

Abstract
Genomic studies of pediatric cancer have primarily focused on specific tumor types or high-risk disease. Here, we used a three-platform sequencing approach, including whole genome (WGS), exome, and RNA sequencing, to examine tumor and germline genomes from 309 prospectively identified children with newly diagnosed (85%) or relapsed/refractory (15%) cancers, unselected for tumor type. Eighty-six percent of patients harbored diagnostic (53%), prognostic (57%), therapeutically-relevant (25%), and/or cancer predisposing (18%) variants. Inclusion of WGS enabled detection of activating gene fusions and enhancer hijacks (36% and 8% of tumors, respectively), small intragenic deletions (15% of tumors) and mutational signatures revealing of pathogenic variant effects. Evaluation of paired tumor-normal data revealed relevance to tumor development for 55% of pathogenic germline variants. This study demonstrates the power of a three-platform approach that incorporates WGS to interrogate and interpret the full range of genomic variants across newly diagnosed as well as relapsed/refractory pediatric cancers.
Keywords: Cancer, genomics, pediatric, DNA, RNA, next generation sequencing
INTRODUCTION
High throughput next generation sequencing (NGS) of pediatric cohorts has provided seminal insights into the genomic landscapes of the major subtypes of childhood cancer. It is now well established that these NGS approaches add significant value by refining or changing cancer diagnoses(1–5), providing prognostic information(3,6), identifying therapeutic targets or markers of therapy resistance(2,3,5,7,8), detecting variants of pharmacogenetic significance(3), and uncovering underlying genetic predisposition(2–5,8,9).
To date, most pediatric NGS studies have focused on patients with high-risk disease, including difficult-to-treat or relapsed/refractory cancers. In many of these studies, patients with newly diagnosed or standard risk cancers are absent or underrepresented. However, many standard risk cancers do not respond or recur following treatment with current best available therapies. Indeed, such is the case for 15 – 20% of acute lymphoblastic leukemia (ALL)(10) or Wilms tumor(11) cases and over 30% of rhabdomyosarcomas(12) or other non-CNS solid tumors. To improve upon these outcomes, it is essential that we comprehensively examine and elucidate the molecular underpinnings of childhood cancer across the full spectrum of presentations.
Each childhood cancer harbors a unique combination of somatic alterations on a background of inherited and de novo germline variants. Moreover, novel diagnostic and prognostic subgroups and the full constellation of genetic drivers are yet to be defined for many rare pediatric cancers. As such, an individual pediatric cancer genome could be described as an “N of 1” case study for which genome-wide analysis can uncover unique molecular drivers and elucidate how individual combinations of somatic and germline variants influence clinical presentation and response to cancer therapy. Therefore, it is essential that comprehensive genomic data are generated and systematically investigated if we are to fully capitalize on the potential of precision medicine across pediatric cancers, including those that are common as well as those that are rare.
Prior studies using distinct sequencing platforms have demonstrated that a genome-wide approach is necessary to enable full discovery of novel driver variants in pediatric cancers(1,5,8,13–15). Towards this end, we developed a three-platform sequencing pipeline that includes whole genome sequencing (WGS), whole exome sequencing (WES) and RNA sequencing (RNA-Seq) of paired tumor and normal samples. This three-platform pipeline has been validated on a retrospective cohort of highly selected high-risk pediatric cancer cases harboring known genomic alterations as shown by classical molecular assays. Furthermore, it greatly improved the accuracy of detection of genomic alterations, obviated the need for validation testing of somatic and germline variants through orthogonal approaches, and facilitated discovery of novel oncogenic processes(14).
Here we present data from “Genomes for Kids” (G4K), a prospective non-therapeutic three-platform sequencing study of 309 pediatric cancer patients, unselected for tumor type or stage, treated at St Jude Children’s Research Hospital. The aims of this study were to: 1) demonstrate the utility of comprehensive whole genome, exome, and transcriptome sequencing of paired tumor and normal samples for patients across the spectrum of standard risk to high risk cancers; 2) show how integrating data from multiple sequencing platforms can elucidate the functional impacts of difficult-to-interpret variants; 3) analyze rare genomic findings in N of 1 cases and show how these findings inform understanding of tumor biology and, when possible, patient care; and 4) discover novel mechanisms driving a diverse array of childhood cancers.
RESULTS
Patient enrollment
From August 2015 to March 2017, 365 pediatric cancer patients were approached for enrollment in G4K (NCT02530658), a non-therapeutic study of three-platform sequencing of paired tumor and normal samples. Three hundred nine (85%) agreed to participate, 53 (15%) declined and three were later removed from the study (Fig. 1A). Race/ethnicity was the only variable that significantly correlated with declining enrollment, with families of black children more likely to decline compared to families of non-Hispanic or Hispanic white children, p<0.001, as we have reported elsewhere(16).
Figure 1. Patient accrual and demographic data.
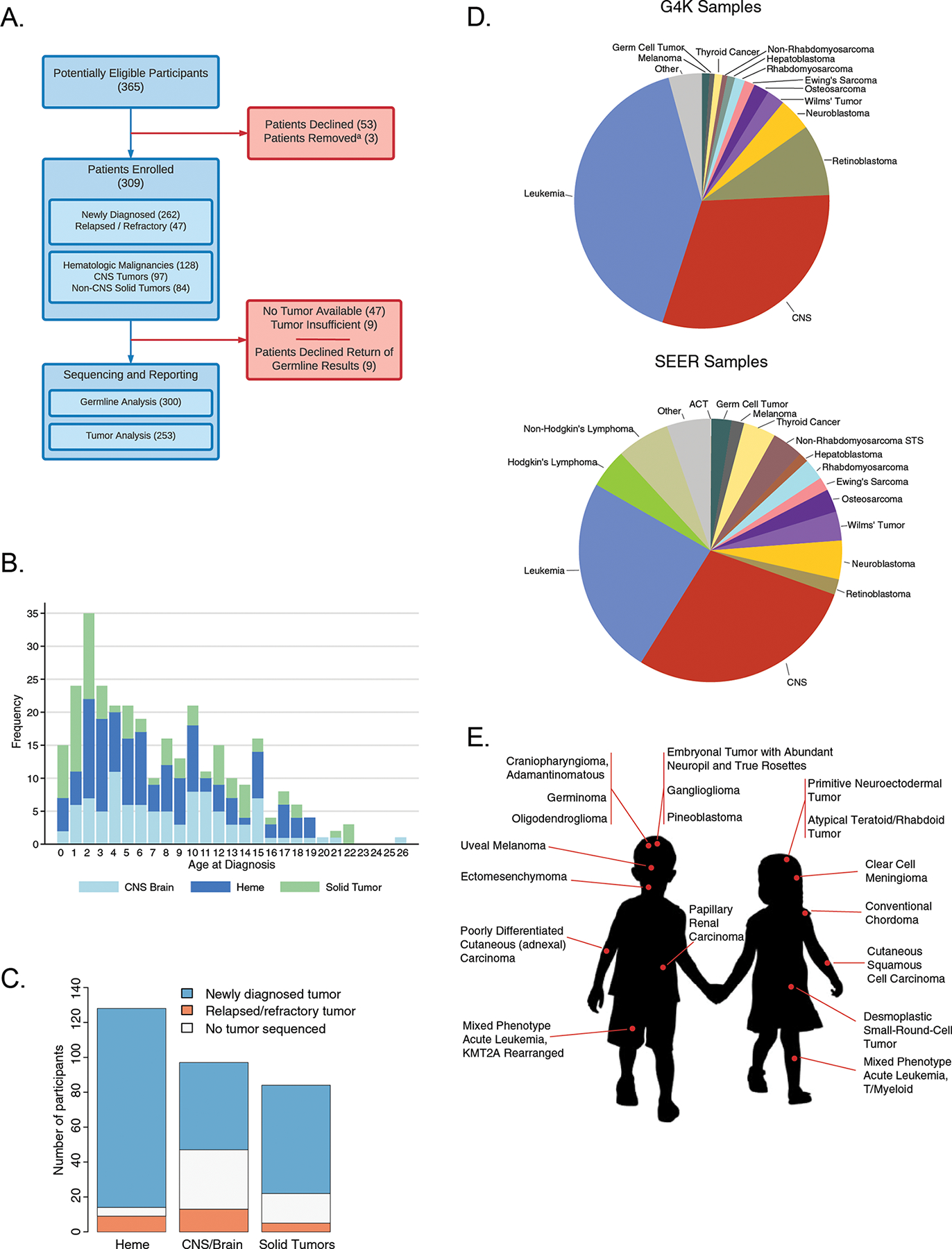
A. Consort diagram depicting patient accrual into G4K. Note: (a) Three patients were removed from the study when pathology revealed no cancer (1); the patient died before a germline sample could be collected (1); or the patient declined return of germline results and there was insufficient tumor for sequencing (1). B. Age distribution of patients. C. Number of patients with newly diagnosed, relapsed or refractory tumors, or no tumor available for sequencing, broken down by major tumor type. D. The distribution of cancer types represented in the G4K cohort (top) compared to the distribution of pediatric cancers in the NCI Surveillance, Epidemiology and End Results (SEER) program (bottom). E. Eighteen rare tumor types found in the G4K cohort present at less than 2 per million children annually in the United States
Forty-seven patients did not undergo tumor biopsy for safety reasons or because biopsy was not considered clinically indicated. Nine patients did not have sufficient tumor DNA or RNA to complete three platform sequencing. Thus, 253 of 309 (82%) enrolled patients had their tumors examined using WGS, WES and RNA-Seq. Nine of 309 patients’ families (3%) declined the return of germline results, leaving 300 who underwent analysis of germline samples using WES and WGS, followed by in depth evaluation and reporting of 156 cancer predisposition genes. Study participants included 166 males and 143 females with an average age at cancer diagnosis of 7.4 years (range: 4 days to 25.7 years, Fig. 1B) (Supplemental Table S1).
At the time of G4K enrollment, 262 patients (85%) had newly diagnosed cancers and 47 (15%) had relapsed or refractory disease (Fig. 1C). The spectrum of cancers in the G4K cohort was similar to that observed in the NCI SEER registry (Fig. 1D), except for Hodgkin and non-Hodgkin lymphoma, which were underrepresented, and leukemia and retinoblastoma, which were overrepresented. One hundred twenty-eight patients (41%) had hematological malignancies of 28 subtypes, 97 (31%) had brain tumors of 27 subtypes, and 84 (27%) had non-CNS solid tumors of 26 subtypes. Forty-five patients (15%) had 18 very rare tumor types, defined here as tumors present in fewer than 2 cases per million annually in the United States (Fig. 1E). Among these 18 tumor types, only craniopharyngioma(17) and mixed phenotype acute leukemia(18) have been studied in enough detail to provide an initial understanding of associated somatic alterations. Thus, the ability to examine the impact of genomic lesions in an N of 1 context is of paramount importance to understand the biologic basis and therapeutic targets in these very rare tumors.
Overview of somatic alterations
Tumor samples were evaluated using 45X PCR-free WGS and 150X WES from DNA and 100 million RNA-Seq reads from total RNA. Paired germline samples were evaluated using both WGS and WES. Twelve hundred genes known to play a role in the pathogenesis of cancer were interrogated using tumor genomic data. Genes were considered in the context of each patient’s tumor type and prioritized for review, as previously described(19). Given the potential effects of structural events and gene fusions on protein expression and function, we reported novel events if there was strong sequence support and the structural event or gene fusion involved genes of potential relevance to cancer. We detected somatic single nucleotide variants (SNVs), small insertions/deletions (indels), loss of heterozygosity (LOH), structural variants (SVs) including fusions, enhancer hijacks, internal tandem duplications (ITDs), and copy number alterations (CNAs) using automated computational pipelines followed by analyst curation(14). Among the SNVs and indels observed on both WGS and WES platforms, 41% had a variant allele fraction (VAF) below 0.2 and 15% had a VAF below 0.1, revealing a large set of sub-clonal variants, including P/LP variants in both SNV and indels, using this approach (Supplemental Figs. S1A, B).
All variants were reviewed by a committee of computational biologists, pathologists, oncologists, geneticists and genetic counselors. Reportable P/LP alterations included SNVs (22%); indels (10%); gross chromosomal losses, gains and LOH (33%); sub-arm copy (focal) number abnormalities (20%) and SVs/gene fusions (44%). Overall, we reported a mean of three pathogenic or likely pathogenic (P/LP) somatically-acquired sequence variants per case (range 0–14) in addition to ploidy alterations, such as gross chromosomal gains and losses and chromosome arm-level regions of copy neutral LOH (CNLOH). Two brain tumors had no reportable P/LP findings despite pathology review indicating adequate tumor tissue. The prevalence of variants deemed P or LP in hematological malignancies, brain tumors, and non-CNS solid tumors, affected by diverse mutational mechanisms, were consistent with other recent pediatric cancer genomic studies(8,14,15,20–22) (Fig. 2A, Supplemental Table S2).
Figure 2. Somatic findings in the 253 analyzed tumors.
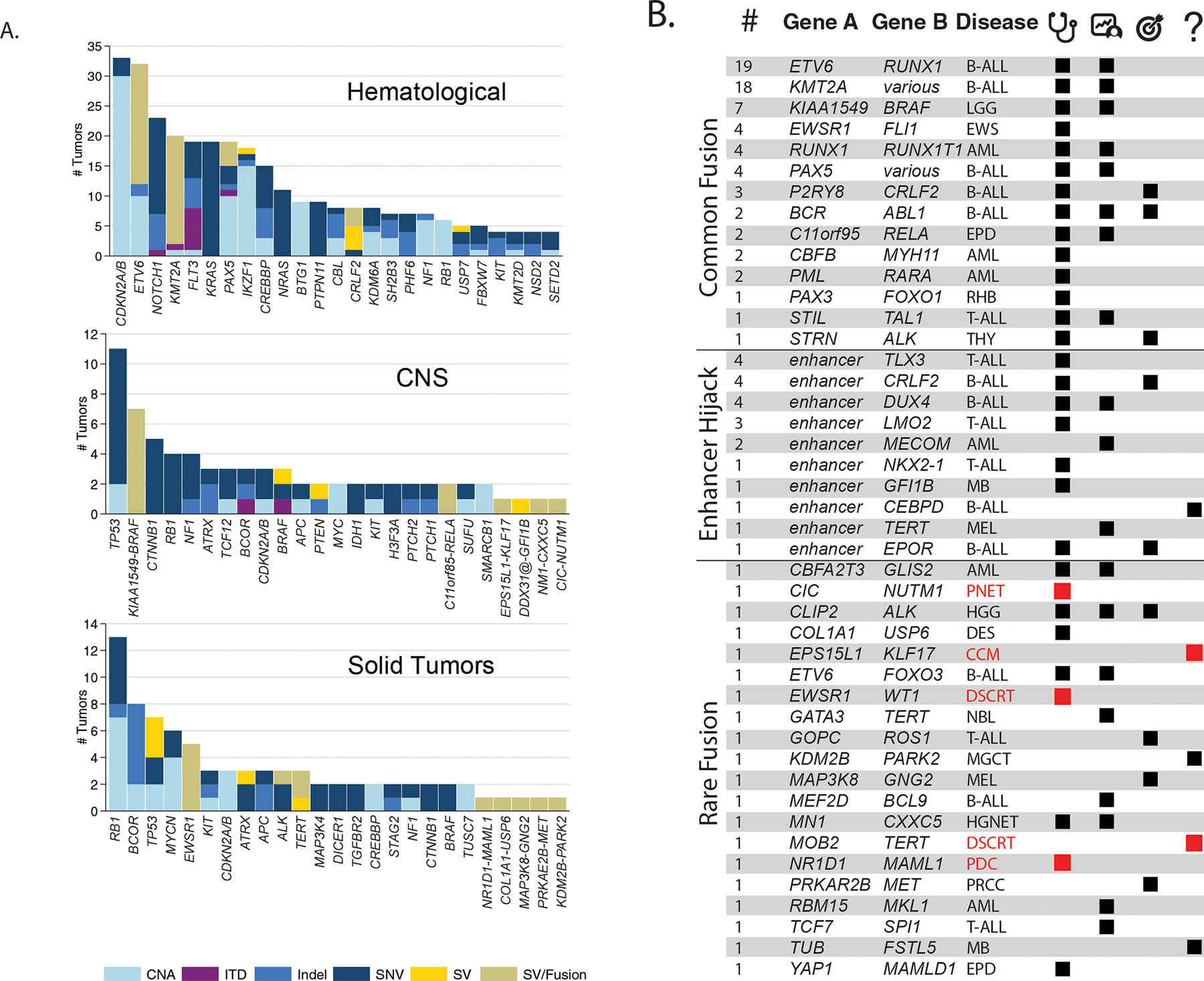
A. Bar charts showing the numbers and relative contributions of mutational mechanisms affecting cancer genes in the tumors analyzed through G4K. The top 25 mutated genes for hematologic, CNS and solid tumors are shown as are gene fusions or enhancer hijack events for singletons in CNS and Solid Tumors. B. Gene fusions and enhancer hijacks detected in G4K samples. Number of samples with a given fusion are indicated in the left most column followed by the genes/loci involved and the diseases in which they were detected (see Supplemental Fig. S3 for schematics depicting the 20 rare fusions). Black or red tiles indicate whether the identified gene fusions or enhancer hijacks have a clear or likely clinical utility, arranged into three columns indicating diagnostic (stethoscope), prognostic (patient chart), and therapeutically-relevant (target) categories. In the right most column (question mark), tiles indicate lesions with an unknown clinical utility, but considered biologically relevant to the tumor. Red disease names and tiles were identified in the rare tumors as shown in Fig. 1E. Disease abbreviations and additional details regarding SV classifications and literature citations can be found in Supplemental Table 2.
Identification of gene fusions, enhancer hijacks, microdeletions
SVs causing gene fusions or enhancer hijacking are important drivers of pediatric cancers(23–25), yet they are challenging to detect by WES as SV breakpoints are most often located in non-coding regions of the genome. Consistent with this finding, we showed previously that inclusion of WGS in the study of pediatric cancer genomes significantly improves the detection of driver gene alterations when compared to WES alone(14). Using the three-platform sequencing approach, we identified in-frame gene fusions in 90 tumors (36%) representing 44 distinct gene-pairings and 34 distinct fusion driver genes of which 23 were diagnostic for a specific cancer type or subgroup. Thirty of the 34 conferred a clear or potential diagnostic, prognostic or therapeutic utility (Fig. 2B, Supplemental Figs. S2 and S3, Supplemental Table S2). For example, brain tumor SJBT030081, originally classified as a high-grade glioma, was found to harbor an MN1-CXCC5 gene fusion and thus reclassified as a CNS high-grade neuroepithelial tumor with MN1 alteration, an entity only recently described(26). Five of the rare fusions shown in Fig. 2B (shown in red) were in patients with very rare tumor types (as shown in Fig. 1E).
In an additional 21 tumors (8%), combined WGS and RNA-Seq analysis enabled detection of 10 distinct enhancer hijacking translocations, wherein an SV brings a transcriptionally active locus into close proximity to an oncogene thereby driving its expression. We correlated outlier oncogene expression with SV breakpoints from WGS data to facilitate the accurate detection of enhancer hijacking events, which ranged from 0–716 kb up or downstream of the target oncogene with outlier expression (see Methods, Supplemental Fig. S4, Supplemental Table S3). Enhancer hijacks included well-characterized events such as IGH@-CRLF2, TCR@-LMO2, IGH@-DUX4, IGH@-EPOR, and DDX31@-GFI1B. In addition, there were less-well-characterized enhancer hijacks such as two instances of CDK6@-MECOM in acute myeloid leukemia, which may have a negative prognostic impact(27), and a novel TLX3-activating translocation in a T-acute lymphoblastic leukemia (T-ALL) sample, as described below (see below, “Disease relevance of novel somatic variants“).
Acute leukemias are susceptible to small microdeletions, resulting from off target recombinase activating gene (RAG)-dependent effects(28,29). Detection of these deletions can be of prognostic significance, as is the case with IKZF1 in B-ALL, where intragenic deletions are associated with a poorer outcome(30). However, intragenic microdeletions can be difficult to detect using WES because exons rarely capture deletion breakpoints and their identification must rely on subtle changes in depth of coverage(31,32). Consistent with this notion, we previously observed that single nucleotide polymorphism (SNP) array or WES have limited power for detecting focal and exon-poor CNAs due to insufficient physical space for robust detection of read-depth changes(14,33). Using WGS data, we detected small intragenic deletions involving as few as one or two exons in cancer-relevant genes in 38 tumors (15%), with all but six being leukemias (Supplemental Table S4). Twenty-six genes were affected, including those expected(28), such as BTG1, CDKN2A, ETV6, RAG1, TCF4, IKZF1, and TP53 among the most commonly involved. Several clinically relevant genes that are less commonly observed as targets of microdeletion, including CREBBP, SH2B3, USP9X, FBXW7, and NR3C2, were also impacted. The majority of these microdeletions (38 of 53 total events; 72%) involved loss of a single gene copy and thus would have been difficult to detect using WES.
Mutation signatures and etiology of pediatric tumors
The whole genome mutational landscape of a tumor records its natural history and reflects the environmental and endogenous exposures that have contributed to tumor initiation and progression(34,35). To elucidate the mutational landscape of pediatric cancers, we evaluated WGS tumor data for the relative proportions of mutation signatures reported by Alexandrov(36) (Supplemental Table S5). In addition to well-known mutational processes such as spontaneous deamination (Signature 1) evident in nearly every patient, Signatures 2 and 13 reflecting activation induced cytidine deaminase (AID) and apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) activity were present in several of B-ALL cases(37,38); Signatures of ultraviolet radiation exposure were present in four B-ALL samples and a cutaneous melanoma (see also(15)) and three tumors exhibited signatures 6 and 15, indicating mismatch repair (MMR) deficiency as well as signature 10, which is associated with mutated DNA polymerase epsilon (POLE). Six tumors harbored high levels of signature 18, thought to arise from DNA damage induced by reactive oxygen species (ROS) with one tumor exhibiting bi-allelic loss of MUTYH, the glycosylase that executes the first step in repair of ROS-induced mutations. The roles of the MMR deficiency, and POLE and ROS signatures in disease pathogenicity are described further below (see “Disease relevance of germline variants”).
Disease relevance of novel somatic variants
Most pediatric cancer genomes harbor a limited number of somatic alterations, many of which are non-recurrent making it difficult to ascertain pathogenicity. Visualization of these variants in the context of large-scale public data was essential to classify some of these challenging N of 1 events. We illustrate this process with two examples. The first case, SJTALL030071, is a T-ALL that harbored a translocation t(5;8)(q35q24) linking an intergenic region on 8q24, 1 Mb distal of MYC, to an intergenic region 29 kb distal to TLX3, a known driver of T-ALL(39). The copy number profile of this tumor showed an 18 Mb duplication of the 8q24->8qter region that did not include MYC (Fig. 3A); but did include the NOTCH MYC enhancer(N-Me)(40) that is known to activate MYC in T-ALL. Indeed, N-Me activity was confirmed by the high level of enhancer RNA and MYC expression in this tumor. The translocation juxtaposed N-Me 29 kb upstream of TLX3 leading to elevated TLX3 expression (Fig. 3B). Moreover, using SNP markers in the DNA at the TLX3 locus, we demonstrated allele-specific expression (ASE) in the RNA, consistent with activation by a cis-acting regulator (Fig. 3C). Consistent with these findings, SJTALL030071 clustered by tSNE analysis among other TLX3-driven T-ALLs from the National Cancer Institute-Therapeutically Applicable Research to Generate Effective Treatments (NCI-TARGET) cohort (Fig. 3D). Altogether, these data suggested mechanistic similarity to known enhancer hijack events linking TLX3 to T-cell receptor enhancer loci(39,41) and accordingly we classified the translocation as LP.
Figure 3. Using multi-omics data to interpret pathogenicity of structural variants.
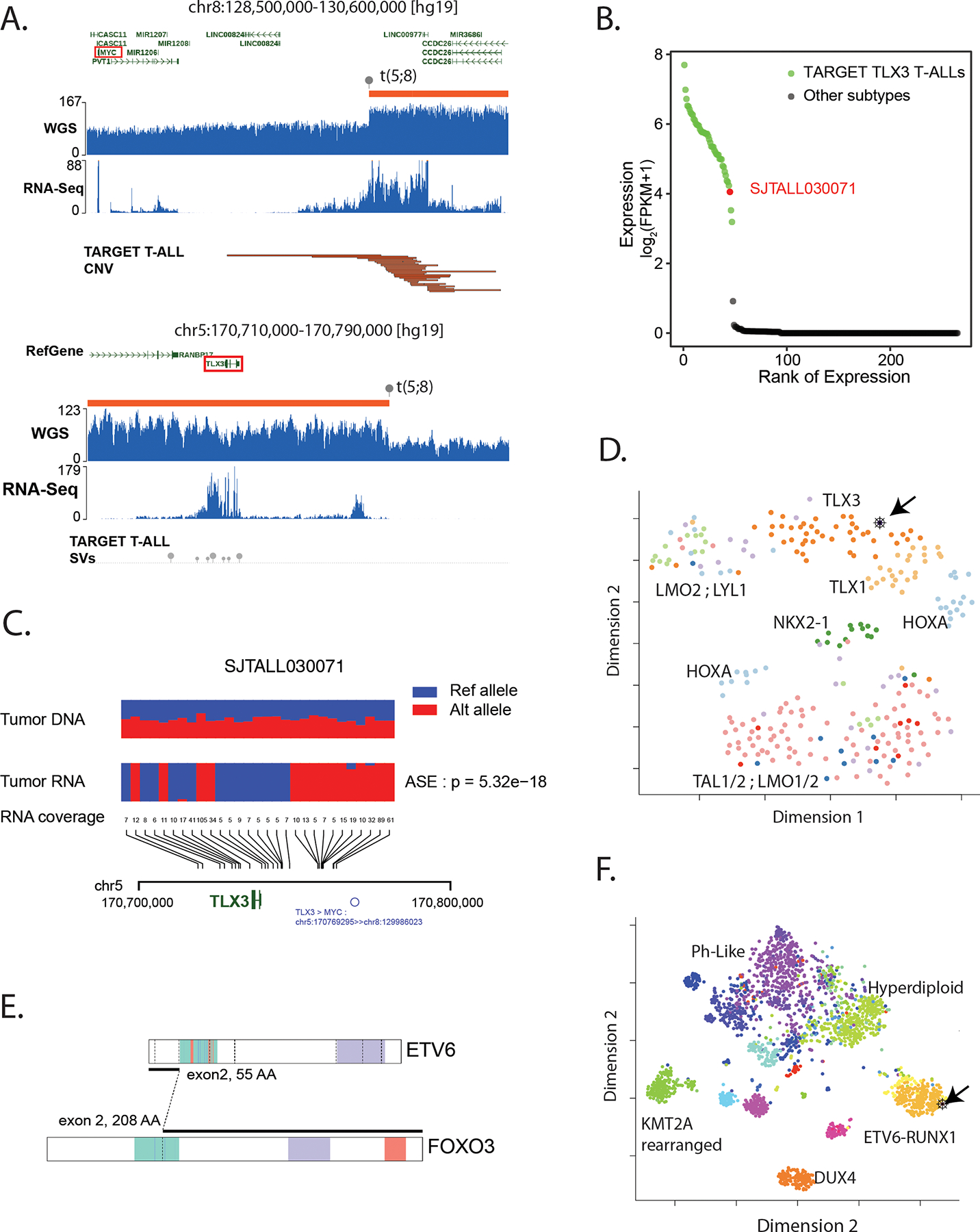
A. GenomePaint plots showing two regions, chr8:128500000–130600000 (top) and chr5: 170710000–170790000 (bottom) [hg19] from SJTALL030071. Both panels consist of the RefSeq gene model in green with MYC and TLX3 highlighted by red boxes; orange bars show regions of copy number gain supported by increased whole genome sequencing coverage plotted as the blue histogram immediately below. Grey lollypops marked t(5;8) indicate the position of the translocation breakpoint. RNA-Seq coverage is shown below the whole genome coverage histogram. Additional data from TARGET is also shown with narrow red bars representing regions of copy number gain and grey lollypops representing structural variant breakpoints surrounding the TLX3 locus. A region or recurrent copy number gain in TARGET samples is adjacent to the chromosome 8 breakpoint in SJTALL030071. Generally high but non-specific RNA-Seq coverage at this locus suggests a region of high transcriptional activity (distal MYC enhancer) is brought into proximity of TLX3 by the translocation. B. A rank-order plot of TALL from TARGET showing expression levels of TLX3 mRNA in a set of TLX3-activated tumors compared to tumors in which TLX3 was not activated. SJTALL030071 TLX3 expression (red dot) groups with the activated set. C. Allele-specific expression of the TLX3 locus in SJTALL030071. The tumor DNA (top row) shows a series of heterozygous alleles in the TLX3 locus (blue and red stacked bars show relative variant allele fraction from WGS data). In the RNA-Seq data (second row) expression of only one allele is observed. RNA coverage, read counts at each allele, are shown numerically (third row). Beneath the read counts, black lines map the locations of the alleles to the chromosome 5 coordinates surrounding TLX3 locus. Beneath the coordinate line, the location of the SJTALL030071 translocation breakpoint is indicated. D. Two-dimensional t-SNE plot of RNA-Seq-derived gene expression data from 264 T-ALL samples(41). Major T-ALL subgroups are indicated on the plot with SJTALL030071 localizing among the TLX3 cluster, as shown by the black arrow. E. Schematic representation of an ETV6-FOXO3 fusion found in SJBALL030052 joining the region N-terminal to the ETV6 sterile alpha motif domain (green) with oligomerization interfaces (red) and ETS domain (purple), with the C-terminal FOXO3 forkhead binding (green), KIX-binding (purple) and transactivation domains (red). F. Two-dimensional t-SNE plot of RNA-Seq-derived gene expression data from 1,988 B-ALL samples(42). Major subgroups are indicated on the plot with SJBALL030052 localizing to the periphery of the ETV6-RUNX1 subgroup, as shown by the black arrow.
The second case, SJBALL030052, is a B-ALL with a complex structural variant that exhibited DNA and RNA evidence of a novel fusion including exon 2 of ETV6 with exon 2 of FOXO3 (Fig. 3E). The impact of this fusion was not initially clear as it could conceivably result in ETV6 loss of function (LOF) or in a gene fusion functionally similar to ETV6-RUNX1. To gain further insights, we compared the RNA-Seq gene expression profiles between our B-ALL sample and the publicly available RNA-Seq profiles from 1,988 other childhood B-ALL cases(42). SJBALL030052 clustered with the ETV6-RUNX1 subgroup (Fig. 3F), suggesting that the fusion impacted gene expression similarly to ETV6-RUNX1. We thus classified the novel fusion as LP, which supported this patient’s clinically determined standard risk classification.
Across all 253 tumors, 65 (25%) harbored a total of 89 genetic alterations that were considered rare or unreported in the tumor type being investigated (Supplemental Table S6). For example, we found AKT1 activating variants in two T-ALL patients, SJTALL030064 and SJTALL030134. AKT1 represents a targetable oncogene in epithelial cancers but it is only very rarely altered in T-ALL and its role and efficacy as a therapeutic target have yet to be investigated in this cancer type. These rare events were often suggestive of unusual mechanisms of activation of an oncogene or signaling pathway. For example, USP9X LOF was found in two cases, and potentially activates JAK/STAT signaling(43). Of these 90 rare or novel events, 23 (26%) were SVs predicted to generate fusions or enhancer hijacks identified by WGS and RNA-Seq analysis. These observations demonstrate that the spectrum of cryptic intergenic events continues to expand through use of these combined non-biased genomic approaches.
Germline cancer predisposing variants
We analyzed 156 cancer predisposition genes using germline WES and WGS data for the 300 patients who consented to return of germline results. This list included 63 genes associated with moderate to high cancer penetrance(9), as well as 93 additional genes, approximately half of which are associated with autosomal recessive cancer predisposing conditions (Supplemental Table S7). We classified variants according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) 2015 germline variant interpretation guidelines(44). Germline P, LP or variants of uncertain significance (VUS) among the 63 moderate/highly penetrant genes were reported back to providers and patients. For the remaining 93 genes, only P or LP variants were included in the final clinical reports.
We identified 58 germline P or LP variants affecting 29 genes in 55 (18%) of 300 patients (Fig. 4A, Supplemental Table S8) and 420 VUS affecting 111 genes in 230 (77%) patients (Supplemental Table S9). The prevalence of P/LP variants ranged from 10% for patients with hematologic malignancies to 40 – 50% for those with retinoblastoma and other solid tumors (Fig. 4B). Thirty-two (55%) of the germline P/LP variants affected genes not generally associated with the patient’s tumor type, such as a germline mutation in WT1 in a patient with B-ALL (SJBALL030057) or a germline mutation in BRCA2 in a patient with a diffuse intrinsic pontine glioma (SJHGG030328, Supplemental Table S8). Therefore, if targeted germline gene panels relevant to the patient’s tumor type had guided testing, these variants might have escaped detection as many of the panels would not have included the affected genes.
Figure 4. Germline variants and assessment of variant pathogenicity based on RNA data.
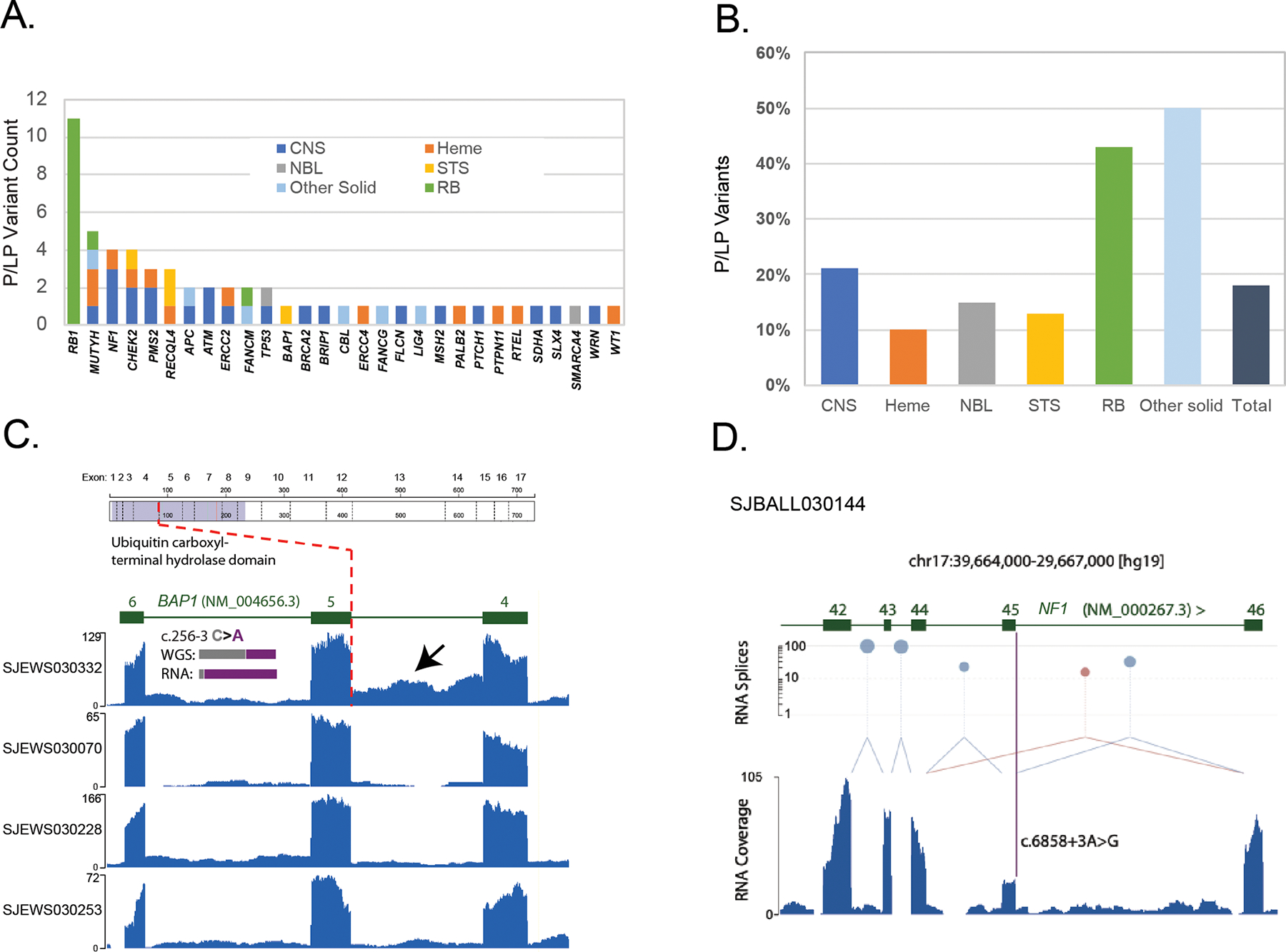
A. Numbers of germline P/LP variants, broken down by gene and tumor type. B. Proportions of germline P/LP variants, broken down by tumor type. C. BAP1 intron 4 retention in SJEWS030332 compared to other G4K Ewing sarcoma cases. Each blue histogram shows hg19-aligned RNA-Seq coverage relative to the BAP1 gene model in green (note that BAP1 is on the negative strand). The position of the exon five splice acceptor mutation is indicated by the red dotted line. Increased read coverage in the SJEWS030332 (bearing a mutation at the −3 position of exon 5) intron relative to the three other samples indicates intron 4 retention (black arrow). Inset histograms show the relative proportion of reference and variant alleles in tumor-derived WGS and RNA-Seq in grey and purple, respectively. Corresponding read counts are WGS: 32G/21T (40% variant allele) and RNA 2G/28T (93% variant allele). Above the RNA coverage plots a schematic of the BAP1 protein is shown with the location of the splice variant leading to protein truncation marked. D. NF1 exon 45 skipping in SJBALL030144. The blue histogram shows RNA-Seq coverage relative to the NF1 gene model in green. Canonical splices are shown as light blue links between exons and a non-canonical splice is shown in mauve. The height of mauve and blue lollypops is proportional to the number of splice junction reads detected plotted on a log scale on the y-axis. The purple bar indicates the position of the NF1 exon 45 splice acceptor mutation. Exon 45 expression is diminished relative to flanking exons and a non-canonical splice linking exons 44 and 46 is observed indicating an exon skipping event.
Disease relevance of novel germline variants
An integral component of our germline variant pathogenicity classification involved simultaneous review of paired tumor-normal data to determine the molecular impacts of germline variants on RNA splicing and expression, tumor mutation signatures and tumor mutation burden (TMB). For example, transcriptome data from Ewing Sarcoma, SJEWS030332, harboring a novel BAP1 intronic variant at the −3 position of exon 5 (NM_004656.3: c.256–3C>A), revealed evidence of intron retention in the tumor relative to other Ewing Sarcoma samples in the study. Fisher’s Exact test showed a significant difference in number of variant-bearing RNA-Seq intronic reads relative to PCR-free tumor WGS (p=0.047) (Fig. 4C). Intron retention, unveiled by the use of tumor RNA data, provided sufficient evidence to classify this novel germline variant as LP.
A patient with B-ALL, SJBALL030144, presented with café au lait macules but no coding mutations to explain this clinical phenotype. Analysis of tumor RNA data revealed that a germline NF1 variant at the +3 position of exon 45 (NM_000267.3: c.6858+3A>G) caused skipping of exon 45, which is predicted to lead to out-of-frame translation of the NF1 protein (Fig. 4D). This variant was classified as LP. The opposite conclusion was obtained for a germline APC variant (NM_000038.5: c.449A>G) in patient SJST030310. Analysis at an external clinical laboratory predicted creation of a de novo splice site with subsequent LOF; however, review of tumor transcriptome data provided no evidence of altered splicing, and we thus classified this variant as a VUS (Supplemental Fig. S5).
TMB and mutation signatures derived from WGS were used to establish the roles of germline variants in generating the molecular phenotypes observed in some tumors. Tumors were classified as hypermutators based on a TMB >10 mutations per Mb and ultra-mutators with >100 mutations per Mb(45). Two cases (SJHGG030335, SJHM030291) harbored germline biallelic PMS2 pathogenic variants leading to tumors that exhibited TMB and mutational signatures consistent with MMR deficiency (Supplemental Table S5). SJHGG030335 harbored >100 mutations per Mb due to an acquired POLE S459F pathogenic somatic variant. The POLE-related mutational signature was also exhibited by this tumor.
Finally, SJST030211, a squamous carcinoma of the lip, exhibited a hypermutator phenotype corresponding to COSMIC signature 11, which is linked to temozolomide treatment (Supplemental Fig. S6A,B). Consistent with this finding, this patient had received prior therapy with temozolomide for a low-grade glioma and had no detectable mutations in MMR genes in the germline or the squamous carcinoma. Thus, the mutator phenotype was caused by therapy, with this information obviating the need for follow-up germline testing for the patient and family.
As more children with cancer undergo gene panel or multiplatform sequencing, an increasing number of pathogenic germline variants are being identified in genes not generally associated with the patient’s tumor type, and in some cases, these variants are associated with adult-onset conditions or autosomal recessive cancer predisposition syndromes(4,5,9,22,46). Nevertheless, it remains poorly understood whether or how these germline variants contribute to the pathogenesis of pediatric cancers. Therefore, we reviewed all germline P/LP variants in the context of each patient’s tumor type to determine whether the germline variant might have played a causal role based on the molecular phenotypes observed in the tumor. We considered a germline variant relevant to development of the child’s tumor if the gene had a known association with the child’s tumor type, or if there was specific molecular evidence supporting a functional consequence of the mutation in the tumor. If neither of these criteria were met, the relevance of the variant in the tumor was considered to be unknown (Supplemental Fig. S7).
Using this scheme, 32 of 58 (55%) germline P/LP variants affecting 15 genes were characterized as relevant to tumor formation (“Disease Related”, Supplemental Table S8, Fig. 5). Most of these genes have known relationships with pediatric cancer, such as RB1 in retinoblastoma (RB), NF1 and PMS2 in glioblastoma (GBM), and PTCH1 in medulloblastoma. Examination of tumor WGS and RNA-Seq data, protein expression and the literature enabled classification of disease relevance in several N of 1 cases, illustrated in the following examples.
Figure 5. The impact of somatic variation in establishing disease relevance of deleterious germline variants.

Each row represents a unique patient. From left to right the columns are as follows: ‘Case ID’, the last 4 digits of the patients’ IDs (compare Supplemental Table 8. Column A). ‘Diagnosis,’ gives the disease code used internally (compare Supplemental Table S8, Column B). Note, matched superscripts indicate mutations in the same patient. ‘Germline Variant’ lists the gene and amino acid change. Germline Testing Indicated, signifies patients whose cancer or other phenotypic characteristics suggested the patient and possibly the family should undergo germline testing (black tiles). ‘Germline Genotype,’ gives the genotype of the variant indicated in the Germline Variant column. ‘Second Hit Category’ gives the genetic configuration of any genetic or epigenetic alterations affecting the remaining wildtype gene copy in the tumor. ‘Molecular Phenotype’ pertains to evidence in the DNA sequence of the tumor as to the activity of the germline variant. Molecular phenotypes included features such as splice aberrations visible in the RNA-Seq data and mutation signatures. Tumor second hits and molecular phenotype were not available for some patients due to absence or inadequate tumor for testing (see Methods). The 5 columns of tiled cells are sorted to group germline mutations by ‘Disease Related’ and then secondarily by ‘Germline Testing Indicated’.
First, we observed a missense germline TP53 variant, (A161T), in patient SJNBL030203 with NBL. Although prior studies have suggested that this variant is damaging with a dominant negative LOF effect(47–49), NBL occurs only very rarely (~1%) in individuals with pathogenic germline TP53 variants(50). Further, like most NBL(51), this patient’s tumor retained the wild-type TP53 gene copy. However, examination of tumor RNA-Seq data revealed expression only of the variant allele (Fig. 6A). Together these data suggest that there is transcriptional silencing of the wild-type gene copy, a mechanism not yet described in NBL, and we considered this germline variant relevant to the child’s tumor.
Figure 6. Evaluation of germline and tumor data to establish disease relevance.
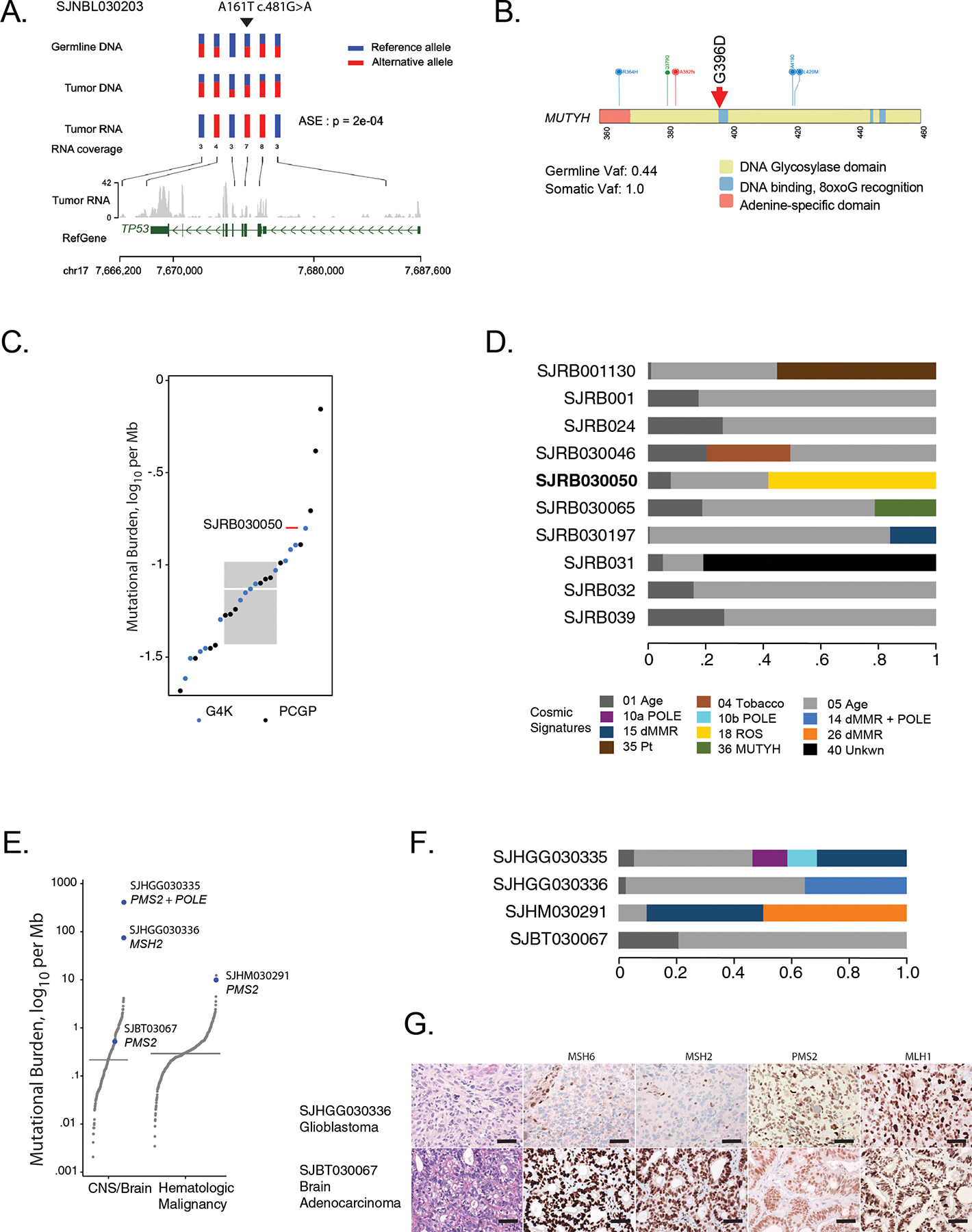
A. P53 A161T is a weakly functional likely pathogenic p53 variant in SJNBL030203. Six markers, including the germline variant C>T at chr17:7578449 (demarked by upside down triangle) are represented by 3 rows of vertical bars. The bars are colored red and blue to show the allele fractions in WGS data. The rows indicating germline and tumor DNA show most positions as heterozygous with both red and blue portions. In the RNA, all markers are mono-colored indicating that only one allele is expressed. At the location of the germline variant (C>T), the bar is completely red, indicating that only the variant allele is expressed, suggesting that the wild-type allele is transcriptionally silenced. B. A pathogenic MUTYH germline founder mutation, G396D, in RB patient SHRB030050. C. The tumor mutation burden (TMB) for SHRB030050, from WGS data, is in upper quartile compared to other RB patients including those from St Jude Cloud. Box includes the 2nd to 3rd quartiles; horizontal bar within box is median. D. Mutation signatures from WGS data from RB patients available in G4K and from the Pediatric Cancer Genome Project in the St Jude Cloud resource (https://www.stjude.cloud/). Sixty percent of tumor mutations in SHRB030050 are attributable to damage by reactive oxygen species (ROS). E. TMB plots of brain and hematologic tumors. The three tumors that are hypermutated (those with >10 mutations/Mb) are labeled with patient ID and the genes that were mutated to cause hypermutation. A fourth brain tumor with a TMB close to median was heterozygous for mutation of PMS2. The two hypermutated patients with PMS2 carried compound heterozygous mutation of the gene. The patient with the highest TMB also carried a S459F mutation of POLE. F. Mutation signatures of the patients in panel (E). G. Immunohistochemical staining for the indicated mismatch repair proteins in the brain tumors of patients SJHGG030336 and SJBT030067. Top. Infiltrative astrocytoma with severe cytologic atypia, mitotic activity, and necrosis; diagnostic of glioblastoma. Immunohistochemistry for mismatch repair proteins exhibited loss of MSH2 and MSH6, but retained staining in stromal elements. Bottom. Gastrointestinal-type adenocarcinoma arising in malignant mixed germ cell tumor. Staining for mismatch repair proteins demonstrated retained expression of MSH2, MSH6, PMS2, and MLH1. All images are 40X magnification; scale bars represent 40 microns.
Patient SJRB030050 with RB harbored a germline MUTYH founder variant (G396D, Fig. 6B) that was rendered homozygous in the tumor through CNLOH. The TMB was in the upper quartile among all RB tumors in the G4K study, as well as a larger set of RB tumors evaluated through the Pediatric Cancer Genome Project (PCGP) (Fig. 6C). Notably, over half of this tumor’s mutations were attributable to the ROS Signature 18 (Fig. 6D). These features indicate that the loss of MUTYH function contributed to the mutational processes in this tumor. As a result, the germline MUTYH variant was classified as relevant to RB development in this patient. In two other patients harboring the same germline MUTYH founder mutation (Supplemental Table S8), the variant was considered of uncertain relevance to disease because there was no loss of the wild-type allele and no ROS mutation signature in their tumors.
Two patients harbored a single heterozygous mutation in one of the MMR genes characteristic of Lynch Syndrome (LS; also known as hereditary non-polyposis colon cancer). Patient SJHGG030336 with GBM harbored a germline MSH2 variant (N653fs) and patient SJBT030067 with an unusual adenocarcinoma of the pineal region of the brain harbored a PMS2 variant (S46I). Neither tumor exhibited mutation or loss of the respective wild-type gene copy. Nevertheless, the MSH2-mutant glioblastoma (SJHGG030336) was hypermutated (Fig. 6E) and exhibited a mutation signature associated with microsatellite instability (MSI, Fig. 6F), consistent with the LS phenotype. Based on the high TMB and mutation signatures, immunohistochemical staining of this child’s tumor was performed, revealing absent MSH2 and MSH6 expression (Fig. 6G, top), and confirming a deficiency in MMR. The germline MSH2 N653fs variant was thus deemed relevant to this child’s GBM. In contrast, the pineal adenocarcinoma in patient SJBT030067 did not exhibit hypermutation (Fig. 6E) or the mutation signature of MSI (Fig. 6F) and it retained PMS2 expression (Fig. 6G, bottom). Therefore, the disease relevance of the germline PMS2 variant to development of this child’s adenocarcinoma remained uncertain.
Finally, patient SJNBL030339 with NBL harbored a pathogenic germline variant in SMARCA4 with focal deletion of the wild-type allele observed in the tumor (Fig. 5). The oncogenic potential of germline SMARCA4 variants is known to predispose to small cell carcinoma of the ovary and less commonly to rhabdoid tumor(52), but it has not been linked to NBL. However, our finding raises the possibility that SMARCA4 represents a target for mutational inactivation in NBL. Consistent with this notion, SMARCA4 is expressed in neural tissues and somatic bi-allelic SMARCA4 variants are present in 1% of NBL tumors(53). Further, the literature includes two cases of NBL with pathogenic germline SMARCA4 variants(4,54). Taken together, these data strengthen the association between germline SMARCA4 variants and predisposition to NBL.
Management implications resulting from genomic findings
Although the G4K study was non-therapeutic by design and thus not intended to match patients to a targeted therapy, we sought to investigate whether genomic findings obtained from three platform sequencing would inform diagnosis, risk stratification, cancer treatment and/or genetic counseling of the patient and family(6,55,56). We deemed variants as therapeutically actionable or potentially actionable if they were classified as P/LP from a somatic perspective and they or their downstream pathways could be targeted(6). To make these determinations, we used available evidence(46,57,58), including U.S. Food and Drug Administration approved targeted therapies, eligibility for treatment on a clinical trial based on NGS results and other professional guidelines or associated information (https://www.oncokb.org/; https://civicdb.org/home).
In total, 218 of 253 patients (86%) who underwent sequencing of paired tumor and normal tissues had at least one finding that was diagnostic (53%), prognostic (57%), therapeutically-targetable (25%), involved in cancer predisposition (18%) or some combination of these features. (Fig. 7A, B; Supplemental Table S10). Among the somatic alterations, the preponderance of targetable abnormalities included gene fusions or hotspot mutations in kinases including BRAF, ALK, MET and ABL1. We also detected several indirectly targetable mutations/fusions affecting genes upstream of the JAK-STAT pathway involving EPOR, CRLF2, SH2B3 and USP9X. Fusions of EPOR and CRLF2 and truncations of SH2B3 and USP9X are associated with Philadelphia chromosome-like B-ALL(57,59), the treatment of which with JAK1/2 inhibitors is under investigation (NCT03117751, NCT02723994).
Figure 7. Clinically actionable findings.
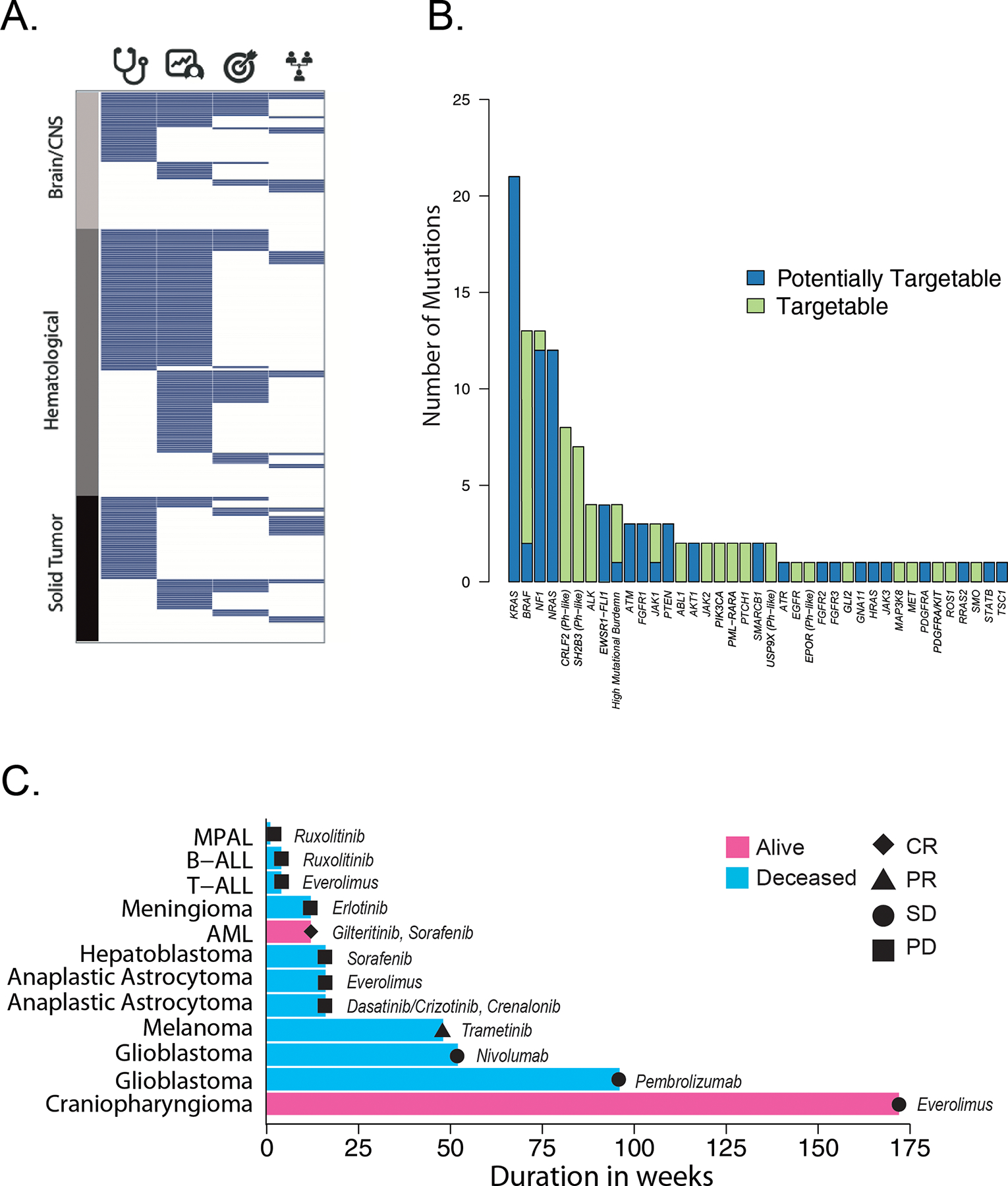
A. Tile plot summarizing clinically actionable findings in the 253 patients who had both tumor and normal tissues sequenced. Each tumor is represented as a row and is grouped according to major tumor type. Columns represent the presence (blue) or absence (white) of a diagnostic (stethoscope), prognostic (patient chart), therapeutically-relevant (target) or cancer predisposing mutation (pedigree). B. Tumors with targetable (Tier 1 and 2, green) or potentially targetable (blue) lesions, categorized by the affected gene, identified by the three-platform sequencing approach. Additional information can be found in Supplemental Tables S2, S10. C. Swimmer plot depicting patients receiving a targeted therapy matched to their tumor genetic lesion. Each bar is one patient, with the disease as labeled. Pink bars, patient is alive; blue bars, patient is deceased. Best response on the targeted therapy is as labeled. CR, complete response; PR, partial response; SD stable disease; PD, progressive disease. The drugs used are labeled adjacent to each bar. See Supplemental Table S11 for further details. MPAL, mixed phenotype acute leukemia; B-ALL, B-acute lymphoblastic leukemia; T-ALL, T acute lymphoblastic leukemia; AML, acute myeloid leukemia.
At the time of diagnosis, the majority of pediatric cancer patients, including those in the G4K study, are placed on IRB-approved clinical trials. However, 78 G4K patients with sequenced tumors presented with relapsed or metastatic tumors for which there was no clinical trial available, or they developed metastatic, relapsed or refractory disease during the course of the study. Thus, approximately one third of the patients were eligible for a change in therapy, including an NGS-directed agent. Thirty-two (41%) of the tumors in this relapsed/refractory group harbored one or more targetable or potentially targetable lesions or a targetable mutation signature. Twelve patients (38%) received a targeted agent matched to their tumor’s genetic lesion or mutational signature (Supplemental Table S11). Five of these 12 patients responded to the genomics-directed therapy (one patient with AML [complete response], one with melanoma [partial response], two with glioblastoma [stable disease, SD], and one with craniopharyngioma [SD]) (Fig. 7C). The two patients whose glioblastomas exhibited mutational signatures consistent with mismatch repair deficiency were treated with checkpoint inhibitors with one of these two surviving for two years under this therapy, while exhibiting no significant side effects. Two patients remain alive at the time of writing: SJAML030286, whose AML harbored a somatic FLT3 internal tandem duplication and was treated with sorafenib and gilteritinib-containing regimens for a total of 12 weeks followed by allogeneic stem cell transplantation; and SJBT030073, whose multiply recurrent craniopharyngioma harbored a somatic PIK3CA mutation and was treated with everolimus for 172 weeks. This patient’s tumor progressed and was subsequently partially resected with sequence analysis of the resected tumor showing loss of the PIK3CA variant.
All participants with P/LP germline variants have undergone genetic counseling and first-degree relatives have been offered clinical evaluation and targeted genetic testing; more distant family members have been encouraged to pursue counseling in their home communities. Although cascade testing is still ongoing, 27 of 77 (35%) tested individuals from 31 families have been found to harbor a cancer predisposing germline variant (Supplemental Table S8) and all have been provided with recommendations for cancer surveillance and when appropriate, risk reducing measures. To date, 19 of 58 germline P/LP variants (33%) have been confirmed as inherited, while 12 (17%) are de novo in origin. The remaining 27 are of unknown inheritance.
Comparison to gene panels
We next sought to determine what proportion of the events found by the three-platform sequencing approach would also be detected by commercially available gene panels. For this analysis we selected four commonly used somatic panels: Foundation One CDx, Foundation One Heme, Oncomine v3, and Oncokids Hotspot. All four panels detect coding SNVs and indels, including exonic ITDs and focal copy number alterations (CNAs). For a defined set of genes, Foundation One CDx detects common intronic translocation breakpoints in DNA and the other panels detect fusion transcripts in cDNA. For comparison of novel gene fusions, we required that the panel include only one partner gene of the fusion pair discovered in our data set (see Methods, “Comparison to gene panels”, Supplemental Table S12–14, for further details).
We observed that 42 – 84% of the evaluable P/LP SNVs/indels/ITDs/fusions/SVs and focal CNAs could have been detected by one or more of the panel designs (Supplemental Tables S12,13). Oncomine, applied to pediatric hematologic malignancy cases, covered the G4K variants least efficiently. This is not surprising since the panel is optimized for evaluation of adult solid tumors. The pediatric-specific Oncokids panel fared better covering 65% and 74% of reported abnormalities for brain/CNS tumors and hematologic malignancies, respectively. The best performing panel was Foundation Heme when applied to pediatric hematologic malignancy cases, covering 84% of reported abnormalities. Confining our analysis to only include clearly targetable mutations (i.e., omitting potentially targetable mutations such as KRAS G12D, EWSR1-FLI1 and others as defined in Supplemental Table S2), panel coverage ranged from 39 to 85% with Oncomine applied to pediatric hematologic malignancies detecting the fewest alterations and Foundation CDx applied to solid and brain tumors detecting the most (Supplemental Table S14).
On a per patient basis, between 36 – 69% of patients had at least one reported mutation through G4K that was not covered by one of these panel designs (Supplemental Tables S13, S14). This number includes abnormalities with a functional impact on the protein but of no known clinical significance. Considering only those variants with clear clinical impact, between 18 – 30% of patients had at least one diagnostic, prognostic, therapeutically relevant or cancer predisposing mutation that was not covered. Interestingly, two of the 12 patients with relapsed or refractory cancers who received a genomics-specified therapy harbored variants that would not have been detected by these panels. These include patient SJMEL030083 with a metastatic melanoma harboring a MAP3K8-GNG2 fusion (treated with the MEK inhibitor trametinib) and patient SJBT030076 whose clear cell meningioma harbored an EPS15L1-KLF17 fusion (treated with the EGFR inhibitor erlotinib) (Supplemental Table S11).
DISCUSSION
Through the G4K study, we performed comprehensive WGS, WES and transcriptome sequencing of tumor and/or paired normal samples from 309 prospective unselected children with cancer to quantify the prevalence and spectrum of genomic variants and evaluate the potential benefits of a three-platform sequencing approach. Several recent reports have focused on cohorts enriched in children with high risk, relapsed and refractory cancers(1,3,8,57,58,60,61). The G4K study intentionally differed from these others in that it was designed to investigate an unbiased cancer cohort, including patients with newly diagnosed standard risk tumors, as well as those with more aggressive forms of disease, to specifically ask whether novel insights into the biology of cancer can emerge from comprehensive sequence analysis of all pediatric cancers.
Incorporation of whole genome DNA sequencing enhanced the identification of clinically relevant lesions that are detected inefficiently or not at all by other genomic platforms. WGS detected inter- and intragenic SV breakpoints, which demarked the location of translocations and large deletions on whole chromosome scales. Translocations that activated oncogenes through enhancer hijacking with chromosomal breakpoints up to more than 750 Kb away from the target oncogene were present in 8% of cases. On a smaller scale, WGS detected intragenic microdeletions affecting one or two exons in 15% of tumors, some of which may not be detectable by exome-based copy number analysis, as shown in our previous study(14). Moreover, the passenger mutations in WGS identified the mutational processes supporting the role of germline lesions in MMR (MSH2, PMS2) and base excision repair genes (MUTYH) that contributed to the pathogenesis of several tumors. Taken together, up to one third of tumors in the G4K study were impacted by oncogenic events detected most sensitively by WGS. Recent results demonstrate that topologically associated chromatin domains(62) can be disrupted by focal copy number variation leading to dysregulated expression of oncogenes and tumor suppressors located in cis, often at long distances from the genomic event(63,64). These data point toward an increasingly important role in the near future for WGS combined with transcriptome sequencing in the clinical evaluation of patient tumor genomes.
Eighty six percent of patients had at least one finding that was diagnostic, prognostic, targetable, or indicating an underlying germline predisposition. One quarter of the tumors analyzed harbored a possible therapeutic target, excluding cases wherein MEK inhibitors would be an option. Benefit from MEK inhibitors, used downstream of mutant KRAS, NRAS and NF1, is more difficult to predict, but including MEK inhibitors, our potentially targetable group would comprise 43% of all tumors tested. While this proportion falls within the range of recent studies(4,5,8), it is notable given that the majority of patients on the G4K study had newly diagnosed standard risk cancers, which might be expected to harbor fewer variants when compared to high risk or relapsed/refractory cases. Potentially targetable abnormalities consisted mostly of JAK/STAT pathway mutations in B-ALL that can be treated with ruxolitinib or other JAK inhibitors; high TMB samples that can be treated with pembrolizumab, ipilimumab or additional check point inhibitors; and kinase fusions including BRAF in low grade glioma, which may be treated with tyrosine kinase or MEK inhibitors. The proportion of patients who respond to these or other therapies remains to be determined and is an active area of investigation.
During the course of the G4K study, 78 patients whose tumors were sequenced presented with or developed metastatic, relapsed or refractory disease with almost half of the tumors harboring potentially targetable lesions. Therapy was changed for 12 patients based on tumor genomic data, with one patient moving on to curative allogeneic stem cell transplantation, and four patients exhibiting prolonged disease stabilization. Recently the Zero Childhood Cancer Program reported on 38 patients receiving a genomics-specified targeted therapy, with 31% demonstrating clinical benefit(22), a proportion similar to ours. Nevertheless, despite favorable clinical outcomes in a limited number of patients, overall responses were modest across both of these studies. Together, these findings highlight that we still do not understand the full spectrum of genomic lesions that drive therapy response in most children with cancer. These findings support our premise that the collection and interrogation of comprehensive genomic information is critical if we are to further push the boundaries of cure.
Revealing an inherited cancer predisposition can impact patient management by informing genetic counseling, enabling familial genetic testing, facilitating implementation of cancer surveillance and cancer risk reducing measures, and directing cancer treatment. Our screening through G4K uncovered germline P/LP variants in 55 patients of whom almost two thirds would not have been detected based on routine clinical indications for genetic testing. Importantly, simultaneous assessment of tumor and germline genomes, including features such as TMB and mutation spectrum, and allele-specific expression, improved variant classification in several cases. Indeed, we found corroborating evidence in the tumor that at least half of the germline P/LP variants we reported were likely to be relevant to the development of the child’s tumor.
Gene panels designed to match patients with currently available targeted therapies offer a rapid turn-around time, low expense, deep sequencing coverage, and sparing of tissue when limited biopsy material is available – all valuable characteristics in the clinical setting. Moreover, the trend toward adapting panels to capture cDNA addresses the need to detect fusion transcripts, allowing for the discovery of novel fusion partners of the more commonly involved oncogenes(65). Thus, for routine standard of care, comprehensive gene panels address the medical need of the majority of patients. Nevertheless, there is much yet to be learned about childhood cancer. Among our set of 12 patients with relapsed or refractory cancers whose therapies were changed based on tumor genomic data, two received therapy as the result of novel alterations that would not have been detected by the any of the panels examined. Further, the relevance of a comprehensive view of the mutational landscape in informing understanding of tumorigenesis and management, even in the diagnostic setting, should not be underestimated. When evaluating the yield of gene panels in the G4K cohort, approximately one in five patients harbored a clinically relevant mutation that we conservatively estimated would have remained undetected using one of the panels examined. For the G4K patients sequenced at diagnosis, it remains to be determined what impact these events will have on overall outcomes.
Despite its potential benefits, comprehensive genomic profiling, such as that reported herein, is currently beyond the scope of most cancer clinical services for a variety of reasons. First, obtaining fresh frozen tissue for three-platform sequencing was not feasible for all patients. For the cancers where only limited biopsy is possible, alternative workflows involving formalin-fixed paraffin embedded samples may need to weigh the benefits of the breadth of WES against the coverage depth of a robust panel design. Second, the infrastructure and computational resources necessary to perform three-platform sequencing are substantial. Although recent innovations in cloud computing and reductions in the costs of NGS instruments and reagents are rapidly making whole genome approaches more affordable, comprehensive genomic profiling will not be standard of care for some time to come. Third, turn-around time is a major challenge to a comprehensive genomics approach in the clinical setting. Throughout the G4K study the entire workflow, from analyte preparation to data analysis, classification, and reporting was under seven weeks for 95% of the cases. Currently turn-around times are under six weeks, and we are investigating improved laboratory and computational methods reduce this still further. Finally, as we recently reported(16), there was a significant overrepresentation of African American patients who declined participation in the G4K study. While all 53 patients were later offered more focused germline testing, only 11 chose to pursue such testing. Thus, disparities exist in the context of cancer genomics and additional efforts are warranted to understand patients’ perspectives and decision making surrounding such testing to enhance future enrollment of diverse populations.
In summary, the G4K study provides evidence that three platform sequencing of tumor and paired normal tissues generates a more detailed picture of the genetic landscape of a tumor, at times revealing clinically relevant information that would go undetected if one used more targeted NGS approaches. As genomic sequencing technologies become less expensive and more widely available, their use will be an important adjunct to gene panels in the evaluation and management of children with newly diagnosed as well as relapsed or refractory cancers.
METHODS
Patient eligibility and accrual
This study was approved by an institutional review board (IRB# Pro00005011) and conducted in accordance with institutional and ethical guidelines. Over 20 months, 918 patients at St. Jude Children’s Research Hospital were assessed for enrollment in the G4K study (NCT02530658). Enrollment criteria consisted of availability of a fresh-frozen tumor sample and a paired normal (i.e., germline) sample. Tumor purity was determined by a pathologist using visual inspection of a hematoxylin & eosin (H & E) stained section of the tumor just adjacent to the portion sent for DNA and RNA extraction. For leukemia samples, tumor purity was determined based on a blast count determined by visual inspection of an H & E stained bone marrow section or by flow cytometric analysis. A tumor purity >40% tumor was preferred in order for sequencing. Tumor purity was >40% in all but 16 cases, where it ranged from 23 – 37%. Nevertheless, sequencing successful in each of these cases.
Three hundred sixty-five patients were eligible to enroll. Patients and their parents met with a research nurse, trained by certified genetic counselors, who introduced the study and answered questions. Patients were provided with written materials describing the study and a copy of the consent form to review. Interested patients were scheduled for an informed consent visit during which the research nurse reviewed key concepts, assessed parent and patient understanding and obtained written informed consent. When possible, patients also met with an oncologist, clinical geneticist or nurse practitioner to undergo collection of a full medical history and completion of a physical examination, and with a genetic counselor who obtained the family history and constructed a three-generation pedigree. A positive family history was defined as presence of at least one first- or second-degree relative with cancer diagnosed before age 50, excluding cervical and non-melanoma skin cancers (see Supplemental Table S15). This same definition was used in our prior report of germline findings from the Pediatric Cancer Genome Project(9) and it is very similar to those in other recent reports(66–68).
Nucleic acid extraction and sequencing
For solid tumors, sample adequacy and tumor cell percentage were assessed using a hematoxylin and eosin-stained section from a block of tumor tissue adjacent to the one from which DNA and RNA were extracted. For leukemias, adequacy was based on bone marrow blast count. Tumor tissue was not available for many patients with RB, craniopharyngioma, optic pathway glioma and diffuse intrinsic pontine glioma due to safety concerns around biopsy (see Supplemental Table S1 for more details).
Nucleic acid extraction, library preparation and sequencing on the Illumina HiSeq 2500/4000 were as described(14) with a single modification. Specifically, in our previous clinical genomics pilot study, WES was performed using the TruSeq DNA LT Sample Prep kit (Illumina Inc., San Diego CA), whereas in the present study it was completed using the Nextera Rapid Capture Exome Kit and the TruSeq Exome kits (both from Illumina). Samples were named according to the following convention: SJ (St. Jude), Disease code (for example RB; disease codes are listed in Supplemental Table S1), patient number, sample type (D1 is first diagnostic sample, D2 is second diagnostic sample, R1 is first relapsed sample, G1 is first germline sample, etc.). Sequencing coverage and other statistics are listed in Supplemental Table S16.
Post-sequencing quality-control (QC) thresholds established during the pilot phase of our clinical service(14), were as follows: for WGS, ≥40% coverage of coding exons at 45X, however ~80% coverage at 30x was acceptable upon the Medical Director’s review. For WES, coverage of coding exons was ideally ≥65% at 45x, but ~80% coverage at 30x was acceptable upon the Medical Director’s review. For RNA-Seq, ideally ≥15% of coding exons were covered at 45x, but 20% at 30x was acceptable upon the Medical Director’s review. Additional sequencing was performed in some samples in order to meet acceptable coverage QC thresholds.
Variant classification and reporting
WGS, WES and RNA-Seq data were aligned and variants called and annotated using previously published algorithms(19,33,69,70) and automated pipeline infrastructure(14). Genome analysts reviewed variant calls and presented each case to a multidisciplinary tumor board consisting of representatives from Pathology, Computational Biology and Oncology. Since our objective was to report SNVs/indels and CNAs from the somatic and germline context, as well as somatically acquired SVs, we adopted a unified reporting nomenclature that encompassed every variant type. As a convention, in referring to regional or focal copy number changes, we use CNA in the somatic context, and CNV in the germline context. We classified germline SNVs and indels according to the ACMG 2015 guidelines(44) and subsequently adopted the same nomenclature for other variant types. We reported germline P, LP and VUS found among 63 high-risk cancer predisposition genes, including those recommended by ACMG v2.0(71) and those for which clinical management recommendations exist or are under development(44), as well as P/LP variants in 93 additional cancer predisposition genes. Germline CNVs were assessed for their functional impact on the gene of interest. If a CNV caused a loss of function, we applied the PVS1 tag(72) and if it was rare or absent from databases such as DGV (http://dgv.tcag.ca/dgv/app/home), we also attached the PM2 tag, allowing us to classify these variants as P/LP in keeping with germline SNV/indel calls.
Somatic variants with a clear or likely impact on the function of a cancer-relevant gene were classified as P or LP, respectively, and those with an uncertain impact were classified as VUS. Somatic variants classified as VUS were not included in clinical tumor reports. When assessing the pathogenicity of somatic SNVs/indels, we considered multiple lines of evidence obtained using functional prediction algorithms and literature mining, as well as recurrence in the PCGP(20) and NCI-TARGET(15) databases, other pediatric cancer mutation data in the St. Jude PeCAN portal(73), and adult cancer data from the Catalogue of Somatic Mutations in Cancer (COSMIC)(74). Whenever possible, we used tumor RNA data to establish the functional impact for novel SVs by seeking evidence of truncation or in-frame gene fusion, and for gene amplification or enhancer hijacks, we used these data to examine gene expression.
We based our assessment criteria for clinical actionability on the system described by ACMG/ASCO/AMP(56,75). For SNVs/indels we called any variant with Tier IA/B or tier IIC as actionable as these comprised diagnostic, prognostic and therapeutically-relevant lesions with a high level of supporting evidence that an oncologist might reasonably act upon. Several lesions that did not meet this stringent threshold were mostly placed in Tier IID and labeled as “potentially actionable” (Supplemental Table S2). For CNAs, we considered Tier 1A/B and 2 variants as actionable(75) and applied these same assessment criteria to SVs.
Bioinformatics analysis
Bioinformatics analysis and variant review were as described(14). Briefly, sequence reads were aligned using BWA(76) for DNA or BWA and STAR via our Strongarm pipeline(77) for RNA. Sequence variants were called using Bambino(69), DNA SVs using CREST(70), RNA SVs including ITD using CICERO(78), a local assembly-based algorithm that integrates RNA-seq read support with extensive annotation for candidate ranking and available online at: https://www.stjude.cloud/tools/rapid_RNA-Seq), and CNVs and allelic imbalance with CONSERTING(33). Variants were annotated using the Medal Ceremony/PeCanPIE pipeline(19) (available online at: https://pecan.stjude.cloud/pie). We estimated TMB by counting all exonic and UTR variants that passed manual review by a genome analyst in addition to high-quality intronic and non-coding somatic variants falling outside of repeat regions. For genome-wide calculations, we used only somatically acquired high quality variants whose Fisher’s Exact Test p-value for somatic origin was <0.05. To calculate mutations per Mb of DNA, we took all high quality somatic variants from WGS and divided by 1445 Mb - the total amount of genome capable of generating a mutation call consisting of Tier 1 (coding exons, splice regions and UTRs): 108,721,345 bp, Tier 2 (potential regulatory regions): 163,138,648 bp, and Tier 3 (intergenic and intronic regions): 1,173,180,850. Tier 4 (repeat regions): 1,448,043,873 were omitted from the calculation since our mutation calling pipeline masks these regions. For mutational burden in exome only regions, we used 108.7 Mb as the denominator - the size of Tier 1 regions listed above. B-ALL and T-ALL 2D t-SNE distributions used data and methods described previously by Gu et al. (2019)(42) and Liu et al. (2017)(41). All additional analyses used standard parameters unless stated otherwise. Gene expression was quantified using the Gencode 75 gene model and HTSeq (version 0.11.1)(79) and normalized between samples using the DESeq2 (version 1.26) variance stabilizing transformation function(80). SigProfilerSingleSample was used to test for the presence and abundance of the COSMIC v3.1 Mutation Signatures(36), as described(45). Briefly, samples with 400 or more mutations (485 samples) were analyzed for 46 of the COSMIC signatures (excluding 23 COSMIC signatures which were rarely detected and manually found to be spurious in each positive case. Samples with fewer than 400 mutations (583 samples) were analyzed for a core set of 13 signatures (1, 2, 3, 5, 7a, 7b, 7c, 7d, 8, 13, 18, 36, and 40) which could be reliably detected in low mutation burden samples and common in pediatric cancers (Supplemental Table S5).
Analysis of putative enhancer hijacks
SJTALL030071 harbored a non-canonical interchromosomal translocation juxtaposing the N-Me (NOTCH MYC enhancer) enhancer to the vicinity of TLX3 caused upregulation of the gene. To investigate further the possibility of activation of TLX3 by somatic regulatory non-coding variants, we analyzed WGS and RNA-seq of this T-ALL to identify cis-activated genes that have outlier expression using cis-X (version 1.4.0) a computational method for discovering regulatory noncoding variants in cancer by integrating whole-genome and transcriptome sequencing data from a single cancer sample(81). cis-X first finds aberrantly cis-activated genes that exhibit allele-specific expression accompanied by an elevated outlier expression. For each gene, outlier high expression of a cancer sample of interest was determined by comparing its expression level to those of reference samples with the same tissue type. A null distribution of ‘leave-one-out (LOO)’ t-statistic score was established using the reference samples. This was then used to determine the false discovery rate (FDR) of the LOO t-statistic score of a cancer sample of interest; those with an FDR < 0.05 were retained as having significant outlier high expression. A minimum of 20 cases is required for this analysis.
Data used for reference expression matrix were obtained from publicly available data on St. Jude Cloud or NCI TARGET to meet the minimum sample size criteria. For T-ALL samples such as SJTALL030071, RNA-seq data from 264 T-ALLs profiled by NCI’s Therapeutically Applicable Research to Generate Effective Treatments (TARGET)(41) was used as the reference expression matrix for evaluation of outlier expression status. For the remaining cancer subtypes that harbor candidate enhancer hijacking events (e.g., medulloblastoma (MB), B-lineage ALL (B-ALL) and acute myeloid leukemia (AML)), reference expression matrix were prepared by querying the relevant cancer diagnosis and selecting File Type “Feature Counts” (pre-calculated using Gencode V31) using the Data Browser of Genomic Platform of the St Jude Cloud(82). Feature counts were subsequently converted to reference FPKM matrix using DEseq2 for calculating outlier high expression status of samples presented in Supplemental Table S3. The AML reference expression matrix did not include therapy-related myeloid neoplasms. The number of samples used to construct the reference expression matrix for each cancer subtype is recorded in Supplemental Table S3.
For case SJTALL030071, we identified 18 genes that exhibit both allele-specific expression and outlier high expression surrounding TLX3 (Supplemental Table S17). None of the genes other the TLX3 play a known role in T-cell development or cancer. There were 29 consecutive SNPs with heterozygous genotypes in tumor DNA around the TLX3 locus exhibiting mono-allelic expression in tumor RNA (see Fig. 3C).
Comparison to gene panels
We selected four commercially available gene panels: Foundation CDx, Foundation Heme, Thermo Fisher Oncomine v3, and CHLA Oncokids in order to represent the breadth and diversity of clinical gene panels. Foundation CDx is a large general-use DNA-based panel, Foundation Heme is a large, combined DNA/RNA panel focused on hematological malignancies, Thermo Fisher Oncomine v3 is a smaller DNA/RNA panel focused on adult cancers and Oncokids is a pediatric-specific version of the Oncomine platform used in support of NCI’s pediatric MATCH trial (see Supplemental Tables 12 footnotes for further details).
All four panels are capable of detecting SNVs/indels, gene fusions and focal CNAs at the gene level. However, gross chromosomal changes cannot be officially reported by these assays. Additionally, complex copy number abnormalities such as chromothripsis and regions of loss of heterozygosity are not reported. Of 253 G4K patients, 75 patients (29.6%), predominantly with heme malignancies, had diagnostic, or prognostic gross chromosomal abnormalities including hyperdiploidy, hypodiploidy and iAMP21. However, an additional karyotype, FISH or microarray test could be run to detect these abnormalities. In order to make a direct comparison of reportable gene-level alterations we omitted gross chromosomal gains and losses from the analysis.
We arrived at a conservative estimate of how many reported and clinically actionable genes are not represented on the exemplar panels as follows:
We included both somatic and germline mutations in the comparison since our combined test reported both.
We used the coding sequence DNA portion of the panel assay for SNVs/indels/ITDs and focal CNAs. We assumed the observed alteration would have been captured and reported at the tumor VAF observed in the G4K patient. This is especially relevant for CNVs as panels generally only report homozygous deletions or multiple copy gains.
We used the RNA panel gene lists combined with intronic sequences designed to capture recurrent translocation breakpoints for fusions, enhancer hijacks and intragenic SVs.
We required only one of the partner genes to be present on the panel for gene fusions. This approach was permissive and enabled a conservative estimate of how many events might be detected by these technologies; we noted in our previous study that detection of gene fusions with low expression such as KMT2A rearrangement and KIAA1549-BRAF benefits from a multiplatform approach(14).
For enhancer hijacks, we were unable to determine if the translocation breakpoints that we observed by WGS were featured on current panel designs. Further, we did not assume outlier expression would be required on the panel data. As such we took the same approach as for gene fusions, requiring only one of the partner loci being present on the panel design.
If a G4K variant was covered by a given panel, it was tallied as detected (Supplemental Table S12). We evaluated all variants within each patient and if one variant was missed by a given panel, the patient was tallied as a miss by that panel (Supplemental Table S13,14).
Visualization of multi-omics data
To examine the impact of somatic variants within tumors, we used visual exploration of aggregated multi-omics data from PCGP, TARGET and other studies(15,20,41,42) using GenomePaint (https://genomepaint.stjude.cloud/).
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Pediatric cancers are driven by diverse genomic lesions and sequencing has proven useful in evaluating high risk and relapsed/refractory cases. We show that combined whole genome, exome, and RNA-sequencing of tumor and paired normal tissues enables identification and characterization of genetic drivers across the full spectrum of pediatric cancers.
ACKNOWLEDGEMENTS:
We thank the patients and families who participated in the G4K study, as well as each of the clinical and research staff who helped to make this study possible. We also gratefully acknowledge David Finkelstein for his assistance in preparing the final Figures.
This work was supported by funding provided by the American Lebanese Syrian Associated Charities and grants R01CA216354 (J Zhang, X Chen, X Zhou) and R01CA216391 (J Zhang, X Zhou) from the National Cancer Institute.
Footnotes
CONFLICTS OF INTEREST:
The authors have no conflicts of interest to disclose.
Software/Code Availability
All software used in this study has been previously published; references are listed throughout the manuscript or are available in St. Jude Cloud. Further, our gene fusion, variant annotation and mutational signature pipelines are available on St. Jude Cloud (https://www.stjude.cloud).
Data availability
All raw and intermediate data are available as a free community resource hosted in St. Jude Cloud: https://www.stjude.cloud under accession number SJC-1004. Study level visualizations are available at https://pecan.stjude.cloud. Germline variants returned to patients during the study have been deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/submitters/506672/).
REFERENCES
- 1.Chang W, Brohl AS, Patidar R, Sindiri S, Shern JF, Wei JS, et al. MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clin Cancer Res 2016;22(15):3810–20 doi 10.1158/1078-0432.CCR-15-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol 2017;19(5):699–709 doi 10.1093/neuonc/now254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oberg JA, Glade Bender JL, Sulis ML, Pendrick D, Sireci AN, Hsiao SJ, et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med 2016;8(1):133 doi 10.1186/s13073-016-0389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol 2016;2(5):616–24 doi 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med 2019;11(1):32 doi 10.1186/s13073-019-0644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marks LJ, Oberg JA, Pendrick D, Sireci AN, Glasser C, Coval C, et al. Precision Medicine in Children and Young Adults with Hematologic Malignancies and Blood Disorders: The Columbia University Experience. Front Pediatr 2017;5:265 doi 10.3389/fped.2017.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin JJ, Riely GJ, Shaw AT. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov 2017;7(2):137–55 doi 10.1158/2159-8290.CD-16-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 2015;314(9):913–25 doi 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 2015;373(24):2336–46 doi 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran TH, Hunger SP. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin Cancer Biol 2020;S1044-579X(20)30218-2 doi 10.1016/j.semcancer.2020.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Brok J, Lopez-Yurda M, Tinteren HV, Treger TD, Furtwangler R, Graf N, et al. Relapse of Wilms’ tumour and detection methods: a retrospective analysis of the 2001 Renal Tumour Study Group-International Society of Paediatric Oncology Wilms’ tumour protocol database. Lancet Oncol 2018;19(8):1072–81 doi 10.1016/S1470-2045(18)30293-6. [DOI] [PubMed] [Google Scholar]
- 12.Pappo AS, Anderson JR, Crist WM, Wharam MD, Breitfeld PP, Hawkins D, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 1999;17(11):3487–93 doi 10.1200/JCO.1999.17.11.3487. [DOI] [PubMed] [Google Scholar]
- 13.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548(7667):297–303 doi 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rusch M, Nakitandwe J, Shurtleff S, Newman S, Zhang Z, Edmonson MN, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun 2018;9(1):3962 doi 10.1038/s41467-018-06485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555(7696):371–6 doi 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howard Sharp KM, Jurbergs N, Ouma A, Harrison L, Gerhardt E, Taylor L, et al. Factors Associated with Declining to Participate in a Pediatric Oncology Next Generation Sequencing Study. JCO Precis Oncol 2020;4:202–11 doi 10.1200/PO.19.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goschzik T, Gessi M, Dreschmann V, Gebhardt U, Wang L, Yamaguchi S, et al. Genomic Alterations of Adamantinomatous and Papillary Craniopharyngioma. J Neuropathol Exp Neurol 2017;76(2):126–34 doi 10.1093/jnen/nlw116. [DOI] [PubMed] [Google Scholar]
- 18.Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018;562(7727):373–9 doi 10.1038/s41586-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edmonson MN, Patel AN, Hedges DJ, Wang Z, Rampersaud E, Kesserwan CA, et al. Pediatric Cancer Variant Pathogenicity Information Exchange (PeCanPIE): a cloud-based platform for curating and classifying germline variants. Genome Res 2019;29(9):1555–65 doi 10.1101/gr.250357.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, et al. The Pediatric Cancer Genome Project. Nat Genet 2012;44(6):619–22 doi 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014;371(11):1005–15 doi 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 2020;26(11):1742–53 doi 10.1038/s41591-020-1072-4. [DOI] [PubMed] [Google Scholar]
- 23.He B, Gao P, Ding YY, Chen CH, Chen G, Chen C, et al. Diverse noncoding mutations contribute to deregulation of cis-regulatory landscape in pediatric cancers. Sci Adv 2020;6(30):eaba3064 doi 10.1126/sciadv.aba3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014;511(7510):428–34 doi 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmerman MW, Liu Y, He S, Durbin AD, Abraham BJ, Easton J, et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov 2018;8(3):320–35 doi 10.1158/2159-8290.CD-17-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehman NL, Usubalieva A, Lin T, Allen SJ, Tran QT, Mobley BC, et al. Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 2019;7(1):42 doi 10.1186/s40478-019-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haferlach C, Bacher U, Grossmann V, Schindela S, Zenger M, Kohlmann A, et al. Three novel cytogenetically cryptic EVI1 rearrangements associated with increased EVI1 expression and poor prognosis identified in 27 acute myeloid leukemia cases. Genes Chromosomes Cancer 2012;51(12):1079–85 doi 10.1002/gcc.21992. [DOI] [PubMed] [Google Scholar]
- 28.Paulsson K, Cazier JB, Macdougall F, Stevens J, Stasevich I, Vrcelj N, et al. Microdeletions are a general feature of adult and adolescent acute lymphoblastic leukemia: Unexpected similarities with pediatric disease. Proc Natl Acad Sci U S A 2008;105(18):6708–13 doi 10.1073/pnas.0800408105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waanders E, Scheijen B, van der Meer LT, van Reijmersdal SV, van Emst L, Kroeze Y, et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet 2012;8(2):e1002533 doi 10.1371/journal.pgen.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009;360(5):470–80 doi 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelhardt KR, Xu Y, Grainger A, Germani Batacchi MG, Swan DJ, Willet JD, et al. Identification of Heterozygous Single- and Multi-exon Deletions in IL7R by Whole Exome Sequencing. J Clin Immunol 2017;37(1):42–50 doi 10.1007/s10875-016-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchuk DS, Crooks K, Strande N, Kaiser-Rogers K, Milko LV, Brandt A, et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One 2018;13(12):e0209185 doi 10.1371/journal.pone.0209185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Gupta P, Wang J, Nakitandwe J, Roberts K, Dalton JD, et al. CONSERTING: integrating copy-number analysis with structural-variation detection. Nat Methods 2015;12(6):527–30 doi 10.1038/nmeth.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips DH. Mutational spectra and mutational signatures: Insights into cancer aetiology and mechanisms of DNA damage and repair. DNA Repair (Amst) 2018;71:6–11 doi 10.1016/j.dnarep.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Hoeck A, Tjoonk NH, van Boxtel R, Cuppen E. Portrait of a cancer: mutational signature analyses for cancer diagnostics. BMC Cancer 2019;19(1):457 doi 10.1186/s12885-019-5677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020;135(1):41–55 doi 10.1182/blood.2019002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 2014;46(2):116–25 doi 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernard OA, Busson-LeConiat M, Ballerini P, Mauchauffe M, Della Valle V, Monni R, et al. A new recurrent and specific cryptic translocation, t(5;14)(q35;q32), is associated with expression of the Hox11L2 gene in T acute lymphoblastic leukemia. Leukemia 2001;15(10):1495–504 doi 10.1038/sj.leu.2402249. [DOI] [PubMed] [Google Scholar]
- 40.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014;20(10):1130–7 doi 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 2017;49(8):1211–8 doi 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 2019;51(2):296–307 doi 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartzman O, Savino AM, Gombert M, Palmi C, Cario G, Schrappe M, et al. Suppressors and activators of JAK-STAT signaling at diagnosis and relapse of acute lymphoblastic leukemia in Down syndrome. Proc Natl Acad Sci U S A 2017;114(20):E4030–E9 doi 10.1073/pnas.1702489114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–24 doi 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017;171(5):1042–56 e10 doi 10.1016/j.cell.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555(7696):321–7 doi 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 47.Freitas AC, Opiniao A, Fragoso S, Nunes H, Santos M, Clara A, et al. Men seeking counselling in a Breast Cancer Risk Evaluation Clinic. Ecancermedicalscience 2018;12:804 doi 10.3332/ecancer.2018.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 2018;50(10):1381–7 doi 10.1038/s41588-018-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A 2003;100(14):8424–9 doi 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 2015;33(21):2345–52 doi 10.1200/JCO.2014.59.5728. [DOI] [PubMed] [Google Scholar]
- 51.Van Maerken T, Vandesompele J, Rihani A, De Paepe A, Speleman F. Escape from p53-mediated tumor surveillance in neuroblastoma: switching off the p14(ARF)-MDM2-p53 axis. Cell Death Differ 2009;16(12):1563–72 doi 10.1038/cdd.2009.138. [DOI] [PubMed] [Google Scholar]
- 52.Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol 2018;35(3):193–8 doi 10.1053/j.semdp.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Bellini A, Bessoltane-Bentahar N, Bhalshankar J, Clement N, Raynal V, Baulande S, et al. Study of chromatin remodeling genes implicates SMARCA4 as a putative player in oncogenesis in neuroblastoma. Int J Cancer 2019;145(10):2781–91 doi 10.1002/ijc.32361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, Wilson CL, Easton J, Thrasher A, Mulder H, Liu Q, et al. Genetic Risk for Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J Clin Oncol 2018;36(20):2078–87 doi 10.1200/JCO.2018.77.8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joseph L, Cankovic M, Caughron S, Chandra P, Emmadi R, Hagenkord J, et al. The Spectrum of Clinical Utilities in Molecular Pathology Testing Procedures for Inherited Conditions and Cancer: A Report of the Association for Molecular Pathology. J Mol Diagn 2016;18(5):605–19 doi 10.1016/j.jmoldx.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19(1):4–23 doi 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pikman Y, Tasian SK, Sulis ML, Stevenson K, Blonquist TM, Apsel Winger B, et al. Matched Targeted Therapy for Pediatric Patients with Relapsed, Refractory, or High-Risk Leukemias: A Report from the LEAP Consortium. Cancer Discov 2021;11(6):1424–39 doi 10.1158/2159-8290.CD-20-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients - The INFORM pilot study. Eur J Cancer 2016;65:91–101 doi 10.1016/j.ejca.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 59.Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017;130(19):2064–72 doi 10.1182/blood-2017-06-743252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen CE, Laetsch TW, Mody R, Irwin MS, Lim MS, Adamson PC, et al. Target and Agent Prioritization for the Children’s Oncology Group-National Cancer Institute Pediatric MATCH Trial. J Natl Cancer Inst 2017;109(5) doi 10.1093/jnci/djw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harttrampf AC, Lacroix L, Deloger M, Deschamps F, Puget S, Auger N, et al. Molecular Screening for Cancer Treatment Optimization (MOSCATO-01) in Pediatric Patients: A Single-Institutional Prospective Molecular Stratification Trial. Clin Cancer Res 2017;23(20):6101–12 doi 10.1158/1078-0432.CCR-17-0381. [DOI] [PubMed] [Google Scholar]
- 62.Szabo Q, Bantignies F, Cavalli G. Principles of genome folding into topologically associating domains. Sci Adv 2019;5(4):eaaw1668 doi 10.1126/sciadv.aaw1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akdemir KC, Le VT, Chandran S, Li Y, Verhaak RG, Beroukhim R, et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet 2020;52(3):294–305 doi 10.1038/s41588-019-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li L, Barth NKH, Pilarsky C, Taher L. Cancer Is Associated with Alterations in the Three-Dimensional Organization of the Genome. Cancers (Basel) 2019;11(12) doi 10.3390/cancers11121886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chmielecki J, Bailey M, He J, Elvin J, Vergilio JA, Ramkissoon S, et al. Genomic Profiling of a Large Set of Diverse Pediatric Cancers Identifies Known and Novel Mutations across Tumor Spectra. Cancer Res 2017;77(2):509–19 doi 10.1158/0008-5472.CAN-16-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goudie C, Coltin H, Witkowski L, Mourad S, Malkin D, Foulkes WD. The McGill Interactive Pediatric OncoGenetic Guidelines: An approach to identifying pediatric oncology patients most likely to benefit from a genetic evaluation. Pediatr Blood Cancer 2017;64(8) doi 10.1002/pbc.26441. [DOI] [PubMed] [Google Scholar]
- 67.Hampel H, Bennett RL, Buchanan A, Pearlman R, Wiesner GL, Guideline Development Group ACoMG, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 2015;17(1):70–87 doi 10.1038/gim.2014.147. [DOI] [PubMed] [Google Scholar]
- 68.Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A 2017;173(4):1017–37 doi 10.1002/ajmg.a.38142. [DOI] [PubMed] [Google Scholar]
- 69.Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics 2011;27(6):865–6 doi 10.1093/bioinformatics/btr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 2011;8(8):652–4 doi 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19(2):249–55 doi 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 72.Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat 2018;39(11):1517–24 doi 10.1002/humu.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 2016;48(1):4–6 doi 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39(Database issue):D945–50 doi 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mikhail FM, Biegel JA, Cooley LD, Dubuc AM, Hirsch B, Horner VL, et al. Technical laboratory standards for interpretation and reporting of acquired copy-number abnormalities and copy-neutral loss of heterozygosity in neoplastic disorders: a joint consensus recommendation from the American College of Medical Genetics and Genomics (ACMG) and the Cancer Genomics Consortium (CGC). Genet Med 2019;21(9):1903–16 doi 10.1038/s41436-019-0545-7. [DOI] [PubMed] [Google Scholar]
- 76.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tian L, Li Y, Edmonson MN, Zhou X, Newman S, McLeod C, et al. CICERO: a versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol 2020;21(1):126 doi 10.1186/s13059-020-02043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11(10):R106 doi 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu Y, Li C, Shen S, Chen X, Szlachta K, Edmonson MN, et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat Genet 2020;52(8):811–8 doi 10.1038/s41588-020-0659-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, Brady SW, et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov 2021;11(5):1082–99 doi 10.1158/2159-8290.CD-20-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and intermediate data are available as a free community resource hosted in St. Jude Cloud: https://www.stjude.cloud under accession number SJC-1004. Study level visualizations are available at https://pecan.stjude.cloud. Germline variants returned to patients during the study have been deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/submitters/506672/).