

Abstract
We analyzed the efficacy and mechanistic interactions of PARP inhibition (PARPi; olaparib) and CDK4/6 inhibition (CDK4/6i; palbociclib or abemaciclib) combination therapy in castration-resistant prostate cancer (CRPC) and neuroendocrine prostate cancer (NEPC) models. We demonstrated that combined olaparib and palbociblib or abemaciclib treatment resulted in synergistic suppression of the p-Rb1-E2F1 signaling axis at the transcriptional and posttranslational levels, leading to disruption of cell-cycle progression and inhibition of E2F1 gene targets, including genes involved in DDR signaling/damage repair, antiapoptotic BCL-2 family members (BCL-2 and MCL-1), CDK1, and neuroendocrine differentiation (NED) markers in vitro and in vivo. In addition, olaparib + palbociclib or olaparib + abemaciclib combination treatment resulted in significantly greater growth inhibition and apoptosis than either single agent alone. We further showed that PARPi and CDK4/6i combination treatment–induced CDK1 inhibition suppressed p-S70-BCL-2 and increased caspase cleavage, while CDK1 overexpression effectively prevented the downregulation of p-S70-BCL-2 and largely rescued the combination treatment–induced cytotoxicity. Our study defines a novel combination treatment strategy for CRPC and NEPC and demonstrates that combination PARPi and CDK4/6i synergistically promotes suppression of the p-Rb1-E2F1 axis and E2F1 target genes, including CDK1 and NED proteins, leading to growth inhibition and increased apoptosis in vitro and in vivo. Taken together, our results provide a molecular rationale for PARPi and CDK4/6i combination therapy and reveal mechanism-based clinical trial opportunities for men with NEPC.
Keywords: PARP inhibitor, CDK4/6 inhibitor, Neuroendocrine differentiation, CRPC, NEPC
Introduction
Prostate cancer is the most common cancer diagnosis in males, with an estimated 191,930 new cases and 33,330 deaths in 2020 in the United States alone, and is the second leading cause of cancer-related death in men (1). Androgen receptor signaling inhibition (ARSI) has significantly improved outcomes for patients with metastatic prostate cancer; however, response is heterogeneous and resistance almost inevitable.
Patients with prostatic adenocarcinomas can present with scattered foci of prostate cancer cells, which demonstrate neuroendocrine differentiation (NED), or can show mixed features of adenocarcinoma and high-grade neuroendocrine prostate cancer (NEPC;2–7). NEPC is an aggressive prostate cancer subtype that commonly originates in castration-resistant prostate adenocarcinoma (CRPC) tumors and is more prevalent following treatment with ARSIs such as abiraterone and enzalutamide (ENZ;5–7). In prostate cancer cells and experimental model systems, a prostatic adenocarcinoma-to-neuroendocrine transition (NET) has been associated with specific genetic alterations in RB1 and TP53, which often involve RB1 deletion or mutation and loss of RB1 function (8–12). Importantly, RB1 alterations, frequently seen in NEPC, have a strong association with poor outcomes in patients with CRPC (8,13). E2F1 protein is a transcription factor that binds preferentially to RB1 in a cell cycle–dependent manner, mediating cell proliferation and apoptosis in a context-dependent fashion (14–16). The activities of E2F family proteins are restricted by the RB1 protein, but are released from RB1 and regulate gene transcription when RB1 is inactivated by CDK/cyclin-mediated RB1 hyperphosphorylation (17,18). E2F proteins control the expression of a multitude of positive-acting factors that are critical for progression through the S phase and the G2–M transition (19).
Activation of CDKs to promote cell-cycle progression plays a critical role across a variety of malignancies, and there are multiple FDA approved CDK inhibitors (20). With increasing understanding of the complexity of transcriptional dysregulation in cancer cells, the use of CDK inhibitors must be considered within the context of the CDK-RB1-E2F1 axis, and establishing which cancers will respond to CDK inhibitors has proved challenging (20,21). A recent report identifies significant genetic events that disrupt the CDK-RB1-E2F1 axis in metastatic breast cancer, including loss of CDKN2A/B; amplified CDK4, CDK6, and CCND1; and RB1 deletion (22). In accordance with this genomic profile, CDK4/6 inhibitors (CDK4/6is) are widely used to treat women with metastatic, hormone receptor–positive, HER2-negative breast cancer (23,24). Reported genetic events that disrupt the CDK-RB1-E2F1 axis in prostate cancer include CDKN2A loss, CCND1 and CDK4 amplification, and RB1 deletion (13,25). However, in contrast with metastatic breast cancer, there are few published preclinical studies and ongoing clinical trials using CDK4/6is in prostate cancer. [Three ongoing clinical trials (NCT02905318, NCT02494921, NCT04408924) and one completed (NCT02059213) are currently listed on the NCI Clinical Trials website.]
NEPC portends a poor prognosis and is difficult to treat with currently available therapies. Thus, there is a significant unmet clinical need for biologically rational treatment strategies that can suppress the NET and conversion to NEPC in men with advanced prostate cancer. We recently showed that PARP inhibitors (PARPis; olaparib and talazoparib) suppress tumor growth and NED via GR-MYCN CDK5-RB1-E2F1 signaling in enzalutamide-induced NEPC (12). In this study we also tested olaparib and dinaciclib, which inhibits CDK2, CDK5, CDK1, and CDK9, in combination experiments in vitro and in vivo, and the results were very promising. Unfortunately, dinaciclib has not met the safety criteria for FDA approval. CDK4/6is, however, are FDA approved for metastatic, hormone receptor–positive, HER2-negative breast cancer, and we hypothesized that PARPis in combination with CDK4/6i would demonstrate therapeutic efficacy with lower cytotoxicity in prostate cancer models. Our rationale for pursuing this combination in experimental and preclinical studies was based on the molecular specificity of clinically available, FDA-approved CDK4/6is and the congruence of the mechanism of action of these agents with our model of NET and the development of NEPC (12). Specifically, we were interested in the potential convergent effects of combined PARPis and CDK4/6i on regulation of the CDK-RB1-E2F1 axis and E2F1 downstream signaling activities in prostate cancer.
Our study herein introduces the novel strategy of combining PARPis and CDK4/6i to induce maximal prostate cancer cell growth inhibition and apoptosis in CRPC and NEPC compared with single-agent therapies in vitro and in vivo. In this study, we also analyzed the underlying molecular mechanisms for this drug combination and identified the molecular determinants and essential pathways that modulate its efficacy for these fatal, treatment-refractory malignancies.
Materials and Methods
Cell lines and reagents
Enzalutamide-sensitive human prostate cancer cell line model C4–2b cells were maintained as described previously (12). NCI-H660 cells were purchased from ATCC and maintained in suggested culture medium. C4–2b was validated by short tandem repeat DNA fingerprint with the AmpFLSTR Identifier PCR Amplification Kit (Thermo Fisher Scientific) in MD Anderson’s Characterized Cell Line Core Facility. Genomic mutation/deletion analysis for all cell lines was performed previously by targeted DNA sequencing. Palbociclib (S1116), abemaciclib (S5716), olaparib (KU-0059436), and enzalutamide (S1250) were purchased from Selleck Chemicals.
RT-qPCR analysis
Total RNA in cells was isolated by TRIzol Reagent (Thermo Fisher Scientific) and reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Quantitative real-time PCR (qPCR) was conducted with Fast SYBR Green Master Mix (Thermo Fisher Scientific) by Mx3005P Real-Time PCR System (Agilent). 2-ΔΔCt method was used to measure the relative quantities of mRNA expression normalized to internal reference gene controls. The qPCR primer sequences of genes are listed in Supplementary Table S1.
Immunoblotting analysis
Immunoblotting analysis was performed as previously described (12). The protein-specific antibodies used are listed in Supplementary Table S2, and the blot images were scanned on a Chemidoc MP imaging system (Bio-Rad).
Flow cytometry
For cell cycle analysis, cells were harvested after drug treatment, stained with propidium iodide, and analyzed on the FACSCanto II flow cytometer (BD Biosciences). PE Annexin V Apoptosis Detection Kit I (BD Biosciences) was used to detect cell apoptosis, and FlowJo software (TreeStar, Inc) was used for quantitative data analysis. In addition, analysis of cleaved caspase-3 by flow cytometry using a Cleaved Caspase-3 Staining Kit (FITC; Abcam) was also performed to determine cell apoptosis after treatment. The experiments were repeated at least three times for statistical analysis.
Plasmid transfection
Cells were seeded 1 day before transfection. Human CDK1 expression vector, Cdc2-HA (Plasmid No. 1888), was obtained from Addgene. Plasmid was transfected into cells using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific). Forty-eight hours after transfection, RNA and protein extracts were prepared for analysis, and biological assays were performed as indicated.
MTS assay
Following the manual instructions, the MTS assay was performed using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) and a microplate reader (BioTek). Briefly, 800 cells per well (C4–2b), 500 cells per well (C4–2b-ENZR), or 1,000 cells per well (NCIH660) were plated in 96-well plate one day before treatment. The cells were treated with the indicated drugs (or vehicle) at specified doses for 48 hours before CellTiter 96 AQueous One Solution Cell Proliferation Assay kit was utilized to determine the viable cells for each treatment. The data from replicate experiments were pooled, plotted, analyzed, and compared for statistical significance between the treatment samples.
Colony-forming assay
C4–2b and C4–2b-ENZR cells were seeded into 6-well plates at 5,000 cells per well and treated with palbociclib or abemaciclib, with or without olaparib, for up to 2 weeks for colony formation. Cell culture medium was replenished with fresh medium containing drugs or DMSO (vehicle control) every 3 days. At the termination of the experiments, colonies were fixed with cold methanol and then stained with 0.5% crystal violet. The number of colonies was imaged and counted with a microscope using NIS-Elements AR 2.30 software (Nikon).
Xenograft models
Aliquots of 2 × 106 NCI-H660 cells in 100 μL of PBS: Matrigel (1:1) were injected subcutaneously to male nude mice. For C4–2b-ENZR cells, a total of 1.5 × 106 cells were mixed with Matrigel and implanted subcutaneously to male nude mice. All tumor-bearing mice were treated with enzalutamide (10 mg/kg/day, every day, oral gavage) until the measured tumor volumes achieved 50mm3. In both tumor models, mice were randomly divided to receive vehicle control [sodium lactate buffer for palbociclib and (2-hydroxypropyl)-b-cyclodextrin for olaparib], olaparib (40 mg/kg/day, 5 days each week, i.p.), palbociclib (100 mg/kg/day, 5 days each week, oral gavage), or combination olaparib + palbociclib for 15 days (C4–2b-ENZR model) or 21 days (NCI-H660 model). Subcutaneous tumors were measured twice a week after initiation of treatment. All animal experiments were conducted in accordance with accepted standards of humane animal care approved by the Institutional Animal Care and Use Committee of MD Anderson Cancer Center (MDACC IACUC).
Statistical analysis
Data were presented as the mean ± SEM. The Wilcoxon rank-sum test was used for data with non-normal distributions or data with small sample sizes such as qRT-PCR analyses, MTS assay, colony assay, and flow cytometry assay.
Results
PARPi and CDK4/6i combination treatment results in significantly increased growth inhibition and apoptosis in prostate cancer cells in vitro.
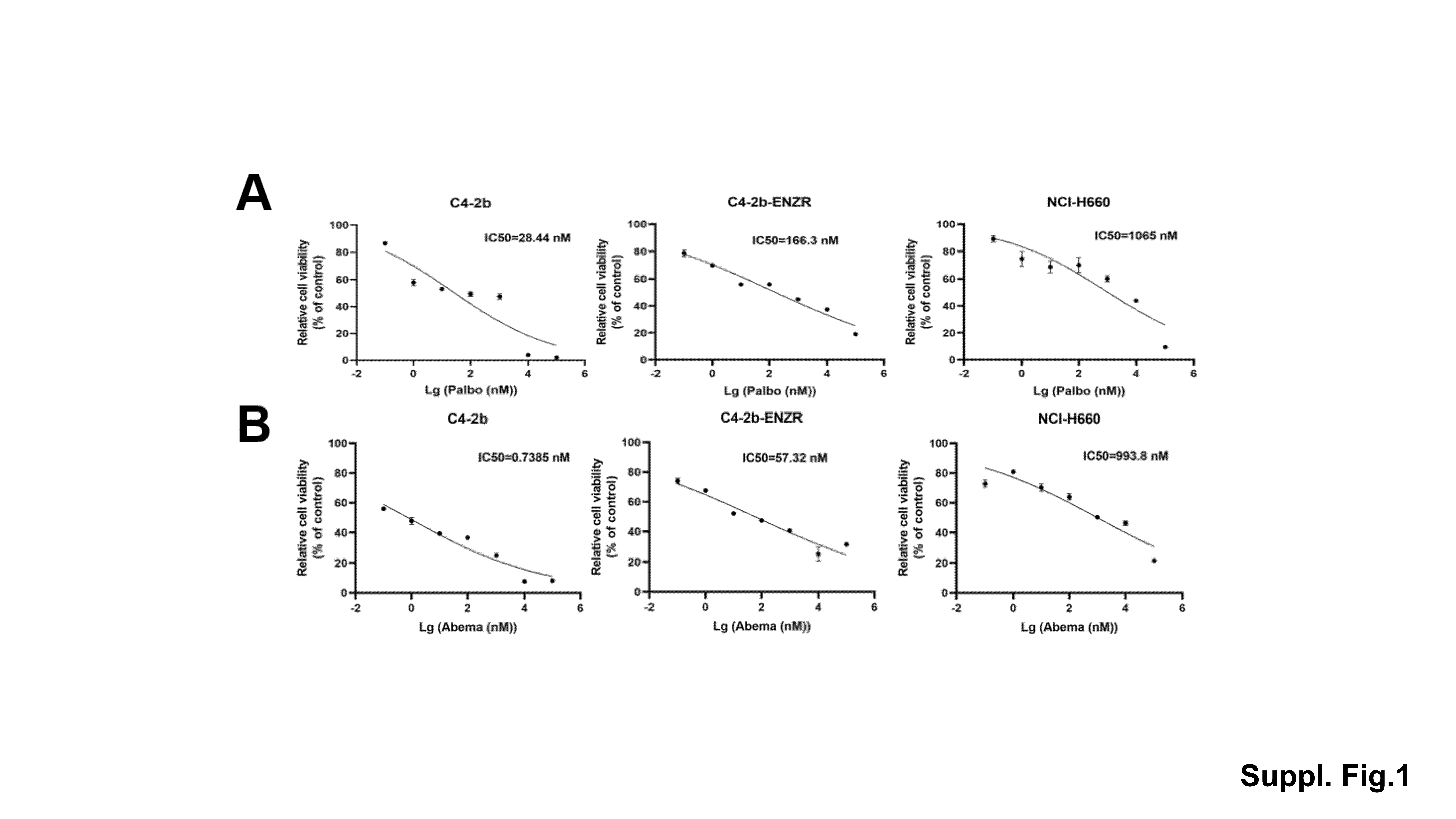
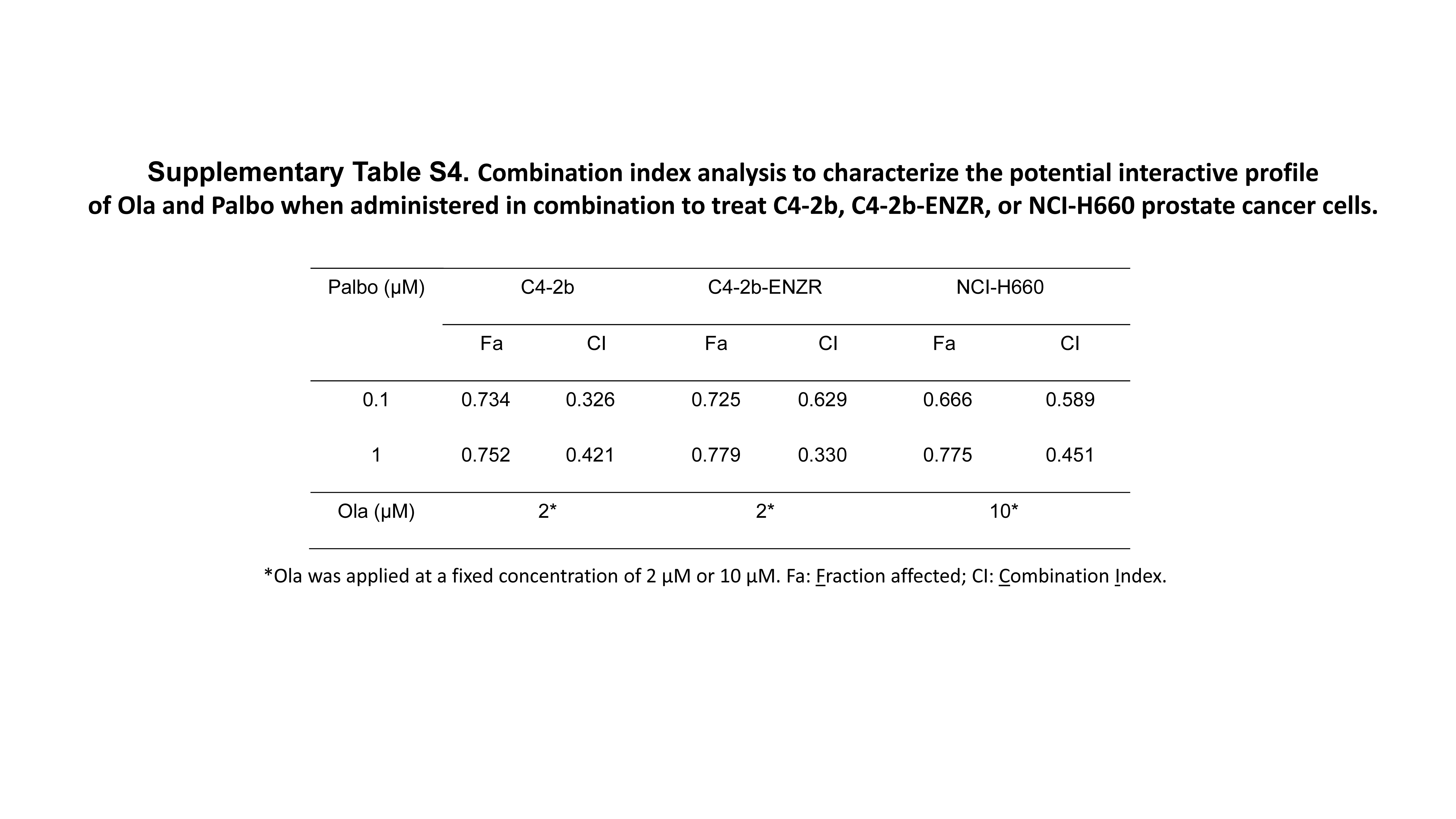
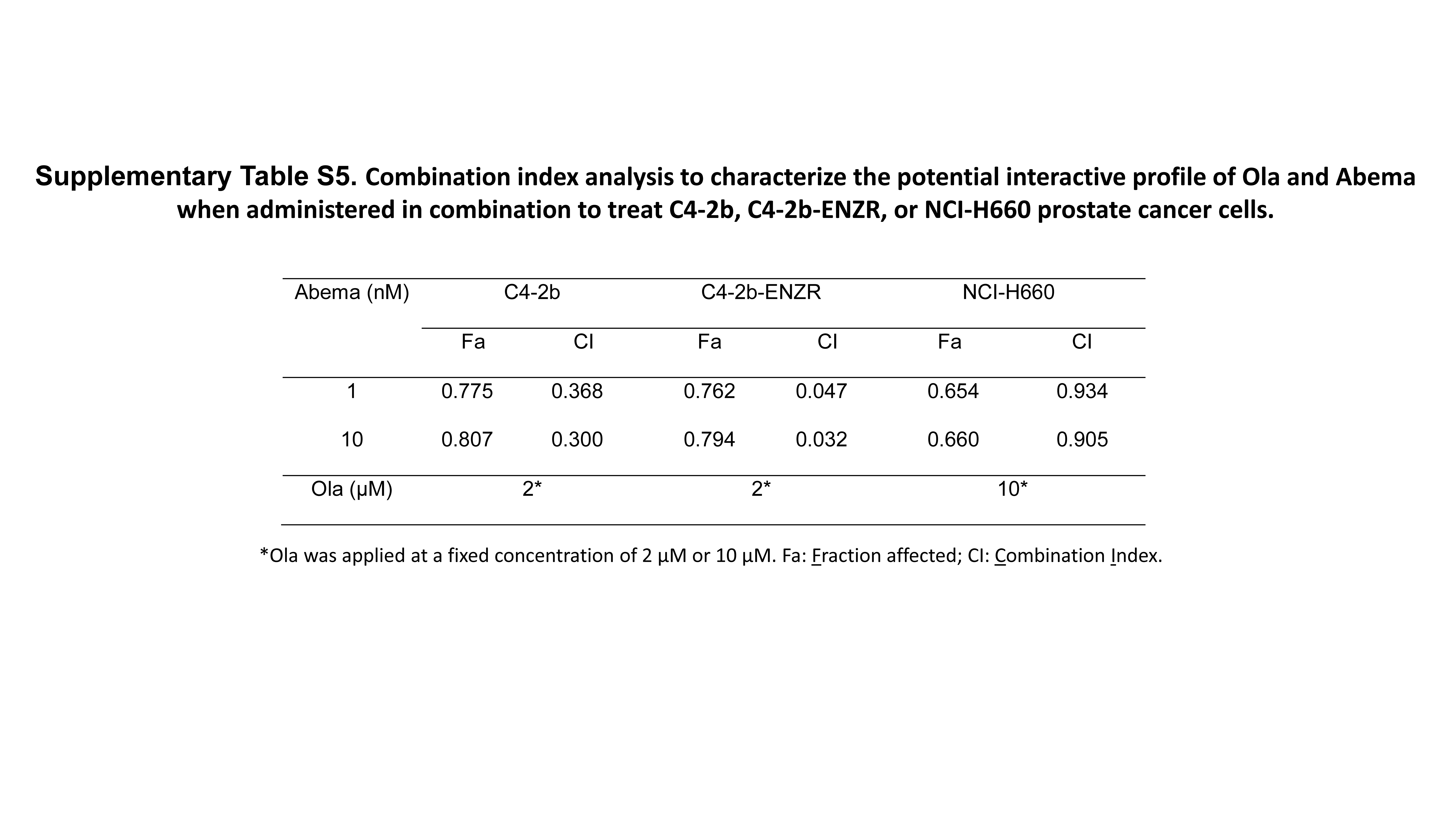
To evaluate the effects of CDK4/6is in prostate cancer cells, we analyzed the response of these cell lines to olaparib, palbociclib, and abemaciclib as single-agent treatments in vitro (Supplementary Table S3 and Supplementary Fig. S1). Compared with single agents, combined treatment with olaparib plus palbociclib or abemaciclib induced significantly greater growth inhibition in these prostate cancer cells (Fig. 1A and B), but this inhibitory effect was progressively reduced in C4–2b, C4–2b-ENZR, and NCI-H660 cells, which correlates with the increasing NED/NEPC phenotypic characteristics of these prostate cancer cell line models (12). Our analysis showed the likelihood of synergistic effects between olaparib + palbociclib or olaparib + abemaciclib compared with either single agent in C4–2b or C4–2b-ENZR cells (ref. 26; Supplementary Tables S4 and S5). In NCI-H660 cells, the analysis showed that olaparib + palbociclib was strongly synergistic (Supplementary Table S4), while olaparib + abemaciclib was only weakly synergistic to suppress cell growth, especially at low abemaciclib concentration (1 nmol/L; Supplementary Table S5). To analyze the underlying mechanisms of this combination treatment strategy, we utilized flow cytometry to determine cell cycle progression and apoptotic activities (sub-G1). As expected, single-agent treatment by palbociclib or abemaciclib arrested prostate cancer cells in the G1 phase, while olaparib arrested cells in the G2-M phase (Fig. 1C). These cell cycle distribution changes were most obvious in C4–2b, reduced in C4–2b-ENZR, and minimal in NCI-H660 cells (Fig. 1C). FACS/cell-cycle analysis of sub-G1 cells demonstrated that olaparib treatment increased the percentage of cells in the sub-G1 phase for all three cell line models compared with control (DMSO; Fig. 1D and E), and single-agent palbociclib or abemaciclib treatment also demonstrated a dose-dependent increase in the percentage of sub-G1 cells compared with control (Fig. 1D and E). Interestingly, olaparib + palbociclib or olaparib + abemaciclib combination treatment increased the percentage of sub-G1 cells to a greater extent than either single-agent treatment in C4–2b, C4–2b-ENZR, and NCI-H660 models (Fig. 1D and E). Taken together, these results indicated that, compared with single-agent treatment, olaparib + palbociclib or olaparib + abemaciclib combination treatment increased cell death, suppressed cell growth, disrupted cell-cycle checkpoints, and increased apoptotic cell death in C4–2b, C4–2b-ENZR, and NCI-H660 cells in vitro; although these drug responses were reduced in NCI-H660 cells.
Figure 1. PARP inhibitor and CDK4/6 inhibitor combination treatment suppresses cell proliferation and induces apoptosis of prostate cancer cells in vitro.

A and B, MTS cell viability assays showing effect of olaparib (Ola), palbociclib (Palbo), abemaciclib (Abema), and olaparib + palbociclib (Ola + Palbo) or olaparib + abemaciclib (Ola + Abema) combination treatment on viable proliferation of C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells. C4–2b and C4–2b-ENZR: olaparib, 2 mmol/L; palbociclib, 100 nmol/L and 1 mmol/L; abemaciclib, 1 nmol/L and 10 nmol/L. NCI-H660: olaparib, 10 mmol/L; palbociclib, 100 nmol/L and 1 mmol/L; abemaciclib, 1 nmol/L and 10 nmol/L. C, Flow cytometry showing effect of olaparib, palbociclib, and abemaciclib treatment on cell cycle distribution in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells. Cells were treated as shown in A and B. D and E, Flow cytometry showing sub-G1 (apoptotic) prostate cancer cells treated by olaparib, palbociclib, abemaciclib, and combination of olaparib + palbociclib or olaparib + abemaciclib. C4–2b and C4–2b-ENZR: olaparib, 2 mmol/L; palbociclib, 10 nmol/L, 100 nmol/L, 1 mmol/L, 10 mmol/L; abemaciclib, 0.1 nmol/L, 1 nmol/L, 10 nmol/L, 100 nmol/L, 1 mmol/L. NCI-H660: olaparib, 10 mmol/L; palbociclib, 10 nmol/L, 100 nmol/L, 1 mmol/L, 10 mmol/L; abemaciclib, 0.1 nmol/L, 1 nmol/L, 10 nmol/L, 100 nmol/L, 1 mmol/L. Data are plotted as the percentage of cells in sub-G1. F, Colony-formation assay of C4–2b and C4–2b-ENZR prostate cancer cells treated with olaparib, palbociclib, abemaciclib, and the combination of olaparib + palbociclib or olaparib + abemaciclib. The results were observed after 14 days of treatment. C4–2b and C4–2b-ENZR: olaparib, 500 nmol/L; palbociclib, 100 nmol/L; abemaciclib, 10 nmol/L. The histogram in the right panel shows mean ± SEM of (clone) counts from at least three assay replicates. G, Quantification analysis of apoptosis and fluorescence intensity of cleaved caspase-3-FITC in PARPi (olaparib), CDK4/6i (palbociclib or abemaciclib), or PARPi + CDK4/6i combination treatment of prostate cancer cells. C4–2b and C4–2b-ENZR: olaparib, 2 mmol/L; palbociclib, 2 mmol/L; abemaciclib, 2 mmol/L; NCI-H660: olaparib, 10 mmol/L; palbociclib, 10 mmol/L; abemaciclib, 10 mmol/L, incubated for 48 and 72 hours, respectively. The data are shown as mean ± SEM of three assay replicates. H, Immunoblots to show the proapoptotic response of prostate cancers treated with combination PARPi and CDK4/6i after 48 hours of incubation. C4–2b and C4–2b-ENZR: olaparib, 2 mmol/L; palbociclib, 2 mmol/L; abemaciclib, 2 mmol/L. NCI-H660: olaparib, 10 mmol/L; palbociclib, 10 mmol/L; abemaciclib, 10 mmol/L. IB signals in respective prostate cancer cells were scanned, quantified, and plotted to show BAX/BCL-2 ratios for the indicated treatments. Densitometric scans of IB bands were quantified and analyzed by ImageJ as previously reported (12), and the relative band intensities expressed as the folds of vinculin (internal IB reference), and indicated below each protein specific IB band image in the figure. For A, B, D, E, F, and G, t tests were used to determine statistical significance of the differences as indicated: ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We further analyzed the growth-inhibitory effects of olaparib + palbociclib or olaparib + abemaciclib combination treatment in C4–2b and C4–2b-ENZR models using colony-formation assays. Compared with single-agent treatment, olaparib + palbociclib or olaparib + abemaciclib induced a significantly greater inhibitory effect on colony growth in these cell models (Fig. 1F). In addition, olaparib + palbociclib or olaparib + abemaciclib, but not single-agent treatment, induced significant apoptotic cell death as determined by flow cytometry–based Annexin V/7-AAD analysis (Fig. 1G; Supplementary Fig. S2). Cleaved caspase-3–FITC analysis supported these results (Fig. 1G). Utilizing immunoblotting, we further analyzed the expression of selective protein markers involved in apoptotic response including BCL-2 family members BAX and BCL-2, cleaved PARP (cPARP), cleaved caspase-9 (CC9), and −3 (CC3) in C4–2b, C4–2b-ENZR, and NCI-H660 cells. The results revealed a substantially increased BAX/BCL-2 ratio, cleaved-PARP, and cleaved caspase-9, and −3 in cells treated with olaparib + palbociclib or olaparib + abemaciclib combinations (Fig. 1H). These results indicate that olaparib + palbociclib or olaparib + abemaciclib combination treatment effectively increases the apoptotic response of prostate cancer cells compared with single-agent treatments. However, whereas the apoptotic response to the olaparib + palbociclib or olaparib + abemaciclib combination is pronounced in C4–2b cells, it is reduced in C4–2b-ENZR, and further reduced in NCI-H660, suggesting an association with NED: increased NED in C4–2b-ENZR and NCI-H660 cells compared with C4–2b cells.
PARPi and CDK4/6i combination therapy leads to widespread inhibition of E2F1 signaling in prostate cancer
Given the role of cyclin D-CDK4/6 in RB protein phosphorylation and the activation of E2F1, which drives gene transcription for G1-S phase transition, we reasoned that E2F1 was involved in the mechanism of action for PARPi + CDK4/6i combination therapy. Thus, we initially analyzed a gene expression dataset available in the GEO database generated from a study of experimental ovarian cancer that examined the treatment effects of olaparib, palbociclib, and olaparib + palbociclib combination strategies (27). Our GSEA and differential gene expression analysis of these data show that in the ovarian cancer models, single-agent olaparib or palbociclib treatment suppressed E2F1 target gene expression, while olaparib + palbociclib combination treatment suppressed E2F1 target gene expression to a greater extent (Supplementary Fig. S3). These results and observations from prostate cancer and pan-cancer cross analysis prompted us to examine the dysregulated E2F1 and E2F1 signaling in C4–2b, C4–2b-ENZR, and NCI-H660 models. Immunoblot and qRT-PCR analysis demonstrated that increasing expression of E2F1 mRNA and protein correlates with increasing NED/NEPC phenotypic characteristics in these prostate cancer cell line models (Fig. 2A and B). In association with increased E2F1, pRB1-Ser780, pRB1-Ser795, and E2F1 NED gene targets, NSE, CHGA, and SYP were also increased as previously reported (12). Immunoblot analysis also showed that olaparib or palbociclib treatment could suppress the pRB1-E2F1 pathway and that olaparib + palbociclib combination treatment enhanced the inhibitory effect on this pathway, including NED proteins NSE, CHGA, and SYP, compared with either single-agent treatment (Fig. 2C). According to published ChIP-seq data in LNCaP prostate cancer cells, the promoters of a selective group of “marker” genes involved in cell-cycle control, including CHEK1, CHEK2, CDC25A, cyclin B1, cyclin B2, and CDK1, are potentially targeted by dysregulated E2F1 signaling (28). These genes are also included in E2F1 target gene public data set(s) and/or have been further validated as being transcriptionally regulated by E2F1. Utilizing qRT-PCR, we confirmed that mRNA expression of these E2F1 target genes (and E2F1) is suppressed by olaparib + palbociclib combination treatment to a significantly greater extent than by olaparib or palbociclib single-agent treatment, in particular in C4–2b-ENZR and NCI-H660 cells (Fig. 2D–J), which is consistent with the enhanced suppression of E2F1 signaling observed in olaparib + palbociclib–treated cells [Fig. 2C; similar, if not more pronounced, results were observed in olaparib + abemaciclib combination treatment, suggesting a general mechanism of action of PARPi + CDK4/6i in prostate cancer cells (Supplementary Fig. S4)]. It is notable that olaparib + CDK4/6i combination treatment showed less efficient inhibition of E2F1 target gene expression (compared with vehicle control) in NCI-H660 NEPC (Fig. 2D–J; Supplementary Fig. S4). This may be explained by the significantly higher baseline expression of E2F1, RB1, and pRB1 in NCI-H660 compared with C4–2b and C4–2b-ENZR models (Fig. 2A and B); this effectively limits the action of olaparib + palbociclib combination treatment, which mechanistically depends on targeting E2F1 and E2F1 signaling through suppression of the pRB1-E2F1 axis (Fig. 2C). This explanation is also worthy of consideration with regard to reduced relative sensitivity of NCI-H660 cells to PARPi + CDK4/6i combination treatment–induced apoptotic response (Fig. 1D, E, and G). Among the E2F1 target gene expressions most strongly inhibited by PARPi + CDK4/6i, CDK1 is especially interesting because CDK1 is essential for cell viability and plays a vital role in DNA damage repair and checkpoint activation during cell response to DNA damage–inducing agents (29–31). Interestingly, CDC25A, which is required for CDK1-cyclin B assembly and CDK1 activation, was also suppressed by olaparib, palbociclib, and olaparib + palbociclib and displayed a significantly enhanced inhibition by olaparib + palbociclib in C4–2b-ENZR (Fig. 2G). Immunoblotting analysis demonstrated that CDC25A and CDK1 protein expression levels in C4–2b, C4–2b-ENZR, and NCI-H660 cells correlate to mRNA expression levels regulated by olaparib, palbociclib, abemaciclib, and olaparib + palbociclib or olaparib + abemaciclib, suggesting regulation of CDC25A and CDK1 protein expression at transcriptional levels in these drug–treated cells (Fig. 2K). Thus, mechanistically, the E2F1 targets CDC25A and CDK1 are maximally inhibited in prostate cancer cell models by olaparib + CDK4/6i combination treatment at the transcriptional (Fig. 2H; Supplementary Fig. S4) and posttranslational levels (Fig. 2K).
Figure 2. PARP inhibitor and CDK4/6 inhibitor combination treatment inhibits E2F1 signaling in prostate cancer through transcriptional and post-translational regulation in prostate cancer cells.

A, Basal expression levels of RB, pRB-Ser780/795, E2F1 proteins, and E2F1 targets including NED markers NSE, CHGA, and SYP in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells. B, E2F1 mRNA expression in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells. C, Effects of PARPi (olaparib), CDK4/6i (palbociclib), and combination olaparib + palbociclib on RB, pRB-Ser780/795, E2F1, and E2F1 signaling targets (NED markers) in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells. C4–2b and C4–2b-ENZR: olaparib, 2 mmol/L; palbociclib, 100 nmol/L and 1 mmol/L. NCI-H660: olaparib, 10 mmol/L; palbociclib, 100 nmol/L and 1 mmol/L. All treatments involved a 48-hour incubation. D–J, qRT-PCR analysis to determine E2F1 and E2F1 target mRNA expression in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells treated with PARPi (olaparib), CDK4/6i(palbociclib), or olaparib + palbociclib combination. K, Immunoblot to show suppression of CDK1 and CDC25 protein expression by PARPi (olaparib), CDK4/6i (palbociclib or abemaciclib), and olaparib + palbociclib or olaparib + abemaciclib combination treatment in prostate cancer cells. Immunoblot signals (A, C, K) were quantified and normalized to vinculin, and the relative band intensities shown below each protein specific IB band image in the figure. For B, D, E, F, G, H, I and J, t tests were used to determine statistical significance of the differences as indicated: ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Apoptotic response of prostate cancer to PARPi + CDK4/6i combination treatment is associated with proapoptotic expression of BCL-2 family proteins
Compared to PARPi or CDK4/6i single-agent treatment, our results demonstrate that combination olaparib + CDK4/6i combination treatment induces greater apoptosis in prostate cancer cells, with progressively reduced responsiveness in C4–2b, C4–2b-ENZR, and NCI-H660 cells (Fig. 1D, E, and G). Consistent with these results, we also showed increased ratios of proapoptotic BCL-2 family protein BAX to anti-proapoptotic protein BCL-2 in these prostate cancer models following treatment with PARPi + CDK4/6i, which was associated with activation and significantly increased levels of downstream apoptotic caspase protein markers (Fig. 1H). We also showed that antiapoptotic BCL-2 family members BCL-2 and/or MCL-1 are upregulated (mRNA and protein) in enzalutamide-resistant C4–2b-ENZR and NCI-H660 NEPC cells (Fig. 3A and B). In addition, compared to PARPi or CDK4/6i single-agent treatments, we also found that proapoptotic BCL-2 member BAX is significantly upregulated in C4–2b and C4–2b-ENZR prostate cancer cells by PARPi and CDK4/6i combination treatment, but not in NCI-H660 NEPC cells (Fig. 1H). Taken together, our results suggest that increased prosurvival BCL-2 protein levels contribute to the relative resistance of C4–2b-ENZR or NCI-H660 cells to the proapoptotic effects of olaparib + CDK4/6i treatment, and, mechanistically, PARPi + CDK4/6i combination–induced apoptosis is limited by the relatively high intrinsic ratio of prosurvival vs proapoptotic BCL-2 family members in these prostate cancer models.
Figure 3. CDK1 overexpression upregulates “prosurvival sensor” p-S70-BCL-2 and rescues prostate cancer cells from PARPi + CDK4/6i combination treatment–induced apoptosis.

A, Immunoblots showing expression of anti-proapoptotic BCL-2 and MCL-1 proteins in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells, which is consistent with BCL-2 and MCL-1 mRNA expression determined by RT-qPCR and shown in B. C, RT-qPCR results show expression of BCL-2 and MCL-1 mRNAs regulated by PARPi (olaparib), CDK4/6i (palbociclib), and olaparib + palbociclib combination treatment in prostate cancer cells. D, Immunoblots showing the CDK1 protein basal expression levels in prostate cancer cell line models. E, Overexpression of exogenous CDK1 protein by transfecting CDK1 expression vectors in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cells. F, Immunoblots showing p-S70-BCL-2 and BCL-2 protein expression in prostate cancer cells treated with olaparib, palbociclib, and olaparib + palbociclib in C4–2b, C4–2b-ENZR, and NCI-H660, with or without CDK1 overexpression by transfection of CDK1 expression vector after 48 hours of treatment. C4–2b and C4–2b-ENZR: olaparib, 2 umol/L; palbociclib, 2 umol/L; or combinations. NCI-H660: olaparib, 10 umol/L; palbociclib, 10 umol/L; or combinations. Immunoblot signals (A, D, E, F) were quantified and normalized to vinculin, and the relative band intensities shown below each protein specific IB band image in the figure. G, Flow cytometry showing effect of olaparib, palbociclib, combination olaparib + palbociclib, and overexpression of CDK1 on cell cycle distribution in C4–2b, C4–2b-ENZR, and NCI-H660 cells. C4–2b and C4–2b-ENZR: olaparib, 2 umol/L; palbociclib, 2 umol/L. NCI-H660: olaparib, 10 umol/L; palbociclib, 10 umol/L. H, Flow cytometry showing sub-G1 (apoptotic) cell distribution with olaparib, palbociclib, or combination olaparib + palbociclib, with or without overexpression of CDK1 in C4–2b, C4–2b-ENZR, and NCI-H660 cells. For B, C, and H, t tests were used to determine statistical significance of the differences as indicated: ns, not significant; * P<0.05, ** P<0.01, *** P<0.001.
Interestingly, published LNCaP prostate cancer cell ChIP-seq data showed the recruitment of E2F1 protein in BCL-2 and MCL-1 gene promoters, indicating E2F1 regulation of these genes (28). To evaluate the potential for E2F1 regulation of BCL-2 and MCL-1 within the context of olaparib + palbociclib combination treatment, we analyzed BCL-2 and MCL-1 mRNA levels by RT-qPCR following olaparib and/or palbociclib treatment. The results showed that, compared to olaparib or palbociclib single-agent treatment, combination olaparib + palbociclib leads to significantly greater downregulation of BCL-2 mRNA expression in C4–2b and C4–2b-ENZR cells and of MCL-1 mRNA expression in C4–2b-ENZR and NCI-H660 prostate cancer cells (Fig. 3C). It is interesting that, while olaparib and palbociclib single-agent treatments upregulated BCL-2 mRNA expression, olaparib + palbociclib combination treatment failed to downregulate BCL-2 mRNA expression in NCI-H660 cells (Fig. 3C), which implies a mechanism other than E2F1 that regulates BCL-2 gene expression in this prostate cancer cell line. Similar observations were shown in response to olaparib + abemaciclib combination treatment, yet the olaparib + abemaciclib combination demonstrated significant downregulation of both BCL-2 and MCL-1 in C4–2b-ENZR and NCI-H660 cells (Supplementary Fig. S4). Together, these results suggest that PARPi + CDK4/6i combination treatment promotes apoptosis through inhibition of E2F1 signaling and downregulation of prosurvival BCL-2 family members in these prostate cancer models.
CDK1 overexpression mediates the survival of PARPi + CDK4/6i–treated prostate cancer cells by upregulation of “prosurvival sensor” p-S70-BCL-2
It is notable that PARPi + CDK4/6i combination treatment ostensibly disrupts the G2 checkpoint through inhibition of CDK1 (an E2F1 target) by suppressing CDK1 expression at both mRNA and protein levels (Fig. 2H and K). Although complex and somewhat conflicting results have been reported regarding the role of CDK1-mediated phosphorylation of BCL-2 family proteins in apoptosis for specific cell types under various experimental conditions (32,33), it has been shown that CDK1 can mediate survival of drug-treated cells through mitosis by phosphorylating BCL-2 at Ser70, which mechanistically depends on the increased binding affinity of p-S70-BCL-2 to its proapoptotic partner (34,35). Thus, we examined the function of the CDK1-p-S70-BCL-2 axis in mediating apoptosis of prostate cancers treated with combination olaparib + palbociclib utilizing CDK1 overexpression in prostate cancer cells transfected with CDK1 expression vector (Fig. 3D and E). We showed that, while combination olaparib + palbociclib suppressed p-S70-BCL-2, this activity was reversed by overexpression of CDK1 in C4–2b, C4–2b-ENZR, and NCI-H660 cells (Fig. 3F). Importantly, we also demonstrated mitigation of G2 arrest (Fig. 3G) and proapoptotic activity (increased sub-G1) following enforced CDK1 expression (Fig. 3H). These results show a functional link between G2 arrest and proapoptotic activities induced by olaparib + palbociclib combination therapy and are consistent with recent reports that show that the proapoptic activities of other potential anti-tumor agents are associated with G2 arrest (36,37). Taken together, our results demonstrate a mechanistic link between CDK1 and BCL-2 in the proapoptotic response of olaparib + palbociclib combination therapy in these prostate cancer models.
Olaparib + palbociclib combination therapy leads to inhibition of E2F1 target genes involved in DNA damage response signaling and DNA repair
E2F1 maintains critical roles in a wide range of cellular processes including cell cycle progression, DNA damage response (DDR) signaling, DNA replication, and DNA repair through transcriptional regulation of specific target genes or direct localization to DNA break sites to facilitate homologous recombination (HR) activity. E2F1 inhibition can lead to compromised DNA repair and DNA replication, leaving unrepaired DNA damage (38–40). Analysis of a published ChIP-seq dataset for E2F1 in LNCaP prostate cancer cells (28) showed that E2F1 may be recruited to the gene promoters and regulate the expression specific genes involved in DDR signaling, DNA repair, and DNA replication in prostate cancer. Utilizing RT-qPCR, we demonstrated reduced expression of putative E2F1 target genes CHEK1 and CHEK2 in prostate cancer cells following PARPi + CDK4/6i combination treatment (Fig. 2E and F; Supplementary Fig. S4D and S4E). We further examined and characterized the expression patterns of these and additional putative E2F1-regulated DDR signaling, DNA replication, and DNA repair genes associated with olaparib + palbociclib or olaparib + abemaciclib combination treatment compared to single-agent treatment in C4–2b, C4–2b-ENZR, and NCI-H660 cells. The results showed greater suppression of nearly all gene-specific mRNA levels by olaparib + palbociclib or olaparib + abemaciclib compared with olaparib, palbociclib, or abemaciclib single-agent treatment for all cell models, and suggest that substantial accumulation of unrepaired DNA damage may accompany this combination treatment (Fig. 4A-H). Interestingly, CDK1 reportedly participates in HR-dependent repair of DNA double-strand breaks (DSB) and phosphorylation and activation of BRCA1 (41–43).
Figure 4. PARP inhibitor + CDK4/6 inhibitor combination treatment leads to downregulation of E2F1 target genes involved in DDR signaling, DNA damage repair, and DNA replication in prostate cancer cells.

A–H, qRT-PCR analysis to determine expression of an array list of genes that are involved in DDR signaling and/or DNA damage repair and/or DNA replication for C4–2b, C4–2b-ENZR and NCI-H660 prostate cancer cells treated with olaparib, palbociclib, abemaciclib, or combination olaparib + palbociclib or olaparib + abemaciclib. The indicated mRNA expressions were plotted and are shown as the folds of their expressions in controls (vehicle treated) for each prostate cancer model, respectively. For statistical analysis, data from treatment replicates were pooled. T tests were used to determine statistical significance of the differences as indicated: ns, not significant; *, P < 0.05, **, P < 0.01, and ***, P < 0.001.
Olaparib + palbociclib combination therapy synergistically suppresses prostate cancer xenograft tumor growth and validates the E2F1 inhibition–mediated mechanistic model
To maintain enzalutamide resistance in C4–2b-ENZR cells during in vitro culture, we included enzalutamide (10 mmol/L) in the culture media (12). To confirm a stable enzalutamide-resistant phenotype for this model in the absence of enzalutamide, we performed clonal growth analysis of C4–2b-ENZR cells with or without enzalutamide treatment for a 2-week period, followed by reseeding in enzalutamide (10 mmol/L)-containing media in vitro. The results showed that C4–2b-ENZR cells maintained enzalutamide resistance for a 2-week period in the absence of enzalutamide, while parental C4–2b cells maintained enzalutamide-sensitive growth inhibition as expected (Supplementary Fig. S5).
We conducted further preclinical studies, which demonstrated that olaparib + palbociclib combination treatment leads to significantly suppressed tumor growth compared with single-agent olaparib or palbociclib treatment in both C4–2b-ENZR and NCI-H660 xenograft models. We observed markedly reduced tumor wet weight and decreased density of viable cells in tumors harvested at the endpoint of treatment from the combination group compared with either single-agent group (Fig. 5A and B). The combination treatment revealed a moderate synergistic interaction between olaparib and palbociclib by Bliss independence analysis [ref. 44; Fig. 5A and B (right)]. Immunoblot analysis of tumor tissue samples confirmed suppression of the pRB-E2F1 axis (E2F1, RB1, and p-S780-RB1) and inhibition of CDK1 expression and CDK1 activation (CCNB1/2, CDC25A, and p-Y15-CDK1/CDK1). These results are consistent with downregulation of p-S70-BCL-2, suppression of antiapoptotic protein MCL-1, and elevation of cleaved caspase-3 in olaparib + palbociclib–treated C4–2b-ENZR and NCI-H660 tumors compared with olaparib or palbociclib single-agent treatment in vivo (Fig. 5C). Further analysis of these tumor samples demonstrated that olaparib + palbociclib combination treatment led to greater suppression of E2F1 and specific E2F1 target mRNA expressions compared with olaparib or palbociclib single agent treatment (Supplementary Fig. S6). Together, our results are consistent with moderate synergistic inhibitory effects of olaparib + CDK4/6i on tumor growth and NED, which involves inhibition of multiple E2F1-driven pathways through direct and indirect mechanisms, including suppression of CDK1 expression and prosurvival BCL-2–mediated functions, as well as suppression of DDR signaling and DNA repair (Fig. 5D).
Figure 5. PARPi + CDK4/6i combination treatment leads to tumor growth inhibition through suppression of p-RB-E2F1 and E2F1 signaling, and suppression of CDK1, p-S70-BCL-2, and antiapoptotic protein MCL-1 in prostate cancer xenograft models compared to PARPi or CDK4/6i single-agent treatment.

A and B, Tumor growth curves and terminal wet weights of C4–2b-ENZR and NCI-H660 xenograft tumors treated with PARPi (olaparib), CDK4/6i (palbociclib), or olaparib + palbociclib combination. Tumor volumes were measured every 3 days. Left, y-axis shows mean tumor volume (mm3), and x-axis shows days of treatment. Treatments were initiated when the tumor volume reached approximately 50mm3. olaparib: 40 mg/kg/days, 5 days/week, i.p.; palbociclib: 100 mg/kg/day, 5 days/week, oral gavage. Right, tumor wet weights at termination. ANOVA analysis was used to determine the statistical significance of tumor volumes as indicated, and t tests were used to determine statistical significance of the differences of tumor wet weights as indicated: ns, not significance, *, P < 0.05; **, for P < 0.01, and ***, for P < 0.001. ^, indicates that the combination treatment revealed a moderate synergistic interaction between olaparib and palbociclib by Bliss independence analysis (44). C, Immunoblot analysis of treated xenograft tumors shows expression of proteins involved in E2F1 signaling and CDK1 activation, as well as antiapoptotic proteins including “prosurvival sensor” p-S70-BCL-2 and MCL-1 as indicated. Immunoblot signals (C) were quantified and normalized to vinculin, and the relative band intensities shown below each protein specific IB band image in the figure. D, Proposed mechanism by which PARPi + CDK4/6i combination treatment suppresses growth and NED markers and induces apoptosis in prostate cancer models in vitro and in vivo.
Discussion
CDK4/6 inhibitors selectively target CDK4 and CDK6 and lead to the suppression of RB phosphorylation and concomitant inhibition of G1-S cell cycle progression, in part through repression of E2F-mediated gene transcription (45). CDK4/6 inhibitors have emerged as a significant advance for cancer treatment with multiple FDA-approved agents in clinical use for the treatment of hormone receptor–positive, HER2-normal breast cancer (46,47). In prostate cancer, there have been relatively few preclinical studies or clinical trials of CDK4/6i completed to date.
In the present study study, we analyzed the effects of olaparib, palbociclib, Abema, olaparib + palbociclib, and olaparib + abemaciclib combination treatments in C4–2b, C4–2b-ENZR, and NCI-H660 prostate cancer cell line models. These models demonstrated increasing NED/NEPC phenotypic characteristics (12). We showed that this increasing NED/NEPC phenotype was associated with a corresponding increase in BCL-2 and/or MCL-1, suggesting dependence of the NED/NEPC phenotype on the antiapoptotic activities of the reduced BAX/BCL-2 ratio. We showed that combination therapy with PARPi and CDK4/6i cooperatively suppressed prostate cancer cell growth and viability, disrupted cell-cycle checkpoints, and induced apoptotic activities in C4–2b, C4–2b-ENZR, and NCI-H660 cells to varying degrees (Fig. 1). Importantly, olaparib + palbociclib or olaparib + abemaciclib treatment resulted in increased proapoptotic protein BAX/antiapoptotic (prosurvival) protein BCL-2 ratios in C4–2b, C4–2b-ENZR, and NCI-H660 cells, further supporting a role for BCL-2 in proapoptotic responses to these drug combinations (Fig. 1H). Previous studies have demonstrated an association between increased G2-M arrest, increased BAX, and increased apoptosis in response to specific experimental therapies (48). Interestingly, in contrast to C4–2b cells, olaparib treatment did not induce G2-M arrest in C4–2b-ENZR or NCI-H660 cells, suggesting that the reduced capacity of this checkpoint function is associated with the development of enzalutamide-resistant CRPC and NEPC (Fig. 1C).
We showed that the E2F1 target gene CDK1 is strongly suppressed by olaparib + palbociclib and olaparib + abemaciclib in C4–2b, C4–2b-ENZR, and NCI-H660 cells (Fig. 2H and K; Supplemental Fig. S4B). It is noteworthy that suppression of CDK1-cyclin B activities, the central regulatory node in the G2 checkpoint, leads to G2-M arrest (49). We demonstrated that overexpression of CDK1 and associated CDK1-mediated BCL-1 phosphorylation at Ser70 could rescue cell viability (which was inhibited by olaparib + palbociclib combination therapy), mitigate G2 arrest, and suppress apoptotic activity (Fig. 3F–H). Thus, increased induction of apoptosis by olaparib + palbociclib or olaparib + abemaciclib, compared to single-agent treatment, is associated with marked suppression of CDK1, disruption of G2-M regulation, and reduced p-S70-BCL-2 in CRPC (C4–2b), enzalutamide-resistant CRPC (C4–2b-ENZR), and NEPC (NCI-H660) cell line models and shows significant therapeutic activity in C4–2b-ENZR and NCI-H660 xenografts (Fig. 5A and B). These results demonstrate a functional link between G2 arrest and apoptosis induced by potential anti-tumor agents and highlight new possibilities for targeting the G2-M checkpoint in anti-cancer therapy, including CDK1 (36,37).
In the present study, we showed that CDK4/6 activities are critical regulators of a functional RB1-E2F1 switch that controls NED through transcriptional regulation of NED gene expression (CHGA, SYP, and NSE) across prostate cancer cell lines (C4–2b, C4–2b-ENZR, and NCI-H660) that represent the transition from prostatic adenocarcinoma to NED and NEPC and harbor functional RB1. Single-agent palbociclib or abemaciclib inhibited NED, yet olaparib + palbociclib and olaparib + abemaciclib demonstrated cooperative, reinforced NED suppression through inhibition of the Rb1-E2F1 pathway and transcriptional regulation of the E2F1 gene in these prostate cancer cell lines (Fig. 2C and D; Supplemental Fig. S4A). We further linked downregulation of the E2F1 target gene CDK1 to the mechanism of action of PARPi + CDK4/6i combination therapy activities (Figs. 3 and 5C; Supplemental Fig. S6C). Our results point to the possibility that E2F1 targets represent a high-yield pool for selecting PARPi + CDK4/6i–predictive biomarkers and potential therapy targets for further studies. Interestingly, the majority of CRPCs harbor functional RB1, and although RB1 alterations are frequent in NEPC, a substantial percentage of these tumors also contain functional RB1 (8,13). The use of CDK4/6is in the treatment of metastatic prostate cancer is under clinical development, but early clinical data in the castrate-sensitive setting suggests that single-agent use is not likely to provide substantial benefit (50), thus biologically rational combination therapy with CDK4/6 inhibitors and other targeted or immune agents is paramount.
In summary, our study defines a novel combination treatment strategy for CRPC and NEPC and demonstrates that combination PARPi and CDK4/6i synergistically promotes suppression of the p-Rb1-E2F1 axis and E2F1 target genes, including CDK1 and NED proteins, leading to growth inhibition and increased apoptosis in vitro and in vivo. Our results provide a molecular rationale for PARPi + CDK4/6i combination therapy and reveal mechanism-based clinical trial opportunities for men with CRPC and NEPC for whom treatments that provide durable responses are greatly needed.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We acknowledge the editorial assistance of Sarah E. Townsend. This research was supported by MD Anderson NCI Prostate Cancer SPORE Grant P50 CA140388, and NCI Cancer Center Support Grant P30 CA16672.
Financial support: This research was supported by MD Anderson NCI Prostate Cancer SPORE Grant P50 CA140388, and NCI Cancer Center Support Grant P30 CA16672.
Footnotes
Conflicts of interest: All authors declare no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate 1999;39(2):135–48. [DOI] [PubMed] [Google Scholar]
- 3.Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol 2014;38(6):756–67 doi 10.1097/PAS.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moch H, Humphrey P, Ulbright T, Reuter V. Neuroendocrine tumours. WHO Classification of Tumours of the Urinary System and Male Genital Organs. 4th ed. Lyon: International Agency for Research on Cancer; 2016. p 172–4. [Google Scholar]
- 5.Palmgren JS, Karavadia SS, Wakefield MR. Unusual and underappreciated: small cell carcinoma of the prostate. Semin Oncol 2007;34(1):22–9 doi 10.1053/j.seminoncol.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013;15(1):1–10 doi 10.1593/neo.121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018;36(24):2492–503 doi 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22(3):298–305 doi 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355(6320):78–83 doi 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355(6320):84–8 doi 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018;362(6410):91–5 doi 10.1126/science.aat5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu B, Li L, Yang G, Geng C, Luo Y, Wu W, et al. PARP Inhibition Suppresses GR-MYCN-CDK5-RB1-E2F1 Signaling and Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer. Clin Cancer Res 2019;25(22):6839–51 doi 10.1158/1078-0432.CCR-19-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019;116(23):11428–36 doi 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin XQ, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin WG Jr. The transcription factor E2F1 is a downstream target of RB action. Mol Cell Biol 1995;15(2):742–55 doi 10.1128/mcb.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett MR, Macdonald K, Chan SW, Boyle JJ, Weissberg PL. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 1998;82(6):704–12 doi 10.1161/01.res.82.6.704. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Li M, Wang H, Agrawal S, Zhang R. Antisense therapy targeting MDM2 oncogene in prostate cancer: Effects on proliferation, apoptosis, multiple gene expression, and chemotherapy. Proc Natl Acad Sci U S A 2003;100(20):11636–41 doi 10.1073/pnas.1934692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chinnam M, Goodrich DW. RB1, development, and cancer. Curr Top Dev Biol 2011;94:129–69 doi 10.1016/B978-0-12-380916-2.00005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14(8):518–28 doi 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015;14(2):130–46 doi 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9(3):153–66 doi 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 21.Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 2019;19(6):326–38 doi 10.1038/s41568-019-0143-7. [DOI] [PubMed] [Google Scholar]
- 22.Paul MR, Pan TC, Pant DK, Shih NN, Chen Y, Harvey KL, et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J Clin Invest 2020;130(8):4252–65 doi 10.1172/JCI129941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curigliano G, Loibl S. CDK4/6 inhibitors in breast cancer: one more step towards reduced mortality. Lancet Oncol 2019. doi 10.1016/S1470-2045(19)30808-3. [DOI] [PubMed] [Google Scholar]
- 24.Gao JJ, Cheng J, Bloomquist E, Sanchez J, Wedam SB, Singh H, et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US Food and Drug Administration pooled analysis. Lancet Oncol 2020;21(2):250–60 doi 10.1016/S1470-2045(19)30804-6. [DOI] [PubMed] [Google Scholar]
- 25.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161(5):1215–28 doi 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70(2):440–6 doi 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 27.Yi J, Liu C, Tao Z, Wang M, Jia Y, Sang X, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019;43:225–37 doi 10.1016/j.ebiom.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNair C, Xu K, Mandigo AC, Benelli M, Leiby B, Rodrigues D, et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest 2018;128(1):341–58 doi 10.1172/JCI93566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecona E, Fernandez-Capetillo O. Targeting ATR in cancer. Nat Rev Cancer 2018;18(9):586–95 doi 10.1038/s41568-018-0034-3. [DOI] [PubMed] [Google Scholar]
- 30.Lemmens B, Hegarat N, Akopyan K, Sala-Gaston J, Bartek J, Hochegger H, et al. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol Cell 2018;71(1):117–28 e3 doi 10.1016/j.molcel.2018.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szmyd R, Niska-Blakie J, Diril MK, Renck Nunes P, Tzelepis K, Lacroix A, et al. Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality. Oncogene 2019;38(7):998–1018 doi 10.1038/s41388-018-0464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blagosklonny MV. Unwinding the loop of Bcl-2 phosphorylation. Leukemia 2001;15(6):869–74 doi 10.1038/sj.leu.2402134. [DOI] [PubMed] [Google Scholar]
- 33.Sakurikar N, Eichhorn JM, Chambers TC. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J Biol Chem 2012;287(46):39193–204 doi 10.1074/jbc.M112.391854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai H, Ding H, Meng XW, Lee SH, Schneider PA, Kaufmann SH. Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance. Cancer Res 2013;73(23):6998–7008 doi 10.1158/0008-5472.CAN-13-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou L, Cai X, Han X, Xu N, Chang DC. CDK1 switches mitotic arrest to apoptosis by phosphorylating Bcl-2/Bax family proteins during treatment with microtubule interfering agents. Cell Biol Int 2014;38(6):737–46 doi 10.1002/cbin.10259. [DOI] [PubMed] [Google Scholar]
- 36.Parida PK, Mahata B, Santra A, Chakraborty S, Ghosh Z, Raha S, et al. Inhibition of cancer progression by a novel trans-stilbene derivative through disruption of microtubule dynamics, driving G2/M arrest, and p53-dependent apoptosis. Cell Death Dis 2018;9(5):448 doi 10.1038/s41419-018-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu CY, Jerry Teng CL, Hung PS, Cheng CC, Hsu SL, Hwang GY, et al. Ovatodiolide isolated from Anisomeles indica induces cell cycle G2/M arrest and apoptosis via a ROS-dependent ATM/ATR signaling pathways. Eur J Pharmacol 2018;819:16–29 doi 10.1016/j.ejphar.2017.09.050. [DOI] [PubMed] [Google Scholar]
- 38.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 2002;16(2):245–56 doi 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res 2012;72(1):13–7 doi 10.1158/0008-5472.CAN-11-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi EH, Kim KP. E2F1 facilitates DNA break repair by localizing to break sites and enhancing the expression of homologous recombination factors. Exp Mol Med 2019;51(9):1–12 doi 10.1038/s12276-019-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004;431(7011):1011–7 doi 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med 2011;17(7):875–82 doi 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 2009;35(3):327–39 doi 10.1016/j.molcel.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Q, Yin X, Languino LR, Altieri DC. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat Biopharm Res 2018;10(2):112–22 doi 10.1080/19466315.2018.1437071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roskoski R Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res 2019;139:471–88 doi 10.1016/j.phrs.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 46.O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 2016;13(7):417–30 doi 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- 47.Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol 2019;37(14):1169–78 doi 10.1200/JCO.18.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo L, Ran R, Yao J, Zhang F, Xing M, Jin M, et al. Se-Enriched Cordyceps militaris Inhibits Cell Proliferation, Induces Cell Apoptosis, And Causes G2/M Phase Arrest In Human Non-Small Cell Lung Cancer Cells. Onco Targets Ther 2019;12:8751–63 doi 10.2147/OTT.S217017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Gooijer MC, van den Top A, Bockaj I, Beijnen JH, Wurdinger T, van Tellingen O. The G2 checkpoint-a node-based molecular switch. FEBS Open Bio 2017;7(4):439–55 doi 10.1002/2211-5463.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmbos PL, Tomlins SA, Daignault S, Agarwal N, Twardowski P, Morgans AK, et al. Clinical outcomes and markers of treatment response in a randomized phase II study of androgen deprivation therapy with or without palbociclib in RB-intact metastatic hormone-sensitive prostate cancer (mHSPC). Journal of Clinical Oncology 2020;38(15_suppl):5573– doi 10.1200/JCO.2020.38.15_suppl.5573. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.