -t 8 -r 500000. Then, we generated a BED file of 10 kbp windows located within the CHM13 centromere assembly. We used the BED file to subset the BAM file, which was subsequently converted into a set of FASTA files. FASTA files contained at least 5 kbp of orthologous sequences from one or more NHP centromere assemblies. Pairs of human and NHP orthologous sequences were realigned using MAFFT (v7.453) and the following command: mafft --maxiterate 1000 --localpair. Sequence divergence was estimated using the Tamura-Nei substitution model84, which accounts for recurrent mutations and differences between transversions and transitions as well as within transitions. Mutation rate per segment was estimated using Kimura’s model of neutral evolution85. In brief, we modeled the estimated divergence (D) is a result of between-species substitutions and within-species polymorphisms; i.e.,
where Ne is the ancestral human effective population size, t is the divergence time for a given human–NHP pair, and μ is the mutation rate. We assumed a generation time of [20, 29] years and the following divergence times: human–macaque = [23e6, 25e6] years, human–orangutan = [12e6, 14e6] years, human–chimpanzee = [4e6, 6e6] years. To convert the genetic unit to a physical unit, our computation also assumes Ne=10,000 and uniformly drawn values for the generation and divergence times.
DATA AVAILABILITY
The complete CHM13 chromosome 8 sequence and all data generated and/or used in this study are publicly available and listed in Supplementary Table 9 with their BioProject, accession #, and/or URL. For convenience, we also list their BioProjects and/or URLs here: complete CHM13 chromosome 8 sequence (PRJNA559484); CHM13 ONT, Iso-Seq, and CENP-A ChIP-seq data (PRJNA559484); CHM13 Strand-Seq alignments (https://zenodo.org/record/3998125); HG00733 ONT data (PRJNA686388); HG00733 PacBio HiFi data (PRJEB36100); testis and fetal brain Iso-Seq data (PRJNA659539); and NHPs [chimpanzee (Clint; S006007), orangutan (Susie; PR01109), and macaque (AG07107) ONT and PacBio HiFi data (PRJNA659034)]. All CHM13 BACs used in this study are listed in Supplementary Table 10 with their accession #s.
CODE AVAILABILITY
Custom code for the SUNK-based assembly method is available at https://github.com/glogsdon1/sunk-based_assembly. All other code is publicly available.
Extended Data
Extended Data Figure 1. Sequence, structure, and epigenetic map of the neocentromeric chromosome 8q21.2 VNTR.
a) Schematic showing the composition of the CHM13 8q21.2 VNTR. This VNTR is comprised of 67 full and 7 partial 12.192 kbp repeats that span 863 kbp in total. The predicted restriction digest pattern is indicated. Each repeat is methylated within a 3 kbp region and hypomethylated within the rest of the sequence. Mapping of CENP-A ChIP-seq data from the chromosome 8 neodicentric cell line known as MS422124,25 (Methods) reveals that approximately 98% of CENP-A chromatin is located within the hypomethylated portion of the repeat. A pairwise sequence identity map across the region indicates a mirrored symmetry within a single layer, consistent with the evolutionarily young status of the tandem repeat. b) PFG Southern blot of CHM13 DNA digested with BmgBI confirms the size and organization of the chromosome 8q21.2 VNTR. Left: EtBr staining; Right: P32-labeled chromosome 8q21.2-specific probe. For gel source data, see Supplementary Fig. 1c,d. c) Copy number of the 8q21 repeat (chr8:85792897–85805090 in GRCh38) throughout the human population. CHM13 is estimated to have 144 total copies of the 8q21 repeat, or 72 copies per haplotype, while GRCh38 only has 26 copies (red data points). Median +/− s.d. is shown.
Extended Data Figure 2. CHM13 chromosome 8 telomeres.
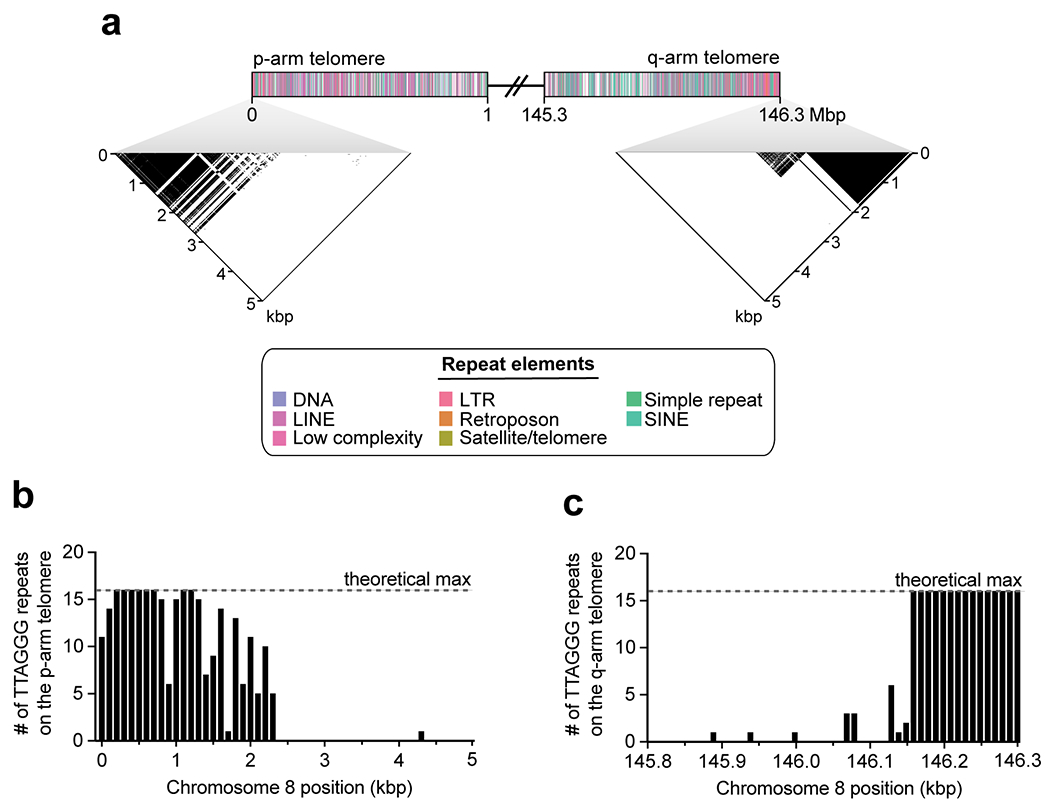
a) Schematic showing the first and last megabase of the CHM13 chromosome 8 assembly. A dot plot of the terminal 5 kbp shows high sequence identity among the last ~2.5 kbp of the chromosome, consistent with the presence of a high-identity telomeric repeating unit. b,c) Number of TTAGGG telomeric repeats in the last 5 kbp of the p- (Panel a) and q- (Panel b) arms in chromosome 8. The p-arm has a gradual transition to pure TTAGGG repeats over nearly 1 kbp, while the q-arm has a very sharp transition to pure TTAGGG repeats that occurs over nearly 300 bp.
Extended Data Figure 3. Genes with improved alignment to the CHM13 chromosome 8 assembly relative to GRCh38.
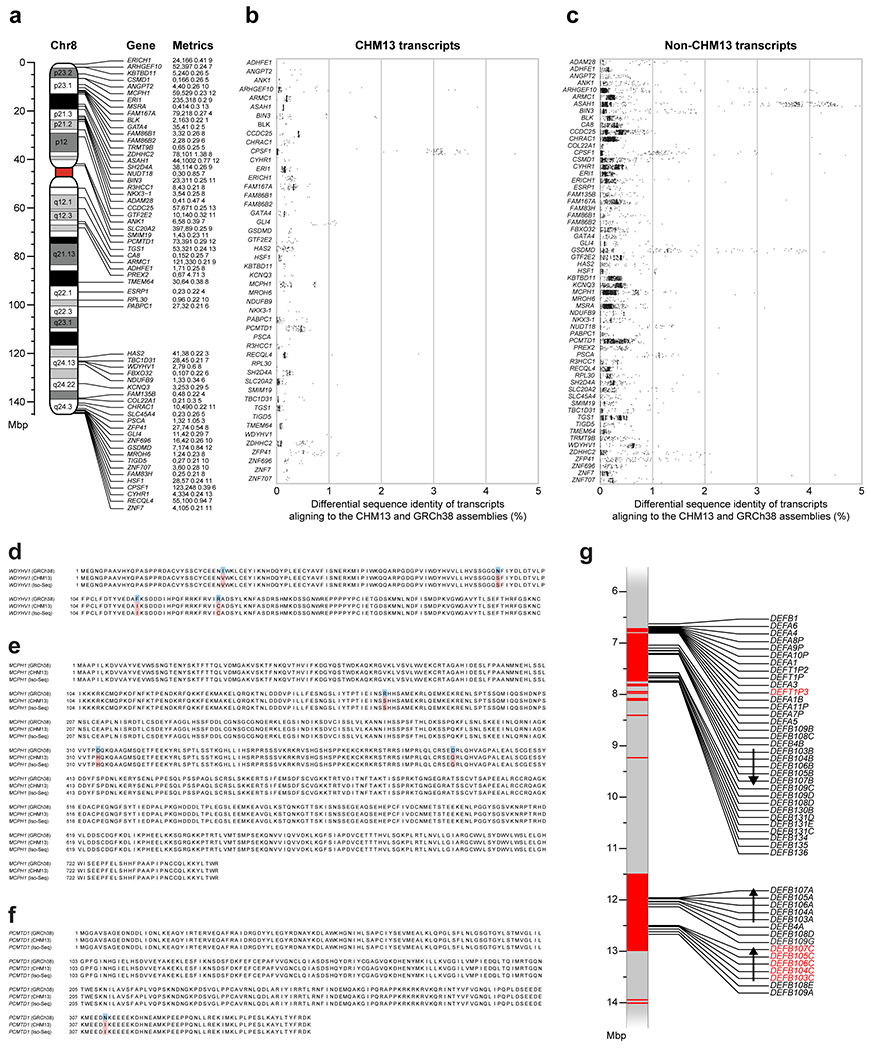
a) Ideogram of chromosome 8 showing protein-coding genes with improved transcript alignments to the CHM13 chromosome 8 assembly relative to GRCh38 (hg38). Each gene is labeled with its name, count of improved transcripts from CHM13 cell line, other tissues, the average percent improvement of non-CHM13 cell line alignments, and the number of tissue sources with improved transcript mappings. b,c) Differential percent sequence identity of transcripts aligning to CHM13 or GRCh38 for b) CHM13 cell line transcripts and c) non-CHM13 cell line transcripts. d-f) Multiple-sequence alignments for a) WDYHV1, b) MCPH1, and c) PCMTD1, all of which have at least 0.1% greater sequence identity of >20 full-length Iso-Seq transcripts to the CHM13 chromosome 8 assembly than to GRCh38 (Methods). For each gene, the GRCh38 annotation is compared to the same annotation lifted over to the CHM13 chromosome 8 assembly, and the substitutions are confirmed by translated predicted ORFs from Iso-Seq transcripts. Matching amino acids are shaded in gray, those matching only the Iso-Seq data are in red, and those different from the Iso-Seq data are in blue. Each substitution in CHM13 relative to GRCh38 has an allele frequency of 0.36 in gnomAD (v3). g) Location of DEFA and DEFB genes in the CHM13 chromosome 8 β-defensin locus. SD regions were identified by SEDEF86, and new paralogs are shown in red. Duplication cassettes are marked with arrows indicating orientation for each copy.
Extended Data Figure 4. Comparison of the CHM13 and GRCh38 β-defensin loci.
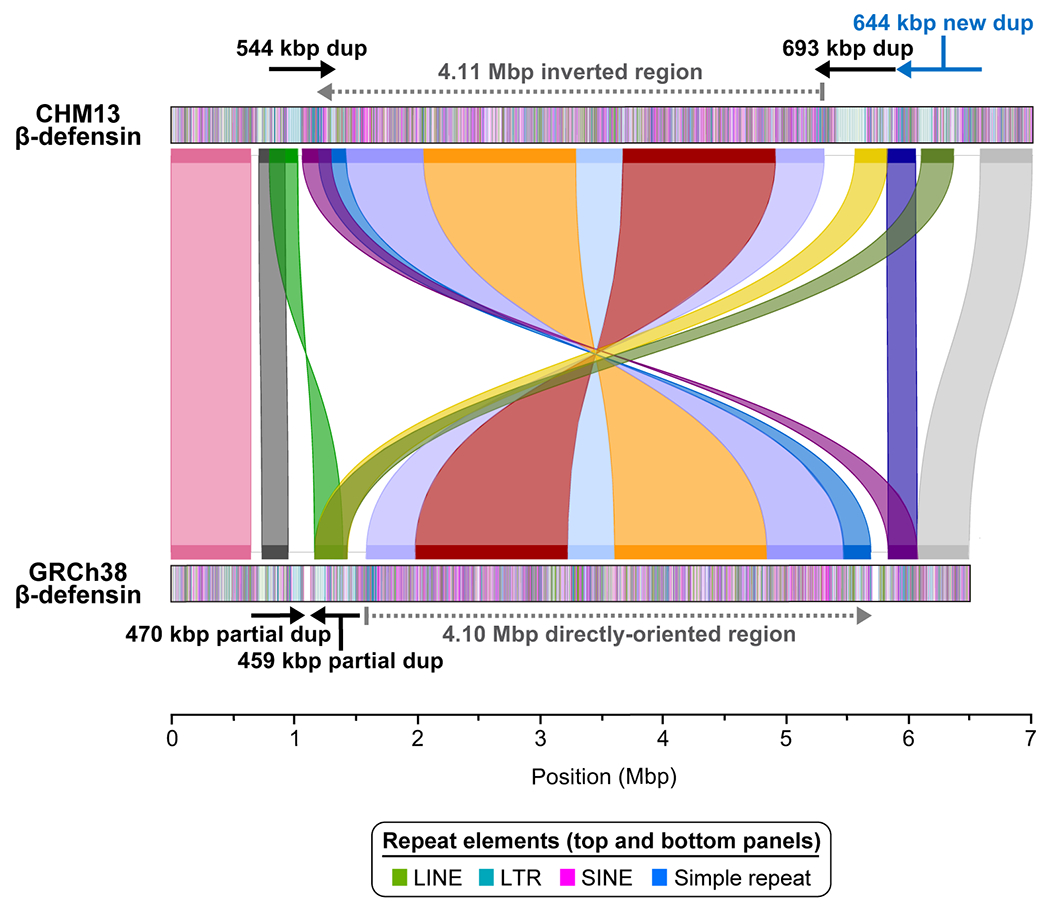
Miropeats comparison of the CHM13 and GRCh38 β-defensin loci identifies a 4.11 Mbp inverted region (dashed gray line) bracketed by proximal and distal segmental duplication (SD; dup) blocks (black and blue arrows) in CHM13. CHM13 also has an additional SD block (blue arrow) relative to the GRCh38. In total, the CHM13 haplotype adds 611.9 kbp of new sequence, of which 602.6 kbp is located within SD blocks and 9.3 kbp is located at the distal edge of the inverted region. Colored segments track blocks of homology between CHM13 and GRCh38.
Extended Data Figure 5. Validation of the CHM13 β-defensin locus, and copy number of the DEFA gene family.
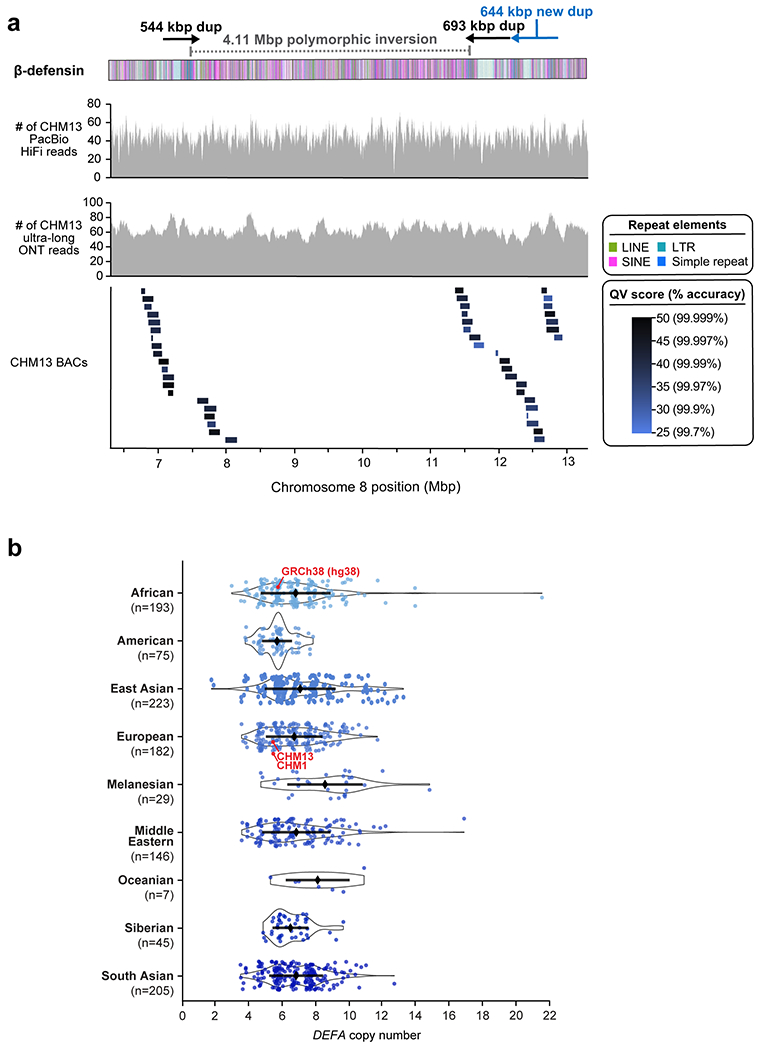
a) Coverage of CHM13 ONT and PacBio HiFi data along the CHM13 β-defensin locus (top two panels). The ONT and PacBio data have largely uniform coverage, indicating it is free of large structural errors. The dip in HiFi coverage near position 10.46 Mbp is due to a G/A bias in HiFi chemistry5. The alignment of 47 CHM13 BACs (bottom panel) reveals that those regions have an estimated QV score >25 (>99.7% accurate). b) Copy number of DEFA [chr8:6976264–6995380 in GRCh38 (hg38)] throughout the human population. Median +/− s.d. is shown.
Extended Data Figure 6. Validation of the CHM13 chromosome 8 centromeric region.
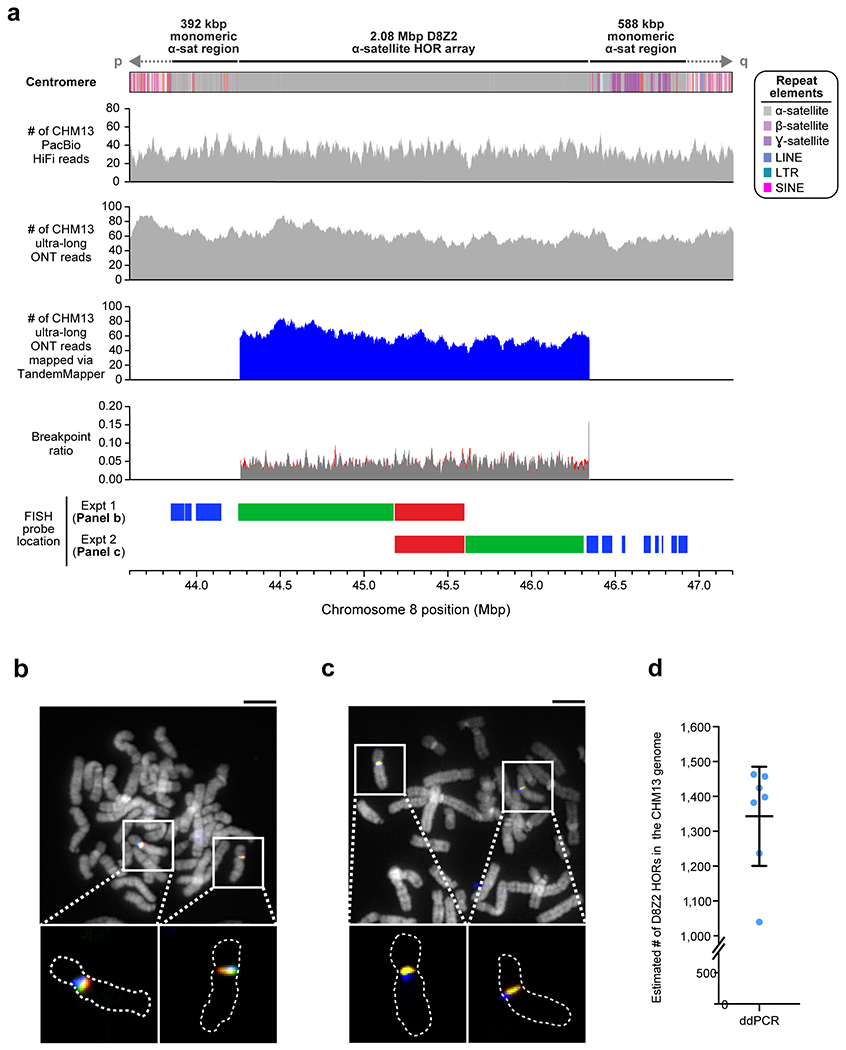
a) Coverage of CHM13 ONT and PacBio HiFi data along the CHM13 chromosome 8 centromeric region (top two panels) is largely uniform, indicating a lack of large structural errors. Analysis with TandemMapper and TandemQUAST53, which are tools that assess repeat structure via mapped reads (third panel) and misassembly breakpoints (fourth panel; red), indicates that the chromosome 8 D8Z2 α-satellite HOR array lacks large-scale assembly errors. Five different FISH probes targeting regions in the chromosome 8 centromeric region (bottom panel) are used to confirm the organization of the α-satellite DNA (Panels b,c). b,c) Representative images of metaphase chromosome spreads hybridized with FISH probes targeting regions within the chromosome 8 centromere (Panel a). Insets show both chromosome 8s with the predicted organization of the centromeric region. d) Droplet digital PCR (ddPCR) of the chromosome 8 D8Z2 α-satellite array indicates that there are 1344 +/− 142 D8Z2 HORs present on chromosome 8, consistent with the predictions from an in silico restriction digest and StringDecomposer42 analysis (Methods). Mean +/− s.d. is shown. Bar = 5 microns. Insets = 2.5× magnification.
Extended Data Figure 7. Sequence, structure, and epigenetic map of human diploid HG00733 chromosome 8 centromeres.
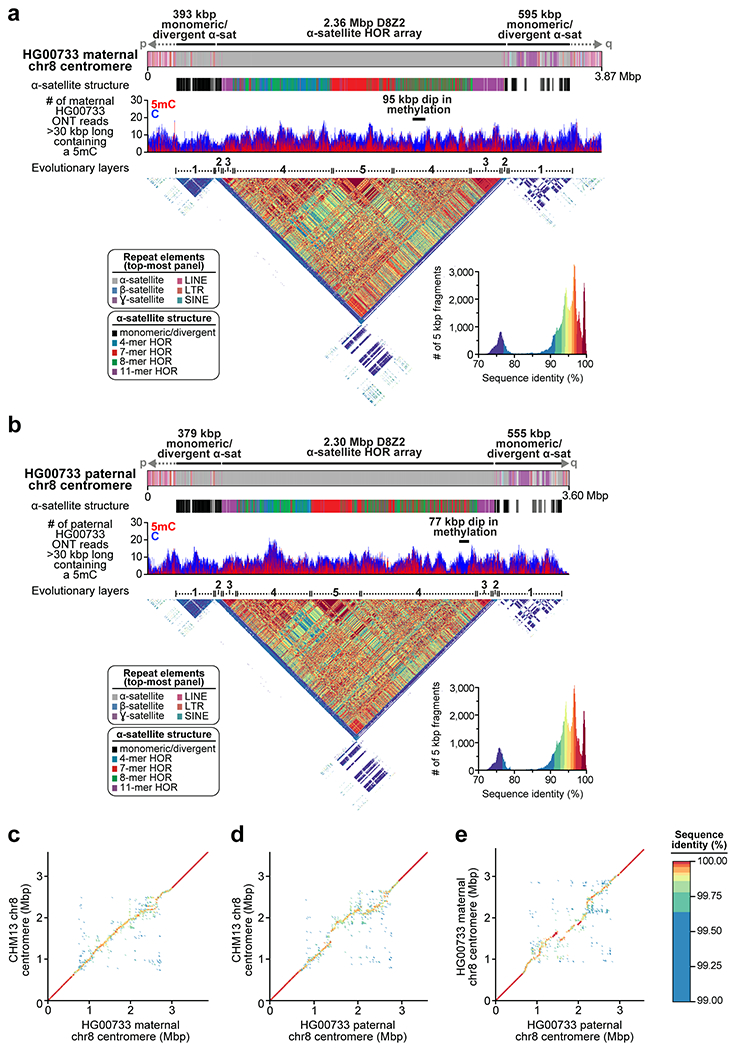
a,b) Repeat structure, alpha-satellite organization, methylation status, and sequence identity heatmap of the a) maternal and b) paternal chromosome 8 centromeric regions from a diploid human genome (HG00733; Supplementary Table 2) shows structural and epigenetic similarity to the CHM13 chromosome 8 centromeric region (Fig. 2a). c-e) Dot plot comparisons between the c) CHM13 and maternal, d) CHM13 and paternal, and e) maternal and paternal chromosome 8 centromeric regions in the HG00733 genome show >99% sequence identity overall, with high concordance in the unique and monomeric α-satellite regions of the centromeres (dark red line) that devolves into lower sequence identity in the α-satellite HOR array, consistent with rapid evolution of this region.
Extended Data Figure 8. Composition, organization, and entropy of the CHM13 D8Z2 α-satellite HOR array.
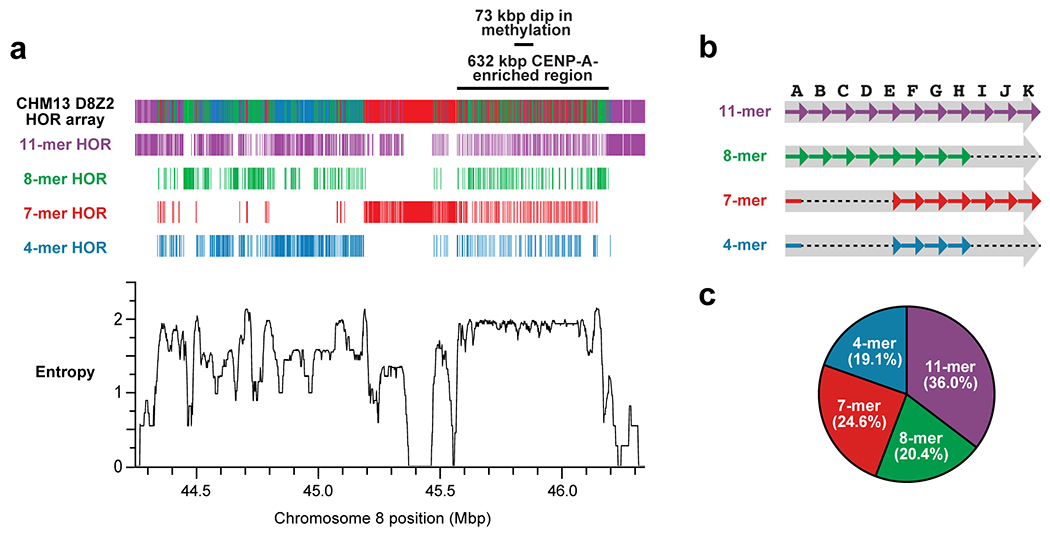
a) HOR composition and organization of the chromosome 8 α-satellite array as determined via StringDecomposer42. The predominant HOR subtypes (4-, 7-, 8-, and 11-mers) are shown, while those occurring less than 15 times are not (see Methods for absolute quantification). The entropy of the D8Z2 HOR array is plotted in the bottom panel and reveals that the hypomethylated and CENP-A-enriched regions have the highest consistent entropy in the entire array. b) Organization of α-satellite monomers within each HOR. The initial monomer of the 4- and 7-mer HORs is a hybrid of the A and E monomers, with the first 87 bp the A monomer and the subsequent 84 bp the E monomer. c) Abundance of the predominant HOR types within the D8Z2 HOR array as determined via StringDecomposer42.
Extended Data Figure 9. Location of CENP-A chromatin within the CHM13 D8Z2 α-satellite HOR array.
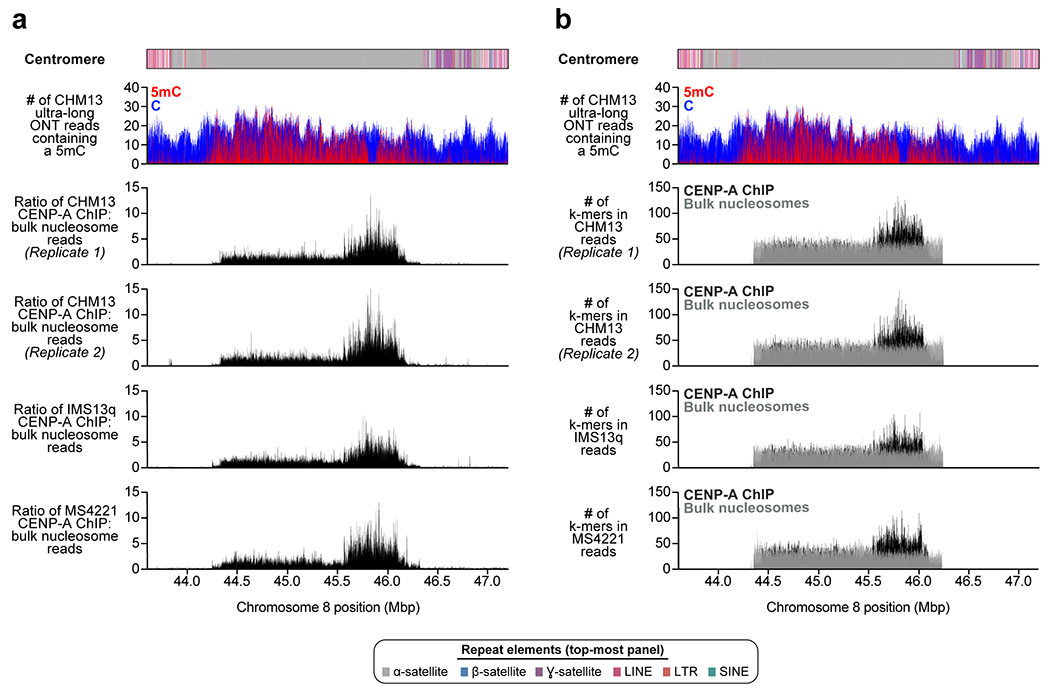
a,b) Plot of a) the ratio CENP-A ChIP to bulk nucleosome reads mapped via BWA-MEM, or b) the number of k-mer-mapped CENP-A ChIP (black) or bulk nucleosome (dark gray) reads (Methods). Shown are two independent replicates of CENP-A ChIP-seq performed on CHM13 cells (top two panels), as well as single replicates of CENP-A ChIP-seq performed on human diploid neocentromeric cell lines (bottom two panels; Methods). While the neocentromeric cell lines have a neocentromere located on either chromosome 13 (IMS13q) or 8 (MS4221)24,25, they both have at least one karyotypically normal chromosome 8 from which centromeric chromatin can be mapped. We limited our analysis to diploid cell lines rather than aneuploid ones to avoid potentially confounding results stemming from multiple chromosome 8 copies that vary in structure, such as those observed in HeLa cells87.
Extended Data Figure 10. Validation of the CHM13 8q21.2 VNTR.
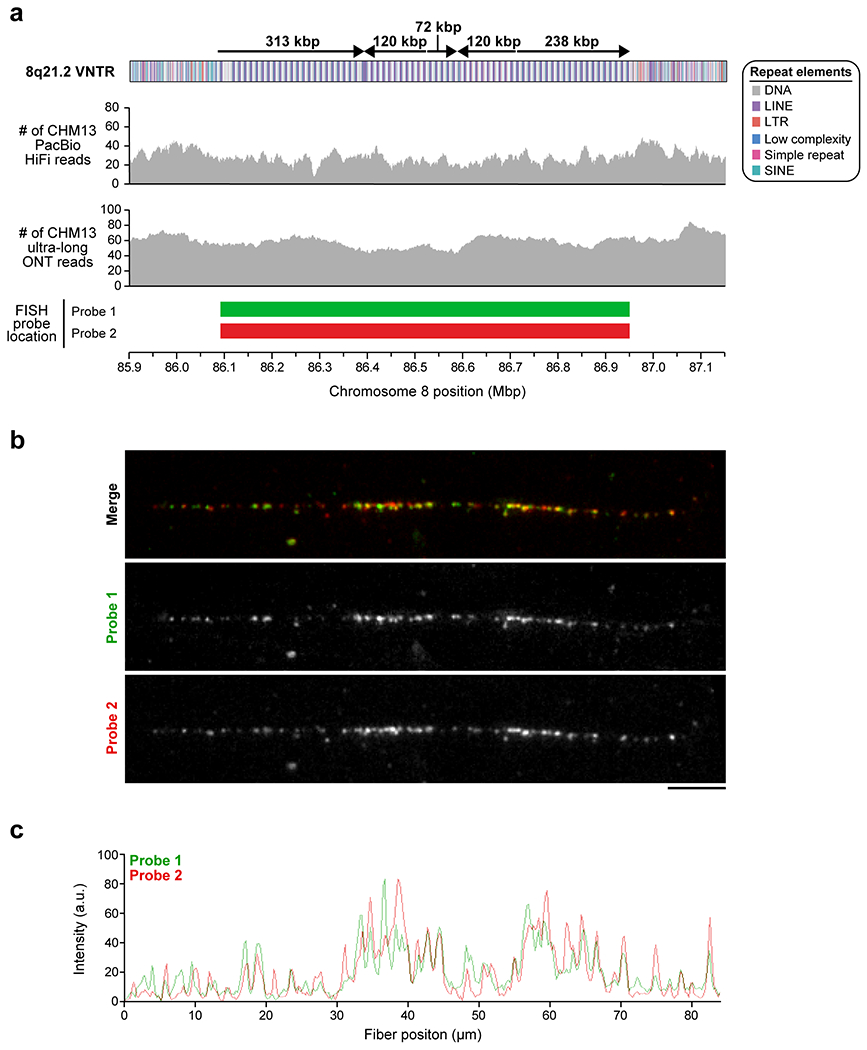
a) Coverage of CHM13 ONT and PacBio HiFi data along the 8q21.2 VNTR (top two panels) is largely uniform, indicating a lack of large structural errors. Two FISH probes targeting the 12.192 kbp repeat in the 8q21.2 VNTR are used to estimate the number of repeats in the CHM13 genome (Panels b,c). b) Representative FISH images of a CHM13 stretched chromatin fiber. Although the FISH probes were designed against the entire VNTR array, stringent washing during FISH produces a punctate probe signal pattern, which may be due to stronger hybridization of the probe to a specific region in the 12.192 kbp repeat (perhaps based on GC content or a lack of secondary structures). This punctate pattern can be used to estimate the repeat copy number in the VNTR, thereby serving as a source of validation. c) Plot of the signal intensity on the CHM13 chromatin fiber shown in Panel b. Quantification of peaks across three independent experiments reveals an average of 63 +/− 7.55 peaks and 67 +/− 5.20 peaks from the green and red probes, respectively, which is consistent with the number of repeat units in the 8q21.2 assembly (67 full and 7 partial repeats). Bar = 5 micron.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Goodwin (CSHL) for sequence data generation; M. Jain (UCSC) and D. Miller (UW) for re-base-calling sequence data; R. Tindell, H. Visse, A. Tornabene, and G. Ellis (UW) for technical assistance; Z. Zhao for computational assistance; F.F. Dastvan (UW) for instrumentation; D. Gordon (UW) for accessioning BACs; G. Bouffard (NHGRI) for accessioning ONT FAST5 data; J.G. Underwood (FHCRC/PacBio) for helpful discussions; and T. Brown (UW) for assistance in editing this manuscript. We acknowledge experimental support from the W. M. Keck Microscopy Center (UW) and the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). This research was supported, in part, by funding from the National Institutes of Health (NIH), HG002385 and HG010169 (EEE); National Institute of General Medical Sciences (NIGMS), F32 GM134558 (GAL); Intramural Research Program of the National Human Genome Research Institute at NIH (SK, AMP, AR); National Library of Medicine Big Data Training Grant for Genomics and Neuroscience 5T32LM012419-04 (MRV); NIH/NHGRI Pathway to Independence Award K99 HG011041 (PH); NIH/NHGRI R21 1R21HG010548-01 and NIH/NHGRI U01 1U01HG010971 (KHM); and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA (VL). EEE is an investigator of the Howard Hughes Medical Institute.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
ADDITIONAL INFORMATION
Supplementary information is available for this paper.
REFERENCES
- 1.International Human Genome Project Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Venter JC et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Alkan C et al. Genome-wide characterization of centromeric satellites from multiple mammalian genomes. Genome Res. 21, 137–145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Nurk S et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res gr.263566.120 (2020) doi: 10.1101/gr.263566.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng H, Concepcion GT, Feng X, Zhang H & Li H Haplotype-resolved de novo assembly with phased assembly graphs. arXiv:2008.01237 [q-bio] (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logsdon GA, Vollger MR & Eichler EE Long-read human genome sequencing and its applications. Nature Reviews Genetics 1–18 (2020) doi: 10.1038/s41576-020-0236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNulty SM & Sullivan BA Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosome Res. 26, 115–138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge Y, Wagner MJ, Siciliano M & Wells DE Sequence, higher order repeat structure, and long-range organization of alpha satellite DNA specific to human chromosome 8. Genomics 13, 585–593 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Hollox EJ, Armour J. a. L. & Barber JCK Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am. J. Hum. Genet 73, 591–600 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollox EJ et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet 40, 23–25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohajeri K et al. Interchromosomal core duplicons drive both evolutionary instability and disease susceptibility of the Chromosome 8p23.1 region. Genome Res 26, 1453–1467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miga KH et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sudmant PH et al. Diversity of human copy number variation and multicopy genes. Science 330, 641–646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falconer E & Lansdorp PM Strand-seq: a unifying tool for studies of chromosome segregation. Semin. Cell Dev. Biol 24, 643–652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders AD, Falconer E, Hills M, Spierings DCJ & Lansdorp PM Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat Protoc 12, 1151–1176 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Rhie A, Walenz BP, Koren S & Phillippy AM Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson JT et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Devriendt K et al. Delineation of the critical deletion region for congenital heart defects, on chromosome 8p23.1. Am J Hum Genet 64, 1119–1126 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giglio S et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am. J. Hum. Genet 71, 276–285 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cantsilieris S & White SJ Correlating multiallelic copy number polymorphisms with disease susceptibility. Hum. Mutat 34, 1–13 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Tyson C et al. Expansion of a 12-kb VNTR containing the REXO1L1 gene cluster underlies the microscopically visible euchromatic variant of 8q21.2. European Journal of Human Genetics 22, 458–463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warburton PE et al. Analysis of the largest tandemly repeated DNA families in the human genome. BMC Genomics 9, 533 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasson D et al. Formation of novel CENP-A domains on tandem repetitive DNA and across chromosome breakpoints on human chromosome 8q21 neocentromeres. Chromosoma 120, 621–632 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Hasson D et al. The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat. Struct. Mol. Biol 20, 687–695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alkan C et al. Organization and evolution of primate centromeric DNA from whole-genome shotgun sequence data. PLoS Comput. Biol 3, 1807–1818 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cacheux L, Ponger L, Gerbault-Seureau M, Richard FA & Escudé C Diversity and distribution of alpha satellite DNA in the genome of an Old World monkey: Cercopithecus solatus. BMC Genomics 17, 916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain M et al. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol 36, 321–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warburton PE et al. Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Current Biology 7, 901–904 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Vafa O & Sullivan KF Chromatin containing CENP-A and α-satellite DNA is a major component of the inner kinetochore plate. Current Biology 7, 897–900 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Smith GP Evolution of repeated DNA sequences by unequal crossover. Science 191, 528–535 (1976). [DOI] [PubMed] [Google Scholar]
- 32.Shepelev VA, Alexandrov AA, Yurov YB & Alexandrov IA The evolutionary origin of man can be traced in the layers of defunct ancestral alpha satellites flanking the active centromeres of human chromosomes. PLOS Genetics 5, e1000641 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexandrov I, Kazakov A, Tumeneva I, Shepelev V & Yurov Y Alpha-satellite DNA of primates: old and new families. Chromosoma 110, 253–266 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Koga A et al. Evolutionary origin of higher-order repeat structure in alpha-satellite DNA of primate centromeres. DNA Res. 21, 407–415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandrov IA, Mitkevich SP & Yurov YB The phylogeny of human chromosome specific alpha satellites. Chromosoma 96, 443–453 (1988). [DOI] [PubMed] [Google Scholar]
- 36.Garg S et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nature Biotechnology 1–4 (2020) doi: 10.1038/s41587-020-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porubsky D et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nature Biotechnology 1–7 (2020) doi: 10.1038/s41587-020-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebert P et al. De novo assembly of 64 haplotype-resolved human genomes of diverse ancestry and integrated analysis of structural variation. bioRxiv 2020.12.16.423102 (2020) doi: 10.1101/2020.12.16.423102. [DOI] [Google Scholar]
ADDITIONAL REFERENCES
- 39.Vollger MR et al. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet (2019) doi: 10.1111/ahg.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huddleston J et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res 24, 688–696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Logsdon GA HMW gDNA purification and ONT ultra-long-read data generation. protocols.io (2020) doi: 10.17504/protocols.io.bchhit36 [DOI] [Google Scholar]
- 42.Dvorkina T, Bzikadze AV & Pevzner PA The string decomposition problem and its applications to centromere analysis and assembly. Bioinformatics 36, i93–i101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain C et al. Weighted minimizer sampling improves long read mapping. Bioinformatics 36, i111–i118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H & Durbin R Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ & Prins P Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porubsky D et al. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics 36, 1260–1261 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Porubsky D et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nature Biotechnology 1–7 (2020) doi: 10.1038/s41587-020-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10, 1784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanders AD et al. Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res. (2016) doi: 10.1101/gr.201160.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghareghani M et al. Strand-seq enables reliable separation of long reads by chromosome via expectation maximization. Bioinformatics 34, i115–i123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mikheenko A, Bzikadze AV, Gurevich A, Miga KH & Pevzner PA TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36, i75–i83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee I et al. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nature Methods 17, 1191–1199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robinson JT et al. Integrative genomics viewer. Nature Biotechnology 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dougherty ML et al. Transcriptional fates of human-specific segmental duplications in brain. Genome Res. 28, 1566–1576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pertea M et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biology 19, 208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shumate A & Salzberg SL Liftoff: an accurate gene annotation mapping tool. bioRxiv 2020.06.24.169680 (2020) doi: 10.1101/2020.06.24.169680. [DOI] [Google Scholar]
- 61.R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing, 2020). [Google Scholar]
- 62.Parsons JD Miropeats: graphical DNA sequence comparisons. Bioinformatics 11, 615–619 (1995). [DOI] [PubMed] [Google Scholar]
- 63.Bergström A et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mafessoni F et al. A high-coverage Neandertal genome from Chagyrskaya Cave. PNAS 117, 15132–15136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mallick S et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meyer M et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prado-Martinez J et al. Great ape genetic diversity and population history. Nature 499, 471–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prüfer K et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hach F et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods 7, 576–577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gymrek M, Golan D, Rosset S & Erlich Y lobSTR: A short tandem repeat profiler for personal genomes. Genome Res 22, 1154–1162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haaf T & Willard HF Chromosome-specific alpha-satellite DNA from the centromere of chimpanzee chromosome 4. Chromosoma 106, 226–232 (1997). [DOI] [PubMed] [Google Scholar]
- 72.Iwata-Otsubo A et al. Expanded satellite repeats amplify a discrete CENP-A nucleosome assembly site on chromosomes that drive in female meiosis. Current Biology 27, 2365–2373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Logsdon GA et al. Human artificial chromosomes that bypass centromeric DNA. Cell 178, 624–639.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ventura M et al. Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome Res gr.124461.111 (2011) doi: 10.1101/gr.124461.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Darby IA In Situ Hybridization Protocols. (Humana Press, 2000). [Google Scholar]
- 76.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 77.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio] (2013). [Google Scholar]
- 78.Ramírez F, Dündar F, Diehl S, Grüning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smit AFA, Hubley R & Green P RepeatMasker Open-4.0. (2013). [Google Scholar]
- 80.Katoh K & Standley DM MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakamura T, Yamada KD, Tomii K & Katoh K Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen L-T, Schmidt HA, von Haeseler A & Minh BQ IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Letunic I & Bork P Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Tamura K & Nei M Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol 10, 512–526 (1993). [DOI] [PubMed] [Google Scholar]
- 85.Kimura M The neutral theory of molecular evolution. (Cambridge University Press, 1983). doi: 10.1017/CBO9780511623486. [DOI] [Google Scholar]
- 86.Numanagic I et al. Fast characterization of segmental duplications in genome assemblies. Bioinformatics 34, i706–i714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Landry JJM et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda) 3, 1213–1224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The complete CHM13 chromosome 8 sequence and all data generated and/or used in this study are publicly available and listed in Supplementary Table 9 with their BioProject, accession #, and/or URL. For convenience, we also list their BioProjects and/or URLs here: complete CHM13 chromosome 8 sequence (PRJNA559484); CHM13 ONT, Iso-Seq, and CENP-A ChIP-seq data (PRJNA559484); CHM13 Strand-Seq alignments (https://zenodo.org/record/3998125); HG00733 ONT data (PRJNA686388); HG00733 PacBio HiFi data (PRJEB36100); testis and fetal brain Iso-Seq data (PRJNA659539); and NHPs [chimpanzee (Clint; S006007), orangutan (Susie; PR01109), and macaque (AG07107) ONT and PacBio HiFi data (PRJNA659034)]. All CHM13 BACs used in this study are listed in Supplementary Table 10 with their accession #s.