

Abstract
BCMA-CD3-targeting bispecific antibodies (BsAb) are a recently developed immunotherapy class which shows potent tumor killing activity in multiple myeloma (MM). Here, we investigated a murine BCMA-CD3-targeting BsAb in the immunocompetent Vk*MYC and its IMiD-sensitive derivative Vk*MYChCRBN models of MM. The BCMA-CD3 BsAb was safe and efficacious in a subset of mice, but failed in those with high-tumor burden, consistent with clinical reports of BsAb in leukemia. The combination of BCMA-CD3 BsAb with pomalidomide expanded lytic T cells and improved activity even in IMiD resistant high-tumor burden cases. Yet, survival was only marginally extended due to acute toxicity and T cell exhaustion, which impaired T cell persistence. In contrast, the combination with cyclophosphamide was safe and allowed for a tempered pro-inflammatory response associated with long-lasting complete remission. Concurrent cytotoxic therapy with BsAb actually improved T cell persistence and function, offering a promising approach to patients with a large tumor burden.
Introduction
While chemotherapeutic agents and molecular medicine are pillars of successful treatment of cancer, the recent clinical development of immunotherapies shows compelling promise in the treatment of many tumor types. In hematologic malignancies, immunotherapies centered upon cytolytic T lymphocytes as drugs, such as CAR-T cells and bispecific T cell engagers (BiTEs) or antibodies (BsAb), are central among these advances. BiTEs and BsAbs are “off-the-shelf” drug therapies that circumvent the need for time consuming and expensive ex vivo manipulation of patients’ cells. These agents often consist of monoclonal antibodies, or single chain variable fragments in the case of BiTEs, engineered with one binding site directed toward a tumor-specific antigen and another against the T lymphocyte activating receptor, CD3-epsilon. BsAbs re-direct T cells to kill tumors by bringing them into physical contact and activating secretion of cytotoxic molecules (1). Due to their novel mode of action, BsAb therapy may provide an effective option for all patients, including those with cytogenetically high risk or heavily pre-treated disease that renders them more resistant to standard of care therapy.
Multiple myeloma (MM) is a largely incurable malignancy caused by monoclonal expansion of plasma cells (PC)s in the bone marrow (BM). Although most MM patients respond well to initial chemotherapy combinations, the disease almost always relapses and eventually becomes resistant to therapy. BsAbs have shown particular efficacy in B cell malignancies, with some of the most promising clinical trials in development for MM with agents targeting the antigen BCMA, encoded by the TNFRSF17 gene (2). There are multiple BCMA/CD3 BsAbs or BiTEs in Phase I clinical trials in heavily pre-treated patients with MM. Preliminary results from these trials show promising efficacy with the majority of patients responding at optimized doses (3–8). Although still under investigation, durability of this therapy in MM is still unclear. Additionally, the mechanisms underpinning the spectrum of responses remain unknown, as do methods to deepen response and prolong remission. Moreover, frequent immune-related adverse events, similar to those observed with CAR-T therapy, are noted, with cytokine release syndrome (CRS) putting patients with greater disease burden at higher risk.
MM creates an immunosuppressive BM TME through a variety of mechanisms (9–12) and its complexity may modulate the efficacy of BsAb MM therapy as it does for AMG-420 (13) and blinatumomab in leukemia therapy (14,15). Many MM standard of care drugs act directly by killing the tumor and remodeling the TME to break cycles of tumor survival. For example, the DNA alkylating agent cyclophosphamide (Cy), in addition to inducing DNA damage, preferentially kills regulatory T cells (T-regs), induces an acute secretory activating phenotype in tumor cells stimulating phagocytic activity and promotes immunogenic tumor cell death (ICD), all of which can boost immunotherapy (16). Furthermore, thalidomide and derivatives (IMiDs), also known as Cereblon (CRBN) modulators, have pleiotropic effects on the immune system that include upregulating cytokine production and potentiating growth and survival of effector T lymphocytes, while also reducing T-regs expansion and function (17,18). The potential ability of IMiDs to augment immunotherapy has recently been highlighted through effective preclinical combinations with anti-MM CAR-T cells in xenograft models (19,20) but caution is warranted given the unpredictable toxicity noted during clinical combination with the anti-PD1 checkpoint inhibitor pembrolizumab (21). Combining BsAb with currently approved drugs for MM is a rational direction forward, if the two approaches provide synergistic effects with non-overlapping toxicity.
Evaluating in vivo mechanisms of MM immunotherapeutic antibodies alone or in combination has been hampered by a lack of appropriate immunocompetent mouse models and murine reagents. While CRBN sequence is highly conserved across species, four amino acid differences in rodent CRBN IMiD binding site impair IMiD response. Specifically, murine CRBN binds to IMiDs but fails to undergo the conformational modification required to induce CK1a, IKZF1 or IKZF3 ubiquitination and degradation (22–24). For this reason, the preclinical evaluation of IMiDs in mice has been difficult and failed to identify their teratogenic activity (25). Furthermore, murine T cells do not release IL-2 following IMiD treatment (22) and we have shown that IMiDs are the only class of drugs active in MM patients but inactive in Vk*MYC mice (26,27). Here, to explore ways to improve the quality and longevity of BsAb therapy in the presence of the native tumor microenvironment we generated a BCMA/CD3 BsAb that binds both human and mouse BCMA with one arm and murine CD3 with the other arm to induce T cell-mediated cytotoxicity. In order to evaluate BsAb combination therapy with IMiDs, we generated two BAC transgenic mouse lines expressing human CRBN and crossed them to the Vk*MYC mouse to generate an immunocompetent mouse model of MM, Vk*MYChCRBN, sensitive to IMiDs. We identify tumor burden and T lymphocyte function-kinetics as determinants of BsAb efficacy. We define mechanisms by which two different standard of care treatments for MM, the IMiD pomalidomide (Pom) and Cy, potentiate BsAb therapy through their dual actions on the tumor and microenvironment. These results highlight the need for additional investigation of the effects of anti-MM standard of care drugs on the patients’ immune system in the context of modern anti-MM immunotherapy combinations.
Results
BCMA is a specific target for immunotherapy in the Vk*MYC model MM
We have previously described the utility of the de novo (aged mice that have spontaneously developed MM) Vk*MYC model of MM and its derivative syngeneic transplantable lines in elucidating the mechanism of therapies that depend upon the microenvironment, including IAP antagonists and checkpoint inhibitors (10,27–32). Here, we establish Vk*MYC as an appropriate model for optimizing T cell based BCMA targeted immunotherapy. Using a previously generated dataset, GSE111921 (33), we determined the expression of Tnfrsf17/BCMA transcript in murine normal PCs, Vk*MYC derived Burkitt’s lymphomas, Balb/c plasmacytomas, de novo Vk*MYC MM, and transplantable Vk*MYC MM lines (Figure 1A). We found that murine Tnfrsf17 is universally expressed in all normal and malignant PCs at significantly higher levels than in lymphoma cells. De novo and transplantable Vk*MYC MM cells shared a similar range in Tnfrsf17 expression. Despite abundant RNA expression, BCMA was barely detectable on the surface of murine MM and plasmacytoma cell lines grown in vitro and in vivo (Figure 1B, upper panel). In contrast, the same human/murine BCMA cross-reactive antibody readily stained the human MM cell line H929. It has been reported that BCMA is commonly shed from normal and malignant PC surface due to direct cleavage of its transmembrane domain by endogenous gamma-secretases (GS) (34,35). Consistently, we detected high levels of soluble BCMA (sBCMA) in the serum of Vk*MYC tumor bearing mice, but not in age-matched controls (Figure 1C). As reported for human MM, the levels of sBCMA correlated with tumor burden, with transplanted mice bearing extra-medullary MM displaying higher sBCMA. Treatment with GS inhibitors stabilized BCMA on the surface of MM cells both in vitro and in vivo (Figure 1B, bottom panel and 1D)
Figure 1. BCMA is a target for bispecific immunotherapy in Vk*MYC MM.
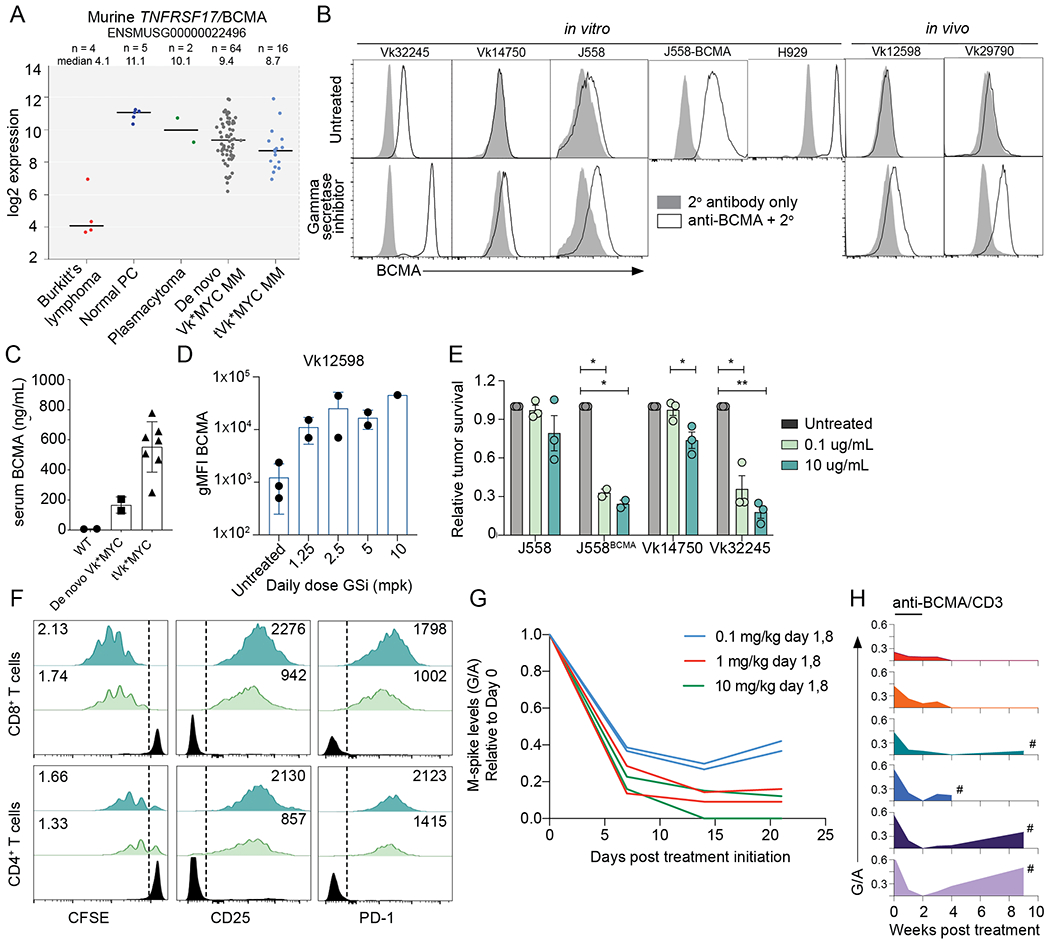
A. RMA summarized expression values of Tnfrsf17/BCMA mRNA of Vk*MYC derived lymphoma cell lines, normal plasma cells (PC), Balb/c plasmacytoma cell lines, Vk*MYC de novo MM and transplantable cell lines (tVK*MYC). Line at median is shown for each group. B. BCMA cell surface staining (white histogram) by FCM of cell lines or primary MM cells with (bottom panel) or without (top panel) GS inhibition (DAPT 1uM, 18 hours for in vitro and LY-411575-I at 5mg/kg for in vivo treatment). The gray histogram depicts negative control staining with secondary antibody only. C. Soluble BCMA levels quantified by ELISA in the serum of moribund tumor bearing or age matched WT control mice. Each symbol represents one untreated mouse, tested in duplicate. D. Surface BCMA quantified by FCM (geometric MFI) of ex vivo CD138+ tumor cells harvested from Vk12598 tumor bearing mice left untreated or treated for 48 hours with the GS inhibitor LY-411575-I at the indicated dose. Each dot represents an individual mouse. E. Tumor cell survival after incubation in vitro with splenocytes and BCMA/CD3-BsAb at two different concentrations, normalized to the untreated conditions. P values determined using multiple comparison T tests with Holm-Sidak adjustment. F. FCM analysis of T cells from killing assay in F, representative of triplicate tests. The proliferation index is in the upper left and the geometric MFI for the indicated markers is presented in the top right corner of both plots. G. M-spike levels (G/A relative to day 0) over time (days) in six de novo Vk*MYC mice treated with increasing doses of anti-BCMA/CD3 BsAb. H. M-spike levels (G/A) over time (weeks) in six de novo Vk*MYC mice treated with 1 mg/kg anti BCMA/CD3 BsAb on day 1,8. Each mouse is represented by a different colored histogram. # shows mice that succumbed to tumor burden.
Murine PC killing in vitro by anti-BCMA/CD3 BsAb is dependent upon BCMA expression levels and BsAb concentration
A mouse BCMA/CD3 IgG1 BsAb antibody, together with the negative control anti-KLH/CD3 BsAb, was generated using the “Electrostatic steering coupled with inter-chain disulfide bond” technology developed at Bristol Myers Squibb (36). For the anti-mCD3 arm, we chose the variable region of the anti-mouse CD3ε antibody 145-2C11; for the anti-mBCMA arm, we used the variable region of an anti-hBCMA antibody that was identified to be cross-reactive to mouse BCMA. The D265A mutation was introduced into the mIgG1-Fc to silence the FcγR effector function (36). SPR analysis confirmed that mouse BCMA/CD3 bispecific binds to mouse BCMA extracellular domain (ECD) with KD=5 nM and mouse CD3εδ complex with KD=169 nM, respectively (Supplemental Figure 1).
Given the variable expression of BCMA by murine PCs, we first sought to evaluate the efficacy of BCMA-targeted therapy in vitro. Tumor cell lines, including the Balb/c plasmacytoma J558, J558 engineered to overexpress BCMA, and two Vk*MYC derived MM cell lines that grow in vitro (Vk14750VITRO and Vk32245VITRO, Table 1) were cultured with whole splenocytes and a titration of BsAb. Here, tumor cell killing was dependent upon both the dose of antibody and the level of tumor BCMA expression (Figure 1E). The parental J558 cell line and Vk14750VITRO cells, with very low BCMA expression, were sensitive to treatment only when 10 μg/ml of BsAb was used. In contrast, J558 overexpressing BCMA and the high BCMA expresser Vk32245VITRO were efficiently killed even at a 100-fold lower antibody concentration. Moreover, consistent with the mechanism of action of BsAbs, anti-BCMA/CD3 treatment of Vk32245VITRO induced a dose dependent CD8+ and CD4+ T cell proliferation and activation (Figure 1F). We noted that in vitro, and subsequently, in vivo, anti-BCMA/CD3 treatment induced higher proliferation of CD8+ T cells than CD4+ T cells that may reflect intrinsic proliferative kinetic differences between cell types (37).
Table 1. Cell lines characteristics.
The genotype and sensitivity to drugs of the cell lines used in this study are reported. MM, E-MM, and VITRO indicate growth restricted to the BM, extra-medullary dissemination, or growth in vitro respectively. For transplantable lines, the time to engraftment, measured as the number of weeks required after transplantation for the M-spike to reach a gamma/albumin ratio = 0.25, indicative of an M-spike = 7 g/L is reported, as well as the median survival, in days, following the detection of a G/A = 0.25. RMA summarized Tnfrsf17 transcript expression is indicated. For cell line growing in vitro, the transduced genes are listed.
| Line | Specie | Strain/Genotype | Growth | Drug sensitivity | Time to G/A 0.25 | Median OS from G/A 0.25 | Tnfrsf17 RMA | Modifications |
|---|---|---|---|---|---|---|---|---|
| Vk12598 | C57BL/6 | Vk*MYC | E-MM | Cy | 5 weeks | 10 days | 50 | n.a. |
| Vk27181 | C57BL/6 | Vk*MYCDLox | E-MM | Cy,Bor | 7 weeks | 20 days | 29 | n.a. |
| Vk29790 | C57BL/6 | Vk*MYCDLox × hC123 × mCRBNhet | MM | Cy, Pom | 14 weeks | 99 days | 54 | n.a. |
| Vk32908 | C57BL/6 | Vk*MYC × hC123 × mCRBNnull | E-MM | Cy, Pom | 4 weeks | 19 days | 208 | n.a. |
| Vk14750VITRO | C57BL/6 | Vk*MYC | VITRO | Pom | n.a. | n.a. | 29 | pWPI-hCRBN |
| Vk32245VITRO | C57BL/6 | Vk*MYC | VITRO | Pom | n.a. | n.a. | 150 | pWPI-hCRBN |
| J558 | Balb/c | WT | VITRO | n.d. | n.a. | n.a. | n.a. | pWPI-mBCMA |
| H929 | Human | WT | VITRO | n.d. | n.a. | n.a. | 1477 | n.a. |
Anti-BCMA/CD3 BsAb is active in vivo against Vk*MYC MM
Next, we sought to investigate the ability of anti-BCMA/CD3 to induce tumor regression in vivo in the aged, de novo Vk*MYC mice, that we believe best model newly diagnosed MM patients and display features of T cell senescence reported in MM patients (38) (Supplemental Figure 2). We chose six mice with a serum gamma/albumin (G/A) ratio on day 0 of 0.5 (corresponding to M-spikes levels of 13.5 g/L) and treated them with anti-BCMA/CD3 at 0.1, 1 or 10 mg/kg on day 1 and 8. M-spike levels, measured weekly over time and compared to initial levels, demonstrated a significant response (defined as >50% reduction in M-spike) in all treated mice, already evident after only one dose of BsAb, which was similarly sustained with a 1 and 10 mg/kg dose (Figure 1G). We therefore selected a dose of 1 mg/kg once per week for two weeks of anti-BCMA/CD3 for all subsequent experiments and treated six Vk*MYC mice with variable levels of tumor burden (G/A on day 0 ranging from 0.2 to 0.6, corresponding to M-spike levels between 5 and 16 g/L). While all mice achieved a complete response (CR, complete disappearance of the M-spike), mice with an initial high tumor burden (G/A >0.5, corresponding to an M-spike > 14 g/L at day 0) rapidly relapsed after treatment discontinuation, whereas mice with a day 0 G/A < 0.4 showed a slower but progressive deepening of the response even after treatment discontinuation, with two of them remaining tumor free for more than a year after treatment (Figure 1H). This initial study indicates a tolerable, rapid, and strong single agent effect of anti-BCMA/CD3 BsAb across aged immunocompetent mice bearing heterogenous BM tumors, with durable protection occurring in a subset of animals.
Anti-BCMA/CD3 BsAb treatment leads to broad T cell activation but its efficacy is variable and affected by both tumor burden and tumor growth kinetics
To evaluate the efficacy of anti-BCMA/CD3 treatment against aggressive and drug resistant transplantable MM, we challenged C57BL/6 wildtype (WT) mice with Vk29790 cell line and initiated treatment when M-spikes reached a G/A > 0.1. Vk29790 is an indolent BM homing tumor cell line with relatively slower time to engraftment than other MM transplantable lines (Table 1). Anti-BCMA/CD3 significantly reduced M-spikes in all treated mice and seven of eight mice achieved and maintained a CR for the duration of the study (16 weeks) (Figure 2A, B). Survival differences were not measured in this experiment due to the indolent nature of this tumor. Immunohistochemistry (IHC) on sections of the BM demonstrated a reduction in IRF4+ MM cells and accumulation of CD3+ T cells already after 72 hours of treatment (Fig. 2C).
Figure 2. The activity of anti-BCMA/CD3 is transient and related to initial tumor burden.
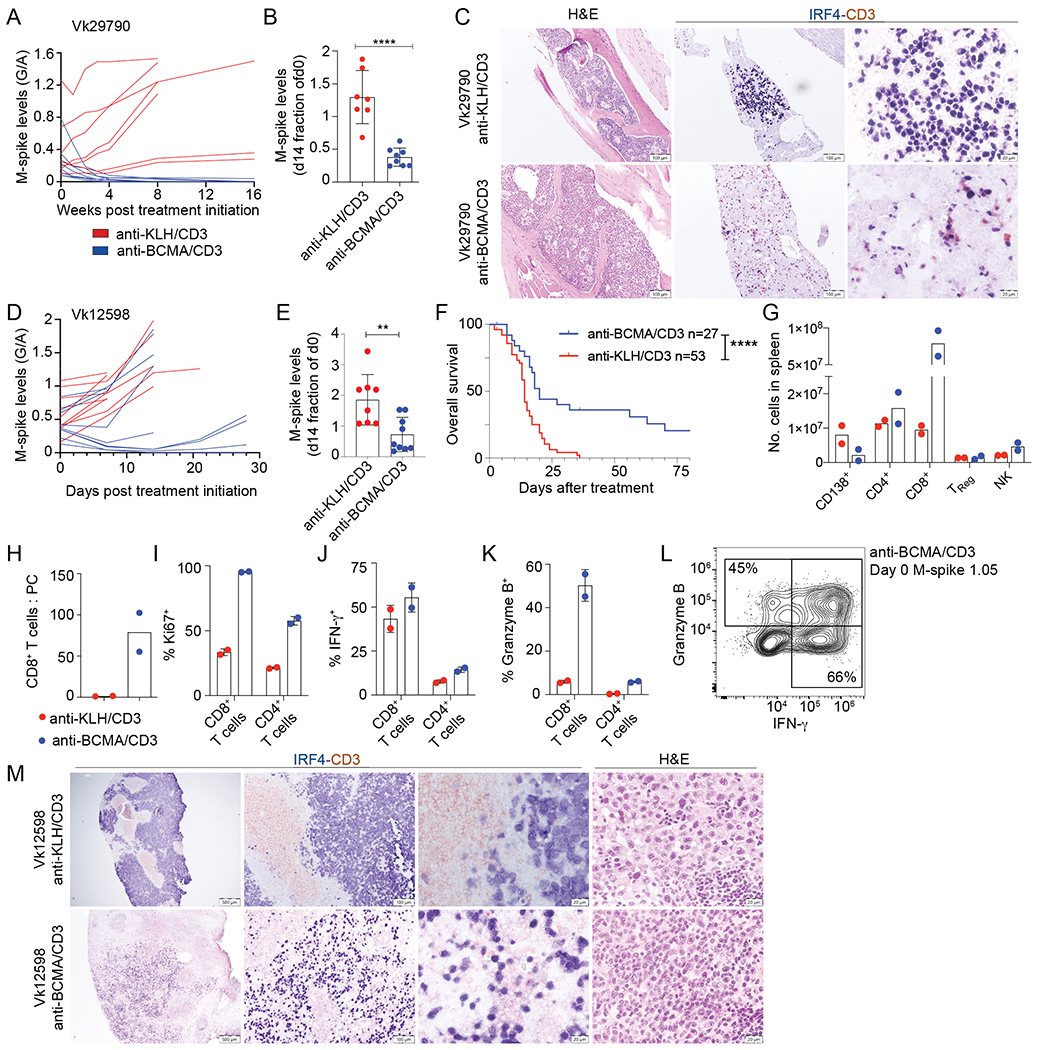
A. M-spike levels (G/A ratio) measured over time in Vk29790 tumor bearing mice treated with control anti-KLH/CD3 BsAb or anti-BCMA/CD3 at 1 mg/kg on day 1,8. B. M-spike levels (% of day 0) in Vk29790 tumor bearing mice measured 14 days after treatment. Each dot represents an M-spike from an individual mouse. Bars show mean M-spike levels with SD. C. IHC staining (surface CD3 in red and nuclear IRF4 in blue) of BM sections from mice treated in A. Scale bar is shown in the bottom right corner. Images are representative of two mice necropsied from each treatment arm. D. M-spike levels (G/A) quantified over time in response to control anti-KLH/CD3 or anti-BCMA/CD3 BsAb given at 1 mg/kg on day 1,8 in Vk12598 tumor bearing mice. E. M-spike levels (% of day 0) in Vk12598 tumor bearing mice measured 14 days after treatment. F. Kaplan-Meier survival plot in days from the initiation of treatment of Vk12598 tumor bearing mice receiving the BsAb or control treatment. P values derived from Mantel-Cox Log-ranked Chi-square test. G-L. FCM analysis of splenocytes from Vk12598 tumor bearing mice harvested three days after treatment initiation as in D. M. Representative CD3 and IRF4 staining by IHC of SPL sections harvested three days after treatment initiation as in D. Unpaired T test P values in B, E, are represented by *. * P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next evaluated the efficacy of anti-BCMA/CD3 treatment using the more aggressive transplantable Vk12598 MM line, where tumor establishes in the BM and expands to extramedullary sites, predominantly in the SPL (Table 1). Again, anti-BCMA/CD3 significantly reduced M-spikes by the end of the two-week treatment and improved overall survival (OS) (Fig. 2D–F). As noted for the de novo mice (Figure 1H), efficacy related to initial tumor burden: only mice with a G/A less than 0.5 on day 0 displayed reduction in M-spike levels. Additionally, most responses were transient with relapses occurring within three weeks of treatment. We next tested whether the addition of GSi to increase BCMA surface levels would increase the efficacy of anti-BCMA/CD3 in high tumor burden situations. We observed that adding a GS inhibitor concurrently, although mildly toxic, only transiently improved BsAb activity (Supplemental Figure 3A) and did not extend survival (Supplemental Figure 3B, C). Three days after treatment, we determined cellular changes in the SPL by flow cytometry (FCM) (Figure 2G–L). Anti-BCMA/CD3 reduced CD138+ tumor cells dramatically increased the number of CD8+ T cells per SPL, resulting in a considerably higher CD8+ T cell to tumor ratio. CD8+ T cells were more proliferative, more likely to produce IFN-γ and granzyme B. Similar increases were observed in IFN-γ+ CD4+ T cells although the total numbers of these cells per SPL did not change (Figure 2I–K). Importantly, while the antibody was less efficacious at high tumor burden, this was not due to lack of activated cytotoxic CD8+ T cells, which were present three days after treatment even in the example case with the highest G/A ratio at day 0 (1.05, corresponding to M-spike of 28 g/L), indicating that the therapy is able to activate T cells regardless of tumor burden (Figure 2L). IHC analysis of SPL sections demonstrated expansion of CD3+ T cells infiltrating the inter follicular space and replacing the IRF4+ tumor cells (Figure 2M).
An immunocompetent hCRBN+ transgenic mouse model mouse model of MM sensitive to IMiDs
To overcome lack of activity of BsAb therapy in high tumor burden setting, we explored combination therapies that could boost T cell function. We hypothesized that IMiD’s immunomodulatory effects on the host and tumor would enhance bispecific activity twofold: in direct clearance of tumor and co-stimulation of the redirected T cells. IMiDs binding to CRBN leads to degradation of IKZF1 and IKZF3 transcription factors, which control the expression of IRF4, essential for PC survival, and inhibit transcription of the IL2 promoter, explaining the co-stimulatory activity of IMiDs in T cells. A few amino acid differences in the mouse CRBN sequence renders them resistant to IMiDs. We generated transgenic mice expressing human CRBN (hCRBN) by microinjecting into pure C57BL/6 pronuclei two bacterial artificial chromosomes (BACs) spanning the entire hCRBN gene and differing in the regulatory regions included (Figure 3A). We obtained a founder line from each BAC, hC123 and hC343, expressing different hCRBN levels (Figure 3B). In order to avoid potential competition for IMiDs binding between the endogenous murine and human CRBN, both hCRBN lines were bred into a murine CRBN−/− background (39). To test for proper IKZF1/3 degradation in response to IMiDs, we treated WT, mCRBN−/−, and the two hCRBN+ founder lines with the more potent IMiD iberdomide (CC-220, at 10 mg/kg) (40), harvested splenocytes after 6 or 24 hours, and monitored IKZF1/3, and CRBN protein levels over time (Figure 3B). We noted that the higher hCRBN expression in hC343 splenocytes correlated to deeper IKZF1/3 downregulation following CC-220 treatment. However, these effects were transient and both proteins returned to baseline levels within 24 hours of treatment. Similarly, we treated hC343 mice with escalating doses of Pom for 6.5 hours and detected a dose dependent decrease in intracellular IKZF1 levels in B and T cells by FCM (Figure 3C). Based of these results, we chose to dose Pom at 50 mg/kg twice per day for subsequent experiments. Moreover, as the line hC343 displayed the higher CRBN levels and strongest IKZF1/3 degradation with IMiD, we selected it for use as the host for subsequent transplantation experiments. We also bred both hC123 and hC343 lines with Vk*MYC mice to generate a new genetically engineered immunocompetent mouse model of MM responsive to IMiDs (referred to as: Vk*MYChCRBN). We obtained two transplantable lines from two independent aged Vk*MYChCRBN MM tumors, displaying different growth characteristics and tropism (Table 1). For both lines, upon engraftment into WT mice Pom delayed tumor progression, while responses were observed in combination with dexamethasone, as seen in human MM (Figure 3D). Overall these data demonstrate that hCRBN expression in vivo is sufficient to induce IMiD responsiveness to murine cells and IMiD sensitivity to murine MM (41). They also confirm that the extent of IKZF1/3 degradation correlates with CRBN levels (41,42).
Figure 3. Pom enhances hCRBN transgenic CD8+ T cells proliferation, cytokine production, and tumor killing induced by anti-BCMA/CD3 in IMiD resistant tumors.
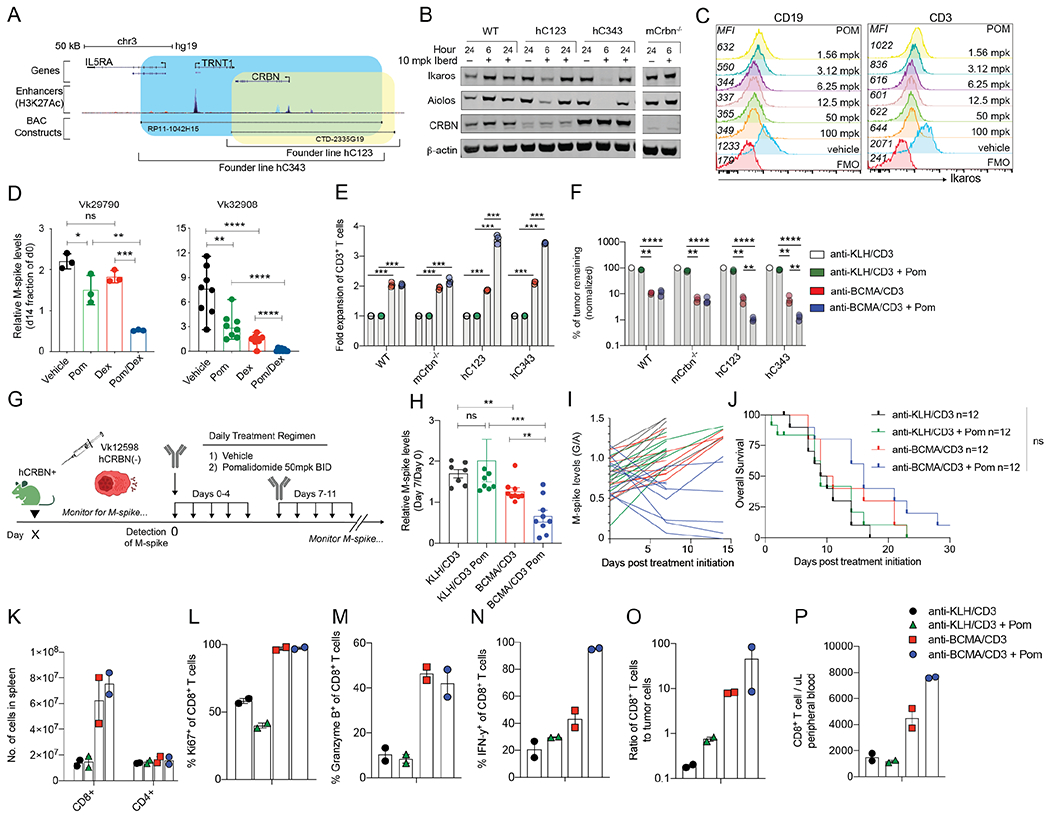
A. Depiction of the location on human chromosome 3 of the two BACs (RP11-1042H15 and CTD-2335G19) chosen to generate hCRBN transgenic mice. Genes included in BAC are shown, then the enhancer regions (identified by H3K27Ac peaks), followed by the span of each BAC (blue hC343, yellow, hC123). B. Western blot of splenocyte protein extracts generated from necropsied mice treated for 6 or 24 hours with 10 mg/kg iberdomide/CC220 and probed with antibodies against the listed proteins. Mouse strains are listed on the top as WT (C57BL/6), hC123, or-343, (representing two different transgenic founder lines expressing hCRBN) and mCrbn−/−. C. Ikaros expression measured by FCM in CD19+ or CD3+ cell surface stained splenocytes harvested from hC343 transgenic mice treated in vivo for 6.5 hours with the indicated dose of Pom. MFI is reported on the left of each histogram D. M-spike levels (% of Day 0) in WT mice bearing the indicated Vk*MYChCRBN MM lines measured two weeks post treatment. Bars indicate mean with SD. E. Fold expansion of splenic T cells of the indicated genotype stimulated ex vivo by Vk32245VITRO MM cells and the indicated BsAb with or without Pom for three days. Bars represent the mean and SEM. Significant differences were tested through multiple comparison T tests with Holms-Sidak adjustment. F. Viability of Vk32245VITRO MM cells surviving tumor killing assay in A, all normalized to the negative control treated condition. Bars in E and F represent the mean and SEM. Significant differences were tested through multiple comparison T tests with Holms-Sidak adjustment. G. Treatment schedule for hC343 recipient mice bearing IMiD-insensitive Vk12598 tumor cells treated with anti-KLH/CD3 or anti-BCMA/CD3 with or without Pom. H. M-spike levels (% of day 0) in Vk12598 tumor bearing mice measured 7 days after treatment in B. Each dot represents an M-spike from an individual mouse. Bars show mean M-spike levels with SD. I. M-spike levels (G/A) were quantified over two weeks. Each line represents an individual mouse. Color of each line corresponds to the treatment groups in B. J. Kaplan-Meier survival plot in days from the initiation of treatment of Vk12598 tumor bearing hCRBN+ mice receiving treatments in B. Differences were tested with Mantel-Cox Log-ranked Chi-square test K. Number of cells per SPL, identified by FCM, of mice (n = two per group) three days after treatment initiation from B. Percentage of CD8+ T cells expressing Ki67 (L), IFN-γ (M), or granzyme B (N), and the ratio of CD8+ T cells to tumor cells (O). P. Number of CD8+ T cells per uL of peripheral blood at necropsy. Unpaired T test *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
IMiDs modulate T cell based BsAb immunotherapy in MM
IMiDs have co-stimulatory effects on human T cells resulting in increased proliferation and cytokine production (18). Here, we cultured T cells from WT, mCRBN−/−, hC123 and hC343 hCRBN+ mice in the presence of Vk32245VITRO target MM cells and BsAb, with or without Pom, and noticed marked T cell expansion in the presence of Pom and hCRBN (Figure 3E), which resulted in significantly more efficient killing of tumor cells (Figure 3F). We also observed the same effects in a parallel assay with purified T cells instead of splenocytes, suggesting this increased tumor killing is due to a direct effect of Pom upon cytolytic T cells (Supplemental Figure 4).
These results prompted us to test if the immunomodulatory properties of Pom could enhance MM cell killing by BsAb treatment in IMiD resistant tumors in vivo, especially in high tumor burden settings, where anti-BCMA/CD3 was not sufficient as a single agent to control tumor growth. We evaluated the combination of anti-BCMA/CD3 treatment with Pom in hC343 hosts transplanted with the hCRBN- IMiD resistant Vk12598 MM cell line (Figure 3G). We initiated two-weeks of treatment with anti-BCMA/CD3 with or without Pom, starting when M-spikes reached a G/A up to 1.2 (M-spike > 30 g/L) indicative of high tumor burden. After one week of treatment, M-spikes were significantly lower upon anti-BCMA/CD3 treatment compared to anti-KLH/CD3, but still higher than day 0 levels, indicating that the treatment only delayed tumor growth (Figure 3H, I). In contrast, the addition of Pom significantly reduced M-spikes below their respective day 0 levels in all treated mice, regardless of initial tumor burden. However, these effects remained transient and did not statistically improve OS of treated mice (Figure 3J).
We performed FCM on SPL cells after three days of treatment to monitor the immunomodulatory, tumor extrinsic effects of IMiDs on T cells during treatment. CD8+ T cell number increased 6- to 7- fold following treatment with anti-BCMA/CD3, with or without Pom (Figure 3K). CD8+ T cells were equally highly proliferative with a similar proportion expressing granzyme B (Figure 3L, M), but a higher proportion expressing IFN-γ with Pom (Figure 3N). CD8+ T cells were also more numerous than MM cells in the SPL and peripheral blood of mice receiving anti-BCMA/CD3 with Pom compared to the single agents (Figure 3O, P). IHC confirmed more effective tumor depletion after three days of anti-BCMA with Pom treatment (Supplemental Figure 5). Overall, in the context of BsAb-directed activation, these data strongly support the use of Pom even in IMiD-resistant tumors to increase the pool of activated IFN-γ-producing T cells in the tumor microenvironment and their migration through the blood.
Tumor intrinsic and extrinsic IMiD activity in combination with anti-BCMA/CD3
Pom boosted BsAb driven T cell activation even in the absence of a tumor cell autonomous direct effect. Next, we sought to determine the additional contribution of Pom’s direct anti-MM activity on T cell function following anti-BCMA/CD3 treatment using in vitro and in vivo methods. Initially, we quantified relative in vitro tumor killing in separate tests, where combinations of WT or hCRBN-expressing tumor cells or splenocytes were co-cultured together with or without suboptimal doses of anti-BCMA/CD3 and Pom. When the tumor was sensitive to Pom (Figure 4A), tumor cell viability, as expected, was reduced to approximately 50% by Pom treatment alone. When tumor cells were co-cultured with WT splenocytes, anti-BCMA/CD3 and Pom, tumor cell survival was further reduced to 32% of untreated. Moreover, the use of hC343 splenocytes with this combination additionally reduced tumor cell viability to 8% of untreated tumor. As expected, Pom boosted T cell proliferation and hCRBN- tumor cell killing only in the presence of hC343, but not WT splenocytes and anti-BCMA/CD3 (Figure 4B).
Figure 4. Tumor-and T cell-autonomous Pom effects enhance and BCMA/CD3 activity.
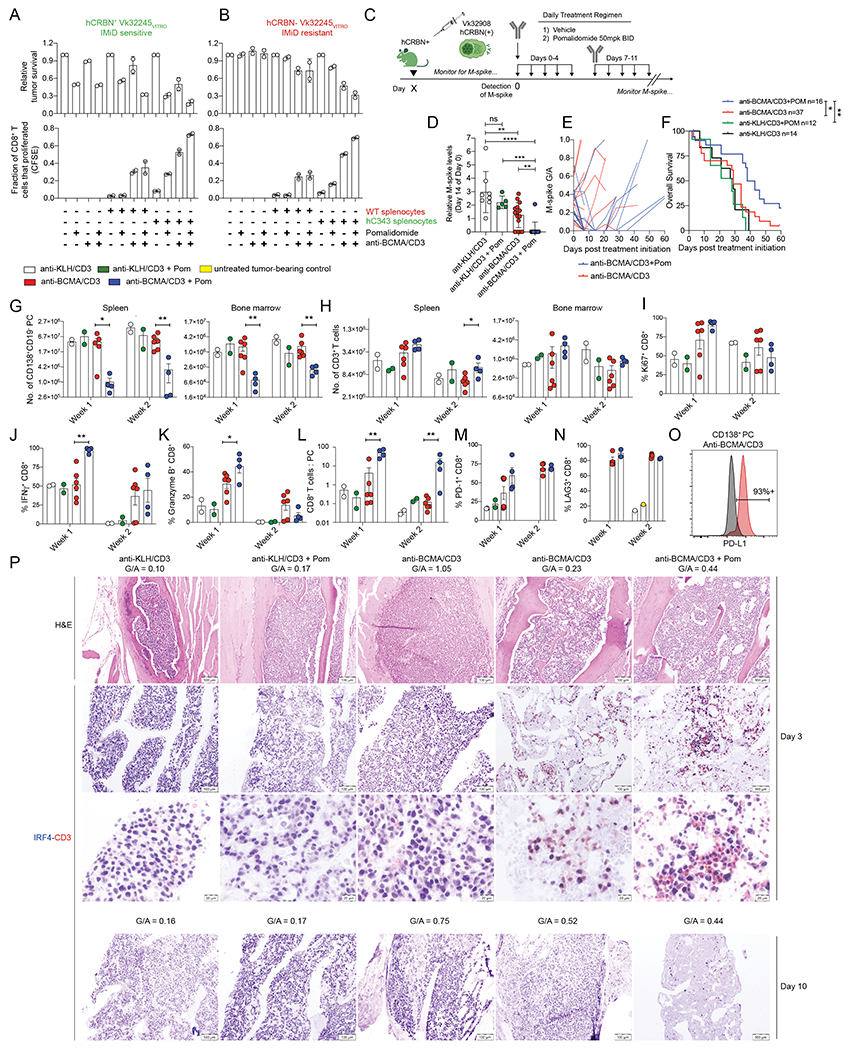
The cell viability by FCM of Vk32245VITRO MM cells transduced with A. hCRBN expressing lentivirus or empty control B. co-cultured in vitro with splenocytes from WT- or hC343 mice and exposed to anti-BCMA/CD3 with or without Pom is shown on top panels, normalized to the untreated conditions. Representative of three independent experiments. Below, the proliferation of CD8+ T cells from the conditions in (A) were quantified by dilution of cytoplasmic CFSE and measured by FCM. Bar graphs indicate the fraction of total cells that proliferated (1 = 100%). Error bars are the SEM of triplicate values. C. Treatment schematic for hC343 recipient mice bearing IMiD sensitive Vk32908 MM cells. D. M-spike levels (% of day 0) in Vk32908 tumor bearing mice measured 14 days after treatment initiation. Each dot represents the M-spike from an individual mouse. Bars show mean with SD. P values derived from unpaired nonparametric T tests. E. M-spike (G/A) of individual mice measured over time after treatment initiation. F. Kaplan-Meier survival plot in days from the initiation of treatment of Vk32908 tumor bearing hCRBN+ mice receiving treatments in (B). P values derived from Mantel-Cox Log-ranked Chi-square test. Bar graphs summarizing Vk32908 tumor cells (G), or T cells (H) quantified by FCM in the SPL and BM of mice in C, three or ten days after treatment initiation. Each dot represents an individual mouse. Bar graphs of the percentage of Ki67+ (I), IFN-γ+ (J) and granzyme B (K) CD8+ splenic T cells as well as the CD8+ T cell to tumor cell ratio in the SPL(L). Percentage of PD-1+ (M), and LAG3+ (N) CD8+ T cells from splenocytes. O. PD-L1 expression by FCM in Vk32908 MM cells from a representative mouse treated with anti-BCMA/CD3, ten days after treatment initiation. Black histogram represents FMO control staining. P. BM sections from mice necropsied three days (1st-3rd row) or ten days (4th row) after treatment initiation in C, stained with H&E or antibodies to CD3 in red and IRF4 in blue. G/A ratio at beginning of treatment is indicated above each image. Two examples of mice treated with anti-BCMA/CD3 are shown: low and high initial M-spike. Scale bar indicates the magnification Unpaired T test *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Having demonstrated a significant synergy in vitro between anti-BCMA/CD3 and Pom when both MM and T cells were IMiD sensitive, we sought to determine if the addition of Pom is able to overcome resistance in vivo, in settings where anti-BCMA/CD3 was not sufficient as a single agent to control tumor growth. We challenged hC343 IMiD-sensitive mice with IMiD-sensitive hCRBN+ Vk32908 MM cell line, and upon tumor engraftment we initiated two-weeks of treatment with anti-BCMA/CD3 and Pom (Figure 4C). Vk32908 is the most aggressive SPL and BM-homing MM cell line with faster engraftment time than other transplantable lines and relatively high BCMA expression (Table 1). Anti-BCMA/CD3 alone slowed tumor growth relative to controls but had no effects on OS. In contrast, the combination of anti-BCMA/CD3 with Pom induced a CR in all treated mice but one and extended OS (Figure 4 D–F). While none of the five mice with a G/A at day 0 > 0.3 (M-spike > 8 g/L) responded to the single agents anti-BCMA/CD3, all but one of the five mice with high tumor burden receiving the combination therapy achieved a CR. However, as we noted with our other transplantable models, tumor depletion was transient, and all mice relapsed within 3-4 weeks. Uniquely to this treatment, three of the 25 mice receiving anti-BCMA/CD3 and Pom, regardless of their day 0 M-spike values, rapidly lost >20% body weight at the third day of treatment, resulting in early mortality, suggesting a therapeutic toxicity of the combination (Supplemental Figure 6A).
By FCM, anti-BCMA/CD3 BsAb transiently decreased MM cell number in the SPL and BM in the presence of low but not high tumor burden. In contrast, the addition of Pom led to a deeper and more prolonged tumor reduction in all treated mice (Figure 4G). T cells rapidly accumulated during the first week of treatment in the SPL and BM of mice treated with anti-BCMA/CD3 BsAb with or without Pom, (Figure 4H) and a similar frequency expressed Ki67 (Figure 4I). Pom increased the frequency of IFN-γ+ and granzyme B+ CD8+ T cells in the first week of treatment with BsAb (Figure 4J, K). However, T cell number contracted during the second week, even when Pom was added, to levels at or below control-treated mice (Figure 4H). Nevertheless, the efficient tumor killing and T cell expansion were reflected by a significantly higher CD8+ T cell to tumor ratio in the SPL of mice treated with anti-BCMA/CD3 and Pom over the single agent during the two weeks (Figure 4L).
Anti-BCMA/CD3 treatment leads to T cell exhaustion
Fewer T cells in the SPL and BM expressed Ki67, IFN-γ and granzyme B in the second week of BsAb treatment. This occurred regardless of whether tumor antigen remained to provide T cell stimulation which led us to hypothesise that in the setting of aggressive tumor, the T cells activated by the BsAb are less functional over time. This prompted us to evaluate additional parameters of T cell functions and exhaustion in response to treatment. PD-1 is upregulated on T cells after activation and its continued expression interrupts T cell signaling. Anti-BCMA, alone and in combination with Pom, induced expression of PD-1 on CD4+ and CD8+ T cells that increased over the two weeks of treatment (Figure 4M, Supplemental Figure 6B). A similar trend was noted for KLRG1, normally expressed on terminally differentiated short lived effector CD8+ T cells (Supplemental Figure 6C) and LAG3, a surface marker associated with activated T cells, among other cell types, with a documented role in CD8+ T cell exhaustion (43) (Figure 4N, Supplemental Figure 6D). The combinatorial upregulation of these markers extending through the weeks of treatment suggests that T cells become functionally exhausted and less responsive to anti-BCMA/CD3 over time, and this is potentiated by Pom. Furthermore, the ligand for PD1, PD-L1, homeostatically expressed in pro-inflammatory milieu and upregulated by IFN-γ, was expressed on nearly all tumor cells during the second week of anti-BCMA/CD3 treatment (example shown in Figure 4O). Finally, we observed an increase in regulatory CD4+ T cells in mice treated with Pom during the second week of treatment (Supplemental Figure 6E, F), which could contribute to suppressing the BsAb response.
IHC identified prominent infiltration of CD3+ T cells in the BM and SPL of anti-BCMA/CD3 treated mice with low, but not high tumor burden at day 0 (Figure 4P and Supplemental Figure 7). However, the majority of T cells expressed IRF4, which we had not found in other models. IRF4 is a nutrient sensitive regulator of gene expression and cell fate in B and T cells. In parallel to B cells, IRF4 induction is linked to the strength of T cell receptor signaling and IRF4 represents an important transcription factor in expansion and differentiation of effector T cells (44). Furthermore, the intensity of IRF4 expression during T cell proliferation correlates with terminal effector differentiation as opposed to self-renewal programs (45). Together with the high expression of exhaustion markers, this suggests that BsAb-activated T cells, in the context of Vk32908 hCRBN+ MM, are hyperactivated, which favors generation of short-lived effectors with potent tumor killing activity but also exhausted cells, and this is potentiated by combination with Pom.
Sustained anti-MM response with anti-BCMA/CD3 and Cy combination therapy
Our data establish that a lower tumor burden is associated with greater efficacy of the BCMA BsAb. Cy is a drug with strong tumoricidal action as a DNA-alkylating agent in tumor cells. We hypothesized that the tumor debulking activity of Cy would potentiate BsAb therapy when used in combination by decreasing the hyperactivation, proliferation and terminal effector T cell differentiation we noted in mice treated with the BsAb alone or with Pom combination. We also recognized that Cy can slow immune cell replication, and act as a lymphodepleting agent, activities that could potentially antagonize BsAb therapy. However, we reasoned that this effect may also target T-regs and therefore may benefit BsAb therapy of aggressive MM. For these reasons, we next tested the combination of concurrent Cy with the anti-BCMA/CD3 antibody in hC343 mice bearing aggressive Vk32908 tumor cells, to allow a direct comparison with Pom combination therapy (Figure 5). We initiated two-weeks of treatment with concurrent anti-BCMA/CD3 and Cy when M-spikes reached a G/A > 0.25 (> 7 g/L). After two weeks of treatment, M-spikes, relative to day zero, were significantly lower upon treatment with Cy compared to control and anti-BCMA/CD3 alone, with the combination inducing a uniform CR in all but one treated mouse (Figure 5A–B). Moreover, while the mice treated with Cy alone relapsed quickly after treatment, the combination with anti-BCMA/CD3 induced longer remission and increased OS (Figure 5B, C). Importantly, no toxicity was observed in the combination treatment arm.
Figure 5. Effective combination of Cy and anti-BCMA/CD3.
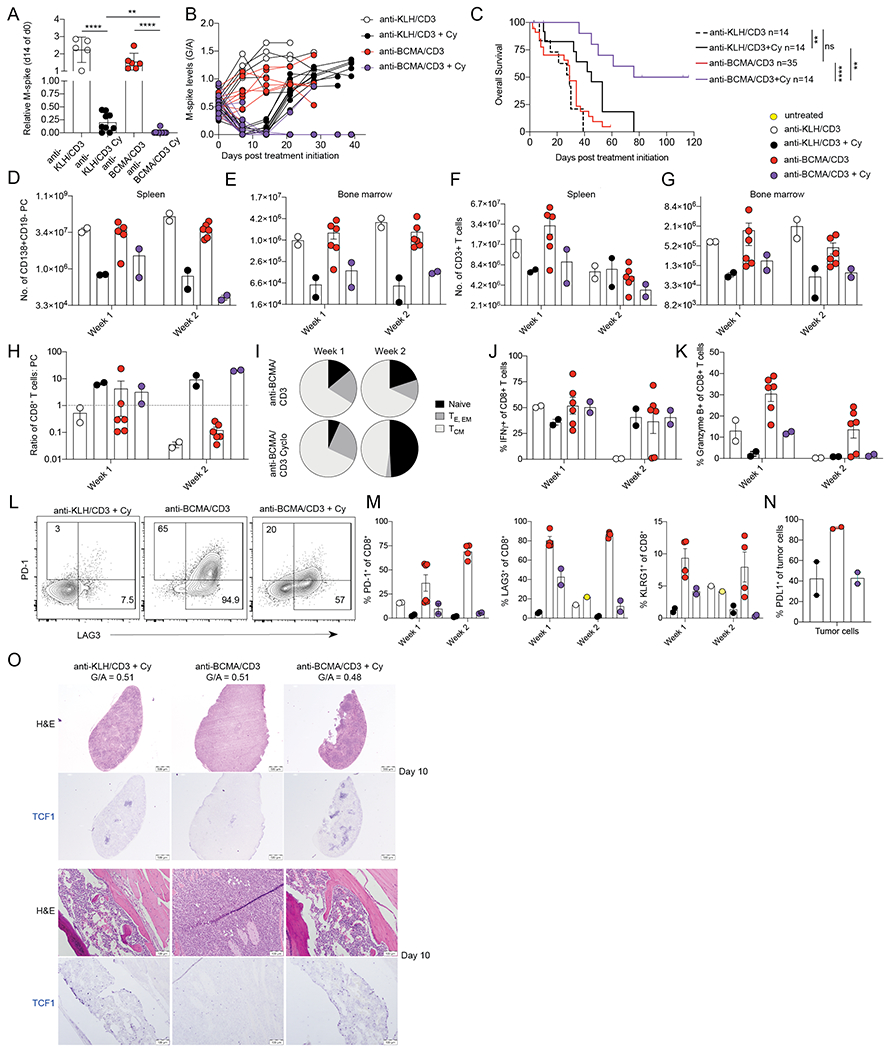
hC343 mice bearing Vk32908 MM cells were treated with anti-KLH/CD3 BsAb or anti-BCMA/CD3 BsAb (1 mg/kg day 1,8) with or without Cy concurrently (100 mg/kg day 1,8). A. M-spike levels (% of day 0) measured 14 days after treatment. Each dot represents an M-spike from an individual mouse. Bars show mean M-spike levels with SD. Unpaired T test *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. B. M-spike levels (G/A) measured weekly for the treatment groups in A. Each dot represents an individual mouse. C. Kaplan-Meier survival plot in days of Vk32908 tumor bearing mice receiving treatments in (A), P values derived from Mantel-Cox Log-ranked Chi-square test. Number of tumor cells (D, E), or T cells (F, G) quantified by FCM in the SPL or BM of mice three or ten days after treatment initiation. Each dot represents an individual mouse. H. CD8+ T cell to tumor cell ratio in the SPL. I. Average frequency of CD8+ T cell memory compartments quantified by FCM (naïve = CD44-CD62L+, effector = CD44+CD62L-KLRG1- or CD44+CD62L-KLRG1+, effector memory = CD44+CD62L-, central memory = CD44+CD62L+). Bar graphs of the frequency of splenic CD8+ T cells expressing IFN-γ (J), or granzyme B (K). L. Example dot plots of CD8+ T cells expression of immediate activation markers LAG3 and PD-1 three days after treatments indicated on top of each graph. (M). Percentage of splenic CD8+ T cells expressing PD-1, LAG3 and KLRG1 N. Percentage of Vk32908 tumor cells expressing PD-L1 ten days after treatment initiation. (O) H&E and TCF1 (blue) staining of SPL (top) and BM (bottom) sections from mice collected ten days after treatment with the indicated drugs. M spike levels (G/A) at day 0 are reported for each mouse.
Upon necropsy, we confirmed superior anti-tumor activity in mice receiving combination treatment (Figure 5D, E). As previously noted, we observed an increase in absolute number of T cells in the SPL and BM with anti-BCMA/CD3, while with Cy, their number decreased (Figure 5F, G). These dynamic cellular changes during treatment resulted in a higher CD8+ T cell to tumor ratio in the combination arm by the end of treatment (Figure 5H). As expected, Cy also decreased the number of T-regs in the TME (Supplemental Fig. 6E, F). Both CD8+ and CD4+ T cells remaining at the end of two weeks of treatment were of naïve and central memory compartments (Figure 5I). A similar frequency of the CD8+ T cells were able to produce IFN-γ and granzyme B as compared to control treatment, indicating a lack of expansion of activated T cells (Figure 5J, K). However, upon closer evaluation of early activation markers, existing CD8+ T cells were still activated by the BsAb in the presence of Cy, as represented by upregulation of LAG3 and (to a lesser degree) PD-1 (Figure 5L–M). In support of the observation of tempered T cell activation, PD-L1 was not upregulated on tumor cells with Cy and anti-BCMA/CD3 treatment, as it was during anti-BCMA/CD3 single agent treatment (Figure 5N). Furthermore, we assessed T cell expression of TCF1 over the course of these treatments. TCF1 is an essential transcription factor in T cell development and in antigen activated CD8+ T cells, it limits effector cell differentiation and promotes self-renewal of memory cells (46–48). The loss of TCF1 promotes terminally differentiated KLRG1-expressing T cells. At the end of treatment, more T cells in the SPL and BM of anti-BCMA/CD3 and Cy treated mice expressed TCF1, while fewer expressed this self-renewal transcription factor from anti-BCMA/CD3 alone (Figure 5O).
Overall, our data suggest that the tumor debulking activity of Cy limits T cell activation by rapidly depleting tumor antigen, thus preventing hyperactivation of T cells and release of IFN-γ, limiting upregulation of PDL1 on tumor cells and preserving anti-tumor functionality. Most of the remaining T cells have a naïve or central memory phenotype, express TCF1, and can be readily activated by anti-BCMA/CD3 BsAb to control relapse in a TME that is depleted of immunosuppressive T-regs.
Combination of anti-BCMA/CD3 and Cy induces long lasting remission and protection
We repeated the immunotherapy combination of anti-BCMA/CD3 and Cy in our widely used Vk12598 transplantable model (Figure 6A). After one week of treatment, M-spikes mimicked the trends noted in the Vk32908 model (Figure 6B–C). Tumors in mice treated with Cy alone grew rapidly after 2 weeks of treatment, whereas the combination with anti-BCMA/CD3 induced a CR resulting in long lasting control and curative remission (Figure 6C, D). To validate the cellular changes underpinning this curative combination, we performed necropsy after three days of treatment and confirmed a more profound decrease in tumor cells in mice receiving Cy (Figure 6E). The number of T cells expanded in the SPL during anti-BCMA/CD3 as observed previously, and Cy largely prevented this. Again, Cy decreased the number of T-regs in the TME (Figure 6E). Due to these dynamic alterations, the CD8+ T cell to tumor cell ratio was highest in the group receiving Cy and anti-BCMA/CD3 (Figure 6F). As noted in Vk32908 in vivo studies, anti-BCMA/CD3 with Cy modulated the frequency of T cell proliferation, IFN-γ and granzyme-B production among CD8+ T cells (Supplemental Figure 8A). In comparison, the addition of bortezomib, another cytotoxic agent, induced only a transient deepening of response to the BsAb alone, without affecting anti-BCMA/CD3 induced T cell expansion and without downregulating T-regs (Supplemental Figure 8B–D). These data confirm the essential tumor debulking activity of Cy and highlight the unexpected improvement in T cell function and composition for successfully controlling MM.
Figure 6. The combination of Cy and anti-BCMA/CD3 is curative and induces long-lasting protection from tumor re-challenge.
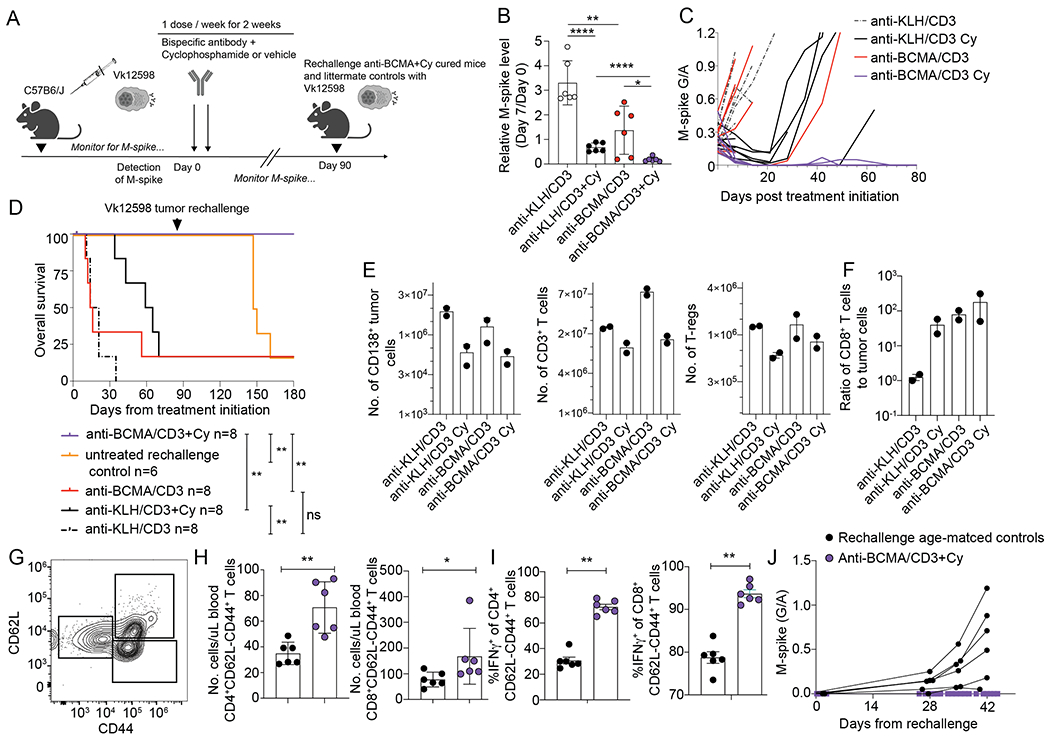
A. Wildtype mice bearing Vk12598 tumors were treated as indicated with control or anti-BCMA/CD3 with or without Cy for two weeks. B. Relative M-spike levels (% of day 0) measured 7 days after the indicated treatment. Each dot represents an M-spike from an individual mouse. Bars show mean M-spike levels with SD. C. M-spike levels (G/A) measured weekly. Each line indicates one mouse. D. Kaplan-Meier survival plot in days from the initiation of treatment of Vk12598 tumor bearing mice receiving treatments in (A). P values derived from Mantel-Cox Log-ranked Chi-square test. Arrow indicates the time at which anti-BCMA/CD3 treated mice were re-transplanted with one million Vk12598 myeloma cells. At the same time, previously untreated and not transplanted age-matched littermate WT mice, were also transplanted with the same pool of Vk12598 cells E. Number of CD138+ tumor cells, CD3+ T cells, and regulatory CD4+ T cells per SPL measured by FCM of samples from necropsied mice three days after treatment initiation of groups in A. F. Ratio of CD8+ T cells to CD138+ tumor cells per mouse G. Representative example of FCM measurement of circulating memory T cells collected from mice in F on day 90, prior rechallenge with Vk12598. H. Quantification of effector memory CD4+ and CD8+ T cells per uL of blood on Day 90, prior to challenge. I. Frequency of IFN-γ producing effector memory CD4+ and CD8+ T cells in the blood of mice prior to challenge with Vk12598 in F. J. M-spike (G/A) after rechallenge of cured or matched WT control mice with Vk12598 cells. P values derived from unpaired T tests. * P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
After nearly three months post-treatment, all of the anti-BCMA/CD3-Cy treated mice remained in CR (Figure 6D). We wanted to test if the combination treatment had provided long term protection through immunosurveillance or if simply had been effective in clearing all of the tumor. We sampled peripheral blood from all of the anti-BCMA/CD3-Cy mice and six littermate naïve age-matched control mice (Figure 6G–I, Day 89). The anti-BCMA/CD3-Cy mice had significantly more circulating memory CD4+ and CD8+ T cells and more of these T cells produced IFN-γ in response to polyclonal ex vivo stimulation (Figure 6G–I). We hypothesize that this is an extended effect of BsAb-mediated T cell expansion or ongoing tumor patrolling, as two of the mice had transient reappearance of a low M-spike around day 50 post-treatment, which was subsequently regressed to none (Figure 6C). These data suggest that the curative combination immunotherapy expands the pool of long-lasting circulating T cells that are licensed to provide ongoing anti-tumor immunosurveillance. To ultimately test our hypothesis that the anti-BCMA/CD3-Cy generated protective immunosurveillance, we challenged the cured mice at day 90 post treatment and the littermate aged-matched controls with the same Vk12598 tumor cell line and monitored weekly for a M-spike (Figure 6A, D, J). The tumor did not grow back in the anti-BCMA/CD3-Cy cured mice but did so in all control mice beginning 4 weeks post-transplant (Figure 6J), providing evidence for ongoing protective anti-tumor immunosurveillance.
Discussion
Bispecific T cell engaging antibodies are novel immunotherapies for MM and show promising clinical results. Their cellular activity induces interrelated changes in the tumor and its microenvironment, containing cytolytic T cells, that determine their efficacy. Here, we model anti-BCMA/CD3 BsAb success and failure in our immunocompetent Vk*MYC and its novel IMiD-responsive derivative Vk*MYChCRBN mouse models. Single agent anti-BCMA/CD3 BsAb induced dramatic anti-MM response in Vk*MYC mice that redirected T cells to lyse the tumor with a pro-inflammatory anti-tumor response. This result was curative in some cases of indolent BM localized tumors representing newly diagnosed MM. However, consistent with clinical results of BsAb in leukemia, the effects of antibody treatment were transient in cases of high tumor burden and associated with loss of T cell functionality.
Treatment of transplantable Vk*MYC with BsAb and Pom caused T cell hyperactivation with increased proliferation, cytokine and cytolytic protein production resulting in more efficient initial tumor depletion even in high tumor burden setting that notably was also seen in IMiD-resistant tumor model. This observation supports the use of IMiDs in combination with BsAb to boost T cell activation in other human cancers, most of which are not directly sensitive to IMiDs. However, we found that with the BsAb alone T cells became less activated and functional over time, permitting universal relapse after treatment ended, and this phenomenon was even more pronounced with the addition of an IMiD. Interestingly, T cell exhaustion was more prominent against the MM line with the higher Tnfrsf17 expression, suggesting that higher levels of tumor antigen, contrary to our in vitro data, may not be beneficial in vivo. Consistently, the addition of GS inhibitors aimed to further increase BCMA expression on MM cells did not improve OS of anti-BCMA/CD3 treated mice.
Given that a lower initial tumor burden was associated with greater efficacy of the BsAb and that expansion of short-lived effector T cells only marginally extended survival, we tested the combination of Cy with anti-BCMA/CD3. Treatment of transplantable Vk*MYC with BCMA BsAb and Cy induced long lasting remission in two models of MM, when the single agents achieved only transient control of the tumor. The superior efficacy of the combination treatment was associated with tempered activation of T cells with self-renewal capacity and a lack of classical immunosuppressive features in the TME, including T-regs. As we have shown that immune activation with IMiDs and suppression with Cy have both positive and negative effects on T cell function when used in combination with BsAb, we speculate that in the clinical development of drug combinations with BsAb it will be important to carefully balance T cell expansion and activation while minimizing exhaustion to maximize therapeutic responses.
We attribute a majority of Cy’s success in combination with the BsAb to our model tumors’ sensitivity to this drug, which is commonly used in combination with other anti-MM drugs in patients with relapsed MM. For those MM patients whose tumors are not sensitive to Cy, we speculate that the addition of other therapies that can induce cytoreduction (e.g., glucocorticoids, proteasome inhibitors), which would need to be precisely tailored to the patient’s tumor, may similarly potentiate immunotherapy.
We were surprised that the BsAb combined with Cy induced long-term protection from relapse, even after tumor re-challenge. This finding presents a contradiction to an accepted mechanism for Cy as a lymphodepleting and generally immunosuppressive agent. We suspect that at an optimized dose, Cy induces ICD of the tumor while increasing the frequency of T cells with self-renewing capacity with opportunities to engage tumor antigen. On the other hand, the lack of long term protection noted after combination treatment with BsAb and bortezomib, a cytotoxic agent also reported to induce ICD, suggests that unique mechanisms of action of Cy, for example T-regs depletion, are required to maximize BsAb efficacy. These mechanisms, combined with the depletion of T-regs in the tumor environment, maximizes the BsAb efficacy and could provide an opportunity to prime a protective response. Future studies are warranted to elucidate how bystander effects may play a role in the superior efficacy of BsAb therapy combinations, including the mechanism of protection in BsAb with Cy, and whether tumor-specific memory T cell immunity is generated in this combination. We conclude that the combination of cytoreductive therapies with BCMA BsAb is an attractive proposal for future trials in MM.
These results warrant future investigation of how to best use BsAb to treat patients whose T cells and microenvironment has undergone many rounds of previous therapies and may have senescent anti-tumor function. Our results show that combining BsAb with chemotherapy or immunomodulators can induce dramatic responses and prolong OS in a highly predictive mouse model of MM, and in the right circumstance can induce long-lasting protective anti-tumor immunity which may be a pre-requisite for cure in patients.
Materials and Methods
Cell lines and reagents.
Human MM and Balb/c plasmacytoma cell lines have been previously described and have been maintained in RPMI 1640 supplemented with 5% FBS and glutamine, without antibiotics (28). The VkMYCVITRO cell lines were generated by culturing permissive transplantable VkMYC cell line in vitro in RPMI-1640 + 5% FBS supplemented with glutamine, penicillin, and streptomycin, “mouse medium”. All cell lines were tested for mycoplasma contamination twice/year using the MycoAlert® kit (Promega) and are periodically validated by copy number polymorphism by PCR. VkMYCVITRO lines expressing human CRBN were generated by lentiviral transduction using human CRBN cDNA subcloned into pWPI lentiviral vector (Addgene plasmid # 12254). Pure populations of EGFP+ transduced cell lines were sorted on a five laser Fortessa (Beckton Dickinson) and purity was checked prior to each experiment on a five laser Cytoflex (Beckman Coulter). The gamma secretase inhibitors DAPT and LY411575-I were obtained from Cayman Chemical; DAPT was solubilized in DMSO and used at 1μM, LY411575-I was freshly suspended in 40% PEG300 + 5% Tween-80 + 45% saline before each in vivo administration. Pomalidomide (Pom) and iberdomide (CC-220), both at > 98% purity were purchased from Pharmablock USA and solubilized in DMSO for in vitro use, or in 0.5% carboxy-methyl-cellulose + 0.25% Tween-80 in water for oral gavage at 50 mg/kg twice per day on day 1-5, 8-12 (Pom), or 10 mg/kg (CC-220). Cyclophosphamide (Cy), dexamethasone and bortezomib were obtained from the clinical pharmacy and solubilized in saline for i.p. injection at 100 mg/kg on day 1 and 8, at 10 mg/kg on day 1-5, 8-12, or at 0.5 mg/kg on day 1,4,8,11, respectively.
Expression & purification of murine BCMA/CD3 and KLH/CD3 BsAb
BsAb was generated by transient co-transfection of four expression plasmids encoding anti-mBCMA heavy/light chain and anti-mCD3 heavy/light chain into Expi293 cells (Thermo Fisher Scientific). Heavy chains (HC) and light chains (LC) were transfected with a DNA ratio (by weight) of 1:1:1:1 for HC_BCMA:HC_CD3:LC_BCMA:LC_CD3. Six days after transfection supernatant was harvested and purified on an AKTA Avant (GE Healthcare). The supernatant was loaded on MabSelect Sure LX (GE Healthcare) column, washed with Phosphate Buffered Saline (PBS), eluted in 100 mM pH 3.6 sodium citrate buffer, neutralized, and dialyzed into PBS. The bispecific quality was examined by intact LC-MS after PNGase F (Prozyme or New Englab Biolabs) deglycosylation. Control KLH/CD3 bsAb was generated following the same protocol as described above.
Gene expression analysis
Gene expression analysis was performed as previously described, using datasets GSE111921 (33).
Mice
All experiments were performed under the approval of Mayo Foundation Institutional Animal Care and Use Committee (IACUC) approval and conformed to all the regulatory Environmental Safety standards. The generation and initial characterization of the Vk*MYC mice (Tg(Igkv3-5*-MYC)#Plbe) and derived transplantable lines has been reported elsewhere (26–28). The original Vk*MYC mice RRID:MMRC_68098_MU) and their derivative lacking LoxP sites, Vk*MYCDLox (RRID: MMRRC_068099-MU) have been deposited to the MMRRC repository, Human CRBN transgenic mice were generated at the Mayo Clinic by microinjection into C57BL/6J pronuclei two independent bacterial artificial chromosomes (BAC): CTD-2335G16 spanning the entire hCRBN gene and RP11-1042H15 extending to include additional regulatory regions. Two founder lines were obtained: hC123 from the former BAC and hC343 from the latter, both capable of germline transmission, detectable by PCR on tail DNA using primers: TAAAGGTGCAGCATGCCAAAC and AGAGCCATTCTGTGTGCATCA. Both founder lines were independently bred into murine CRBNnull background (39) and to Vk*MYC or Vk*MYCDLox mice to generate Vk*MYChCRBN mice. De novo mice were aged and monitor for tumor burden by serum protein electrophoresis as previously described (33). M-spikes were quantified by calculating the ratio of densitometric values of the M-spike and albumin bands (G/A), assuming albumin at a standard value of 27 g/L (49). Transplantation details are provided in Supplemental methods. Transplanted mice were enrolled in drug studies when their M-spike was detected with a G/A >0.1 depending on the aggressiveness of the line. Mice were randomized to different treatment arms, stratified by the size of their M-spikes. Unless specified, treatment duration was 12 days.
Detection of sBCMA
Soluble BCMA was detected in the serum of VK*MYC collected from moribund mice at the time of necropsy and from age matched WT mice using the Mouse BCMA DuoSet ELISA kit (R&D System) using manufacturer’s instruction and diluting Vk*MYC sera 1:500.
Flow cytometry
Single cell suspensions of cell lines or ACK-lysed tissue preparations from necropsy of mice were stained using the antibodies listed in the supplemental methods. Samples were analyzed using a five-laser CytoFlex LX (Beckman Coulter).
FCM based killing assay to monitor redirected killing of myeloma cells with CD3-BCMA antibody
We isolated splenocytes from mice aged 8-12 weeks old and lysed RBCs with ACK buffer. Then we stained splenocytes with cell trace violet or CFSE following manufacturer’s instructions (Molecular Probes). We used J558, J558-BCMA or VkMYCVITRO cell lines as targets which we harvested from exponential growing in vitro cultures and stained with SNARF (Molecular probes) following manufacturer’s instructions or anti-CD138. We diluted and titrated antibodies and drugs in complete T cell medium. The splenocytes, target cells, and antibodies were added together in a round bottom 96-well plate and cultured in a humidified incubator at 37C for 72 hours then harvested for FCM analysis. In experiments with purified T cells, T cells were isolated from splenocytes using a bead-based T cell negative selection kit (Stemcell Technologies).
Supplementary Material
Statement of Significance.
BCMA-targeted therapy induces deep but transient clinical responses. We developed an immunocompetent IMiD-sensitive GEMM, and show that IMiDs potentiate T cell activation increasing short-term efficacy of anti-BCMA/CD3 BsAb but exacerbate T cell exhaustion. Suprisingly, by reducing tumor burden and depleting regulatory T cells, cyclophosphamide prevents BsAb-induced T cell exhaustion and promotes long-term MM control.
Acknowledgements
This work was supported by grants from the National Institute of Health (RO1CA234181, U54CA224018 and CA186781). We are grateful to Dr. Anjali Rajadhyaksha for sharing the Crbn knock out mice, which were shipped to us by Dr. Perlie Epling-Burnette. We would like to thank Dr. Isaac Boss for helpful discussion, as well as Sochilt Brown, Kennedy Todd and Zachary Hammond for experimental assistance. Experimental schemes created with BioRender.com.
Footnotes
Declaration of Interests
M.C and P.L.B have rights to the intellectual property of Vk*MYC, hCRBN, Vk*MYChCRBN mice and derivative transplantable lines and received royalties from the licensing of hCRBN mice. N.B. was an employee of Bristol-Myers Squibb. M.L.W, B.C, F.W, and P.S are currently employed by Bristol Myers Squibb.
References
- 1.Chesi M, Fonseca R. Antibodies Create Killer Bonds in Myeloma. Cancer Cell 2017;31(3):305–7 doi 10.1016/j.ccell.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Shah N, Aiello J, Avigan DE, Berdeja JG, Borrello IM, Chari A, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of multiple myeloma. J Immunother Cancer 2020;8(2) doi 10.1136/jitc-2020-000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costa LJ, Wong SW, Bermúdez A, de la Rubia J, Mateos M- V, Ocio EM, et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019;134(Supplement_1):143- doi 10.1182/blood-2019-122895. [DOI] [Google Scholar]
- 4.Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Evaluation of AMG 420, an anti-BCMA bispecific T-cell engager (BiTE) immunotherapy, in R/R multiple myeloma (MM) patients: Updated results of a first-in-human (FIH) phase I dose escalation study. Journal of Clinical Oncology 2019;37(15_suppl):8007- doi 10.1200/JCO.2019.37.15_suppl.8007. [DOI] [Google Scholar]
- 5.Garfall AL, Usmani SZ, Mateos M-V, Nahi H, van de Donk NWCJ, San-Miguel JF, et al. Updated Phase 1 Results of Teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020;136(Supplement 1):27- doi 10.1182/blood-2020-138831. [DOI] [Google Scholar]
- 6.Harrison SJ, Minnema MC, Lee HC, Spencer A, Kapoor P, Madduri D, et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (bispecific T-cell engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020;136(Supplement 1):28–9 doi 10.1182/blood-2020-134063. [DOI] [Google Scholar]
- 7.Lesokhin AM, Levy MY, Dalovisio AP, Bahlis NJ, Solh M, Sebag M, et al. Preliminary Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of Subcutaneously (SC) Administered PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020;136(Supplement 1):8–9 doi 10.1182/blood-2020-133355.32614959 [DOI] [Google Scholar]
- 8.Rodriguez C, D’Souza A, Shah N, Voorhees PM, Buelow B, Vij R, et al. Initial Results of a Phase I Study of TNB-383B, a BCMA × CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020;136(Supplement 1):43–4 doi 10.1182/blood-2020-139893. [DOI] [Google Scholar]
- 9.Gorgun GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013;121(15):2975–87 doi 10.1182/blood-2012-08-448548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura K, Kassem S, Cleynen A, Chretien ML, Guillerey C, Putz EM, et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018;33(4):634–48 e5 doi 10.1016/j.ccell.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood 2018;132(16):1689–94 doi 10.1182/blood-2018-01-825265. [DOI] [PubMed] [Google Scholar]
- 12.Kawano Y, Zavidij O, Park J, Moschetta M, Kokubun K, Mouhieddine TH, et al. Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J Clin Invest 2018;128(6):2487–99 doi 10.1172/JCI88169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J Clin Oncol 2020;38(8):775–83 doi 10.1200/JCO.19.02657. [DOI] [PubMed] [Google Scholar]
- 14.Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 2017;31(10):2181–90 doi 10.1038/leu.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feucht J, Kayser S, Gorodezki D, Hamieh M, Doring M, Blaeschke F, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016;7(47):76902–19 doi 10.18632/oncotarget.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swan D, Gurney M, Krawczyk J, Ryan AE, O’Dwyer M. Beyond DNA Damage: Exploring the Immunomodulatory Effects of Cyclophosphamide in Multiple Myeloma. Hemasphere 2020;4(2):e350 doi 10.1097/HS9.0000000000000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother 2009;58(7):1033–45 doi 10.1007/s00262-008-0620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haslett PA, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med 1998;187(11):1885–92 doi 10.1084/jem.187.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Walter M, Urak R, Weng L, Huynh C, Lim L, et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin Cancer Res 2018;24(1):106–19 doi 10.1158/1078-0432.CCR-17-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Works M, Soni N, Hauskins C, Sierra C, Baturevych A, Jones JC, et al. Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T cell Function against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol Cancer Ther 2019;18(12):2246–57 doi 10.1158/1535-7163.MCT-18-1146. [DOI] [PubMed] [Google Scholar]
- 21.Gormley NJ, Pazdur R. Immunotherapy Combinations in Multiple Myeloma-Known Unknowns. N Engl J Med 2018;379(19):1791–5 doi 10.1056/NEJMp1803602. [DOI] [PubMed] [Google Scholar]
- 22.Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol 2014;21(9):803–9 doi 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- 23.Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014;512(7512):49–53 doi 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kronke J, Hurst SN, Ebert BL. Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 2014;3(7):e941742 doi 10.4161/21624011.2014.941742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parman T, Wiley MJ, Wells PG. Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nat Med 1999;5(5):582–5 doi 10.1038/8466. [DOI] [PubMed] [Google Scholar]
- 26.Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012;120(2):376–85 doi 10.1182/blood-2012-02-412783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008;13(2):167–80 doi 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med 2016;22(12):1411–20 doi 10.1038/nm.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calcinotto A, Brevi A, Chesi M, Ferrarese R, Garcia Perez L, Grioni M, et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nature communications 2018;9(1):4832 doi 10.1038/s41467-018-07305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest 2015;125(7):2904 doi 10.1172/JCI82646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guillerey C, Nakamura K, Pichler AC, Barkauskas D, Krumeich S, Stannard K, et al. Chemotherapy followed by anti-CD137 mAb immunotherapy improves disease control in a mouse myeloma model. JCI Insight 2019;5 doi 10.1172/jci.insight.125932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vuckovic S, Minnie SA, Smith D, Gartlan KH, Watkins TS, Markey KA, et al. Bone marrow transplantation generates T cell-dependent control of myeloma in mice. J Clin Invest 2019;129(1):106–21 doi 10.1172/JCI98888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chesi M, Stein CK, Garbitt VM, Sharik ME, Asmann YW, Bergsagel M, et al. Monosomic loss of MIR15A/MIR16-1 is a driver of multiple myeloma proliferation and disease progression. Blood Cancer Discov 2020;1(1):68–81 doi 10.1158/0008-5472.bcd-19-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nature communications 2015;6:7333 doi 10.1038/ncomms8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghermezi M, Li M, Vardanyan S, Harutyunyan NM, Gottlieb J, Berenson A, et al. Serum B-cell maturation antigen: a novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017;102(4):785–95 doi 10.3324/haematol.2016.150896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang F, Tsai JC, Davis JH, Chau B, Dong J, West SM, et al. Design and characterization of mouse IgG1 and IgG2a bispecific antibodies for use in syngeneic models. MAbs 2020;12(1):1685350 doi 10.1080/19420862.2019.1685350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol 2002;168(4):1528–32 doi 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 38.Cooke RE, Gherardin NA, Harrison SJ, Quach H, Godfrey DI, Prince M, et al. Spontaneous onset and transplant models of the Vk*MYC mouse show immunological sequelae comparable to human multiple myeloma. J Transl Med 2016;14:259 doi 10.1186/s12967-016-0994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajadhyaksha AM, Ra S, Kishinevsky S, Lee AS, Romanienko P, DuBoff M, et al. Behavioral characterization of cereblon forebrain-specific conditional null mice: a model for human non-syndromic intellectual disability. Behav Brain Res 2012;226(2):428–34 doi 10.1016/j.bbr.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matyskiela ME, Zhang W, Man HW, Muller G, Khambatta G, Baculi F, et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J Med Chem 2017. doi 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- 41.Fink EC, McConkey M, Adams DN, Haldar SD, Kennedy JA, Guirguis AA, et al. Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 2018;132(14):1535–44 doi 10.1182/blood-2018-05-852798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011;118(18):4771–9 doi 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15(8):486–99 doi 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol 2013;14(11):1155–65 doi 10.1038/ni.2710. [DOI] [PubMed] [Google Scholar]
- 45.Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, et al. Asymmetric PI3K Signaling Driving Developmental and Regenerative Cell Fate Bifurcation. Cell Rep 2015;13(10):2203–18 doi 10.1016/j.celrep.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 2009;15(7):808–13 doi 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci U S A 2010;107(21):9777–82 doi 10.1073/pnas.0914127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010;33(2):229–40 doi 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaias J, Mineau M, Cray C, Yoon D, Altman NH. Reference values for serum proteins of common laboratory rodent strains. J Am Assoc Lab Anim Sci 2009;48(4):387–90. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.