

Abstract
Rationale:
A previous analysis found significantly higher lung function in the US paediatric cystic fibrosis (CF) population compared to the UK with this difference apparently decreasing in adolescence and adulthood. However, the cross-sectional nature of the study makes it hard to interpret these results.
Objectives:
To compare longitudinal trajectories of lung function in children with CF between the US and UK and to explore reasons for any differences.
Methods:
We used mixed effects regression analysis to model lung function trajectories in the study populations. Using descriptive statistics, we compared early growth and nutrition (height, weight, BMI), infections (P.aeruginosa, S.aureus) and treatments (rhDnase, hypertonic saline, inhaled antibiotics).
Results:
We included 9,463 children from the US and 3,055 children from the UK with homozygous F508del genotype. Lung function was higher in the US than in the UK when first measured at age six and remained higher throughout childhood. We did not find important differences in early growth and nutrition, or P.aeruginosa infection. Prescription of rhDNase and hypertonic saline was more common in the US. Inhaled antibiotics were prescribed at similar levels in both countries, but Tobramycin was prescribed more in the US and Colistin in the UK. S.aureus infection was more common in the US than the UK.
Conclusions:
Children with CF and homozygous F508del genotype in the US had better lung function than UK children. These differences do not appear to be explained by early growth or nutrition, but differences in the use of early treatments need further investigation.
Introduction
Cystic fibrosis (CF) is a serious, multi-organ inherited disease characterised by pulmonary infections and progressively declining lung function. Most people with CF die prematurely from their disease through respiratory failure1,2. In the 1960s median survival in the United Kingdom (UK) was estimated to be below 10 years of age3. Over the past decades, outcomes have improved due to multidisciplinary care, nutritional support and new treatments, such that half of the babies born with CF in the UK and the United States (US) today can be expected to live at least in to their late forties 2,4–6.
Previous international comparisons of outcomes in people with CF have highlighted the impact of different healthcare practices and approaches to treatment, and have contributed to improvements in care for people with CF. For example, comparisons of nutritional, pulmonary and survival outcomes between the US and Canadian CF populations provided evidence for high-fat, high-calorie diets for people with CF7,8.
A previous cross-sectional study comparing the 2010 US and UK CF populations suggested better lung function in children in the US compared to those in the UK. After adjustment for a set of potential demographic and clinical confounders, the authors estimated a difference in percent of predicted forced expiratory volume in one second (%FEV1) of 7.62 percentage points (95% CI: 6.24 to 9.00) between children in the US and UK under the age of 12. This difference seemed to be smaller in adolescence and disappeared by age 30 9. However, due to the cross-sectional nature of the study, the age trends demonstrated could not be interpreted as average trends for individuals over time. The narrowing of the US-UK gap with increasing age was therefore difficult to interpret, conflating potentially different rates in lung function decline between populations with cohort effects and survivor bias later in adulthood. The aim of this study was therefore to compare lung function trajectories, i.e. lung function at age 6 years and rates of lung function decline, between the US and UK populations. Secondary aims were to explore potential reasons for any differences demonstrated. To ensure comparability of populations we restricted our analysis to the paediatric populations to reduce the impact of survivor bias, and to people who were homozygous F508del – the most common CF genotype - to reduce differences in case-mix.
Methods
Study Design
We carried out retrospective longitudinal analyses of lung function in comparable cohorts of children with cystic fibrosis aged ≥6 to <18 years and with homozygous F508del genotype captured in the US Cystic Fibrosis Foundation Patient Registry (CFFPR) and UK Cystic Fibrosis Registry.
Data Sources
We utilised the US CFFPR and the UK CF Registry. The current CFFPR includes data from 1986 onwards on over 50,000 people with CF and is estimated to capture about 84% of the current US CF population. Since 2003, CF care centres have been encouraged to enter information from all clinical encounters at CF Foundation Care Centre Network facilities 10. Current CFF guidelines are for people with CF to be seen quarterly for routine care 11.
In the UK, it is recommended that care teams submit annual encounter data to the Registry from a clinic visit approximately 12 months after the previous entry and when the patient is clinically stable. Records date back to the 1990s and are estimated to capture approximately 99% of the current CF population12. See supplementary material section 1 for more details.
Setting and Participants
We included children aged ≥6 to <18 with homozygous F508del genotype with data recorded in the CFFPR or UK CF Registry between January 2003 and December 2014 and with at least one lung function measurement in that time period. Restricting the analysis to children with CF who were homozygous for F508del reduced the differences in case-mix between the two countries. The dates were chosen to ensure comparability between populations as it preceded licensing of Orkambi (Lumacaftor/Ivacaftor) in the US. For individuals who had a transplant, observations were censored at transplant.
We excluded individuals who did not have complete data on sex, year of birth and age at diagnosis.
Outcomes and covariates
Our main outcome of interest was lung function measured by percent of predicted forced expiratory volume in one second (%FEV1) based on GLI reference equations 13 and the rate of lung function decline.
Secondary outcomes were time-varying indicators of growth and nutrition (height, weight, BMI; raw values and z-scores using the CDC reference populations in both countries and additionally UK-WHO reference population in the UK), prescribed chronic medications (rhDNase, hypertonic saline), common inhaled antibiotics (Tobramycin, Colistin, Aztreonam) and common infections (P. aeruginosa, S. aureus) between birth and age 18 for the study population. More information on secondary outcomes and when they began collection can be found in supplementary material section 1 table 1.
Table 1:
Details of the datasets and study populations in the US and UK, respectively.
| US Data | UK Data | |
|---|---|---|
| Sample size | 9,463 | 3,055 |
| Number of FEV1 measures per person (median [IQR], [full range]) | 24 [11 - 40], [1 −199] | 4 [2 - 7], [1 - 17] |
| Number of individuals with one FEV1 measure (%) | 251 (2.7%) | 398 (13.0%) |
| Number of FEV1 measures per person per year (median [IQR]) | 4 [3 −6] | 1 [1 - 1] |
| Time in years between first and last FEV1 measurement included in the study per person (median [IQR]) * | 5.5 [2.7 - 8.4] | 4.9 [2.2 - 7.5] |
| Informative drop-out during Study Period (%) | ||
| Lung Transplants | 142 (1.5%) | 27 (0.9%) |
| Deaths | 263 (2.8%) | 70 (2.3%) |
| Sex (%) | ||
| Female | 4,625 (48.9%) | 1,457 (47.7%) |
| Male | 4,838 (51.1%) | 1,598 (52.3%) |
| Year of birth (median) [IQR] | 1996 [1991-2002] | 1996 [1991 - 2002] |
| Race (%) | ||
| Non-Caucasian | 384 (4.1%) | 71 (2.3%) |
| Caucasian | 9,083 (95.9%) | 2,984 (97.7%) |
| Age at Diagnosis in years (median [IQR]) | 0.3 [0.0, 1.2] | 0.2 [0.0, 1.0] |
| Diagnosis by newborn screening (%) | 1,079 (11.4%) | 501 (16.3%) |
| %FEV1 at age 6 (mean (sd)) | 92.96 (17.7) | 88.17 (16.9) |
Excluding individuals with only one FEV1 measurement
To adjust for remaining potential differences in case-mix, we included the following baseline covariates in the models used: sex (binary: male/female; reference level: female), year of birth (continuous, centred at 1997), and age at diagnosis (continuous).
Statistical Analysis
Using all the collected data on our study population from age 6 up to (excluding) age 18 (encounter data in the US and annual review data in the UK), we developed models for the longitudinal trajectories of lung function to compare estimated population-level mean lung function and population-level mean lung function decline by age between the US and UK paediatric populations.
We fitted a series of models of different complexity to the US and UK study populations. We fitted models using linear 14,15, quadratic and cubic polynomials of age, and models including flexible functions of age 16,17 using natural cubic splines with one knot at age 12, two knots at ages 8 and 14, 5 knots with one knot every two years (8, 10, 12, 14 and 16 years) and 11 knots – one every year. All models included sex, year of birth and age at diagnosis as covariates; year of birth and age at diagnosis entered the models linearly. We did not consider any interactions between functions of age and the covariates (for exploratory plots see supplementary material section 2).
To appropriately capture the correlation between repeated measurements within an individual overtime, we initially included random intercepts in the models and then added: (1) random slope (only in the model with a linear function of age), (2) random slope and exponential correlation function (only in the model with a linear function of age), (3) exponential correlation function and (4) linear correlation function. Within each country all the model fits were compared using the log-likelihood, Akaike information criterion (AIC) and Bayesian information criterion (BIC); the best fitting models are those with the highest log-likelihood, lowest AIC and lowest BIC. For more details see supplementary material section 3.
To explore reasons for any differences in lung function trajectories we compared indicators of growth and nutrition, treatments, and infections cross-sectionally from birth to age 18 between the two study populations. This included records collected prior to 2003 on individuals in the study population. Observations remained censored at lung transplant. We summarised height, weight and BMI by their median, 25th and 75th percentile at each year of age for all individuals in the study population for whom data were available at that age. For treatments and infections we presented the proportion of individuals with at least one record of receiving the treatments/having the infections before age 6, before age 12 and before age 18 years; as well as the median, 25th and 75th percentile, minimum and maximum of the age at first recorded treatment/infection.
We stratified these comparisons by year of birth (before and after 1997) as post 1997 data will be more complete with regards to treatments and infections and may give a better representation of healthcare practices.
We used R packages nlme18 and splines19 for data analysis and ggplot220 and the dns() function from the JMbayes21 package for visualisations; the supplementary material used knitr22,23.
Robustness test
To assess the generalisability of our results to more recent paediatric CF populations, we repeated our analysis using the best fitting models applied to the recent cohort born after 1997.
Ethical Considerations
NHS research ethics approval (Huntingdon Research Ethics Committee 07/Q0104/2) was granted for the collection of data into the UK database. The Cystic Fibrosis Trust database committee approved the use of anonymised data in this study. The use of CFFPR data for this project was reviewed by Advarra and determined to be exempt from human subjects’ review (Protocol #: 00043631).
Results
Study populations
There were 9,463 and 3,055 individuals in the US and UK study population, respectively (see supplementary material section 4 for the derivation of the study populations). In the US individuals had a median of 24 lung function measurements, with a median of 4 [IQR: 3-6] measures per individual per year (Table 1). In the UK, each individual had a recorded review measurement approximately once a year, with an average of 4 lung function measurements during the study period. The average lung function for those observed at age 6 was 93 (standard deviation: 17.7) in the US and 88.2 (standard deviation: 16.9) %FEV1 in the UK. The populations were generally comparable with regards to their demographic characteristics (Table 1, supplementary material section 5).
Estimated lung function and lung function decline
In both countries, the estimated population-level mean lung function between ages 6 and 18 and the covariate effects were very similar across all the different models (supplementary material section 6). The best fitting model for lung function in both countries was the model with a linear term for age with random intercept, random slope, and exponential correlation function. In the US the model including age using a spline with 5 knots, random intercept and exponential correlation function gave only a marginally worse fit, therefore we report results for both models.
Population mean lung function was higher in the US study population throughout childhood (Figure 1). Based on our model that included a linear function of age, lung function declined at a faster rate in the UK than in the US population (−1.61 per year (95% CI: −1.72 to −1.50) in the UK compared to −1.41 (95% CI: −1.47, −1.36) in the US); children in the UK lost on average an additional 0.20 percentage points in %FEV1 per year (95% CI 0.08 to 0.32) compared to the US. Based on the model that included age using a spline with 5 knots there was an indication of a faster rate of decline in the UK at almost all ages but the evidence was not clear (Figure 1, supplementary material section 8).
Figure 1:
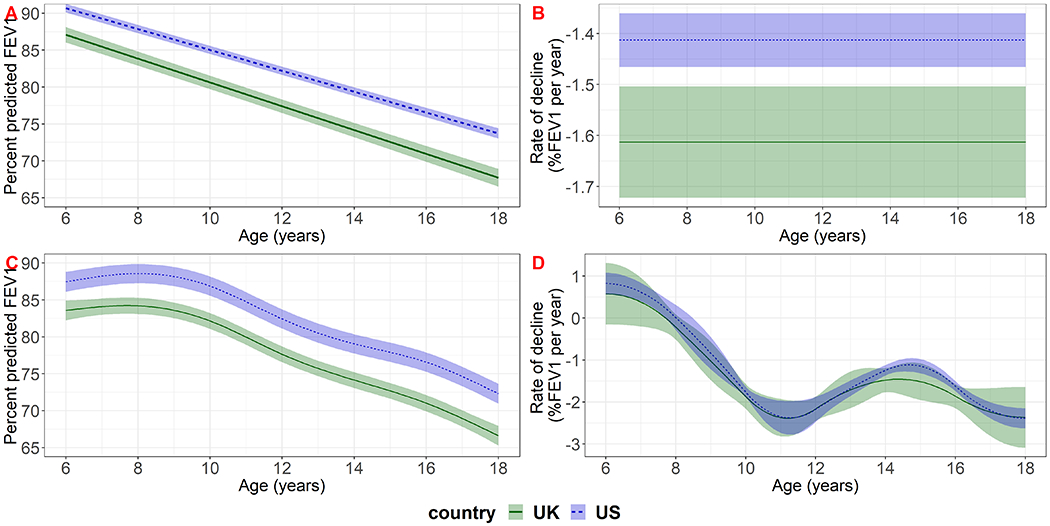
Estimated population-level mean lung function (panels A and C) and lung function decline (panels B and D) in the UK and US study populations for reference covariate values (female, born in 1997, diagnosed at birth). The top row (panels A and B) shows results based on the model that included a linear term for age with random intercept and slope and exponential correlation function; the bottom row (panels C and D) shows results based on the model that included age using a spline with 5 knots with random intercept and exponential correlation function.
Based on the model with a linear term for age and covariate reference levels (female, born in 1997, diagnosed at birth) the estimated difference between the US and UK populations at ages 6, 12 and 17 was 3.60 (95% CI 2.40 to 4.80), 4.80 (95% CI 3.38 to 6.22) and 5.80 (95% CI 3.98, 7.62) % FEV1, respectively. Based on the model including age using a spline model with 5 knots, the difference between the two countries was 3.89 (95% CI 2.00, 5.77), 4.78 (95% CI 3.10 to 6.47) and 5.69 (95% CI 3.97 to 7.41) % FEV1 at ages 6, 12 and 17, respectively. The US-UK gap was estimated to increase over calendar time by 0.13 percent predicted per year of birth (95% CI 0.003 to 0.25) based on the model with a linear term for age, and 0.12 percent predicted per year (95% CI −0.01 to 0.25) based on the model including age using a spline with 5 knots (Table 2). In the UK, males had on average 2.50 percentage points (95% CI: 1.32 to 3.67) higher %FEV1 than females, whereas in the US, the difference was 1.42 percentage points (95% CI: 0.70 to 2.14) (based on the model with a linear term for age; results were similar for the model that included age using a spline with 5 knots). The difference between the US and UK populations may therefore be less for males than females (see supplementary material section 7 for estimated differences between the US and UK for selected sets of covariate values).
Table 2:
Estimated covariate effects (95% confidence intervals) based on the model that included a linear term for age with random intercept, random slope and exponential correlation function and the model that included age using a spline with 5 knots, random intercept and exponential correlation function.
| US | UK | |||
|---|---|---|---|---|
| Linear function | Spline | Linear function | Spline | |
| Sex= male | 1.42 (0.70, 2.14) | 1.80 (1.04, 2.55) | 2.5 (1.33, 3.7) | 2.71 (1.49, 3.93) |
| Age at diagnosis | 0.59 (0.43, 0.74) | 0.57 (0.42, 0.72) | 0.62 (0.30, 0.95) | 0.6 (0.28, 0.92) |
| Year of birth | 0.37 (0.30, 0.43) | 0.42 (0.35, 0.48) | 0.24 (0.13, 0.35) | 0.3 (0.19, 0.41) |
Indicators of growth and nutrition, treatments and infections
Children in the US study population were on average lighter and shorter than children in the UK population during childhood and into adolescence. (Figure 2, supplementary material section 9).
Figure 2:
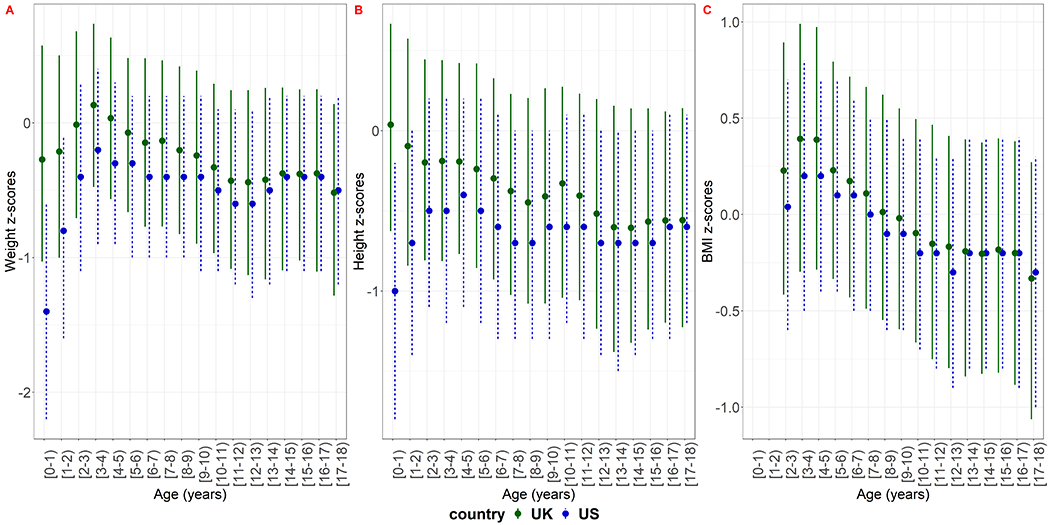
Cross-sectional summary measures of weight, height and BMI z-scores of the study population based on the CDC reference population in the US and the UK. The dots show the median and the lines the interquartile range.
Rates of infection with P. aeruginosa were similar in both countries but methicillin sensitive and methicillin resistant S. aureus infections were more common in the US (supplementary material section 11).
A larger proportion of the study population was treated with rhDNase and hypertonic saline in the US compared to the UK. In the post 1997 birth cohort, 73% were treated with rhDNase before age 6 in the US and 96% before age 18 compared to 20% and 86%, respectively, in the UK (supplementary material section 10). Hypertonic saline was prescribed for 76% of the study population before age 18 in the post 1997 birth cohort in the US versus 50% in the UK (supplementary material section 10). Inhaled antibiotics were prescribed at slightly higher levels before age 6 in the US but prescribed at similar levels at later ages (supplementary material section 10). However, there was a big difference in the inhaled antibiotic of choice between the two countries. Tobramycin was the most prescribed inhaled antibiotic in the US, whereas Colistin was prescribed most in the UK (supplementary material section 10).
Robustness test
In both countries lung function decline was less steep in the population born after 1997 compared to the whole study population. The estimated gap in absolute level of lung function was comparable to the estimates derived from the whole population (supplementary material section 12).
Discussion
We assessed longitudinal lung function trajectories in comparable populations of children in the US and UK using data from national registries and found that lung function was better in US children over the entire age range from 6 up to age 18 years. For example, based on the best fitting longitudinal model, females in the US were estimated to have approximately 3.60 (95%CI: 2.40 to 4.80) and 5.80 (95% CI 3.88-7.62) percentage points higher %FEV1 than females in the UK at age 6 and 17, respectively. The gap was slightly smaller for males. Children in the US with CF may have a slower rate of decline in lung function compared to UK children, but the strength of evidence for this was sensitive to modelling choices and was less clear in more recent cohorts born after 1997.
Strengths and Limitations
A major strength of this study is the use of high quality, longitudinal data with broad national coverage in each country and the application of statistical methods suitable for longitudinal data analysis which enabled us to compare population level mean lung function trajectories across childhood. We fitted a range of models to reduce the possibility that estimated differences in lung function were due to sub-optimal model fit in the populations. Our main results were consistent across models.
A further strength is the reduction of differences in case-mix between the two populations through the restriction to the F508del homozygous population and the adjustment for important covariates. We also reduced the influence of survivor bias by only considering the paediatric populations. However, this does mean that our results may not be generalisable to the whole CF population and further studies are needed to understand whether the differences we found also exist for different genotypes.
The previous study by Goss et al. 9 accounted for differences in data collection methods between registries by merging the US and UK datasets into a single data set and matching a single US clinic visit to a UK annual visit based on month of calendar year. In this study we did not try to harmonise the data sets but rather used identical inclusion/exclusion criteria and analysis methods. By including all available data points, we can more precisely estimate trends in lung function and rates of decline, but this may lead to bias due to seasonality or time between clinic visits (supplementary material section 1). However, we do not expect this to have a major impact on our results as seasonality has been shown to have a negligible effect on lung function15,24. The difference in the number of data points between the two populations is also unlikely to have significantly impacted our results based on preliminary findings from a parallel investigation of different analysis strategies applied to the US CFFPR. The potential impacts of these strategies on estimating lung function decline have been reported in abstract form from a Cystic Fibrosis Foundation workgroup25. The findings show that lung function trajectory estimates were similar if using all available encounter-level %FEV1 data, median quarterly data or median annual data. It has been noted that in the UK CF population, the average best measured %FEV1 per year is significantly higher (approximately 4 percentage points) than the annual review %FEV1 that we used in this study2,26. Best FEV1 has only been collected in the UK CF Registry since 2012 and therefore we could not conduct a full comparison of this measure between the two countries. However, a cross-sectional description of annual vs best %FEV1 in the UK and average-per-year vs best %FEV1 in the US, showed similar differences in the two countries and indicated that an analysis using best %FEV1 would likely return similar results to our findings (see supplementary material section 13).
Although we adjusted for a number of baseline covariates, a limitation remains the potential difference in case-mix, especially due to socio-economic factors. These have been shown to impact outcomes in CF14,27,28 but due to differences in measuring socio-economic status between countries and differences in data collection between registries, it was not possible to adjust for these. It has previously been suggested that the US CFFPR may not capture the full spectrum of socio-economic conditions as the UK CF Registry does. A sensitivity analysis of this assumption in relation to the study by Goss et al, however did not alter their results significantly and is therefore not likely to have a major impact on our findings29. A related open question is whether individuals not captured in the registries differ significantly from those who are. If this is the case, then any results from analyses of the registry population may not be generalisable to the population as a whole. Therefore, when interpreting the findings from our study, we need to be mindful that about 16% of the US CF population is not captured by the CFFPR and in the UK it is only since 2012 that 99% have been captured.
A further limitation is the generalisability of these results to the CF population in the era of new CFTR modulators. In future studies, it will be important to assess whether the observed differences have widened with the earlier introduction of Orkambi (Lumacavtor/Ivacavtor) in the US compared to the UK and whether we can expect the difference to decrease in the next generation when both countries will have access to CFTR modulators, including the triple combination therapy, for almost the whole population. However, the fast-paced development and the variation in introduction and eligibility criteria for CFTR modulator therapies across different countries poses additional difficulties for conducting cross-country comparison analyses of registry data.
Comparison with previous studies
Our overarching finding, that the paediatric homozygous F508del population in the US had higher lung function than this population in the UK, corroborates the results from the cross-sectional study by Goss et al. However, the longitudinal nature of our study enabled us to compare the population-level mean lung function trajectories throughout childhood. We showed that the gap between the two populations was sustained and may even be increasing during childhood and adolescence. Our findings are in contrast to Goss et al. who had previously found that cross-sectionally the gap in lung function between the US and UK appeared to be decreasing in adolescence and disappeared at age 30. Their findings are likely due to cohort effects and could be explained by our result that the gap between the two countries may be widening with calendar time.
Implications for clinical practice
The sustained gap in lung function between the US and UK which may be increasing with age and calendar time is a major concern for the UK CF community. As suboptimal nutritional status early in life is associated with lower pulmonary function30, we compared anthropomorphic measures between the two study populations. Children in our UK study population had on average better early nutritional status than children in the US study population. If anything, this would portend, if all other things were equal, that lung function in the UK would be better than in the US. In other words, the factors contributing to the difference in lung function between the two populations may be responsible for an even greater difference than just the difference reported in our study.
Additional factors such as differential use of medications between the countries may impact lung function31,32. We found big differences in the proportion of children receiving rhDNase and hypertonic saline with much more aggressive treatment at younger ages in the US compared to the UK. In the pivotal study, once daily rhDNase reduced pulmonary exacerbations by 28% and improved FEV1 by 5.8% compared to placebo32 but a recent study in the UK found that rhDNase may only improve lung function in individuals with reduced FEV1 at baseline33. In both studies, however, children under 5 years of age were excluded and there remains a paucity of data on the use of rhDNase in pre-school children. Although overall levels of prescribed inhaled antibiotics were similar between the countries, US children were almost entirely treated with Tobramycin. Whereas in the UK, Colistin was used almost exclusively, especially in children under the age of 6. These results raise the question whether the earlier prescription of muco-active agents in the US led to improved lung function at age 6, or whether differences between the US and UK in early antibiotic use, both nebulised and oral34, may have an impact on lung function. Further work will be needed to investigate this and other potential reasons for the observed differences. This will include investigating whether there are differences in lung function between the healthy populations in the US and the UK. We will also need to understand the generalisability of these findings to the whole CF population and the implications for long term outcomes including survival or time to lung transplant.
Conclusion
Using patient registry data to conduct international comparisons of health among people with CF is challenging due to variation in data collection methods but may highlight differences in health care and health outcomes between countries. In our comparative longitudinal analysis of children with CF, we found significant differences in lung function between the US and UK. Children in the US had higher lung function at age six which was sustained throughout childhood and adolescence. The rate of decline in lung function with age may also be slower in the US. Further work will need to investigate possible reasons for the gap in lung function between the US and the UK at age 6 including a more detailed analysis of treatment patterns.
Supplementary Material
Key Messages.
What is the key question?
Was there a difference in the longitudinal trajectories of lung function in children with CF between the US and UK during 2003-2014?
What is the bottom line?
Children with cystic fibrosis and homozygous F508del genotype in the UK had persistently worse lung function than those in the US.
Why read on?
This finding does not appear to be explained by differences in case-mix or early growth or nutrition, but there are differences in the use of early treatments between the two countries which will need further investigation.
Acknowledgments
The authors would like to thank the people with CF, care providers, and clinic coordinators at CF Centres throughout the United States and United Kingdom for their contributions to the CF Foundation Patient Registry and the UK CF Registry. Additionally, we would like to thank the Cystic Fibrosis Foundation and the Cystic Fibrosis Trust for the use of registry data to conduct this study. We would also like to thank Dr. Nicholas Simmonds and Prof. Steve Cunningham for critically reading the manuscript and providing helpful feedback.
Sources of Support:
DKS, SBC, DTR were supported by the Strategic Research Centre “CF-EpiNet: Harnessing data to improve lives” funded by the Cystic Fibrosis Trust. DTR is funded by the MRC on a Clinician Scientist Fellowship (MR/P008577/1). RDS was supported by grants from the Cystic Fibrosis Foundation (SZCZES18AB0) and NIH/NHLBI (R01 HL141286). CHG was supported by grants from the Cystic Fibrosis Foundation, the NIH (UM1 HL119073, P30 DK089507, U01 HL114589, UL1 TR000423) and the FDA (R01 FD003704). RHK is supported by a UKRI Future Leaders Fellowship ((MR/S017968/1).
The funding sponsors did not contribute to the design and conduct of the study; collection, analysis, or interpretation of the data; or preparation, review, approval, or decision to submit the manuscript for publication.
Footnotes
Some of the data were presented at the 2019 North American Cystic Fibrosis Conference.
References
- 1.Cystic Fibrosis Foundation. 2018 Patient Registry Annual Data Report; 2019. Accessed September 9, 2020. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/#:~:text=The%20Patient%20Registry%20Annual%20Data,and%20pulmonary%20therapies%20and%20transplantation.
- 2.UK Cystic Fibrosis Registry. Annual Data Report 2019; 2020. Accessed September 9, 2020. https://www.cysticfibrosis.org.uk/the-work-we-do/uk-cf-registry/reporting-and-resources
- 3.Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax. 1991;46(12):881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cystic Fibrosis Foundation. 2019 Cystic Fibrosis Foundation Patient Registry Highlights; 2020. Accessed September 9, 2020. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/#:~:text=The%20Patient%20Registry%20Annual%20Data,and%20pulmonary%20therapies%20and%20transplantation.
- 5.Keogh RH, Szczesniak R, Taylor-Robinson D, Bilton D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J Cyst Fibros. 2018;17(2):218–227. doi: 10.1016/j.jcf.2017.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacKenzie T, Gifford AH, Sabadosa KA, et al. Longevity of Patients With Cystic Fibrosis in 2000 to 2010 and Beyond: Survival Analysis of the Cystic Fibrosis Foundation Patient Registry. Ann Intern Med. 2014;161(4):233–241. doi: 10.7326/M13-0636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corey M, McLaughlin FJ, Williams M, Levison H. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol. 1988;41(6):583–591. doi: 10.1016/0895-4356(88)90063-7 [DOI] [PubMed] [Google Scholar]
- 8.Goss CH, Sykes J, Stanojevic S, et al. Comparison of Nutrition and Lung Function Outcomes in Patients with Cystic Fibrosis Living in Canada and the United States. Am J Respir Crit Care Med. 2018;197(6):768–775. doi: 10.1164/rccm.201707-15410C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goss CH, MacNeill SJ, Quinton HB, et al. Children and young adults with CF in the USA have better lung function compared with the UK. Thorax. 2015;70(3):229–236. doi: 10.1136/thoraxjnl-2014-205718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knapp EA, Fink AK, Goss CH, et al. The Cystic Fibrosis Foundation Patient Registry. Design and Methods of a National Observational Disease Registry. Ann Am Thorac Soc. 2016;13(7):1173–1179. doi: 10.1513/AnnalsATS.201511-781OC [DOI] [PubMed] [Google Scholar]
- 11.Clinical Practice Guidelines for Cystic Fibrosis Committee. Clinical Practice Guidelines for Cystic Fibrosis; 2016. https://www.cff.org/Care/Clinical-Care-Guidelines [Google Scholar]
- 12.Taylor-Robinson D, Archangelidi O, Carr SB, et al. Data Resource Profile: The UK Cystic Fibrosis Registry. Int J Epidemiol. 2018;47(1):9–10e. doi: 10.1093/ije/dyx196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40(6):1324–1343. doi: 10.1183/09031936.00080312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor-Robinson DC, Smyth RL, Diggle PJ, Whitehead M. The effect of social deprivation on clinical outcomes and the use of treatments in the UK cystic fibrosis population: a longitudinal study. Lancet Respir Med. 2013;1(2):121–128. doi: 10.1016/S2213-2600(13)70002-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qvist T, Schlüter DK, Rajabzadeh V, et al. Seasonal fluctuation of lung function in cystic fibrosis: A national register-based study in two northern European populations. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2019;18(3):390–395. doi: 10.1016/j.jcf.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szczesniak RD, McPhail GL, Duan LL, Macaluso M, Amin RS, Clancy JP. A semiparametric approach to estimate rapid lung function decline in cystic fibrosis. Ann Epidemiol. 2013;23(12):771–777. doi: 10.1016/j.annepidem.2013.08.009 [DOI] [PubMed] [Google Scholar]
- 17.Szczesniak RD, Su W, Brokamp C, et al. Dynamic predictive probabilities to monitor rapid cystic fibrosis disease progression. Stat Med. 2020;39(6):740–756. doi: 10.1002/sim.8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinheiro J, Bates D, DebRoy S, et al. Nlme: Linear and Nonlinear Mixed Effects Models; 2020. Accessed March 10, 2020. https://CRAN.R-project.org/package=nlme [Google Scholar]
- 19.Wang W, Yan J. Splines2: Regression Spline Functions and Classes.; 2018. Accessed March 10, 2020. https://CRAN.R-project.org/package=splines2 [Google Scholar]
- 20.Wickham H, Chang W, Henry L, et al. Ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics.; 2020. Accessed March 10, 2020. https://CRAN.R-project.org/package=ggplot2 [Google Scholar]
- 21.Rizopoulos D JMbayes: Joint Modeling of Longitudinal and Time-to-Event Data under a Bayesian Approach.; 2020. Accessed March 10, 2020. https://CRAN.R-project.org/package=JMbayes [Google Scholar]
- 22.Xie Y. knitr: A General-Purpose Package for Dynamic Report Generation in R version 1.28 from CRAN. Published 2020. Accessed May 13, 2020. https://rdrr.io/cran/knitr/ [Google Scholar]
- 23.Xie Y. Dynamic Documents with R and Knitr. 2nd ed. Chapman and Hall/CRC; 2015. https://yihui.org/knitr/ [Google Scholar]
- 24.Collaco JM, McGready J, Green DM, et al. Effect of Temperature on Cystic Fibrosis Lung Disease and Infections: A Replicated Cohort Study. PLOS ONE. 2011;6(11):e27784. doi: 10.1371/journal.pone.0027784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szczesniak R, Su W, Andrinopoulou ER, Afonso PM, Fernandez GS, Fink A. Lung function decline in cystic fibrosis: assessment of the impact of data and modeling choices and assumptions on clinical interpretations. In: Vol 55(S2). Paediatric Pulmonology; 2020:93–94. doi: 10.1002/ppul.25089 [DOI] [Google Scholar]
- 26.Hoo ZH, Curley R, Campbell MJ, Walters SJ, Wildman MJ. The importance of data issues when comparing cystic fibrosis registry outcomes between countries: Are annual review FEV1 in the UK only collected when subjects are well? J Eval Clin Pract. 2018;24(4):745–751. doi: 10.1111/jep.12967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schechter MS, Shelton BJ, Margolis PA, Fitzsimmons SC. The association of socioeconomic status with outcomes in cystic fibrosis patients in the United States. Am J Respir Crit Care Med. 2001;163(6):1331–1337. doi: 10.1164/ajrccm.163.6.9912100 [DOI] [PubMed] [Google Scholar]
- 28.Oates GR, Schechter MS. Socioeconomic status and health outcomes: cystic fibrosis as a model. Expert Rev Respir Med. 2016;10(9):967–977. doi: 10.1080/17476348.2016.1196140 [DOI] [PubMed] [Google Scholar]
- 29.Taylor-Robinson DC, Schechter MS, Smyth RL. Comparing cystic fibrosis outcomes across the pond. Thorax. 2015;70(3):203–204. doi: 10.1136/thoraxjnl-2014-206393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eh Y, H Q, D B. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. The Journal of pediatrics. doi: 10.1016/j.jpeds.2012.08.040 [DOI] [PubMed] [Google Scholar]
- 31.Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354(3):229–240. doi: 10.1056/NEJMoa043900 [DOI] [PubMed] [Google Scholar]
- 32.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331(10):637–642. doi: 10.1056/NEJM199409083311003 [DOI] [PubMed] [Google Scholar]
- 33.Newsome SJ, Daniel RM, Carr SB, Bilton D, Keogh RH. Investigating the effects of long-term dornase alfa use on lung function using registry data. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2019;18(1):110–117. doi: 10.1016/j.jcf.2018.08.004 [DOI] [PubMed] [Google Scholar]
- 34.Hurley MN, Fogarty A, McKeever TM, Goss CH, Rosenfeld M, Smyth AR. Early Respiratory Bacterial Detection and Antistaphylococcal Antibiotic Prophylaxis in Young Children with Cystic Fibrosis. Ann Am Thorac Soc. 2018;15(1):42–48. doi: 10.1513/AnnalsATS.201705-376OC [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.