

Abstract
Background:
Immunotherapies mediate the regression of human tumors through recognition of tumor antigens by immune cells that trigger an immune response. Mutations in the RAS oncogenes occur in about 30% of all cancer patients. These mutations play an important role in both tumor establishment and survival and are commonly found in hotspots.
Purpose:
Discovering T cell receptors (TCR) that recognize shared mutated RAS antigens presented on major histocompatibility complex (MHC) class I and class II molecules are thus promising reagents for “off-the-shelf” adoptive cell therapies (ACT) following insertion of the TCR into lymphocytes.
Experimental Design:
In this ongoing work, we screened for RAS antigen recognition in tumor-infiltrating lymphocytes (TIL) or by in-vitro stimulation (IVS) of peripheral blood lymphocytes (PBLs). TCRs recognized mutated RAS were identified from the reactive T cells. The TCRs were then reconstructed and virally transduced into PBLs and tested.
Results:
Here, we detect and report multiple novel TCRs sequences that recognize non-synonymous mutant RAS hotspot mutations with high avidity and specificity and identify the specific class-I and II MHC restriction elements involved in the recognition of mutant RAS.
Conclusions:
The TCR library directed against RAS hotspot mutations described here recognize RAS mutations found in about 45% of the Caucasian population and about 60% of the Asian population and represent promising reagents for “off-the-shelf” adoptive cell therapies (ACT).
Keywords: T cell, TCR, RAS hotspot mutation
Introduction
Most approaches to cancer immunotherapy, including adoptive cell therapy (ACT), rely on T cell target-specific recognition (1–3). Neoantigens, which are encoded by mutated genes that exist only in cancer cells can be a good target for T cells (4–6). Administration of tumor-infiltrating lymphocytes (TILs) enriched with T cells that recognize tumor-specific neoantigens can mediate long term objective responses in patients with various metastatic cancers, including complete remissions, that are dependent on specific T cell receptor (TCR) recognition of the MHC-cancer antigen complex (6–17). Although the majority of cancer antigens are unique and specific only to the patient in which they are found (18), a relatively small group of mutations occur at specific amino acid positions in genes that play a critical role in tumorigenesis, referred to as hotspot or driver mutations. Hotspots mutations in members of the RAS family represent proteins that are involved in the transduction of signals that are essential for tumor establishment and survival and therefore are likely to be clonally expressed in all tumor cells present in the cancer patient (19,20).
The incidence of RAS mutations varies with cancer histology; however they are found in about 30% of all cancer patients (20–23). Up to 94% of pancreatic cancers and up to 45% of colon cancer express a RAS mutation(19–21,23–28). The human RAS super-family proteins (KRAS/NRAS/HRAS) have an identical amino acid sequence in positions 1 through 86 (20,25), and over 99% of all mutations in this gene family occur at positions 12, 13 or 61 (COSMIC database (29)) within the domains responsible for GTP dephosphorylation into GDP, resulting in constitutive activation of the RAS protein (20–23,25,28,30). Since these are gain of function mutations, one mutated allele expressed at normal levels is sufficient to support cancer growth and establishment (22,31,32). Mutations in RAS predominantly occur in KRAS (85%), but also in NRAS or HRAS (11 and 4% respectively). The most common mutations in KRAS are G12D (35%), G12V (24%), G13D (13%), G12C (12%), G12A (6%), G12S (5%), G12R (3%) (22). While a small molecule targeting the RASG12C mutation is currently being evaluated in patient clinical trials, there is no available treatment for other more common RAS hotspot mutations, such as G12D or G12V (33–35).
Previously, we have shown that ACT with TIL targeting a KRAS neoepitope recognized in the context of the HLA-C*08:02 restriction element expressed by approximately 3% of the patient population, resulted in durable tumor regression in a patient with metastatic colon cancer, who is still alive and disease-free over four years later (17). In most patients bearing RAS mutant tumors, screening of TIL has failed to result in the identification of mutant RAS-specific T cells, which is likely due to the low frequency of such T cells (18).
The administration of peripheral blood lymphocytes (PBLs) virally transduced with genes encoding for TCRs or chimeric antigen receptors (CARs) can mediate regression of established cancers (36–40). Although specific TCRs recognizing RAS hotspot mutations could potentially be used in ACT as an “off-the-shelf” reagent in an allogeneic setting for patients whose tumors express the relevant hotspot mutation and appropriate MHC restriction element, there are few RAS hotspot mutation-reactive TCRs isolated from TIL reported in the literature (41,42). The use of a patient’s PBL, which will be genetically engineered to express a specific anti-RAS TCR has the potential advantage of generating highly enriched tumor-reactive cells in a less differentiated state than cells present in TIL. Here we describe the identification of multiple CD4 and CD8 T cell reactivities targeting RAS hotspot antigens by TIL screening and in vitro stimulation (IVS) of PBLs from cancer patients harboring a RAS mutation in their tumor. We report the TCR sequences and cognate MHC restrictions of a validated TCR library targeting products of RAS mutations with little to no reactivity against wild-type RAS. This library has the potential to treat about 45% of the Caucasian population and about 60% of the Asian population in the United States bearing RAS mutatant cancers.
Materials and Methods
Cell media and reagents
The media and buffer: RPMI-1640, DMEM, AIM-V, phosphate-buffered saline (PBS), OPTI-MEM. Media supplementary materials: Pen Strep (penicillin and streptomycin), L-glutamine, HEPES buffer saline, gentamicin, MEM non-essential amino acid and 2-mercaptoethanol (all purchased from Gibco, Thermo Fisher), Ethylenediaminetetraacetic acid (EDTA) (Corning), Human AB serum (Valley Biomedical, Inc), Fetal bovine serum (FBS) (Sigma-Aldrich), recombinant Interleukin 2 (IL-2) (Prometheus), recombinant Interleukin 4 (IL-4) (Peprotech), recombinant Granulocyte-macrophage colony-stimulating factor GM-CSF (Leukine).
RPMI complete media was comprised of RPMI-1640 supplemented with 5% human AB serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 25 mM HEPES, and 10 μg/ml gentamicin.
Dendritic cell (DC) media was comprised of RPMI complete media supplemented with 800 IU/ml GM-CSF, and 200 U/ml IL-4.
Tissue culture media was comprised of RPMI 1640 supplemented with 10% FBS, non-essential amino acid, 1mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 10 μg/ml gentamicin and 55 uM 2-mercaptoethanol.
Cell line media was comprised of DMEM containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 12.5 mM HEPES, 110mg/ml sodium pyruvate and 2 mM L-glutamine.
T cell media was comprised of a 1:1 mix of RPMI-1640 and AIM-V supplemented with 5% human AB serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 12.5 mM HEPES, 10 μg/ml gentamicin and 5% human serum. Unless otherwise noted, TIL were grown in T cell media supplemented with 3000 IU/ml IL2 andPBLs were grown in T cell media supplemented with 300 IU/ml IL2.
FACS buffer was comprised of sterile PBS, 1% FBS, and 2 mM EDTA.
Freezing media was comprised of 90% FBS and 10% DMSO.
All media were 0.22 μm-filtered.
Antibodies, flow cytometry, and cell sorting
The anti-mouse TCRβ (mTCR)- PerCp-Cy5.5, CD8a-eFluor 450, were purchased from eBioscience. CD3-APC-Cy7, CD4- PE/FITC/APC, 4–1BB- APC/PE, CD8- PE-Cy7/FITC, OX40- FITC/PE-Cy7, CD62L-PE, and CD45RO-APC/BV421 were purchased from BD (Becton Dickinson Biosciences). CD3-Alexa Fluor 700, CD62L-PE-Cy7, and the live/dead stains- DAPI/PI were purchased from Biolegend.
For FACS sample preparation, cells were harvested and washed with FACS buffer. Cells were resuspended in FACS buffer at a concentration of 1e6–50e6 cells/ml and extracellular fluorescent-conjugated primary antibodies were added and mixed in. After a 20–60 min incubation at 4oC, which was also protected from light exposure, the samples’ cells were washed and resuspended with FACS buffer.
The flow cytometry assays were performed on FACSCanto I/II (BD Biosciences) and the acquired data were analyzed with FlowJo software (TreeStar). Cells were sorted to: Live/CD3+, separated for CD4+ or CD8+, TEMRA/ TEM/TCM based on sorting out CD62L+/CD45RO- (T naïve cells). Cocultures were sorted for enrichment or into single reactive cells based on 41BB+/OX40+ (both double and single-positive cells), Live/CD3+, separated for CD4+ or CD8+ by SH800S/MA900 instrument (Sony Biotechnology) or by FACSAria II (BD).
Construction of genes, peptides, and in vitro transcription
The amino acid sequences from peptides used are shown in Supplementary Fig 1. ME sequence predictions were generated for RAS mutated sequences (NetMHC/4.0) and ME (9–12 amino acids short peptides) predicted to attach to common MHC-I or to multiple MHCs. Peptides with predicted binding affinity percentage ranks of less than 0.5 are generally denoted as strong binders while peptides with ranks of between 0.5 and 2 are denoted as weak binders. Long peptides (LP) were 24–25-mer, designed with the mutated amino acid in the middle and 12 normal amino acids upstream and downstream. LP were ordered for RAS hotspots mutations found in more than 0.5% of RAS mutated cancer patients (G12D, G12V, G12C, G12A, G12S, G13D, G13R, G13V, Q61R, Q61L, Q61K, and Q61H) and the equivalent WT (G12, G13, Q61). All peptides were ordered from GenScript or JPT and were HPLC purified (>90%). For tandem minigenes (TMG) construction, each RAS hotspot mutation and the equivalent WT minigene encoding the same LP sequences were composed sequentially into one gene (Supplementary Fig 1.) and cloned into the pcRNA2SL-GFP plasmid. Full length (FL) RAS genes encoding to the mutated or wild type KRAS were synthesized and cloned into pcRNA2SL-GFP using EcoRI and BamHI. Gene synthesis and cloning was done by GenScript. For mRNA in vitro transcription (IVT), we used mMessage mMachine T7 Ultra kit (Thermo Fisher Scientific) and all procedures in the manufacturer instructions were followed. Briefly, the pcRNA2SL constructed plasmids were linearized with Not-I restricted enzyme, followed by ethanol precipitation. Next, we used 1 μg of linearized DNA to generate IVT mRNA. The mRNA was ethanol precipitated and resuspended at 1 μg/μl.
Generation of tumor-infiltrating lymphocytes (TIL)
TILs were generated as described elsewhere (18,43) and are further explained at Supplementary Materials and Methods.
Generation of antigen-presenting cells
Immature dendritic cells (DC) were generated in the laboratory using a standard protocol of adherence method. Apheresis samples were thawed, washed, set to 5–10e6 cells/ml with AIM-V media (Life Technologies), and 1.75–2 e8 viable cells were incubated in T175 flasks (Corning Inc.) at 37oC. After 2 hours, the flasks were washed 2–3 times vigorously with sterile PBS to collect non-adherent cells for T cell sorting. For the adherent cells, 30 ml DC media were added, and cells were incubated at 37ºC, 5% CO2. On day 4 or 5, cells were harvested and used or frozen for further use. DCs were seeded into low-attachment 96-, 12-, or 6-well plates for peptide loading with 0.1–10 μg/μl peptide for 2–12 hours at 37ºC, 5% CO2. Alternatively, mRNA transfection with TMG or FL was done with mMessengerMAX reagent (Life Technologies) according to the manufacturer’s instructions, and cells were incubated for 8–20 hours at 37oC, 5% CO2. Before the coculture experiment (for analysis/sorting/IVS), DCs were harvested by washing twice with 0.9mM ethylenediaminetetraacetic (EDTA) in PBS. DCs were resuspended in T cell media at a concentration of 2.5e5–1e6 cells/ml.
Memory T cell preparation
Nonadherent cells (from the antigen-presenting cells protocol) were spun down, resuspended in T cell medium, and rested overnight at 37°C, 5% CO2. Cells were prepared and sorted (as described above) for T memory (TEMRA/TEM/TCM) cells. Sorted CD4 or CD8 T memory cells were collected, counted, and washed. T cells were resuspended in T cell medium containing 60 ng/ml IL-21 at a concentration of 2e6 cells/ml.
IVS to memory T cells.
This method is published in detail elsewhere (41) and is further explained at Supplementary Materials and Methods.
Coculture screening assays: IFN-γ ELISpot, ELISA assays, and flow cytometry for OX40 and 41BB activation markers staining
DCs or cell line cells were used as target cells, with 3e4–1e5 cells/well in 96-well plates. 2e4–5e4 cells/well of effector T cells were used in 96-well plates. All cocultures were performed in T cell media in the absence of exogenously added cytokines. Phorbol 12-myristate 13-acetate: ionomycin mixture (eBioscience) or CD28/CD3 Dynabeads (Thermo Fisher Scientific) were used as a positive control. In HLA-blocking assays, target cells were incubated with 20–50 μg/mL blocking antibodies for 2 hours at 37oC, 5% CO2, and then effector cells were added and incubated for 12–18 h. IFN-γ ELISpot assays were performed in MultiScreen-IP filter plates (EMD Millipore). Each plate was pretreated with 50 μl 70% ethanol/well for < 2 min, washed three times with PBS and then coated with 10 μg/ml of an IFN-γ capture antibody (60 μl/well, clone: 1-D1K, Mabtech, diluted in PBS) overnight at 4°C. At the day of coculture, each plate was washed three times with PBS and blocked with complete media without IL-2 for 2 h at room temperature. After overnight coculture (18–24 h), the cells were harvested and transferred into a round-bottom 96-well plate for flow cytometry staining and analysis. Each ELISpot plate was washed five times with PBS containing 0.05% Tween 20 (MP Biomedicals) and then incubated for 2 h with 1 μg/ml, 0.22 μm-filtrated anti-human-IFN-γ detection antibody (clone: 7-B6–1, Mabtech, 100 μl/well, diluted in PBS + 0.5% fetal bovine serum (FBS)). Next, each plate was washed five times with PBS containing 0.05% Tween 20 and incubated for 1 h with streptavidin-ALP (Mabtech, 100 μl/well, 1:3,000 diluted with PBS + 0.5% FBS), followed by three washes with PBS and development with 0.45-μm-filtered BCIP-NBT substrate solution (KPL, 100 μl/well) for 5–15 min. The reaction was stopped by rinsing thoroughly with cold tap water. After plates completely dried, each ELISpot plate was scanned and counted using an ImmunoSpot plate reader and associated software (Cellular Technologies Limited). The harvested cells from the coculture ELISpot wells prior to IFNγ spot development were spun down and the media discarded. Cells were resuspended in the remaining liquid and stained for flow cytometry and surface expression of 41BB and OX40 in Live/CD3+, CD4+ or CD8+, and mTCR+ (when iTCR was tested in the experiment) was assessed using BD FACSCantoI or BD FACSCantoII (BD Biosciences). All flow cytometry data were analyzed using FlowJo software (TreeStar Inc). Cell media were analyzed to IFN-γ by human IFN-gamma Duoset ELISA (R&D Biosystem) and completed as the protocol instructed.
Reactivity was defined by a higher percentage of cells either: expressing 4–1BB and/or OX40 or demonstrating an increased number of spots in the IFN-γ ELISpot when the RAS mutated antigen was tested in comparison to the WT and DMSO. Reactivity could be in the gene (FL/TMG), the LP levels, or in both.
Single-cell sorting and single-cell RT-PCR
T cells were sorted to single-cell cultures by identifying their reactivity as reflected by the expression of 41BB/OX40 above the background to target cells (single or double-positive). As described earlier (44,45). Cells were sorted into a 96-well plate containing RT-PCR buffer (CellsDirect One-Step qRT-PCR kit, Thermo Fisher Scientific), with each well containing one primer to the Cα and Cβ regions (1.2 µM for these gene-specific RT primers) and multiple Vα and Vβ primers (0.6 µM) in 10 µl volume. Cycling conditions were: 50oC for 15 min, 95oC for 2 min, and 18 cycles of 95oC for 15 s, 55oC for 30 s, 72oC for 1 min, 4oC. The second PCR was performed separately for TCRα and TCRβ chains. Platinum II Hot-Start PCR master mix (2X) was used for both PCRs for a total of 25 ul volume that included 2.5 µl of the RT-PCR mix with several nested primers (0.6 µM each) for either TCRα or TCRβ targeting the extended CDR3 regions of both chains. The PCR programs were: 95oC for 7 min, 5 cycles of 95oC for 15 s, 65oC for 15 s, and 72oC for 30 s, followed by 5 cycles of 95oC for 15s, 60oC for 15 s, and 72oC for 30 s. This was followed by 40 cycles of 95oC for 15 s, 65oC for 15 s, and 72oC for 30 s, then incubation at 72oC for 7 min, and finally incubation at 4oC. The PCR products were purified and sequenced by Sanger methods within internally nested Cα and Cβ region primers (Beckmann Coulter).
TCR reconstruction, subcloning into a retroviral vector
The Sanger sequencing method produced sequencing data that contained the 3’ end of the variable region and the full CDR3 region of matching TCRα and TCRβ genes. The data were analyzed using IMGT/V-Quest tool (http://www.imgt.org/IMGT_vquest/vquest). The pairing methods used herein are described elsewhere (16,46,47) and are further explained at Supplementary Materials and Methods. The full iTCR construct was cloned into a pMSGV1 retroviral vector.
T cell transduction
T cell stimulation was done by thawing autologous or healthy donor apheresis nonadherent cells (from the antigen-presenting cells protocol), resuspended in T cell medium and rested overnight at 37°C, 5% CO2. Cells were harvested and stimulated by concentrating 3.75e6 cells/mL in T cell media containing 50 ng/ml soluble OKT3 (Miltenyi Biotec) and 300 IU/ml IL-2 (Chiron), and every 2 ml (7.5e6 cells) were seeded in a single well of a 24-well plate for 2 days. Retroviral supernatants were generated (44) by seeding 2 ml of 6e5 cells/ml (cell line medium) HEK-293GP packaging cell line in 6-well poly-D-lysine-coated plates. After 24 hours, the media was changed to 2 ml cell line media without antibiotic. HEK293 GP were co-transfected with 2 μg/well pMSGV-1 cloned plasmid and 1.4 μg/well envelope-encoding plasmid RD114 using Lipofectamine 2000 (Life Technologies). Retroviral supernatants were collected 48 hours later and then centrifuged onto Retronectin-coated (20 μg/ml; Takara) non-tissue culture-treated 6-well plates at 2000 G for 2 h at 32°C. Stimulated T cells were then washed, concentrated (2e6 per well at 0.5e6 cells/ml in IL-2 containing T cell media), and spun onto the retrovirus plates for 10 min at 1500 RPM with the acceleration and brake set at 1. After 12–48 hours of incubation at 37°C, 5% CO2, the cells were removed from the plates and further cultured in rIL-2-containing T cell media. GFP and mock transduction controls were included in transduction experiments. The TCR transduction efficacy was confirmed by using mouse TCRβ antibody 8–16 days after day of stimulation. The transduced TCR was tested in reactivity and avidity experiments.
TCR survey and deep sequencing
TCR-Vβ deep sequencing was performed by immunoSEQ (Adaptive Biotechnologies) on genomic DNA isolated from peripheral blood T cells and frozen tumor tissues. T cell numbers in sequenced samples ranged from ~2e4 to 1e6 cells. TCRβ chain clonality and productivity were analyzed using immunoSEQ Analyzer 3.0 (Adaptive Biotechnologies). Only productive TCR rearrangements were used in the calculations of TCR frequencies.
Patient-derived xenografts (PDX)
Fresh tumor tissues from patients were mechanically separated into fragments of 2 mm in dimension. One fragment was then implanted subcutaneously at the flank of an NSG mouse using a 20-gauge needle. Tumor growth was monitored weekly. PDX were harvested when their sizes were greater than 1 cm in dimension.
Tumor cell line derived from PDX
Freshly harvested PDX were cut into small fragments and placed in GentleMACS c-tubes containing 20 ml of tissue culture media. The tumor cells were then mechanically dissociated by GentleMACS dissociator (Miltenyi Biotec) using the “mouse implanted tumor 1.01” program. The resulting cell suspension was run through a 100 μM cell strainer and washed once with culture media before being placed in tissue culture flasks. Media were refreshed every 3–7 days and cells were split when confluence reached 70%. The PDX derived tumor cell lines were characterized as explain in more details at Supplementary Materials and Methods.
Coculture of PDX derived tumor cells with neoantigen specific TCR transduced T cells
One day prior to coculture, tumor cells were placed into a 96-well plate in concentration of 1e5 cells in 200 μl of culture media per well. On the day of assay, 100 μl of fresh culture media with or without peptides added. 1e5 T cells in 100 μl of culture media were added to the tumor cells. The coculture was incubated at 37ºC, 5% CO2 for 16 h. Supernatants from the coculture wells were collected from IFNg ELISA and nonadherent cells were harvested for surface 41BB and OX40 markers detection by FACS.
Results
We prospectively tested TILs from 20 gastrointestinal (GI) cancer patients enrolled in clinical trials at the NCI Surgery Branch over the last 2 years whose tumors expressed RAS mutations. The general scheme for identifying and validating TCRs targeting RAS hotspot mutations is shown in Figure 1. Briefly, to identify T cell reactivity against RAS hotspot mutations, patient TILs were cocultured with antigen-presenting cells (APC) either transfected with in-vitro-transcribed (IVT) RNAs or pulsed with 24 or 25-mer long peptides (LP) flanked on either side of the hotspot mutation with 11 or 12 residues from the normal RAS protein sequence (Supplementary Fig 1A). The IVT RNAs encoded either the full-length (FL) mutant, the FL wild type (WT) KRAS gene, tandem minigenes (TMG) encoding 12 of the RAS hotspot mutations or corresponding wild type residues (Supplementary Fig 1A and 1B). Generally, LPs were more efficient at detecting CD4 reactivities and transfected IVT RNA was more efficient for the detection of reactive CD8 T cells (18). To test CD8 T cell reactivity, we also used a library of predicted minimal epitope (ME) peptides from the RAS mutations for common MHC-I using the prediction algorithm NetMHC 4.0 (DTU Bioinformatics) (Supplementary Fig 1C). As positive controls, we took advantage of an HLA-A*11*01-restricted mouse TCR recognizing RASG12D and the human HLA-A*11:01-restricted TCR recognizing RASG12V that our group previously reported (41,48) (Supplementary Fig 2.). Reactivity was tested by IFN-γ secretion and by 41BB and OX40 coreceptors upregulation. Although, we always tested and gated for both 41BB and OX40 upregulation, as expected 41BB upregulation usually detects CD8 reactivity and OX40 upregulation usually detects CD8 reactivity.
Fig 1. Identification and validation of RAS hotspot mutation-reactive T cells from cancer patients with RAS mutations.
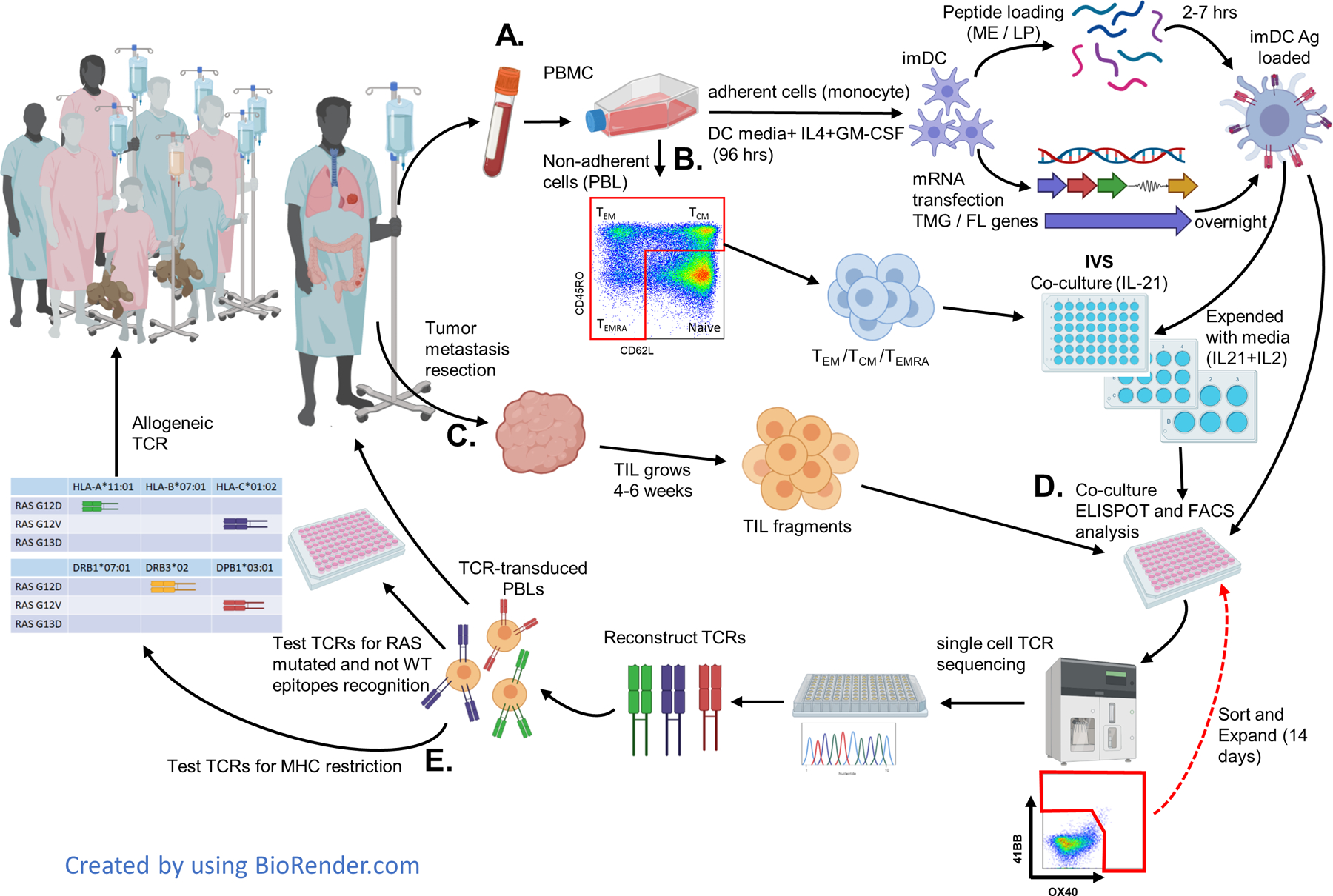
A. Patient apheresis is collected. The blood is separated into adherent cells that are used to generate immature DC (imDC). The imDC are loaded with short predicted minimal epitopes (ME) or with long peptides (LP) or are transfected with mRNA of the full length (FL) gene of the specific RAS mutation or with tandem minigenes (TMG) containing all the common (more than 0.5%) RAS mutations. These Ag-loaded DC are used for activating and testing T cells in all stages. B. Non-adherent cells (PBL) are sorted into CD4 or CD8 T cells (CD3), central/effector memory (TCM/TEM), or effector (TEMRA) cells (excluding naïve T cells) by using CD45RO and CD62L. This subset is used for in vitro stimulation (IVS) with RAS Ag-loaded DC by coculture for three days with IL-21 and then expanded with media containing IL-21+IL-2 every two days. Cells are restimulated and sorted for high expression of 41BB/OX40 and rapidly expanded (REP) for another 14 days. C. Tumor metastasises are resected and cut into 24 fragments. Tumor-infiltrating lymphocytes (TIL) collected from each fragment are expanded in media containing IL-2 until there are enough cells for testing (usually about 4–6 weeks). D. T cells are tested for reactivity to RAS mutations by IFN-γ ELISpot and 41BB/OX40 flow cytometry assays. The positive cultures are sorted to enrich the RAS reactive cells and then single cells are sequenced. The TCRs are reconstructed (iTCR) using mouse constant TCR elements to enhance the iTCR α/β pairing. The iTCR are transduced into autologous/healthy donor PBLs and tested for specific RAS-mutated and WT reactivity. Specific RAS-mutated TCRs with good avidity could then be used to treat the patient (as neo-antigen treatment). E. After identifying the MHC restriction, the TCR could potentially be used to treat other patients having the same RAS mutation and MHC. All TCRs are saved in a library as prepared GMP vectors.
Identification of anti-RAS T cell reactivity from TIL
As an example of this approach, 24 tumor fragments dissected from a metastatic colon cancer deposit from patient 4391 that expressed the KRASG12V mutation were initially cultured in the presence of IL-2 as previously described (49). Lymphocytes generated from these fragments were subsequently cocultured with autologous dendritic cells (DC) loaded with RAS peptides/IVT RNA (Fig. 2). One TIL fragment (F1) demonstrated reactivity against RASG12V based on the upregulation of 4–1BB and OX40 (Fig 2 A-B) and by enzyme-linked immunosorbent spot (ELISpot) measuring IFN-γ secretion levels (Fig 2C).
Fig 2. TCR recognized RASG12V hot spot mutation discovered by TIL screening.
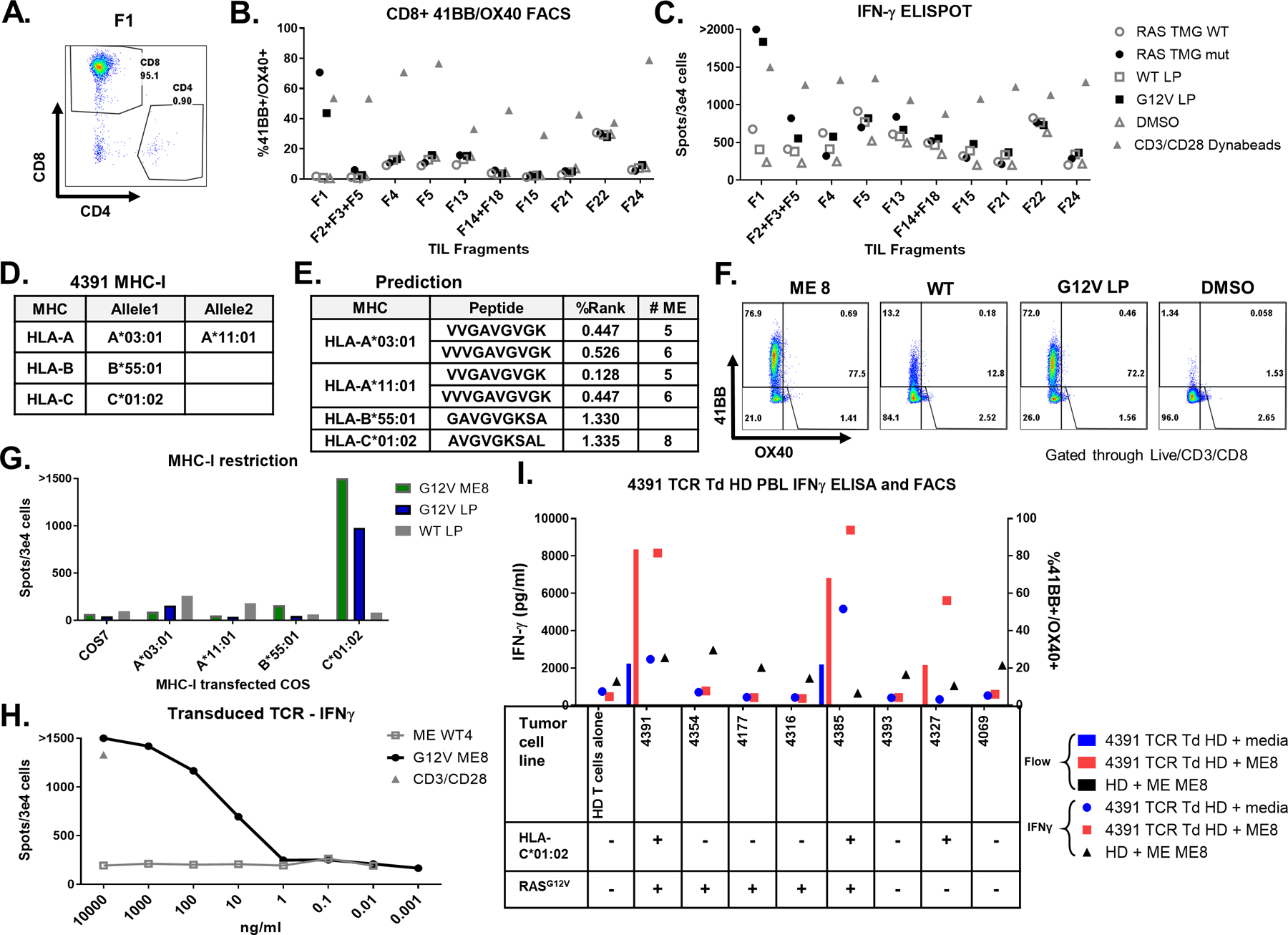
TIL fragments were screened for reactivity against autologous DC transfected with RAS TMG (WT/Mutated) or loaded with 24-mer long peptide (RASG12V/ RASWT). Coculture with DC loaded with DMSO was used as a negative control. Anti-CD28/CD3 beads were used as a positive control. A. Most fragments, after gating on the live/CD3+ cells and including fragment 1 (F1), primarily contained CD8+ cells. B. Fragment reactivity tested in flow cytometry assays measured upregulation of 41BB+ and OX40+ or C. by IFN-γ ELISpot. D. Table containing the patient 4391 MHC-I restriction elements. E. Table with the best-predicted RASG12V minimal epitopes (low % rank) to bind to the patient MHCs by the NetMHC 4.0. F. To determine which ME is recognized by TIL F1, cells were cocultured with autologous DC preloaded with RASG12V ME8, with LP (RASG12V/ RASWT), or with DMSO at an equivalent concentration. Reactivity was determined by upregulation of 41BB and OX40 surface markers in live/CD3+/CD8+ by flow cytometry. G. For testing the MHC-I restriction element, TIL F1 were cocultured with COS7 cells pre-transfected with patient’s class I HLA DNA plasmids and loaded with LP (RASG12V/RASWT) or ME8. Reactivity was determined by IFN-γ ELISpot. One TCR was identified from patient 4391 TIL fragment F1 after single-cell sequencing. This TCR was virally transduced to healthy donor (HD) PBLs. H. The TCR avidity was tested in the transduced cells by coculture with DC loaded with different concentrations of RASG12V/RASWT 9-mer ME8 or the equivalent WT sequence WT4. The cells were tested via IFN-γ ELISpot, with anti-CD28/CD3 beads used as a positive control. I. The TCR specificity and tumor recognition tested by coculturing the TCR transduced healthy donor (HD) cells with different tumor cell lines with or without (+/−) RASG12V and HLA-C*01:02. Tumor cell lines were loaded with RASG12V ME8, or not peptide-loaded (media). Unloaded tumor cell lines expressed their native peptidomes (including RASG12V). The reactivity was tested by IFN-γ ELISA and by flow cytometry assay for 41BB/OX40 surface marker upregulation in live/CD3+/mTCR+/CD8+ cells. Untransduced HD loaded with ME8 was used as a negative control.
A prediction algorithm (NetMHCpan4.0) was then used to identify candidate MEs from RASG12V potentially able to bind to the HLA class I restriction elements (REs) present in patient 4391(Fig 2D). In addition to the VVGAVGVGK and VVVGAVGVGK peptides, previously shown to be recognized in the context of HLA-A*11:01 (48), the AVGVGKSAL peptide (ME8) was predicted to be a weak binder to C*01:02 and the peptide GAVGVGKSA was predicted to bind weakly to HLA-B*55:01 (Fig 2E). When F1 was tested against DCs loaded with RASG12V MEs (listed in Supplementary Fig 3A), reactivity was found against ME8 (Fig 2F and Supplementary Fig 3B). Reactivity was also detected against the RASG12V LP but not the RASWT LP (Fig 2F). To identify the restriction element responsible for presentation of the RAS ME8 peptide, F1 TILs were cocultured with peptide-pulsed COS7 cells that had been transfected individually with plasmids to encoding the patient’s HLA class I alleles. The results demonstrated that COS7 cells expressing HLA-C*01:02 but not any of the additional HLA REs expressed by patient 4391 were recognized by TIL F1 T cells that had been pulsed with the ME8 peptide or the RASG12V LP (Fig. 2G). To evaluate the avidity of the recognition, TIL F1 was cocultured with autologous DCs loaded with different peptide concentrations. Recognition of the ME8 peptide by TIL F1 was seen in concentrations as low as 10 ng/ml based on IFN-γ ELISpot (Supplementary Fig 3C). To identify TCR(s) reactive with the RASG12V C*01:02 epitope, F1 T cells that had been stimulated with DC transfected with the mutated TMG, were sorted for single-cell sequencing which revealed one functional TCR. The TRA and TRB chain sequences (TCR data in Supplementary Table 6.) were cloned into the MSGV1 retroviral vector which was then used to generate transient retroviral supernatants that used to virally transduced into healthy donor PBLs. The resulting bulk population was then evaluated for it’s ability to recognize DCs pulsed with the ME8 and corresponding WT peptides. Based on IFN-γ ELISpot, the TCR recognized the mutated ME loaded cells at concentrations as low as 10 ng/ml, similar to the minimum concentration recognized by TIL F1 T cells (Fig 2H). Although the transduced cells were not separated into CD4 and CD8, the reactivity appeared to be at least partially CD8 coreceptor dependent as shown by FACS analysis of the gated populations (Supplementary Fig 3D-E). Although the TCR was isolated from TILs, there was a potential risk that the TCR recognizing the synthetic peptides/TMG will not recognize the tumor or will cross-react to an unknown antigen expressed in normal tissues. In an attempt to address this potential issue, 4391-TCR-transduced cells were tested for recognition against multiple tumor cell (TC) lines that either did or did not express HLA*C01:02 or RASG12V. TCR-transduced cells only recognized xenograft cell lines from patients 4391 and 4385, both of which expressed HLA-C*01:02 and RASG12V, but not 6 additional cell lines that lacked expression of either RASG12V or HLA-C*01:02. Pulsing the 4391 and 4385 TC lines with the ME8 peptide further enhanced recognition of the lines by 4391-TCR transduced T cells, and in addition led to recognition of 4327, a colorectal TC line that expressed HLA-C*01:02 but not the RASG12V mutation (Fig 2I and Supplementary Fig 3F-G).
Similar analyses were carried out in other patients with tumors bearing RAS mutations. Screening of TIL fragment cultures from patient 4385 TIL demonstrated that Fragment 11 also recognized RASG12V in the context of HLA-C*01:02, (Supplementary Fig 4A-I). The single dominant TCR identified from the reactive fragment was then cloned into the MSGV1 recombinant retroviral construct, and transient retroviral supernatants used to transduce healthy donor PBLs. The results of a peptide titration assay demonstrated that the TCR from patient 4385 possessed a similar avidity as the TCR isolated from 4391 (Supplementary Fig. 4E-M). Additionally, patient 4373 TIL Fragment 8 recognized the RASG12D (Supplementary Fig. 5A), and PBLs transduced with TCR-2 isolated as described above were reactive to RASG12D in the context of HLA-A*11:01 (Supplementary Fig. 5B). Peptide titration assays indicated that TCR recognized the RASG12D 10-mer VVVGADGVGK at a minimal concentration of 1 ng/ml but recognized the 9-mer VVGADGVGK at approximately 1,000 fold higher concentration (Supplementary Fig. 5C-D). Lastly, CD4 reactivity against RASG13D was detected in patient 4400 TIL fragments (Supplementary Fig A-C). From six isolated TCRs, two specifically recognized RASG13D (Supplementary Fig 6D-F) in the context of HLA-DQA1*05:01/HLA-DQB1*03:01 (Supplementary Fig 7A-B) at a minimum concentration of 1 ng/ml (Supplementary Fig 7C-F). Overall screening of TIL fragments from 20 patients with tumors bearing RAS mutations for anti-RAS reactivity demonstrated reactivity in 25% (5/20) of the screened patients and in 2.2% (8/370) of total fragments screened. (Supplementary Table 1).
Identification of anti-RAS T cell receptors from PBL
Recently we demonstrated that reactivities that were not readily detected by screening TIL could be detected by stimulating patient’s PBL using DC presenting the mutated antigens (41,50,51). This in vitro stimulation (IVS) was required due to the low frequency of neoantigen-specific T cells in the blood. Based on studies of the sequence of the V-β chain of human TCR, these reactive T cells can be as rare as 0.001% in PBL and can be below the threshold of detection of our reactivity test (51). We used this method to search for additional RAS specific TCRs directed against non-synonymous RAS mutations from patients’ PBL. Initial screening of TIL fragments from colorectal cancer patient 4360 whose tumor bore a RASG12V failed to demonstrate reactivity against the mutated RAS long peptide or minigene antigen (Supplementary Fig. 9). PBL (non-adherent PBMCs) collected by apheresis were subject to IVS by a method (Fig 1B) involving electronic sorting of the TCM, TEM, and TEMRA populations of the patient’s bulk PBL (41). Sorted T cells were cocultured with autologous DC loaded with RAS antigens for 14 days and screened for reactivity by flow cytometry following antigen stimulation (Fig 3A). T cells upregulating 41BB and OX40 costimulatory molecules following RASG12V LP IVS that had been sorted and subjected to a rapid expansion protocol (REP) and were then tested for reactivity. CD4 reactivity against the mutated LP and lesser reactivity against the wild type peptide were seen (Fig 3B-C). Following IVS with the RASG12V LP recognized DC loaded with the mutant RASG12V LP down to a concentration of 100 pg/ml. although the RASWT LP was recognized at a minimal concentration of 10 ng/ml, Reactivity against RASG12V LP was higher in all concentrations except 10µg/ml, indicating specificity for the mutant neoepitope (Fig 3D-E).
Fig 3. TCR discovered by PBL in vitro stimulation (IVS).
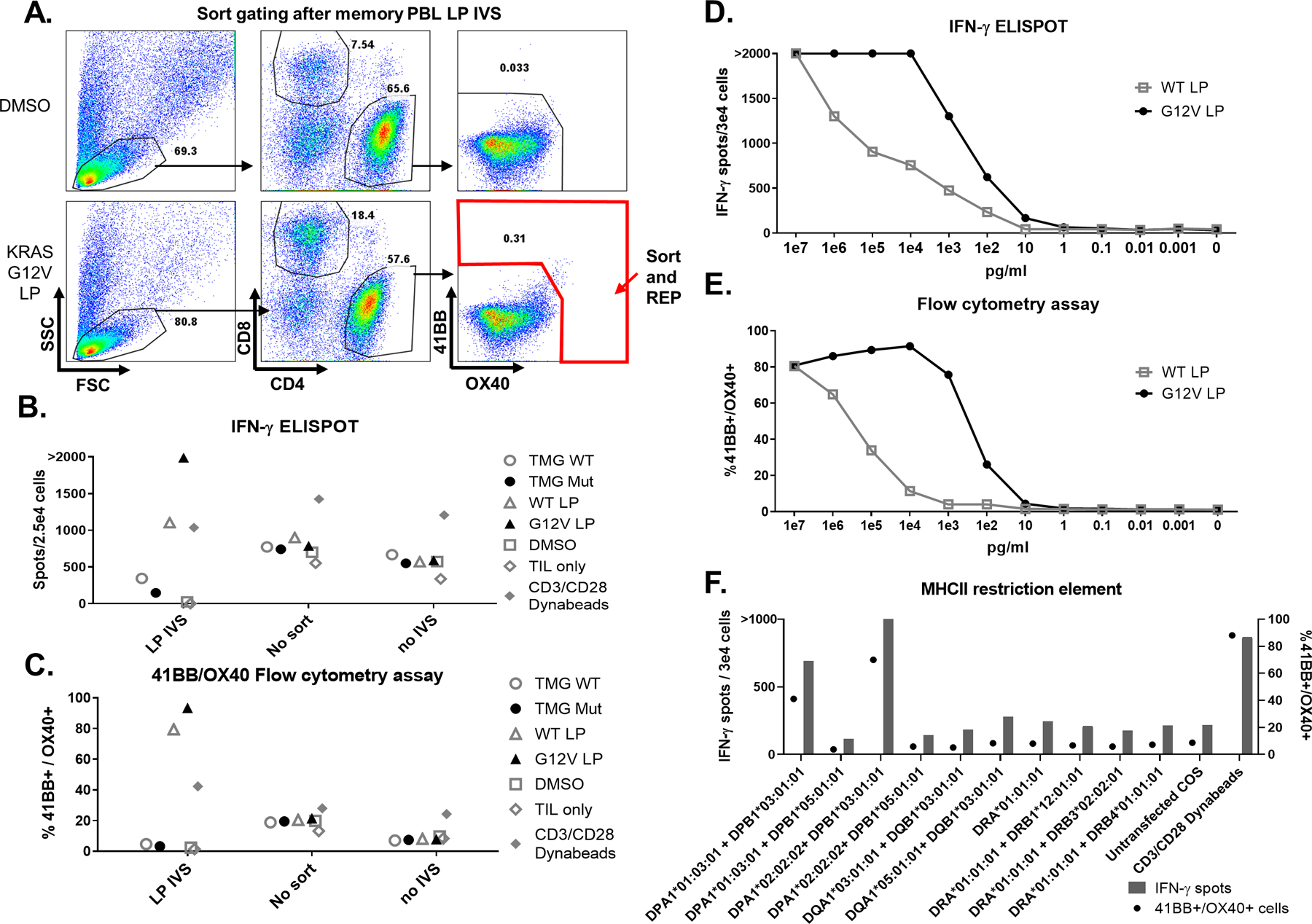
PBL were sorted into CD4 or CD8 memory and effector T cells and in vitro stimulated (IVS) with DC loaded with RASG12V LP. After the stimulation period, T cells were restimulated with the same target cells. A. Flow cytometry assay dot plot showing the gating strategy in which CD4 cells were sorted for high expression of OX40 and 41BB surface markers following RASG12V LP IVS, with DMSO used as a negative control. B-C. The sorted cells were rapidly expanded (REP) following the IVS protocol (LP IVS), and were tested for reactivity against RAS by coculturing with DC transfected with WT/Mut TMG or loaded with RASG12V/RASWT LP or an equivalent amount of DMSO. Cells that were stimulated but didn’t sort (no sort) or unstimulated cells (no stimulation) were used as a control for the IVS. T cell only and with anti-CD28/CD3 beads used as negative and positive controls, respectively. After overnight coculturing, cells were analyzed for B. IFN-γ ELISpot and C. 41BB/OX40 surface marker upregulation in the live/CD3+/CD4+ gated population by flow cytometry analysis. D-E. CD4 cells after RASG12V LP IVS (including one REP after sorting) were tested for avidity to RASG12V. The cells were cocultured with DC loaded with RASG12V/ RASWT LP at various concentrations. After overnight coculturing, cells were analyzed via B. IFN-γ ELISpot and C. 41BB/OX40 surface marker upregulation in the live/CD3+/CD4+ gated population by flow cytometry analysis. F. The MHC-II restriction element recognized by 4360 CD4 PBL after RASG12V LP IVS was determined via IFN-γ ELISpot (left axis and bars) and 41BB/OX40 flow cytometry assay (right axis and circles). The cells were cocultured with COS7 transfected with DNA plasmids containing the different combinations of the patient’s MHC-II α and β chains and loaded with RASG12V LP.
In order to identify the MHC-II restriction elements recognized by the reactive cells, the cells following LP IVS were cocultured with COS7 cell lines transfected with DNA plasmids encoding the MHC-II α and β chains of patient 4360 and loaded with RASG12V LP. Results showed that the recognition was restricted to DPB1*03:01. Interestingly, reactivity was seen when DPB1*03:01 was co-transfected with either DPA1*01:03 or DPA1*02:02 (Fig 3F). In the US, about 20% of the Caucasian population has the DPB1*03:01 allele (Allele Frequency Net Database - allelefrequencies.net). Reactive cells were sorted and single-cell sequencing resulted in the identification of two candidate reactive TCRs (TCR1 and TCR2). Deep sequencing of four tumor fragments (Adaptive Biotechnologies) showed that the TCR1 (CDR3 β chain) sequence was detected in only one of the tumor fragments at the frequency of 5.7×10−5 (specific TCR in total TCRs), whereas TCR2 was not detected in any of the tested tumor fragments (Fig 4A). These two TCRs were tested in a reporter system (see Materials and Methods and (45)). The TCRs were either IVT RNA-transfected or retrovirally-transduced into Jurkat cells containing the Luciferase reporter gene under the regulation of NFAT and cocultured with DCs loaded with the RASG12V or RASWT LP. Luciferase activity showed that TCR1 specifically recognized the RASG12V mutated peptide whereas TCR2 had higher specificity to the RASWT peptide (Fig 4B-C). These results were supported by carrying out peptide titration assays of autologous PBLs that were enriched for CD4 or CD8 cells, followed by transduction of the 4360 TCRs (Fig 4D-K). These results showed that TCR1 is highly specific against the RASG12V LP-loaded autologous DC, and indicated that this reactivity was partly CD4 coreceptor dependent (Fig 4D-G). Overall, by using the PBL IVS method, mutated RAS-specific reactivity was found in 3 of 7 patients in whom this reactivity was not previously detected by TIL fragment screening (Supplementary Table 2).
Fig 4. 4360 TCRs recognized RAS hotspot mutation: Validation, avidity and reactivity.
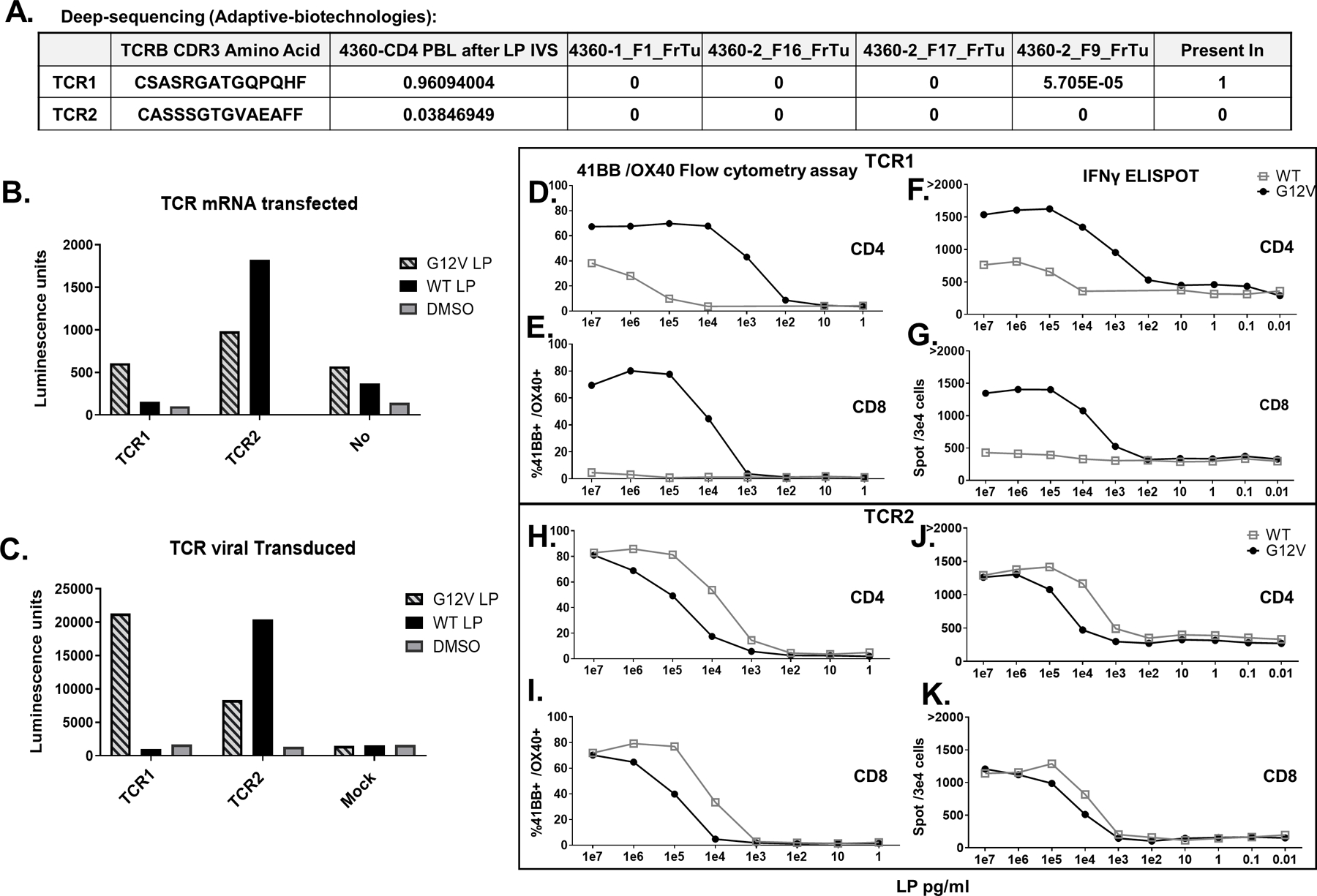
A-K. Two TCRs (TCR1, TCR2) identified from patient 4360 CD4 PBL after LP IVS single-cell sequencing. A. Deep sequencing (Adaptive Biotechnologies) of DNA extracts from four tumor fragments (FrTu) revealed TCR1 existed in one of these fragments (5.7 repeats in 100,000 cells) while TCR2 did not exist in any. B. Both TCRs were mRNA transfected or C. virally transduced into a Jurkat-CD4-NFAT-Luciferase cell line and then cocultured with DC loaded with RASG12V/ RASWT LP or the equivalent amount of DMSO. Luciferase activity was measured and presented in Luminescence units. D-G. TCR1 and H-K. TCR2 were virally transduced into patient 4360 PBLs. The cells enriched to CD3+/CD4+ cells D, F, H, and J or enriched to CD3+/CD8+ E,G,I, and K. The transduced cells were cocultured with DC loaded with different concentration of RASG12V/ RASWT LP. TCR avidity in live/CD3+/mTCR+ cells was analyzed by D, E, H, I. flow cytometry for 41BB/OX40 surface marker upregulation and F, G, J, K. IFN-γ ELISpot.
Our current clinically relevant and validated TCRs recognizing a RAS mutation in high avidity are presented here in tables: one for CD8 (Table 1) and for CD4 (Table 2) cell recognition. This library of TCRs against RAS mutations has the potential for use as off-the-shelf reagents for patient treatment. Among patients having RAS mutated cancers, at least one TCR (CD4 or CD8) is available for use in treatment for about 45% of the Caucasian and about 60% of the Asian populations in the US. Further data regarding receptors’ sequences and antigens are also available (Supplementary Table 6).
Table 1. Validated CD8 TCRs recognizing RAS hotspot mutations, available for allogeneic treatment.
CD8 TCR table showing columns (from left to right): the hotspot mutation recognized by the TCR and the percentage of this mutation among RAS-mutated cancer patients. Number of TCRs discovered from the same patient recognizing the same target. The HLA-I restriction each TCR recognizes. Percentage of the allele frequency (taken from Allelefrequencies.net) and calculation of the percentage of the individuals in the Caucasian or Asian populations (*). Percentage of patients eligible for treatment, calculated by multiplying the frequency of individuals that have the allele with the frequency of this RAS mutation (as in the first column) with the sum of percentages from all TCRs (the bottom of this column). ME sequence recognized by the TCR. Patient number. The method used to find the TCR (TIL screen/PBL IVS). Patient cancer diagnosis in which TCR was found. Reference if the TCR has been published before and, if so, where.
| RAS mutation | # TCRs discovered | HLA - restriction | % of individuals that have the allele (Allele Frequency) | % Patients eligible for treatment. *Asian population | Minimal epitopes | patient (no.) | Method | Cancer diagnosis | Reference |
|---|---|---|---|---|---|---|---|---|---|
| KRAS p.G12D (35%) | 1 | C*08:02 | 8% (4.2%) | 2.8 | GADGVGKSA | 3995 | TIL screening | Colon | (17) |
| 4 | C*08:02 | 8% (4.2%) | 2.8 | GADGVGKSA (3 TCRs)\GADGVGKSAL (1 TCR) | 4095 | TIL screening | Colon | (17) | |
| 1 | A*11:01 | 14% (7%), *>30% in Asian population | 4.9 *>10.5 | VVVGAGGDGK | 4373 | TIL screening | Colon | ||
| KRAS p.G12V (24%) | 1 | A*11:01 | 14% (7%), *>30% in Asian population | 3.36 *>7.2 | VVGAVGVGK | 4148 | PBL IVS | Endometrial | (41) |
| 1 | C*01:02 | 5.3% (2.6%), *>30% in Asian population | 1.27 *>7.2 | AVGVGKSAL | 4391 | TIL screening | Colon | ||
| 1 | C*01:02 | 5.3% (2.6%), *>30% in Asian population | 1.27 *>7.2 | AVGVGKSAL | 4394 | TIL screening | Colon | ||
| 1 | C*01:02 | 5.3% (2.6%), *>30% in Asian population | 1.27 *>7.2 | AVGVGKSAL | 4385 | TIL screening | Colon | ||
| 12.4 *>27.5 |
Table 2. CD4 validated TCR recognizing RAS hotspot mutation, available for allogeneic treatment.
CD4 TCR table showing columns (from left to right): the hotspot mutation recognized by the TCR and the percentage of this mutation among RAS-mutated cancer patients. Number of TCRs discovered from the same patient recognizing the same target. The HLA-II restriction each TCR recognizes. Percentage of the allele frequency (taken from Allelefrequencies.net) and calculation of the percentage of the individuals in the Caucasian or subpopulations (*). Percentage of patients eligible for treatment, calculated by multiplying the frequency of individuals that have the allele with the frequency of this RAS mutation (as in the first column) with the sum of percentages from all TCRs (the bottom of this column). Patient number. The method used to find the TCR (TIL screen/PBL IVS). Patient cancer diagnosis in which TCR was found. Reference if the TCR has been published before and, if so, where.
| RAS mutation | # TCRs discovered | HLA - restriction | % of individuals that have the allele (Allele Frequency) | % Patients eligible for treatment. | patient (no.) | Method | Cancer diagnosis | Reference |
|---|---|---|---|---|---|---|---|---|
| KRAS p.G12D (35%) | 6 | DRB3*02 | 32% (*16.6% USA San Francisco Caucasian) | 11.2 | 4238 | PBL IVS | Colon | (41,51) |
| KRAS p.G12V (24%) | 1 | DRB1*07:01 | 25.5% (14.5) | 6.12 | 4148 | TIL screening | Endometrial | (52) |
| 1 | DPB1*03:01 | 20% (10) | 4.8 | 4360 | PBL IVS | Colon | ||
| 1 | DRB1*01:01 | 18% (9) | 4.32 | 4304 | TIL screening | Colon | ||
| KRAS p.G13D (13%) | 2 | DQA1*05:01/DQB1*03:01 | 35.1% (19.1%) | 4.55 | 4400 | TIL screening | Colon | |
| KRAS p.G12R (3%) | 1 | DQA1*05:05(05:01)/DQB1*03:01 | 35.1% (19.1%) | 1.05 | 4268 | TIL screening | Colon | |
| 1 | DRB5*01 | 35% (*18% USA San Francisco Caucasian) | 1.05 | 4270 | TIL screening | Pancreatic | ||
| 33.09 |
Discussion
ACT using autologous TIL can mediate objective responses in about half of patients with metastatic melanoma, including a quarter with durable complete responses (7,9). As described elsewhere, this method involves the excision of a metastatic tumor and culture of tumor fragments in the presence of IL-2 to enable the growth of TIL. After adequate growth occurs, these TIL are tested for recognition of tumor antigens by either coculture with autologous DC pulsed with peptide pools or transfection with mRNA encoding mutations identified from the whole-exome sequencing of the patient’s tumor. Using this method, we found T cell reactivity in about 80% of patients with metastatic cancers across multiple histologies, that include melanoma, colon, pancreas, ovary, and breast (4,8,13,15,18). However, this process can fail to identify reactivities to antigens recognized by a low percentage of T cells.
RAS mutations can potentially serve as good targets for immunotherapies since the mutations are usually in hotspot mutations and are common among cancer patients with various types of cancer (19–28). We have previously detected T cell reactivity against RAS hotspot driver mutations and identified the accompanying TCR from reactive TILs from 3 patients (17,52). We also showed that administration of TIL targeting a KRAS mutation could induce the regression of metastatic cancer in a patient with colon cancer who is now living disease-free over four years later (17).
Here, we are describing an efficient and sensitive means to identify anti-RAS reactivities using two methods established in our lab. Unlike our previous screening of TIL fragments against all somatic mutations, here the TIL screening and stimulations were performed specifically against the known RAS hotspot mutations by using available and validated reagents specifically for RAS mutations. By using this method, we found that 25% (5/20) of newly screened patients had TIL reactivity to a RAS hotspot mutation (Supplementary Table 1.).
A second method, employing a PBL IVS approach, enabled the identification and isolation of additional RAS reactive T cells that were present at very low frequencies in metastatic cancer patients’ PBL (41). Using the PBL IVS method, we were able to identity RAS reactive T cells in 43% (3/7) of the patients tested. Notably, these reactivities to RAS were not found by screening TIL (Supplementary Table 2.). An advantage of identifying TCRs from PBL is that it does not require invasive procedures to harvest patients’ tumor lesions and thus it also helps reduce the time needed to produce a cell product for treatment (50).
We previously showed that administration of PBL retrovirally transduced with TCRs targeting MART-1 or gp100 melanocyte/melanoma antigens can mediate regression in patients with metastatic melanoma (53). In addition, we demonstrated that the transduction of a TCR targeting the NY-ESO-1, a cancer germline antigen, could mediate regression of metastatic synovial cell sarcomas (54,55). An advantage of using PBL transduced with TCRs is the ability to create less differentiated effector cells. This contrasts with the senescent state of antigen reactive TILs where the effector cells have been repetitively stimulated by antigen in vivo and have lost much of their proliferative and effector potential.
Since all T cells underwent negative selection in the thymus, TCRs discovered from human T cells are less likely to recognize patient self-antigens. However, IVS method has a risk of producing de novo reactivity that did not previously exist in vivo and might generate T cells with TCRs that recognized normal as well as tumor cells. To address this risk, the cells used for IVS were solely memory cells. These cells had already been stimulated in-vivo and likely differentiated and proliferated following infection or malignancy. Addition to the higher chance of finding a reactive cell, it is less likely that de novo reactivity will be generated by using this IVS approach. Using our methods, the TCRs were discovered using TIL or memory T cells that by definition developed naturally by the patient in vivo. This approach is, therefore, less likely to recognize self-antigens compared to the use of normal healthy donors as described by others (56,57). The TCRs identified through IVS of PBL from patient 4360 failed to recognize autologous APC pulsed with RAS wild type peptides. In addition, through deep sequencing of tumor fragments we detected mutant RAS-reactive TCR1 at low concentrations, further supporting that the IVS we performed did not introduce a de novo reactivity but rather enriched the RAS-reactive TILs that pre-existed. Our data showed that PBLs expressing anti-mutant RAS TCRs specifically recognized not only the autologous patient-derived tumor cell line but also allogenic tumor cell lines with matching MHC restriction and RAS mutation, which suggests that these TCRs can potentially be used as an “off-the-shelf” reagents for immunotherapy (Fig 3D). Further studies are needed to demonstrate their ability to treat tumor cells in vivo.
The library of TCRs presented here are those that were identified to have strong avidity to mutant RAS and little to no self or RAS wild type recognition. We focused on RASG12D/RASG12V mutations because they are the most common; however we also successfully isolated TCRs targeting less common RASG13D and RASG12R. Taken together, among cancer patients whose tumor cells contain a RAS mutation (about 30% of all cancer patients), about 45% of the Caucasian and about 60% of the Asian populations in the US could potentially benefit from the RAS specific TCRs library we have developed. In on-going research, we are attempting to identify additional TCRs that can be used to treat additional populations. We have recently received permission to treat patients with the allogeneic anti-mutant RAS TCRs from the TCR library described in this paper under the NCI clinical protocol (NCI-18-C-0049) and are making all TCR sequences we have identified in this report now widely available.
Supplementary Material
Statement of significance.
Approximately 30% of all cancers comprising the most common malignancies possess a RAS hotspot mutation. In this work, we characterize a library of TCRs directed against mutant RAS epitopes that can potentially be used for the evaluation of TCR-based adoptive cell transfer (ACT) therapies in the 45–60% of patients wheres tumor bears RAS mutations.
Acknowledgments
The authors thank the NCI Surgery Branch TIL laboratory for growing TILs. The authors thank Arnold Mixon and Shawn Farid for their assistance with FACS. The authors thank to Alicia A. Livinski NIH Library Writing Center for manuscript editing assistance.
Financial Support: National cancer institute (NCI) and Cooperative R&D Agreements (CRADA), Kite Pharma.
Footnotes
Conflict of Interest Disclosure Statement: The authors declare no potential conflicts of interest.
References
- 1.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. science reviews 2015;348(6230):62–8 doi 10.1126/science.aaa4967 %J Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohaan MW, Wilgenhof S, Haanen JBAG. Adoptive cellular therapies: the current landscape. Virchows Archiv 2019;474(4):449–61 doi 10.1007/s00428-018-2484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nature Reviews Immunology 2020;20(11):651–68 doi 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol 2017;18(3):255–62 doi 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nature Medicine 2015;21(1):81–5 doi 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 6.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014;344(6184):641–5 doi 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011;17(13):4550–7 doi 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013;19(6):747–52 doi 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol 2016;34(20):2389–97 doi 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besser MJ, Itzhaki O, Ben-Betzalel G, Zippel DB, Zikich D, Kubi A, et al. Comprehensive single institute experience with melanoma TIL: Long term clinical results, toxicity profile, and prognostic factors of response. Mol Carcinog 2020;59(7):736–44 doi 10.1002/mc.23193. [DOI] [PubMed] [Google Scholar]
- 11.Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma: Intent-to-Treat Analysis and Efficacy after Failure to Prior Immunotherapies. Clinical Cancer Research 2013;19(17):4792–800 doi 10.1158/1078-0432.Ccr-13-0380. [DOI] [PubMed] [Google Scholar]
- 12.Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin Cancer Res 2018;24(22):5562–73 doi 10.1158/1078-0432.CCR-18-0573 %J Clinical Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 2014;20(13):3401–10 doi 10.1158/1078-0432.CCR-14-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. science 2017;356(6334):200–5 doi 10.1126/science.aak9510 %J Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 2018;24(6):724–30 doi 10.1038/s41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350(6266):1387–90 doi 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 2016;375(23):2255–62 doi 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov 2019;9(8):1022–35 doi 10.1158/2159-8290.CD-18-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serebriiskii IG, Connelly C, Frampton G, Newberg J, Cooke M, Miller V, et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun 2019;10(1):3722 doi 10.1038/s41467-019-11530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016;129(7):1287–92 doi 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Molecular Cancer Research 2015;13(9):1325–35 doi 10.1158/1541-7786.Mcr-15-0203. [DOI] [PubMed] [Google Scholar]
- 22.Stolze B, Reinhart S, Bulllinger L, Frohling S, Scholl C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep 2015;5:8535 doi 10.1038/srep08535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kodaz H, Osman K, Muhammet B, Hacioglu, Bulent E, Cagnur EK, Ilhan H, et al. Frequency of RAS Mutations (KRAS, NRAS, HRAS) in Human Solid Cancer. Eurasian Journal of Medicine and Oncology 2017. doi 10.14744/ejmo.2017.22931. [DOI] [Google Scholar]
- 24.Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491(7424):399–405 doi 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72(10):2457–67 doi 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lennerz JK, Stenzinger A. Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist 2015;20(4):e8–e9 doi 10.1634/theoncologist.2014-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi MH, Mejlænder-Andersen E, Manueldas S, El Jellas K, Steine SJ, Tjensvoll K, et al. Mutation analysis by deep sequencing of pancreatic juice from patients with pancreatic ductal adenocarcinoma. BMC Cancer 2019;19(1):11 doi 10.1186/s12885-018-5195-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murugan AK, Grieco M, Tsuchida N. RAS mutations in human cancers: Roles in precision medicine. Semin Cancer Biol 2019;59:23–35 doi 10.1016/j.semcancer.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019;47(D1):D941–d7 doi 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith MJ, Neel BG, Ikura M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proceedings of the National Academy of Sciences 2013;110(12):4574–9 doi 10.1073/pnas.1218173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krasinskas AM, Moser AJ, Saka B, Adsay NV, Chiosea SI. KRAS mutant allele-specific imbalance is associated with worse prognosis in pancreatic cancer and progression to undifferentiated carcinoma of the pancreas. Mod Pathol 2013;26(10):1346–54 doi 10.1038/modpathol.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grant AD, Vail P, Padi M, Witkiewicz AK, Knudsen ES. Interrogating Mutant Allele Expression via Customized Reference Genomes to Define Influential Cancer Mutations. Scientific Reports 2019;9(1):12766 doi 10.1038/s41598-019-48967-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patricelli MP, Janes MR, Li L-S, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery 2016;6(3):316–29 doi 10.1158/2159-8290.Cd-15-1105. [DOI] [PubMed] [Google Scholar]
- 34.Porru M, Pompili L, Caruso C, Biroccio A, Leonetti C. Targeting KRAS in metastatic colorectal cancer: current strategies and emerging opportunities. Journal of Experimental & Clinical Cancer Research 2018;37(1):57 doi 10.1186/s13046-018-0719-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575(7781):217–23 doi 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 36.Hughes MS, Yu YYL, Dudley ME, Zheng Z, Robbins PF, Li Y, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther 2005;16(4):457–72 doi 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (New York, NY) 2006;314(5796):126–9 doi 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England journal of medicine 2013;368(16):1509–18 doi 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosati S, Parkhurst M, Hong Y, Zheng Z, Feldman S, Rao M, et al. A novel murine T-cell receptor targeting NY-ESO-1. Journal of immunotherapy (Hagerstown, Md : 1997) 2014;37 doi 10.1097/CJI.0000000000000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33(6):540–9 doi 10.1200/jco.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun 2019;10(1):449 doi 10.1038/s41467-019-08304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iiizumi S, Ohtake J, Murakami N, Kouro T, Kawahara M, Isoda F, et al. Identification of Novel HLA Class II-Restricted Neoantigens Derived from Driver Mutations. Cancers (Basel) 2019;11(2) doi 10.3390/cancers11020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dudley ME, Gross CA, Somerville RP, Hong Y, Schaub NP, Rosati SF, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol 2013;31(17):2152–9 doi 10.1200/JCO.2012.46.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, et al. Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res 2016;4(9):734–43 doi 10.1158/2326-6066.CIR-16-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paria BC, Levin N, Lowery FJ, Pasetto A, Deniger DC, Parkhurst MR, et al. Rapid Identification and Evaluation of Neoantigen-reactive T-Cell Receptors From Single Cells. Journal of Immunotherapy 2020;Publish Ahead of Print doi 10.1097/cji.0000000000000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 2006;66(17):8878–86 doi 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014;124(5):2246–59 doi 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol Res 2016;4(3):204–14 doi 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 2003;26(4):332–42 doi 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest 2019;129(3):1109–14 doi 10.1172/JCI123791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. The Journal of Clinical Investigation 2020;130(11) doi 10.1172/JCI134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018;3(19) doi 10.1172/jci.insight.122467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009;114(3):535–46 doi 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29(7):917–24 doi 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(5):1019–27 doi 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016;352(6291):1337–41 doi 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 57.Ali M, Foldvari Z, Giannakopoulou E, Böschen ML, Strønen E, Yang W, et al. Induction of neoantigen-reactive T cells from healthy donors. Nat Protoc 2019;14(6):1926–43 doi 10.1038/s41596-019-0170-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.