

Abstract
We report a three generation family in which five members, three females and two males, demonstrate a 44 bp deletion (1164–1207del44) in the MECP2 gene associated with Rett syndrome, leading to a truncation of the C-terminus of the protein. Two of the three females and both males do not meet RTT criteria whereas the youngest female has classic RTT. Both males demonstrated a clear pattern of progressive involvement including dystonia. The transmitting females do not demonstrate features of RTT as a result of unbalanced X chromosome inactivation (XCI) and were only identified as carriers following the evaluation of the affected males and the girl with classic RTT. As such, accurate assessment of the precise frequency of MECP2 mutations in carrier females with mild cognitive impairment or borderline cognitive function will be under-represented unless an affected offspring is recognized. Strategies for accurate diagnosis in such instances should be considered carefully.
Keywords: MECP2, Mutation, X chromosome inactivation, Phenotype-genotype, Rett syndrome, Male, Dystonia
Introduction
Rett syndrome is a neurodevelopmental disorder, predominantly appearing in young girls and first described in 1966 by Andreas Rett [1], and is characterized by cognitive impairment, communication dysfunction, stereotypic movement disorder, and growth failure. RTT has an incidence varying from 0.43–0.71/10,000 females in France [2] to 1.09/10,000 females in Australia [3]. Diagnosis is based on consensus clinical criteria [4]. Mutations were identified in the methyl-CpG-binding protein 2 gene (MECP2) in 1999 [5]. More recent reports indicates that approximately 95% individuals meeting these criteria will have one of the more than 200 mutations identified in MECP2 [6, 7]. Eight common point mutations (60%), 3′ frameshift mutations (8–10%), or large deletions (7–9%) account for the majority of affected individuals [6-8]. Mutations may also be seen in males and females who do not meet the RTT consensus criteria [6, 9, 10].
In most instances (>99.9%), RTT is a sporadic disorder. Nonetheless, recurrences within families may occur [11]. In such families, the mother is the transmitting parent and, due to favorable X-chromosome inactivation (XCI) of the abnormal chromosome, does not demonstrate features of RTT. In addition, males in these families tend to present with a severe encephalopathy in infancy [12]. Few families in which males demonstrate less severe involvement have been described [13]. We have recently evaluated five members, three females and two males, in a three generation family, all of whom demonstrate a 44 bp deletion (1164–1207del44). This deletion occurs in a known mutation ‘hotspot’ and has been described previously in RettBase (www.chw.edu.au) in more than thirty females in association with several phenotypes including classic and atypical (congenital variant, preserved speech variant, and forme fruste) RTT and in at least one male variant. Similar deletions beginning at nucleotide 1164 have been described in another three individuals. In the present family, two of the three females and both males do not meet RTT criteria whereas the youngest female has classic RTT. This report describes this family in detail and provides the molecular underpinning for the variable clinical presentations. The two older females demonstrate no evidence of progressive features unlike the males who appear to be following a course of progressive decline.
Family description
The family (Fig. 1) described below was evaluated as part of the NIH-funded Rare Disease Natural History study (registered at www.clinicaltrials.gov; #NCT00299312). The protocol and consent form for this study have been approved by the Institutional Review Board for the University of Alabama at Birmingham. Each affected family member has the same MECP2 mutation, which is an intragenic deletion in exon 4, 1164–1207del44 that causes a truncation of the protein in the C-terminal domain. In individuals meeting consensus criteria for RTT, mutations in this region have been associated with a less severe phenotype. Each affected member had a complete evaluation in accordance with the study protocol. All dataforms were completed including two severity scales, the Clinical Severity Score (CSS) and the Motor-Behavioral Assessment (MBA) [7]. In our RTT natural history study, the mean values for those with classic RTT (N=529) are CSS=23.5 and MBA=50.5. For individuals with 3′ frameshift mutations leading to a truncation of the C-terminus of the MeCP2 protein in our natural history study (N=42), CSS=21.2 and MBA=49.2.
Fig. 1.
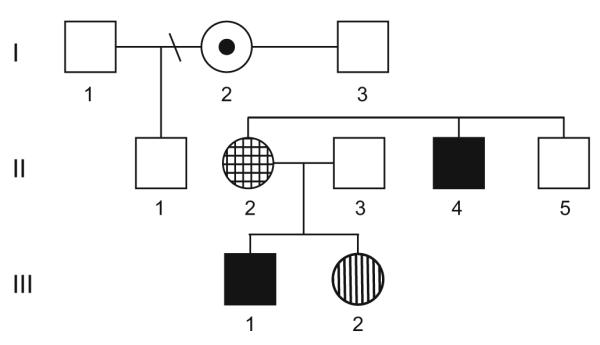
The family pedigree: The five individuals with MECP2 mutations are depicted in this three generation pedigree. The female in generation I represented by the bulls-eye has cognitive delay and lacks the clinical features of RTT; the female in generation II represented by the checkerboard pattern has cognitive delay and aberrant behavior and lacks the clinical features of RTT; the female in generation III represented by the vertical stripes has typical features of RTT; and both males have cognitive delay and a pattern of progressive motor impairments that do not reflect the clinical features of RTT
Family member I:2 is a 52 year old female for whom limited developmental history is available. However, no evidence of specific developmental delays or loss of previously acquired skill is known. She does not meet any RTT consensus criteria and only constipation of the supportive criteria. In addition to the mutation in MECP2, she has polycystic kidney disease and currently requires hemodialysis. She completed education through junior high school and is able to conduct activities of daily living. Psychometric testing at the University of Michigan in 2005 revealed a full scale IQ of 59. She currently lives in a group home in order to assist with her on-going medical management. Her two sons, II:1 and II:4, are described as being in good health. The former is regarded as having a learning disability whereas the latter has no known medical or neurodevelopmental problems. Neither of these males has been examined directly. On assessment, her general physical and neurological examinations were unremarkable. Her CSS was 1 and MBA was 3 (low scores indicating minimal severity).
Family member II:2 is a 25 year old female whose early development was considered to be normal without regression of previously acquired skills. She completed junior high school. She also does not meet any RTT consensus or supportive criteria. However, she has a history of significant behavioral problems related to poor judgment, disinhibition, promiscuity, and compulsive prevarication (mythomania). She has polycystic kidney disease as well. Although she can manage her personal care, she has been unable to manage activities associated with independent living or care for her children adequately such that they were placed in foster care and subsequently adopted. She currently resides in the group home with her mother on whom she relies for emotional support. When tested in early 2009, her full scale IQ was 61 (verbal: 66 and performance: 60). Adaptive behavior assessment was extremely low at 58. On assessment, her general physical and neurological examinations were unremarkable. CSS was 1 and MBA score was 2.
Family member II:4 is a 23 year old male with progressive neurodevelopmental problems. He was born at term with no known problems. However, his development was quite delayed. He rolled over at 5 months, but did not come to sit or crawl until 24 months. He pulled to stand at 12 months and lost this at age 5 years, and he walked with support at 36 months and lost this at age 11 years. He held a bottle and reached for a toy at 10 months losing both at age 14 years. He transferred at 36 months, had a pincer grasp, and finger fed at age 5 years, losing all three at age 10 years. He cooed at 3 months and smiled at 5 months and still does so. He babbled at 15 months and lost this at an unknown age. He spoke in single words at 36 months, but lost the ability to speak at age 7 years. He gestured and pointed for wants at age 5 years and lost both at age 10 years. He fixed and followed at 5 months and followed a command at an unknown age and retains both. He has always liked being held and was never visually or aurally inattentive. Seizures began at age 2 years, but are no longer present. Breathholding was noted at 14 months. Hyperventilation has never been present. Constipation and gastroesophageal reflux have been a problem since age 3 years. At age 10 years, he was noted to have repetitive hand flicking movements, but these are no longer present and no other stereotypies typical for RTT have been noted. Scoliosis was noted at age 7 years, becoming severe at 13 years despite bracing. A feeding gastrostomy was placed at age 15 years and a tracheostomy was performed for ventilatory failure during sleep at age 18 years. He is now ventilator dependent although he will breathe independently for a brief time. He was described as hypotonic and ataxic during early childhood; dystonia and increased muscle tone (rigidity) have been progressively more severe since then, necessitating treatment with an intrathecal baclofen pump at age 13. From age 13 to the present, botulinum toxin, phenol injections, and orthopedic surgeries were performed to address progressive dystonia and contractures. Recurrent torticollis has also recently been treated with botulinum toxin. At present, he is alert, anxious, and interactive with prolonged eye contact. Visual fixation and following are normal. He neither verbalizes nor vocalizes and has no stereotypic movements. His scoliosis is greater than 60° with right scapular prominence and rotation to the right. Muscle mass is reduced. Strength is normal and muscle stretch reflexes could not be obtained. He has no purposeful hand use and is non-ambulatory. He has marked dystonia of his upper extremities and at the ankles; contractures are present at the knees and ankles. Head circumference has shown a mild deceleration in growth rate (10th to < 2nd percentile) after age 5 months. While he has demonstrated a pattern of progressive motor difficulties with evident regression, most skills were lost after age 5 years. CSS was 34 and MBA score was 61. Overall, he does not fulfill the consensus criteria for RTT, but has displayed features at various points in time such as stereotypic movements that are RTT-like.
Family member III:1 is an 8 year old male who is following the same neurodevelopmental trajectory as his uncle (II:3). Pregnancy, labor and delivery, and early developmental progress were not regarded as remarkable. However, by age 2 months deceleration in the rate of head growth was noted. Early gross motor skills developed appropriately but by age 6 years he could no longer come to a sit and by 7 years could no longer crawl. He never pulled to a stand or walked independently, but did learn to walk with support and can still do so with significant help. Early fine motor skills including reaching for a toy, transferring, and finger feeding appeared to be on time; however, feeding pattern was always abnormal and a pincer grasp never developed. Cooing, babbling, and words never appeared and while he was attentive, he never responded to commands. Social interaction was always appropriate. Seizures and periodic breathing have not occurred. He was noted to have transient hand wringing and periodically rub his nose in a repetitive fashion. He has not demonstrated a regression of any previously acquired fine motor skills, but has failed to progress. Head circumference has shown a moderate deceleration in growth rate (50th to the 10th percentile) after age 2 months. Since age 6, he has had progressive bilateral tremors, dystonia of the left upper extremity requiring botulinum toxin, and spasticity of the femoral adductors requiring tenotomy. On assessment, he was alert and interactive and gave good eye contact. He vocalized, but did not verbalize. Breathing pattern was normal. He did engage in rubbing his nose periodically. He was hypotonic in his extremities and axial muscles. Strength, bulk, and muscle stretch reflexes were normal. He reached for objects with his right hand using a raking-type grasp. He sat with support, but would not walk with support. He had a variety of involuntary movements including tremor, myoclonus, and severe dystonia of his left upper extremity. While seated on a narrow base, he had marked titubation. He responded appropriately to light touch. His hands were cool, but his feet were warm. He has a mild degree of scoliosis. CSS was 20 and MBA score was 40. Overall, he does not fulfill the consensus criteria for RTT, but does have features such as stereotypic movements that are RTT-like.
Family member III:2 is a 6 year old female who meets RTT consensus criteria. She was born at term with no known problems. Other than poor feeding initially, early development was regarded as normal. She sat, crawled, walked, and developed fine motor hand skills at appropriate ages. She used single words by age 12 months and phrases by 18 months. She did have a brief period of visual and aural inattention between age 15 and 17 months. However, at age 3 years she began to regress with respect to these skills and at age 6 years demonstrated hand stereotypies including hand squeezing and finger flicking. She has had no seizures, periodic breathing, or bruxism, but constipation has been a problem since age 2 years, self-abusive behavior in the form of biting since age 3 years, and GE-reflux since age 4 years. She has difficulty going to sleep and awakens frequently throughout the night. On assessment, she was alert and interactive with good eye contact. She smiled and had intermittent laughter. She vocalized, but did not verbalize and was noted to hold her breath briefly and intermittently. Hand stereotypies consisted of hand mouthing on the left and patting herself on her body. Muscle tone, bulk, strength, and muscle stretch reflexes were normal. Gegenhalten was noted in the upper extremities. She reached for objects with either hand using a pincer grasp. Gait was performed reasonably well on a narrow base, but was semi-purposeful as she would not follow specific directions and tended to wander. She had a mild degree of generalized tremor and significant truncal rocking while at rest. Skin temperature and response to light touch were normal. No scoliosis was noted. CSS was 14 and MBA score was 27. In our natural history study, both scores are well below the mean values (N=529; CSS=23.5 and MBA=50.5) for those with classic RTT.
Molecular testing MECP2 molecular testing of each family member identified the same 44 base pair deletion beginning at nucleotide 1164 (1164–1207del44), This mutation is predicted to lead to a frameshift in translation of MeCP2 beginning at lysine 408, followed by insertion of twelve unrelated amino acids before encountering a termination codon (pK408fs12X). Each female had testing for X-chromosome inactivation (XCI) through the Greenwood Genetic Center clinical diagnostic laboratory using standard methodology for the androgen receptor locus. Family member I:2 had 89:11 distribution, family member II:3 had 77:23 distribution, and family member III:2 had 59:41 distribution. The results correlate with the phenotype noted in each female. The two older females with favorable XCI do not meet criteria for RTT despite their inability to be completely independent. The youngest female with random XCI meets criteria for RTT and expresses a clinical severity pattern that is less than the mean value for those in our natural history study (N=42; CSS=21.2 and MBA=49.2) with C-terminal truncations. No evidence for mosaicism or X-chromosome aneuploidy was detected in the males.
Discussion
RTT is a genetic disorder whose occurrence is sporadic in most individuals, more than 99.5% resulting from a de novo mutation in MECP2. Extended family pedigrees of the type described here are rare. As such, the present family provides important insights into the varied effects of a given mutation depending on gender and XCI. The specific 44 bp deletion in this family has been described previously in females with classic and atypical RTT phenotypes. This mutation occurs in a known ‘hotspot’ for deletions (8.8% in the North American database of 1928 individuals) and may confer milder involvement [7, 8]. As noted above, RettBase lists individuals with both classic and milder (forme fruste and preserved speech) RTT variants. In previously reported multiplex families, the males have had a severe infantile encephalopathy such that assessing the clinical progression in them over an extended period was often not possible [9, 10]. The lone male previously reported with this deletion was described as manifesting RTT [14]. His mother carried the same mutation, but was described as normal. Her XCI testing revealed a 95:5 distribution. In the present family, the opportunity to observe two generations of males with a much milder phenotype has been quite revealing. Both males demonstrated a clear pattern of progressive involvement beginning with normal early development and subsequent motor and language delays that became apparent after the first six months followed by the onset of dystonia in mid-childhood and in the older male, loss of motor skills, progression of dystonia, and gradual increase in muscle tone to rigidity and joint contractures at present. Although both males do not fulfill diagnostic criteria for RTT, each has had features seen in RTT in the form of deceleration in the rate of head growth and transient presence of stereotypic hand movements. Therefore, in the absence of a normal X chromosome as in females with RTT, it is not surprising that MECP2 mutations in males are likely to result in progressive dysfunction.
In females, the transmitting parent (or parents as in this pedigree), as a result of unbalanced XCI, is typically not recognized until a daughter meeting criteria for RTT is born. The present family presents an additional level of complexity in that c-terminal mutations have been associated with somewhat milder involvement. The most vexing problem is that accurate assessment of the precise frequency of MECP2 mutations is likely to be under-represented among females with mild cognitive impairment or borderline cognitive function unless an affected offspring is identified. Strategies for accurate diagnosis in such instances should be considered carefully.
Acknowledgement
This study was supported by NIH grants RR019478 and IDDRC grant HD38985, and funds from the International RTT Foundation and Civitan International Research Center. The authors acknowledge the gracious participation and provision of information by the families of the reported participants. The CSS and MBA data were provided by Hye-Seung Lee, PhD, at the Data Technology Coordinating Center at the University of South Florida. Dr. Mary Lou Oster-Granite, Health Scientist Administrator at NICHD, provided invaluable guidance, support, and encouragement for this Rare Disease initiative.
Contributor Information
Kimberly Augenstein, Neuromuscular and Rehabilitation Associates of Northern Michigan, Traverse City, MI, USA.
Jane B. Lane, University of Alabama at Birmingham, 1530 3rd Avenue South, CIRC 320E, Birmingham, AL 35294-0021, USA
Antony Horton, International Rett Syndrome Foundation, Cincinnati, OH, USA.
Carolyn Schanen, Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, DE, USA.
Alan K. Percy, University of Alabama at Birmingham, 1530 3rd Avenue South, CIRC 320E, Birmingham, AL 35294-0021, USA
References
- 1.Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wiener Medizinische Wochenschrift. 1966;116:723–6. [PubMed] [Google Scholar]
- 2.Bienvenu T, et al. The incidence of Rett syndrome in France. Pediatr Neurol. 2006;34(5):372–5. doi: 10.1016/j.pediatrneurol.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Laurvick CL, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148(3):347–52. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 4.Hagberg B, et al. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6(5):293–7. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- 5.Amir R, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Percy AK, et al. Rett syndrome: North American database. J Child Neurol. 2007;22(12):1338–41. doi: 10.1177/0883073807308715. [DOI] [PubMed] [Google Scholar]
- 7.Neul JL, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in rett syndrome. Neurology. 2008;70:1313–21. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bebbington A, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70(11):868–75. doi: 10.1212/01.wnl.0000304752.50773.ec. [DOI] [PubMed] [Google Scholar]
- 9.Moog U, et al. Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2) Eur J Paediatr Neurol. 2003;7(1):5–12. doi: 10.1016/s1090-3798(02)00134-4. [DOI] [PubMed] [Google Scholar]
- 10.Kankirawatana P, et al. Early progressive encephalopathy in boys and MECP2 mutations. Neurology. 2006;67(1):164–6. doi: 10.1212/01.wnl.0000223318.28938.45. [DOI] [PubMed] [Google Scholar]
- 11.Schanen C, et al. A new Rett syndrome family consistent with X-linked inheritance expands the X chromosome exclusion map. American Journal Human Genetics. 1997;61:634–41. doi: 10.1086/515525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schanen C, Francke U. A severely affected male born into a Rett syndrome kindred supports X-linked inheritance and allows extension of the exclusion map. Am J Genetics. 1998;63:267–9. doi: 10.1086/301932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Budden SS, Dorsey HC, Steiner RD. Clinical profile of a male with Rett syndrome. Brain Dev. 2005;27(Suppl 1):S69–71. doi: 10.1016/j.braindev.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Dayer AG, et al. MECP2 mutant allele in a boy with Rett syndrome and his unaffected heterozygous mother. Brain Dev. 2007;29(1):47–50. doi: 10.1016/j.braindev.2006.06.001. [DOI] [PubMed] [Google Scholar]