

Abstract
Microphthalmia, coloboma, and aniridia are congenital ocular phenotypes with a strong genetic component but often unknown cause. We present a likely causative novel variant in MAB21L1, c.152G>T p.(Arg51Leu), in two family members with microphthalmia and aniridia, as well as novel or rare compound heterozygous variants of uncertain significance, c.184C>T p.(Arg62Cys)/c.-68T>C, and c.658G>C p.(Gly220Arg)/c.*529A>G, in two additional probands with microphthalmia, coloboma and/or cataracts. All variants were predicted damaging by in silico programs. In vitro studies of coding variants revealed normal subcellular localization but variable stability for the corresponding mutant proteins. In vivo complementation assays using the zebrafish mab21l2Q48Sfs*5 loss-of-function line demonstrated that while overexpression of wild-type MAB21L1 mRNA compensated for the loss of mab21l2, none of the coding variant mRNAs produced a statistically significant rescue, with p.(Arg51Leu) showing the highest degree of functional deficiency. Dominant variants in a close homolog of MAB21L1, MAB21L2, have been associated with microphthalmia and/or coloboma and repeatedly involved the same Arg51 residue, further supporting its pathogenicity. The possible role of p.(Arg62Cys) and p.(Gly220Arg) in microphthalmia is similarly supported by the observed functional defects, with or without an additional impact from non-coding MAB21L1 variants identified in each patient. This study suggests a broader spectrum of MAB21L1-associated disease.
Keywords: MAB21L1, aniridia, microphthalmia, coloboma, rescue
INTRODUCTION
Developmental ocular disorders have complex genetic etiologies due to the intricate and tightly controlled genetic networks involved in eye development (Skalicky et al., 2013). Microphthalmia, anophthalmia and coloboma (MAC) are rare congenital malformations of the eye involving a small eye, absence of an eye, and gap in ocular structures, respectively (Gregory-Evans, Williams, Halford, & Gregory-Evans, 2004; Verma & Fitzpatrick, 2007). Over 80 genes have been published in association with MAC phenotypes (Reis & Semina, 2015). However, about 50% of patients lack a confirmed genetic diagnosis, suggesting novel genes have yet to be discovered (Plaisancie, Calvas, & Chassaing, 2016).
Aniridia is a panocular disorder with its primary feature being partial or complete absence of the iris, but also including lens opacities, glaucoma, keratopathy, foveal and optic nerve hypoplasia, strabismus, ptosis, and fibrosis syndrome (Hall, Williamson, & FitzPatrick, 2019; Hingorani, Hanson, & van Heyningen, 2012; Lim, Kim, & Kim, 2017). Up to 90% of cases with aniridia can be explained by loss-of-function mutations in the PAX6 gene (Hingorani et al., 2012); rarely, disruption of FOXC1, PITX2, and other genes have been identified as causative (Hall et al., 2019; Hingorani et al., 2012). However, there is a small portion of the aniridia population that remains genetically unexplained.
The Male-Abnormal 21-Like gene MAB21L2 is a recently identified factor involved in human MAC-spectrum disorders, where both dominant and recessive missense alleles have been recognized as causative in eight unrelated families (Aubert-Mucca et al., 2020; Deml et al., 2015; Horn et al., 2015; Patel et al., 2018; Rainger et al., 2014). In four out of seven dominant families, the pathogenic variant is a missense allele affecting residue 51 of the resulting protein. Similarly, a mouse Mab21l2 model with a heterozygous p.(Arg51Cys) mutant allele (identical to two of the affected human patients (Horn et al., 2015; Rainger et al., 2014)) further demonstrated the importance of this residue, resulting in defects in early ocular development including rudimentary and mispositioned optic cup, undetectable optic stalk, abnormalities of the retinal pigment epithelium, and failure to induce a lens placode (Tsang, Guo, Chan, Huang, & Chow, 2018). Additional null/loss-of-function animal models have displayed MAC-spectrum defects in mice (Yamada, Mizutani-Koseki, Koseki, & Takahashi, 2004) and zebrafish (Deml et al., 2015; Gath & Gross, 2019; Hartsock, Lee, Arnold, & Gross, 2014; Wycliffe et al., 2020).
MAB21L1, a closely related family member to MAB21L2, has also been implicated in human disease. Homozygous MAB21L1 variants have been reported in six unrelated families exhibiting cerebello-oculo-facio-genital syndrome, with five of these variants resulting in premature truncation of the protein (Bruel et al., 2017; Rad et al., 2019). Ocular abnormalities included corneal dystrophy/opacities, nystagmus, strabismus, dry eye, pigment granularity, retinal degeneration, optic atrophy, buphthalmos and cataracts (Bruel et al., 2017; Rad et al., 2019). A null mouse model for Mab21l1 likewise exhibits embryonic ocular defects, though more severe than the published human phenotype. Abnormalities include microphthalmia, malformed retina and retinal pigment epithelium, along with aphakia, thickened cornea and absent iris (Yamada et al., 2003).
The precise protein function(s) of the MAB21L family are unknown. A possible role in transcriptional regulation has been suggested (Baldessari, Badaloni, Longhi, Zappavigna, & Consalez, 2004) with nuclear localization (Mariani et al., 1999) and a mild affinity for nucleic acid shown in vitro (de Oliveira Mann, Kiefersauer, Witte, & Hopfner, 2016; Rainger et al., 2014).
Here we report three families with unique MAB21L1 variants exhibiting ocular phenotypes including MAC-spectrum and aniridia. Functional studies of the proteins associated with coding variants revealed differences from wild-type MAB21L1. Thus, this study suggests a phenotypic expansion for MAB21L1-associated human disease.
MATERIALS AND METHODS
Editorial Policies and Ethical Considerations
Human studies conformed to the US Federal Policy for the Protection of Human Subjects and were approved by the Children’s Hospital of Wisconsin Institutional Review Board and the Sydney Children’s Hospitals Network Research Ethics Committee, with written informed consent obtained from all participating individuals and/or their legal representatives.
Human DNA screening and in silico variant analyses
The MAB21L1 variant in Individual 1 was initially identified through clinical exome sequencing using the previously published protocol (Guillen Sacoto et al., 2020) and matched to the study through the Matchmaker Exchange GeneMatcher node (Philippakis et al., 2015; Sobreira, Schiettecatte, Valle, & Hamosh, 2015) followed by enrollment and research exome sequencing and analysis; the MAB21L1 variants in Individuals 2 and 3 were identified through research exome sequencing and analysis using previously described methods (Deml et al., 2016; Ma et al., 2020). Variants in known ocular genes were ruled out in all three families. All variants were confirmed via Sanger sequencing of the MAB21L1 coding region by amplification of a 1405-bp product using the flanking primers F-5’-CCGAAAGGCATTTTTGATCC-3’, R-5’-TCCGCTTCCCCTACTTTTTC-3’ and also internal primers F-5’-AGATCACGCCGGCCTTTA-3’, R-5’-ACCCAGGCGTCGCTCTC-3’. PCR amplicons were sequenced as previously described (Deml et al., 2015) or through Functional Biosciences™ DNA Sequencing Services (Madison, WI, USA). Parental samples were analyzed using the same protocol. To determine relative positions of variants identified in Individual 2, the amplified 1405-bp product was cloned into pCR®II-TOPO® plasmid, which was followed by sequencing of 32 independent clones; 18 clones contained c.184C>T p.(Arg62Cys) allele and wild-type 5’ UTR sequence and 14 clones contained c.-68T>C 5’ UTR variant and wild-type coding region sequence indicating their trans configuration.
The following information was collected for the identified variants: frequency in the general ethnically matched population using gnomAD (Karczewski et al., 2020), predicted effect on protein function and amino acid conservation (via dbNSFP (X. Liu, Jian, & Boerwinkle, 2013) accessed through Ensembl Variant Effect Predictor (VEP) (McLaren et al., 2016)) and nucleotide conservation (using UCSC Genome Browser (Kent et al., 2002)) with results obtained for the following tools- SIFT (Sim et al., 2012), Polyphen2 (Adzhubei et al., 2010), MutationTaster (Schwarz, Cooper, Schuelke, & Seelow, 2014), MutationAssessor (Reva, Antipin, & Sander, 2011), FATHMM-MKL (Shihab et al., 2014), CADD PHRED (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019), REVEL (Ioannidis et al., 2016), GERP++RS (Davydov et al., 2010) and PhyloP100wayAll (Pollard, Hubisz, Rosenbloom, & Siepel, 2010). Reported score ranges and pathogenic criteria for the above programs are as follows: for CADD PHRED, scores over 20 and 30 have been found to be the top 1% and 0.1% most damaging variants in the genome, respectively (Kircher et al., 2014); for REVEL, the range is 0 to 1 with 75.4% of disease-causing variants having a score > 0.5 (Ioannidis et al., 2016)); for nucleotide conservation, GERP++RS scores range from −12.3 to 6.17 (max, and most conserved) and PhyloP100way vertebrate scores range −20.0 to 10.003 (max, and most conserved). To highlight conservation, nucleotide alignments were generated using UCSC Multiz Alignments of 100 Vertebrates; protein alignments were generated with Kalign multiple sequence alignment tool using the following sequences: human MAB21L1 (NP_005575.1), human MAB21L2 (NP_006430.1), mouse Mab21l1 (NP_034880.1), chicken Mab21l1 (NP_989864.1), zebrafish mab21l1 (NP_694506.2) and C. elegans mab-21 (NP_497940.2).
Additional in silico analyses were performed for the MAB21L1 UTR variants (c.-68T>C and c.*529A>G) to assess possible functional impacts. To determine changes to minimum free energy and RNA secondary structure, both 5’ UTR and 3’ UTR DNA sequences carrying wild-type or variant alleles were submitted to the RNAfold 2.4.17 Webserver, accessed through The Vienna RNA Websuite 2.0 (Lorenz et al., 2011). To assess changes to microRNA target prediction, wild-type and variant 3’ UTR sequence was submitted to MicroSNiPer (Barenboim, Zoltick, Guo, & Weinberger, 2010) with a minimum seed length constraint of 7 base pairs. In addition, the PolymiRTS Database 3.0, a database of human SNPs affecting predicted mRNA target sites (Bhattacharya, Ziebarth, & Cui, 2014), was searched for the 3’ UTR variant, rs1775984, to determine overlapping predicted miRNA sites. PolymiRTS calculated the strength of the predicted miRNA site and provided a conservation score, which was determined based on the number of vertebrate genomes in which the miRNA site is present, and context+ score change (a ranking of miRNA target predictions) (Garcia et al., 2011). To determine variant effects on RNA binding protein (RBP) binding sites, wild-type and variant 5’ UTR and 3’ UTR sequences were submitted to RBPmap version 1.1 (Paz, Kosti, Ares, Cline, & Mandel-Gutfreund, 2014) to identify RBP motifs. Predicted motifs containing the affected nucleotide were assessed for lost/gained interactions between wild-type or mutant sequence and RBPs.
In silico protein modeling was executed for the MAB21L1 wild-type and MAB21L1-p.(Arg51Leu), MAB21L1-p.(Arg62Cys) and MAB21L1-p.(Gly220Arg) proteins. The wild-type MAB21L1 crystal structure has been previously solved and thus the full-length MAB21L1 structure pdb file (PDB ID: 5EOG) (de Oliveira Mann et al., 2016) was utilized (RCSB Protein Data Bank). To predict changes to the structure invoked by the three missense mutations, the altered protein sequence was submitted to I-TASSER (Yang & Zhang, 2015) and the corresponding pdb file downloaded (predicted model #1). Files were uploaded into PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC) where labeled images of protein structures were created. Alpha helices and beta sheets were named as previously described (de Oliveira Mann et al., 2016).
Western blot, immunofluorescence and ELISA experiments
For protein expression experiments, MAB21L1 wild-type and mutant constructs for the p.(Arg51Leu), p.(Arg62Cys) and p.(Gly220Arg) were developed. To do so, an N-terminal FLAG-tagged human MAB21L1 (NM_005584.4) clone in a pEZ-M11 vector was obtained (GeneCopoeia™, Rockville, MD, USA). To generate mutants, site-directed mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). HPLC purified primers were as follows: for c.152G>T p.(Arg51Leu), s-5’-GAGAGCTGATGAACAGCGGCTCCTGCACT-3’, as-5’-AGTGCAGGAGCCGCTGTTCATCAGCTCTC-3’; for c.184C>T p.(Arg62Cys), s-5’-GCCCTCGTAGCAATTGTCCATCTCGTTGAGAGA-3’, as-5’-TCTCTCAACGAGATGGACAATTGCTACGAGGGC-3’; and for c.658G>C p.(Gly220Arg), s-5’-GAGCTCTGCTTGCGGGCCAAGGAGTGG-3’, as-5’-CCACTCCTTGGCCCGCAAGCAGAGCTC-3’. Transformed colonies were selected and plasmids isolated using Invitrogen™ PureLink™ Quick Plasmid Miniprep Kit (ThermoFisher Scientific, Waltham, MA, USA) and sequenced with the following primers: F-5’-CAGCCTCCGGACTCTAGC-3’, R-5’-TAATACGACTCACTATAGGG-3’.
Then, 2.5μg of N-terminal FLAG-tagged MAB21L1 wild-type and mutant constructs, were transfected into Human Lens Epithelial (HLE-B3) cells (ATCC®, Manassas, VA, USA) using Invitrogen™ Lipofectamine™ 2000 Transfection Reagent (ThermoFisher Scientific) in Opti-MEM (ThermoFisher Scientific). Cells were cultured in Gibco™ Minimal Essential Medium (MEM) (ThermoFisher Scientific) with 20% Fetal Bovine Serum (Millipore Sigma, St. Louis, MO, USA), 1X L-glutamine (ThermoFisher Scientific) and 1X Sodium Pyruvate (ThermoFisher Scientific). 48 hours post-transfection, cells were collected in 1X Phosphate Buffered Saline (PBS). To obtain whole-cell lysates for Western blot and ELISA analysis, pelleted cells were resuspended in 1% Triton™ X-100 (Millipore Sigma, Burlington, MA, USA) with 100X protease inhibitor (Millipore Sigma).
For Western blot, samples were denatured by adding 4X Laemmli sample buffer (Bio-Rad Laboratories, Inc., Hercules, CA, USA), boiled at 95°C for 5 minutes, and run on a 10% Criterion™ Tris-HCl Precast Gel (Bio-Rad Laboratories, Inc.). Membranes were incubated with mouse 1:1000 anti-FLAG (Millipore Sigma) or 1:1000 rabbit anti-βactin for normalization (GeneTex, Irvine, CA, USA) overnight, and the following day with corresponding secondary antibody, either 1:2000 goat anti-mouse HRP conjugate (ThermoFisher Scientific) or 1:2000 goat anti-rabbit HRP conjugate (ThermoFisher Scientific). For detection, SuperSignal™ West Pico or Femto Maximum Sensitivity chemiluminescent substrates were used (ThermoFisher Scientific).
For ELISA semi-quantitative protein expression analysis, a DYKDDDDK-Tag Detection ELISA Kit (Cayman Chemical, Ann Arbor, Michigan, USA) was used. Biological samples (from 3 separate transfections) were run in duplicate. Data was analyzed using elisaanalysis.com with a 4-parameter regression analysis and plotted in GraphPad Prism 9 (GraphPad, San Diego, CA, USA). Statistical significance was determined using an unpaired samples t-test and a P value < 0.05.
Immunofluorescence experiments were used to determine protein localization. The transfection protocol was as described above with 7.5μg wild-type and mutant N-terminally tagged MAB21L1 constructs into HLE-B3 cells. Cells were fixed with 1:1 methanol/acetone permeabilized with 1% Triton X-100 and blocked with 10% donkey serum in 1X PBS, followed by an overnight incubation with 1:100 mouse anti-FLAG primary antibody at 4°C. The next day cells were incubated with 1:1000 donkey anti-mouse Alexa Fluor 488 secondary antibody (ThermoFisher Scientific) and stained with Invitrogen™ 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) (ThermoFisher Scientific).
Animal Care and Use
The care and use of zebrafish has been approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin in compliance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals, the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals, and Guide for the Care and Use of Laboratory Animals. All breeding and housing were conducted as previously described (Y. Liu & Semina, 2012). Developmental stages were determined as previously described by hours post fertilization (hpf) and morphology (Kimmel, Ballard, Kimmel, Ullmann, & Schilling, 1995).
RNA Complementation Assay
For mRNA complementation assays, MAB21L1 wild-type and mutant (p.(Arg51Leu), p.(Arg62Cys) and p.(Gly220Arg)) constructs were developed. To do so, a human MAB21L1 (NM_005584.4) clone in a pCR®II-TOPO® vector was used to generate mutants by performing site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies). HPLC purified primers for the MAB21L1 mutant constructs are as described above. Transformed colonies were selected, plasmids isolated using Invitrogen™ PureLink™ Quick Plasmid Miniprep Kit (ThermoFisher Scientific) and sequenced with the following primers: F-5’- GTAAAACGACGGCCAG -3’, R-5’-CAGGAAACAGCTATGAC-3’. Plasmids were then linearized with the restriction enzyme HindIII (Millipore Sigma) and DNA clean and concentrate kit was used as needed (ZymoResearch, Irvine, CA, USA). mRNA was synthesized from the linearized plasmid using the Invitrogen™ mMESSAGE mMACHINE™ T7 Transcription Kit (ThermoFisher Scientific) and the Invitrogen™ Poly(A) Tailing Kit (ThermoFisher Scientific). mRNA was purified using an RNA Clean & Concentrator Kit (ZymoResearch) or phenol-chloroform purification. Concentration and purity were measured using a NanoDrop™ 1000 Spectrophotometer (ThermoFisher Scientific) and integrity was assessed with agarose gel electrophoresis. 400pg of wild-type or mutant mRNA was injected using a Drummond Nanoject II instrument (Drummond Scientific, Broomall, PA, USA), into embryos from a mab21l2 c.141_153del p.(Gln48Serfs*5) heterozygous cross (Deml et al., 2015). At 24-hpf, when the mutant phenotype is clearly observable in homozygous mab21l2Q48rfs*5 embryos, injected offspring were examined for the presence or absence of the previously described ocular phenotype and the proportion of normal embryos was determined. Graphs were created using GraphPad Prism 9. Statistical significance was determined using an unpaired samples t-test and a P value of < 0.05.
RESULTS
Identification of MAB21L1 variants in three families with congenital ocular disease
Examination of exome sequencing data in genetically unexplained families with MAC phenotypes identified several new variants of interest within the MAB21L1 gene, including a likely causative heterozygous missense variant in one family with dominant transmission (Individuals 1A and 1B) and four additional variants of uncertain significance in two unrelated families, with one coding (missense) and one non-coding allele in trans in each affected proband.
Individual 1A is a 3-year-old female child from the Dominican Republic (Black/Hispanic ancestry) with bilateral microphthalmia, aniridia, microcornea, microspherophakia, and nystagmus; she is otherwise non-dysmorphic and developmentally normal. Clinical sequencing and deletion/duplication analysis of PAX6, FOXC1, PITX2 and CYP1B1, as well as a clinical microphthalmia/anophthalmia gene panel did not reveal causative variants. Whole exome sequencing identified a heterozygous missense variant in MAB21L1, c.152G>T p.(Arg51Leu) (Figure 1, Table 1) and repeat research exome analysis confirmed this and did not identify any other pathogenic variants. The variant was predicted to be damaging by 5/5 programs and combined scores from REVEL (0.682) and CADD PHRED (29.6) also supported pathogenicity (Table 1). The arginine residue at position 51 is highly conserved with a GERP++RS score of 5.66 and a PhyloP score of 7.78; the identified variant is novel (not present in ~250,000 alleles in the general population). Family history revealed that her father (Individual 1B) also exhibits bilateral microphthalmia, aniridia, ectopia lentis, and microcornea; research exome analysis and Sanger sequencing identified the same heterozygous c.152G>T p.(Arg51Leu) variant in the father and wild-type MAB21L1 alleles in the unaffected mother (Figure 1B). Interestingly, variants affecting the same conserved Arg51 residue in a close homolog of MAB21L1, MAB21L2, are known to cause dominant MAC (Deml et al., 2015; Horn et al., 2015; Rainger et al., 2014), further supporting the causality of the identified MAB21L1 c.152G>T p.(Arg51Leu) allele.
Figure 1. MAB21L1 variant details.
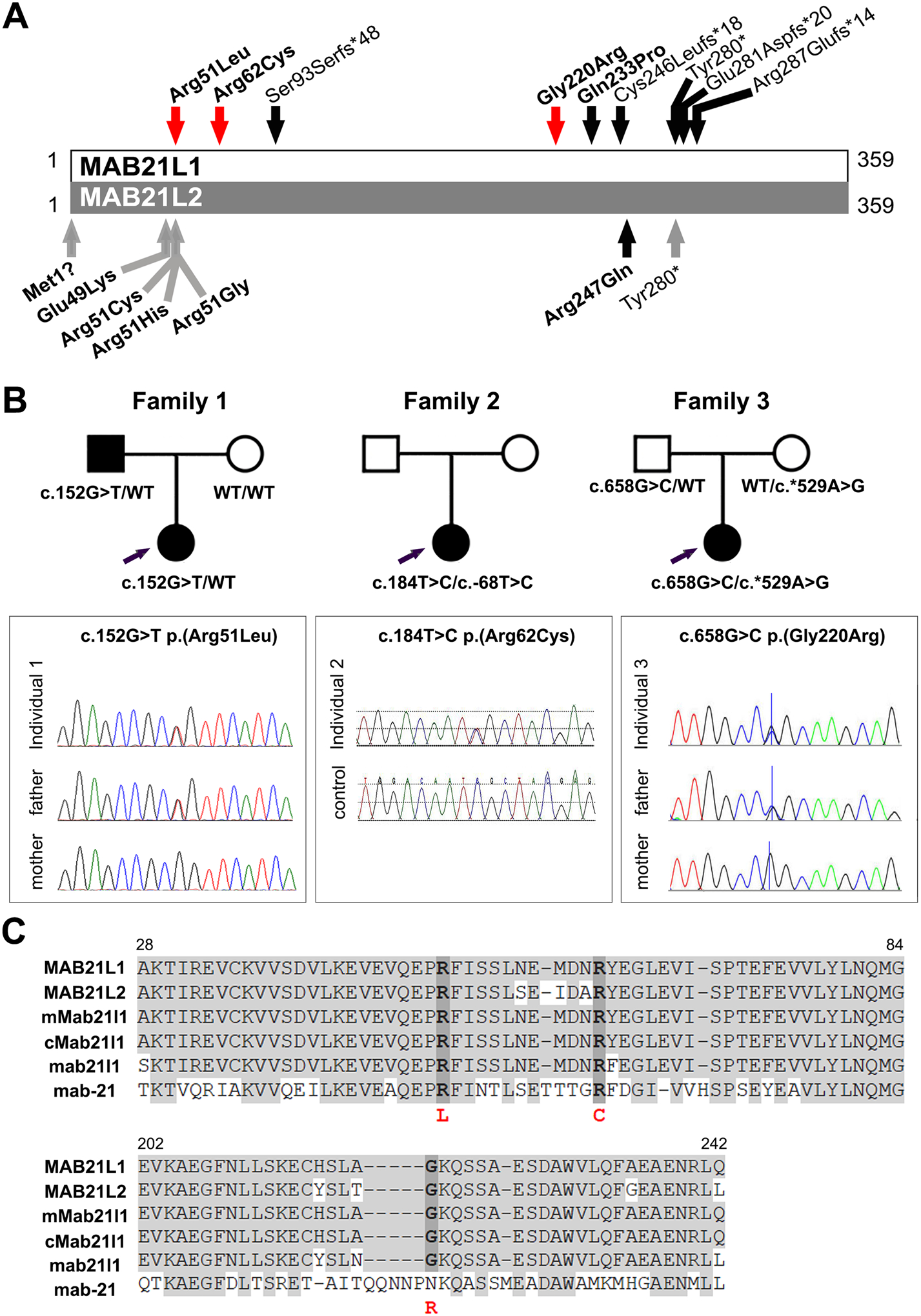
A. Schematic of MAB21L1 (top) and MAB21L2 (bottom) proteins. Newly identified heterozygous MAB21L1 coding variants are indicated by a red arrow; previously reported MAB21L1 and MAB21L2 recessive variants are indicated with a black arrow; previously reported MAB21L2 dominant variants are indicated with a gray arrow. Missense variants are bolded. B. Pedigrees for Families 1, 2 and 3 indicating MAB21L1 genotype and Sanger sequencing traces for the identified coding variants. C. Alignment of MAB21L1 and related proteins showing conservation at and around the Arg51Leu, Arg62Cys and Gly220Arg variants. Identical amino acids are shaded in grey; positions of variant amino acids are indicated with dark grey. Human MAB21L1 (NP_005575.1), human MAB21L2 (NP_006430.1), mouse Mab21l1 (NP_034880.1), chicken Mab21l1 (NP_989864.1), zebrafish mab21l1 (NP_694506.2) and C. elegans mab-21 (NP_497940.2) are shown.
Table 1:
Summary of new and previously reported MAB21L1 variants.
| ID | Age; ancestry | DNA changea | Protein change | gnomADb | Predicted Effectc | Phenotype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | PP | MT | MA | FM | REV | CADD | GERP | Phy | RNA fold | Micro | RBP | Eye | Other | ||||||
| HETEROZYGOUS AND COMPOUND HETEROZYGOUS VARIANTS | |||||||||||||||||||
|
Individual 1A (this report) |
3 y; Black/Hispanic | c.152G>T | p.Arg51Leu | 0/~250,000 | D | D | D | D | D | 0.68 | 29.6 | 5.66 | 7.78 | - | - | - | B microphthalmia, microcornea, aniridia, microspherophakia, nystagmus | NR | |
|
Individual 1B (this report) |
25 y; Black/Hispanic | c.152G>T | p.Arg51Leu | 0/~250,000 | D | D | D | D | D | 0.68 | 29.6 | 5.66 | 7.78 | - | - | - | B microphthalmia, microcornea, aniridia, ectopia lentis | NR | |
|
Individual 2 (this report) |
6 m; South Asian | c.184C>T | p.Arg62Cys | 2/30,616 | T | D | D | D | D | 0.59 | 31 | 5.66 | 9.87 | - | - | - | L microphthalmia, coloboma; R cataract | NR | |
| c.-68T>C | - | 0/~31,000 | - | - | - | - | D | - | - | 3.36 | 1.01 | U | - | A | |||||
|
Individual 3 (this report) |
2 y; White | c.658G>C | p.Gly220Arg | 1/112,548 | T | D | D | D | D | 0.45 | 26.2 | 5.76 | 7.80 | - | - | - | B microphthalmia, coloboma | NR | |
| c.*529A>G | - | 715/15,432 | - | - | - | - | D | - | - | 5.24 | 2.20 | U | A | A | |||||
| HOMOZYGOUS VARIANTS | |||||||||||||||||||
| Bruel et al. 2017 | 7 y; Algerian | c.735dup | p.Cys246Leufs | 0/~250,000 | Premature truncation | - | - | - | Corneal dystrophy, buphthalmos, strabismus, nystagmus | FD, SA, GD, CM | |||||||||
| Rad et al. 2019: Fam 1 | 5 y, 7 y, 17 y 7 m; Persian | c.841del | p.Glu281Aspfs | 0/~250,000 | Premature truncation | - | - | - | Corneal dystrophy, strabismus, nystagmus | FD, SA, AL, GD, CM | |||||||||
| Rad et al. 2019: Fam 2 | 26 m; Persian | c.698A>C | p.Gln233Pro | 0/~280,000 | D | T | D | T | D | 0.33 | 25.2 | 5.76 | 8.0 | - | - | - | Corneal dystrophy, nystagmus, retinal dystrophy, optic atrophy | FD, SA, CM | |
| Rad et al. 2019: Fam 3 | 2 y; Lebanese Shia | c.279_286 del | p.Ser93Serfs | 0/~250,000 | Premature truncation | - | - | - | Corneal dystrophy | FD, AL, GD, CM | |||||||||
| Rad et al. 2019: Fam 4 | 18 y 9 m; Turkish | c.859del | p.Arg287Glufs | 0/~250,000 | Premature truncation | - | - | - | Corneal dystrophy, nystagmus | FD, SA, GD, CM | |||||||||
| Rad et al. 2019: Fam 5 | 10 y, 16 y, 6 m, 12 m; Turkish | c.840C>G | p.Tyr280* | 0/~250,000 | Premature truncation | - | - | - | Corneal dystrophy, nystagmus | FD, SA, AL, GD, CM | |||||||||
RefSeq NM_005584.4;
gnomAD frequency for the relevant population is indicated;
programs utilized: PP- PolyPhen; MT- Mutation Taster; MA- Mutation Assessor; FM- FATHMM-MKL; REV-REVEL; CADD- CADD PHRED; GERP- GERP++ RS; Phy- PhyloP; Micro- MicroSNiPer & PolymiRTS; RBP- RBPmap;
A altered; AL absent labia majora; B bilateral; CM cerebellar malformation; D damaging; FD facial dysmorphism; GD global development delay; L left; NR none reported; R right; SA scrotal agenesis; T tolerated; U unaffected ; - not applicable or not assessed.
Individual 2, a 6-month-old South Asian (Indian) female diagnosed with microphthalmia and optic disc coloboma in the left eye and isolated congenital cataract in the right eye, was found to have a heterozygous missense variant in MAB21L1, c.184C>T p.(Arg62Cys) (Figure 1, Table 1). No systemic abnormalities were noted. The amino acid substitution was predicted to be likely damaging by 4/5 prediction programs and had REVEL (0.585) and CADD PHRED (31) scores suggesting pathogenicity (Table 1). Again, the arginine residue at position 62 was found to be highly conserved with a GERP++RS score of 5.66 and a PhyloP score of 9.87 (Table 1). However, the variant was found to be present in the general population, albeit at a very low frequency (2/30,616 alleles, 0.0065% in the ethnically matched South Asian population). Additionally, the patient was found to have a novel (0/~31,000 alleles) heterozygous non-coding variant in the 5’ untranslated region (UTR) of the MAB21L1 gene, c.-68T>C. The patient’s parents were unaffected, but unavailable for further testing; however, the variant alleles were determined to be positioned in trans through PCR amplification, cloning and sequencing of multiple copies of the region encompassing both alleles.
Individual 3 is a 2-year-old white (Australian) female patient diagnosed with bilateral colobomatous microphthalmia and also identified to have a heterozygous missense variant in MAB21L1, c.658G>C p.(Gly220Arg) (Figure 1, Table 1). The amino acid substitution was predicted to be likely damaging by 4/5 programs and had a CADD PHRED score of 26.2, suggesting pathogenicity (Table 1); the REVEL score was 0.45 (~25% of pathogenic variants have a score below 0.5 (Ioannidis et al., 2016)). The glycine residue at position 220 was found to be highly conserved with a GERP++RS score of 5.76 and a PhyloP score of 7.80. The variant was found to be present in 1/112,548 alleles in the ethnically matched European (non-Finnish) population (0.00178%) in gnomAD. Additionally, the patient was found to have a heterozygous non-coding variant in the 3’ UTR of the MAB21L1 gene, c.*529A>G, which is present in 715/15,432 alleles in European (non-Finnish) population (4.6%; 18 homozygotes reported). Both parents are unaffected; examination of parental samples identified the coding variant in the father’s sample and the non-coding allele in the mother, indicating trans configuration for these alleles in the patient.
In silico analysis of coding and non-coding alleles
For in silico modeling of the wild-type and mutant MAB21L1 proteins, iTASSER and Pymol software were utilized (Supp. Figure S1). The wild-type protein structure is described as two-lobed, containing an N-terminal and C-terminal lobe, with an alpha-helix spine (α1) spanning the two; the N-terminal lobe is surface-accessible and contains a subdomain with structural homology to the catalytic nucleotidyltransferase (NTase) core domain of cGAS (de Oliveira Mann et al., 2016). The arginine 51 residue is located just outside of the α1 spine; the arginine 62 residue is located in the same linker region between α1 and β1 and the glycine residue at position 220 is also found in a linker region between β8 and β9.
Comparing the p.(Arg51Leu) predicted structure to the established wild-type, there appears to be a loss of α3 in the N-terminal lobe. For p.(Arg62Cys), the most notable difference was the loss of β8 and β9 within the NTase core subdomain of the N-terminal lobe. There is also a subtle distortion of the α1 spine. Interestingly, previous work in co-crystallizing MAB21L1 with a CTP moiety found that CTP interacted directly with the Arg62 residue in a positively charged pocket, denoted the ‘ligand binding pocket’ (de Oliveira Mann et al., 2016). While no nucleotidyltransferase catalytic activity has been determined for this protein or its family members (de Oliveira Mann et al., 2016; Rainger et al., 2014), changes to this residue and the positively charged side chain may affect its ability to form ligand interactions. Finally, comparing the p.(Gly220Arg) predicted structure to wild-type, there was a gain of a small α-helix between β8 and β9 of the NTase core domain near the N-terminal lobe as well as a downward shift in the position of α8. These identified structural deviations have the possibility to translate to functional effects such as altered interactions.
To investigate non-coding variants, in silico analyses were conducted to assess the effects on the upstream open-reading frame (uORF), RNA secondary structure and minimum free energy, miRNA target sites and RNA binding protein (RBP) motifs. No effect on the uORF (for the c.-68T>C variant) and no secondary structure changes (for either the c.-68T>C or c.*529A>G) were predicted, with little or no effect on minimum free energy (−156.90 kcal/mol for wild-type 5’ UTR, −156.90 kcal/mol for c.-68T>C; −239.64 kcal/mol for wild-type 3’ UTR, −239.94 kcal/mol for c.*529A>G) in comparison to corresponding wild-type sequences. Because miRNAs typically bind to sites within the 3’ UTR, potential target sites were assessed in wild-type and the variant 3’ UTR sequence (Grimson et al., 2007). This revealed several target sites that were either disrupted or created by the c.*529A>G variant (Supp. Table S1). In addition, several RBP motifs in both the 5’ and 3’ UTR were predicted to be affected: c.-68T>C disrupted a potential SRSF3 motif and created a RBM6 motif; c.*529A>G disrupted predicted motifs for HNRNPL, IGF2BP2, RBM41 and SRSF3 RNA binding factors and generated a new sequence expected to bind SRSF5 (Supp. Table S2). These RNA-binding proteins (or their closely related family members) have demonstrated important roles in RNA-regulation (including splicing, export, stability, polyadenylation and translation) (Cao, Mu, & Huang, 2018; Jain, Wordinger, Yorio, & Clark, 2012; Oberdoerffer et al., 2008; Rothrock, House, & Lynch, 2005; Sutherland, Rintala-Maki, White, & Morin, 2005; Twyffels, Gueydan, & Kruys, 2011; Zhong, Wang, Han, Rosenfeld, & Fu, 2009; Zhou et al., 2020); in addition, SRSF3 and HNRNPL showed associations with ocular disease, mainly glaucoma (Jain et al., 2012; Schmitt et al., 2020). Finally, in silico evaluations using FATHMM-MKL predicted deleterious effects and GERP++RS and PhyloP indicated nucleotide conservation for both 5’ and 3’UTRs (Table 1; Supp. Figure S2). Therefore, while the mechanisms remain unclear, it is possible the non-coding variants in Individuals 2 and 3 could be contributing to disease through disruption of miRNA and RBP target sites, and thus regulatory activities upon the MAB21L1 mRNA.
Expression and localization of MAB21L1 wild-type and mutant proteins
The stability and localization of wild-type and mutant proteins were tested by expressing FLAG-tagged WT, p.(Arg51Leu), p.(Arg62Cys) and p.(Gly220Arg) constructs in human lens epithelial cells. To assess the stability of the recombinant protein, ELISA and Western blot were used (Figure 2A, B). Both MAB21L1-p.(Arg62Cys) and p.(Gly220Arg) exhibited reduced protein levels compared with wild-type, while MAB21L1-p.(Arg51Leu) conversely showed an increase in protein level, suggesting increased stability for this mutant. Quantification using ELISA confirmed these observations and determined that p.(Arg62Cys) and p.(Arg51Leu) levels were significantly different from wild-type levels (P=0.012 and P=0.0074, respectively); however, the p.(Gly220Arg) decrease was not statistically significant (P=0.068) (Figure 2B).
Figure 2. Functional analyses of MAB21L1 and variant proteins.
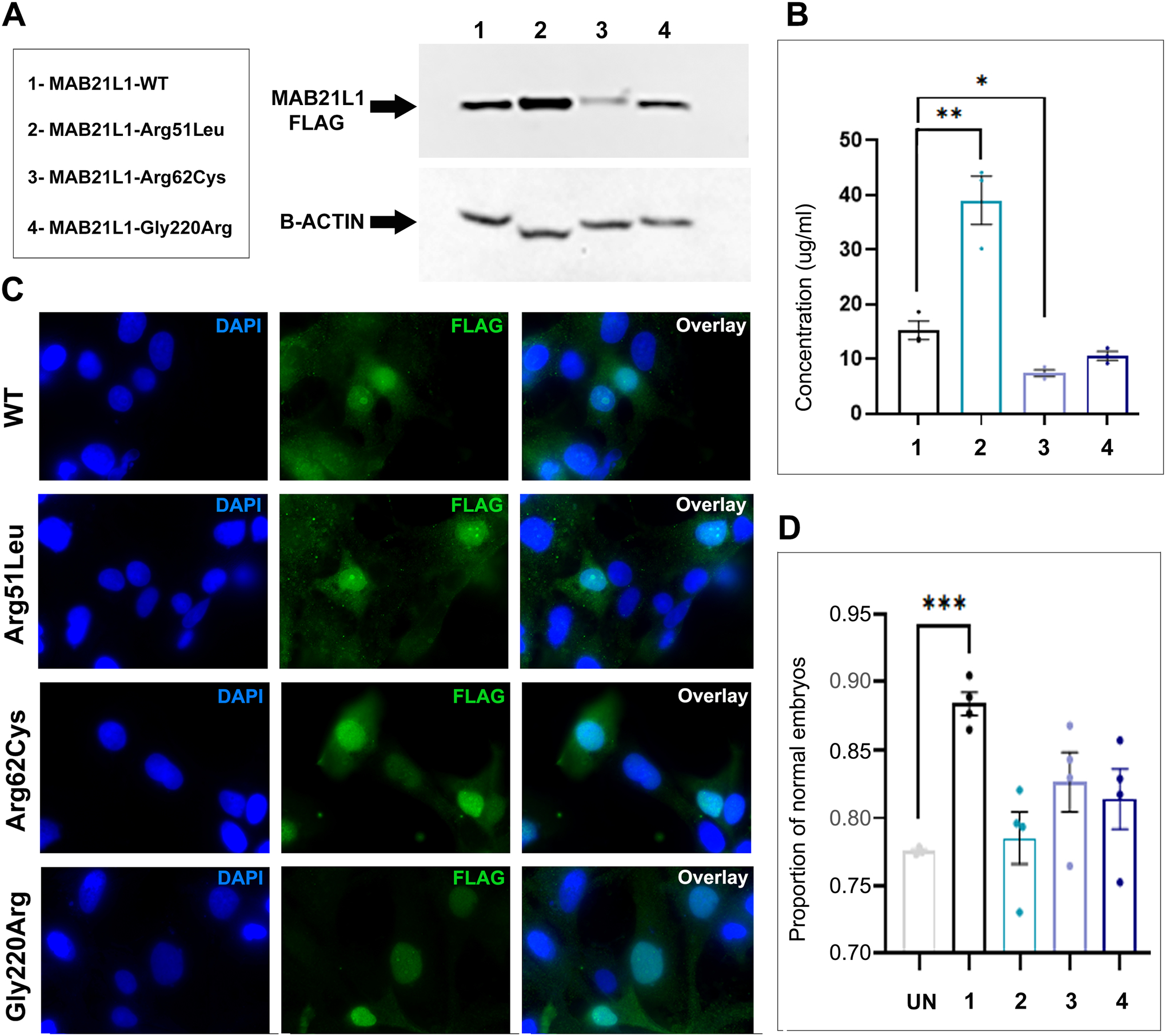
A. Western Blot. Western blot analysis of N-terminal FLAG tagged MAB21L1 wild-type and Arg51Leu, Arg62Cys and Gly220Arg variants. Constructs were expressed in HLE-B3 cells. β-actin was used as a loading control. The proteins correspond to their expected molecular weight (~41kDa MAB21L1 and ~42kDa β-actin). B. ELISA. N-terminal FLAG tagged MAB21L1 wild-type and Arg51Leu, Arg62Cys, and Gly220Arg variants were transfected into HLE-B3 cells. Cell lysates were assessed for FLAG-tagged protein expression; protein levels of Arg51Leu and Arg62Cys were found to be significantly affected. C. Immunocytochemistry. N-terminal FLAG tagged MAB21L1 wild-type and variants were transfected into HLE-B3 cells and stained for FLAG (green) and DAPI (blue; cell nuclei). Wild-type and variant proteins can be found within the cell nucleus, indicating no disruption in localization. D. In vivo complementation assays. Proportion of phenotypically normal embryos at 24-hpf in progeny of heterozygous mab21l2Q48Sfs*5 crosses injected with wild-type or variant Arg51Leu, Arg62Cys or Gly220Arg MAB21L1 mRNA. Statistical significance is indicated by asterisks; * P≤0.05 ** P≤0.01 *** P≤0.001; error bars indicate SEM. UN: uninjected; 1-MAB21L1-WT; 2-MAB21L1-Arg51Leu; 3-MAB21L1-Arg62Cys; 4-MAB21L1-Gly220Arg.
Next, we investigated the localization of the wild-type and mutant protein within the cell. Previous studies have found the Mab21l1 protein localizes to the cell nucleus (Mariani et al., 1999). Similarly, our immunofluorescence experiments showed wild-type MAB21L1 protein within cell nuclei. For all three mutant proteins, no disruption of nuclear localization was seen (Figure 2C). To note, for both wild-type and mutants, staining was detected in the cytoplasm along with the nucleus. The cytoplasmic staining may be a result of overexpression of our transfected constructs or possibly an indication of nuclear-cytoplasmic shuttling.
MAB21L1 wild-type and mutant mRNA complementation assays in zebrafish mab21l2Q48Sfs*5 mutant
To further investigate the effect of the variants, in vivo mRNA complementation assays were performed by injecting mRNA encoding for either MAB21L1 wild-type or the p.(Arg51Leu), p.(Arg62Cys) or p.(Gly220Arg) variant proteins in equal amounts into 1–4-cell zebrafish embryos generated by mab21l2Q48Sfs*5 heterozygous crosses. The mab21l2Q48Sfs*5 line (Deml et al., 2015) was used in this experiment because: 1) homozygous mab21l2-deficient embryos have an obvious and fully-penetrant ocular phenotype (microphthalmia with small/absent lens) beginning at 24-hpf (Deml et al., 2015); 2) the underlying mutation causes loss-of-function of mab21l2; 3) injections of mRNA encoding for wild-type MAB21L2 protein were shown to rescue the mutant phenotype (Deml et al., 2015); 4) MAB21L/mab21l proteins are extremely conserved with 94.4% identity between human MAB21L1 and MAB21L2 and 98% identity between human/zebrafish MAB21L1/mab21l1 or MAB21L1/mab21l2 (Supp. Figure S3), suggesting a high degree of functional redundancy.
In accordance with Mendel’s principles, heterozygous mab21l2Q48Sfs*5 parents are expected to produce 75% phenotypically normal embryos comprising a mix of heterozygous (p.Gln48Serfs*5/+) (50%) and wild-type (+/+) (25%) genotypes and 25% homozygous (p.Gln48Serfs*5/p.Gln48Serfs*5) fish that exhibit the ocular phenotype (Deml et al., 2015). Consistent with this, 77.56%±0.27% (number of clutches=3; total number of embryos per clutch: 131, 75 and 40) of uninjected progeny of heterozygous crosses had normal eyes. In comparison to uninjected embryos, wild-type MAB21L1 mRNA injection (number of clutches=4; total number of embryos per clutch: 36, 105, 73 and 37) resulted in a significant increase in phenotypically normal embryos (88.38%±1.7%; P=0.0001). This suggests human wild-type MAB21L1 is able to functionally substitute for mab21l2 and rescue the effects of its deficiency (Figure 2D). In contrast, when MAB21L1 p.(Arg51Leu)-encoding mRNA (number of clutches=4; total number of embryos per clutch: 49, 92, 52 and 39), p.(Arg62Cys)-encoding mRNA (number of clutches=4; total number of embryos per clutch: 102, 53, 129 and 70), or p.(Gly220Arg)- encoding mRNA (number of clutches=4; total number of embryos per clutch: 146, 14, 93 and 93) was injected, 78.52%±3.8% (P=0.6922), 82.62±4.4% (P= 0.1100) or 81.40%±4.4% (P=0.2030) of embryos, correspondingly, were phenotypically normal, indicating no statistically significant rescue in comparison to uninjected control had occurred. The lack of rescue was strongest for p.(Arg51Leu). Similarly, mRNA encoding for MAB21L2 Arg51 variant, p.(Arg51Gly), was unable to rescue the mab21l2Q48Sfs*5 phenotype (Deml et al., 2015).
DISCUSSION
In this study, we present four individuals with unique heterozygous coding MAB21L1 variants, p.(Arg51Leu), p.(Arg62Cys), and p.(Gly220Arg), exhibiting microphthalmia in all, along with variable aniridia, coloboma, microcornea, lens defects (microspherophakia, cataracts) and nystagmus. Previously, homozygous MAB21L1 variants (mainly truncations) in humans have been associated with a syndromic disorder including ocular features, primarily corneal dystrophy/opacities and nystagmus with variable additional eye anomalies, as well as facial dysmorphism, genital abnormalities, and cerebellar hypoplasia (Bruel et al., 2017; Rad et al., 2019). The data presented in this report suggest that heterozygous missense variants in MAB21L1 may also be disease-causing and extend the associated disease spectrum.
All three missense variants were identified in patients with microphthalmia. The c.152G>T p.(Arg51Leu) variant in individuals 1A and 1B was also associated with aniridia and represents the strongest likely causative allele identified in our study, with high functional predictions and complete absence in the general population; in vitro and in vivo studies identified normal localization but higher stability and inability to functionally rescue mab21l2 deficiency. In addition, mutations affecting the conserved Arg51 residue represent the most common cause of dominant disease in a closely related protein, MAB21L2 (Deml et al., 2015; Horn et al., 2015; Rainger et al., 2014). The p.(Arg62Cys) and p.(Gly220Arg) variants identified in individuals 2 and 3, respectively, were additionally associated with coloboma; both missense variants demonstrated high functional predictions and inability to effectively rescue mab21l2 deficiency but since one variant was inherited from an unaffected parent and both variants, though ultra-rare, were present in the general population, they are currently classified as variants of uncertain significance.
In silico analyses predicted some local structural alterations of variant MAB21L1 proteins. The MAB21L1 structure has two lobes, an N-terminal and C-terminal, connected by a long alpha-helix spine. The N-terminal lobe is largely surface-accessible and contains several positively charged residues, along with a subdomain structurally similar to the NTase domain of cyclic GMP-AMP synthase (cGAS) (de Oliveira Mann et al., 2016), which is involved in the recognition of cytosolic nucleic acid and subsequent production of 2’,3’-cGAMP (Ablasser et al., 2013; Gao et al., 2013). Notably, all three affected residues, Arg51, Arg62 and Gly220, are in or near the N-terminal lobe upon folding. Arg51 is involved in the formation of salt bridges with residue Glu115, and likely important for protein stabilization (de Oliveira Mann et al., 2016). Arg62 was found to be involved in binding CTP in a denoted ‘ligand-binding pocket’ outside of the NTase domain (de Oliveira Mann et al., 2016). While it is unclear whether CTP is a true physiological ligand for MAB21L1, this may highlight the importance of this region for forming interactions and the involvement of Arg62. Finally, Gly220 is located near the N-terminal lobe upon folding, in a linker region between β8 and β9. Upon mutation to arginine, a gain of an alpha helix is noted, however, it is unclear what the functional consequences of this might be.
Further examination of the identified coding variants by in vitro studies demonstrated normal subcellular localization but variable stability for the corresponding proteins; p.(Arg62Cys) and p.(Gly220Arg) MAB21L1 proteins showed reduced protein levels (statistically significant for p.(Arg62Cys)), while p.(Arg51Leu) demonstrated a significantly higher level of protein compared to wild-type. There are conflicting reports on how the variants at Arg51 affect the stability of MAB21L proteins. The MAB21L1 crystal structure has been solved and revealed that Arg51 participates in salt-bridge formation and stabilization of loop structures in the protein, suggesting mutations to this residue would affect stabilization of the protein (de Oliveira Mann et al., 2016). Cyclohexamide protein stability assays for p.(Arg51Gly) in MAB21L2 revealed reduced stability in comparison to wild-type (Deml et al., 2015). Similarly, thermal shift assays for p.(Arg51Cys) in MAB21L1 had reduced melting points compared to wild-type, suggesting decreased stability (de Oliveira Mann et al., 2016). Conversely, tetracycline protein stability assays for p.(Arg51Cys) and p.(Arg51His) in MAB21L2 indicated increased protein stability (Rainger et al., 2014). The increased stability of p.(Arg51Leu) reported here may further contribute to the pathogenic effects of this variant. For example, since crystallization data suggest that oligomerization may be part of normal MAB21L1 function (de Oliveira Mann et al., 2016), the variant protein may form nonfunctional but more stable complexes with wild-type MAB21L1, thus exerting a dominant-negative effect. Another possibility is that increased stability may lead to abnormal persistence of downstream signaling activity; persistent phospho-ERK signaling has been suggested as a possible pathogenic mechanism for similar MAB21L2 mutants that exhibited increased stability (Rainger et al., 2014).
Finally, mRNA complementation assays were conducted to test the efficiency of MAB21L1 wild-type and mutant mRNAs in rescuing a zebrafish mab21l2 mutant phenotype. While overexpression of wild-type MAB21L1 mRNA compensated for the loss of mab21l2 and rescued the mutant eye phenotype, similar experiments with mRNA encoding for each of the three mutant proteins failed to produce a statistically significant rescue effect, with p.(Arg51Leu) showing the highest degree of functional deficiency. The observed functional deficiencies of the identified coding variants support their involvement in the corresponding ocular disorders. The presence of the p.(Arg62Cys) and p.(Gly220Arg) variants in the general population (though at ultra-rare frequencies) and an unaffected parent in one family could be explained by incomplete penetrance. In general, incomplete penetrance has been noted for several genes involved in developmental ocular anomalies. While pathogenic variants in OTX2 are an established cause of MAC-spectrum, review of the literature identified that a number of these variants were inherited from unaffected parents (Schilter et al., 2011). Even more striking, variants in TEK associated with primary congenital glaucoma were shown to be inherited from an unaffected parent in all families where both parents were tested (Souma et al., 2016). At the same time, both individuals carrying these variants were found to have additional non-coding MAB21L1 alleles in trans in either the 5’ UTR (Individual 2; c.-68T>C) or 3’ UTR (Individual 3; c.*529A>G). 5’ UTR variants have the potential to affect translational regulation via impacting the binding of the preinitiation complex, altering recruitment of RNA-binding proteins (and thus subsequent events such as RNA capping, splicing or polyadenylation), or disrupting upstream open reading frames (Steri, Idda, Whalen, & Orru, 2018). 3’ UTR variants can affect polyadenylation signals, RNA stability and localization, miRNA binding sites, and recruitment of other RNA-binding proteins involved in translational regulation (Steri et al., 2018). In silico analyses predicted that both the 5’ and 3’ UTR variants identified here affect RBP motifs and the 3’UTR allele may have a further effect on miRNA target sites, suggesting both variants could result in aberrant regulation of the MAB21L1 mRNA. Therefore it is possible that the non-coding sequence variations affecting the second MAB21L1 allele in patients carrying p.(Arg62Cys) and p.(Gly220Arg) missense variants contribute to the observed phenotypes in a bi-allelic manner. Further evidence via identification of other similar families will be needed.
The possible connection of MAB21L1 with MAC-spectrum in humans is plausible given the broad expression of this factor in the developing ocular structures in animal models. A Mab21l1 null mouse displays a severe developmental ocular phenotype consistent with MAC-spectrum (Yamada et al., 2003). Homozygous embryos fail to form a lens vesicle and exhibit aphakia, malformed retina, and abnormally thick cornea, culminating in severe microphthalmia, disorganized retinal lamination and highly abnormal anterior structures including absent lens and iris in adults (Yamada et al., 2003). Expression of Mab21l1 overlaps the developmental pattern of its close homolog, Mab21l2 (Yamada et al., 2003; Yamada et al., 2004), whose deficiency is also associated with severe ocular defects in mouse (Tsang et al., 2018; Yamada et al., 2004) and zebrafish models (Deml et al., 2015; Gath & Gross, 2019; Hartsock et al., 2014; Wycliffe et al., 2020). Dominant and recessive MAB21L2 variants have been linked with MAC phenotypes (Aubert-Mucca et al., 2020; Deml et al., 2015; Horn et al., 2015; Patel et al., 2018; Rainger et al., 2014), with heterozygous alleles affecting the Arg51 residue in four of seven previously published dominant families (c.151C>G p.(Arg51Gly), c.151C>T p.(Arg51Cys) (in two unrelated families) and c.152G>A p.(Arg51His)) and a nearby amino acid at position 49 in another family (c.145G>A p.(Glu49Lys)) (Deml et al., 2015; Horn et al., 2015; Rainger et al., 2014). Additionally, two presumed loss-of-function dominant alleles were identified in two unrelated families, c.1A>C p.(Met1?) and c.840C>G p.(Tyr280*) (Aubert-Mucca et al., 2020; Patel et al., 2018) and one recessive allele was identified in MAB21L2, c.740G>A p.(Arg247Gln) (Rainger et al., 2014).
The link between MAB21L1 and aniridia is also consistent with prior knowledge. The most common cause of aniridia is pathogenic variants in the PAX6 gene, accounting for up to 90% of cases (Hingorani et al., 2012), with FOXC1, PITX2 and a few other genes explaining some of the remaining cases (Hall et al., 2019; Hingorani et al., 2012) but still leaving about 5–10% of aniridia genetically unexplained. Interestingly, in Caenorhabditis elegans, a mab-18 mutant (a PAX6 orthologue) was found to have a very similar phenotype to a mab-21 mutant (a MAB21L orthologue), both affecting sensory ray formation of the tail (Baird, Fitch, Kassem, & Emmons, 1991). Further, both Mab21l1 and Mab21l2 have previously been suggested as downstream targets of Pax6 in mice. In small-eye (sey) homozygous embryos, in situ hybridization identified a significant reduction in Mab21l1 expression in both the surface ectoderm and optic vesicles during ocular development, while expression of Mab21l2 in the same tissues was unchanged (Yamada et al., 2003); at the same time, in heterozygous Pax6lacZ/+mice (St-Onge, Sosa-Pineda, Chowdhury, Mansouri, & Gruss, 1997), Mab21l2 was found to be up-regulated in lens tissue implying that Mab21l2 expression may be normally repressed via Pax6 in the lens (Wolf et al., 2009). Pax6 binding sites were identified in the regulatory regions of Mab21l2 (Wolf et al., 2009), which further corroborates this interaction and suggests a direct effect. Furthermore, while variants in PAX6 are typically connected with aniridia, a small number of bilateral and unilateral microphthalmia/coloboma cases have been identified, with or without aniridia (Williamson & FitzPatrick, 2014). The missense variants reported in this manuscript suggest a role for MAB21L1 in microphthalmia, aniridia, and coloboma in humans, similar to the PAX6 spectrum. Thus, this study provides additional support for the likely involvement of MAB21L1 and PAX6 in the same pathway. Further genetic screening for MAB21L1 variants in a wide spectrum of ocular disorders will help to define its role in human eye development and disease.
Supplementary Material
Supp. Table 1. Predicted miRNA target effects for the c.*529A>G variant.
Supp. Table 2. Predicted RNA binding protein (RBP) effects with non-coding variation.
Supp. Figure 1. In silico protein modeling. Protein structures for the solved MAB21L1 wild-type (5EOG) (de Oliveira Mann et al., 2016) (A,C) and the I-TASSER predicted mutants (Arg51Leu B,D; Arg62Cys E; Gly220Arg F). Amino acids (aa) of interest are highlighted as follows: aa51 orange, aa62pink, aa220 yellow. Alpha helices are purple, beta sheets green, linker regions blue. Red arrows (B,D,E,F) indicate observed structural changes upon mutation.
Supp. Figure 2. Multi-species sequence comparisons of MAB21L1 non-coding regions. Nucleotide alignments for 5’UTR (A) and 3’UTR (B) are shown using data generated by UCSC Multiz Alignments of 100 Vertebrates. Identical nucleotides are shaded in grey; positions of variant nucleotides are indicated in bold; non-coding variants in Individuals 2 (A) and 3 (B) are shown in red.
Supp. Figure 3. Alignment of full-length MAB21L1 and related proteins. Identical amino acids are shaded in grey; positions of variant amino acids are indicated in bold and variants identified in Individuals 1a, 1b, 2 and 3 are shown in red. Note high conservation between different vertebrate MAB21L1/Mab21l1/mab21l1 proteins as well as with its close human homolog MAB21L2 and C. elegans mab-21. Human MAB21L1 (NP_005575.1), human MAB21L2 (NP_006430.1), mouse Mab21l1 (NP_034880.1), chicken Mab21l1 (NP_989864.1), zebrafish mab21l1 (NP_694506.2) and C. elegans mab-21 (NP_497940.2).
ACKNOWLEDGEMENTS
The authors gratefully acknowledge patients and their families for their participation in research studies. This work was supported by the National Eye Institute grants [R01EY025718 to E.V.S., T32EY014537 to S.E.S.] and by the Children’s Research Institute Foundation at Children’s Hospital of Wisconsin (to E.V.S.).
Funding information:
This work was supported by the National Eye Institute grants [R01EY025718 to E.V.S., T32EY014537 to S.E.S.] and by the Children’s Research Institute Foundation at Children’s Hospital of Wisconsin (to E.V.S.).
Grant numbers:
NEI R01EY025718 and T32EY014537
Footnotes
Conflict of interest statement:
Adi Reich AR is an employee of GeneDx, Inc.; the other authors have no conflict of interest to disclose.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material. Variants reported in the manuscript have been submitted to LOVD https://www.lovd.nl/MAB21L1 (IDs: 0036029, 00336030 and 00336031).
WEB RESOURCES
gnomAD Browser: https://gnomad.broadinstitute.org/
Ensembl Variant Effect Predictor: http://uswest.ensembl.org/Tools/VEP
UCSC Genome Browser: https://genome.ucsc.edu/
Kalign: https://www.ebi.ac.uk/Tools/msa/kalign
ViennaRNA Web Services: http://rna.tbi.univie.ac.at/
MicroSNiPer: http://vm24141.virt.gwdg.de/services/microsniper/
PolymiRTS Database 3.0: http://compbio.uthsc.edu/miRSNP/
RBPmap: http://rbpmap.technion.ac.il/index.html
FATHMM-MKL: http://fathmm.biocompute.org.uk/fathmmMKL.htm
RCSB Protein Data Bank: www.rcsb.org
I-TASSER: https://zhanglab.ccmb.med.umich.edu/I-TASSER/
ELISA Analysis: elisaanalysis.com
REFERENCES
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, … Hornung V (2013). cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature, 498(7454), 380–384. doi: 10.1038/nature12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7(4), 248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert-Mucca M, Pernin-Grandjean J, Marchasson S, Gaston V, Habib C, Meunier I, … Plaisancie J (2020). Confirmation of FZD5 implication in a cohort of 50 patients with ocular coloboma. Eur J Hum Genet. doi: 10.1038/s41431-020-0695-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird SE, Fitch DH, Kassem IA, & Emmons SW (1991). Pattern formation in the nematode epidermis: determination of the arrangement of peripheral sense organs in the C. elegans male tail. Development, 113(2), 515–526. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/1782863 [DOI] [PubMed] [Google Scholar]
- Baldessari D, Badaloni A, Longhi R, Zappavigna V, & Consalez GG (2004). MAB21L2, a vertebrate member of the Male-abnormal 21 family, modulates BMP signaling and interacts with SMAD1. BMC Cell Biol, 5(1), 48. doi: 10.1186/1471-2121-5-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenboim M, Zoltick BJ, Guo Y, & Weinberger DR (2010). MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum Mutat, 31(11), 1223–1232. doi: 10.1002/humu.21349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Ziebarth JD, & Cui Y (2014). PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res, 42(Database issue), D86–91. doi: 10.1093/nar/gkt1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel AL, Masurel-Paulet A, Riviere JB, Duffourd Y, Lehalle D, Bensignor C, … Thevenon J (2017). Autosomal recessive truncating MAB21L1 mutation associated with a syndromic scrotal agenesis. Clin Genet, 91(2), 333–338. doi: 10.1111/cge.12794 [DOI] [PubMed] [Google Scholar]
- Cao J, Mu Q, & Huang H (2018). The Roles of Insulin-Like Growth Factor 2 mRNA-Binding Protein 2 in Cancer and Cancer Stem Cells. Stem Cells Int, 2018, 4217259. doi: 10.1155/2018/4217259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, & Batzoglou S (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol, 6(12), e1001025. doi: 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Mann CC, Kiefersauer R, Witte G, & Hopfner KP (2016). Structural and biochemical characterization of the cell fate determining nucleotidyltransferase fold protein MAB21L1. Sci Rep, 6, 27498. doi: 10.1038/srep27498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deml B, Kariminejad A, Borujerdi RH, Muheisen S, Reis LM, & Semina EV (2015). Mutations in MAB21L2 result in ocular Coloboma, microcornea and cataracts. PLoS Genet, 11(2), e1005002. doi: 10.1371/journal.pgen.1005002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deml B, Reis LM, Lemyre E, Clark RD, Kariminejad A, & Semina EV (2016). Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur J Hum Genet, 24(4), 535–541. doi: 10.1038/ejhg.2015.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, … Patel DJ (2013). Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell, 153(5), 1094–1107. doi: 10.1016/j.cell.2013.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DM, Baek D, Shin C, Bell GW, Grimson A, & Bartel DP (2011). Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol, 18(10), 1139–1146. doi: 10.1038/nsmb.2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gath N, & Gross JM (2019). Zebrafish mab21l2 mutants possess severe defects in optic cup morphogenesis, lens and cornea development. Dev Dyn, 248(7), 514–529. doi: 10.1002/dvdy.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory-Evans CY, Williams MJ, Halford S, & Gregory-Evans K (2004). Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet, 41(12), 881–891. doi: 10.1136/jmg.2004.025494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, & Bartel DP (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell, 27(1), 91–105. doi: 10.1016/j.molcel.2007.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillen Sacoto MJ, Tchasovnikarova IA, Torti E, Forster C, Andrew EH, Anselm I, … Juusola J (2020). De Novo Variants in the ATPase Module of MORC2 Cause a Neurodevelopmental Disorder with Growth Retardation and Variable Craniofacial Dysmorphism. Am J Hum Genet, 107(2), 352–363. doi: 10.1016/j.ajhg.2020.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall HN, Williamson KA, & FitzPatrick DR (2019). The genetic architecture of aniridia and Gillespie syndrome. Hum Genet, 138(8–9), 881–898. doi: 10.1007/s00439-018-1934-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsock A, Lee C, Arnold V, & Gross JM (2014). In vivo analysis of hyaloid vasculature morphogenesis in zebrafish: A role for the lens in maturation and maintenance of the hyaloid. Dev Biol, 394(2), 327–339. doi: 10.1016/j.ydbio.2014.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani M, Hanson I, & van Heyningen V (2012). Aniridia. Eur J Hum Genet, 20(10), 1011–1017. doi: 10.1038/ejhg.2012.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn D, Prescott T, Houge G, Braekke K, Rosendahl K, Nishimura G, … Spranger J (2015). A Novel Oculo-Skeletal syndrome with intellectual disability caused by a particular MAB21L2 mutation. Eur J Med Genet, 58(8), 387–391. doi: 10.1016/j.ejmg.2015.06.003 [DOI] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, … Sieh W (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet, 99(4), 877–885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Wordinger RJ, Yorio T, & Clark AF (2012). Spliceosome protein (SRp) regulation of glucocorticoid receptor isoforms and glucocorticoid response in human trabecular meshwork cells. Invest Ophthalmol Vis Sci, 53(2), 857–866. doi: 10.1167/iovs.11-8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, & Haussler D (2002). The human genome browser at UCSC. Genome Res, 12(6), 996–1006. doi: 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, & Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev Dyn, 203(3), 253–310. doi: 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet, 46(3), 310–315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HT, Kim DH, & Kim H (2017). PAX6 aniridia syndrome: clinics, genetics, and therapeutics. Curr Opin Ophthalmol, 28(5), 436–447. doi: 10.1097/ICU.0000000000000405 [DOI] [PubMed] [Google Scholar]
- Liu X, Jian X, & Boerwinkle E (2013). dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat, 34(9), E2393–2402. doi: 10.1002/humu.22376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, & Semina EV (2012). pitx2 Deficiency results in abnormal ocular and craniofacial development in zebrafish. PloS one, 7(1), e30896. doi: 10.1371/journal.pone.0030896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, & Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol, 6, 26. doi: 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Yousoof S, Grigg JR, Flaherty M, Minoche AE, Cowley MJ, … Jamieson RV (2020). Revealing hidden genetic diagnoses in the ocular anterior segment disorders. Genet Med, 22(10), 1623–1632. doi: 10.1038/s41436-020-0854-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani M, Baldessari D, Francisconi S, Viggiano L, Rocchi M, Zappavigna V, … Consalez GG (1999). Two murine and human homologs of mab-21, a cell fate determination gene involved in Caenorhabditis elegans neural development. Hum Mol Genet, 8(13), 2397–2406. doi: 10.1093/hmg/8.13.2397 [DOI] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, … Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol, 17(1), 122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, & Rao A (2008). Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science, 321(5889), 686–691. doi: 10.1126/science.1157610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Khan AO, Alsahli S, Abdel-Salam G, Nowilaty SR, Mansour AM, … Alkuraya FS (2018). Genetic investigation of 93 families with microphthalmia or posterior microphthalmos. Clin Genet, 93(6), 1210–1222. doi: 10.1111/cge.13239 [DOI] [PubMed] [Google Scholar]
- Paz I, Kosti I, Ares M Jr., Cline M, & Mandel-Gutfreund Y (2014). RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res, 42(Web Server issue), W361–367. doi: 10.1093/nar/gku406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, … Rehm HL (2015). The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat, 36(10), 915–921. doi: 10.1002/humu.22858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisancie J, Calvas P, & Chassaing N (2016). Genetic Advances in Microphthalmia. J Pediatr Genet, 5(4), 184–188. doi: 10.1055/s-0036-1592350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, & Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res, 20(1), 110–121. doi: 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad A, Altunoglu U, Miller R, Maroofian R, James KN, Caglayan AO, … Schmidts M (2019). MAB21L1 loss of function causes a syndromic neurodevelopmental disorder with distinctive cerebellar, ocular, craniofacial and genital features (COFG syndrome). J Med Genet, 56(5), 332–339. doi: 10.1136/jmedgenet-2018-105623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainger J, Pehlivan D, Johansson S, Bengani H, Sanchez-Pulido L, Williamson KA, … FitzPatrick DR (2014). Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. Am J Hum Genet, 94(6), 915–923. doi: 10.1016/j.ajhg.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LM, & Semina EV (2015). Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res C Embryo Today, 105(2), 96–113. doi: 10.1002/bdrc.21097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res, 47(D1), D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, & Sander C (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res, 39(17), e118. doi: 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothrock CR, House AE, & Lynch KW (2005). HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J, 24(15), 2792–2802. doi: 10.1038/sj.emboj.7600745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilter KF, Schneider A, Bardakjian T, Soucy JF, Tyler RC, Reis LM, & Semina EV (2011). OTX2 microphthalmia syndrome: four novel mutations and delineation of a phenotype. Clin Genet, 79(2), 158–168. doi: 10.1111/j.1399-0004.2010.01450.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt HM, Johnson WM, Aboobakar IF, Strickland S, Gomez-Caraballo M, Parker M, … Stamer WD (2020). Identification and activity of the functional complex between hnRNPL and the pseudoexfoliation syndrome-associated lncRNA, LOXL1-AS1. Hum Mol Genet, 29(12), 1986–1995. doi: 10.1093/hmg/ddaa021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods, 11(4), 361–362. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Mort M, Cooper DN, Day IN, & Gaunt TR (2014). Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum Genomics, 8, 11. doi: 10.1186/1479-7364-8-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, & Ng PC (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res, 40(Web Server issue), W452–457. doi: 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalicky SE, White AJ, Grigg JR, Martin F, Smith J, Jones M, … Jamieson RV (2013). Microphthalmia, anophthalmia, and coloboma and associated ocular and systemic features: understanding the spectrum. JAMA Ophthalmol, 131(12), 1517–1524. doi: 10.1001/jamaophthalmol.2013.5305 [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, & Hamosh A (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat, 36(10), 928–930. doi: 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, … Young TL (2016). Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest, 126(7), 2575–2587. doi: 10.1172/JCI85830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, & Gruss P (1997). Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature, 387(6631), 406–409. doi: 10.1038/387406a0 [DOI] [PubMed] [Google Scholar]
- Steri M, Idda ML, Whalen MB, & Orru V (2018). Genetic variants in mRNA untranslated regions. Wiley Interdiscip Rev RNA, 9(4), e1474. doi: 10.1002/wrna.1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland LC, Rintala-Maki ND, White RD, & Morin CD (2005). RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem, 94(1), 5–24. doi: 10.1002/jcb.20204 [DOI] [PubMed] [Google Scholar]
- Tsang SW, Guo Y, Chan LH, Huang Y, & Chow KL (2018). Generation and characterization of pathogenic Mab21l2(R51C) mouse model. Genesis, 56(11–12), e23261. doi: 10.1002/dvg.23261 [DOI] [PubMed] [Google Scholar]
- Twyffels L, Gueydan C, & Kruys V (2011). Shuttling SR proteins: more than splicing factors. FEBS J, 278(18), 3246–3255. doi: 10.1111/j.1742-4658.2011.08274.x [DOI] [PubMed] [Google Scholar]
- Verma AS, & Fitzpatrick DR (2007). Anophthalmia and microphthalmia. Orphanet J Rare Dis, 2, 47. doi: 10.1186/1750-1172-2-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson KA, & FitzPatrick DR (2014). The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet, 57(8), 369–380. doi: 10.1016/j.ejmg.2014.05.002 [DOI] [PubMed] [Google Scholar]
- Wolf LV, Yang Y, Wang J, Xie Q, Braunger B, Tamm ER, … Cvekl A (2009). Identification of pax6-dependent gene regulatory networks in the mouse lens. PLoS One, 4(1), e4159. doi: 10.1371/journal.pone.0004159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wycliffe R, Plaisancie J, Leaman S, Santis O, Tucker L, Cavieres D, … Valdivia LE (2020). Developmental delay during eye morphogenesis underlies optic cup and neurogenesis defects in mab21l2u517 zebrafish mutants. Int J Dev Biol. doi: 10.1387/ijdb.200173lv [DOI] [PubMed] [Google Scholar]
- Yamada R, Mizutani-Koseki Y, Hasegawa T, Osumi N, Koseki H, & Takahashi N (2003). Cell-autonomous involvement of Mab21l1 is essential for lens placode development. Development, 130(9), 1759–1770. doi: 10.1242/dev.00399 [DOI] [PubMed] [Google Scholar]
- Yamada R, Mizutani-Koseki Y, Koseki H, & Takahashi N (2004). Requirement for Mab21l2 during development of murine retina and ventral body wall. Dev Biol, 274(2), 295–307. doi: 10.1016/j.ydbio.2004.07.016 [DOI] [PubMed] [Google Scholar]
- Yang J, & Zhang Y (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res, 43(W1), W174–181. doi: 10.1093/nar/gkv342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XY, Wang P, Han J, Rosenfeld MG, & Fu XD (2009). SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol Cell, 35(1), 1–10. doi: 10.1016/j.molcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Lin Z, Wang Y, Li M, Wang L, … Li P (2020). Emerging Roles of SRSF3 as a Therapeutic Target for Cancer. Front Oncol, 10, 577636. doi: 10.3389/fonc.2020.577636 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Table 1. Predicted miRNA target effects for the c.*529A>G variant.
Supp. Table 2. Predicted RNA binding protein (RBP) effects with non-coding variation.
Supp. Figure 1. In silico protein modeling. Protein structures for the solved MAB21L1 wild-type (5EOG) (de Oliveira Mann et al., 2016) (A,C) and the I-TASSER predicted mutants (Arg51Leu B,D; Arg62Cys E; Gly220Arg F). Amino acids (aa) of interest are highlighted as follows: aa51 orange, aa62pink, aa220 yellow. Alpha helices are purple, beta sheets green, linker regions blue. Red arrows (B,D,E,F) indicate observed structural changes upon mutation.
Supp. Figure 2. Multi-species sequence comparisons of MAB21L1 non-coding regions. Nucleotide alignments for 5’UTR (A) and 3’UTR (B) are shown using data generated by UCSC Multiz Alignments of 100 Vertebrates. Identical nucleotides are shaded in grey; positions of variant nucleotides are indicated in bold; non-coding variants in Individuals 2 (A) and 3 (B) are shown in red.
Supp. Figure 3. Alignment of full-length MAB21L1 and related proteins. Identical amino acids are shaded in grey; positions of variant amino acids are indicated in bold and variants identified in Individuals 1a, 1b, 2 and 3 are shown in red. Note high conservation between different vertebrate MAB21L1/Mab21l1/mab21l1 proteins as well as with its close human homolog MAB21L2 and C. elegans mab-21. Human MAB21L1 (NP_005575.1), human MAB21L2 (NP_006430.1), mouse Mab21l1 (NP_034880.1), chicken Mab21l1 (NP_989864.1), zebrafish mab21l1 (NP_694506.2) and C. elegans mab-21 (NP_497940.2).