

Abstract
Purpose:
FGFR1 overexpression has been associated with endocrine resistance in ER+ breast cancer. We found FGFR1 localized in the nucleus of breast cancer cells in primary tumors resistant to estrogen suppression. We investigated a role of nuclear FGFR1 on gene transcription and antiestrogen resistance.
Experimental Design:
Tumors from patients treated with letrozole were subjected to Ki67 and FGFR1 IHC. MCF7 cells were transduced with FGFR1(SP-)(NLS) to promote nuclear FGFR1 overexpression. FGFR1 genomic activity in ER+/FGFR1-amplified breast cancer cells ± FOXA1 siRNA or ± the FGFR tyrosine kinase inhibitor (TKI) erdafitinib was examined by ChIP-Seq and RNA-Seq. The nuclear and chromatin-bound FGFR1 interactome was investigated by Mass Spectrometry (MS).
Results:
High nuclear FGFR1 expression in ER+ primary tumors positively correlated with post-letrozole Ki67 values. Nuclear FGFR1 overexpression influenced gene transcription and promoted resistance to estrogen suppression and to fulvestrant in vivo. A gene expression signature induced by nuclear FGFR1 correlated with shorter survival in the METABRIC cohort of patients treated with antiestrogens. ChIP-Seq revealed FGFR1 occupancy at transcription start sites, overlapping with active transcription histone marks. MS analysis of the nuclear FGFR1 interactome identified phosphorylated RNA-Polymerase II and FOXA1, with FOXA1 RNAi impairing FGFR1 recruitment to chromatin. Treatment with erdafitinib did not impair nuclear FGFR1 translocation and genomic activity.
Conclusions:
These data suggest nuclear FGFR1 contributes to endocrine resistance by modulating gene transcription in ER+ breast cancer. Nuclear FGFR1 activity was unaffected by FGFR TKIs, thus supporting the development of treatment strategies to inhibit nuclear FGFR1 in ER+/FGFR1 overexpressing breast cancer.
Keywords: FGFR1, breast cancer, endocrine resistance, tyrosine kinase inhibitors
Statement of Translational Relevance:
We show herein evidence to support an unconventional role for FGFR1 in gene transcription regulation in ER+ breast cancer. Nuclear FGFR1 induces a gene expression profile promoting resistance to antiestrogens. Further, nuclear FGFR1 translocation and activity are not impaired by FGFR1 TKIs that inhibit canonical FGFR1 signaling pathway. These findings support development of pharmacological inhibitors to repress this novel receptor function in ER+/FGFR1 overexpressing breast cancer with a therapeutic intent.
Introduction
FGFR1 belongs to the family of Fibroblast Growth Factor Receptors, comprising four highly conserved transmembrane receptor tyrosine kinases (RTKs) FGFR1–4, and another membrane-associated receptor lacking the intracellular domain (FGFR5 or FGFRL1) (1). FGFRs are activated upon binding of ligands (fibroblast growth factors, FGFs) to their extracellular domain (2) followed by receptor dimerization and phosphorylation of C-terminal tyrosines. These phosphorylated tyrosines dock several adaptor proteins that induce activation of downstream signaling pathways, including RAS/RAF/MEK/ERK, PI3K/AKT, PLCγ and STATs (3–6).
The genomic locus of FGFR1, 8p11–12, is amplified in various cancer types, including breast, lung, ovarian and bladder tumors (7–10). FGFR1 gene amplification has been identified in about 15% of patients with ER+ breast cancers (11). We have previously shown that FGFR1 gene amplification is associated with resistance to estrogen suppression in a cohort of patients with ER+ breast cancer treated with the aromatase inhibitor letrozole (12).
The oncogenic role of FGFR1 is generally the result of genomic aberrations, such as gene amplification, activating mutations, gene fusions, or by dysregulated autocrine/paracrine signaling, involving FGF ligands (13). In addition to the canonical intracellular signaling function of membrane-bound FGFR1, there is experimental evidence that FGFR1 can localize in the nucleus of cancer cells (14,15). The role of nuclear FGFR1 has been investigated in the context of neuronal development. In embryonic stem cells (ESCs), nuclear FGFR1 has been shown to interact with chromatin, alone or in association with the nuclear receptors RXR (Retinoid X Receptor) or NR4A1 (also known as Nur77 or nerve growth factor IB), thereby promoting gene transcription associated with developmental pathways (16). Furthermore, in human medulloblastoma cells, FGFR1 interacts with CREB-Binding Protein (CBP) and ribosomal S6 kinase 1 (RSK1) in the nucleus. In this ternary complex, FGFR1 induces CBP-mediated transcription by releasing CBP from RSK1 inhibition and directly activating CBP (17).
There is a growing body of evidence showing that RTKs, in addition to their signal transduction function as membrane-bound receptors, can also be found in the nucleus (18). For example, it was recently reported that the Insulin Receptor (IR) associates with several gene promoters, thus regulating gene expression profiles involved in classic insulin-related functions (19). Genomic binding studies recently identified a direct involvement of HER2 (ERBB2) in gene transcription regulation in HER2-amplified breast cancers (20). Other studies showed that EGFR binds to promoters of genes involved in cancer cell proliferation and regulates their expression (21).
FGFR1 overexpression has been associated with resistance to antiestrogen therapy in ER+ breast cancers (22,23). We previously showed that estradiol deprivation in patients resulted in a significant increase in both cytosolic and nuclear FGFR1 levels in ER+/FGFR1-amplified breast cancer cells in primary tumors (24). In this study, we now report abundant levels of FGFR1 in nuclear and chromatin-bound fractions from ER+/FGFR1-amplified PDXs as well as a correlation between nuclear localization of FGFR1 and resistance to estrogen suppression in ER+ primary tumors in patients. Hence, we have examined whether nuclear FGFR1 has a potential causal role in endocrine resistance. To investigate a transcriptional function of nuclear FGFR1, we analyzed genome-wide chromatin occupancy of FGFR1 by chromatin immunoprecipitation followed by next-generation sequencing (ChIP-Seq) and FGFR1-associated gene expression profiles by RNA sequencing (RNA-seq) in ER+/FGFR1-amplified breast cancer cells. Taken together, these analyses identified a functional role for nuclear FGFR1 in transcriptional regulation, driving gene expression associated with antiestrogen resistance and poor patient outcome.
Materials and Methods
Cell lines and inhibitors:
MCF-7 (ATCC® HTB-22™), CAMA1 (ATCC® HTB-21™), and MDA-MD-134-VI (ATCC® HTB-23™) human breast cancer cells were obtained from the American Type Culture Collection (ATCC) and maintained in ATCC-recommended media supplemented with 10% FBS (Gibco) and 1x antibiotic/antimycotic (Gibco) at 37°C in a humidified atmosphere of 5% CO2 in air. Cell lines were authenticated by ATCC prior to purchase by the short tandem repeat (STR) method. All experiments were performed <2 months after thawing early passage cells. Mycoplasma testing was conducted for each cell line before use. Fulvestrant, tamoxifen and erdafitinib were purchased from Selleck Chemicals.
Xenograft studies:
Mouse experiments were approved by the UT Southwestern Institutional Animal Care and Use Committee (IACUC). Female ovariectomized athymic mice (Envigo) were implanted with a 14-day-release 17β-estradiol pellet (0.17 mg; Innovative Research of America). The following day, 1 × 107 MCF-7EV or MCF-7FGFR1(SP-)(NLS) cells suspended in PBS and Matrigel at 1:1 ratio were injected subcutaneously (s.c.) into the right flank of each mouse. Approximately 4 weeks later, mice bearing tumors measuring ≥150 mm3 were randomized to treatment with vehicle (control) or fulvestrant (5 mg per week; s.c.). Tumor diameters were measured with caliper every four days and tumor volume in mm3 was calculated with the formula: volume = width2 × length/2. After 4 weeks, tumors were harvested and snap-frozen in liquid nitrogen or fixed in 10% neutral buffered formalin followed by embedding in paraffin. Four-micron paraffinized sections were used for IHC using ERα (Santa Cruz Biotechnology #8002) antibody. Sections were scored by an expert pathologist (P.G.E) blinded to treatment arm.
Immunohistochemistry:
For IHC analysis, samples were prepared as previously described (24). Briefly, formalin-fixed paraffin-embedded (FFPE), 4-μm tissue sections were deparaffinized. Antigen retrieval was performed with citrate buffer pH 6. Endogen peroxidase was blocked and protein block was applied. Sections were then incubated with FGFR1 ab10646 (Abcam) antibody at 1:2000 overnight at 4°C. The visualization system Envision (Agilent Technologies, Santa Clara, CA). DAB was used as chromogen (Agilent Technologies) and hematoxylin was applied as counterstain. Slides were digitally acquired using an AxioScan Z1 slide scanner (Carl Zeiss) at 20x. Automated semiquantitative scoring was performed on whole slide images using QuPath software (25). Color deconvolution stains were set for hematoxylin and DAB. Cell segmentation was determined on hematoxylin. An object classifier was trained to define tumor and stroma compartments. Percentage of FGFR1 positive cells over total tumor cell and H-score were calculated with the positive cell detection algorithm according to the nuclear DAB optical densities mean. Each selected region was visually assessed for correct performance of the quantification algorithm. Ki67 expression in biopsies from patients treated with letrozole prior to surgery was measured by automated quantitative immunofluorescence, as previously described (12,26). Studies involving patients’ samples were conducted in accordance with the Declaration of Helsinki and approved by the Vanderbilt-Ingram Cancer Center Institutional Review Board. All patients provided a written informed consent.
ChIP and reChIP assays:
Cells cultured in 15-cm dishes were washed with PBS once and incubated with serum free DMEM media containing 1% formaldehyde, for 10 min at room temperature. Crosslinking was quenched with glycine. Plates were next washed with ice-cold PBS three times and cells scraped off the plate with ice-cold PBS with protease inhibitor tablet (Mini, EDTA-free Protease Inhibitor Cocktail – ROCHE – Cat no. 04693159001). Cells were centrifuged at 720 xg for 10 min at 4°C and resuspended in 1.5 ml sonication buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0–8.1) with protease inhibitor. After rotation at 4°C for 10 min, chromatin was sonicated using a Bioruptor Plus sonication device (Diagenode) at 4°C. Sonicated chromatin was adjusted to a final concentration of 200 mM NaCl and incubated at 65°C overnight. The following day, the chromatin was incubated with RNAse A for 30 min at 37°C, followed by Proteinase K for 1 h at 42°C. DNA was purified using QIAquick PCR Purification Kit (Cat 28106). DNA shearing was checked, with an average fragment size of 200–750 bp. To conduct chromatin immunoprecipitation (ChIP), the sonicated chromatin was eluted with ChIP dilution buffer (0.01% SDS buffer, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl, 16.7 mM Tris-HCl, pH 8.0–8.1) with protease inhibitors and precleared with Gammabind G Sepharose beads (GE Healthcare, Cat no. 17-0885-01) previously washed three times with ChIP-dilution buffer and preblocked in 0.5% BSA at 4°C for 1 h. The precleared chromatin was incubated at 4°C overnight with primary antibody. The following day, Gammabind G Sepharose beads were added to the antibody pulldowns for 2 h at 4°C. Next, beads were washed once in Buffer I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 150 mM NaCl and protease inhibitors, pH 8.0–8.1), once in Buffer II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl, and protease inhibitors), once in Buffer III (0.25 M LiCl, 1% NP40, 1% Na-deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0–8.1) and twice in TE buffer, pH 8.0–8.1. Complexes were eluted off the beads in Elution Buffer (0.1 M NaHCO3, 1%SDS) at 65°C for 10 min, twice, in a thermomixer. Eluates from the antibody pulldowns and sonicated chromatin (as input controls) were adjusted to 200 mM NaCl and incubated at 65°C overnight. The following day, sequential incubations with RNAse A for 30 min at 37°C, and Proteinase K for 1 h at 42°C were performed. DNA was purified using QIAquick PCR Purification Kit. ChIPed and input DNA was analyzed by qRT-PCR or used to construct sequencing libraries. For ChIP-qPCR, the enrichment was shown as percent of input. ChIP-qPCR results were reproduced in two or more independent experiments. We used the following primary antibodies for ChIP experiments: rabbit anti-FGFR1 (Abcam, ab10646), normal rabbit IgG (Cell Signaling Technology, 2729), rabbit anti-histone H3 acetyl K27 (Abcam, ab4729), rabbit anti-histone H3 trimethyl K4 (Abcam, ab8580), mouse anti-Rpb1 CTD 4H8 (Cell Signaling Technology, 2629), mouse anti-HNF-3α (Santa Cruz Biotechnology, sc-101058). For ChIP-qPCR, primer pair sequences were designed based on the nucleotide sequences identified by ChIP-Seq peaks and derived from UCSC Genome Browser. The identified sequence was input in BLAST (Basic Local Alignment Search Tool) to design valuable pair sequences. Primer pair sequences are indicated in Supplementary Table S1.
For reChIP, ChIP with rabbit anti-FGFR1 or normal rabbit IgG was performed as described above but after the last TE wash, beads were incubated with reChIP elution buffer (10 mM Tris pH 7.6, 1 mM EDTA, 2% SDS, 20 mM DTT) and heated at 37°C for 30 min (19). Samples were centrifuged at 1000 xg x 1 min and the supernatant was diluted 20-fold in ChIP dilution buffer containing protease inhibitors. Next, samples were split and incubated with rabbit IgG or mouse anti-HNF-3α or mouse anti-Rpb1 CTD 4H8 at 4°C overnight. The reChIP samples were collected, washed, and eluted as described above for ChIP. The enrichment value was measured by qRT-PCR and shown as percent (%) of input of the first round of ChIP.
ChIP-Sequencing analysis:
Cells were cultured in 15-cm dishes and kept for 48 h in IMEM/10% Charcoal Stripped Serum (CSS) prior to apply the ChIP protocol described above. To test the effect of FGF2 and erdafitinib on FGFR1 genomic distribution, CAMA1 cells were cultured for 24 h in serum free DMEM media, prior to treatment with FGF2 (R&D Systems, 233-FB) (5 ng/ml, 3 h) ± erdafitinib (250 nM, 3 h). All ChIP-Seq in CAMA1 cells experiments were conducted on two biological replicates. ChIP-Seq experiments in MCF7FGFR1(SP-)(NLS) cells were conducted on three biological replicates. Libraries from 5–10 ng ChIP DNA were prepared using KAPA Hyper Library Preparation Kit. ChIPed DNA was quantified on the Qubit® 4 Fluorometer (Invitrogen). Samples were end repaired, 3’ ends adenylated and barcoded with multiplex adapters, followed by size selection with Ampure XP beads and PCR amplification. Samples were next quantified by Qubit and validated on the Agilent Tapestation 4200, normalized and pooled, and then run on the Illumina NextSeq 500 using 75 cycle SBS v2.5 reagents. Reads were aligned to human reference genome (hg38) using Bowtie2(v2.2.3) (27) with default parameters. Low quality reads and duplicate reads were removed from aligned files using “samtools view -bh -F 0×04 -q 10“ (28) and “Picard MarkDuplicates.jar” (https://github.com/broadinstitute/picard) commands. Model-based Analysis of ChIP-Seq (MACS) (29) software tool (v.2.1.2) was used to identify TF bound regions from the ChIP-seq data. MACS uses 200-bp as the fragment length and each read was shifted by 100-bp to identify candidate peaks with significant tag enrichment. Default parameters of MACS were applied using an FDR cutoff of 5% to select the peaks for further analysis. For ChIP-Seq experiments in CAMA1 cells, only peaks reproduced in both replicates were considered relevant and used for further analysis. For experiments in MCF7FGFR1(SP-)(NLS) cells only peaks reproduced in at least two replicates were considered for further analysis. For motif analysis, 200-bp around the peak region was used. De novo motif discovery analysis was performed using findMotifsGenome module in homer (v4.9). We used bdgcmp executable available in MACS software tools to calculate the background signal from the input sample and subtract it from the ChIP signal. We used the “ppois” method to calculate the normalized signal. To plot sequencing read densities relative to specific positions in the genome, we used annotatePeaks function available in Homer tools (30). This function takes the peak coordinates, tags directories as input and extends each tag by their estimated ChIP-fragment length, and calculates ChIP fragment coverage represented in per base pairs per peak. Heatmaps were plotted using signal density in the 5000-bp region around each peak. Density plots around TSS were plotted using ngsplot tool (31). GREAT (http://great.stanford.edu/public/html/) was used to identify the potential biological functions of the FGFR1 DNA binding sites with default parameters. All the processed ChIP-Seq data generated for this manuscript are included in Supplementary Table S2.
RNA-Sequencing analysis:
CAMA1 cells were cultured in 10-cm dishes in either IMEM/10% CSS for 48 h or 24 h in serum free DMEM media, prior to treatment with DMSO or FGF2 (5 ng/ml, 6 h). Cells were harvested and RNA was purified using a RNA purification kit (Maxwell, Promega). Each experiment was conducted in three biological replicates. Samples were run on the Agilent Tapestation 4200 to ensure use of only high quality RNA (RIN Score ≥8). The Qubit fluorometer was used to determine sample concentration prior to preparation of libraries. One μg of total DNAse treated RNA was then prepared with the TruSeq Stranded mRNA Library Prep Kit from Illumina. Poly-A RNA was purified and fragmented before strand specific cDNA synthesis. cDNAs were then a-tailed and indexed adapters were ligated. After adapter ligation, samples were PCR amplified and purified with AmpureXP beads, then validated again on the Agilent Tapestation 4200. Before being normalized and pooled, samples were quantified by Qubit then run on the Illumina NextSeq 500 using V2.5 reagents. RNA-seq reads were mapped to human genome (hg38) and junctions were identified using tophat(v2.1.2). RNA-seq expression counts for each gene were quantified using featureCounts module from Subread package (v.1.6.3). Differential expression analysis was performed using DESeq2 (v.1.24.0) R package. Binding and Expression Target Analysis (BETA) v1.0.7, with the BETA-plus protocol, was used to evaluate significant association between FGFR1-bound genomic loci in MCF7FGFR1(SP-)(NLS) and differentially expressed genes in MCF7EV vs MCF7FGFR1(SP-)(NLS) cells. BETA was also used to evaluate significant association between FGFR1-bound genomic loci and FGF2-induced DEGs in CAMA1 cells. GSEA analysis were performed using a JAVA GSEA 3.0 program. All the processed RNA-Seq data generated for this manuscript are included in Supplementary Table S3.
Gene expression analysis.
RNA was extracted and purified from cells or xenografts using Maxwell RSC simplyRNA Cells Kit or simplyRNA Tissue Kit (Promega Corporation). cDNA was generated using iScript cDNA Synthesis Kit (Bio-Rad). qPCR was performed with a cDNA equivalent of 50 ng RNA, 1 mmol/L each of the forward and reverse primers, and SYBR Green PCR Master Mix (Applied Biosystems), using a QuantStudio 3 Real-Time PCR System Machine (Applied Biosystems). All primers were purchased from QIAGEN: GAPDH (PPH00150F) PGR (PPH01007F), CCND1 (PPH00128F), VEGFA (PPH00251C), TFF1 (PPH00998C), PDZK1 (PPH08038E), CDK4 (PPH00118F), CDK12 (PPH05712A), DUSP1 (PPH00406A), FTO (PPH16000B), JUNB (PPH00179A), GREB1 (PPH20761F), MYC (PPH00100B), FOS (PPH00094A), BRD2 (PPH09948A). Ct (threshold cycle) values were determined in triplicate samples by subtracting the target gene Ct from the GAPDH Ct; 2−ΔΔCt was used to determine the expression of selected mRNAs relative to GAPDH.
Immunoprecipitation and western blot analysis:
Cells were seeded in 15-cm dishes and after three washes with ice-cold PBS, harvested in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, and 1% Triton X-100) supplemented with protease inhibitors (Mini, EDTA-free Protease Inhibitor Cocktail – ROCHE) and phosphatase inhibitor (PhosSTOP; ROCHE). Lysates were sonicated using a Bioruptor Plus sonication device (Diagenode) at 4°C, at maximum power for 10 min with 30 sec on and 30 sec off twice. Lysates were incubated with DNase I (Thermo Fisher Scientific) and Benzonase (Sigma-Aldrich) at 37°C for 10 min, followed by centrifugation at 20,000 xg at 4°C for 10 min. Protein concentration in the supernatants was determined using Pierce BCA Protein Assay Reagents (Thermo Fisher). Lysates were incubated overnight with primary antibodies at 4°C and antibody pulldowns were captured by incubation with Protein G dynabeads for 2 h at 4°C. Beads were washed and the immune complexes eluted from the beads following manufacturer’s instructions (Dynabeads Protein G Immunoprecipitation Kit – Thermo Fisher). Eluted proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk at room temperature for 1 h and then incubated overnight with primary antibodies at 4°C, followed by incubation with HRP-conjugated rabbit or mouse secondary antibodies for 1–2 h at room temperature. Protein bands were detected with an enhanced chemiluminescence substrate (Pierce ECL Western Blotting substrate, Thermo Fisher). The following primary antibodies were used for immunoprecipitation: rabbit anti-FGFR1 (Abcam, ab10646), mouse anti-FLAG M2 (Sigma-Aldrich), mouse anti-Rpb1 CTD 4H8 (Cell Signaling Technology, 2629), rabbit Phospho-Rpb1 CTD (Ser2) (E1Z3G) (Cell signaling technology, 13499), mouse anti-CDK7 (CST, 2916), mouse anti-CDK9 (Santa Cruz Biotechnology, sc-13130), mouse anti-HNF-3α (Santa Cruz Biotechnology, sc-101058), normal rabbit IgG (CST, 2729), mouse IgG2b isotype control (CST, 53484). The following primary antibodies were used for immunoblotting: rabbit anti-FGFR1 (Abcam ad76464), mouse anti-FLAG M2 (Sigma-Aldrich), mouse anti-Rpb1 CTD 4H8 (Cell Signaling Technology, 2629), rabbit Phospho-Rpb1 CTD (Ser2) (E1Z3G) (Cell signaling technology, 13499), mouse anti-CDK7 (CST, 2916), mouse anti-CDK9 (Santa Cruz Biotechnology, sc-13130), mouse anti-HNF-3α (Santa Cruz Biotechnology, sc-101058), rabbit anti-α/β Tubulin (CST, 2148), rabbit anti-AIF (CST, 5318), mouse anti-Lamin A/C (CST, 4777), rabbit anti-Histone H3 (CST, 4499), rabbit anti-phospho-FGF Receptor (Tyr653/654) (CST, 3471), rabbit anti-phospho-FRS2α (Tyr436) (CST, 3861), rabbit anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/204) (CST, 4370), rabbit anti-p44/42 MAPK (Erk1/2)(137F5) (CST, 4695), mouse anti-β-actin (CST, 3700). For immunoprecipitation followed MS, CAMA1FGFR13XFLAG cells underwent subcellular fractionation according to the manufacturer’s protocol (Subcellular Protein Fractionation Kit for Cultured Cells, CST #78840). Soluble nuclear and chromatin-bound fractions were combined, sonicated and nuclease-treated as described above and used for immunoprecipitation with mouse anti-FLAG M2 (Sigma-Aldrich) and mouse IgG2b isotype control (CST, 53484) antibodies. For subcellular fractionation assays, CAMA1 and MDA-MB-134 cells were cultured for 24 h in serum free DMEM media, prior to treatment with FGF2, 5 ng/ml ± erdafitinib 250 nM for 3 h.
Mass Spectrometry:
Immunoprecipitated proteins were separated by SDS-PAGE. Excised Gel band samples were digested overnight with trypsin (Promega) following reduction and alkylation with DTT and iodoacetamide (Sigma–Aldrich). Digests underwent solid-phase extraction cleanup with an Oasis MCX plate (Waters) followed by run on an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 um i.d., 75-cm long EasySpray column (Thermo). As previously described (32), the mass spectrometer operated in positive ion mode. We used source voltage of 2.2 kV and ion transfer tube temperature of 275°C. Orbitrap acquired MS scans at 120,000 resolution; MS/MS spectra, up to 10, were obtained in the ion trap using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 20 sec after an ion was selected for fragmentation. Proteome Discoverer v2.2 (Thermo) was used to analyze raw MS data. Sequest HT was used to identify peptides from UniProt human protein database. Carbamidomethylation of Cys and oxidation of Met were set as a fixed and variable modifications, respectively. We applied a false-discovery rate (FDR) cutoff of 1% for all peptides. We used three replicates of FLAG-immunoprecipitates and two replicates for IgG-immunoprecipitates. Only the proteins present in all three FLAG pulldown replicates and absent in IgG pulldown replicates or with a fold enrichment in FLAG over IgG >10 were considered as positive hits. Proteins with less than two peptide hits were not included.
Immunofluorescence.
Cells were seeded in MatTek 35 mm dishes (MatTek Life Sciences) and after 48 h, fixed with 4% PFA for 30 min. Cells were next incubated in 0.25% Triton X-100 in PBS for 5 min, then in blocking buffer (10% BSA in PBS) for 60 min, followed by an overnight incubation with an FGFR1 antibody (Abcam cat #76464) at 4 °C. The following day, cells were incubated with a goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher Scientific, cat #A-11008) for 60 min, followed by DAPI staining. Vectashield antifade mounting medium (Vector Laboratories, cat #H-1000–10) was used as mounting solution. Images were collected with a fully automated inverted microscope (DMI8, Leica microsystems). Quantification of nuclear fluorescence was performed with the software ImageJ.
FGFR1 fluorescent in situ hybridization (FISH) analysis.
Four-μm tissue sections were mounted on charged slides and hybridized overnight with the SPEC FGFR1:CEN8 Dual Color Probe (ZytoVision, catalog# Z-2072-200). Briefly, deparaffinization, protease treatment and washes were performed as per standard protocols. After this pretreatment, the slides were denatured in the presence of 10 μL of the probe for 6 min at 72°C, and hybridized at 37°C overnight in StatSpin (Thermobrite, Abbott Molecular, Inc.). Post-hybridization saline-sodium citrate washes were performed at 72°C and the slides were then stained with DAPI before analysis. Tumor tissue was scanned for amplification hot spots under 20x magnification (Olympus BX60 Fluorescent microscope). If the FGFR1 signals were homogeneously distributed, then random areas were used for counting the signals. Images for cell counting were captured with a 100x oil immersion objective using Cytovision software. Twenty to sixty tumor cell nuclei were individually evaluated by counting green FGFR1 and orange centromere 8 signals. The FGFR1:CEN8 ratio and the average FGFR1 copy number for each specimen were calculated next. Based on HER2 guidelines (33), samples were considered to be FGFR1 amplified under one of the following conditions: a) FGFR1:CEN8 ratio ≥2.0; b) average number of FGFR1signals ≥6.
Clonogenic assay.
Cells (5 × 104/well) were seeded in triplicate in 10% DMEM-FBS in 6-well plates and treated with 0.5–10 nM fulvestrant or with increasing doses of erdafitinib or tamoxifen. Seven days later, when control wells reached 50–70% confluence, monolayers were fixed and stained with 20% methanol/80% water/0.5% crystal violet for 20 min, washed with water, and dried. Photographic images of the plates were obtained. The crystal violet was solubilized with 20% acid acetic and the image intensity of the monolayers was quantified by spectrophotometric detection at 490 nm using a plate reader (GloMax®-Multi Detection System, Promega).
PDX-derived cell sorting:
HCI-011 PDXs established in ovariectomized SCID/beige mice were harvested and dissociated using digestion buffer [125 μg/mL DnaseI (#LS002138, Worthington), 10 μg/mL Dispase (#LS02109, Worthington), 500 μg/mL Collagenase 3 (#LS004182, Worthington), and 5x triple antibiotics (#15240–062, Invitrogen) in CnT-PR media (Cellntec)]. Dissociated single cells were washed with ice-cold PBS, stained with APC anti-human CD298 Antibody (Biolegend, #341706) for 30 min at 4°C, and fixed with 1% formaldehyde for 15 min at 4°C. FACS analysis was performed on the FACSAria Fusion flow cytometer (BD Biosciences). APC+ cells were collected, washed with ice-cold PBS twice and then resuspended in sonication buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0–8.1) with protease inhibitors.
Plasmid and viral transfection:
3XFLAG-FGFR1-expressing lentiviral constructs were generated in the pLX302 Gateway vector (Open Biosystems). To generate stably transduced CAMA1 cells, 4 μg of the 3XFLAG-FGFR1-pLX302 construct were cotransfected with 3 μg psPAX2 and 1 μg pMD2G envelope plasmid into 293FT cells using FuGENE 6 (Promega). 293FT growth media was changed 24 h post-transfection; virus-containing supernatants were harvested 48 and 72 h post-transfection, passed through a 0.45-mm filter, diluted 1:4, and applied to target cells with 8 μg/mL polybrene (Sigma Aldrich). Virus-producing cells were selected in 1 μg/mL puromycin. pCDNA3.1(–)FGFR1(SP-)(NLS) has been previously described (34). FGFR1(SP-)(NLS) genomic sequence was cloned from pCDNA3.1(–)FGFR1(SP-)(NLS) into pLVX-M-puro plasmid by restriction enzyme cloning, to generate the pLVX-M-puro-FGFR1(SP-)(NLS) plasmid. pLVX-M-puro empty vector and pLVX-M-puro-FGFR1(SP-)(NLS) plasmid were cotransfected with 3 μg psPAX2 and 1 μg pMD2G envelope plasmid into 293FT cells as described above. Virus-containing supernatants from 293FT cells were collected as described above and applied to MCF-7 cells with 8 μg/mL polybrene (Sigma Aldrich). Virus-producing cells were selected in 1 μg/mL puromycin. The mutant FGFR1 D432N was generated from wild type human FGFR1 in pLX302 using site-directed mutagenesis (Q5 Site-Directed mutagenesis kit, New England Biolabs) using the primer 5′-GGTGTCTGCTAACTCCAGTGC-3′, and its reverse complement. Plasmid was cotransfected with psPAX2 and pMD2G envelope plasmids into 293FT to generate lentiviral particles as described above. FGFR1 was knocked out using pX458 (Addgene plasmid # 48138). Briefly, two sgRNAs that target FGFR1 were separately cloned into pX458 as previously described (35). The plasmids were then co-transfected into MCF-7. Forty-eight hours post-transfection, top 10% of the GFP-positive single MCF-7 cells were sorted into 96-well plates to grow FGFR1 knockout clones. The knockout of FGFR1 were validated by Western blot. The primers used are reported in Supplementary Table S1.
siRNA transfection:
Silencer Select pre-designed siRNAs targeting FGFR1, FOXA1 and SP1 were purchased from Ambion. Cells were reverse-transfected with siRNAs of interest using lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific) as per manufacturer’s instructions. Twenty-four h post-transfection, the culture media was replaced with IMEM/10% CSS. Seventy-two h post-transfection, the ChIP or RNA extraction protocols described above were applied. Simultaneously, transfected cells were harvested in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, and 1% Triton X-100) for protein extraction.
Statistical analysis:
Log-rank Mantel-Cox test, two-tailed Mann-Whitney test, unpaired t-test, multiple t-test analysis, two-way ANOVA with Sidak’s multiple comparisons test or Wilcoxon matched-pairs test were performed as indicated in figure legends. All data quantification and the statistical analysis described above were performed using Prism (v8).
Data availability
The datasets generated and analyzed during the current study are available in the GEO repository, code GSE148313 and can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148313
All the analyzed ChIP-Seq and RNA-Seq data can be found in Supplementary material as Supplementary Tables S2, S3.
Results
Nuclear FGFR1 promotes antiestrogen resistance
We performed immunohistochemistry (IHC) and fluorescent in situ hybridization (FISH) of FGFR1 in 155 biopsies from women with operable ER+ breast cancer who received letrozole for 10–21 days before surgery (in trial NCT00651976) (Supplementary Table S4) (Figure S1A) (12,24). In this published trial, response to estrogen suppression with letrozole was defined on the basis of post-treatment Ki67 levels [sensitive: natural log (ln) of post-letrozole Ki67 ≤1.0 or ≤2.4% tumor cells; intermediate: ln=1.1–1.9 or 2.5–7.3% tumor cells; resistant: (ln ≥2.0 or ≥7.4% tumor cells)]. Nineteen (19/155, 12%) tumors exhibited FGFR1 amplification, defined as an FGFR1:CEN8 ratio ≥2.0 (Supplementary Table S4) (Figure S1B). IHC revealed that, in addition to faint FGFR1 cytosolic staining, FGFR1 was clearly localized in breast cancer cell nuclei (Figure 1A). We observed similar FGFR1 localization in three ER+/FGFR1-amplified PDXs (Figures S1C and S1D). Subcellular fractionation of the PDX lysates revealed that, in addition to the expected localization in the plasma membrane, a substantial fraction of FGFR1 was detected in nuclear fractions (Figure S1E). ER+/FGFR1-amplified CAMA1 and MDA-MB-134 human breast cancer cell fractions also showed nuclear and chromatin-bound FGFR1 (Figure S1F).
Figure 1. Nuclear FGFR1 promotes antiestrogen resistance.
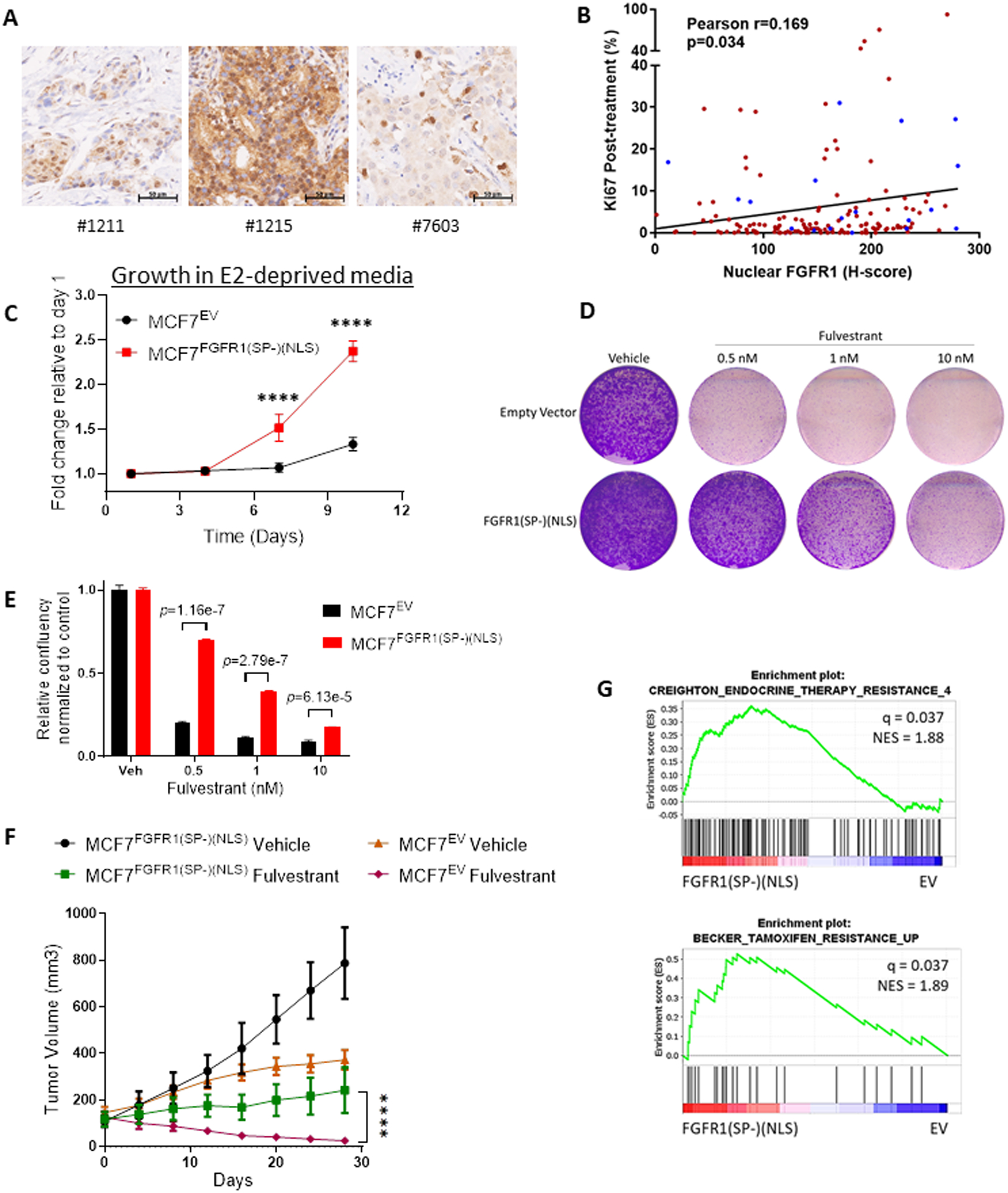
(A) Sections of FFPE of ER+/FGFR1-amplified breast primary tumors were subjected to IHC with an FGFR1 antibody as described in the methods. (B) Plot showing direct correlation between nuclear FGFR1 H-Score, measured by IHC, and post-treatment Ki67 in 155 ER+/HER2- breast cancer biopsies from women treated with pre-operative letrozole for 10–21 days in the NCT00651976 trial (12,24). In blue, samples exhibiting FGFR1 amplification (n=19/155), defined as an FGFR1:CEN8 ratio ≥2.0. (C) MCF-7EV and MCF-7FGFR1(SP-)(NLS) cells were seeded in 6-well plates in estrogen-deprived media. Media was replenished every 72 hours. Monolayers were stained with crystal violet on days 1, 4, 7 and 10. Quantification of the integrated intensity values as fold change normalized to day 1 (Sidak’s multiple comparisons test, two-way ANOVA, **** p<0.0001). (D) MCF-7EV and MCF-7FGFR1(SP-)(NLS) cells were seeded in 6-well plates in full media and treated with vehicle (DMSO) or fulvestrant. After seven days, monolayers were stained with crystal violet. (E) Quantification of the integrated intensity values as fold change normalized to vehicle-treated controls (Multiple Student’s t-test). (F) MCF-7EV and MCF-7FGFR1(SP-)(NSL) xenografts were established in ovariectomized athymic mice implanted with a s.c. 14-day release, 0.17-mg 17β-estradiol pellet. Once tumors established, mice were randomized to treatment with vehicle or fulvestrant (5 mg/week). Each data point represents the mean tumor volume in mm3 ± SEM (n=8 per arm, ****p < 0.0001; Two-way ANOVA). (G) GSEA of RNA-Seq data for MCF7FGFR1(SP-)(NLS) versus MCF7EV, showing enrichment of gene sets associated with antiestrogen resistance in MCF7FGFR1(SP-)(NLS) cells.
We next correlated FGFR1 localization with response to estrogen suppression. Nuclear FGFR1 levels in these tumors positively correlated with post-letrozole Ki67 values (Figures 1B, p=0.034 and S2A, p=0.0055). Although nuclear FGFR1 levels tended to be higher in FGFR1-amplified compared to non FGFR1-amplified tumors, this difference was not statistically significant (Figure S2B, p=0.096). Further, analysis of only those tumors without FGFR1-amplification (n=136) revealed that high nuclear FGFR1 correlated with higher on-treatment Ki67 (Figure S2C, p=0.027), suggesting that nuclear FGFR1 protein levels are associated with antiestrogens resistance regardless of FGFR1 gene copy number. We evaluated the distribution of nuclear FGFR1 H-score according to the Ki-67 defined groups. Nuclear FGFR1 H-Score values were borderline higher in resistant samples (Figure S2D, p=0.056). We next divided the cohort in two groups based on a median nuclear FGFR1 H-Score of 156.77. We found that 21/78 (27%) tumors with nuclear FGFR1 H-Score above the median were resistant (Figure S2E, Chi-Square test = 0.04).
Next, to explore a causal association of nuclear FGFR1 with antiestrogen resistance, we transduced MCF-7 cells with an FGFR1(SP-)(NLS) plasmid (34) (Figure S2F). The lack of an N-terminal Signal Peptide (SP-) sequence and the presence of a strong Nuclear Localization Signal (NLS) (PKKKRKV) are intended to prevent membrane localization of the resulting protein, while enforcing its nuclear localization. Indeed, the levels of FGFR1 in the nucleus of MCF-7 cells were increased upon transfection with FGFR1(SP-)(NLS), as shown by subcellular fractionation assay (Figure S2G) and immunofluorescence analysis (Figure S2H). MCF7FGFR1(SP-)(NLS) cells exhibited higher estrogen-independent growth (Figure 1C) and reduced sensitivity to fulvestrant (Figures 1D, E) compared to control cells. We next implanted MCF7EV and MCF7FGFR1(SP-)(NLS) cells in ovariectomized athymic mice. In agreement with the in vitro findings, MCF7FGFR1(SP-)(NLS) xenografts exhibited higher estrogen-independent growth compared to control tumors (Figure 1F). Further, while fulvestrant treatment induced tumor regression in control tumors, MCF7FGFR1(SP-)(NLS) xenografts did not regress (Figure 1F). Gene Set Enrichment Analysis (GSEA) of the 3,014 differentially expressed genes (DEGs) in MCF7FGFR1(SP-)(NLS) vs MCF7EV cells (shown in figure S2I) also revealed enrichment of gene sets associated with resistance to endocrine therapy and to tamoxifen (Figure 1G and Supplementary Table S3), suggesting that nuclear FGFR1 overexpression induces a transcriptional profile causal to antiestrogen resistance in breast cancer. Previous studies reported that Granzyme B cleaves FGFR1 at Asp432, producing a C-terminal fragment that translocates to the nucleus (15). Hence, we transduced MCF7 cells with an FGFR1 D432N mutant, previously reported as resistant to Granzyme B cleavage (15), to investigate a potential effect on antiestrogen resistance. Nuclear fractions of MCF7FGFR1 D432N cells showed markedly higher expression of both full-length (~140 KDa) and the cleaved form (~55 KDa) of FGFR1 (Figure S3A), suggesting that Granzyme B does not contribute to FGFR1 cleavage in these cells. Finally, we observed that overexpression of FGFR1 wild-type, FGFR1 D432N and FGFR1(SP-)(NLS) in MCF7 cells resulted in enhanced estradiol-independent growth and reduced sensitivity to fulvestrant and tamoxifen (Figures S3B–E).
Nuclear FGFR1 occupies chromatin at transcription start sites of genes associated with antiestrogen resistance
We next examined a role of nuclear FGFR1 in transcriptional regulation in breast cancer. To study chromatin occupancy by FGFR1, we performed ChIP-Seq in CAMA1 cells cultured for 48 h in estradiol-deprived media, which identified 4412 high-confidence binding sites. (Figure 2A). De novo consensus motif discovery analysis of the FGFR1 DNA-binding sites revealed GC-rich motifs among the top enriched motifs (Figures 2B and S4A).
Figure 2. Nuclear FGFR1 occupies chromatin at transcription start sites of genes associated with antiestrogen resistance.
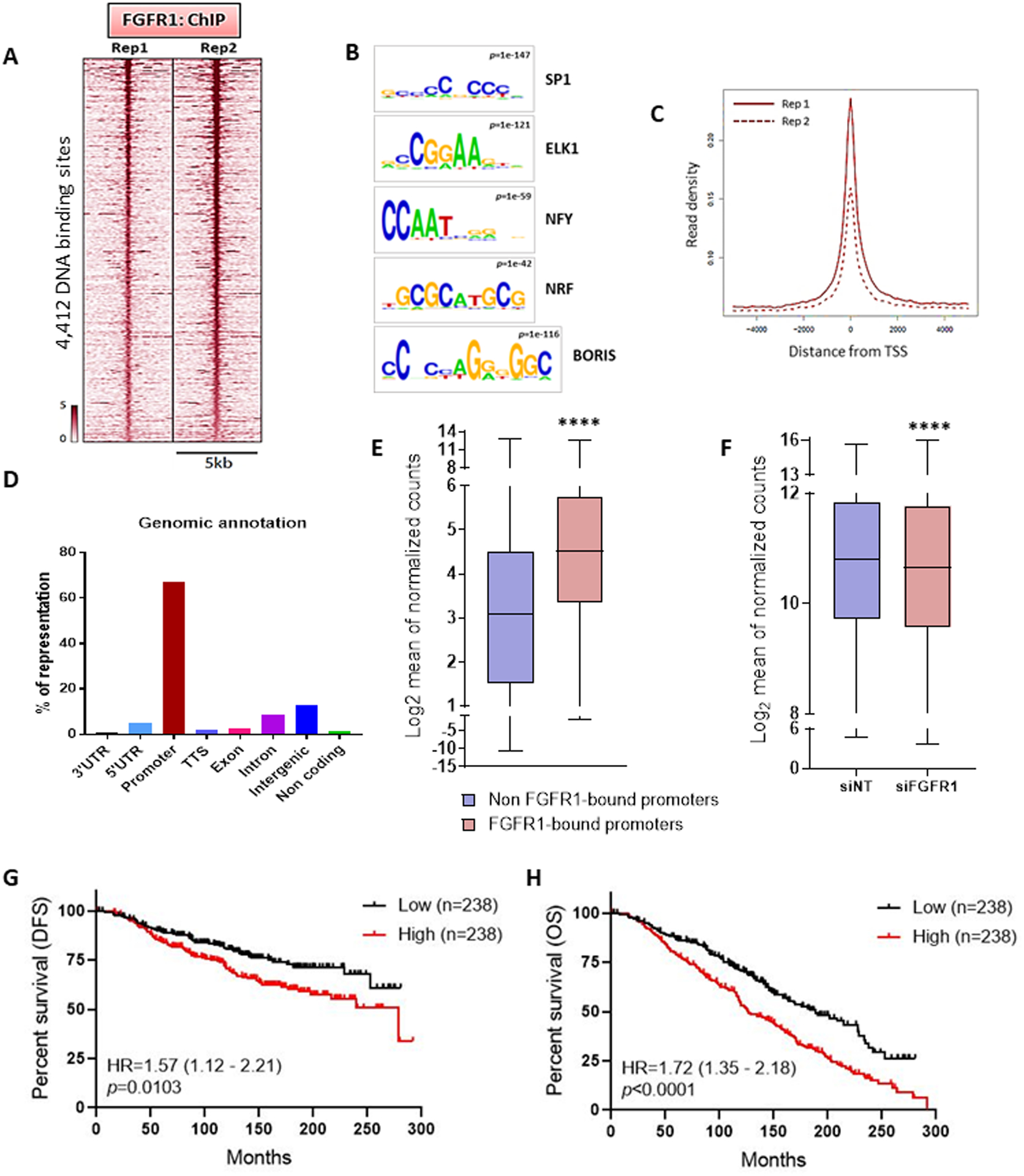
(A) Heatmap showing ChIP-Seq read densities around the FGFR1 bound regions in CAMA1 cells, in two replicates. The x-axis represents read densities within 5-kb region around the peak summit; the y-axis represents each predicted binding site. Cells were cultured for 48 h in IMEM/10% CSS. (B) Top consensus motifs, identified by de novo motif analysis, at genomic loci bound by FGFR1 identified in A. Statistical significance expressed as p value for each motif is shown. (C) Plot representing the density of the FGFR1 distribution around the Transcription Start Sites (TSS) in the two ChIP-Seq replicates. (D) Genomic annotations for the FGFR1 binding sites identified by ChIP-Seq showing enrichment of promoter regions. (E) Expression level of genes whose promoter is bound by FGFR1 (red) (2704 genes) versus gene expression of all other genes (blue) (10,452 genes). Data are derived from RNA-Seq on CAMA1 cells cultured for 48 h in IMEM/10% CSS (**** p<0.0001, Mann-Whitney test). (F) Gene expression of genes whose promoter is bound by FGFR1 in CAMA1 cells upon siRNA-mediated FGFR1 knockdown (n=3) (**** p<0.0001, Two-tailed Wilcoxon test). (G) Disease Free Survival (DFS) and Overall Survival (H) of the METABRIC cohort of 950 ER+/HER2- breast cancer patients treated with antiestrogens as a function of the FGFR1-associated polygenic score (Lowest Quartile vs Highest Quartile). The signature score was calculated by GSVA (77).
Most mammalian promoters (i.e., regions upstream the transcription start sites) are enriched with GC-rich sequences, also named CpG islands (CGIs) (36,37). Also, many human active promoters are enriched with SP1, Nrf-1, E2F and ETS transcription factor-binding sites motifs, all containing CGIs (38,39). Consistent with the predominance of GC-rich motifs identified (Figures 2B and S4A), the majority of FGFR1 binding sites were near the transcription start site (TSS) (Figure 2C), with 67% of ChIP-Seq peaks occurring at gene promoters (Figure 2D). Based on the nucleotide sequence of FGFR1-bound genomic loci identified by ChIP-Seq (Figure S5A), we next designed specific primer pair sequences to validate FGFR1 occupancy at nine selected promoter regions in CAMA1 (Figure S5B) and MDA-MB-134 cells (Figure S5C) by ChIP-qPCR. We further examined the binding of FGFR1 to the same promoter regions in tumor epithelial cells isolated by FACS from disaggregated HCI-011 PDXs (Figure S5D). For this, we performed ChIP-qPCR on the isolated tumor epithelial cells and confirmed enrichment of FGFR1 at similar promoter regions examined in CAMA1 and MDA-MB-134 cells (Figure S5E). These results suggest that FGFR1 binds to gene promoters and support a role for FGFR1 in transcriptional regulation.
RNA-Seq analysis of CAMA1 cells cultured for 48 h in estrogen-deprived media revealed that the average expression of genes whose promoter was bound by FGFR1 was significantly higher than the expression of all other genes (Figure 2E; Mann-Whitney, p<0.0001), implying that FGFR1 target genes are actively transcribed. Silencing of FGFR1 with siRNA significantly reduced the expression of genes whose promoters is bound by FGFR1 (Figure 2F, Wilcoxon test p<0.0001).
We intersected the list of 2901 genes whose promoter was bound by FGFR1, as identified by ChIP-Seq, with the 2048 genes downregulated by FGFR1 siRNA in CAMA1 cells (Figure S6A). A polygenic risk score was generated based on the 561 overlapping genes (Figure S6A and Supplementary Table S5). Analysis of 212 ER+/HER2- primary breast tumors in TCGA with known 8p11.23 copy number, where the FGFR1 locus lies, showed that the signature score was enriched in cancers with 8p11.23 amplification, compared to all other samples not harboring 8p11.23 copy gain or amplification (Figure S6B; t-test, p=0.039). In the METABRIC cohort of 1,294 ER+/HER2- breast cancers, the signature score was enriched in tumors exhibiting 8p11.23 amplification, compared to cancers without FGFR1 copy gain or amplification (Figure S6C; t-test, p=0.038). These results suggest that FGFR1-induced transcriptional signature is statistically enriched in primary breast tumors harboring FGFR1 gene amplification. We next investigated whether the FGFR1-associated risk score correlated with antiestrogen resistance. In the METABRIC cohort of ER+ breast cancers treated with antiestrogens (n=950), the risk score correlated with a worse DFS (Figure 2G; Log rank p=0.0103, HR=1.57, 95% CI 1.12 – 2.21, median 278.4 vs not reached) and OS (Figure 2H; Log rank p<0.0001, HR=1.72, 95% CI 1.35 – 2.18, median 126.5 vs 188.2 months). Taken together, these findings further suggest a role for nuclear FGFR1 in resistance to endocrine therapy.
FGFR1 associates with RNA Polymerase II and promotes gene expression
To further dissect the function of FGFR1 in transcriptional regulation, we sought to uncover which components of the transcriptional machinery physically interact with nuclear FGFR1. For this purpose, we transduced CAMA1 cells with an FGFR1–3XFLAG lentiviral vector. Next we immunoprecipitated FLAG from mixed nuclear and chromatin fractions of CAMA1FGFR1−3XFLAG cells followed by mass spectrometry (MS) of the antibody pulldowns. Among the proteins recovered in the nuclear FGFR1 interactome, we found several RNA Polymerase II (Pol II) subunits (Figures 3A and 3B) (Supplementary Table S6). RNA Pol II plays a pivotal role in gene transcription, that also requires other proteins critical for RNA polymerase recruitment to promoters, transcriptional initiation, and elongation (40). These findings were in line with enrichment of FGFR1 binding near transcription start sites (shown in Figure 2C), which are also the sites where Pol II is recruited for transcription initiation. We confirmed the FGFR1-Pol II association by co-precipitation with an FGFR1 antibody followed by Pol II western blot analysis of CAMA1 and MDA-MB-134 cell lysates (Figures 3C and S7A) and of HCI-011 PDX lysates (Figure S7B). FGFR1-Pol II co-precipitation was observed when we performed sonication and nuclease treatment, allowing the release of more soluble Pol II.
Figure 3. FGFR1 associates with RNA Polymerase II and promotes gene expression.
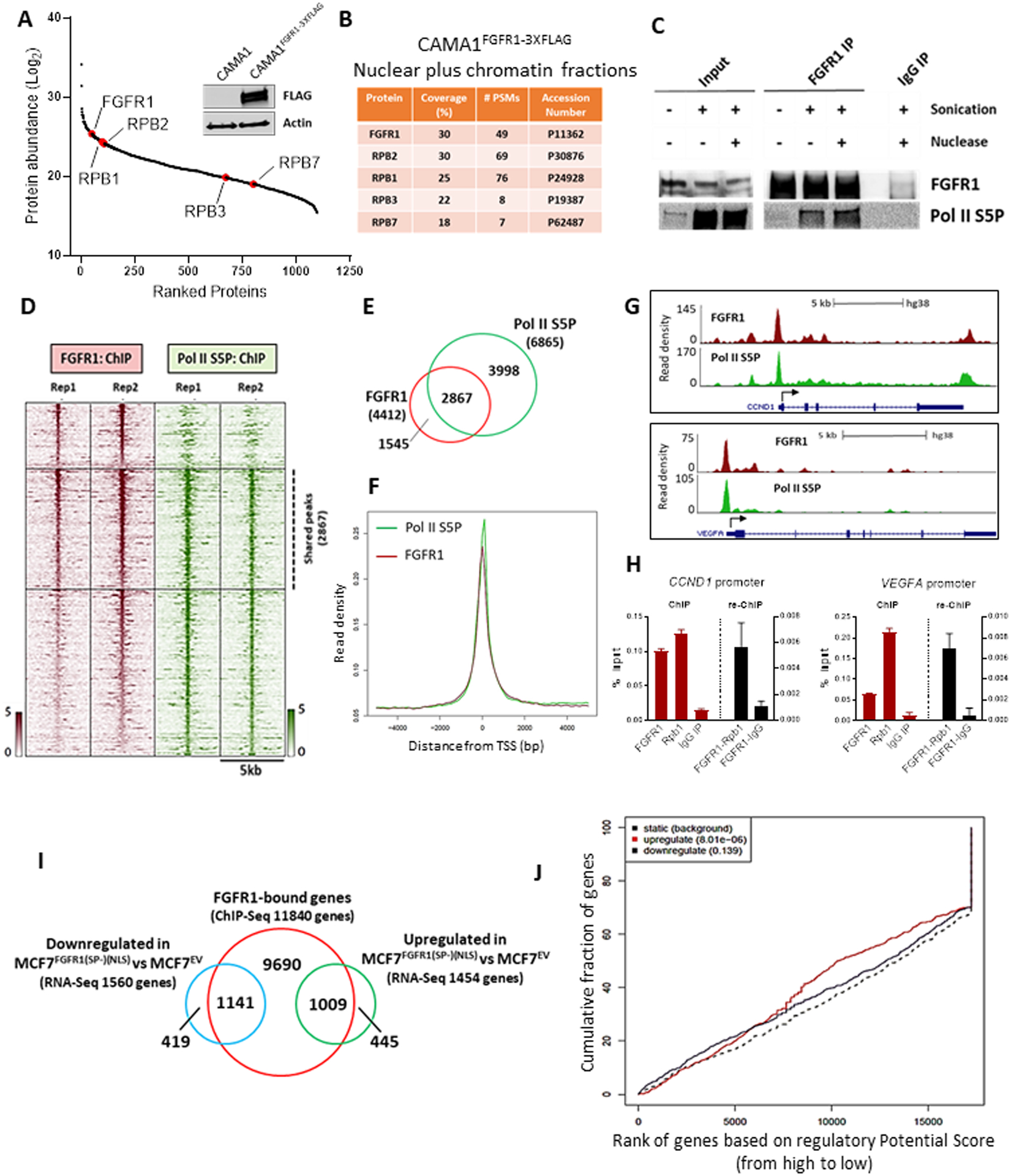
(A) FLAG-immunoprecipitation followed by MS was performed on sonicated and nuclease-treated mixed nuclear and chromatin fractions of CAMA1FGFR13XFLAG cells. FGFR1 and RNA Polymerase II subunits are highlighted (red). The western blot on the right shows FLAG-FGFR1 overexpression in CAMA1FGFR13XFLAG cells. (B) List of RNA Polymerase II subunits that co-precipitate with FLAG-FGFR1. Coverage (%) indicates the percentage of the protein sequence that was covered by the identified peptides. #PSMs indicates the number of Peptide Spectrum Matches or the number of spectra assigned to peptides that contributed to inference of the protein. (C) Co-precipitation of FGFR1 and RNA Polymerase phosphorylated on Serine residue 5 (Pol II S5P), with or without sonication and nuclease treatment. (D) Heatmaps of ChIP-Seq read densities around the FGFR1 (red) and Pol II S5P bound regions (green) in CAMA1 cells. (E) Venn Diagram and (F) density plots related to FGFR1- and Pol II S5P-ChIP-Seq shown in D. (G) Distribution of FGFR1 and Pol II S5P binding peaks at the VEGFA and CCND1 promoters (UCSC genome browser). (H) ChIP-reChIP assay performed by sequential ChIP-qPCR with a FGFR1 antibody followed by an antibody against Pol II S5P (Rpb1) or normal rabbit IgG (control), at the VEGFA and CCND1 promoters. Enrichment values expressed as percent (%) of input. (I) Venn Diagram of FGFR1-bound genes by ChIP-Seq in MCF7FGFR1(SP-)(NLS) cells (red circle), and genes upregulated (green) and downregulated (blue) in MCF7FGFR1(SP-)(NLS) vs MCF7EV cells. (J) Prediction of activating or repressing transcription function of FGFR1 by Binding and Expression Target Analysis (BETA). ChIP-Seq and RNA-Seq data from I were integrated. Genomic regions bound by FGFR1 are predicted to regulate the expression of upregulated genes, but not of downregulated genes. p values indicate the significance of the associations compared to background non-regulated genes.
FGFR1 interacted with Pol II phosphorylated on Ser5 and Ser2 of the C-terminal domain (CTD) of the largest Pol II subunit, RPB1 (Pol II S5P, Pol II S2P) (Figures 3C and S7A–C). Phosphorylation of Ser5 and Ser2 on the CTD are post-translational modifications required for transcription initiation and elongation, respectively (41,42). MS analysis of CAMA1FGFR1−3XFLAG pulldowns also revealed proteins known to be directly involved in Pol II phosphorylation and subsequent transcriptional activity, such as CDK7 and CDK9 (43) (Supplementary Table S6). We next confirmed the association of FGFR1 with CDK7-CDK9 in CAMA1 and MDA-MB-134 cells by co-precipitation with an FGFR1 antibody (Figure S7D). The binding of FGFR1 with Pol II S5P, Pol II S2P, CDK7 and CDK9 was also confirmed by immunoprecipitation assays with Pol II S5P, Pol II S2P, CDK7 and CDK9 antibodies, followed by western blot analysis for FGFR1, in CAMA1 cells (Figure S7E). Comparison of the genomic coordinates of FGFR1 occupancy with data from previously published ChIP-Seq datasets for transcription factors (44) identified RNA Polymerase II subunit B1, RPB1 (POLR2A), as one of the top hits (Figure S8A). Using ChIP-Seq to evaluate the distribution of Pol II S5P across the CAMA1 cells genome, we found that 2867/4412 (65%) FGFR1 binding peaks overlapped with Pol II S5P binding sites (Figures 3D and 3E), with distribution around similar locations near the TSS (Figures 3F, 3G and S8B), further suggesting that FGFR1 genomic distribution occurs at actively transcribed loci. ChIP-reChIP assays, with sequential FGFR1 and Pol II S5P antibodies, confirmed FGFR1-Pol II S5P association at selected gene promoters in CAMA1 (Figures 3H and S8C) and MDA-MB-134 cells (Figure S8D). In addition, by RT-qPCR we confirmed that silencing FGFR1 by siRNA in CAMA1 cells resulted in downregulation of selected FGFR1 target genes (Figure S8E). Taken together, these findings suggest that FGFR1 is part of a chromatin-associated Pol II complex engaged in active transcription.
Using MCF7FGFR1(SP-)(NLS) cells, we next investigated the effect of exogenous nuclear FGFR1 in gene transcription. By ChIP-Seq, we identified 24,475 FGFR1 DNA binding sites in MCF7FGFR1(SP-)(NLS) cells, corresponding to 11,840 genes (Figure 3I). We intersected the FGFR1-bound genes with the differentially regulated transcripts (FDR<0.05) in MCF7FGFR1(SP-)(NLS) vs MCF7EV cells (shown in Figure S2G), finding that 1,009 of the 1,454 upregulated genes and 1,141 of the 1,560 downregulated genes were also bound by FGFR1 at a genomic level (Figure 3I). To examine whether nuclear FGFR1 directly affects gene expression in MCF7FGFR1(SP-)(NLS) cells, we used Binding and Expression Target Analysis (BETA) platform, a computational program which integrates ChIP-Seq and RNA-Seq data (45). Results from this analysis suggested a direct role for genomic-bound FGFR1 in activating gene expression (p=8.01e-06; Figure 3J). Conversely, there was no significant association between FGFR1-bound loci and repressed genes (p=0.139; Figure 3J). Next, to further demonstrate a direct role for genomic-bound FGFR1 in activating gene transcription, we first silenced the expression of FGFR1 using CRISPR/Cas9 in MCF7 cells (MCF7FGFR1 KO) (Figure S8F). Next, we rescued the expression of FGFR1(SP-)(NLS), FGFR1 D432N and wild-type FGFR1 (Figure S8G). By RT-qPCR, we demonstrated that re-expression of FGFR1(SP-)(NLS), FGFR1 D432N and wild-type FGFR1 in MCF7FGFR1 KO cells induced the expression of selected FGFR1 target genes (Figure S8H). GSEA of the 1,009 genes upregulated in MCF7FGFR1(SP-)(NLS) and bound by FGFR1 at a genomic level (shown in Figure 3I) revealed a strong enrichment of estrogen response early (FDR = 2.29e-44) and late (FDR = 6.43e-33) genes (Figure S9A), suggesting that nuclear FGFR1 induces an ERα-associated transcriptional profile that may contribute to endocrine resistance. By RT-qPCR, we confirmed the upregulation of canonical ERα-regulated genes in MCF7FGFR1(SP-)(NLS) cells compared to control cells (Figure S9B). We hypothesized that FGFR1 may regulate the expression of canonical ERα-regulated genes independent of ERα transcription. To support this, we performed RT-qPCR analysis of xenografts treated with fulvestrant (Figure 1F). In both MCF7EV and MCF7FGFR1(SP-)(NLS) xenografts, fulvestrant treatment suppressed ERα protein expression by IHC (Figure S9C). Expression of canonical ERα target genes was downregulated in fulvestrant-treated MCF7EV xenografts but not in fulvestrant-treated MCF7FGFR1(SP-)(NLS) xenografts (Figure S9D), suggesting that nuclear FGFR1 induces the expression of an ERα-associated signature independent of ERα transcription.
Nuclear FGFR1 overlaps with active transcription histone marks
To examine if FGFR1 is associated with active gene transcription, we first superimposed the genomic coordinates of FGFR1-binding sites in CAMA1 cells with those from publicly available ChIP-Seq datasets for histone marks (44). This analysis revealed a strong enrichment for active transcription histone marks, such as H3K4me3 and H3K27ac (Figure S10A). H3K4me3 is commonly enriched around TSS of actively transcribed genes (46). Similarly, H3K27 acetylation is a marker of active regulatory elements and found at both promoters and enhancers (47). We next analyzed the genomic distribution of H3K27ac in CAMA1 cells by ChIP-Seq. Notably, 3961/4175 (94.8%) of FGFR1 peaks overlapped with H3K27ac, suggesting that FGFR1 binding to DNA occurs at genomic loci that are enriched for promoters of actively transcribed genes (Figures 4A and 4B). We also investigated the distribution of the H3K4me3 histone mark in CAMA1 cells. In agreement with the finding of FGFR1 enrichment at TSSs/promoter regions (shown in Figures 2C, D), we identified a strong overlap between FGFR1 and H3K4me3 (4055/4265 sites or 95%), implying that FGFR1 is occupying sites engaged in transcription initiation (Figures 4C and 4D). Taken together, our findings suggest that FGFR1 is recruited to genomic regions where epigenetic modifications, such as H3K27 acetylation and H3K4 trimethylation, co-occur in order to promote a chromatin state accessible for active transcription complexes at promoter regions (Figure 4E).
Figure 4. Nuclear FGFR1 overlaps with active transcription histone marks.
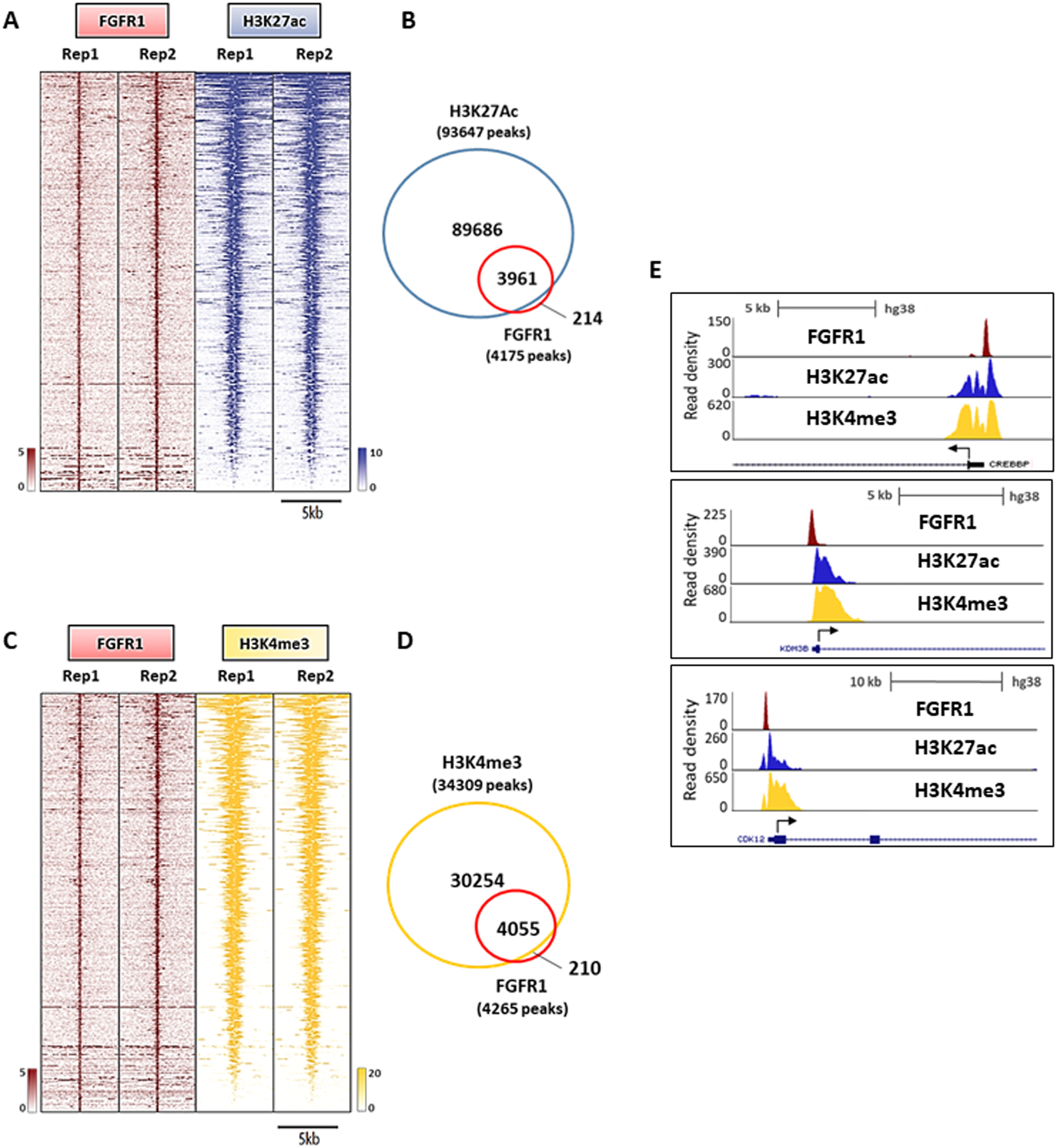
(A) Heatmaps of ChIP-Seq read densities around the FGFR1-bound regions (red) and areas of increased acetylation of H3K27 (H3K27ac, blue) in CAMA1 cells. Two replicates for each antibody are shown. (B) Venn diagram of FGFR1 ChIP-Seq peaks and H3K27ac regions. (C) Heatmaps of ChIP-Seq read densities around the FGFR1-bound regions (red) and areas of increased trimethylation of H3K4 (H3K4me3; yellow) in CAMA1 cells. Two replicates for each antibody are shown. (D) Venn diagram of FGFR1 ChIP-Seq peaks and H3K4me3 regions. (E) Distribution of FGFR1 binding peaks, H3K27ac and H3K4me3 histone marks at the CREEBP, KDM4B and CDK12 promoters (UCSC genome browser).
FOXA1 mediates FGFR1 recruitment to chromatin
We next sought to identify mechanisms regulating FGFR1 recruitment to chromatin. To this end, we further examined the list of FGFR1-interacting proteins identified by MS of FLAG antibody pulldowns from mixed nuclear and chromatin bound CAMA13XFLAG-FGFR1 cell fractions (Supplementary Table S6). Our attention was captured by FOXA1 (Forkhead Box A1) (Figure 5A), a master regulator that modifies chromatin accessibility for transcription factors (48). FOXA1 has been shown to be required for estrogen receptor binding to chromatin and to induce antiestrogen resistance in ER+ breast cancer (49,50). FOXA1 also influences the genomic binding of other transcription factors, such as the androgen receptor in prostate cancer, contributing to castration resistance in these tumors (51). FOXA1 is required for the survival of ER+ breast cancer cells, as demonstrated by Dependency Map Project (https://depmap.org/portal/) (52) (Figure S11A), and has the ability to promote the association of transcription factors with chromatin (53). Thus, we sought to determine whether FOXA1 affects FGFR1 transcriptional function.
Figure 5. FOXA1 mediates FGFR1 recruitment to chromatin.
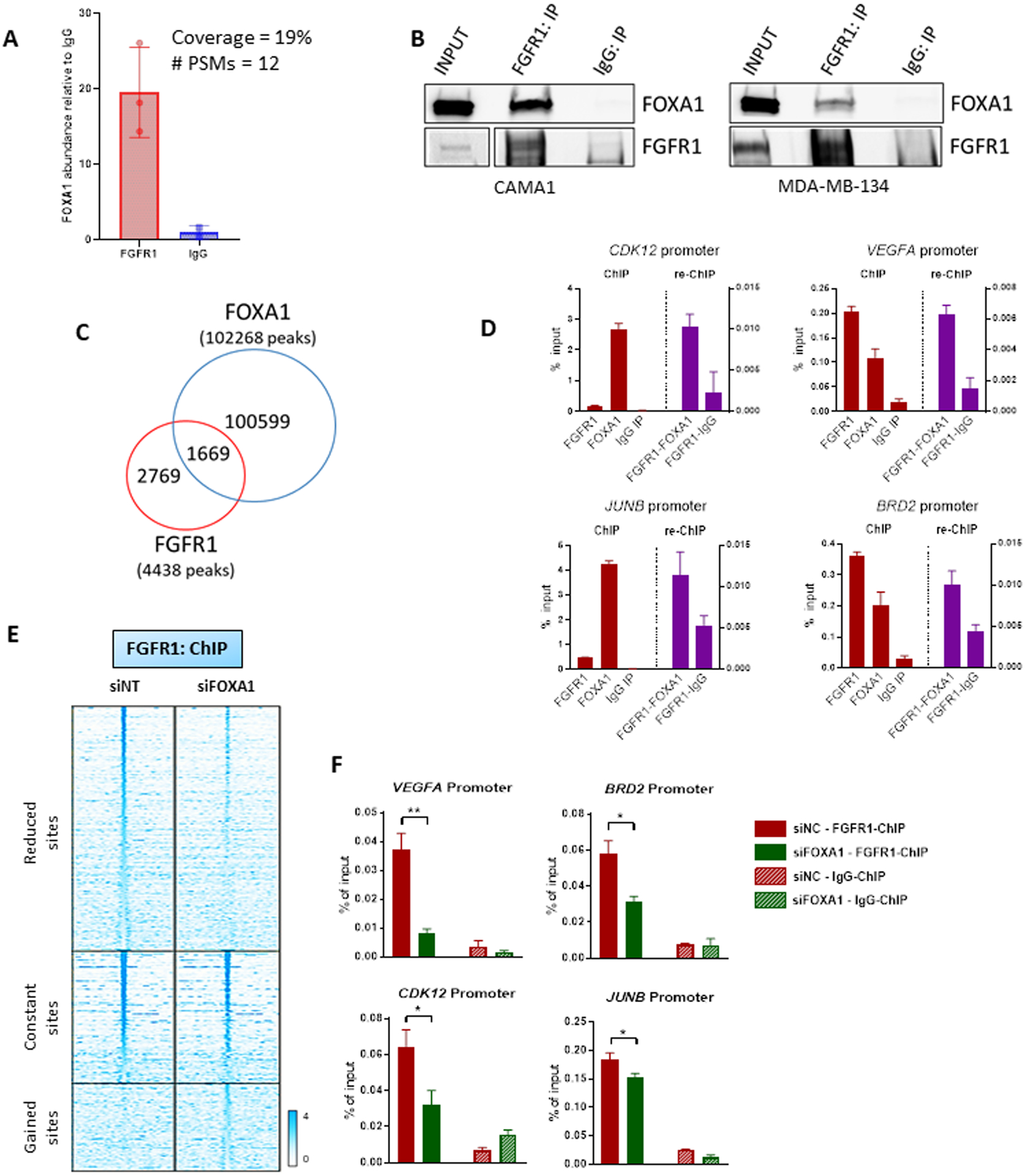
(A) Immunoprecipitation of FLAG-FGFR1 followed by mass spectrometry was conducted on mixed nuclear plus chromatin-bound fractions of CAMA13XFLAG-FGFR1 cells. The plot shows the enrichment of FOXA1 in the FLAG-FGFR1-bound fraction compared to IgG control. (B) Co-precipitation of FGFR1 and FOXA1, followed by western blot analysis, in CAMA1 (left panel) and MDA-MB-134 (right panel) cells. (C) Venn diagram of FGFR1 peaks (red circle), identified by ChIP-Seq (in Figure 2A) and FOXA1 DNA binding loci identified by ChIP-Seq (blue circle) in CAMA1 cells. (D) ChIP-reChIP assay performed by sequential ChIP-qPCR with a FGFR1 antibody followed by an antibody against FOXA1 or normal rabbit IgG, at the BRD2, CDK12, VEGFA and JUNB promoters. Enrichment values expressed as percent (%) of input. (E) Heatmaps of ChIP-Seq read densities around the FGFR1 bound regions in CAMA1 cells transfected with Non Targeting (siNT) or FOXA1 siRNAs. Twenty-four h post-transfection, dishes were replenished with IMEM/10% CSS and cells were collected 48 h later for CHIP. (F) ChIP-qPCR confirmation of reduced FGFR1 binding at selected genomic loci, VEGFA (t test, p=0.0071), BRD2 (p=0.0145), CDK12 (p=0.0114), JUNB (p=0.0252) promoters, upon siRNA-mediated FOXA1 knockdown in CAMA1 cells. Enrichment values expressed as percent (%) of input.
Co-immunoprecipitation of CAMA1 and MDA-134 cell lysates with an FGFR1 antibody followed by FOXA1 western showed association of FOXA1 with FGFR1 (Figures 5B and S11B). We next investigated the FOXA1 genomic distribution in CAMA1 cells by ChIP-Seq. We found that 1,669/4,438 (38%) of FGFR1 DNA binding sites were shared with FOXA1 (Figures 5C and S11C). ChIP-reChIP experiments, conducted by sequential chromatin immunoprecipitation of FGFR1 and FOXA1, confirmed that these two proteins are both present in the transcriptional complex binding at selected genomic loci in CAMA1 and MDA-MB-134 cells (Figures 5D and S11D). Finally, we silenced FOXA1 in CAMA1 cells using siRNA. Notably, 52% of FGFR1 binding peaks were reduced or lost upon FOXA1 suppression (Figure 5E). Using ChIP-qPCR, we confirmed that FGFR1 recruitment to target genomic loci was significantly reduced upon FOXA1 knockdown in CAMA1 and MDA-MB-134 cells (Figures 5F and S11E). Expression of FGFR1 target genes was reduced by siFOXA1 as measured by RT-qPCR (Figure S11F). Of note, FOXA1 knockdown did not have any effect on FGFR1 expression or protein levels (Figure S11G). Altogether, these data suggest a role for FOXA1 in regulating FGFR1 recruitment to chromatin and, consequently, its transcriptional activity.
In addition, since the de novo motif analysis of the FGFR1 DNA binding sites (shown in Figure 2B) identified SP1 as the top enriched motif, we investigated whether SP1 influenced the genomic distribution of FGFR1. We silenced SP1 expression by siRNA in CAMA1 cells (Figure S12A). ChIP-qPCR assays revealed that siRNA-mediated SP1 knockdown did not inhibit FGFR1 recruitment to gene promoters (Figure S12B) identified by FGFR1 ChIP-Seq (shown in Figure 2A and listed in Supplementary Table S2). Further, SP1 knockdown enhanced the FGFR1 binding at CCND1, CDK12, JUNB and PPP6R3 promoters (Figure S12B). Concordant with these results, the expression of these genes was increased upon SP1 knockdown (Figure S12C). These results may suggest a scenario in which SP1 competes with FGFR1 to bind to selected genomic loci, with SP1 having a potential repressive action on FGFR1 recruitment to chromatin and consequent transcriptional activity.
Nuclear FGFR1 activity is enhanced by FGF2 but not affected by receptor tyrosine kinase blockade
Previous studies have shown that growth factors such as EGF and insulin can induce nuclear trafficking of EGFR and Insulin Receptor (IR), respectively, enhancing their transcriptional activity (19,21). FGF2 and FGFR1 have been shown to co-localize in the nucleus of pancreatic cancer cells, promoting an invasive phenotype (14). In contrast to FGFR1, FGF2 contains an NLS peptide sequence, which may promote importin β-mediated translocation of FGFR1 into the nucleus (54). Thus, we examined if growth factor stimulation influences FGFR1 transcriptional function in breast cancer cells. Stimulation with FGF2 increased FGFR1 levels in subcellular nuclear fractions of CAMA1 cells (Figure S13A) as well as the FGFR1-Pol II S5P association measured by co-immunoprecipitation followed by immunoblot analysis (Figure S13B). To further characterize the effect of FGF2 on FGFR1-mediated transcription, we first performed RNA-Seq analysis of FGF2-stimulated CAMA1 cells. This analysis identified 7,923 differentially expressed (DEGs) as a function of FGF2 treatment (FDR <0.05) (Figure 6A). Integration of the RNA-Seq data and FGFR1 ChIP-Seq results from FGF2-stimulated CAMA1 cells (shown in figure S13C) with the BETA platform (45) suggested that FGFR1 DNA binding sites have a significant role in promoting FGF2-induced transcription (Figure 6B, p=0.029). Conversely, there was not a statistical association between FGFR1 DNA binding sites and FGF2-repressed genes (p=0.392). We also employed the Genomic Regions Enrichment of Annotations Tool (GREAT) (55) to interpret the biological functions of FGFR1 DNA binding sites identified by ChIP-Seq in FGF2-treated CAMA1 cells. Consistent with the BETA platform, genes associated with FGFR1 DNA binding sites were highly enriched for signatures of cellular response to growth factor stimulation and activation of the FGFR pathway (Figure 6C and Supplementary Table S7).
Figure 6. Nuclear FGFR1 activity is enhanced by FGF2 but not affected by receptor tyrosine kinase blockade.
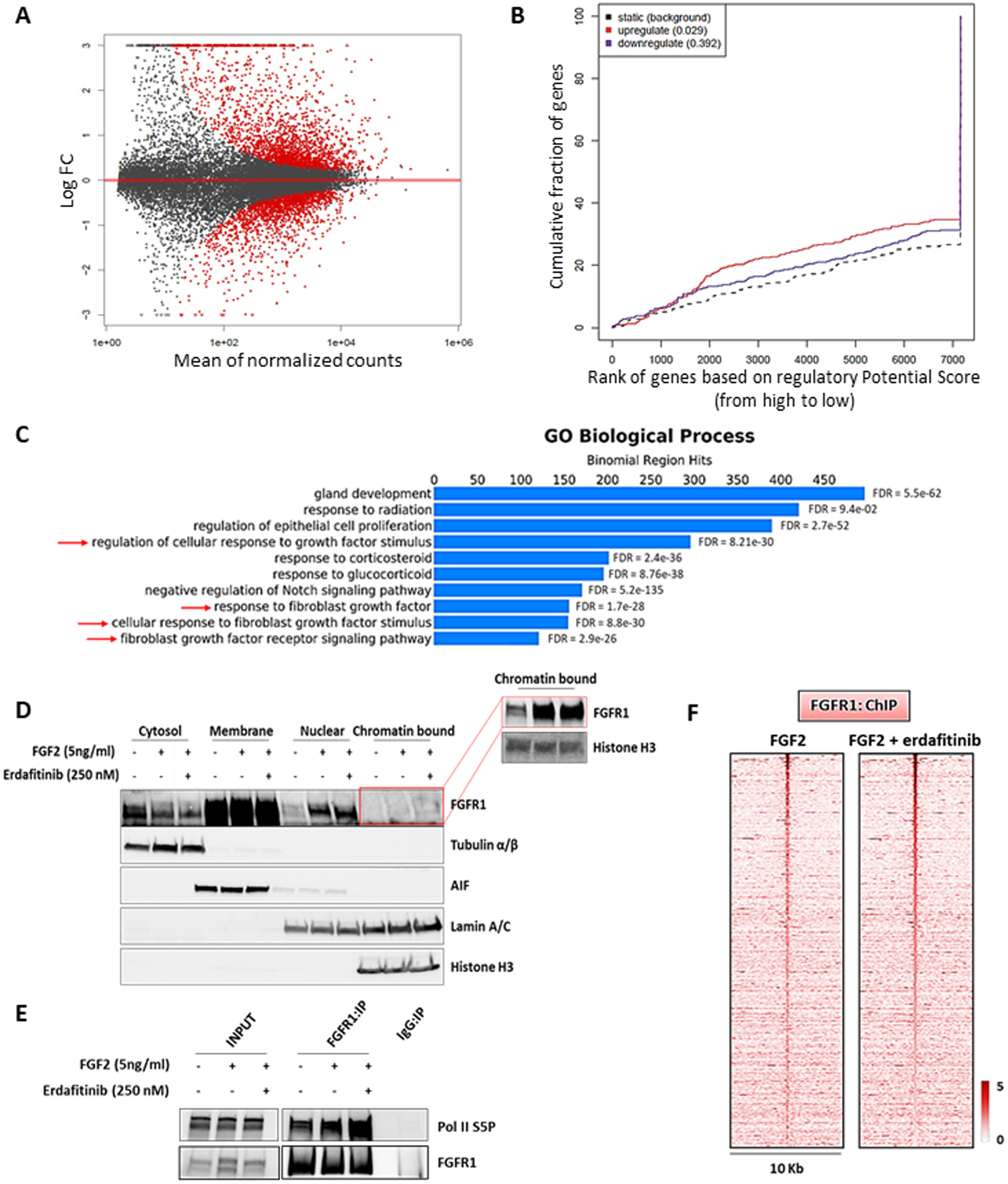
(A) MA plot of M (log ratio) versus A (log mean average) normalized counts, showing differentially expressed genes with p <1e-5 in response to FGF2 (5 ng/ml, 6 h). (B) Prediction of activating or repressing transcription function of FG2-stimulated FGFR1 by BETA platform. FGFR1 ChIP-Seq results from CAMA1 cells stimulated with FGF2 (5 ng/ml, 3 h) and RNA-Seq data from A were integrated. Genomic regions bound by FGFR1 are predicted to modulate expression of genes upregulated upon FGF2 stimulation, but not downregulated genes; p values indicate the significance of the associations compared to background non-regulated genes. (C) GREAT analysis the FGFR1 DNA binding sites identified by FGFR1 ChIP-Seq in CAMA1 cells stimulated with FGF2 (5 ng/ml, 3 h). Top ten enriched Gene Ontology (GO) Biological Processes and binomial FDR values are shown. (D) Subcellular fractionation on CAMA1 cells, showing FGFR1 in cytosolic, membrane, soluble nuclear and chromatin bound compartments, after treatment with FGF2 (5 ng/ml, 3h) ± erdafitinib (250 nM, 3h). Red box shows longer exposure. (E) Co-precipitation of FGFR1 and Pol II S5P from sonicated and nuclease-treated lysates of CAMA1 cells ± FGF2 (5 ng/ml, 3h) ± erdafitinib (250 nM, 3h). (F) Heatmaps of ChIP-Seq read densities around the FGFR1-bound regions in CAMA1 cells treated with FGF2 (5 ng/ml, 3h) ± erdafitinib (250 nM, 3h).
The effects of FGF2 on nuclear translocation and transcriptional regulation suggested a causal link with the FGFR1 tyrosine kinase activity. Thus, we investigated the effect of the pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib (56). Treatment with erdafitinib abolished FGFR1, FRS2α and ERK phosphorylation/activation (Figure S13D) but did not abrogate the FGF2-induced increase in FGFR1 levels in CAMA1 (Figure 6D) and MDA-MB-134 (Figure S13E) cell nuclear fractions. Further, treatment with erdafitinib did not affect the FGF2-induced association of FGFR1 with Pol II S5P association (Figures 6E and S13F) nor the FGFR1 genomic distribution as shown by ChIP-Seq of CAMA1 cells (Figures 6F and S13G). Finally, treatment with erdafitinib did not impair growth of MCF7FGFR1(SP-)(NLS) cells, compared to MCF7EV cells (Figure S13H), suggesting the FGFR1 activated TK is not causally associated with the receptor’s activity in the nucleus.
Discussion
FGFR signaling has multifaceted roles in normal physiology and development, such as embryogenesis, organ development, tissue repair, remodeling, angiogenesis and metabolism (57–60). Gene rearrangements, activating tyrosine kinase domain mutations, and fusions in FGFR genes are oncogenic drivers in several cancer types, such as breast, bladder, and biliary duct tumors (61–63). The canonical FGFR pathway is triggered by activation of the receptor’s tyrosine kinase at the plasma membrane followed by induction of signaling nodes, primarily RAS/RAF/MEK/ERK and PI3K/AKT. However, recent work has shown the presence of FGFR receptors, such as FGFR2, in the nucleus of cancer cells where it may affect gene transcription (64). In medulloblastoma cells, nuclear FGFR1 has been shown to bind CREB-Binding Protein (CBP) and modulate its transcriptional activity (17). We found FGFR1 abundance in the nucleus of breast cancer cells in ER+ primary tumors and PDXs (Figures 1A, S1C and S1E). Therefore, we investigated herein the role of nuclear FGFR1, which may shed light on mechanisms by which aberrant FGFR signaling drives cancer progression and its therapeutic implications.
FGFR1 overexpression has been shown to induce antiestrogen resistance (12,22,23). In line with these reports, we found that high nuclear levels of FGFR1 are associated with a lower response to estrogen suppression in ER+ tumors in patients (Figures 1B and S2A). This correlation between nuclear FGFR1 levels and resistance to estrogen suppression was independent of FGFR1 gene amplification (Figure S2C). We acknowledge that other genomic and non-genomic alterations, not investigated in this clinical cohort may have influenced the results of the correlative analysis reported in Figure 1B. However, in agreement with our clinical findings, MCF7 cells transduced with an FGFR1 expression vector containing a strong nuclear localization sequence exhibited robust growth in the absence of estrogen and, when established as xenografts in ovariectomized mice, were relative resistant to the ER antagonist fulvestrant (Figures 1C–F). In these cells, genomic-bound FGFR1 induced the expression of estrogen receptor early and late genes sets (Figure S9A), suggesting that nuclear FGFR1 contributes to endocrine resistance promoting the ERα-associated transcriptional profile. Further, a gene signature score based on the gene promoters bound by FGFR1 using an approach previously used to predict genes near ERα binding sites in breast cancer (65), was associated with antiestrogen resistance in the METABRIC cohort of patients with ER+ breast cancer treated with antiestrogens (Figures 2G, H). Further suggesting a clinical significance of our findings, the FGFR1-induced polygenic risk score also correlated with FGFR1 copy number alterations in two large datasets of patients, TCGA and METABRIC (Figures S6B, C).
Unlike the findings from Chioni et al (15), nuclear fractions of MCF7 cells transduced with an FGFR1 D432N mutant exhibited higher expression of both full-length (~140 KDa) and the cleaved form (~55 KDa) of FGFR1 (Figure S3A). In agreement with these results, neither GZMB knockdown by siRNA, nor treatment with recombinant human Granzyme B (rhGrB), altered nuclear levels of full-length and cleaved FGFR1 in MCF7, CAMA1 and MDA-MB-134 cells (data not shown). These results suggest that Granzyme B does not contribute to FGFR1 cleavage in these cells.
Mapping of FGFR1 genomic occupancy in ER+/FGFR1-amplified breast cancer cells by ChIP-Seq revealed marked enrichment at TSS/promoter regions (Figures 2C, D). Supporting a role of FGFR1 recruitment in active gene transcription, FGFR1 coupled with phosphorylated RNA-Polymerase II (Figures 3C, S7A–C). This hypothesis was further strengthened by the overlap of FGFR1 with histone marks H3K27ac and H3K4me3 (Figures 4A, C), representative of promoters of actively transcribed genes. These findings are in line with previous studies reporting FGFR1 localization to nuclear speckles, which are sites of active transcription, and colocalization with RNA Pol II in TE671 human medulloblastoma cells (66). To the best of our knowledge, this is the first report describing the genome-wide distribution of FGFR1 in cancer.
The interaction of FGFR1 with CDK7, CDK9, Cyclin H, CTR9, and MED family proteins, revealed by MS (Figure S7D; Supplementary Table S6), further supported the notion that FGFR1 is part of a complex that regulates gene transcription. FGFR1 does not harbor a bona fide DNA binding domain. Thus, it is likely that FGFR1 is tethered to promoters through interaction with other proteins, presumably general transcription factors. We also showed that FOXA1 plays a significant role in the recruitment of FGFR1 to chromatin. FOXA1 orchestrates transcription factor binding to chromatin and drives gene expression that results in endocrine therapy resistance in cancer (67,68). FOXA1 pioneering function has been shown to be independent on steroids and hormone signaling (53) but can be affected by mitogenic signals (65). Also, since FOXA1 is currently considered undruggable, understanding how extracellular and intracellular signals modulate FOXA1-FGFR1 interaction may provide rationale to test new therapeutic strategies to inhibit FGFR1 transcriptional function.
Our findings provide new insight into the biology and oncogenic functions of RTKs. For example, nuclear EGFR has been associated with resistance to the anti-EGFR antibody cetuximab (69). EGFR nuclear translocation has been associated with resistance to chemotherapy and ionizing radiation in lung cancer cells, primarily by modulating the DNA repair process (70). Nuclear HER2 was reported to be involved in resistance to the HER2 antibody trastuzumab in HER2+ breast cancer cells (71). More recently, Hancock et al. reported an unexpected role of the nuclear insulin receptor (INSR) as a transcription factor that induces gene expression associated with insulin-related functions and insulin resistance (19). Previous studies showed that ligand activation of membrane-bound FGFR1 can induce its migration to the nucleus, mainly by importin β family proteins (14,54). There is also evidence that Sec61 α family proteins can mediate translocation of newly synthesized FGFR1 from the endoplasmic reticulum to the nucleus (72). A complete understanding of the factors regulating these mechanisms could identify new therapeutic targets in order to suppress FGFR1 nuclear localization and FGFR1-induced gene expression.
FGFR TKIs have been clinically effective against cancers harboring FGFR alterations acting as dominant oncogenic drivers, such as FGFR3 mutations or fusions in urothelial carcinoma, or FGFR2 fusions in cholangiocarcinoma (73,74). Even though testing has been more limited in patients with FGFR1-amplified breast cancer, FGFR TKIs have not shown any meaningful clinical activity against these tumors (75,76). The reasons for this failure may be attributed to various reasons. First, the inhibitors that have been tested are not specific and exhibit non-FGFR associated toxicities that limit their dosing (1). Second, it is not clear to what extent FGFR1-amplified tumors depend on canonical FGFR signaling. Third, there is not agreement regarding the optimal threshold of copy number to be used to stratify patients enrolled in clinical trials. Results from the study showed herein may shed light on an unconventional role of FGFR1 in these tumors, different from the classical RTK-induced signaling and, as such, have translational implications for drug development. For example, treatment with erdafitinib, a pan-FGFR TKI currently in clinical trials in breast cancer, did not affect nuclear FGFR1 levels and its genomic distribution (Figures 6D, F), nor growth of cells overexpressing nuclear FGFR1 (Figure S13H), ultimately suggesting that FGFR1 transcriptional activity is not influenced by therapeutic inhibition of the FGFR1 tyrosine kinase. We speculate this could explain at least in part the low activity of FGFR TKIs against FGFR1 overexpressing tumors. Taken together, these data provide a rationale for the development of targeted therapies that inhibit nuclear FGFR1 function in ER+ breast cancer.
Supplementary Material
Acknowledgements
We thank the assistance of the UTSW Proteomics Core. We acknowledge Professor Yonghao Yu (Department of Biochemistry at UT Southwestern Medical Center) for sharing the pCDNA5-zz-FLAG plasmid. We thank Professor Michal Stachowiak (State University of New York) for sharing the pCDNA3.1(-)FGFR1(SP-)(NLS) plasmid. We thank Professor Alana Welm (University of Utah) for sharing the HCI-003 and HCI-011 PDXs. We received the following financial support to conduct this work: UTSW Simmons Cancer Center P30 CA142543, CPRIT RR170061 (C.L.A.), NCI Breast SPORE P50 CA098131, Vanderbilt-Ingram Cancer Center P30 CA68485, Susan G. Komen Breast Cancer Foundation SAB1800010 (C.L.A.), Breast Cancer Research Foundation (C.L.A.), NCI R01CA224899 (C.L.A. and A.B.H.), Susan G. Komen Postdoctoral Fellowship PDF17487926 (K.M.L.), Associazione Italiana per la Ricerca sul Cancro (AIRC) Investigator Grant IG2018-21339 (R.B.) and AIRC My First Grant MFGA2018-21505 (L.F.).
Footnotes
Conflicts of Interest: A.B.H. receives research grant support from Takeda and has received a travel grant from Puma Biotechnology, Inc.; A.G.Z. receives research grant support from Pfizer; receives travel expenses support from Pfizer and Novartis; has consultory role for Novartis and AstraZeneca; C.L.A receives or has received research grants from Pfizer, Lilly and Takeda; holds minor stock options in Provista and Y-TRAP; serves or has served in an advisory role to Novartis, Merck, Lilly, Daiichi Sankyo, Taiho Oncology, OrigiMed, Puma Biotechnology, Immunomedics, AstraZeneca, and Sanofi; and reports scientific advisory board remuneration from the Susan G. Komen Foundation.
References
- 1.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer [Internet]. 2010;10:116–29. Available from: http://www.nature.com/articles/nrc2780 [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, et al. Crystal Structure of a Ternary FGF-FGFR-Heparin Complex Reveals a Dual Role for Heparin in FGFR Binding and Dimerization. Mol Cell [Internet]. 2000;6:743–50. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097276500000733 [DOI] [PubMed] [Google Scholar]
- 3.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev [Internet]. 2005;16:139–49. Available from: https://linkinghub.elsevier.com/retrieve/pii/S135961010500002X [DOI] [PubMed] [Google Scholar]
- 4.Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, et al. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature [Internet]. 1992;358:678–81. Available from: http://www.nature.com/articles/358678a0 [DOI] [PubMed] [Google Scholar]
- 5.Klint P Signal transduction by fibroblast growth factor receptors. Front Biosci [Internet]. 1999;4:d165. Available from: http://www.bioscience.org/1999/v4/d/klint/list.htm [DOI] [PubMed] [Google Scholar]
- 6.Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene [Internet]. 2000;19:3309–20. Available from: http://www.nature.com/articles/1203650 [DOI] [PubMed] [Google Scholar]
- 7.Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, et al. FGFR1 Emerges as a Potential Therapeutic Target for Lobular Breast Carcinomas. Clin Cancer Res [Internet]. 2006;12:6652–62. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-06-1164 [DOI] [PubMed] [Google Scholar]
- 8.Yang W, Yao Y-W, Zeng J-L, Liang W-J, Wang L, Bai C-Q, et al. Prognostic value of FGFR1 gene copy number in patients with non-small cell lung cancer: a meta-analysis. J Thorac Dis [Internet]. 2014;6:803–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24977006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorringe KL, Jacobs S, Thompson ER, Sridhar A, Qiu W, Choong DYH, et al. High-Resolution Single Nucleotide Polymorphism Array Analysis of Epithelial Ovarian Cancer Reveals Numerous Microdeletions and Amplifications. Clin Cancer Res [Internet]. 2007;13:4731–9. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-07-0502 [DOI] [PubMed] [Google Scholar]
- 10.Simon R, Richter J, Wagner U, Fijan A, Bruderer J, Schmid U, et al. High-throughput tissue microarray analysis of 3p25 (RAF1) and 8p12 (FGFR1) copy number alterations in urinary bladder cancer. Cancer Res [Internet]. 2001;61:4514–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11389083 [PubMed] [Google Scholar]
- 11.Comprehensive molecular portraits of human breast tumours. Nature [Internet]. 2012;490:61–70. Available from: http://www.nature.com/articles/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV., et al. Genomic profiling of ER + breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med [Internet]. 2017;9:eaai7993. Available from: https://stm.sciencemag.org/lookup/doi/10.1126/scitranslmed.aai7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer [Internet]. 2017;17:318–32. Available from: http://www.nature.com/articles/nrc.2017.8 [DOI] [PubMed] [Google Scholar]
- 14.Coleman SJ, Chioni A, Ghallab M, Anderson RK, Lemoine NR, Kocher HM, et al. Nuclear translocation of FGFR 1 and FGF 2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol Med [Internet]. 2014;6:467–81. Available from: https://onlinelibrary.wiley.com/doi/10.1002/emmm.201302698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chioni A-M, Grose R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J Cell Biol [Internet]. 2012;197:801–17. Available from: https://rupress.org/jcb/article/197/6/801/36836/FGFR1-cleavage-and-nuclear-translocation-regulates [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terranova C, Narla ST, Lee Y-W, Bard J, Parikh A, Stachowiak EK, et al. Global Developmental Gene Programing Involves a Nuclear Form of Fibroblast Growth Factor Receptor-1 (FGFR1). van Wijnen A, editor. PLoS One [Internet]. 2015;10:e0123380. Available from: https://dx.plos.org/10.1371/journal.pone.0123380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang X, Stachowiak EK, Dunham-Ems SM, Klejbor I, Stachowiak MK. Control of CREB-binding Protein Signaling by Nuclear Fibroblast Growth Factor Receptor-1. J Biol Chem [Internet]. 2005;280:28451–62. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925820565447 [DOI] [PubMed] [Google Scholar]
- 18.Carpenter G, Liao H-J. Receptor Tyrosine Kinases in the Nucleus. Cold Spring Harb Perspect Biol [Internet]. 2013;5:a008979–a008979. Available from: http://cshperspectives.cshlp.org/lookup/doi/10.1101/cshperspect.a008979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hancock ML, Meyer RC, Mistry M, Khetani RS, Wagschal A, Shin T, et al. Insulin Receptor Associates with Promoters Genome-wide and Regulates Gene Expression. Cell [Internet]. 2019;177:722–736.e22. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867419302132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redmond AM, Omarjee S, Chernukhin I, Le Romancer M, Carroll JS. Analysis of HER2 genomic binding in breast cancer cells identifies a global role in direct gene regulation. Mantovani R, editor. PLoS One [Internet]. 2019;14:e0225180. Available from: https://dx.plos.org/10.1371/journal.pone.0225180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin S-Y, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol [Internet]. 2001;3:802–8. Available from: http://www.nature.com/articles/ncb0901-802 [DOI] [PubMed] [Google Scholar]
- 22.Drago JZ, Formisano L, Juric D, Niemierko A, Servetto A, Wander SA, et al. FGFR1 Amplification Mediates Endocrine Resistance but Retains TORC Sensitivity in Metastatic Hormone Receptor–Positive (HR + ) Breast Cancer. Clin Cancer Res [Internet]. 2019;25:6443–51. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-19-0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res [Internet]. 2010;70:2085–94. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-09-3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, et al. Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER + Breast Cancer. Clin Cancer Res [Internet]. 2017;23:6138–50. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-17-1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep [Internet]. 2017;7:16878. Available from: http://www.nature.com/articles/s41598-017-17204-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med [Internet]. 2002;8:1323–8. Available from: http://www.nature.com/articles/nm791 [DOI] [PubMed] [Google Scholar]
- 27.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods [Internet]. 2012;9:357–9. Available from: http://www.nature.com/articles/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics [Internet]. 2009;25:2078–9. Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol [Internet]. 2008;9:R137. Available from: http://genomebiology.biomedcentral.com/articles/10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell [Internet]. 2010;38:576–89. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097276510003667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen L, Shao N, Liu X, Nestler E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics [Internet]. 2014;15:284. Available from: http://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vilcaes AA, Chanaday NL, Kavalali ET. Interneuronal exchange and functional integration of synaptobrevin via extracellular vesicles. Neuron [Internet]. 2021;109:971–983.e5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0896627321000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. Arch Pathol Lab Med [Internet]. 2018;142:1364–82. Available from: http://meridian.allenpress.com/aplm/article/142/11/1364/103146/Human-Epidermal-Growth-Factor-Receptor-2-Testing [DOI] [PubMed] [Google Scholar]
- 34.Peng H, Moffett J, Myers J, Fang X, Stachowiak EK, Maher P, et al. Novel Nuclear Signaling Pathway Mediates Activation of Fibroblast Growth Factor-2 Gene by Type 1 and Type 2 Angiotensin II Receptors. Mumby M, editor. Mol Biol Cell [Internet]. 2001;12:449–62. Available from: https://www.molbiolcell.org/doi/10.1091/mbc.12.2.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc [Internet]. 2013;8:2281–308. Available from: http://www.nature.com/articles/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet [Internet]. 2007;8:424–36. Available from: http://www.nature.com/articles/nrg2026 [DOI] [PubMed] [Google Scholar]
- 37.Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res [Internet]. 2012;22:2399–408. Available from: http://genome.cshlp.org/cgi/doi/10.1101/gr.138776.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev [Internet]. 2011;25:1010–22. Available from: http://genesdev.cshlp.org/cgi/doi/10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landolin JM, Johnson DS, Trinklein ND, Aldred SF, Medina C, Shulha H, et al. Sequence features that drive human promoter function and tissue specificity. Genome Res [Internet]. 2010;20:890–8. Available from: http://genome.cshlp.org/cgi/doi/10.1101/gr.100370.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahn S Structure and mechanism of the RNA polymerase II transcription machinery. Nat Struct Mol Biol [Internet]. 2004;11:394–403. Available from: http://www.nature.com/articles/nsmb763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buratowski S Progression through the RNA Polymerase II CTD Cycle. Mol Cell [Internet]. 2009;36:541–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097276509007849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komarnitsky P Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev [Internet]. 2000;14:2452–60. Available from: http://www.genesdev.org/cgi/doi/10.1101/gad.824700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol [Internet]. 2015;16:167–77. Available from: http://www.nature.com/articles/nrm3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res [Internet]. 2019;47:D729–35. Available from: https://academic.oup.com/nar/article/47/D1/D729/5193328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc [Internet]. 2013;8:2502–15. Available from: http://www.nature.com/articles/nprot.2013.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet [Internet]. 2007;39:311–8. Available from: http://www.nature.com/articles/ng1966 [DOI] [PubMed] [Google Scholar]
- 47.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature [Internet]. 2015;518:317–30. Available from: http://www.nature.com/articles/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of Compacted Chromatin by Early Developmental Transcription Factors HNF3 (FoxA) and GATA-4. Mol Cell [Internet]. 2002;9:279–89. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097276502004598 [DOI] [PubMed] [Google Scholar]
- 49.Fu X, Pereira R, De Angelis C, Veeraraghavan J, Nanda S, Qin L, et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc Natl Acad Sci [Internet]. 2019;116:26823–34. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1911584116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet [Internet]. 2011;43:27–33. Available from: http://www.nature.com/articles/ng.730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson JLL, Hickey TE, Warren AY, Vowler SL, Carroll T, Lamb AD, et al. Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene [Internet]. 2014;33:5666–74. Available from: http://www.nature.com/articles/onc2013508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell [Internet]. 2017;170:564–576.e16. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867417306517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glont S-E, Chernukhin I, Carroll JS. Comprehensive Genomic Analysis Reveals that the Pioneering Function of FOXA1 Is Independent of Hormonal Signaling. Cell Rep [Internet]. 2019;26:2558–2565.e3. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2211124719302074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reilly JF, Maher PA. Importin β–Mediated Nuclear Import of Fibroblast Growth Factor Receptor. J Cell Biol [Internet]. 2001;152:1307–12. Available from: https://rupress.org/jcb/article/152/6/1307/45828/Importin-βMediated-Nuclear-Import-of-Fibroblast [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol [Internet]. 2010;28:495–501. Available from: http://www.nature.com/articles/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perera TPS, Jovcheva E, Mevellec L, Vialard J, De Lange D, Verhulst T, et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol Cancer Ther [Internet]. 2017;16:1010–20. Available from: http://mct.aacrjournals.org/lookup/doi/10.1158/1535-7163.MCT-16-0589 [DOI] [PubMed] [Google Scholar]
- 57.Lan T, Morgan DA, Rahmouni K, Sonoda J, Fu X, Burgess SC, et al. FGF19, FGF21, and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab [Internet]. 2017;26:709–718.e3. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413117305569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oladipupo SS, Smith C, Santeford A, Park C, Sene A, Wiley LA, et al. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc Natl Acad Sci [Internet]. 2014;111:13379–84. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1324235111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ono K, Kita T, Sato S, O’Neill P, Mak S-S, Paschaki M, et al. FGFR1-Frs2/3 Signalling Maintains Sensory Progenitors during Inner Ear Hair Cell Formation. Cheah KSE, editor. PLoS Genet [Internet]. 2014;10:e1004118. Available from: https://dx.plos.org/10.1371/journal.pgen.1004118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao M, Ross JT, Itkin T, Perry JM, Venkatraman A, Haug JS, et al. FGF signaling facilitates postinjury recovery of mouse hematopoietic system. Blood [Internet]. 2012;120:1831–42. Available from: https://ashpublications.org/blood/article/120/9/1831/30912/FGF-signaling-facilitates-postinjury-recovery-of [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov [Internet]. 2017;7:252–63. Available from: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-16-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pal SK, Rosenberg JE, Hoffman-Censits JH, Berger R, Quinn DI, Galsky MD, et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov [Internet]. 2018;8:812–21. Available from: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-18-0229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell [Internet]. 2018;34:427–438.e6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1535610818303684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee JE, Shin S-H, Shin H-W, Chun Y-S, Park J-W. Nuclear FGFR2 negatively regulates hypoxia-induced cell invasion in prostate cancer by interacting with HIF-1 and HIF-2. Sci Rep [Internet]. 2019;9:3480. Available from: http://www.nature.com/articles/s41598-019-39843-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature [Internet]. 2012;481:389–93. Available from: http://www.nature.com/articles/nature10730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Somanathan S, Stachowiak EK, Siegel AJ, Stachowiak MK, Berezney R. Nuclear matrix bound fibroblast growth factor receptor is associated with splicing factor rich and transcriptionally active nuclear speckles. J Cell Biochem [Internet]. 2003;90:856–69. Available from: http://doi.wiley.com/10.1002/jcb.10672 [DOI] [PubMed] [Google Scholar]
- 67.Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci [Internet]. 2016;113:E6600–9. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1612835113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 Translates Epigenetic Signatures into Enhancer-Driven Lineage-Specific Transcription. Cell [Internet]. 2008;132:958–70. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867408001189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene [Internet]. 2009;28:3801–13. Available from: http://www.nature.com/articles/onc2009234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liccardi G, Hartley JA, Hochhauser D. EGFR Nuclear Translocation Modulates DNA Repair following Cisplatin and Ionizing Radiation Treatment. Cancer Res [Internet]. 2011;71:1103–14. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-10-2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cordo Russo RI, Béguelin W, Díaz Flaqué MC, Proietti CJ, Venturutti L, Galigniana N, et al. Targeting ErbB-2 nuclear localization and function inhibits breast cancer growth and overcomes trastuzumab resistance. Oncogene [Internet]. 2015;34:3413–28. Available from: http://www.nature.com/articles/onc2014272 [DOI] [PubMed] [Google Scholar]
- 72.Stachowiak MK, Fang X, Myers JM, Dunham SM, Berezney R, Maher PA, et al. Integrative nuclear FGFR1 signaling (INFS) as a part of a universal ?feed-forward-and-gate? signaling module that controls cell growth and differentiation. J Cell Biochem [Internet]. 2003;90:662–91. Available from: http://doi.wiley.com/10.1002/jcb.10606 [DOI] [PubMed] [Google Scholar]
- 73.Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med [Internet]. 2019;381:338–48. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1817323 [DOI] [PubMed] [Google Scholar]
- 74.Goyal L, Shi L, Liu LY, Fece de la Cruz F, Lennerz JK, Raghavan S, et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion–Positive Intrahepatic Cholangiocarcinoma. Cancer Discov [Internet]. 2019;9:1064–79. Available from: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-19-0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with Dovitinib (TKI258): Preclinical and Clinical Data in Breast Cancer. Clin Cancer Res [Internet]. 2013;19:3693–702. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-13-0190 [DOI] [PubMed] [Google Scholar]
- 76.Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord J-P, Hidalgo M, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Stu. J Clin Oncol [Internet]. 2017;35:157–65. Available from: https://ascopubs.org/doi/10.1200/JCO.2016.67.2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics [Internet]. 2013;14:7. Available from: http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available in the GEO repository, code GSE148313 and can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148313
All the analyzed ChIP-Seq and RNA-Seq data can be found in Supplementary material as Supplementary Tables S2, S3.