

Abstract
The metabolic complexity and flexibility commonly observed in brain tumors, especially glioblastoma, is fundamental for their development and progression. The ability of tumor cells to modify their genetic landscape and adapt metabolically, subverts therapeutic efficacy and inevitably instigates therapeutic resistance. In order to overcome these challenges and develop effective therapeutic strategies targeting essential metabolic processes, it is to necessary to identify the mechanisms underlying heterogeneity and define metabolic preferences and liabilities of malignant cells. In this review we will discuss metabolic diversity in brain cancer and highlight the role of cancer stem cells in regulating metabolic heterogeneity. We will also highlight potential therapeutic modalities targeting metabolic vulnerabilities and examine how intercellular metabolic signaling can shape the tumor microenvironment.
Keywords: Glioma, tumor microenvironment (TME), metabolism, cell communication, tumor initiating cells, slow-cycling cells, immune cells
Introduction
Glioblastoma (GBM) is the most common and most lethal form of brain cancer [1]. GBM tumors are notorious and devastating in large part because of the two specific disease features. GBM tumor cells invade extensively throughout brain tissue. Additionally, subsets of tumor cells develop a hyper-aggressive, cancer stem cell (CSC) phenotype, enabling them to resist and survive the effects of radiological [2], chemical [3], immunological [4], and putatively magnetic [5] therapies. Today’s clinical standard of care integrates gross neurosurgical resection with adjuvant chemoradiation [6]. This strategy effectively targets the primary tumor mass and eliminates a fraction of highly proliferative tumor cells, but leaves behind residual, infiltrative CSCs that evade treatment and ultimately serve as the seeds of disease recurrence and patient mortality. Targeting CSCs alone is unlikely sufficient, but should be in combination with conventional therapies. Therefore, current treatments do not adequately account for the complexity and adaptability of GBM. Because of their propensity to drive tumor recurrence, targeting CSCs has become an attractive, yet challenging possibility.
GBM tumors establish an ecosystem defined by heterogeneity under the conditions of nutritional scarcity associated with tumor progression and treatment. At the histological level, a single GBM tumor is comprised of intertwined tumor populations along with non-transformed neural and immune cells in dynamic and reciprocal states of interaction with one another and with their growth environment. Further, within the cancerous populations, GBM tumors become enriched with multiple CSC lineages [7,8], each uniquely capable of tissue invasion, chemotherapeutic resistance, and tumorigenesis [9]. Consequently, introducing a selective pressure in the form of a single or dual agent treatment effectively ablates a fraction of the total disease represented by the treatment sensitive cancerous and non-cancerous populations. However, treatment resistant CSC lineages expand into the void left behind by this initial ablation, driving disease recurrence and hastening patient mortality. Relatively recently, the field has turned its attention to the metabolism of GBM tumors. Unlike the classic view of Warburg metabolism [10] in which tumor cells were exclusively dependent on glucose fermentation for energy generation, it is increasingly recognized that GBM CSCs, unlike co-mingled non-stem tumor cells, have the propensity to metabolize multiple nutritional substrates [11–15].
The concept of metabolic dependency has been gaining traction within the field [16]. This concept suggests that because certain tumor cell populations are exclusively dependent on specific metabolic substrates, revealing these dependencies naturally reveals points of disease vulnerability. The caveat here is that we currently do not know how adaptable one CSC lineage is compared to the numerous others within a given GBM tumor; however, even modest adaptability will enable CSCs to out-compete less plastic tumor cells with clear metabolic dependencies. Thus, in parallel with developing tools to counteract chemotherapeutic resistance, it will become critical to identify and elucidate the mechanisms responsible for CSC metabolic adaptability. Here, we will address this concern according to the following conceptual frames: i) We will discuss the complexity and diversity of metabolism in brain cancer and highlight the role CSCs play in driving heterogeneity and plasticity at the metabolic level. ii) We will address the implications of CSC metabolism in treatment resistance and the potential therapeutic strategies aiming at exploiting metabolic vulnerabilities of brain tumor cells. iii) Lastly, we will discuss how interactive cellular metabolism serves to shape the tumor microenvironment.
Metabolic heterogeneity/plasticity in GBM cancer stem cells
GBM represents a prototypical example of a highly heterogeneous tumor with multiple distinct molecular subclasses [17–19]. More recent approaches using single cell RNA-sequencing have found that combinations of several molecular subclasses can be found within the same tumor [7,20,8,21]. This indicates that molecular subclasses likely correspond to different functional states of GBM cells or cell populations, rather than a global transcriptional program applicable to the entire tumor. This cellular diversity of GBM underpins a functional heterogeneity on multiple levels, including cell plasticity and adaptability, therapy resistance, and metabolism. The profound resilience to therapy and high recurrence rates of GBM are a direct consequence of this intrinsic heterogeneity.
Progression and recurrence of GBM are due to residual cancer cells that escape treatment of the primary tumor, by physical displacement from the main tumor site, and acquisition of resistance to chemo/radiotherapy. Diffuse infiltration and therapy resistance are linked, and both are considered hallmarks of GBM CSCs [2,3,22–25]. Indeed, GBM CSCs are considered a prime suspect for tumor recurrence, as these cells retain the highest potential for initiating tumor growth [24,26]. This is supported by many studies demonstrating CSCs as a candidate population for initiating tumor recurrence across multiple brain cancers [22,27–29]. GBM CSCs produce non-stem cancer cell progenies via asymmetric division, with only CSCs capable of propagating tumor formation [25], although there is evidence that CSCs are more reflective of a cell state rather than a clonal entity [30,31]. In a genetically engineered mouse model of GBM, therapy-resistant CSCs were identified as drivers of long-term tumor growth via the generation of a rapidly growing transient population of cells [24]. GBM CSCs are further considered as a source of intratumoral clonal heterogeneity [7,20,25].
Cancer stem cells:
Since their initial discovery [27,32,33], much progress has been made to support that GBM CSCs are key drivers of tumor progression and recurrence [2,3,22–25,34–37]. Similar to their non-transformed counterparts, CSCs retain the ability to self-renew, and to generate more differentiated progenies [24,25,38–40]. In contrast to normal stem cells, CSCs are capable of adapting to different niches in their environment [41], which enables them to move throughout the brain without compromising their stem cell identity. Hence, GBM CSCs can leave the main tumor site and thus escape surgical excision. Due to their resilience to chemo- and radiotherapy [2,3,22,24], GBM CSCs are inaccessible to virtually all therapeutic efforts. Their capability for initiating tumor growth enables these cells to generate tumor recurrences after treatment.
Four critical niches have been defined in GBM that are linked to pathways of stemness and tumor progression: the hypoxic, perivascular, invasive and acidic niches [42,43]. Molecular regulators of stemness have been identified within hypoxic and perivascular niches, e.g. HIF1/2 [38,44], VEGF [45], integrins [46,47], and recently signaling pathways affecting stemness within the invasive niche are also emerging [21]. Patient MRI studies indicate changes in GBM metabolic states between primary and recurrent tumors that are linked to changes in the microenvironment [48]. An acidic extracellular pH is a pathological feature of the brain tumors microenvironment. The GSCs niche is particularly acidic due to the accumulation of acidosis products of the glycolytic metabolism. Acidic stress has been shown to support a GSC phenotype [49] and promote a mesenchymal differentiation of GSCs cultured under acidic conditions, leading to increased therapeutic resistance [50]. Further, an acidic pH increased VEGF expression thus supporting tumor progression in glioblastoma [51]. How crosstalk between CSCs and their niche affects CSC metabolism, and how this contributes to GBM progression and recurrence has yet to be resolved [52].
Three characteristics of GBM contribute to tumor recurrence: firstly, the diffuse infiltration of these tumors prevents complete resection and therefore residual GBM cells remain after surgery. Secondly, therapy resistance enables some residual GBM cells to survive chemo- and radiotherapy. Thirdly, expansion of therapy refractory GBM cell clones results in tumor recurrence. Below, we will briefly discuss how these three characteristics are ingrained in GBM CSCs, and how tumor recurrence is connected to metabolic plasticity in these cells.
Studies have shown that migration and proliferation are inversely correlated in gliomas [53,54]. This has been termed the ‘go-or-grow hypothesis’ [55]. Using label-retention paradigms, we have demonstrated that slower proliferating GBM cells are indeed more invasive than more rapidly proliferating cells from the same patient-derived culture [22,56]. Slower proliferating GBM cells are also more capable of initiating tumor growth and are more resistant to chemotherapy [22,56]. Hence, these cells constitute a population of GBM CSCs. Similar metabolic gene expression profiles between recurrent patient tumors and slower proliferating GBM CSCs support that these cells may drive tumor recurrence [22]. A recent study profiling single cells from GBM patients by RNA-Seq identified divergent molecular signatures of invasive cells and the tumor mass, and that only 1.6% of infiltrating GBM cells were actively proliferating, compared to 7.7% of tumor core cells [57].
CSCs are more resistant to therapy and can subsequently give rise to larger clusters of expanding clones that constitute recurrent tumors [3,22,24,38,46,58,59]. In order to initiate recurrence, residual cancer cells need to propagate and generate sufficient progenies to produce a new tumor mass. GBM recurrences occur most frequently in close proximity to the resection cavity, but radical resection of the entire tumor-containing hemisphere also results in contralateral recurrence [60,61]. This indicates that locally invasive cells may proliferate in response to the trauma of debulking surgery, and thus generate a faster growing recurrence than more distant invasive cells. How proliferation is re-initiated in infiltrating GBM cells is unknown, but intravital imaging studies suggest this may occur in response to ‘injury’ to the tumor. In these studies, ablation of GBM cells resulted in increased proliferation of GBM CSCs connected through tumor microtubes [23,62]. GBM recurrence is a complex process driven by CSCs that are maintained by cell-intrinsic molecular alterations and metabolic adaptability to a changing tumor microenvironment (TME) [22,24,39]. How CSCs adapt to a changing TME over time and space as GBM progresses, is treated, and recurs is still obscure. The transcriptomes from recurrent GBM highlight different metabolic signatures than those from primary tumors [22]. This indicates that the tumor cells (or cell populations) driving recurrence are metabolically different to the cells driving the growth of the primary tumor. Whether this is a cell-autonomous feature, or whether this is caused by an adaptation of the tumor cells to TME changes caused by first line treatment remains to be determined.
Metabolic support of GBM CSCs:
According to the Warburg hypothesis [63], tumorigenesis is partly driven by an impairment of mitochondrial function and oxidative phosphorylation (OxPhos). These alterations result in the Warburg effect, which is characterized by cancer cells generating most of their energy from glucose fermentation, i.e. aerobic glycolysis, with a limited ability to perform nutrient oxidation [64]. This metabolic reprogramming is thought to be an adaptation mechanism of rapidly growing tumor cells to cover their increasing energy demands. Recent studies have demonstrated residual activity of mitochondrial function in GBM cells [65–67], suggesting that some of these cells might utilize mitochondrial OxPhos (Fig. 1). GBM CSCs display increased invasion, chemoresistance, and metabolic profiles that are different from non-stem GBM cell populations and engage metabolic pathways that overlap with those found in recurrent GBM [3,22]. Additionally, (epi)genetic changes in GBM reinforce metabolic reprogramming in these tumors (reviewed in [68–70]). This is particularly notable for IDH1-mutant tumors, where the oncometabolite 2-hydroxyglutarate inhibits chromatin remodeling and DNA methylation. In IDH1 wild type tumors, EGFR signaling has been shown to induce translocation of the metabolic enzyme PKM2 to the nucleus, where it causes histone acetylation and activation of oncogene expression [71]. Crosstalk between metabolites and epigenetic alterations is therefore a potential driver of stemness programs that enables GBM CSCs to maintain a higher degree of plasticity. For instance, higher α-ketoglutarate levels affect stem cell plasticity through promoting histone demethylation [72].
Figure 1: Metabolic differences between cancer stem cells and non-stem cancer cells.
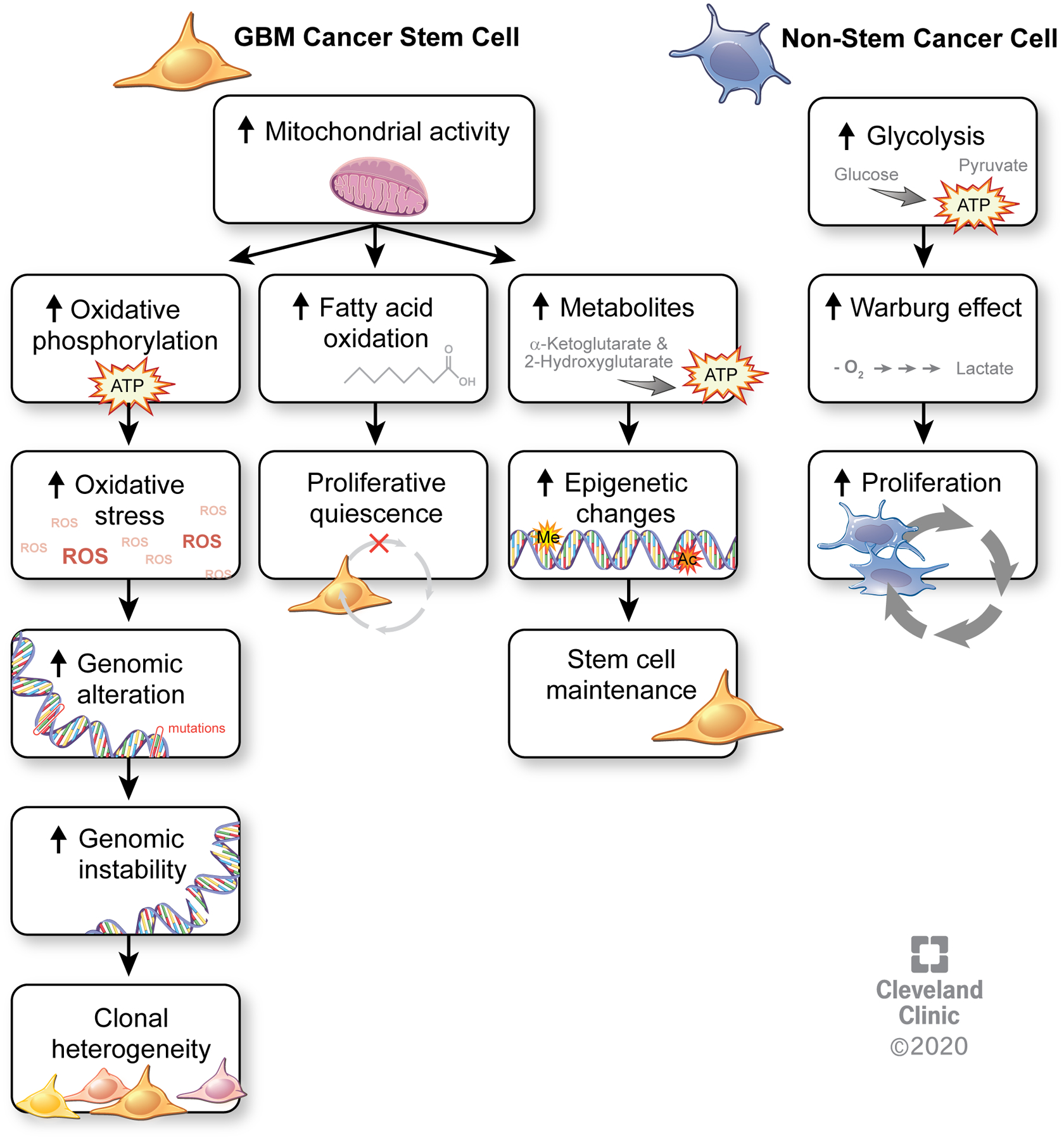
Increased mitochondrial activity in GBM CSCs results in increased oxidative phosphorylation, leading to elevated oxidative stress from reactive oxygen species that promote genomic alterations and instability that ultimately lead to clonal heterogeneity. Increased fatty acid oxidation is linked to the relative proliferative quiescence of GBM CSCs. Other metabolites, such as the levels of α-ketoglutarate and 2-hydroxyglutarate promote epigenetic alterations that contribute to stem cell maintenance. By contrast, non-stem cancer cells primarily depend on aerobic glycolysis that results in the Warburg effect and drives increased proliferation in these cells.
Most studies investigating metabolism in GBM (and in solid tissue cancers in general) have focused on the tumor mass, showing a large dependency on aerobic glycolysis in cancer cells, also known as the Warburg effect. More recently, reports have recognized metabolic heterogeneity in GBM and other cancers (including melanoma, lung and pancreatic cancer) [22,66,73–75]). One report found that functional mitochondrial oxidative phosphorylation is maintained in GBM, suggesting that these tumors may not exclusively rely on aerobic glycolysis [66]. In line with these findings, we have recently shown that invasive GBM cells rely on different metabolic pathways than cells constituting the tumor mass [22]. Less invasive, mass forming GBM cells predominantly utilize glucose to meet their energy demands, whereas infiltrating GBM CSCs rely on oxidative phosphorylation and lipid metabolism. These findings further underline the functional divergence of tumor mass and infiltrating GBM cells.
In this context it is noteworthy that non-transformed, quiescent adult neural stem cells (NSCs) require high levels of fatty acid oxidation, and that activation of NSCs is accompanied by a metabolic switch to lipogenesis [76]. Similarly, in pluripotent stem cells, lipogenesis is necessary to maintain pluripotency [77]. Importantly, changes in availability of lipid metabolites (malonyl-CoA) are sufficient to convert quiescent NSCs into actively proliferating stem cells. GBM CSCs rely on fatty acid oxidation and oxidative phosphorylation and are slower dividing than their non-stem counterparts, which indicates that metabolic pathways between quiescent NSCs and GBM CSCs may be conserved [22,65,76,78]. A recent study by Duman and colleagues supports this notion, demonstrating that GBM cells require fatty acid oxidation to proliferate, which is dependent on acyl-CoA transport. Blocking acyl-CoA transport resulted in increased survival of tumor-bearing mice in this study [79]. Our study demonstrated that slower proliferating GBM CSCs contain increased numbers of mitochondria compared to non-stem cancer cells [22]. The contributions of mitochondria to the function of NSCs and neurogenesis are only just emerging (reviewed in [80]).
Metabolic targeting in Glioblastoma (exploiting metabolic vulnerabilities)
Cancer cell plasticity, which is essentially the ability of tumor cells to adjust and adapt to a changing microenvironment, defines their survival and proliferation. For instance, the acquisition of a CSC phenotype was recently proposed to be phenotypic and inducible state which is acquired in response to environmental cues [30]. These cues consist of endogenous factors mainly the stromal cells and the metabolic environment in the brain [39] or exogenous factors such as therapeutic interventions (surgery, chemotherapy, radiation). Understanding tumor cell plasticity that allows GBM to adapt and proliferate in response to environmental pressure, including nutrients, acidity and hypoxia, is paramount for identifying metabolic vulnerabilities and therefore effective targeted therapies that could be integrated into traditional treatment modalities such as chemoradiation.
A highly heterogeneous genetic and phenotypic landscape in GBM, complicates treatment efforts and particularly those focused on targeting genetic drivers such as EGFR, PDGFR and others. One alternative strategy that is less explored in the clinic, is to target the altered metabolism of tumor cells. Fast proliferating tumor cells have an increased requirement for nutrients. Further, both rapidly dividing, and slow cycling brain tumor cells need to adapt and survive environmental pressure in the brain, including scarce nutrients, acidity and hypoxia. Consequently, metabolic reprogramming, a defining feature in the genomic landscape of high-grade gliomas [81,82], is pivotal for allowing brain tumor cells to adapt and proliferate in their environment. While clearly advantageous to the tumor, such reprogramming does however create metabolic vulnerabilities that could be therapeutically explored. Overall there are four predominant strategies that could be employed to target tumor metabolism either by depleting extrinsic nutrients from the tumor milieu, preventing the uptake of extrinsic nutrients by tumor cells, blocking the biosynthesis of endogenous metabolites that are essential for cell signaling and tumor growth or promoting the toxic accumulation of intermediary metabolites (Fig. 2).
Figure 2: Therapeutic strategies to target brain tumors’ metabolism.
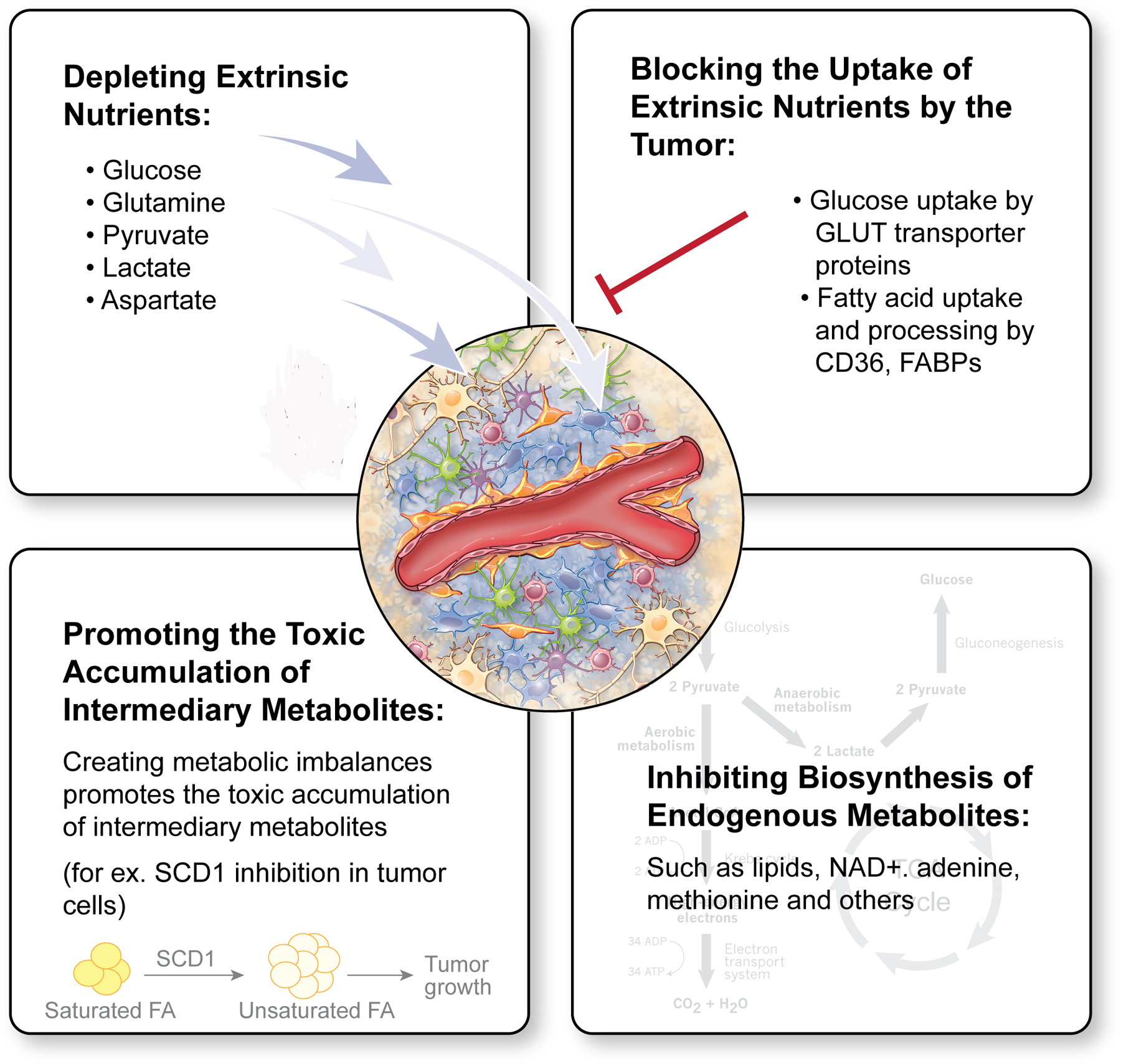
Metabolic targeting in brain tumors can be divided into four major strategies: depletion of extrinsic nutrients from the tumor milieu, blocking the uptake of extrinsic nutrients by tumor cells, inhibiting the biosynthesis of essential endogenous metabolites, or promoting the toxic accumulation of intermediary metabolites.
Availability of extrinsic nutrients:
The availability of bioenergetic substrates, primarily glucose, glutamine, pyruvate, lactate and aspartate in the tumor environment can modulate metabolic activities in tumor cells thereby affecting ATP production, biosynthesis of macromolecules as well as the regulation of redox state [83,84]. An increased uptake of glucose and glutamine is a metabolic hallmark of cancer cells [85,86]. Solid tumors in general rely heavily on glycolysis rather than oxidative phosphorylation to generate ATP, which could be exploited to image brain tumors with radiolabeled [18F]deoxyglucose positron emission tomography (PET). Since GBM cells including GSCs can avidly take up glucose[87], it was proposed that reducing circulating glucose levels, which can be achieved through a ketogenic diet (KD), can impact GBM tumor growth and sensitize to conventional therapeutics [88]. Unlike GBM cells, normal brain cells are endowed with a metabolic flexibility which allows them to process ketone bodies when glucose is depleted [89]. Therefore, a KD would, in theory leave GBM tumors particularly vulnerable to hypoglycemia. It is worth noting that the core idea of GBM tumors being unable to process ketone bodies has been challenged in one study reporting that in two rodent glioma models, ketone bodies were oxidized by GBM cells and the normal brain at a similar rate [90]. Despite some preclinical evidence of enhanced therapeutic efficacy in animal models of glioma under KD [91–93,88], clinical studies available to date indicate a modest therapeutic efficacy when a KD is combined with other therapies in recurrent GBM [94]. The limited efficacy of such approaches may be explained by the metabolic heterogeneity and plasticity observed in these tumors, which contain cells (i.e. slow-cycling) maintaining functional and enhanced oxidative phosphorylation activities enabling metabolism of a variety of alternative nutrients such as fatty acid [22]. In another example, in the absence of glucose GBM cells can also rely of glutamine metabolism by increasing the activity of glutamate dehydrogenase (GDH) in order to sustain proliferation [95]. In fact, the combination of a calorie-restricted KD with a glutamine antagonist, 6-diazo-5-oxo-L-norleucine (DON), achieved a superior therapeutic effect in two murine GBM models [96]. Nevertheless, it is also possible that calorie-restriction in itself, regardless of the diet, could contribute to the observed prolonged survival as it has been previously shown [97,98]. Similarly, a combination of a KD with GDH inhibitors could be further explored.
While tumor cells generally scavenge nutrients from their environment in order to sustain their proliferation, the uptake of few extrinsic factors such as ascorbate can result in cancer-specific cytotoxicity in various tumor types including GBM [99]. GBM and non-small cell lung cancer (NSCLC) cells are particularly sensitive to high levels of ascorbate due to an altered redox-active iron metabolism which mediates toxicity and sensitizes to chemoradiation [99]. A high pharmacological dose of ascorbate in combination with radiation and temozolomide is currently being tested in a phase II clinical trial in GBM patients (NCT02344355).
Uptake of extrinsic nutrients:
Although a substantial body of research in the cancer field in general, including in gliomas, has focused on actively targeting glycolysis, success has been fairly limited [83]. The glucose analog 2-deoxy glucose (2-DG) competes with glucose uptake thus reducing glycolysis. However, clinical exposure to high levels 2-DG leads to significant toxicity albeit some tumor response, thus limiting its clinical use [83]. It was shown that GBM CSCs can compete for glucose uptake through an increased expression of the high affinity glucose transporter type 3 (Glut3) which is necessary for GBM CSCs proliferation and tumorigenicity [87]. Different GLUT inhibitors WZB117, indinavir and ritonavir showed some efficacy in limiting proliferation of cultured GBM and GBM CSCs, yet GLUT1 inhibition is less effective in glioma models [100–102] and is likely to cause systemic toxicity due to the ubiquitous expression of GLUT1.
Emerging evidence suggest that, in addition to glucose, other metabolic substrates such as acetate [67] and fatty acids [65,22], can also support glioma growth thus challenging the idea that restricting glucose uptake is sufficient to block tumor growth. For instance, NMR spectroscopy studies in brain tumor patients suggest that less than 50% of glucose is utilized for acetyl-CoA production [103]. Therefore, the ability of brain tumors to scavenge nutrients from their environment warrants further investigation. Common lipids include fatty acids, the main component of most lipids, triglycerides, phospholipids and cholesterol. The brain is rich in lipids [104], therefore the crosstalk between normal brain structures, such as astrocytes or myelin sheaths and the tumor cells can affect brain tumor metabolism. For instance, polyunsaturated fatty acids released by astrocytes can activate peroxisome proliferator-activated receptor γ (PPARγ) in metastatic melanoma and breast cancer cells thus promoting brain metastasis [105]. Further, it is conceivable that elevated levels of circulating fatty acids could also support oncogenic signaling and GBM proliferation. While fatty acids are generally synthesized from excess glucose, they can also be actively taken up from the blood or endogenously synthesized through de novo fatty acids biosynthesis. Common dietary fatty acids, such as Palmitic Acid (PA; C16:0) which serves as a precursor to longer fatty acids, are capable of penetrating the blood-brain-barrier and can be rapidly taken up by the brain [106–111]. Further, systemically administered PA is taken up by GBM tumors and converted into lipid droplets in a patient-derived xenograft mouse model of GBM [112]. Although the mechanistic details of fatty acids uptake by brain tumors are poorly defined, several proteins involved in fatty acids cellular processing are upregulated in GBM. CD36 is a scavenger transmembrane protein which mediates fatty acids uptake, is particularly enriched in GSCs cultures and is necessary for GBM tumor initiation and progression [11]. Following their uptake, fatty acids are transported into different cell compartments through fatty acid binding proteins (FABP). FABP7 was found to be enriched in GBM CSCs and glioma slow-cycling cells [113] and its increased expression negatively correlates with glioma patient survival [22,114,115]. Further, FABP7 promotes proliferation and invasion in GBM [22,116,117]. In the absence of brain-penetrant inhibitors of proteins such as FABP7, CD36 and others, it is unclear whether preventing fatty acids uptake or binding is a viable therapeutic option for brain tumors.
Biosynthesis of endogenous metabolites:
Aberrant oncogenic pathways play a fundamental role in metabolic reprogramming of cancer cells. Exploiting unique metabolic dependencies rather than oncogenic drivers is an attractive therapeutic strategy that could potentially present new treatment options for brain tumors.
Increased lipid synthesis is one of the metabolic hallmarks of cancer [118], and indeed, fatty acids are particularly abundant in GBM [119–121]. Gliomas have increased levels of lipids and an upregulated lipid metabolism [121]. Guo et al. uncovered one of the key molecular mechanisms that promote lipid metabolism in GBM and showed that EGFR/PI3K/Akt signaling pathway, a major oncogenic driver in GBM, activates SREBP, a master transcriptional regulator of de novo lipid synthesis [122,123]. De novo synthesis of fatty acid is essential for membrane synthesis and lipid signaling molecules and therefore a limiting factor for cell proliferation. The therapeutic targeting of Fatty Acid Synthase (FASN) which catalyzes the synthesis of PA is currently being explored in a randomized phase 2 clinical trial in patients with recurrent high-grade astrocytoma using the FASN inhibitor TVB-2640 in combination with Bevacizumab (NCT03032484). The transcriptional activity of SREBP is also upregulated following lipid or oxygen retrieval in cultured GBM cells [124]. Further, SREBP-1 is activated under ER stress in GSCs and in turn promotes the expression of Stearoyl-CoA Desaturase 1 (SCD1) [112]. SCD1 is essential for regulating ER stress through the synthesis of unsaturated fatty acids. Therefore, the dependence of GBM CSCs on fatty acid desaturation presents an exploitable vulnerability to target GBM. Indeed, we showed that a small molecule inhibitor of SCD1 can achieve a strong therapeutic effect in GBM xenograft mouse models [112]. Given the role of SCD1 is regulating ER stress and supporting self-renewal of GBM CSCs [112], its upregulation is also linked to therapeutic resistance to temozolomide in GBM [125].
Fatty acids are catabolized by fatty acid oxidation (FAO; also referred to as β-oxidation). Inhibiting FAO with etomoxir in cultured glioma cells depletes NADPH levels and promote ROS-mediated cell death [126]. The therapeutic efficacy of etomoxir was also confirmed in a syngeneic mouse model of malignant glioma whereby animals treated with the FAO inhibitor exhibited prolonged survival [65]. While FAO inhibitors have not yet been tested in a clinical setting, efforts to measure FAO in glioma patients using 18F-FPIA [(18)F-fluoro-pivalic acid] PET/MRI are currently ongoing (NCT04097535). Fatty acids are first converted into acyl-CoA and subsequently oxidized into Acetyl-CoA. Acyl-CoA-binding protein (ACBP) which binds to acyl-CoAs (catalyzed into acetyl-CoA) was recently proposed as a druggable target in GBM due to its supporting role in FAO [79].
In addition to limiting glucose levels or blocking glucose uptake, one alternative therapeutic strategy to target glycolysis, is to activate pyruvate dehydrogenase (PDH) by targeting its molecular inhibitor, pyruvate dehydrogenase kinase (PDK). This could be achieved using the PDK inhibitor dichloroacetate (DCA) [127]. Another glycolysis inhibitor 3-bromo-2-oxopropionate-1-propyl ester (3-BrOP) was reported to effectively target GSCs particularly under hypoxic conditions [128].
Nicotinamide adenine dinucleotide (NAD+), a central enzyme in many cellular and biochemical processes is synthesized through the conversion of nicotinamide by Nicotinamide phosphoribosyltransferase (NAMPT) or through an alternate salvage pathway whereby Nicotinic acid is converted to NAD+ by phosphoribosyltransferase (Naprt1). Mutant IDH1 gliomas inhibit NAPRT activity and therefore rely primarily on NAMPT mediated synthesis of NAD+, which creates a particular vulnerability to NAD+ depletion achieved through NAMPT inhibitors [129].
Chromosomal deletions are the most frequent somatic genetic alteration in neoplastic cells such as malignant gliomas. Due to the chromosomal proximity of metabolic and tumor suppressor genes, these deletions generally lead to the inactivation of tumor suppressors, in few instances, they could also promote metabolic vulnerabilities. The glycolytic enzyme enolase is encoded by two redundant genes ENO1 and ENO2. The former resides on chromosome 1p36 locus (along with tumor suppressor genes) which is homozygously deleted in 1–5% of GBM. Therefore, this subset of GBM tumors are particularly vulnerable to therapeutic targeting of ENO2 [130]. Homozygous deletion of the chromosome 9p21 locus (harbors CDKN2A gene) is observed in over 50% of GBM. This chromosome locus also contains the gene encoding methylthioadenosine phosphorylase (MTAP) which metabolizes methylthioadenosine (MTA) to adenine and methionine. Loss of MTAP results in the accumulation of MTA which in turn inhibits the activity of protein arginine methyltransferase PRMT5 and renders MTAP-deficient cancer cells particularly vulnerable to inhibitors of PRMT5 [131,132].
Promoting the toxic accumulation of intermediary metabolites as a therapeutic intervention:
Targeting metabolic vulnerabilities and inhibiting the synthesis of metabolites that are limiting for glioma proliferation could be effective in limiting tumor growth. However, the development of compensatory metabolic networks to alternatively support tumor growth could limit therapeutic efficacy. One alternative treatment paradigm exploits the upregulated metabolic pathways in glioma in order to create metabolic imbalances and promote the toxic accumulation of intermediary metabolites. Identifying intermediary metabolites that promote cytotoxicity upon intracellular accumulation could offer new therapeutic avenues. For instance, by blocking the desaturase activity of SCD1, but not upstream enzymes involved in de novo fatty acid synthesis, leads to the toxic accumulation of saturated fatty acids in GBM CSCs [112].
Glioma cells residing in hypoxic niches, as well as rapidly dividing cells rely on glutathione synthesis in order to maintain redox homeostasis and counteract a heightened redox stress. Depleting intracellular glutathione levels leads to an upregulated ROS levels thus rendering tumor cells particularly vulnerable to oxidative or genotoxic stress. For instance, the ability of GBM to hijack glutamine could be hindered by glutaminase inhibitors such as CB-839 that effectively depletes glutamate and glutathione in IDH-mutant gliomas [133]. The combination of CB-839 with radiation and temozolomide is currently being tested in a phase 1b clinical trial in patients with IDH-mutant low-grade glioma (NCT03528642). An increased expression of the cystine/glutamate exchange transporter SLC7A11 (also known as xCT), essential for cystine uptake and glutathione synthesis, confers protection against oxidative stress in gliomas and other cancers [134]. Further, SLC7A11 is reportedly upregulated in EGFR amplified GBM [135] and supports EGFR-mediated tumorigenesis in EGFR amplified glioma [136]. Overall the ability of gliomas to increase glutathione synthesis serves as a defense mechanism against oxidative stress and aptly, the overexpression of SLC7A11 is associated with increased stem cell properties and chemoresistance in GBM [137]. While effective brain permeable inhibitors of glutathione metabolism are yet to be identified, effectively blocking glutathione metabolism creates a metabolic imbalance caused by ROS accumulation and therefore presents a vulnerability that could be targeted to increase oxidative stress and enhance the outcome of genotoxic glioma therapy.
Metabolic interplay and communication in GBM microenvironment
As discussed above, GBM are metabolically dynamic entities that rewire and adapt their metabolic properties, which are controlled autonomously or regulated by the interactions of tumor cells with other cells in the microenvironment. It is now well recognized that GBM cell energetics strongly dictate the metabolic landscape of the tumor microenvironment supporting tumor development and growth. Consequently, fully understanding the complex metabolic interactions in GBM tumor microenvironment not only will provide critical insight into tumor progression, but also identify new therapeutically exploitable vulnerabilities.
The tumor microenvironment is a complex network of diverse cellular compartments where tumor cells interact with a variety of non-neoplastic cells including immune cells, which represent key components of the tumor milieu. The metabolic specificities of GBM can determine fates and functions of neoplastic cells but also of immune cells creating specific niches, which play critical roles in anti-tumor responses, immunosuppression, tumor growth and response to treatment [138–141]. This section will focus on the immune microenvironment and its metabolic features and interactions with GBM cells. GBM-infiltrating immune cells typically include macrophages, but also T lymphocytes (mostly CD4+, CD8+ and regulatory), natural killer (NK) cells, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs) and neutrophils [138,142–144]. Notwithstanding the presence of immune cells in GBM, the tumor microenvironment is globally immunosuppressive. Immune evasion and metabolic reprogramming are now well recognized hallmarks of cancer and are considered to be functionally linked. Multiple studies reported that immune cells also possess defined metabolic characteristics and requirements that regulate their immunological function and contribute to the overall tumor metabolic heterogeneity in GBM [145–149]. Below we will discuss the metabolic reprogramming that specifically regulate functions of tumor-associated immune cells and how these mechanisms may be modulated by the tumor metabolic activities thus contributing to disease progression.
Regulation of the anti-tumor response by metabolic competition:
Histological infiltration of effector CD8+ T lymphocytes in GBM tumors has been well documented [150]. However, in order to be activated and exert their antitumor function, naïve T cells must undergo metabolic reprogramming shifting from oxidative mechanisms, which support immunosurveillance and quiescence, to aerobic glycolysis-based metabolism requiring massive increase in nutrient uptake such as glucose to support their rapid growth and cytotoxic function [151]. NK cells are highly effective cytotoxic lymphocytes in the innate immune response and are also involved in tumor killing processes via production of IFNg and cytotoxic molecules such as granzyme B. Similar to T cells, NK cells rely mainly on oxidative phosphorylation under resting conditions but undergo metabolic shift increasing glycolytic flux upon prolonged activation [152]. Neutrophils that can exhibit an anti-tumor effect [153] are also strongly committed to glycolysis controlling their functionality [154,155]. Despite the presence of these cytotoxic immune cells in the tumor microenvironment of GBM, the overall immune response is inefficient. This is due in part to the suppressive action of glioma-produced cytokines, but also to the specific metabolic activities of tumor cells. Tumor glycolytic activity has been associated with poor prognostic and restricted tumor infiltration and activation of T cells [156]. Indeed, high glycolytic activities of GBM cells coupled with inconsistent vasculature within the tumor mass can engender nutrient depletion for immune cells impairing their tumoricidal function. Additionally, tumor metabolism generates metabolic byproducts such as lactate, which can be toxic for tumor targeting immune cells.
Dendritic cells are professional antigen-presenting cells involved in T cell activation, however they can also promote immune tolerance specifically when in the tumor microenvironment where they have been reported to exhibit impaired ability to trigger response and promote immunosuppression [157–159]. Though the precise mechanisms underlying the impaired function of tumor associated DCs are not well understood, it has been suggested that metabolic regulation might be implicated [160–162]. The glycolytic pathway is a key metabolic regulator of DCs, with increased glycolysis being necessary for early activation and survival [163]. A recent study showed that rapidly growing cancer cells can impose accumulation of adenosine promoting oxidative phosphorylation and decrease of glycolysis in DCs, thereby contributing to their tolerigenic state [160]. Abnormal fatty acid accumulation via activation of lipid biosynthetic and uptake pathways has also been observed in tumor associated DCs, further speculating the role of metabolism in promoting their immunosuppressive properties [164–166].
Overall, GBM cells show capacities to shape their environmental surroundings imposing metabolic stresses and challenges for protective immune cells, resulting in immune escape and promoting disease progression.
Pro-tumorigenic metabolic processes:
MDSCs are one of the most abundantly recruited and expanded myeloid lineage cells in the GBM microenvironment and are defined by their potent immunosuppressive properties on antigen-activated CD8+ T cells and NK cells [167,168]. Their suppressive activities mainly result from their metabolic characteristics and involve amino acid metabolism and oxidative stress [168]. MDSCs can deplete L-arginine, L-cysteine or tryptophan, amino acids that are essential for T cell activation and expansion [169–173]. Tryptophan can also induce the expansion of immunosuppressive regulatory T cells [172,173]. MDSCs’ metabolism induces the production of reactive oxygen species which are toxic to T cells, therfore inhibiting their activation and proliferation [168]. Enhanced tricarboxylic acid (TCA) activity and fatty acid oxidation have been noted and demonstrated to regulate the suppressive properties of tumor infiltrative MDSCs [174]. This suggests a potentially reduced sensitivity of MDSCs to tumor-imposed glucose restriction with maintenance of their immunosuppressive function while under metabolically challenging conditions. Regulatory T cells (Treg) also appear tolerant to tumor-imposed glucose competition, as they mainly rely on fatty acid oxidation rather than glycolysis facilitating survival and immunosuppressive effect [175,176]. Importantly, hypoxia and metabolic waste such as lactate or kynurenine secreted by tumor and other stromal cells promote Treg phenotypic expansion and immunosuppressive action via PD-L1 induction [177,178].
By far the most dominant tumor immune infiltrates within GBM are macrophages, which are functionally and phenotypically plastic [179]. Macrophages exhibit multi-dimensional activation states integrating environmental signals in a stimulus-specific fashion producing various functional outcomes. Classically activated pro-inflammatory macrophages (M1) promote anti-tumoral responses, whereas immunoregulatory (M2) macrophages are pro-tumorigenic [179]. Polarization status of macrophages is associated with distinct metabolic pathways. Pro-Inflammatory macrophages engage in glycolysis whereas the M2 immunosuppressive phenotype is associated with increased oxidative phosphorylation [180–182]. Lactate has been shown to polarize macrophages to an immunosuppressive and tumor-promoting state [183]. The active TCA cycle in these macrophages is supported by glutamine metabolism but also lipid metabolism [184]. Upregulation of fatty acid uptake, fatty acid synthase and oxidation via lysosomal lipolysis is essential for M2 activation and immunosuppression [185,186]. It also worth noting that GBM tumors also contain macrophages with mixed state that reside between classical M1/M2 polarization [187].
Together, these observations demonstrate the changes that take place in the microenvironment of GBM and the mechanisms of immunometabolic dysregulation linked to cancer cell metabolism, immunosuppression and tumor growth.
Metabolic support from the tumor microenvironment:
GBM cells metabolically evolve and adapt as tumor progresses. This may reflect an evolutionary advantage giving rise to metabolic heterogeneity. This heterogeneity generates a diversity of metabolic niches marked by specific cellular composition and heterotopic metabolic communications across cell types. The studies discussed above emphasize how glioma metabolism can shape the tumor microenvironment; however, this hetero-cellular metabolic remodeling can be multi-directional where the tumor microenvironment also affects tumor cell behavior and metabolic fitness and adaptability. Several examples illustrate how proliferating cancer cells take advantage of their surroundings to satisfy their metabolic requirements. Metabolic coupling between tumor cells and stromal/immune cells can be essential for cancer growth via providing critical signals and nutrients. Such metabolic interactions have been described in multiple cancers. For instance, the non-neoplastic compartment secretes alanine, which support metabolic activities of pancreatic cancer cells [188]. In another example, fatty acids released by adipocytes provide necessary substrates supporting the metabolism of metastatic ovarian cancer cells [189]. This hetero-cellular metabolic symbiosis has been described in pancreatic and prostate cancers with cancer-associated fibroblasts (CAFs) secreting exosomes containing nutrient cargoes including a variety of fatty acid, amino acid and TCA metabolites [190,191]. Symbiotic transfer of entire organelles has also been reported with the transfer of mitochondria from stromal cells to lung cancer cells [192]. Such metabolic interconnections in GBM have yet to be evidently defined and remain to be further investigated and understood. For example, learning more about the metabolic interactions between glioma cells and macrophages would be critical. Macrophages are the most represented tumor infiltrating cells and demonstrate metabolic adjustments, such as enhanced fatty acid biosynthesis and trafficking [184], which may participate in regulating the metabolism of glioma cells, especially treatment-resistant slow-cycling CSCs, which were shown to rely on lipid metabolism [56,22].
Despite some overlap in metabolic requirements between GBM cells and immune infiltrates, which represent a potential therapeutic challenge, targeting key immune-tumor cell metabolic interdependencies may reveal lethal vulnerabilities holding great therapeutic promises. Will the targeting of this hetero-cellular metabolic relationships provide viable targets with tractable therapeutic window remains to be demonstrated? Treatment modalities should be finely tuned and consider the symbiotic or competitive nature of these interactions between tumor cells and supportive or suppressive immune populations.
Cancer initiation and progression as well as treatment resistance are not exclusively dependent on intrinsic properties of the cancer cells but rather rely on the contribution of a multitude of cellular and molecular components creating a complex tumor microenvironment. GBM cells manipulate and shape their microenvironment and can for example, affect immune cells metabolism leading to anergy or death of anti-tumor immune cells and stimulation of immunosuppressive phenotype further supporting tumor growth. However stromal/immune cells are also pivotal in the maintenance of the network that feeds and sustains glioma cells, undoubtedly essential for tumor initiation and progression. Undermining this equilibrium may define novel therapeutic approaches. However, even though much progress has been made in unraveling the metabolic characteristic of GBM and its microenvironment(s), there is much to be determined about metabolic communications and dependencies and how these vulnerabilities can be exploited and translated into viable therapeutic strategies.
Concluding remarks
The GBM TME is extremely complex and heterogeneous with a diversity of neoplastic and non-neoplastic cells exhibiting specific metabolic needs and regulation and defining anatomical compartments or niches. The nature of these niches constantly evolves and can be influenced by (epi)genetic alterations, anti-cancer treatments or additional extrinsic factors such as oxygen or nutrients availability (Fig. 3). All these interconnected factors shaping the TME regulate tumor initiation, progression, treatment response and disease recurrence.
Figure 3: Features of the tumor microenvironment in glioblastoma contributing to tumor initiation, progression and recurrence.
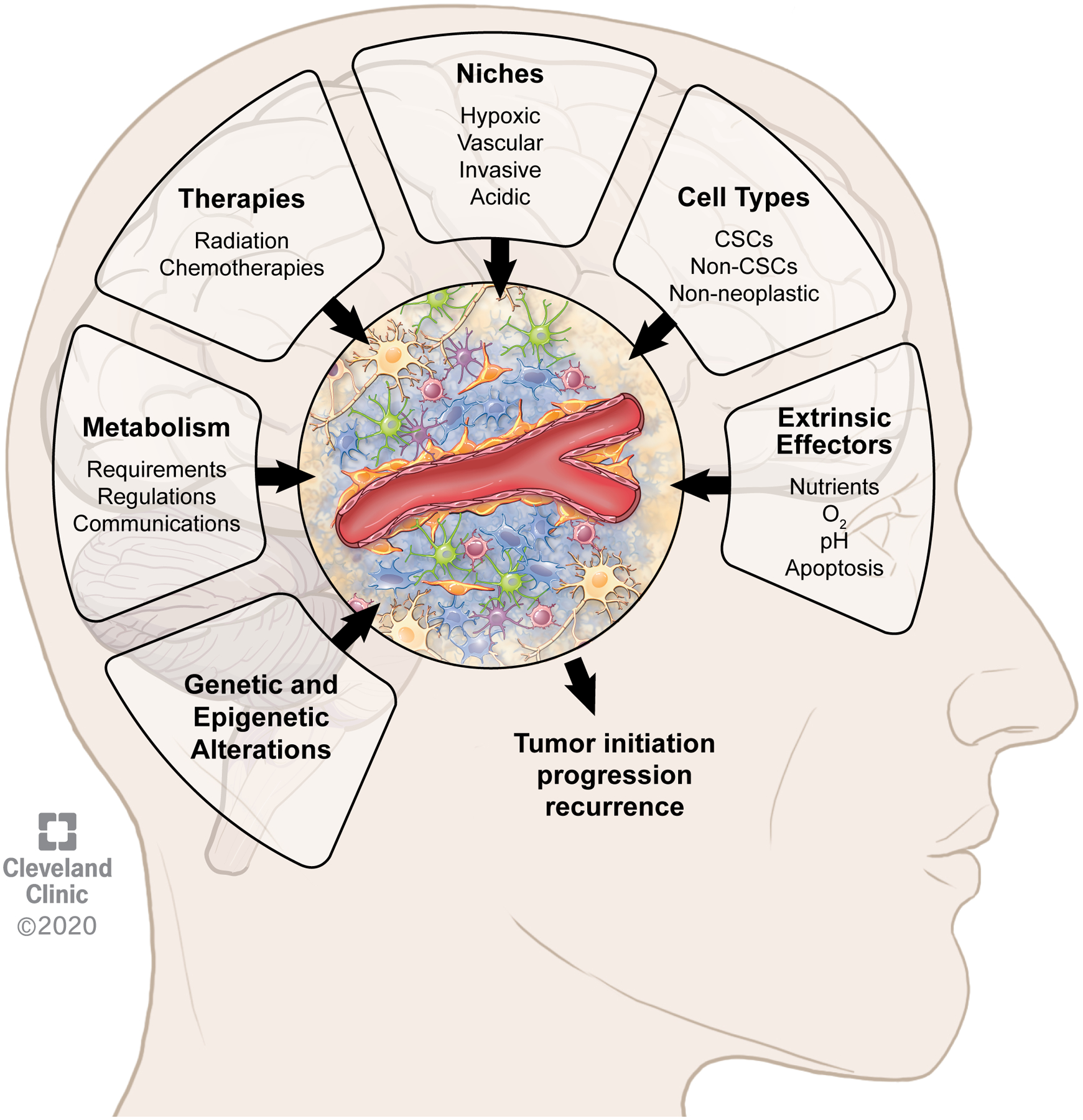
The composition and regulation of the tumor microenvironment is dependent on multiple interconnected factors including in a non-hierarchical order metabolism, therapies, niches, cell types, (epi)genetic alterations and additional extrinsic effectors such as nutrients, oxygen (O2), pH and apoptosis.
This review specifically underlines the complexity and diversity of cancer metabolism in GBM and examines how understanding metabolic heterogeneity is essential to the development of applicable and efficient metabolism-based therapeutic modalities.
Genetic aberrations, which are fundamental drivers of tumorigenesis in GBM, are also associated with metabolic adaptations allowing cancer cells to survive and proliferate in varied tumor microenvironments. This metabolic flexibility drives heterogeneity in cancer metabolism. One point that was not specifically discussed in this review and that should be taken into account when studying metabolic heterogeneity are the metabolic changes in respect to the different molecular subtypes of GBM that have been associated with distinct microenvironment also marked by specific immune constituents and metabolism [82,81,19,193,17,77,194,195].
It is also important to note the challenges in drawing conclusions on cancer metabolism when cells are studied under conditions that do not mimic the tumor environment, including for example high level of nutrients like glucose, high O2 concentration, controlled pH and utilization of serum. Models need to be improved to better recapitulate the metabolic restraints of the tumor microenvironment. Ameliorating our analytical toolbox will enable us to probe deeper into our investigations of tumor metabolism and increase our understanding of the multi-faceted metabolic dependencies in GBM.
Targeting the altered metabolism in malignant brain tumors provides exciting and promising avenues to tackle this morbid and incurable disease. A new arsenal of therapeutics that engage metabolic vulnerabilities could be effective especially if integrated into traditional treatment modalities such as DNA damaging interventions or immunotherapy. Yet, there are several challenges that remain to be addressed and thoroughly investigated. An increased understanding of the brain tumor microenvironment, as well as genetic alterations that promotes an altered metabolism in brain tumors will certainly guide effective therapeutic strategies. Further, a better understanding of tumor cell plasticity as well as various metabolic switches and rewiring of metabolic pathways which allow to bypass the inhibited pathways, would certainly help thwart therapeutic resistance. Identifying metabolic inhibitors that effectively penetrate brain tumors without eliciting systemic toxicity is paramount for their clinical application. High degree of heterogeneity in genetic alterations identified in cancer represents a major contributor of treatment resistance, however limiting treatment design and clinical decision-making strictly on genome-based tumor characterization holds inherent limitations to prevent outgrowth of resistant cancer cell populations and improve patient outcome. Current metabolism-based experimental therapies, which are predominantly based on the existence of oncogenic alterations, have not translated into compelling clinical results. Metabolic synthetic lethality has emerged as an alternative concept of metabolic targeting of cancer that appears to be more challenging than originally anticipated [196,197]. Since tumor metabolic alterations and (epi)genetic alterations are interactional, identifying metabolism-related synthetic lethality may help overcome resistance to therapies based on tumor cell genetic alterations. For example, lysyl oxidase (LOX) has been identified as potential metabolic synthetic lethal interactor in PTEN-deficient GBM as LOX inhibition impairs tumor growth by limiting the expression of SPP1 by tumor associated macrophages [198]. Additionally, characterizing and understanding the adaptive tumor landscape’s characteristics, marked by specific criteria such as oxygen availability, nutrient deprivation, pH status may help overcoming these limitations. Indeed, the TME can impose selection forces generating a metabolic heterogeneity and promoting emergence and/or acquisition of phenotypic attributes contributing to treatment resistance, disease progression and recurrence regardless of tumor cell genetic alterations [197]. Metabolic heterogeneity offers tumor cells adaptive mechanisms to face their hostile and very competitive environment and escape therapy. The role of the intimate interplay between tumor metabolism and therapy resistance begins to be appreciated however it remains to be fully understood. Development of relevant patient-based (pre)clinical models integrating genetic drivers, cell lineage of origin, types of therapy, niche specificities and metabolism is absolutely required to accurately explore and understand cancer cell metabolic vulnerabilities and enhance drug response.
Acknowledgements
CEB is funded by American Brain Tumor Association (ABTA) Discovery Grant supported by the Uncle Kory Foundation and Dana-Farber/Harvard Cancer Center (DF/HCC) SPORE in brain cancer DRP Award.
FAS is funded by MRC grant MR/S007709/1.
LPD is funded by Florida Center for Brain Tumor Research & Accelerate Brain Cancer Cure (P0058849), ABTA Discovery Grant (DG1800014), Pray for Dominic St Baldrick’s Research Grant (638733) and UFHCC Cancer Therapeutics & Host Response Research grant (00096885).
References
- 1.Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, Nishikawa R, Rosenthal M, Wen PY, Stupp R, Reifenberger G (2015) Glioma. Nat Rev Dis Primers 1:15017. doi: 10.1038/nrdp.2015.17 [DOI] [PubMed] [Google Scholar]
- 2.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444 (7120):756–760 [DOI] [PubMed] [Google Scholar]
- 3.Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D, Nabilsi NH, Kladde MP, Suslov O, Brabletz S, Brabletz T, Reynolds BA, Steindler DA (2013) The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5 (8):1196–1212. doi: 10.1002/emmm.201302827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, Drappatz J, O’Rourke DM, Wong M, Hamilton MG, Finocchiaro G, Perry J, Wick W, Green J, He Y, Turner CD, Yellin MJ, Keler T, Davis TA, Stupp R, Sampson JH, investigators AIt (2017) Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 18 (10):1373–1385. doi: 10.1016/S1470-2045(17)30517-X [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, Di Meco F, Lieberman F, Zhu JJ, Stragliotto G, Tran D, Brem S, Hottinger A, Kirson ED, Lavy-Shahaf G, Weinberg U, Kim CY, Paek SH, Nicholas G, Bruna J, Hirte H, Weller M, Palti Y, Hegi ME, Ram Z (2017) Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 318 (23):2306–2316. doi: 10.1001/jama.2017.18718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for R, Treatment of Cancer Brain T, Radiotherapy G, National Cancer Institute of Canada Clinical Trials G (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352 (10):987–996. doi: 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 7.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A 110 (10):4009–4014. doi: 10.1073/pnas.1219747110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344 (6190):1396–1401. doi: 10.1126/science.1254257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinartz R, Wang S, Kebir S, Silver DJ, Wieland A, Zheng T, Kupper M, Rauschenbach L, Fimmers R, Shepherd TM, Trageser D, Till A, Schafer N, Glas M, Hillmer AM, Cichon S, Smith AA, Pietsch T, Liu Y, Reynolds BA, Yachnis A, Pincus DW, Simon M, Brustle O, Steindler DA, Scheffler B (2017) Functional Subclone Profiling for Prediction of Treatment-Induced Intratumor Population Shifts and Discovery of Rational Drug Combinations in Human Glioblastoma. Clin Cancer Res 23 (2):562–574. doi: 10.1158/1078-0432.CCR-15-2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324 (5930):1029–1033. doi: 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, Wu Q, Flavahan W, Levison B, Johansen ML, Schmitt D, Neltner JM, Huang P, Ren B, Sloan AE, Silverstein RL, Gladson CL, DiDonato JA, Brown JM, McIntyre T, Hazen SL, Horbinski C, Rich JN, Lathia JD (2014) Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 32 (7):1746–1758. doi: 10.1002/stem.1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, Gu Y, Lum KM, Masui K, Yang H, Rong X, Hong C, Turner KM, Liu F, Hon GC, Jenkins D, Martini M, Armando AM, Quehenberger O, Cloughesy TF, Furnari FB, Cavenee WK, Tontonoz P, Gahman TC, Shiau AK, Cravatt BF, Mischel PS (2016) An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 30 (5):683–693. doi: 10.1016/j.ccell.2016.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi Y, Kim LJY, Prager BC, Flavahan WA, Liu X, Singer M, Hubert CG, Miller TE, Zhou W, Huang Z, Fang X, Regev A, Suva ML, Hwang TH, Locasale JW, Bao S, Rich JN (2017) Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci 20 (5):661–673. doi: 10.1038/nn.4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung J, Kim LJ, Wang X, Wu Q, Sanvoranart T, Hubert CG, Prager BC, Wallace LC, Jin X, Mack SC, Rich JN (2017) Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2 (10). doi: 10.1172/jci.insight.90019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T, Andrews N, Schmoll Massari M, Patel J, Amin K, Vuong A, Jimenez-Pascual A, Kubilis P, Garrett TJ, Moneypenny C, Pacak CA, Huang J, Sayour EJ, Mitchell DA, Sarkisian MR, Reynolds BA, Deleyrolle LP (2018) Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. doi: 10.15252/embj.201798772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bi J, Chowdhry S, Wu S, Zhang W, Masui K, Mischel PS (2020) Altered cellular metabolism in gliomas - an emerging landscape of actionable co-dependency targets. Nat Rev Cancer 20 (1):57–70. doi: 10.1038/s41568-019-0226-5 [DOI] [PubMed] [Google Scholar]
- 17.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Cancer Genome Atlas Research N (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17 (1):98–110. doi: 10.1016/j.ccr.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, Kratz A, Wefers AK, Huang K, Pajtler KW, Schweizer L, Stichel D, Olar A, Engel NW, Lindenberg K, Harter PN, Braczynski AK, Plate KH, Dohmen H, Garvalov BK, Coras R, Holsken A, Hewer E, Bewerunge-Hudler M, Schick M, Fischer R, Beschorner R, Schittenhelm J, Staszewski O, Wani K, Varlet P, Pages M, Temming P, Lohmann D, Selt F, Witt H, Milde T, Witt O, Aronica E, Giangaspero F, Rushing E, Scheurlen W, Geisenberger C, Rodriguez FJ, Becker A, Preusser M, Haberler C, Bjerkvig R, Cryan J, Farrell M, Deckert M, Hench J, Frank S, Serrano J, Kannan K, Tsirigos A, Bruck W, Hofer S, Brehmer S, Seiz-Rosenhagen M, Hanggi D, Hans V, Rozsnoki S, Hansford JR, Kohlhof P, Kristensen BW, Lechner M, Lopes B, Mawrin C, Ketter R, Kulozik A, Khatib Z, Heppner F, Koch A, Jouvet A, Keohane C, Muhleisen H, Mueller W, Pohl U, Prinz M, Benner A, Zapatka M, Gottardo NG, Driever PH, Kramm CM, Muller HL, Rutkowski S, von Hoff K, Fruhwald MC, Gnekow A, Fleischhack G, Tippelt S, Calaminus G, Monoranu CM, Perry A, Jones C, Jacques TS, Radlwimmer B, Gessi M, Pietsch T, Schramm J, Schackert G, Westphal M, Reifenberger G, Wesseling P, Weller M, Collins VP, Blumcke I, Bendszus M, Debus J, Huang A, Jabado N, Northcott PA, Paulus W, Gajjar A, Robinson GW, Taylor MD, Jaunmuktane Z, Ryzhova M, Platten M, Unterberg A, Wick W, Karajannis MA, Mittelbronn M, Acker T, Hartmann C, Aldape K, Schuller U, Buslei R, Lichter P, Kool M, Herold-Mende C, Ellison DW, Hasselblatt M, Snuderl M, Brandner S, Korshunov A, von Deimling A, Pfister SM (2018) DNA methylation-based classification of central nervous system tumours. Nature 555 (7697):469–474. doi: 10.1038/nature26000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, Zoppoli P, Ling S, Rao AA, Grifford M, Cherniack AD, Zhang H, Poisson L, Carlotti CG Jr., Tirapelli DP, Rao A, Mikkelsen T, Lau CC, Yung WK, Rabadan R, Huse J, Brat DJ, Lehman NL, Barnholtz-Sloan JS, Zheng S, Hess K, Rao G, Meyerson M, Beroukhim R, Cooper L, Akbani R, Wrensch M, Haussler D, Aldape KD, Laird PW, Gutmann DH, Network TR, Noushmehr H, Iavarone A, Verhaak RG (2016) Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 164 (3):550–563. doi: 10.1016/j.cell.2015.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, Dewitt J, Gritsch S, Perez EM, Gonzalez Castro LN, Lan X, Druck N, Rodman C, Dionne D, Kaplan A, Bertalan MS, Small J, Pelton K, Becker S, Bonal D, Nguyen QD, Servis RL, Fung JM, Mylvaganam R, Mayr L, Gojo J, Haberler C, Geyeregger R, Czech T, Slavc I, Nahed BV, Curry WT, Carter BS, Wakimoto H, Brastianos PK, Batchelor TT, Stemmer-Rachamimov A, Martinez-Lage M, Frosch MP, Stamenkovic I, Riggi N, Rheinbay E, Monje M, Rozenblatt-Rosen O, Cahill DP, Patel AP, Hunter T, Verma IM, Ligon KL, Louis DN, Regev A, Bernstein BE, Tirosh I, Suva ML (2019) An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178 (4):835–849 e821. doi: 10.1016/j.cell.2019.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Xu SL, Duan JJ, Yi L, Guo YF, Shi Y, Li L, Yang ZY, Liao XM, Cai J, Zhang YQ, Xiao HL, Yin L, Wu H, Zhang JN, Lv SQ, Yang QK, Yang XJ, Jiang T, Zhang X, Bian XW, Yu SC (2019) Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat Neurosci 22 (1):91–105. doi: 10.1038/s41593-018-0285-z [DOI] [PubMed] [Google Scholar]
- 22.Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T, Andrews N, Schmoll Massari M, Patel J, Amin K, Vuong A, Jimenez-Pascual A, Kubilis P, Garrett TJ, Moneypenny C, Pacak CA, Huang J, Sayour EJ, Mitchell DA, Sarkisian MR, Reynolds BA, Deleyrolle LP (2018) Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J 37 (23). doi: 10.15252/embj.201798772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, Huang L, Ratliff M, Karimian Jazi K, Kurz FT, Schmenger T, Lemke D, Gommel M, Pauli M, Liao Y, Haring P, Pusch S, Herl V, Steinhauser C, Krunic D, Jarahian M, Miletic H, Berghoff AS, Griesbeck O, Kalamakis G, Garaschuk O, Preusser M, Weiss S, Liu H, Heiland S, Platten M, Huber PE, Kuner T, von Deimling A, Wick W, Winkler F (2015) Brain tumour cells interconnect to a functional and resistant network. Nature 528 (7580):93–98. doi: 10.1038/nature16071 [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488 (7412):522–526. doi: 10.1038/nature11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan X, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, Guilhamon P, Lee L, Kushida MM, Pellacani D, Park NI, Coutinho FJ, Whetstone H, Selvadurai HJ, Che C, Luu B, Carles A, Moksa M, Rastegar N, Head R, Dolma S, Prinos P, Cusimano MD, Das S, Bernstein M, Arrowsmith CH, Mungall AJ, Moore RA, Ma Y, Gallo M, Lupien M, Pugh TJ, Taylor MD, Hirst M, Eaves CJ, Simons BD, Dirks PB (2017) Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 549 (7671):227–232. doi: 10.1038/nature23666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vescovi AL, Galli R, Reynolds BA (2006) Brain tumour stem cells. Nat Rev Cancer 6 (6):425–436 [DOI] [PubMed] [Google Scholar]
- 27.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63 (18):5821–5828 [PubMed] [Google Scholar]
- 28.Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, Kushida M, Head R, Morrissy S, Zhu X, Aviv T, Voisin V, Clarke ID, Li Y, Mungall AJ, Moore RA, Ma Y, Jones SJ, Marra MA, Malkin D, Northcott PA, Kool M, Pfister SM, Bader G, Hochedlinger K, Korshunov A, Taylor MD, Dirks PB (2014) Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 26 (1):33–47. doi: 10.1016/j.ccr.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baysan M, Woolard K, Cam MC, Zhang W, Song H, Kotliarova S, Balamatsias D, Linkous A, Ahn S, Walling J, Belova GI, Fine HA (2017) Detailed longitudinal sampling of glioma stem cells in situ reveals Chr7 gain and Chr10 loss as repeated events in primary tumor formation and recurrence. Int J Cancer 141 (10):2002–2013. doi: 10.1002/ijc.30887 [DOI] [PubMed] [Google Scholar]
- 30.Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S, Brons NHC, Leite S, Sauvageot N, Sarkisjan D, Seyfrid M, Fritah S, Stieber D, Michelucci A, Hertel F, Herold-Mende C, Azuaje F, Skupin A, Bjerkvig R, Deutsch A, Voss-Bohme A, Niclou SP (2019) Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun 10 (1):1787. doi: 10.1038/s41467-019-09853-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siebzehnrubl FA, Reynolds BA, Vescovi A, Steindler DA, Deleyrolle LP (2011) The origins of glioma: E Pluribus Unum? Glia 59 (8):1135–1147. doi: 10.1002/glia.21143 [DOI] [PubMed] [Google Scholar]
- 32.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA (2002) Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39 (3):193–206 [DOI] [PubMed] [Google Scholar]
- 33.Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414 (6859):105–111 [DOI] [PubMed] [Google Scholar]
- 34.Lee, Kotliarova, Kotliarov, Li, Su, Donin, Pastorino, Purow, Christopher, Zhang, Park, Fine (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9 (5):391–403. doi: 10.1016/j.ccr.2006.03.030 [DOI] [PubMed] [Google Scholar]
- 35.Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, De Filippis L, Mangiola A, Maira G, Anile C, De Bonis P, Reynolds BA, Pasquale EB, Vescovi AL (2012) The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 22 (6):765–780. doi: 10.1016/j.ccr.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444 (7120):761–765 [DOI] [PubMed] [Google Scholar]
- 37.Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, Cuartas I, Raventos C, Martinez-Ricarte F, Poca MA, Garcia-Dorado D, Lahn MM, Yingling JM, Rodon J, Sahuquillo J, Baselga J, Seoane J (2010) TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer cell 18 (6):655–668. doi: 10.1016/j.ccr.2010.10.023 [DOI] [PubMed] [Google Scholar]
- 38.Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, Rivers CS, Modrusan Z, Nacu S, Guerrero S, Edgar KA, Wallin JJ, Lamszus K, Westphal M, Heim S, James CD, VandenBerg SR, Costello JF, Moorefield S, Cowdrey CJ, Prados M, Phillips HS (2010) A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 17 (4):362–375. doi:S1535-6108(10)00065-6[pii] 10.1016/j.ccr.2009.12.049 [DOI] [PubMed] [Google Scholar]
- 39.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN (2015) Cancer stem cells in glioblastoma. Genes Dev 29 (12):1203–1217. doi: 10.1101/gad.261982.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jimenez-Pascual A, Hale JS, Kordowski A, Pugh J, Silver DJ, Bayik D, Roversi G, Alban TJ, Rao S, Chen R, McIntyre TM, Colombo G, Taraboletti G, Holmberg KO, Forsberg-Nilsson K, Lathia JD, Siebzehnrubl FA (2019) ADAMDEC1 Maintains a Growth Factor Signaling Loop in Cancer Stem Cells. Cancer Discov 9 (11):1574–1589. doi: 10.1158/2159-8290.CD-18-1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shingu T, Ho AL, Yuan L, Zhou X, Dai C, Zheng S, Wang Q, Zhong Y, Chang Q, Horner JW, Liebelt BD, Yao Y, Hu B, Chen Y, Fuller GN, Verhaak RG, Heimberger AB, Hu J (2017) Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat Genet 49 (1):75–86. doi: 10.1038/ng.3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lathia JD, Heddleston JM, Venere M, Rich JN (2011) Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell stem cell 8 (5):482–485. doi: 10.1016/j.stem.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vander Linden C, Corbet C (2019) Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front Oncol 9:159. doi: 10.3389/fonc.2019.00159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, Wolter M, Sommerlad D, Henze AT, Nister M, Reifenberger G, Lundeberg J, Frisen J, Acker T (2010) A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133 (Pt 4):983–995. doi: 10.1093/brain/awq042 [DOI] [PubMed] [Google Scholar]
- 45.Oka N, Soeda A, Inagaki A, Onodera M, Maruyama H, Hara A, Kunisada T, Mori H, Iwama T (2007) VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun 360 (3):553–559 [DOI] [PubMed] [Google Scholar]
- 46.Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, Rich JN (2010) Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 6 (5):421–432. doi: 10.1016/j.stem.2010.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haas TL, Sciuto MR, Brunetto L, Valvo C, Signore M, Fiori ME, di Martino S, Giannetti S, Morgante L, Boe A, Patrizii M, Warnken U, Schnolzer M, Ciolfi A, Di Stefano C, Biffoni M, Ricci-Vitiani L, Pallini R, De Maria R (2017) Integrin alpha7 Is a Functional Marker and Potential Therapeutic Target in Glioblastoma. Cell Stem Cell 21 (1):35–50 e39. doi: 10.1016/j.stem.2017.04.009 [DOI] [PubMed] [Google Scholar]
- 48.Stadlbauer A, Oberndorfer S, Zimmermann M, Renner B, Buchfelder M, Heinz G, Doerfler A, Kleindienst A, Roessler K (2019) Physiologic MR imaging of the tumor microenvironment revealed switching of metabolic phenotype upon recurrence of glioblastoma in humans. J Cereb Blood Flow Metab:271678X19827885. doi: 10.1177/0271678X19827885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN (2011) Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18 (5):829–840. doi: 10.1038/cdd.2010.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teng J, da Hora CC, Kantar RS, Nakano I, Wakimoto H, Batchelor TT, Chiocca EA, Badr CE, Tannous BA (2017) Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol 19 (6):820–832. doi: 10.1093/neuonc/now253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu L, Fukumura D, Jain RK (2002) Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem 277 (13):11368–11374. doi: 10.1074/jbc.M108347200 [DOI] [PubMed] [Google Scholar]
- 52.Thomas TM, Yu JS (2017) Metabolic regulation of glioma stem-like cells in the tumor micro-environment. Cancer Lett 408:174–181. doi: 10.1016/j.canlet.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME (2007) Molecular targets of glioma invasion. Cellular and molecular life sciences : CMLS 64 (4):458–478. doi: 10.1007/s00018-007-6342-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giese A, Loo MA, Tran N, Haskett D, Coons SW, Berens ME (1996) Dichotomy of astrocytoma migration and proliferation. International journal of cancer Journal international du cancer 67 (2):275–282. doi: [DOI] [PubMed] [Google Scholar]
- 55.Xie Q, Mittal S, Berens ME (2014) Targeting adaptive glioblastoma: an overview of proliferation and invasion. Neuro Oncol 16 (12):1575–1584. doi: 10.1093/neuonc/nou147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, Olson S, Gabrielli B, Osborne G, Vescovi A, Reynolds BA (2011) Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 134 (Pt 5):1331–1343. doi: 10.1093/brain/awr081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, Li G, Chang SD, Connolly ID, Li Y, Barres BA, Gephart MH, Quake SR (2017) Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep 21 (5):1399–1410. doi: 10.1016/j.celrep.2017.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN (2009) Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15 (6):501–513. doi: 10.1016/j.ccr.2009.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hitomi M, Deleyrolle LP, Mulkearns-Hubert EE, Jarrar A, Li M, Sinyuk M, Otvos B, Brunet S, Flavahan WA, Hubert CG, Goan W, Hale JS, Alvarado AG, Zhang A, Rohaus M, Oli M, Vedam-Mai V, Fortin JM, Futch HS, Griffith B, Wu Q, Xia CH, Gong X, Ahluwalia MS, Rich JN, Reynolds BA, Lathia JD (2015) Differential connexin function enhances self-renewal in glioblastoma. Cell Rep 11 (7):1031–1042. doi: 10.1016/j.celrep.2015.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dandy W (1928) Removal of right cerebral hemisphere for certain tumours with hemiplegia. JAMA 90:823–825 [Google Scholar]
- 61.Vogelbaum MA (2012) Does extent of resection of a glioblastoma matter? Clin Neurosurg 59:79–81. doi: 10.1227/NEU.0b013e31826b2e75 [DOI] [PubMed] [Google Scholar]
- 62.Weil S, Osswald M, Solecki G, Grosch J, Jung E, Lemke D, Ratliff M, Hanggi D, Wick W, Winkler F (2017) Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol 19 (10):1316–1326. doi: 10.1093/neuonc/nox070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. Journal of General Physiology 8 (6):519–530. doi: 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11 (5):325–337. doi: 10.1038/nrc3038 [DOI] [PubMed] [Google Scholar]
- 65.Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R, Stoll EA (2017) Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol 19 (1):43–54. doi: 10.1093/neuonc/now128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, Cai L, Good L, Tu BP, Hatanpaa KJ, Mickey BE, Mates JM, Pascual JM, Maher EA, Malloy CR, Deberardinis RJ, Bachoo RM (2012) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15 (6):827–837. doi: 10.1016/j.cmet.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159 (7):1603–1614. doi: 10.1016/j.cell.2014.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong Z, Cui H (2019) Epigenetic modulation of metabolism in glioblastoma. Semin Cancer Biol 57:45–51. doi: 10.1016/j.semcancer.2018.09.002 [DOI] [PubMed] [Google Scholar]
- 69.Agnihotri S, Zadeh G (2016) Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol 18 (2):160–172. doi: 10.1093/neuonc/nov125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Venneti S, Thompson CB (2017) Metabolic Reprogramming in Brain Tumors. Annu Rev Pathol 12:515–545. doi: 10.1146/annurev-pathol-012615-044329 [DOI] [PubMed] [Google Scholar]
- 71.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z (2011) Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480 (7375):118–122. doi: 10.1038/nature10598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB (2015) Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518 (7539):413–416. doi: 10.1038/nature13981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE, DeBerardinis RJ (2016) Metabolic Heterogeneity in Human Lung Tumors. Cell 164 (4):681–694. doi: 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M (2010) A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141 (4):583–594. doi: 10.1016/j.cell.2010.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF (2014) Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514 (7524):628–632. doi: 10.1038/nature13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knobloch M, Pilz GA, Ghesquiere B, Kovacs WJ, Wegleiter T, Moore DL, Hruzova M, Zamboni N, Carmeliet P, Jessberger S (2017) A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep 20 (9):2144–2155. doi: 10.1016/j.celrep.2017.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, Barthel F, Cho HJ, Lin YH, Satani N, Martinez-Ledesma E, Zheng S, Chang E, Sauve CG, Olar A, Lan ZD, Finocchiaro G, Phillips JJ, Berger MS, Gabrusiewicz KR, Wang G, Eskilsson E, Hu J, Mikkelsen T, DePinho RA, Muller F, Heimberger AB, Sulman EP, Nam DH, Verhaak RGW (2017) Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 32 (1):42–56 e46. doi: 10.1016/j.ccell.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL (2014) Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9 (1):349–365. doi: 10.1016/j.celrep.2014.08.056 [DOI] [PubMed] [Google Scholar]
- 79.Duman C, Yaqubi K, Hoffmann A, Acikgoz AA, Korshunov A, Bendszus M, Herold-Mende C, Liu HK, Alfonso J (2019) Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab 30 (2):274–289 e275. doi: 10.1016/j.cmet.2019.04.004 [DOI] [PubMed] [Google Scholar]
- 80.Khacho M, Harris R, Slack RS (2019) Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat Rev Neurosci 20 (1):34–48. doi: 10.1038/s41583-018-0091-3 [DOI] [PubMed] [Google Scholar]
- 81.Cancer Genome Atlas Research N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455 (7216):1061–1068. doi: 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, Network TR (2013) The somatic genomic landscape of glioblastoma. Cell 155 (2):462–477. doi: 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vander Heiden MG, DeBerardinis RJ (2017) Understanding the Intersections between Metabolism and Cancer Biology. Cell 168 (4):657–669. doi: 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou W, Wahl DR (2019) Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers (Basel) 11 (9). doi: 10.3390/cancers11091231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim JW, Dang CV (2006) Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 66 (18):8927–8930. doi: 10.1158/0008-5472.CAN-06-1501 [DOI] [PubMed] [Google Scholar]
- 86.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18 (1):54–61. doi: 10.1016/j.gde.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB (2013) Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16 (10):1373–1382. doi: 10.1038/nn.3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martuscello RT, Vedam-Mai V, McCarthy DJ, Schmoll ME, Jundi MA, Louviere CD, Griffith BG, Skinner CL, Suslov O, Deleyrolle LP, Reynolds BA (2016) A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clin Cancer Res 22 (10):2482–2495. doi: 10.1158/1078-0432.CCR-15-0916 [DOI] [PubMed] [Google Scholar]
- 89.Seyfried TN, Flores R, Poff AM, D’Agostino DP, Mukherjee P (2015) Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett 356 (2 Pt A):289–300. doi: 10.1016/j.canlet.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 90.De Feyter HM, Behar KL, Rao JU, Madden-Hennessey K, Ip KL, Hyder F, Drewes LR, Geschwind JF, de Graaf RA, Rothman DL (2016) A ketogenic diet increases transport and oxidation of ketone bodies in RG2 and 9L gliomas without affecting tumor growth. Neuro Oncol 18 (8):1079–1087. doi: 10.1093/neuonc/now088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stafford P, Abdelwahab MG, Kim DY, Preul MC, Rho JM, Scheck AC (2010) The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab (Lond) 7:74. doi: 10.1186/1743-7075-7-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN (2007) The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab (Lond) 4:5. doi: 10.1186/1743-7075-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lussier DM, Woolf EC, Johnson JL, Brooks KS, Blattman JN, Scheck AC (2016) Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer 16:310. doi: 10.1186/s12885-016-2337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rieger J, Bahr O, Maurer GD, Hattingen E, Franz K, Brucker D, Walenta S, Kammerer U, Coy JF, Weller M, Steinbach JP (2014) ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int J Oncol 44 (6):1843–1852. doi: 10.3892/ijo.2014.2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ (2009) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69 (20):7986–7993. doi: 10.1158/0008-5472.CAN-09-2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, Kondakci G, Narain NR, Kiebish MA, Bronson RT, Arismendi-Morillo G, Chinopoulos C, Seyfried TN (2019) Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun Biol 2:200. doi: 10.1038/s42003-019-0455-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mukherjee P, El-Abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN (2002) Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br J Cancer 86 (10):1615–1621. doi: 10.1038/sj.bjc.6600298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang YS, Wang FR (2013) Caloric restriction reduces edema and prolongs survival in a mouse glioma model. J Neurooncol 114 (1):25–32. doi: 10.1007/s11060-013-1154-y [DOI] [PubMed] [Google Scholar]
- 99.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, Sandhu S, Carlisle TL, Smith MC, Abu Hejleh T, Berg DJ, Zhang J, Keech J, Parekh KR, Bhatia S, Monga V, Bodeker KL, Ahmann L, Vollstedt S, Brown H, Shanahan Kauffman EP, Schall ME, Hohl RJ, Clamon GH, Greenlee JD, Howard MA, Schultz MK, Smith BJ, Riley DP, Domann FE, Cullen JJ, Buettner GR, Buatti JM, Spitz DR, Allen BG (2017) O2(−) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 31 (4):487–500 e488. doi: 10.1016/j.ccell.2017.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ojelabi OA, Lloyd KP, Simon AH, De Zutter JK, Carruthers A (2016) WZB117 (2-Fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) Inhibits GLUT1-mediated Sugar Transport by Binding Reversibly at the Exofacial Sugar Binding Site. J Biol Chem 291 (52):26762–26772. doi: 10.1074/jbc.M116.759175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Azzalin A, Nato G, Parmigiani E, Garello F, Buffo A, Magrassi L (2017) Inhibitors of GLUT/SLC2A Enhance the Action of BCNU and Temozolomide against High-Grade Gliomas. Neoplasia 19 (4):364–373. doi: 10.1016/j.neo.2017.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, Kitanaka C (2015) Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 6 (2):651–661. doi: 10.18632/oncotarget.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C, Madden C, Mathews D, Pascual JM, Mickey BE, Malloy CR, DeBerardinis RJ (2012) Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed 25 (11):1234–1244. doi: 10.1002/nbm.2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.O’Brien JS, Sampson EL (1965) Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res 6 (4):537–544 [PubMed] [Google Scholar]
- 105.Zou Y, Watters A, Cheng N, Perry CE, Xu K, Alicea GM, Parris JLD, Baraban E, Ray P, Nayak A, Xu X, Herlyn M, Murphy ME, Weeraratna AT, Schug ZT, Chen Q (2019) Polyunsaturated Fatty Acids from Astrocytes Activate PPARgamma Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. doi: 10.1158/2159-8290.CD-19-0270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Katz R, Hamilton JA, Pownall HJ, Deckelbaum RJ, Hillard CJ, Leboeuf RC, Watkins PA (2007) Brain uptake and utilization of fatty acids, lipids & lipoproteins: recommendations for future research. J Mol Neurosci 33 (1):146–150 [DOI] [PubMed] [Google Scholar]
- 107.Hamilton JA, Brunaldi K (2007) A model for fatty acid transport into the brain. J Mol Neurosci 33 (1):12–17 [DOI] [PubMed] [Google Scholar]
- 108.Mitchell RW, Hatch GM (2011) Fatty acid transport into the brain: of fatty acid fables and lipid tails. Prostaglandins Leukot Essent Fatty Acids 85 (5):293–302. doi: 10.1016/j.plefa.2011.04.007 [DOI] [PubMed] [Google Scholar]
- 109.Yamazaki S, DeGeorge JJ, Bell JM, Rapoport SI (1994) Effects of pentobarbital on incorporation of plasma palmitate into rat brain. Anesthesiology 80 (1):151–158 [DOI] [PubMed] [Google Scholar]
- 110.Grange E, Deutsch J, Smith QR, Chang M, Rapoport SI, Purdon AD (1995) Specific activity of brain palmitoyl-CoA pool provides rates of incorporation of palmitate in brain phospholipids in awake rats. J Neurochem 65 (5):2290–2298 [DOI] [PubMed] [Google Scholar]
- 111.Smith QR, Nagura H (2001) Fatty acid uptake and incorporation in brain: studies with the perfusion model. J Mol Neurosci 16 (2–3):167–172; discussion 215–121. doi: 10.1385/JMN:16:2-3:167 [DOI] [PubMed] [Google Scholar]
- 112.Pinkham K, Park DJ, Hashemiaghdam A, Kirov AB, Adam I, Rosiak K, da Hora CC, Teng J, Cheah PS, Carvalho L, Ganguli-Indra G, Kelly A, Indra AK, Badr CE (2019) Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Reports 12 (4):712–727. doi: 10.1016/j.stemcr.2019.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Morihiro Y, Yasumoto Y, Vaidyan LK, Sadahiro H, Uchida T, Inamura A, Sharifi K, Ideguchi M, Nomura S, Tokuda N, Kashiwabara S, Ishii A, Ikeda E, Owada Y, Suzuki M (2013) Fatty acid binding protein 7 as a marker of glioma stem cells. Pathol Int 63 (11):546–553. doi: 10.1111/pin.12109 [DOI] [PubMed] [Google Scholar]
- 114.Kaloshi G, Mokhtari K, Carpentier C, Taillibert S, Lejeune J, Marie Y, Delattre JY, Godbout R, Sanson M (2007) FABP7 expression in glioblastomas: relation to prognosis, invasion and EGFR status. J Neurooncol 84 (3):245–248. doi: 10.1007/s11060-007-9377-4 [DOI] [PubMed] [Google Scholar]
- 115.Liang Y, Bollen AW, Aldape KD, Gupta N (2006) Nuclear FABP7 immunoreactivity is preferentially expressed in infiltrative glioma and is associated with poor prognosis in EGFR-overexpressing glioblastoma. BMC Cancer 6:97. doi: 10.1186/1471-2407-6-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liang Y, Diehn M, Watson N, Bollen AW, Aldape KD, Nicholas MK, Lamborn KR, Berger MS, Botstein D, Brown PO, Israel MA (2005) Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci U S A 102 (16):5814–5819. doi: 10.1073/pnas.0402870102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mita R, Coles JE, Glubrecht DD, Sung R, Sun X, Godbout R (2007) B-FABP-expressing radial glial cells: the malignant glioma cell of origin? Neoplasia 9 (9):734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Beloribi-Djefaflia S, Vasseur S, Guillaumond F (2016) Lipid metabolic reprogramming in cancer cells. Oncogenesis 5:e189. doi: 10.1038/oncsis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Srivastava NK, Pradhan S, Gowda GA, Kumar R (2010) In vitro, high-resolution 1H and 31P NMR based analysis of the lipid components in the tissue, serum, and CSF of the patients with primary brain tumors: one possible diagnostic view. NMR Biomed 23 (2):113–122. doi: 10.1002/nbm.1427 [DOI] [PubMed] [Google Scholar]
- 120.Gopal K, Grossi E, Paoletti P, Usardi M (1963) Lipid Composition of Human Intracranial Tumors: A Biochemical Study. Acta Neurochir (Wien) 11:333–347 [DOI] [PubMed] [Google Scholar]
- 121.Guo D, Bell EH, Chakravarti A (2013) Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol 2 (3):289–299. doi: 10.2217/cns.13.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, Zhu S, Kofman AA, Bensinger SJ, Yong WH, Vinters HV, Horvath S, Watson AD, Kuhn JG, Robins HI, Mehta MP, Wen PY, DeAngelis LM, Prados MD, Mellinghoff IK, Cloughesy TF, Mischel PS (2009) EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2 (101):ra82. doi: 10.1126/scisignal.2000446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, Babic I, Tanaka K, Dang J, Iwanami A, Gini B, Dejesus J, Lisiero DD, Huang TT, Prins RM, Wen PY, Robins HI, Prados MD, Deangelis LM, Mellinghoff IK, Mehta MP, James CD, Chakravarti A, Cloughesy TF, Tontonoz P, Mischel PS (2011) An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov 1 (5):442–456. doi: 10.1158/2159-8290.CD-11-0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, Chakravarty P, East P, Dankworth B, Alibhai D, Harris AL, Schulze A (2015) SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 34 (40):5128–5140. doi: 10.1038/onc.2014.439 [DOI] [PubMed] [Google Scholar]
- 125.Dai S, Yan Y, Xu Z, Zeng S, Qian L, Huo L, Li X, Sun L, Gong Z (2017) SCD1 Confers Temozolomide Resistance to Human Glioma Cells via the Akt/GSK3beta/beta-Catenin Signaling Axis. Front Pharmacol 8:960. doi: 10.3389/fphar.2017.00960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M (2011) Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta 1807 (6):726–734. doi: 10.1016/j.bbabio.2010.10.022 [DOI] [PubMed] [Google Scholar]
- 127.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2 (31):31ra34. doi: 10.1126/scitranslmed.3000677 [DOI] [PubMed] [Google Scholar]
- 128.Yuan S, Wang F, Chen G, Zhang H, Feng L, Wang L, Colman H, Keating MJ, Li X, Xu RH, Wang J, Huang P (2013) Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells 31 (1):23–34. doi: 10.1002/stem.1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, He T, Shi X, Gerszten RE, Zhang Y, Yeh JJ, Curry WT, Zhao D, Sundaram S, Nigim F, Koerner MVA, Ho Q, Fisher DE, Roider EM, Kemeny LV, Samuels Y, Flaherty KT, Batchelor TT, Chi AS, Cahill DP (2015) Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 28 (6):773–784. doi: 10.1016/j.ccell.2015.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, Lee MA, Hu B, Hu J, Sahin E, Ong D, Fletcher-Sananikone E, Ho D, Kwong L, Brennan C, Wang YA, Chin L, DePinho RA (2012) Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488 (7411):337–342. doi: 10.1038/nature11331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, Ho S, Yue Y, Liu Y, Yan-Neale Y, Yang G, Lin F, Yin H, Gao H, Kipp DR, Zhao S, McNamara JT, Sprague ER, Zheng B, Lin Y, Cho YS, Gu J, Crawford K, Ciccone D, Vitari AC, Lai A, Capka V, Hurov K, Porter JA, Tallarico J, Mickanin C, Lees E, Pagliarini R, Keen N, Schmelzle T, Hofmann F, Stegmeier F, Sellers WR (2016) Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351 (6278):1208–1213. doi: 10.1126/science.aad5944 [DOI] [PubMed] [Google Scholar]
- 132.Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, Bielski CM, Scott JM, Dennis C, Cowley GS, Boehm JS, Root DE, Golub TR, Clish CB, Bradner JE, Hahn WC, Garraway LA (2016) MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351 (6278):1214–1218. doi: 10.1126/science.aad5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, Sarosiek KA, Briggs KJ, Robbins AK, Sewastianik T, Shareef SJ, Olenchock BA, Parker SJ, Tateishi K, Spinelli JB, Islam M, Haigis MC, Looper RE, Ligon KL, Bernstein BE, Carrasco RD, Cahill DP, Asara JM, Metallo CM, Yennawar NH, Vander Heiden MG, Kaelin WG Jr. (2018) Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175 (1):101–116 e125. doi: 10.1016/j.cell.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Savaskan NE, Eyupoglu IY (2010) xCT modulation in gliomas: relevance to energy metabolism and tumor microenvironment normalization. Ann Anat 192 (5):309–313. doi: 10.1016/j.aanat.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 135.Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, Schaffer P, Leprivier G, Sorensen PH (2019) Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci U S A 116 (19):9433–9442. doi: 10.1073/pnas.1821323116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, Wakimoto H, Yoshikawa M, Seishima R, Iwasaki Y, Morikawa T, Abe S, Takao A, Shimizu M, Masuko T, Nagane M, Furnari FB, Akiyama T, Suematsu M, Baba E, Akashi K, Saya H, Nagano O (2016) The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(−). Cancer Res 76 (10):2954–2963. doi: 10.1158/0008-5472.CAN-15-2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Aboody KS (2017) SLC7A11 Overexpression in Glioblastoma Is Associated with Increased Cancer Stem Cell-Like Properties. Stem Cells Dev 26 (17):1236–1246. doi: 10.1089/scd.2017.0123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B (2017) Immune microenvironment of gliomas. Lab Invest 97 (5):498–518. doi: 10.1038/labinvest.2017.19 [DOI] [PubMed] [Google Scholar]
- 139.Boussiotis VA, Charest A (2018) Immunotherapies for malignant glioma. Oncogene 37 (9):1121–1141. doi: 10.1038/s41388-017-0024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gieryng A, Kaminska B (2016) Myeloid-derived suppressor cells in gliomas. Contemp Oncol (Pozn) 20 (5):345–351. doi: 10.5114/wo.2016.64592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Elliott LA, Doherty GA, Sheahan K, Ryan EJ (2017) Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front Immunol 8:86. doi: 10.3389/fimmu.2017.00086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, James CD, Molinaro A, Phillips JJ (2012) Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One 7 (8):e43339. doi: 10.1371/journal.pone.0043339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H (2012) The brain tumor microenvironment. Glia 60 (3):502–514. doi: 10.1002/glia.21264 [DOI] [PubMed] [Google Scholar]
- 144.Wainwright DA, Nigam P, Thaci B, Dey M, Lesniak MS (2012) Recent developments on immunotherapy for brain cancer. Expert Opin Emerg Drugs 17 (2):181–202. doi: 10.1517/14728214.2012.679929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Biswas SK, Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11 (10):889–896. doi: 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- 146.Biswas SK, Mantovani A (2012) Orchestration of metabolism by macrophages. Cell Metab 15 (4):432–437. doi: 10.1016/j.cmet.2011.11.013 [DOI] [PubMed] [Google Scholar]
- 147.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42 (3):419–430. doi: 10.1016/j.immuni.2015.02.005 [DOI] [PubMed] [Google Scholar]
- 148.Pearce EL, Pearce EJ (2013) Metabolic pathways in immune cell activation and quiescence. Immunity 38 (4):633–643. doi: 10.1016/j.immuni.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ghesquiere B, Wong BW, Kuchnio A, Carmeliet P (2014) Metabolism of stromal and immune cells in health and disease. Nature 511 (7508):167–176. doi: 10.1038/nature13312 [DOI] [PubMed] [Google Scholar]
- 150.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, Grau S, Hiraoka N, Eckstein V, Ecker RC, Korff T, von Deimling A, Unterberg A, Beckhove P, Herold-Mende C (2011) Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res 17 (13):4296–4308. doi: 10.1158/1078-0432.CCR-10-2557 [DOI] [PubMed] [Google Scholar]
- 151.Siska PJ, Rathmell JC (2015) T cell metabolic fitness in antitumor immunity. Trends Immunol 36 (4):257–264. doi: 10.1016/j.it.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, Finlay DK (2014) mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol 193 (9):4477–4484. doi: 10.4049/jimmunol.1401558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11 (8):519–531. doi: 10.1038/nri3024 [DOI] [PubMed] [Google Scholar]
- 154.Azevedo EP, Rochael NC, Guimaraes-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, Palhano FL, Foguel D (2015) A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem 290 (36):22174–22183. doi: 10.1074/jbc.M115.640094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ (2015) Metabolic requirements for neutrophil extracellular traps formation. Immunology 145 (2):213–224. doi: 10.1111/imm.12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, Peoples MD, Li L, Karpinets T, Huang L, Malu S, Creasy C, Leahey SE, Chen J, Chen Y, Pelicano H, Bernatchez C, Gopal YNV, Heffernan TP, Hu J, Wang J, Amaria RN, Garraway LA, Huang P, Yang P, Wistuba II, Woodman SE, Roszik J, Davis RE, Davies MA, Heymach JV, Hwu P, Peng W (2018) Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab 27 (5):977–987 e974. doi: 10.1016/j.cmet.2018.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Apetoh L, Locher C, Ghiringhelli F, Kroemer G, Zitvogel L (2011) Harnessing dendritic cells in cancer. Semin Immunol 23 (1):42–49. doi: 10.1016/j.smim.2011.01.003 [DOI] [PubMed] [Google Scholar]
- 158.Dong H, Bullock TN (2014) Metabolic influences that regulate dendritic cell function in tumors. Front Immunol 5:24. doi: 10.3389/fimmu.2014.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL (2015) Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol 194 (7):2985–2991. doi: 10.4049/jimmunol.1403134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, Fairhurst AM, Connolly JE (2015) High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol 194 (11):5174–5186. doi: 10.4049/jimmunol.1303316 [DOI] [PubMed] [Google Scholar]
- 161.Pearce EJ, Everts B (2015) Dendritic cell metabolism. Nat Rev Immunol 15 (1):18–29. doi: 10.1038/nri3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, Park Y, Jones DP, Chappert P, Davoust J, Weiss DS, Virgin HW, Ron D, Pulendran B (2014) Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 343 (6168):313–317. doi: 10.1126/science.1246829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115 (23):4742–4749. doi: 10.1182/blood-2009-10-249540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, Artyomov MN, Jones RG, Pearce EL, Pearce EJ (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15 (4):323–332. doi: 10.1038/ni.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, Knight SC, Padhya T, McCaffrey TV, McCaffrey JC, Antonia S, Fishman M, Ferris RL, Kagan VE, Gabrilovich DI (2010) Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med 16 (8):880–886. doi: 10.1038/nm.2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR, Glimcher LH (2015) ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161 (7):1527–1538. doi: 10.1016/j.cell.2015.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Won WJ, Deshane JS, Leavenworth JW, Oliva CR, Griguer CE (2019) Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 3 (2):47–65. doi: 10.15698/cst2019.02.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12 (4):253–268. doi: 10.1038/nri3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70 (1):68–77. doi: 10.1158/0008-5472.CAN-09-2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Martinez FO, Gordon S, Locati M, Mantovani A (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177 (10):7303–7311. doi: 10.4049/jimmunol.177.10.7303 [DOI] [PubMed] [Google Scholar]
- 171.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL (1999) Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 189 (9):1363–1372. doi: 10.1084/jem.189.9.1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Grohmann U, Bronte V (2010) Control of immune response by amino acid metabolism. Immunol Rev 236:243–264. doi: 10.1111/j.1600-065X.2010.00915.x [DOI] [PubMed] [Google Scholar]
- 173.Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD (2007) The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol 179 (8):5041–5053. doi: 10.4049/jimmunol.179.8.5041 [DOI] [PubMed] [Google Scholar]
- 174.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, Rodriguez PC, Ochoa AC (2015) Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res 3 (11):1236–1247. doi: 10.1158/2326-6066.CIR-15-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC (2014) The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20 (1):61–72. doi: 10.1016/j.cmet.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC (2011) Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186 (6):3299–3303. doi: 10.4049/jimmunol.1003613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J (2008) Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol 38 (9):2412–2418. doi: 10.1002/eji.200838318 [DOI] [PubMed] [Google Scholar]
- 178.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211 (5):781–790. doi: 10.1084/jem.20131916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, Bhat KP, Curran MA, de Groot JF, Heimberger AB (2019) Immune biology of glioma associated macrophages and microglia: Functional and therapeutic implications. Neuro Oncol. doi: 10.1093/neuonc/noz212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, Bosca L (2010) Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185 (1):605–614. doi: 10.4049/jimmunol.0901698 [DOI] [PubMed] [Google Scholar]
- 181.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496 (7444):238–242. doi: 10.1038/nature11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4 (1):13–24. doi: 10.1016/j.cmet.2006.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513 (7519):559–563. doi: 10.1038/nature13490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Biswas SK (2015) Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 43 (3):435–449. doi: 10.1016/j.immuni.2015.09.001 [DOI] [PubMed] [Google Scholar]
- 185.Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr., Artyomov MN, Pearce EL, Pearce EJ (2014) Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15 (9):846–855. doi: 10.1038/ni.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Park J, Lee SE, Hur J, Hong EB, Choi JI, Yang JM, Kim JY, Kim YC, Cho HJ, Peters JM, Ryoo SB, Kim YT, Kim HS (2015) M-CSF from Cancer Cells Induces Fatty Acid Synthase and PPARbeta/delta Activation in Tumor Myeloid Cells, Leading to Tumor Progression. Cell Rep 10 (9):1614–1625. doi: 10.1016/j.celrep.2015.02.024 [DOI] [PubMed] [Google Scholar]
- 187.Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19 (1):20–27. doi: 10.1038/nn.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, Asara JM, Evans RM, Cantley LC, Lyssiotis CA, Kimmelman AC (2016) Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536 (7617):479–483. doi: 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17 (11):1498–1503. doi: 10.1038/nm.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, Alvarez H, Gupta S, Maiti SN, Cooper L, Peehl D, Ram PT, Maitra A, Nagrath D (2016) Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 5:e10250. doi: 10.7554/eLife.10250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Achreja A, Zhao H, Yang L, Yun TH, Marini J, Nagrath D (2017) Exo-MFA - A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab Eng 43 (Pt B):156–172. doi: 10.1016/j.ymben.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Spees JL, Olson SD, Whitney MJ, Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103 (5):1283–1288. doi: 10.1073/pnas.0510511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, Cancer Genome Atlas Research N (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17 (5):510–522. doi: 10.1016/j.ccr.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chen Z, Hambardzumyan D (2018) Immune Microenvironment in Glioblastoma Subtypes. Front Immunol 9:1004. doi: 10.3389/fimmu.2018.01004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Quinones A, Le A (2018) The Multifaceted Metabolism of Glioblastoma. Adv Exp Med Biol 1063:59–72. doi: 10.1007/978-3-319-77736-8_4 [DOI] [PubMed] [Google Scholar]
- 196.Zecchini V, Frezza C (2017) Metabolic synthetic lethality in cancer therapy. Biochim Biophys Acta Bioenerg 1858 (8):723–731. doi: 10.1016/j.bbabio.2016.12.003 [DOI] [PubMed] [Google Scholar]
- 197.Vander Linden C, Corbet C (2020) Reconciling environment-mediated metabolic heterogeneity with the oncogene-driven cancer paradigm in precision oncology. Semin Cell Dev Biol 98:202–210. doi: 10.1016/j.semcdb.2019.05.016 [DOI] [PubMed] [Google Scholar]
- 198.Chen P, Zhao D, Li J, Liang X, Li J, Chang A, Henry VK, Lan Z, Spring DJ, Rao G, Wang YA, DePinho RA (2019) Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 35 (6):868–884 e866. doi: 10.1016/j.ccell.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]