

Abstract
Silicosis is a lethal pneumoconiosis for which no therapy is available. Silicosis is a global threat and more than 2.2 million people/year are exposed to silica in the US. The initial response to silica is mediated by innate immunity. Phagocytosis of silica particles by macrophages is followed by recruitment of mitochondria to phagosomes, generation of mitochondrial Reactive-Oxygen-Species (ROS), and cytokine (IL-1β, TNF-α, IFN-β) release. In contrast to lipopolysaccharide (LPS), the metabolic remodeling of silica-exposed macrophages is unclear. This study contrasts mitochondrial and metabolic alterations induced by LPS and silica on macrophages and correlates them with macrophages viability and cytokine production, central to the pathogenesis of silicosis. Using high-resolution respirometer and liquid chromatography-high resolution mass spectrometry (LC-HRMS), we determined the effects of silica and LPS on mitochondrial respiration, and changes in central carbon metabolism of murine macrophage cell lines RAW 264.7 and IC-21. We show that silica induces metabolic reprogramming of macrophages. Silica, as well as LPS, enhances glucose uptake and increases aerobic glycolysis in macrophages. In contrast to LPS, silica affects mitochondria respiration, reducing complex-I and enhancing complex-II activity, to sustain cell viability. These mitochondrial alterations are associated, in silica, but not in LPS-exposed macrophages, with reductions of tricarboxylic-acid cycle (TCA) intermediates, including succinate, itaconate, glutamate, and glutamine. Furthermore, in contrast to LPS, these silica-induced metabolic adaptations do not correlate with IL-1β or TNF-α production, but with the suppressed release of IFN-β. Our data highlights the importance of CII-activity, and TCA-remodeling to macrophage survival and cytokine-mediated inflammation in silicosis.
INTRODUCTION
Crystalline silica is one of the most abundant minerals on Earth. More than 230 million individuals around the world, and more than 2 million workers in the United States, predominantly in construction and mining occupations, are exposed to silica every year [1–3]. Inhalation of crystalline silica leads to the development of silicosis, a progressive pneumoconiosis characterized by lung inflammation and fibrosis, for which no specific therapy is available. Silicosis is associated with an increased risk of tuberculosis, lung cancer, chronic obstructive pulmonary disease (COPD), kidney disease, and autoimmune disease, and these health risks remain elevated even after silica exposure has ceased [2, 4–6].
Macrophages are fundamental to the pathogenesis of silicosis. Similar to live bacteria, silica particles are phagocytosed by macrophages into phagosomes. Subsequently, macrophages activate pattern recognition receptors (PRRs) and the NLRP3 inflammasome, and release cytokines such as Interleukin-1 beta (IL-1β), Tumor Necrosis Factor-alpha (TNF-α), and Interferons, key mediators of the pathogenesis of silicosis [7]. In contrast to bacteria, silica particles cannot be degraded, and the persistent activation results in an increased NADPH oxidase (Phox) activation, and mitochondrial Reactive Oxygen Species (ROS) production that ultimately leads to macrophage cell death and release of silica particles that perpetuate inflammation [2, 8–10]. Activated macrophages exhibit altered immuno-metabolism, intracellular metabolic changes that govern immune effector mechanisms, such as cytokine secretion. For example, lipopolysaccharide (LPS)-stimulated macrophages demonstrate enhanced glycolysis, and increased secretion of IL-1β and TNF-α [11–13]. Inhibiting glycolysis or blocking succinate dehydrogenase (SDH), an enzyme that links glycolysis and the electron transport chain (ETC), in LPS-stimulated macrophages inhibits IL-1β, but not TNF-α, proving that the metabolic pathway is specifically involved in cytokine production [11].
Despite decades of research, the nature of the immune-metabolic response induced by silica in macrophages, its contribution to cytokine specification, and the pathogenesis of silicosis are not well understood. In addition, recent studies indicate that the macrophages’ ontogeny, resident in the lung versus recruited from bone marrow in response to injury, determines differences in the in vivo metabolic response to LPS stimuli [14–16]. Therefore, as an initial step to characterize the difference between the silica- and LPS- induced metabolic effects on macrophages, we adopted an in vitro system using well-characterized macrophage cell lines that differ on the cytokine production in response to these agents, and applied state-of-the-art techniques, such as high-resolution respirometry and liquid chromatography-high resolution mass spectrometry (LC-HRMS), to determine the effects of silica on mitochondrial respiration, and the changes in central carbon metabolism of silica-exposed macrophages. Specifically, we characterized the metabolic reprogramming of silica-stimulated macrophages, in terms of alteration in glycolysis, TCA cycle, mitochondrial respiration, and ETC activity, to correlate their contribution to cytokine specification. Using macrophage cell lines, we reported significant differences in immuno-metabolism in the macrophages’ response to silica and LPS. We found that, similar to LPS exposure, silica increases glucose uptake and enhances the glycolysis in macrophages. In contrast to LPS, which induces accumulation of TCA intermediates such as citrate, succinate, and itaconate, silica negatively affects the TCA activity, reducing the intracellular amount of TCA intermediates succinate and itaconate below control levels. Silica also induces specific ETC adaptations: enhanced mitochondrial complex II (CII) activity and downregulation of mitochondrial complex I (CI) activity via reductions of the CI specific protein Ecsit, that are required for macrophage survival to silica-induced cytotoxicity. These metabolic responses correlated with the observed differences in macrophage production of inflammatory cytokines in response to LPS and silica. Thus, although RAW 264.7 macrophages enhanced IL-1β and TNF-α production in response to LPS or silica, only LPS-stimulated RAW 264.7 macrophages exhibited HIF-1α stabilization under normoxic conditions, activation of the NLRP3 inflammasome and malonylation of GAPDH in a time-related manner that preceded the release of IL-1β and TNF-α. In addition, we observed differences in the RAW 264.7 macrophages accumulation of itaconate that correlate, inversely, with the transcription and release of interferon-beta (IFN-β) in response to LPS or silica. Despite intrinsic limitations associated with in vitro studies and the use of murine macrophages cell lines, the present studies provide valuable information as to how metabolic pathways determine macrophage immune metabolism in response to silica exposure. Further studies to determine therapeutic targets are warranted to investigate macrophage immune-metabolic responses to silica in vivo.
METHODS.
Cell culture.
The mouse macrophage cell lines RAW 264.7 and IC-21 were purchased from the American Type Culture Collection (Rockville, MD) and were cultured in DMEM medium (Gibco BRL, Rockville, MD) or RPMI 1640 (ATCC, Rockville, MD) respectively, supplemented with 10% fetal bovine serum, 100Uml/1 of penicillin G and 100 mg/ml/1 streptomycin and grown at 37°C in 5% CO2.
Silica Particles, antibodies, and reagents
Crystalline silica was isolated from environmentally relevant sources and characterized at the National Institute of Occupational Safety and Health (NIOSH, Pittsburgh PA) using a Multi-Cyclone Sampling Array (MCSA) as previously described [10, 17]. α-quartz; average size, 1.7 μm was also obtained from U.S. Silica (Berkeley Springs, WV). Particles were subjected to sedimentation, according to Stokes’ law, acid hydrolyzed and baked overnight (200°C, 16 h) to inactivate endotoxin contamination.
Primary antibodies: Anti-HIF-1α (cat.# NB100-449), anti-GAPDH load control (cat# MA5-15738), were obtained from Thermo Fisher; Total OXPHOS Rodent WB Antibody cocktail (cat#ab110413), anti-Ecsit antibody (cat# ab21288), from Abcam; anti IL-1β/IL-1F2 Antibody (cat# AF-201-NA) from R&D System Inc, anti-caspase-1 p10 (M-20) (cat# SC-514) and A/G plus agarose beads (cat# sc-2003) and normal mouse IgG (cat# sc-2025) from Santa Cruz Biotechnology, anti b-actin (cat# 4967) from Cell Signaling Technology, anti malonyl lysine (cat# PTM-901) from PTM Biolabs, anti-GAPDH (cat# MAB374) from EMO Millipore Corp USA.
Secondary antibodies: anti-goat IgG (cat# ab6741) from Abcam, Anti-mouse IgG HRP-linked antibody (cat#7076) and anti-rabbit IgG HRP linked antibody (cat# 7074S) from Cell Signaling; ATP Synthase (cat# MA1-930) and anti-mouse Alexa 488 (cat# A11029) from Invitrogen, Anti-rabbit Cy3 (cat# 111-164-144) from Jackson Immuno.
Lipopolysaccharides from Escherichia coli 0111:B4 (cat# L4391-1MG), L-Lactate Dehydrogenase (L-LDH) from rabbit muscle (cat# 10127230001), and Sodium L-Lactate (cat# 71718) were purchased from Millipore-sigma (St Louis, MO, USA).
Pierce™ RIPA Buffer (cat# PI89900), Halt™ Protease and Phosphatase Inhibitor and Propidium Iodide-1.0 mg/mL Solution in Water (cat# P3566) were from Thermo Fisher. The D-glucose (U-13C6, 99%, cat# CLM-1396-0) was purchased from Cambridge Isotope Laboratories (Andover, MA, USA). Annexin V-FITC Apoptosis Kit Plus (cat # K201-100) was purchased from Biovision.
Detection of cell death
Cell death was analyzed by evaluating (flow cytometry) Annexin V binding and the exclusion of Propidium iodide by macrophages, as previously described [18].
Lactate Dehydrogenase assay.
Lactate dehydrogenase (LDH) in the supernatant of exposed cells was measured using a commercial lactate dehydrogenase release assay (Sigma-Aldrich, TOX7) according to the manufacturer’s instructions. The absorbance was measured spectrophotometrically at a wavelength of 490 nm, using Cytation 5 Cell Imaging Multi-Mode Reader Biotek.
Lactate Assay.
Lactate concentration in the supernatant of exposed cells was assessed using a modification of the commercially available lactate assay (Sigma-Aldrich, TOX7). The reaction is based on the oxidation of lactate by lactate dehydrogenase (LDH). Cell supernatant was incubated with an equal volume of LDH (f.c. 1M), 1x LDH assay cofactor preparation, and LDH Assay Dye Solution (cat#L2277). Absorbance was acquired spectrophotometrically at a wavelength of 490 nm, using Cytation 5 Cell Imaging Multi-Mode Reader Biotek.
ETC Assays.
Respirometric Experiments.
An Oxygraph O2k Polarographic instrument (Oroboros Instruments, Innsbruck, Austria), equipped with a Clark-type electrode for high-resolution respirometry, was used to measure oxygen fluxes and concentrations. Dulbecco’s Modified Eagle’s medium (DMEM) without glucose (2.1 mL) was added to each chamber and equilibrated for 20 minutes before the addition of cell suspension (2.5 × 106cells/ml) (prepared as described above) into the Oxygraph chambers (sealed from the atmosphere) at 37°C. Oxygen turnover was examined by the addition of succinate (final concentration, f.c., 10 mM).
Substrate-uncoupler-inhibitor titration (SUIT) assay.
SUIT protocol was used for the analysis of oxidative phosphorylation, to study respiratory control in a sequence of coupling and pathway control states induced by multiple titrations. After equilibration of the cells into the chamber the substrates, inhibitors, or uncoupler were added according to the following protocol: rotenone (f.c. 0.5 μM) to inhibit complex I, succinate (f.c., 10 mM) to stimulate complex II, ADP (f.c. 1mM) to measure the total respiratory capacity, Carbonyl cyanide m-chlorophenyl hydrazone CCCP (f.c. 0.05 μM) to determine the state of coupling of complex III, IV, V, antimycin A (f.c. 2.5 μM) to inhibit complex III (Sigma Aldrich). Hydrogen Peroxidase (H2O2) flux was assessed simultaneously using the H2O2-sensitive probe Amplex UltraRed, by the addition of Amplex Red (f.c. 10 μM) and Horseradish-Peroxidase (HRP) (f.c. 1U/ml) (Sigma Aldrich) following the variation of the resorufin concentration while performing the SUIT assay protocol. Additions by gas-tight syringe into the sealed Oxygraph chambers. Respirometric data analysis was carried out with DatLab 7 software provided by Oroboros.
Complex I Assay
Mitochondrial Complex I activity was measured spectrophotometrically (DU-530; Beckman Coulter). Mitochondrial pellets were lysed in 0.05mM potassium/phosphate buffer (0.05mM, pH 8) containing NADH (10.0 mM), K3Fe(CN)6 (25mM), a phospholipid supplement (0.005% n-Dodecyl-β-d-Maltopyranoside from Anatrace), Antimycin A (2.5mM), and 320 μg protein from the cell pellet by following the oxidation of NADH at 420 nm initiated by K3Fe(CN)6 at 30–38 C. Data were acquired for 100 seconds after initiation of the reaction.
Complex II Assay
Mitochondrial Complex II activity was measured spectrophotometrically (DU-530; Beckman Coulter), using a modification of the commercially available Complex II Enzyme Activity Microplate assay Kit (colorimetric) (Abcam, cat# ab109908). Proteins were extracted in PBS and detergent, then lysed in complex II activity buffer, lipid/phospholipid mix, 2,6-dichloroindophenol (DCIP) and Ubiquinone-2, and succinate as provided by the kit and 300 μg protein from the cell pellet. The reaction was followed by measuring the decrease in absorbance at 600nm for 100 sec at room temperature. The activity of complex II was expressed as mAbs/min/μg protein.
Protein immunoprecipitation and western blotting.
A total of 10×106 cells/condition were lysed with 1 ml of ice-cold Pierce™ RIPA Buffer with protease inhibitors and disrupted by repeated aspiration through a 21 gauge needle. Protein concentration was measured using BCA protein assay (cat# 23225, Thermo Fisher) and normalized across samples prior to immunoprecipitation. 500 μl of lysate was pre-cleared with 1.0 μg of appropriate control IgG (normal mouse) and 20 μl A/G PLUS agarose, for 30 minutes at 4°C. For GAPDH immunoprecipitation, 100–500 μg of total cellular protein were incubated with 2 μg of primary antibody for 1 hour at 4°C, then added 20 μl A/G beads overnight and incubated overnight at 4°C on a rocker platform. Immunoprecipitates were collected by centrifugation at 1000xg for 5 minutes at 4°C, the liquid was removed and the pellets were washed 4 times with PBS. Immune complexes were eluted by adding 40 μl of 1x electrophoresis sample buffer, boiled for 5 minutes at 95°C, and analyzed by SDS-PAGE.
Protein samples from cultured cells were prepared by lysis with RIPA buffer with protease inhibitors. Protein concentration was determined using the BCA protein assay (cat# 23225, Thermo Fisher). 20–50 μg protein samples were separated by SDS-PAGE and transferred onto a PVDF membrane via iBlot (Invitrogen) transfer. Membranes were blocked in 5% non-fat milk and incubated with primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. Membranes were probed with the respective antibodies and visualized using SuperSignal West Femto Maximum sensitivity substrate (cat# 34095, Thermo Scientific) and Amersham Imager 600.
Immunofluorescence staining.
RAW 264.7 macrophages were plated on coverslips, allowed to grow overnight, and stimulated with LPS (1 ng/ml) or Silica (50 μg/cm2) with or without priming with LPS (1 ng/ml) for 24h. Cells were fixed with 2% paraformaldehyde in PBS for 15 minutes at 4 degrees. Cells were permeabilized with 0.1% Triton X-100 in PBS with .5% BSA for 15 min. Cells were blocked with 2% normal goat serum for 45 minutes and stained overnight for primaries for ECSIT (ab21288, Abcam) with ATP Synthase(MA1-930, Invitrogen) and HIF-1 alpha (NB100-449, Novus) with ATP Synthase. Corresponding secondary antibodies anti-rabbit Cy3 (111-164-144, Jackson Immuno) and anti-mouse Alexa 488 (A11029, Invitrogen) were added for one hour. Nuclei were stained with Hoechst (B2883, Sigma) 1mg/100ml dH20 for one minute, washed in PBS, and mounted in gelvatol. Large area scan images were obtained on Nikon A1 confocal microscope with NIS Elements v4.4 at 60x magnification.
Isolation of mitochondria.
Mitochondria were isolated from control, silica, or silica plus LPS stimulated RAW 264.7 macrophages using a Mitochondria Isolation Kit for Cultured Cells (cat# 89874, Thermo Fisher) following manufacturer instructions.
Enzyme-Linked Immunosorbents Assay
Cytokines concentration in cell supernatants were measured using ELISA kits for mouse IL-1β, TNF-α and IFN-β, according to the manufacturer’s instruction, Mouse IL-1β ELISA MAX™ Standard (cat# 432601) or Mouse TNF-α ELISA MAX™ Standard (cat#430902), both purchased by Biolegend. Mouse IFN-beta DuoSet ELISA (cat#DY8234-05) was purchased by Bio-Techne. Optical density values were measured at a wavelength of 450 nm, using Cytation 5 Cell Imaging Multi-Mode Reader Biotek.
RNA analysis.
Total cellular RNA was isolated from cells using TRIZOL reagent (Invitrogen) following the manufacturer’s instructions. The concentration of total RNA was quantified at an absorbance of 260nm. Real-time qPCR was performed on RNA by an RT-PCR using TaqMan RNA-to Ct 1-Step Kit (Applied Biosystems), according to the manufacturer’s instructions, using 500 ng of RNA as a starting material, in a total reaction volume of 10 μL PCR conditions were following: 15 min 48°C, 10 min 95°C, followed by 40 cycles with 15 sec at 95°C and 1 min at 60°C in the ABI 7300 real-time PCR system. The qRT-PCR was performed on RNA using probes specific for TNF-α (IDMm00443258_m1), IL-1β (ID Mm00434228_m1), and IFN-β (Mm00439552_s1) (Thermo Fisher). The GUSB (ID Mm01197698_m1) or GAPDH (Mm9999915_g1) (Thermo Fisher) was used for normalization; fold change was calculated using the equation described in the last. The ΔCt was calculated subtracted the Ct of GUSB or GAPDH from the Ct of the gene of interest. ΔΔCt was calculated by subtracting the ΔCt of the reference sample from ΔCt of Control sample. Fold change was generated using the equation 2−ΔΔ.
Metabolic assay.
Stable isotope labeling.
13C tracer analysis and high-resolution mass spectrometry were used to discern subtle shifts in energy substrate metabolism in specific reactions through the detection and quantification of metabolite isotopologues. RAW 264.7 macrophages (1×106/well 6-wells plate) were activated with LPS (10 ng/ml) or silica (50 μg/cm2) dissolved in DMEM without glucose, supplemented with 12C- or U13C-labeled glucose (4.5 mg/l), 5% Fetal Bovine Serum dialyzed. Experiments were conducted n = 6/condition. Controls included vehicle control and the 12C glucose control for each treatment condition.
Untargeted liquid chromatography-high resolution mass spectrometry (LC-HRMS). Sample preparation.
Metabolic quenching and polar metabolite extraction were performed using ice-cold 80% methanol/0.1% formic acid at a ratio of 500 μL per 1×106 adherent cells. An internal standard mix containing (D3)-creatinine and (D3)-alanine, (D4)-taurine, and (D3)-lactate (Sigma-Aldrich) was added to the sample lysates at a final concentration of 100 μM. After 3 minutes of vortexing, the supernatant was cleared of protein by centrifugation at 16,000 × g. Cleared supernatant (3 μL) was subjected to online separation and analysis. LC-HRMS Method. Analyses were performed by untargeted LC-HRMS. Briefly, samples were injected via a Thermo Vanquish UHPLC and separated over a reversed-phase Thermo HyperCarb porous graphite column (2.1×100mm, 3μm particle size) maintained at 55°C. For the 20 minute LC gradient, the mobile phase consisted of the following: solvent A (water/0.1% FA) and solvent B (ACN/0.1% FA). The gradient was the following: 0–1min 1% B, increase to 15%B over 5 minutes, increasing to 98%B over 5 minutes, and held at 98%B for five minutes before equilibration to starting conditions. The Thermo ID-X tribrid mass spectrometer was operated in positive and negative ion mode, scanning in Full MS mode (2 μscans) from 100 to 800 m/z at 70,000 resolution with an AGC target of 2 × 105. Source ionization setting was 3.0 kV/2.4kV spray voltage for positive and negative mode, respectively. Source gas parameters were 45 sheath gas, 12 auxiliary gas at 320°C, and 8 sweep gas. Calibration was performed prior to analysis using the PierceTM FlexMix Ion Calibration Solution (Thermo Fisher Scientific). Alignment and peak area integration were then extracted manually using Quan Browser (Thermo Fisher Xcalibur ver. 2.7). Atomic percent enrichment was calculated using the established Mass isotopomer multi-ordinate spectral analysis (MIMOSA) method to remove the natural 13C abundance background [19].
Transmission electron microscopy (TEM) analysis
At one, two, and four hours after exposure, cells were fixed in 2.5% glutaraldehyde in phosphate-buffered saline (PBS) and post-fixed in 1% osmium tetroxide in PBS, dehydrated through a graded series of alcohols, and embedded in Epon (Energy Beam Sciences, Agawam, MA). Thin (70-nm) sections were cut using a Reichert Ultracut S (Leica, Deerborn, MI), mounted on 200-mesh copper grids, and counterstained with 2% aqueous uranyl acetate for seven minutes and 1% aqueous lead citrate for two minutes. Observation was with a JEOL 1011 transmission electron microscope (Peabody, MA). After TEM images were collected, they were formatted using Adobe Photoshop for brightness and contrast. In addition, during slide preparation, a silica particle could create stretching in the epon resin during the slicing sequence, and when stretching was severe the silica particle could fall out of the resin. During TEM imaging, any areas where the silica particles fell out will show as bright white, causing difficulty in image focusing. These images were corrected by recoloring the white areas back to the color of the silica particles. Importantly, this may result in a slight increase in the silica particle size for the areas that were recolored. In the images at higher magnification, these areas are labeled.
Statistics.
The results are presented as mean ± SD from at least three experiments, and statistical analyses were performed using the Student t-test or one-way ANOVA corrected for multiple comparisons, using Prism software (version 7, GraphPad Software Inc). The statistical significance of differences was set at p<0.05.
RESULTS
Crystalline silica and low dose LPS enhance the glycolysis without affecting macrophage viability.
To study the effect of silica on glycolysis, we utilized RAW 264.7 macrophages, which we have previously shown release high concentrations of TNF-α and IL-1β in response to silica while experiencing low cytotoxicity to a wide range of concentrations of silica particles [8]. Using these macrophages, we show that both low dose LPS (1 ng/ml) that did not alter macrophage viability, as well as high dose concentrations (10 ng/ml), that induced a significant amount of cell necrosis with associated release of lactate dehydrogenase (LDH), were as effective as non-cytotoxic doses of silica (ranging from 10–50 μg/cm2) in promoting the release of significant amount of lactate in RAW 264.7 macrophages, over a period of 24h (Figure 1A). Addition of silica to low dose LPS-primed RAW 264.7 macrophages induced a significantly greater increase in the release of lactate compared to cells treated with silica or LPS alone (Figures 1B, C). Under these experimental conditions, RAW 264.7 macrophages exposed to silica (50 μg/cm2) or LPS (10 ng/ml) alone experienced low levels of necrosis, and less than 30% of the cells exhibited propidium iodide uptake 24h after exposure (Figure 1D, E). In contrast, the combination of silica and LPS induced significant necrosis, with 70% of the cells up taking propidium iodine 24h after exposure (Figure 1D, E).
Figure 1. Crystalline silica and low dose LPS enhance glycolysis without affecting macrophage viability.
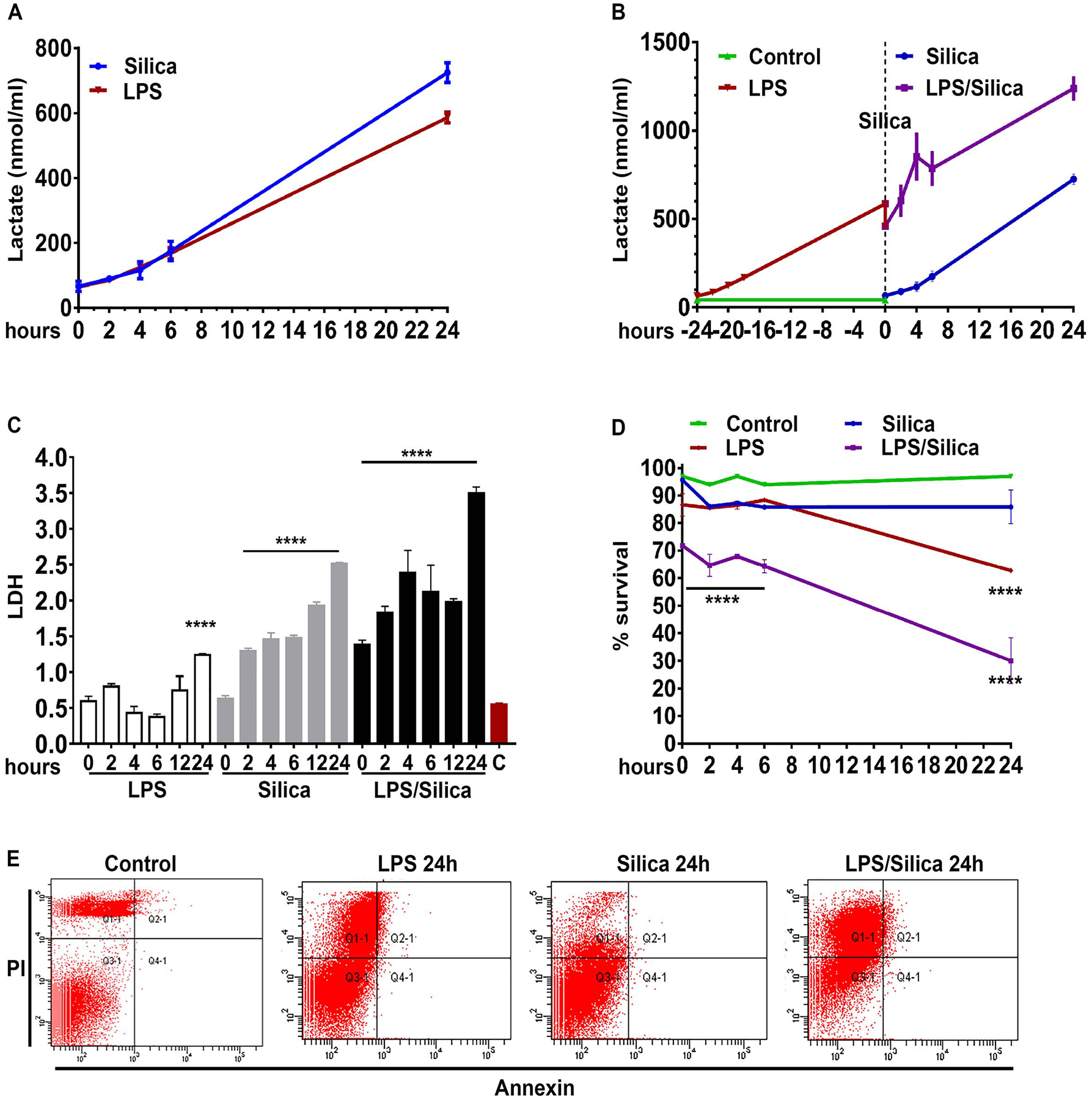
RAW 264.7 macrophages were exposed to LPS (1 ng/ml) or silica (50 μg/cm2) or primed with LPS (1 ng/ml for 24h) and then exposed to silica (50 μg/cm2) and studied at time points of T0, 2h, 4h, 6h, 24h. The cell supernatants were examined for the concentration of released (A–B) lactate and (C) LDH. (D–E) Annexin V and Propidium iodide were used to analyze cell survival by flow cytometry: (D) graph summarizing the survival of cells treated with LPS (10 ng/ml) or silica (50 μg/cm2) or primed with LPS (1 ng/ml for 24h) and then exposed to silica (50 μg/cm2). (E) Gating strategy to differentiate cell death by early apoptosis (Annexin V+PI−), late apoptosis (Annexin V+PI+), or necrosis (Annexin V−/PI). Data were normalized by equalizing the protein concentration or equating the actual number of live cells used to perform the experiments. N=3 **** p<0.0001
Silica remodels ETC-complexes activity.
In response to live bacteria, but not LPS, macrophages experience mitochondrial respiratory change adaptations that contribute to their anti-microbial response [12]. These effects require the phagocytosis of bacteria into a phagosome, are metabolically demanding, and contribute to cytokine production [12]. Similar to bacteria, silica particles are phagocytosed by macrophages into phagosomes and attract mitochondria to these organelles (Supplemental Figure 1) [10]. Using a high-resolution respirometer, we evaluated the effects of silica on mitochondrial respiration as well as the activity, and integrity of the ETC in RAW 264.7 macrophages. To evaluate the contribution of CII in the respiration of silica-activated macrophages, LPS or silica-exposed RAW 264.7 macrophages were stimulated with succinate, the CII substrate, and the oxygen flux was subsequently recorded. When healthy untreated cells were stimulated with succinate, the oxygen flux through the ETC physiologically increased (Figure 2A). This physiologic response, identified 2h after exposure, was significantly augmented in cells treated with silica and, although it peaked 6h after exposure when the oxygen flux increased threefold, it lasted for the 24 h experiment (2h 2.265 ± 0.04950 p<0.0001; 4h 2.265 ± 0.03536 p<0.0001; 6h 2.850 ± 0.7212 p<0.0001; 24h 2.095 ± 0.1344 p<0.0001). In contrast, RAW 264.7 macrophages exposed to LPS only showed a modest, although significant, and transitory, achieving maximal response by 6h (1.929 ± 0.06757, p<0.01), increase in mitochondrial respiration in response to succinate, even in the presence of silica (Figure 2B). LPS/silica treatment induced a statistically significant increase in oxygen consumption only after 4–6 h after exposure (4h 1.970 ± 0.08485 p<0.01; 6h 1.905 ± 0.04950 p<0.01) (Figure 2B). The comparison between LPS and silica treatment at every time point during the study, confirmed that silica-induced a statistically significantly greater increase oxygen consumption at 2h (1.486 ± 0.0005459 LPS vs 2.265 ± 0.04950 silica, p<0.01), 4h (1.634 ± 0.08633 LPS vs 2.265 ± 0.03536 silica p<0.05), 6h (1.929 ± 0.06757 LPS vs 2.850 ± 0.7212 silica p<0.001), 24h (1.144 ± 0.05383 LPS vs 2.095 ± 0.1344 silica, p<0.0001). Compared to silica treatments, priming with LPS, induced a statistically significant increase in oxygen turnover at 2h (2.265 ± 0.04950 silica vs 1.670 ± 0.08485 LPS/Silica, p<0.05), 6h (2.850 ± 0.7212 silica vs 1.905 ± 0.04950 LPS/Silica, p<0.001) and 24h (2.095 ± 0.1344 silica vs 1.310 ± 0.01414 LPS/Silica, p<0.01) (Figure 2B).
Figure 2. Silica remodels ETC-complexes activity.
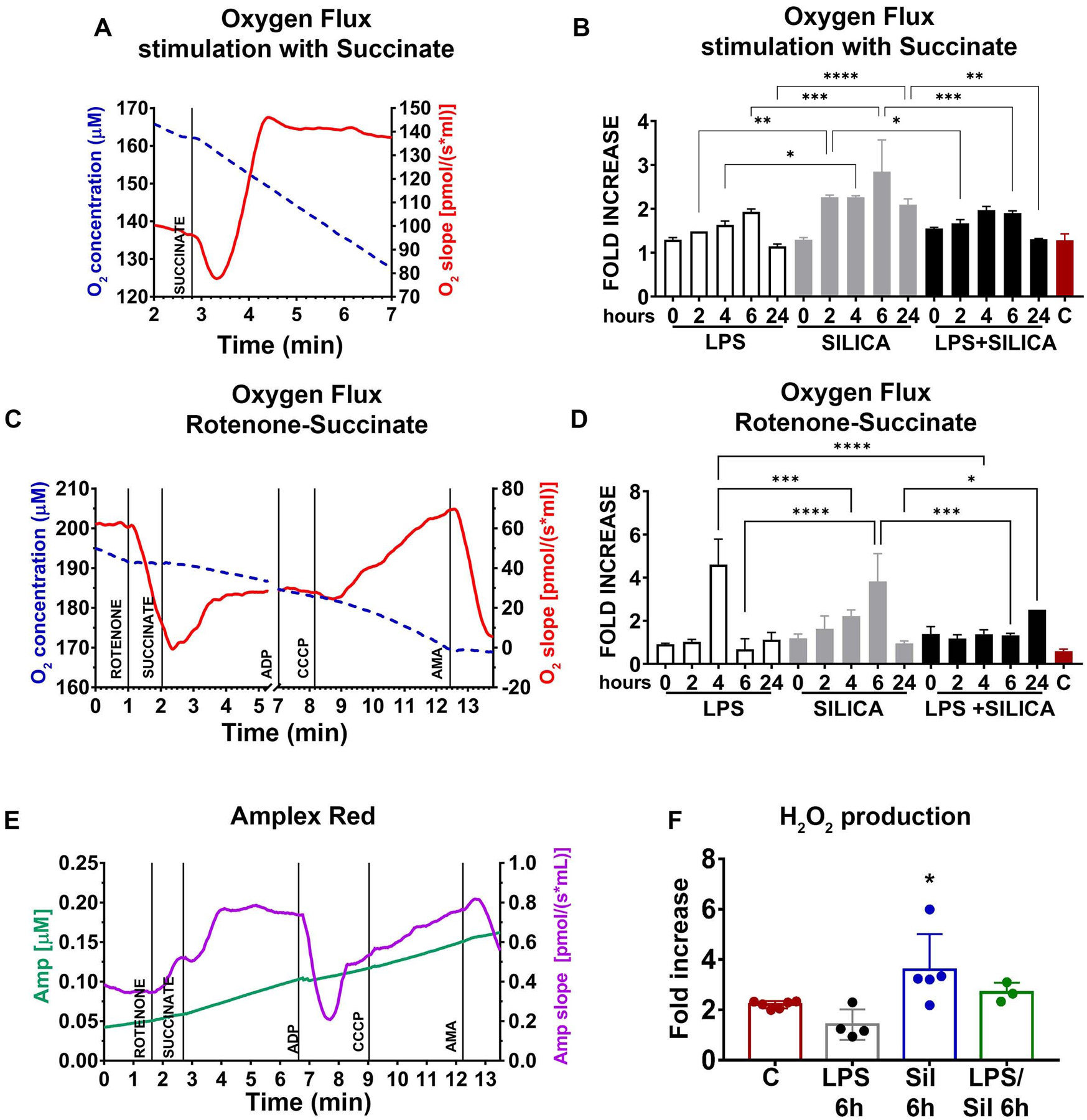
RAW 264.7 macrophages were exposed to LPS (1 ng/ml) or silica (50 μg/cm2) or primed with LPS (1 ng/ml for 24h) and then exposed to silica (50 μg/cm2) at time points of T0, 2h, 4h, 6h, 24h. RAW 264.7 macrophages (5×106/chamber) were allowed to equilibrate in the chamber for about 10 minutes prior to measuring oxygen flow. Default respirometric settings of block temperature 37°C; stir bar speed 400 rpm, and data recording every 2s were used. Oxygen turnover (oxygen concentration, left y-axis, oxygen flux, right y-axis) by treated RAW 264.7 macrophages was followed after addition of (A) succinate (final concentration 10 mM) or (C) rotenone (f.c. 0.5 μM), succinate (f.c. 10 mM), ADP (f.c. 2.5 mM), CCCP (f.c. 0.05 μM) and antimycin A (f.c. 2.5 μM). After the addition of each compound, cells were allowed to come to equilibrium. [Since the variation in oxygen concentration (broken traces) are difficult to appreciate because it is very little, the slope of the oxygen concentration is also shown (solid trace), which represents the negative derivative of the oxygen concentration]. Variations in oxygen flux for each condition were measured as a ratio between the value of the O2 slope at the equilibrium after treatment with succinate (with or without prior inhibition of complex I with rotenone), and the value of O2 slope at the baseline. (B) Fold increase of oxygen flux after stimulation with succinate. (D) Fold increase of oxygen flux after the inhibition of CI with rotenone and stimulation of CII with succinate. (E) The production of H2O2 has been assessed following the slope (purple trace) of the concentration of the fluorescent Amplex Red assay product resorufin (green trace) while performing a SUIT assay protocol (see Experimental section). Variations in H2O2 for each condition were measured as the ratio between the value of the slope at the equilibrium after the inhibition of CI with rotenone and the stimulation of CII with succinate, the slope at the equilibrium at the baseline (F) Fold increase of H2O2 production after inhibition of CI with rotenone and stimulation of CII with succinate. N=4 * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001, versus control.
To assess the production of ROS and oxygen consumption by CII in the forward direction (toward complex V, ATPase enzyme), we performed a substrate-uncoupler-inhibitor titration (SUIT) assay, wherein pre-treated RAW 264.7 macrophages were incubated with CI inhibitor (rotenone), before stimulation with CII substrate succinate, (see Experimental section). The SUIT assay performed on resting cells showed a decrease in O2 flux after inhibition of CI, followed by a slight recovery of the O2 consumption and flux resulting from stimulation of CII with succinate. Subsequently, the uncoupler increased the O2 slope above the baseline, while antimycin A, blocking CIII, cytochrome c reductase, and suppressed mitochondrial respiration (Figure 2C). The sequential treatment of silica exposed RAW 264.7 macrophages with CI inhibitor and CII substrate confirmed the aforementioned synergistic effect of silica on the enzymatic effect of CII as these cells exhibited a significant time-dependent increase of O2 consumption, that although identified at 2h, it peaked at 4–6h exposure (4h 2.221 ± 0.2787 p<0.01; 6h 3.824 ± 1.286 p<0.0001), proving a substantial rise in respiration only through complex II (Figure 2D). LPS induced a statistically significant increase of oxygen flux only at 4h (4.605 ± 1.176 p<0.0001), while priming with LPS before silica exposure induced an enhancement of oxygen turnover only after 24h (2.514 p<0.001) (Figure 2D).
The comparison between LPS and silica with or without LPS-priming at every time point during the study, confirmed that LPS increases the oxygen flux at 4h (4.605 ± 1.176 LPS vs 2.221 ± 0.2787 silica p<0.001; 4.605 ± 1.176 LPS vs 1.375 ± 0.2051 LPS/silica p<0.0001) (Figure 2D). However, silica increased the oxygen flux at 6h compared to LPS and LPS/silica (0.6788 ± 0.4884 LPS vs 3.824 ± 1.286 silica p<0.0001; 1.320 ± 0.09899 LPS/Silica vs 3.824 ± 1.286 silica p<0.001), while priming with LPS before silica increased the oxygen flux after 24h co-exposure compared to 24h silica exposure(0.9500 ± 0.1131 silica vs 2.514 LPS/Silica, p<0.05) (Figure 2D).
The integrity of the mitochondrial membrane was also assessed by the addition of the uncoupler. Silica itself did not uncouple the ETC even in the presence of priming, as shown by the increase of O2 flux after the addition of CCCP (Supplemental Figure 2). In contrast, LPS markedly damaged the mitochondrial membrane, as shown by the absence of alteration in the O2 curve after the addition of CCCP (Supplemental Figure 2).
Subsequently, we then examined the CII-mediated production of hydrogen peroxide (H2O2) by Amplex Red while subjecting the RAW 264.7 macrophages to the SUIT assay protocol. CI inhibition followed by CII stimulation in resting RAW 264.7 macrophages caused an immediate increment of H2O2 generation, similar to the one observed above during the proton leak state. Addition of ADP, inducing CII-linked oxidative phosphorylation (OXPHOS), reduced H2O2 production. Furthermore, the uncoupling of the system rebooted the production of H2O2 that was ultimately suppressed by CIII inhibition (Figure 2E). Compared to LPS treated cells, silica-exposed RAW 264.7 macrophages showed significantly higher production of H2O2 6h after silica exposure, confirming the increased O2 consumption through CII (Figure 2F). These findings suggest that silica itself acts within the cells to enhance the activity of mitochondria complex II.
Silica inhibits CI activity in part by reducing ECSIT expression.
In addition to the high-resolution respirometry, we measured, via the enzymatic assays, the individual activity of CI and CII of silica exposed RAW 264.7 macrophages. Previously, we have shown that in RAW 264.7 macrophages, silica induces a time-dependent decrease in the expression of 39-kDa CI subunit, and subsequently, CI enzymatic activity was downregulated [8]. In agreement with our previous data, we found that silica downregulates CI activity significantly in a time-dependent fashion with a lower activity after 6h exposure, while the non-toxic concentration of LPS (1 ng/ml) did not interfere with the enzyme function of this mitochondrial complex (Figure 3A). No difference was identified in CII activity between LPS and/or silica exposed macrophages (average mean 0.446 ± 0.0325) at baseline (Figure 3B). Compared to control-treated cells at baseline (baseline 0.4465 ± 0.0325), LPS induced a transient, but non-statistically significant difference in enzymatic activity of complex II, observed at 4–18h after exposure. In contrast, silica significantly enhanced the enzymatic activity of CII as early as 2h (0.622± 0.0492 p<0.01) (Figure 3B). This statistically significant difference persisted during the 24h exposure time as follows: 4h 0.667 ± 0.110 p<0.001; 6h 0.659 ± 0.0692 p<0.001; 18h 0.627 ± 0.0609 p<0.001, and 24h 0.639 ± 0.101 p<0.001 (Figure 3B). The comparison between LPS and silica treatment at every time point during the study, confirmed that silica-induced a statistically significantly greater increase of CII activity at 2h (0.482 ± 0.0448 LPS vs 0.622± 0.0492 silica, p<0.05), 6h (0.527 ± 0.0371 LPS vs 0.659 ± 0.0692 silica p<0.05), 18h (0.502 ± 0.0853 LPS vs 0.627 ± 0.0609 silica, p<0.05), and 24h (0.422 ± 0.0696 LPS vs 0.639 ± 0.101 silica, p<0.001) (Figure 3B). The difference between these groups at 4h was noted but did not achieve statistical significance.
Figure 3. Silica inhibits CI activity in part by reducing Ecsit expression.

RAW 264.7 macrophages were exposed to LPS (1 ng/ml) or Silica (50 μg/cm2) or primed with LPS (1 ng/ml for 24h) and then exposed to Silica (50 μg/cm2) and examined as a function of time 0 to 24h. (A, B). Mitochondria pellets were isolated from RAW 264.7 macrophages, and complex I and complex II enzymatic activity were analyzed spectrophotometrically and measured as mAbs/min/μg proteins (See Experimental section). n=4, * p<0.05, ** p<0.01, *** p< 0.001, **** p<0.0001 LPS vs silica, ## p<0.01. ### p<0.001 versus baseline. (C, D) Mitochondria were isolated from RAW 264.7 macrophages exposed to LPS (C) or silica (D) for the indicated times, and Ecsit (37 kDa subunit) abundance was determined by western blot while using Complex IV subunit MTCO1 as markers for mitochondria and GAPDH expression as a loading control. (E) Confocal microscopy of RAW 264.7 macrophages stimulated for 4 hours with LPS (1 ng/ml) or Silica (50 μg/cm2) in the presence or absence of LPS priming. Mitochondria are visualized by staining with ATPase (red), DNA into the nuclei of cells with Hoechst (blue), followed by the analysis of Ecsit (green). Scale bar 10 microns. (F) Graph illustrating the abundance of ESCIT in LPS or silica exposed RAW 264.7 macrophages. Data, expressed as the mean ± SEM, is representative of three different experiments.
Compared to LPS or silica treatments, priming with LPS prior to silica exposure, induced a statistically significant increase in enzymatic activity of CII only 18h (0.606 ±0.0188 LPS/silica vs 0.4465 ± 0.0325 baselines, p< 0.01) and 24h post-exposure (0.628 ± 0.0456 LPS/silica vs 0.4465 ± 0.0325 at baseline p<0.001) (Figure 3B). These findings reproduced the results of the respirometry assay, confirming a key role of CII in the mitochondrial response of macrophages to silica.
CI is the largest of the five ETC complexes in the mitochondrial membrane; it consists of 44 core and accessory subunits [20–22]. Among the 14 factors involved in this assembly process, Evolutionarily Conserved Signaling Intermediate In Toll Pathway (Ecsit) has a key role in the regulation of the stability and activity of the CI and subsequently the mitochondrial function [23]. To understand the mechanism that drives the decreased enzymatic activity of CI in silica-exposed RAW 264.7 macrophages, we studied the expression of Ecsit in the mitochondria of LPS or silica-exposed cells by western blotting. In contrast to LPS which enhances Ecsit expression as a function of time (Figure 3C), mitochondrial protein derived from silica-exposed cells showed a time-dependent decreased abundance of Ecsit 6h after silica exposure (Figure 3D). This finding was confirmed by direct visualization of the protein using immunofluorescence, wherein cells treated with silica with or without LPS priming exhibit a significantly decreased content of Ecsit overtime with a peak after 4 h exposure (Figure 3E–F). In contrast, cells treated with LPS did not show a substantial decrease in Ecsit protein and demonstrated increased expression of Ecsit after 4 hours exposure (Figure 3E–F).
The importance of mitochondrial Complex II activity on macrophage survival to silica.
Macrophages differ greatly in their response to silica, and previously we reported that the RAW 264.7 and IC-21 macrophage cell lines, which reproduce the effects of silica on primary mouse macrophages derived from BALB/c and C57BL/6J mice, respectively [18, 24], differ significantly in their viability and cytokine response to silica [8]. Thus, while RAW 264.7 macrophages release TNF-α and exhibit a low level of cell cytotoxicity in response to silica, IC-21 macrophages do not release TNF-α and experience universal cell death, even at low concentrations of silica particles [8]. Therefore, to better understand the role of mitochondrial complex II on macrophage survival, we compared the effect of silica particles on the enzymatic activity of complex II on RAW 264.7 and IC-21 macrophages.
Our studies on cell viability demonstrated that while silica-induced significant cell cytotoxicity and LDH release in IC-21 macrophages, LPS does not significantly impact these parameters 6–24 h after exposure (Figure 4A, B). Importantly, the addition of silica to IC-21 macrophages primed with a non-toxic concentration of LPS resulted in a synergistic effect that reduced the cell survival to 5% at 24h post-exposure (Figure 4A, B).
Figure 4. The importance of mitochondrial Complex II activity on macrophage survival in response to silica.
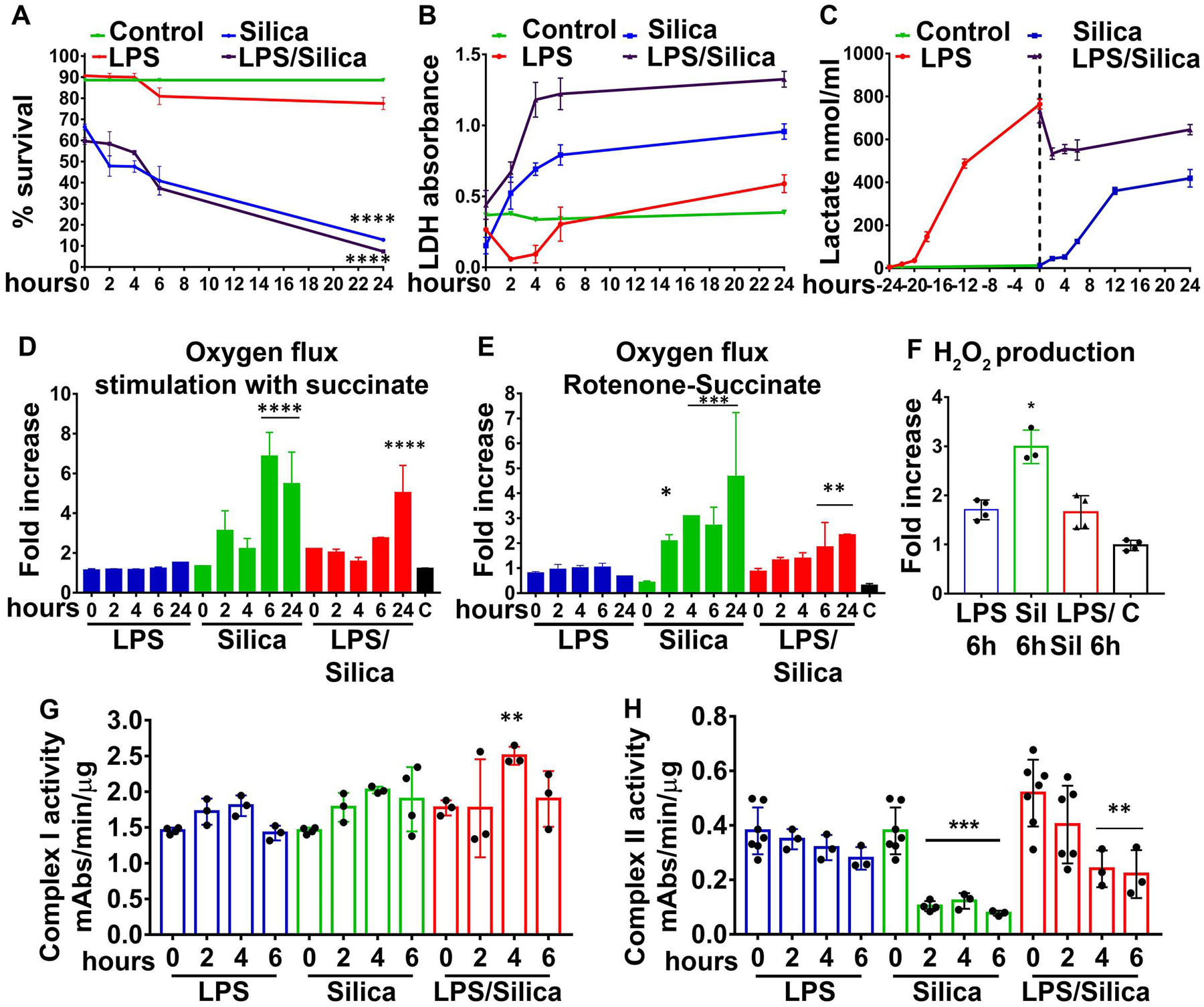
IC-21 macrophages were exposed to LPS (1 ng/ml) or Silica (50 μg/cm2), or primed with LPS (1ng/ml for 24h) and then exposed to Silica (50 μg/cm2) for up to 24h. (A) Annexin V and Propidium iodide were used to analyze cell survival by flow cytometry: graph summarizing the survival of cells treated as above. (B, C) The supernatants of cells were examined for released LDH and Lactate. (D–F) Respirometric analysis: IC-21 macrophages (2.5×106/chamber) were allowed to equilibrate in the chamber for about 10 minutes prior to measuring oxygen flow. Default respirometric settings of block temperature 37°C; stir bar speed 400 rpm, and data recording every 2s were used. Oxygen turnover by treated IC-21 macrophages was followed after addition of (D) succinate 10 mM or (E) Rotenone (f.c. 0.5 μM) succinate (f.c. 10 mM), ADP (f.c. 2.5 mM), CCCP (f.c. 0.05 μM) and antimycin A (f.c. 2.5 μM). After the addition of each compound, cells were allowed to come to equilibrium. Variations in oxygen flux for each condition were measured as a ratio between the value of the O2 slope at the equilibrium after treatment with succinate (with or without previous treatment with rotenone), and the value of O2 slope at the baseline. (D) Fold increase of oxygen flux after stimulation with succinate. (E) Fold increase of oxygen flux after the inhibition of CI with rotenone and stimulation of CII with succinate. (F) The production of H2O2 has been assessed following the concentration of the fluorescent Amplex Red assay product resorufin while performing a SUIT assay protocol. Variations in H2O2 for each condition were measured as the ratio between the value of the slope at the equilibrium after the inhibition of CI with rotenone and the stimulation of CII with succinate and the value of the slope at the baseline. N=3 * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001, versus control. (G, H) Mitochondria pellets were isolated from IC-21 macrophages, and CI and CII enzymatic activity were measured spectrophotometrically. and measured as mAbs/min/μg proteins N=3 **** p<0.0001 versus control.
Under these experimental conditions, LPS-stimulated IC-21 macrophages enhanced the release of lactate to a greater extent than IC-21 macrophages exposed to silica (Figure 4C). In addition, in contrast to the data described above for RAW 264.7 macrophages, stimulation of CII with succinate, in the presence or absence of rotenone-inhibition of CI, does not affect the oxygen flux, nor the mitochondrial ROS production in LPS treated IC-21 macrophages (Figure 4D, E, F). In contrast to LPS, silica particles induced significant increases in the oxygen flux and mitochondrial ROS production in IC-21 macrophages (Figure 4D, E, F) suggesting that the enzymatic activity of CI was not affected by silica exposure on IC-21 macrophages (Figure 4G). Conversely, CII activity was significantly inhibited in IC-21 macrophages by silica alone as early as 2 h post-exposure, even after LPS priming (Figure 4H)
Silica and LPS exert similar effects on glucose uptake and glycolysis but differ on the effects on the TCA cycle in macrophages.
LPS-stimulated macrophages exhibit immuno-metabolic regulation leading to perturbations of glycolysis and the TCA cycle that contribute to cytokine specification [11, 25–29]. To investigate the effects of silica on glucose metabolism and the TCA cycle activity of RAW 264.7 macrophages, we fed cells with 13C6-glucose, and conducted a stable isotope tracer analysis using high-resolution mass spectrometry to discern subtle shifts in energy substrate metabolism, through the detection and quantification of metabolite isotopologues from control, non-cytotoxic dose of silica (50 μg/cm2), or LPS (10 ng/ml) exposed macrophages (Supplemental figure 3). Our LC-HRMS analysis revealed that when compared to control cells, both LPS, as well as silica, induce an increase in the uptake and phosphorylation of glucose, as illustrated by intracellular enrichment in glucose-6-phosphate and glyceraldehyde-3-phosphate (Figures 5A–C). Isotopologues analysis shows that compared to control cells, both silica-, as well as LPS-exposed RAW 264.7 macrophages exhibit greater, but comparable, enrichment in intracellular levels of pyruvate and lactate (Figure 5D–F). However, LPS treated RAW 264.7 macrophages demonstrate a greater ratio (0.38) of conversion of pyruvate into lactate when compared to control (0.29) or silica exposed (0.33) macrophages leading to a greater intracellular (Figures 1&5) and extracellular (supplemental Figure 4) enrichment in lactate.
Figure 5. Silica and LPS exert similar effects on glucose uptake and glycolysis in macrophages.
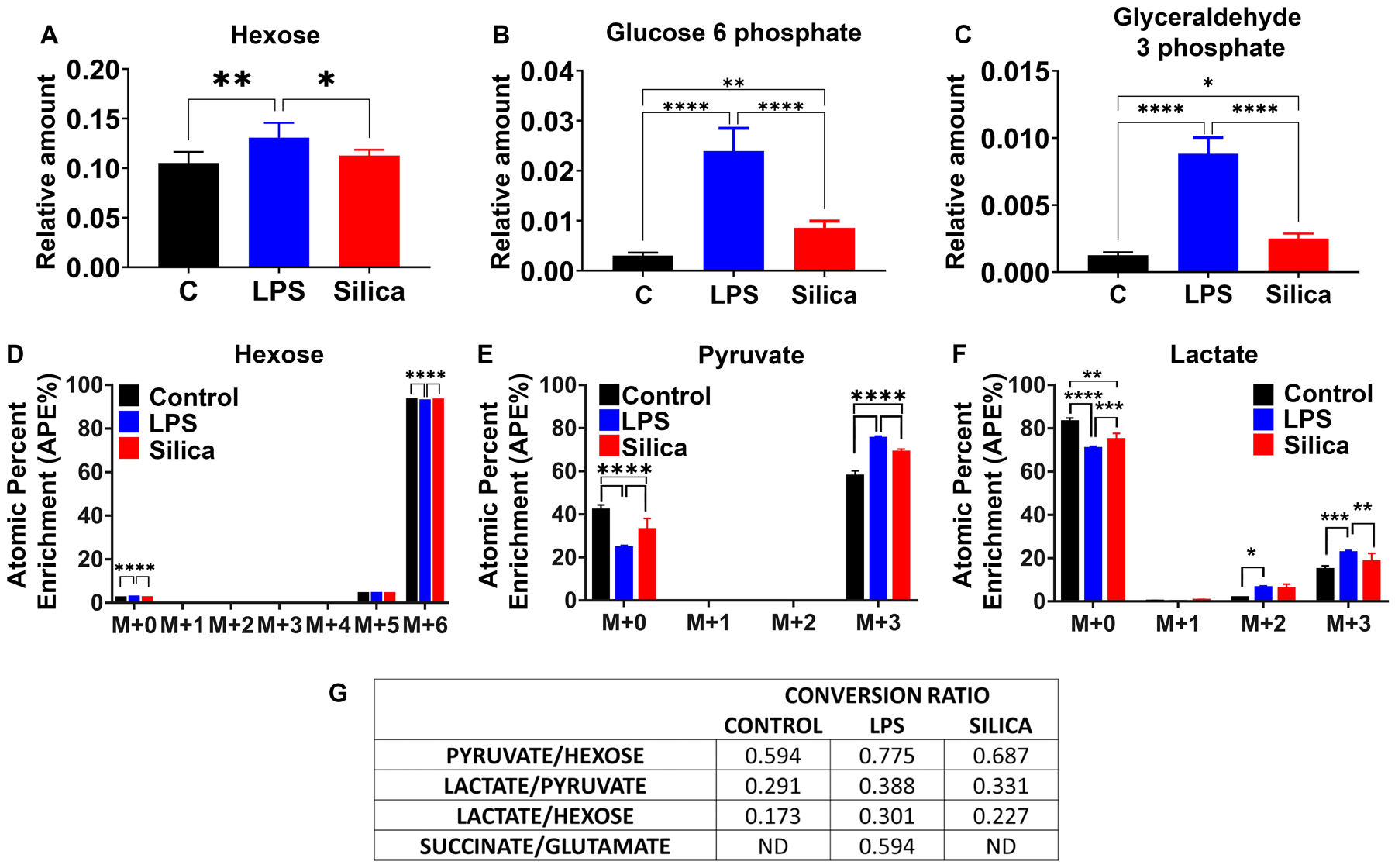
RAW 264.7 macrophages were treated with LPS (10 ng/ml) or Silica (50 μg/cm2) or left untreated for 6 h. (A–C) Intracellular relative amount of hexose uptake and glycolytic metabolites glucose-6-phosphate and glyceraldehyde-3-phosphate. (D–F) 13C6 uptake and intracellular pyruvate and lactate enrichment were determined by LC-HRMS. Atomic percent enrichment was calculated using the Mass isotopomer multi-ordinate spectral analysis (MIMOSA) method. Data are the mean ± SEM of 6 samples N=6 * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001, control vs LPS vs Silica. (G) Table representing the conversion ratio of selected metabolites calculated dividing the total enrichment (sum of absolute number M+1 to M+6) of the product by the total enrichment of the reactant.
Compared to control or silica-treated RAW 264.7 macrophages, LPS induces an increase in the intracellular relative amount of the TCA cycle intermediates glutamine, succinate, fumarate, malate, and itaconate while maintaining the levels of citrate, alpha-ketoglutarate, and glutamate (Figure 6A–H). In addition, LPS stimulated cells show enrichment of glutamine, proline, and arginine, derived from these intermediates (Figure 6G, I, L). In contrast, to control or LPS-exposed macrophages, intracellular levels of citrate are not detectable in silica-exposed macrophages (Figure 6A), and these silica-exposed macrophages exhibit significant reduction, compared to control or LPS-stimulated cells, in the intracellular levels of alpha-ketoglutarate, itaconate, succinate, fumarate, malate, glutamine, and glutamate (Figure 6A–H).
Figure 6. Silica and LPS affect TCA cycle in differential manner.
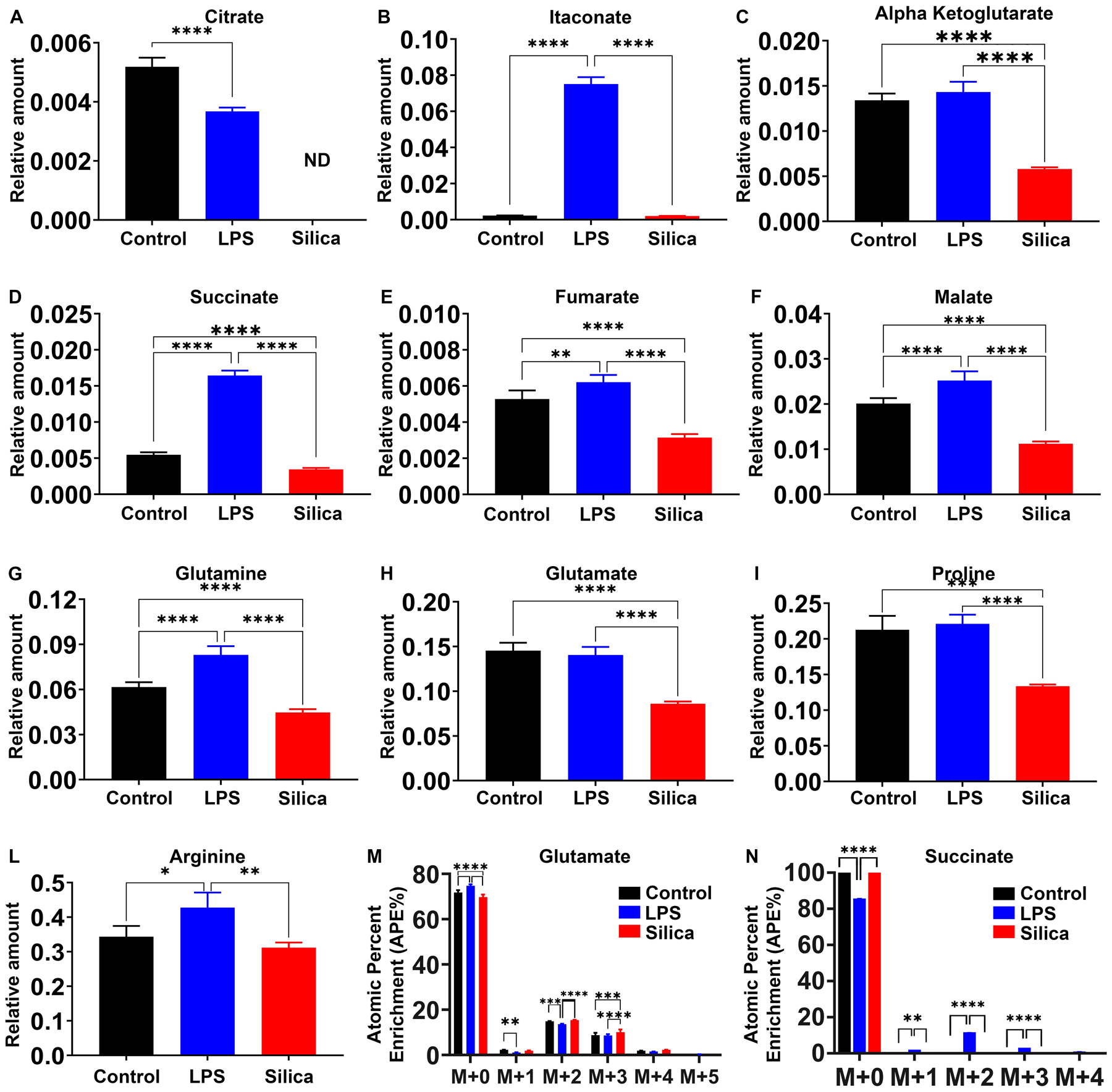
RAW 264.7 macrophages were treated with LPS (10 ng/ml) or Silica (50 μg/cm2) or left untreated for 6 h. (A–L) Intracellular relative amount of TCA cycle (A–F) metabolites and (G–L) amino acids. (M, N) Changes in succinate and glutamate atomic percent enrichment at steady state determined by LC-HRMS. Atomic percent enrichment was calculated using the Mass Isotopomer Multi-Ordinate Spectral Analysis (MIMOSA) method. Data are the mean ± SEM of 6 samples N=6 * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001, control vs LPS vs silica.
Our data show that both LPS, as well as silica, influence the enzymatic activity of SDH, an enzyme that plays a fundamental role in both glycolysis and ETC (Figure 2D and 3B). Glutamine-dependent anapleurosis is the principal source of succinate, although the ‘GABA (γ-aminobutyric acid) shunt’ pathway also has a role in LPS-stimulated macrophages. Therefore, we measured the conversion of glutamate into succinate. LPS treated macrophages show increased rates (0.59) of glutamate conversion to succinate (Figure 5G). This conversion was not significantly altered by silica, and isotopologues analysis showed that while the baseline glutamate enrichment was comparable among control, LPS-, or silica- treated cells (M+0 to M+4); a higher enrichment of succinate was detected at M+2 only in LPS-exposed macrophages (Figure 6M, N).
LPS, but not Silica exposure, induces stabilization of HIF-1α, activation of NLRP3 inflammasome, and release of IL-1β.
As we described in the introduction, LPS-stimulated macrophages exhibit altered intracellular metabolic changes that specify immune effector mechanisms such as cytokine secretion. To determine whether these changes also take place in silica exposed macrophages we correlated the metabolic changes observed in RAW 264.7 macrophages in response to silica with the production of IL-1β, TNF-α, and INF-β, three cytokines that have been implicated in the pathogenesis of silicosis [30]. In bone marrow-derived macrophages LPS enhances succinate levels, leading to succinate-mediated stabilization of HIF-1α, activation of NLRP3 inflammasome, and IL-1β production [11, 13, 31, 32]. In this work, we find that in contrast to LPS, silica reduced the intracellular levels of succinate below the baseline levels observed in unstimulated RAW 264.7 macrophages (Figure 6D). To assess the correlation between the SDH activity, the level of succinate, and expression of HIF-1α, we stimulated RAW 264.7 macrophages with silica for 6 hours, with or without priming with LPS (1 ng/ml for 24h). Giving the well-known sensitivity of HIF-1α gene to oxygen tension, we documented the ability of silica or LPS to stabilize HIF-1α expression under either normoxic (O2=20%) or hypoxic (O2 less than 5%) conditions. Silica as well as LPS, promoted the time-related, stabilization of HIF-1α protein under normoxic conditions as demonstrated via immunoblotting (Figure 7A upper panel), and confirmed by immunofluorescence and quantitative analysis of nuclear localization. (Figure 7B–C).
Figure 7. LPS, but not Silica exposure, induces stabilization of HIF-1α, activation of NLRP3 inflammasome, and release of IL-1β.

RAW 264.7 macrophages were stimulated with LPS (1 ng/ml) for 24h, silica (50 μg/cm2) for 6h, with or without priming, or left untreated. Cells were incubated in normoxia (20% Oxygen) or hypoxia (<5% oxygen). (A) Proteins were isolated from RAW 264.7 macrophages exposed as described and HIF-1α (110 kDa subunit) abundance, Pro-Caspase 1, Caspase 1 (10 kDa), and IL-1β (37 kDa) were determined by western blot while using β-actin expression as a loading control. (B) Confocal microscopy of RAW 264.7 macrophages stimulated for 6 hours with LPS (1 ng/ml) or Silica (50 μg/cm2) in the presence or absence of priming. DNA into the nuclei of cells is visualized with Hoechst, cytoskeleton filaments were visualized with actin antibody (white), followed by the analysis of HIF-1α (red). Scale bar 10 microns. (C) Graph illustrating the abundance of HIF-1α in LPS or silica exposed RAW 264.7 macrophages. Data, expressed as the mean ± SEM, is representative of 3 different experiments. (D) IL-1β mRNA expression levels quantified by RT–PCR in RAW 264.7 macrophages exposed to LPS or Silica or LPS and Silica, for the indicated time (0, 6, 24h). (E) ELISA of IL-1β release in the supernatant by RAW 264.7 macrophages stimulated as indicated. Data are the mean ± SEM of 6 samples N=6 * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001, versus control, † p<0.0001 versus silica treated.
However, these changes on HIF-1α protein expression, that were accentuated by hypoxia, were associated with activation of the NLRP3 inflammasome, documented by the cleavage of the pro-caspase 1 into low molecular weight (p10) mature caspase 1, and the fragmentation and release of IL-1β only in LPS stimulated RAW 264.7 macrophages (Figure 7A middle panel). Yet, silica stimulated RAW 264.7 macrophages demonstrated enhanced IL-1β mRNA and release of IL-1β peptide in culture in the absence of pro-caspase 1 activation (Figure 7D, E) suggesting that silica-induced IL-1β production could take place independent of succinate associated-HIF-1α mediated NLRP3 inflammasome activation.
Malonylation of GAPDH correlates with TNF-α production in LPS, but not in Silica exposed macrophages.
The citrate-derived metabolite malonyl-CoA induces the malonylation of multiples proteins including glyceraldehyde-3-phosphate dehydrogenase (GAPDH) specifically on Lysine 213. In resting cells, GAPDH sequesters the TNF-α mRNA, blocking its translation. Upon LPS stimulation, GAPDH undergoes malonylation, a reaction that facilitates the release of TNF-α mRNA for transcription and subsequent secretion [33]. In our study, we find that LPS, as well as silica, stimulated macrophages to release TNF-α in a dose-dependent manner (Figure 8A). Compared to non-stimulated RAW 264.7 macrophages, silica, as well as LPS, enhanced GAPDH and TNF-α mRNA expression (Figure 8B). However, immunoprecipitation of GAPDH from protein lysates obtained from silica or LPS stimulated RAW 264.7 macrophages identified GAPDH malonylation, in a manner that proceeded TNF-α release, exclusively in LPS exposed macrophages (Figure 8C).
Figure 8. Malonylation of GAPDH correlates with TNF-α production in LPS-treated, while decreased itaconate levels correlate with decreased IFN-β in silica-exposed macrophages.
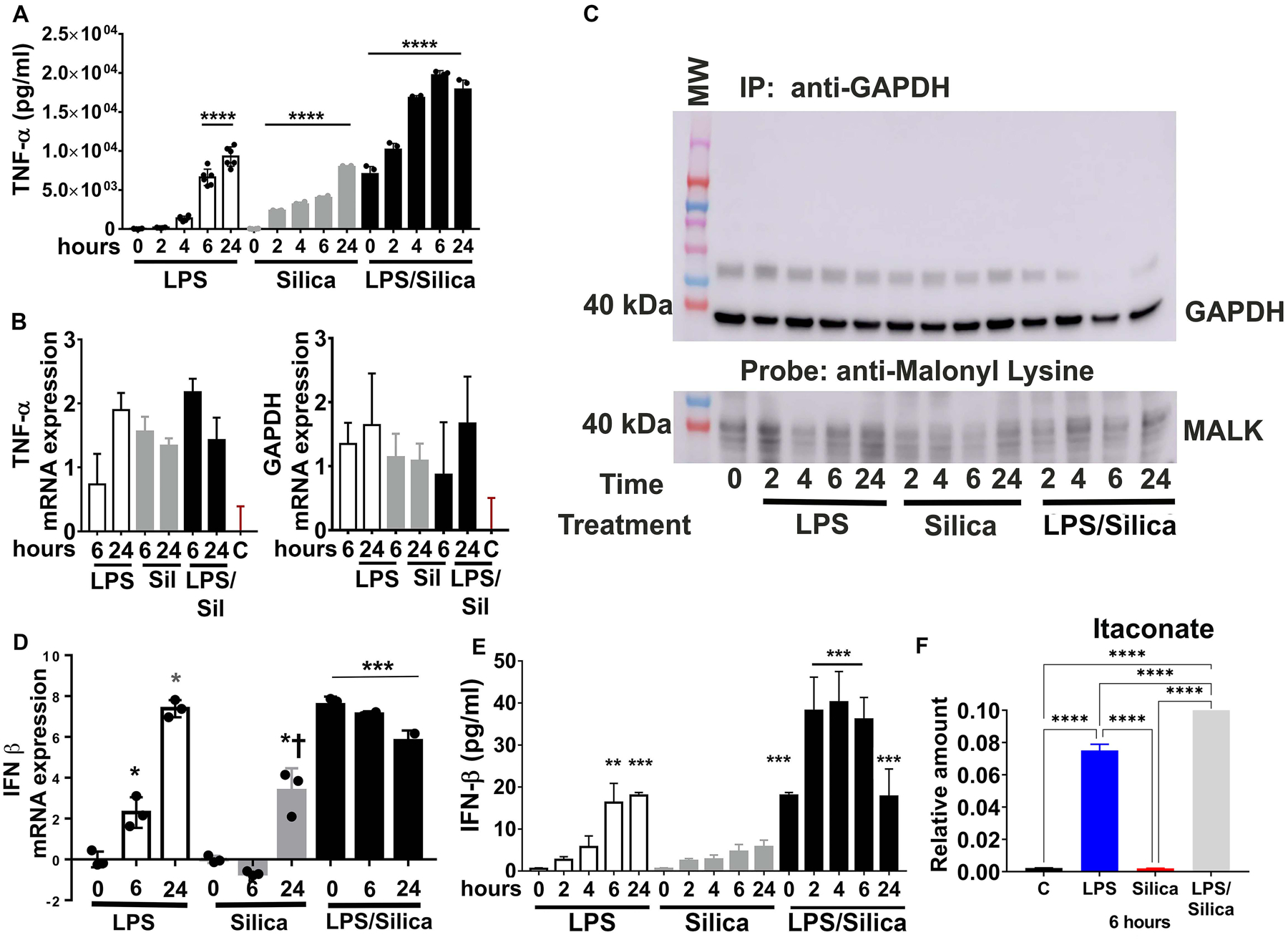
(A–C) RAW 264.7 macrophages were stimulated with LPS (1 ng/ml) or silica (50 μg/cm2) with or without priming and examined at timepoints up to 24h. (A) Cell supernatants were examined for TNF-α secretion using ELISA. (B) TNF-α and GAPDH mRNA expression levels quantified by RT–PCR. (C) Immunoprecipitated GAPDH from RAW 264.7 macrophages treated as indicated. Samples probed with an anti-malonyl lysine (anti malk) antibody (lower panel). GAPDH expression in the immunoprecipitated (upper panel) samples was also examined. (D, E) RAW 264.7 macrophages were treated with LPS (10 ng/ml) or silica (50 μg/cm2) with or without priming. (D) IFN-β mRNA expression levels quantified by RT–PCR in RAW 264.7 macrophages treated as described, for the indicated time. (E) ELISA of IFN-β released in supernatant shows that cells exposed to silica or LPS secrete the same amount of cytokine, but silica inhibits the release in primed cells. (F) The intracellular relative amount of itaconate at a steady-state is determined by LC-HRMS. Atomic percent enrichment was calculated using the Mass Isotopomer Multi-Ordinate Spectral Analysis (MIMOSA) method. Data are the mean ± SEM of 6 samples N=6 * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001.
Decreased itaconate levels correlate with decreased IFN-β in silica exposed Macrophages.
The anti-inflammatory features of the TCA metabolite itaconate have been described in LPS treated macrophages. Endogenous itaconate regulates succinate levels and limits IL-1β production in LPS activated macrophages [34, 35]. Our data show that LPS-activated RAW 264.7 macrophages increase the intracellular amount of itaconate, while silica-activated macrophages reduce the concentrations of this TCA intermediate below control levels (Figure 6B–8F). Thus, LPS (10 ng/ml) significantly stimulates the transcription of IFN-β in macrophages during 24h exposure, while silica (50 μg/cm2) significantly reduces the transcription and release of this mediator during the first 12h, and increased levels for IFN- β mRNA were only observed at 24h post-exposure (Figure 8D). However, the increased levels in IFN-β mRNA 24h after silica exposure were significantly (p<0.05) lower than those observed in LPS treated cells at the same time (Figure 8D). Release of IFN- β by RAW 264.7 macrophages parallels the increase in mRNA with statistically significant levels of the cytokine detected by ELISA 6–24h after LPS, but not in silica stimulated macrophages. LPS-priming of macrophages prior to silica exposure reconstitute IFN-β mRNA expression and the ability of RAW 264.7 macrophages to secrete this cytokine (Figure 8D, E). The addition of silica to LPS primed RAW 264.7 macrophages restores the ability of RAW 264.7 macrophages to release IFN-β in a synergistic manner (Figure 8E).
DISCUSSION
The main finding of the current work is that in contrast to the prevalent view, respirable, sterile, crystalline silica alone is capable of inducing an innate immune response without requiring previous macrophage activation by LPS. This silica-induced activation of macrophages is different from the LPS-induced, since they affect differently the CII of ETC, which plays a crucial role in macrophages survival and silica-induced inflammatory response.
In contrast to LPS, very little data are available regarding the metabolic reprogramming of macrophages in silica-induced inflammation and subsequent development of silicosis. The purpose of our study is to elucidate critical aspects of silica-induced fibrotic inflammation, comparing the metabolic effect of low dose LPS, to silica with or without priming with LPS.
LPS-activated macrophages switch their metabolism from OXPHOS to aerobic glycolysis, “Warburg effect” [36], which consists of increased uptake of glucose, glycolysis rate, and subsequent secretion of lactate, in conjunction with a reduced level of OXPHOS via the TCA cycle [11]. In our study, we found that non-toxic concentrations of LPS, as well as silica, induce increased uptake of hexoses, increased relative amount of intracellular glycolytic metabolites, and augmented the relative amount of both intra- and extracellular lactate. Moreover, priming of macrophages with LPS before silica exposure enhanced cytotoxicity and the observed effect of LPS alone.
In a recent study, Garaude et al. reported that following phagocytosis of live bacteria by bone marrow-derived macrophages (BMDM) showed alterations in the assembly of the ETC super-complexes, consisting of reduced CI activity, and enhanced CII abundance and activity [12]. These findings were not observed when BMDMs were stimulated with LPS [12]. Consistent with this work, our study demonstrates that shortly after macrophages engulf silica particles, they form phagosomes and recruit mitochondria to these structures (Supplemental Figure 1). Concomitant to these ultrastructural findings, silica-exposed macrophages remodel the activity of ETC and increase mitochondrial oxygen flux through CII, 2 h post-exposure, even after the inhibition of CI with rotenone (Figure 2). In contrast to silica, LPS only induces a transitory enhancement on mitochondrial respiration in the absence of mitochondrial CI inhibition by rotenone (Figure 2). The measurement of mitochondrial ROS production also revealed that silica engulfment is more efficient than LPS in the induction of mitochondrial H2O2 release from the activated macrophages. Priming of macrophages with LPS before silica treatment did not enhance the effect of silica on CII, nor the production of mitochondrial ROS (Figure 2). The analysis of the kinetic enzymatic activity of CI and CII also confirmed that while LPS did not alter CI activity, it only slightly enhanced CII activity, 4–6 h post-exposure, in contrast to silica that significantly decreased CI activity while simultaneously enhancing CII activity (Figure 3).
The most commonly observed CI dysfunctions are explained by alterations in CI subunits critical for its assembly. Ecsit is a CI subunit that plays a critical role in both the CI assembly stability and the metabolic activity of the complex in macrophages [21, 23, 37]. This confers an essential role for Ecsit in the antibacterial response of macrophages, due to its involvement in the recruitment of mitochondria around the site of intracellular bacteria [38], and its regulation of mitochondrial ROS production from the ETC [23]. In our study, the downregulation of Ecsit occurs in silica-, but not in LPS, exposed macrophages in a time-dependent manner, supporting the notion that this protein plays an essential role in the modulation of CI activity in response to silica.
Using IC-21 macrophages, that we previously demonstrated generate greater amount of mitochondrial ROS, experience higher levels of cell death, and secrete lower amount of TNF-α than RAW 264.7 macrophages in response to silica [8], we further show evidence supporting the notion that in the absence of a functional CI, the enhanced activity of mitochondrial CII is fundamental to preserve mitochondrial respiration and the survival of macrophages in response to silica. Thus, in IC-21 macrophages CI activity is preserved (Figure 4) and in contrast to RAW 264.7 macrophages, silica exposure rapidly diminishes, as early as 2h, the enzymatic activity of mitochondrial CII (Figure 4) and oxygen flux proceeds via mitochondrial CI activity leading to greater amount of mitochondrial ROS generation that we previously show contributes to cardiolipin oxidation and cell death [8]
Our data also support the concept that although both LPS, as well as silica, stimulate aerobic glycolysis, they affect the TCA cycle differently. An important difference observed between LPS- and silica-stimulated macrophages in the activity of the TCA cycle is the accumulation of succinate, which acts as a potent pro-inflammatory signal [11, 13]. LPS-induced increases in succinate can be explained by several mechanisms, including the inhibition of SDH, as well as replenishment of succinate from glutamine through the anapleurosis of alpha-ketoglutarate, or through the GABA shunt [11]. In contrast, silica induces an overall intracellular depletion of TCA metabolites and amino acids, including succinate, alpha-ketoglutarate, fumarate, glutamine, glutamate (Figures 5 and 6). These data suggest that in contrast to LPS, silica enhances the enzymatic activity of mitochondrial CII, demonstrated here as enhanced CII-mediated mitochondrial respiratory, in the absence of a functional CI (Figure 2). This CII activity enhances TCA dynamics with consumption, rather than accumulation, of succinate. Similarly, our data show that silica does not enhance the conversion of glutamate into succinate observed in LPS exposed macrophages (Figure 5).
Activated macrophages exhibit altered immuno-metabolism: intracellular metabolic changes governing immune effector mechanisms such as cytokine secretion. Similar to LPS, silica-treated RAW 264.7 macrophages release TNF-α and IL-1β in a dose-dependent manner (Figures 7 & 8). However, studies in LPS-stimulated macrophages show that these metabolic changes contribute to the production of TNF-α and IL-1β by different mechanisms leading to cytokine specification [11]. Thus, inhibiting glycolysis or blocking SDH in LPS-stimulated macrophages inhibits IL-1β, but not TNF-α, production [11]. In macrophages, succinate is an inflammatory signal that contributes to the activation of NLRP3 inflammasome [13, 32]. These effects, described in LPS-stimulated macrophages, are mediated via the stabilization of HIF-1α that subsequently induces transcription of HIF-1α target genes such as IL-1β [11, 13, 31, 32]. In the current work, we find that silica stimulated RAW 264.7 macrophages demonstrate stabilization of HIF-1α, and enhanced transcription and release of IL-1β (Figure 7). However, in contrast to LPS, these silica-induced changes occur in the presence of decreased levels of intracellular succinate (figure 6) and are unrelated to NLRP3 inflammasome activation as these RAW 264.7 macrophages do not show caspase1 activation in response to silica particles alone (Figure 7). Importantly, previous reports demonstrating the ability of silica to activate the NLRP3 inflammasome were conducted in LPS-primed macrophages [7, 30, 39].
The role of TNF-α in the pathogenesis of silicosis is well recognized [40]. However, the mechanisms specifying TNF-α production by silica exposed macrophages are not entirely understood. Recent information shows that macrophages secretion of TNF-α and interferons in response to LPS is mediated, in part, via malonylation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [33, 41, 42]. White et al. identified a role for GAPDH as a noncanonical RNA-binding protein [43]. In resting macrophages, GAPDH binds to and suppresses the translation of inflammatory mRNAs, including IFN-β and TNF-α [43]. GAPDH binds NAD+ to retain mRNAs silent. LPS induced malonylation of GAPDH requires the separation of NAD+ from the enzymatic domain to facilitate protein malonylation on lysine 213 [33]. However, in this work, we only documented lysine 213 malonylation on protein precipitates from LPS exposed RAW 264.7 macrophages, in a manner that precedes the transcription and release of TNF-α (figure 8). These data agree with previous observations linking the silica-induced TNF-α generation to NADPH- and mitochondrial ROS mediated NF-κB activation and TNF-α mRNA transcription [38, 44, 45].
The anti-inflammatory properties of itaconate, a TCA cycle metabolite generated through the decarboxylation of cis-aconitate, a product of citrate, have also been described in LPS-activated macrophages [46]. LPS-induced accumulation of itaconate, mainly due to the isocitrate dehydrogenase impairment, can regulate the SDH activity, thereby decreasing the inflammatory response, and also regulate the IFN-β secretion [35, 47]. In this study, we observed that, while the intracellular level of itaconate is significantly augmented in LPS activated macrophages, the same does not occur after silica engulfment, and consequently this directly correlates with the transcription and release of interferon-beta (IFN-β) in response to either LPS or silica (figure 8).
Previous in vivo studies, demonstrated that macrophage ontogeny plays a crucial role in their immune-metabolic response [15]. Specifically, increased glycolysis- and decreased TCA cycle- driven metabolism upon bacterial activation is observed in macrophages recruited to the lung, while stable or minimal alteration of glycolysis and TCA cycle enzymes, and increased fatty acid oxidation is observed in resident alveolar macrophages. [14–16]. A limitation to the current studies is that the data were procured with the use of macrophage cell lines. RAW 264.7 and IC-21 cells, which are derived from resident peritoneal macrophages, demonstrate baseline rates of glycolysis, phospholipid content, and fatty acid oxidation that are representative of human monocyte derived macrophages [8], as well as those observed in primary alveolar macrophages from their corresponding mouse strains, C57BL/6 or BALB/c respectively [8, 48]. Therefore, further in vivo studies are required to fully comprehend the difference in the immune-metabolic response to inhaled crystalline silica of alveolar- and interstitial- macrophages, as well as monocyte recruited from the peripheral circulation into the lung in response to silica exposure.
In summary, the present work indicates that upon internalization into phagolysosomes, respirable crystalline silica drives a metabolic adaptation of macrophages consisting of increased uptake of glucose, increased glycolysis, accompanied by an increased lactate secretion, at the expense of the OXPHOS, which becomes sustained only by an increased CII activity, while CI activity is reduced. Given the role of CII as a component of both the TCA cycle and the ETC, its activity becomes a key regulator of macrophages’ survival. Silica also modulates the TCA cycle, where not only the succinate level is measured below baseline, but also the total intracellular level of all key TCA cycle intermediates and amino acids examined are decreased probably as a result of high demand and consumption. In contrast to LPS, these silica-induced metabolic adaptations do not correlate with IL-1β, TNF-α, production but with the suppressed release of IFN-β. Further studies are needed to validate this concept and to better understand the mechanisms that induce the release of inflammatory cytokines after the phagocytosis of silica into macrophages.
Supplementary Material
Key Points:
Crystalline silica induces an immune-metabolic response in RAW 264.7 macrophages.
Silica induces macrophages activation without requiring LPS-priming.
Mitochondrial ComplexII is crucial for macrophages response and survival to silica.
Fundings:
Funder Agency HHS - NIH - NHLBI Grant # 1R01HL114795 Author Luis A Ortiz
Funder Agency HHS - NIH - NHLBI Grant # 1R01HL110344 Author Luis A Ortiz
Funder Agency HHS - NIH - NIH Office of the Director (OD) Grant # S10OD023402 Author Stacy G Wendell
Funder Agency HHS - NIH - NIEHS Grant # ES015859 Author L A Ortiz
REFERENCES
- 1.Gottesfeld P, Reid M, and Goosby E, Preventing tuberculosis among high-risk workers. The Lancet. Global health, 2018. 6(12): p. e1274–e1275. [DOI] [PubMed] [Google Scholar]
- 2.Leung CC, Yu ITS, and Chen W, Silicosis. The Lancet, 2012. 379(9830): p. 2008–2018. [DOI] [PubMed] [Google Scholar]
- 3.Anderson SE, et al. , Biological effects of inhaled hydraulic fracturing sand dust. VIII. Immunotoxicity. Toxicology and applied pharmacology, 2020. 408: p. 115256–115256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Giuseppe M, et al. , Systemic inhibition of NF-kappaB activation protects from silicosis. PloS one, 2009. 4(5): p. e5689–e5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO, Elimination of silicosis, . GOHNET news letter,, 2007: p. 1–20. [Google Scholar]
- 6.Biological effects of inhaled hydraulic fracturing sand dust. IX. Summary and significance. Toxicol Appl Pharmacol, 2020. 409: p. 115330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cassel SL, et al. , The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A, 2008. 105(26): p. 9035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fazzi F, et al. , TNFR1/phox interaction and TNFR1 mitochondrial translocation Thwart silica-induced pulmonary fibrosis. J Immunol, 2014. 192(8): p. 3837–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tschopp J and Schroder K, NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nature Reviews Immunology, 2010. 10(3): p. 210–215. [DOI] [PubMed] [Google Scholar]
- 10.Mischler SE, et al. , Differential activation of RAW 264.7 macrophages by size-segregated crystalline silica. Journal of Occupational Medicine and Toxicology, 2016. 11(1): p. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tannahill GM, et al. , Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature, 2013. 496(7444): p. 238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garaude J, et al. , Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol, 2016. 17(9): p. 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mills EL, et al. , Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell, 2016. 167(2): p. 457–470 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang L, et al. , Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. Journal of experimental medicine, 2018. 215(4): p. 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mould KJ, et al. , Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. American journal of respiratory cell and molecular biology, 2017. 57(3): p. 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pisu D, et al. , Dual RNA-Seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell reports, 2020. 30(2): p. 335–350. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mischler SE, et al. , A multi-cyclone sampling array for the collection of size-segregated occupational aerosols. J Occup Environ Hyg, 2013. 10(12): p. 685–93. [DOI] [PubMed] [Google Scholar]
- 18.Gambelli F, et al. , Phosphorylation of tumor necrosis factor receptor 1 (p55) protects macrophages from silica-induced apoptosis. The Journal of biological chemistry, 2004. 279(3): p. 2020–2029. [DOI] [PubMed] [Google Scholar]
- 19.Alves TC, et al. , Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab, 2015. 22(5): p. 936–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenzie M and Ryan MT, Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life, 2010. 62(7): p. 497–502. [DOI] [PubMed] [Google Scholar]
- 21.Guerrero-Castillo S, et al. , The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metabolism, 2017. 25(1): p. 128–139. [DOI] [PubMed] [Google Scholar]
- 22.Nouws J, et al. , Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain, 2011. 135(1): p. 12–22. [DOI] [PubMed] [Google Scholar]
- 23.Carneiro FRG, et al. , An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages. Cell Reports, 2018. 22(10): p. 2654–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fazzi F, et al. , TNFR1/phox interaction and TNFR1 mitochondrial translocation Thwart silica-induced pulmonary fibrosis. Journal of immunology (Baltimore, Md. : 1950), 2014. 192(8): p. 3837–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infantino V, et al. , The mitochondrial citrate carrier: a new player in inflammation. Biochem J, 2011. 438(3): p. 433–6. [DOI] [PubMed] [Google Scholar]
- 26.O’Neill LA, A critical role for citrate metabolism in LPS signalling. Biochem J, 2011. 438(3): p. e5–6. [DOI] [PubMed] [Google Scholar]
- 27.Mills EL and O’Neill LA, Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol, 2016. 46(1): p. 13–21. [DOI] [PubMed] [Google Scholar]
- 28.Angajala A, et al. , Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front Immunol, 2018. 9: p. 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills EL, Kelly B, and O’Neill LAJ, Mitochondria are the powerhouses of immunity. Nat Immunol, 2017. 18(5): p. 488–498. [DOI] [PubMed] [Google Scholar]
- 30.Hornung V, et al. , Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol, 2008. 9(8): p. 847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corcoran SE and O’Neill LA, HIF1alpha and metabolic reprogramming in inflammation. J Clin Invest, 2016. 126(10): p. 3699–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills E and O’Neill LA, Succinate: a metabolic signal in inflammation. Trends Cell Biol, 2014. 24(5): p. 313–20. [DOI] [PubMed] [Google Scholar]
- 33.Galvan-Pena S, et al. , Malonylation of GAPDH is an inflammatory signal in macrophages. Nat Commun, 2019. 10(1): p. 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills EL, et al. , Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature, 2018. 556(7699): p. 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lampropoulou V, et al. , Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell metabolism, 2016. 24(1): p. 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warburg O, Wind F, and Negelein E, The Metabolism of Tumors in the Body. J Gen Physiol, 1927. 8(6): p. 519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogel RO, et al. , Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev, 2007. 21(5): p. 615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West AP, et al. , TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature, 2011. 472(7344): p. 476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dostert C, et al. , Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (New York, N.Y.), 2008. 320(5876): p. 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piguet PF and Vesin C, Treatment by human recombinant soluble TNF receptor of pulmonary fibrosis induced by bleomycin or silica in mice. Eur Respir J, 1994. 7(3): p. 515–8. [DOI] [PubMed] [Google Scholar]
- 41.Galvan-Pena S and O’Neill LA, Metabolic reprograming in macrophage polarization. Front Immunol, 2014. 5: p. 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meiser J, et al. , Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J Biol Chem, 2016. 291(8): p. 3932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White MR and Garcin ED, The sweet side of RNA regulation: glyceraldehyde-3-phosphate dehydrogenase as a noncanonical RNA-binding protein. Wiley Interdiscip Rev RNA, 2016. 7(1): p. 53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scarfi S, et al. , Ascorbic acid pre-treated quartz stimulates TNF-alpha release in RAW 264.7 murine macrophages through ROS production and membrane lipid peroxidation. Respir Res, 2009. 10: p. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik E and Dixit VM, Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med, 2011. 208(3): p. 417–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michelucci A, et al. , Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A, 2013. 110(19): p. 7820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cordes T, et al. , Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem, 2016. 291(27): p. 14274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phinney DG, et al. , Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun, 2015. 6: p. 8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.