

Abstract
For CASP14, we developed deep learning-based methods for predicting homo-oligomeric and hetero-oligomeric contacts and used them for oligomer modeling. To build structure models, we developed an oligomer structure generation method that utilizes predicted inter-chain contacts to guide iterative restrained minimization from random backbone structures. We supplemented this gradient-based fold-and-dock method with template-based and ab initio docking approaches using deep learning-based subunit predictions on 29 assembly targets. These methods produced oligomer models with summed Z-scores 5.5 units higher than the next best group, with the fold-and-dock method having the best relative performance. Over the eight targets for which this method was used, the best of the five submitted models had average oligomer TM-score of 0.71 (average oligomer TM-score of the next best group: 0.64), and explicit modeling of inter-subunit interactions improved modeling of six out of 40 individual domains (ΔGDT-TS > 2.0).
Introduction
Hetero and homo-oligomeric states of proteins are critical to their function1–3. Many computational methods have been developed to predict oligomer structures4–9, but good performance has required matching oligomer template structures or utilization of experimental data, and accurate subunit structures10,11. Protein inter-chain contact predictions have been utilized for oligomer modeling12–14, but accuracy has been limited due to the limited predicted contact accuracy and the lack of efficient modeling methods.
In this CASP, we aimed to improve oligomer modeling performance by 1) developing deep learning-based inter-chain contact prediction methods for both homo-oligomeric and hetero-oligomeric complexes, 2) modeling entire complex structures from scratch guided by predicted intra-chain distances and inter-chain contacts when available, and 3) taking advantage of the recent progress in tertiary structure modeling15–18 for oligomer template search.
Methods
Overall pipeline
We used three different approaches to generate oligomer structures depending on the information available as depicted in Figure 1. We first generated multiple sequence alignments (MSA) by HHblits19 searches against UniRef3020 and metagenomic databases provided by JGI21 (step 0). Inter-chain contacts were predicted using GREMLIN22,23 or deep learning techniques (described in next section) based on MSAs (step 1, details are described in the next section). We also searched for oligomer templates based on sequence similarity using HHsearch19 and structure similarity using TM-align24 (step 2).
Figure 1.
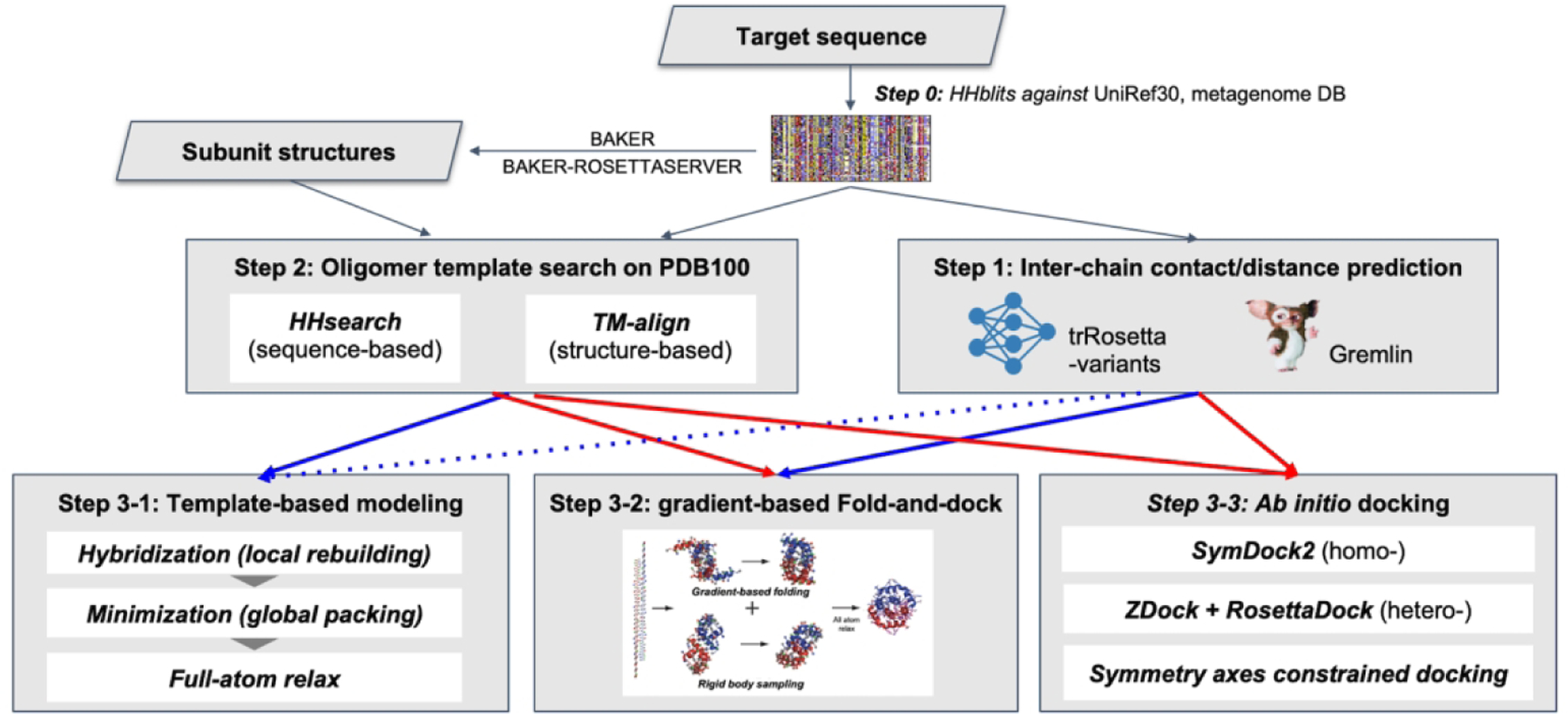
The oligomer structure modeling procedure used by the BAKER-experimental group
Based on these results, oligomer models were generated using one of three approaches: template-based modeling (step 3–1), gradient-based fold and dock (step 3–2), or ab initio docking (step 3–3). The approaches taken for each target are summarized in Table 1. Details for steps 1–3 are provided in the following sections.
Table 1.
Summary of modeling strategies and performances
| Target | Difficulty | Inter-chain contact† | Modeling method | Model 1 | Best out of 5 (based on Z-score) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Z-score‡ | ICS | TM-score (oligo) | Z-score§ | ICS | TM-score (oligo) |
||||
| H1036¶ | medium | No | Template | 0.84 | 0.68 | 0.70 | 1.17 | 0.72 | 0.71 |
| H1036v0¶ | medium | No | Template | 0.93 | 0.27 | 0.69 | 0.92 | 0.27 | 0.69 |
| H1045 | medium | No | Template | 1.05 | 0.71 | 0.87 | 1.35 | 0.77 | 0.87 |
| H1047 | hard | Yes (G) | Ab initio | 1.35 | 0.04 | 0.39 | 1.54 | 0.04 | 0.38 |
| H1060v1 | medium | No | Ab initio | 0.94 | 0.06 | 0.31 | 1.09 | 0.08 | 0.31 |
| H1060v2 | medium | No | Template | 0.38 | 0.09 | 0.86 | 0.34 | 0.10 | 0.88 |
| H1060v3 | medium | No | Template | 0.43 | 0.01 | 0.75 | 0.85 | 0.12 | 0.84 |
| H1060v4 | medium | No | Template | 0.74 | 0.22 | 0.75 | 0.93 | 0.21 | 0.73 |
| H1060v5 | medium | Yes (G) | Template | 1.52 | 0.48 | 0.95 | 1.67 | 0.50 | 0.95 |
| H1065 | hard | Yes (DL) | Fold-and-dock | 1.74 | 0.40 | 0.79 | 1.82 | 0.40 | 0.79 |
| H1072 | medium | Yes (DL) | Fold-and-dock | 0.10 | 0.04 | 0.34 | 0.28 | 0.03 | 0.37 |
| H1081v0 | medium | No | Ab initio | 1.46 | 0.35 | 0.97 | 1.59 | 0.35 | 0.97 |
| H1097 | medium | Yes (T) | Fold-and-dock | 2.13 | 0.44 | 0.73 | 1.99 | 0.44 | 0.74 |
| T1032 | easy | No | Template | 1.08 | 0.38 | 0.69 | 1.10 | 0.40 | 0.68 |
| T1034 | medium | No | Template | −0.81 | 0.00 | 0.17 | −0.38 | 0.00 | 0.23 |
| T1038 | hard | No | Ab initio | −0.58 | 0.00 | 0.17 | 0.36 | 0.01 | 0.20 |
| T1048 | medium | Yes (DL) | Fold-and-dock | 3.09 | 0.50 | 0.59 | 4.29 | 0.58 | 0.83 |
| T1052 | easy | No | Template | 0.63 | 0.51 | 0.69 | 0.72 | 0.51 | 0.69 |
| T1054 | hard | No | Ab initio | 0.13 | 0.00 | 0.44 | 0.81 | 0.00 | 0.52 |
| T1061 | hard | No | Template | 1.85 | 0.15 | 0.64 | 1.97 | 0.17 | 0.69 |
| T1070 | hard | No | Template | 0.94 | 0.06 | 0.31 | 2.10 | 0.10 | 0.37 |
| T1078 | medium | No | Ab initio | 0.19 | 0.00 | 0.54 | 2.50 | 0.25 | 0.67 |
| T1080 | hard | Yes (DL) | Fold-and-dock | 1.92 | 0.12 | 0.55 | 2.60 | 0.13 | 0.61 |
| T1083 | medium | Yes (DL) | Fold-and-dock | 1.48 | 0.23 | 0.63 | 1.60 | 0.23 | 0.63 |
| T1084 | medium | Yes (DL) | Fold-and-dock | 2.17 | 0.81 | 0.92 | 2.20 | 0.84 | 0.91 |
| T1087 | medium | Yes (DL) | Fold-and-dock | 2.27 | 0.36 | 0.79 | 2.86 | 0.36 | 0.79 |
| T1099v0 | medium | No | Template | −0.23 | 0.03 | 0.24 | 0.22 | 0.02 | 0.45 |
| T1099v1 | medium | No | Template | 0.15 | 0.00 | 0.55 | 0.27 | 0.00 | 0.55 |
| T1099v2 | medium | No | Template | 0.75 | 0.13 | 0.60 | 0.89 | 0.16 | 0.60 |
G: GREMLIN; DL: Deep learning-based methods; T: Partial templates
Calculated on model 1 submissions
Calculated on all model submissions
Having a completely wrong prediction for the antigen-antibody interface
Step 1: Inter-chain contact prediction using trRosetta-homo and trRosetta-discont
For homo-oligomer targets, we developed a deep learning-based homo-oligomer contact prediction method called trRosetta-homo to predict inter-chain contacts from MSAs generated by searching sequence databases. trRosetta-homo (Figure 2A) is based on a 2D residual convolution network having the same architecture as the original trRosetta16 except for the last layer. It was trained to predict not only intra-chain distances and orientations but also inter-chain contacts at a 12Å Cβ-Cβ distance threshold so that the network could distinguish inter-chain coevolution signals from intra-chain signals. The input features for the network are derived from MSAs, including 1) one-hot-encoded amino acid sequence of the query protein, 2) position-specific frequency matrix, 3) positional entropy, and 4) coevolution couplings derived from the inverse of the shrunk covariance matrix. The network was trained on 6,932 homo-oligomer structures from the original trRosetta training set16. High-probability GREMLIN contacts which were not made within the monomer were also treated as potential inter-chain contacts. The predicted inter-chain contacts for homo-oligomers were converted to the Rosetta bounded restraints (contact probability > 0.95) or sigmoidal restraints (0.5 < contact probability < 0.95)25 (shapes shown in Figure 2A) and were used to guide the overall sampling process and to select final models. For homo-oligomers having more than two subunits, each restraint was treated as an ambiguous restraint: distances were evaluated for the relevant residue pair over all pairs of chains, and the constraint score was taken for the one best matching the restraint.
Figure 2.
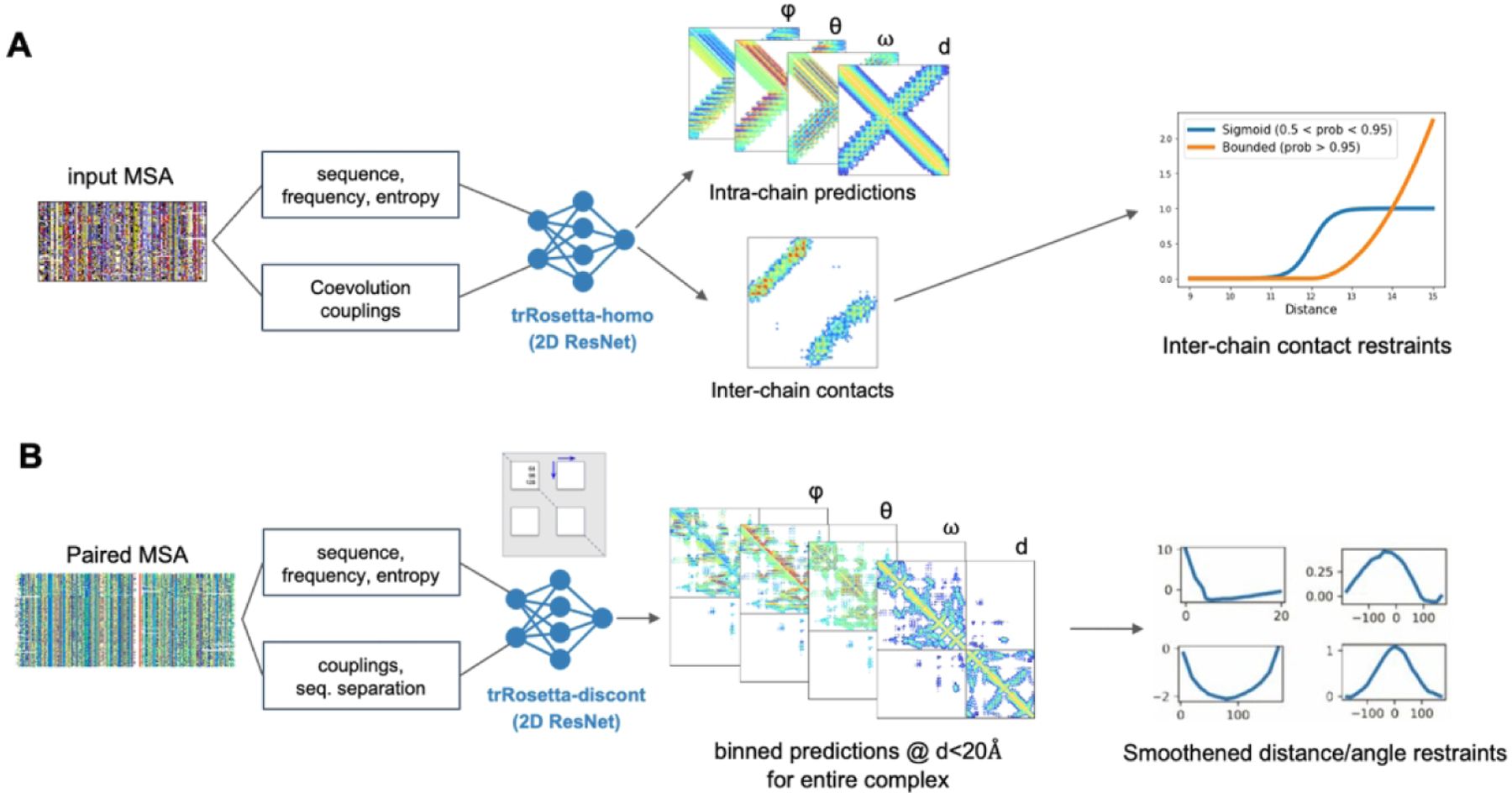
Deep learning-based residue pairwise interaction prediction for (A) homo-oligomers (trRosetta-homo) and (B) hetero-oligomers (trRosetta-discont)
For hetero-oligomer targets, we developed a modified version of trRosetta called trRosetta-discont to predict oligomer structures based on paired alignments (Figure 2B). To extract coevolutionary signals between two proteins forming a hetero complex, the sequences from the corresponding MSAs must be properly paired12. For H1047 and H1065, which are protein complexes present in bacteria, we deployed a simple sequence pairing strategy relying on the fact that genes encoding interacting proteins tend to be co-located on the same operon in the prokaryotic genome. First, we collected MSAs for both proteins forming a complex by performing sequence searches against UniProtKB/TrEMBL26 and metagenomic and metatranscriptomic sets from JGI21. Next, assuming that UniProt Accession IDs and JGI’s IMG/M IDs are serially assigned in the genome or a contig, we paired all sequences from the two MSAs satisfying ΔID≤10 into one. The resulting paired alignments were cleaned at 95% sequence identity and 75% coverage cutoffs. For both H1047 and H1065 the majority of the sequences in the final MSA came from JGI. This approach could only be applied to these two targets.
During training of trRosetta-discont, long proteins over 300 residues in length were trimmed by randomly selecting two non-intersecting sequence fragments; input MSAs and target distance and orientation maps were cropped accordingly. The discontinuity in the resulting sequence was communicated to the network through the sequence separation feature which was first calculated from the non-trimmed sequence and then cropped in the same way as other network inputs and outputs. Despite the network being trained on single protein chains, we deployed its ability to make inferences on discontinuous sequence fragments to the target H1065. We treated each of the proteins in the hetero-complex as an individual sequence fragment and increased the sequence separation feature by adding 500 to approximate a chain break (this number was not optimized) to the inter-chain regions of this feature map. Predicted residue-residue distances and orientations were then used to recreate the 3D structure model of the complex.
Step 2: Oligomer template search based on sequence and structure similarity
HHsearch19,27 and TM-align24 were used to detect oligomer templates from the PDB100 database based on not only sequence similarity but also structure similarity to the subunit structures predicted by BAKER or BAKER-ROSETTASERVER group. For homo-oligomers, using HHsearch, up to five oligomer templates in the given oligomer state were selected according to their ranks among the top100 HHsearch hits. In addition to the sequence-based oligomer templates, up to five oligomer templates were chosen purely based on the structural similarity to the given subunit models using TM-align. Among the selected hits from both sequence- and structure-based searches, those having similar subunit structures to the given model (TM-score > 0.5) were chosen as final oligomer templates to build complex structures. For hetero-oligomers, we identified HHsearch hits to the same PDB ID for both subunits of the hetero-oligomer and ranked these based on the HHsearch ranking and structural similarity to the subunit (TM-score > 0.5).
Step 3–1: Template-based complex modeling
The Rosetta hybridization protocol28 was used to refine oligomer models starting from initial complex structures generated by superposing subunit structures to one of the detected templates. During the hybridization process, local regions were rebuilt by recombining the secondary structure segments with detected templates and inserting fragments in the centroid representation. The overall structures were further optimized by relaxing full-atom structures using Rosetta FastRelax29. The intra-chain restraints derived from trRosetta16 were applied during the entire model building process, and inter-chain restraints were also applied if there were predicted inter-chain contacts from either GREMLIN or deep learning-based methods. The whole process was symmetry-aware for homo-oligomers. Total 500 structures were sampled by running the independent template-based modeling protocol, and five models having the lowest Rosetta REF2015 energy30 (with inter-chain contact restraints if applicable) were selected after clustering.
Step 3–2: Gradient-based fold-and-dock
Even with reasonable subunit structures and inter-chain contact predictions to guide overall conformational search, small local inaccuracies at the interface can hinder generating correct oligomer structures with ab initio docking31. Moreover, as proteins interact with other proteins, their lowest free-energy backbone conformations can shift in response to their partners, complicating typical docking after folding approaches. A “fold-and-dock” method32 was developed to overcome this limitation, but it is quite computationally expensive as it employs Monte Carlo fragment assembly trajectories.
For CASP14, we developed a fold-and-dock approach using gradient-based energy minimization to sample structures instead of fragment assemblies. As depicted in Figure 3A, this approach has two stages. In the first low-resolution stage with the Rosetta centroid level representation, oligomer conformations are sampled by alternating gradient-based folding and low-resolution docking starting from a conformation with random backbone torsion angles. Gradient-based folding employs L-BFGS (Limited memory Broyden–Fletcher–Goldfarb–Shanno algorithm) minimization against the Rosetta centroid energy function supplemented with intra-chain restraints derived from trRosetta and inter-chain restraints derived from predicted contacts using either GREMLIN, trRosetta variants (trRosetta-homo or trRosetta-discont depending on complex type), or partial oligomer templates. To optimize orientation between subunits, low-resolution docking was used with a centroid level scoring function consisting of Motif Dock Score9, clash terms (quadratic penalties for overlaps)33, and inter-chain restraints.
Figure 3.
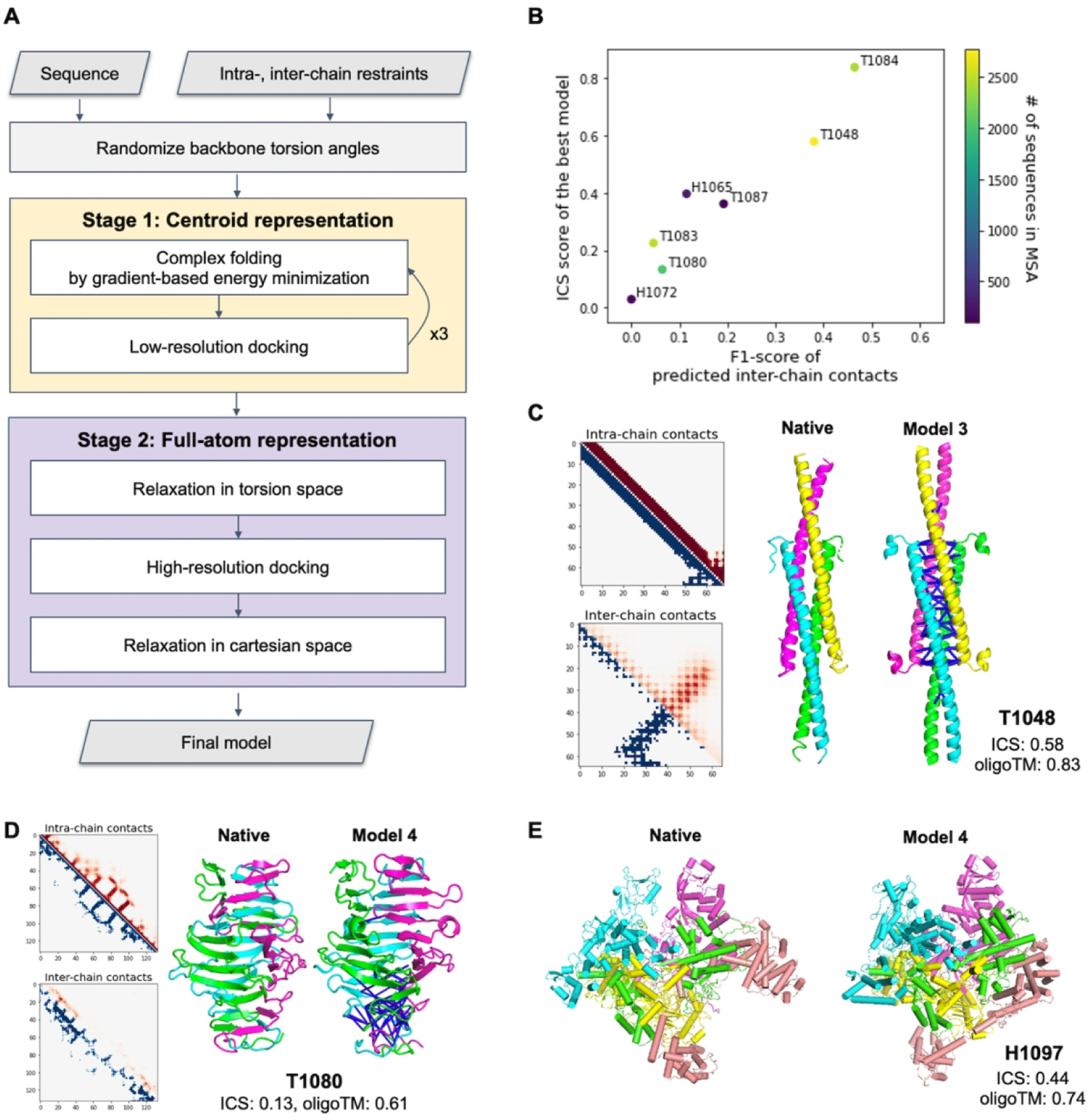
Performance of the gradient-based fold-and-dock method. A) Schematic outline of the fold-and-dock procedure consisting of two stages: repetitive folding and docking in centroid representation followed by full-atom docking and relaxation. B) Correlation between the quality of predicted inter-chain contacts and that of modeled interfaces. C–D) Examples of successful predictions using gradient-based fold-and-dock methods with predicted inter-chain contacts. Predicted intra-chain distances and inter-chain contacts are shown in the upper diagonal (colored in red) of 2D maps while those from native structures are shown in the lower diagonal (colored in blue). The correctly predicted inter-chain contacts are shown as blue lines in the model structures. Both native and model structures are colored by chains. E) Native and the best prediction submitted as model 4 for H1097.
In the second stage, side chains are built into the backbone conformations, and the overall structures are relaxed in torsion space using Rosetta FastRelax29. High-resolution docking followed by full-atom relaxation in Cartesian space is then performed to refine overall complex structures further. A total of 150 structures were generated in independent trajectories, and five models having the lowest Rosetta energy with inter-chain restraints were selected after clustering. For homo-oligomer targets, symmetry was considered during the entire process.
We used this gradient-based fold-and-dock approach when there were no oligomer templates, but inter-chain contacts were predicted with high confidence based on MSAs. We also utilized this method to predict complex structures when subunits were highly intertwined with each other and detected templates had many insertions and deletions that made it hard to predict oligomer structures using the template-based approach. The codes for the gradient-based fold-and-dock method are available at https://github.com/RosettaCommons/trRosetta2. It requires about an hour per oligomer model having 500 residues using a single CPU core.
Step 3–3: Ab Initio docking-based approach
When there were neither oligomer templates nor predicted contacts with high confidence for the target protein, oligomer structures were predicted using ab initio docking with subunit structures predicted by BAKER-ROSETTASERVER or BAKER group. SymDock28 was employed to predict symmetric homo-oligomer structures, while ZDOCK7 and RosettaDock9 were used to predict hetero-oligomers. For the targets having symmetric subunits, the symmetry axes of homo-oligomer subunits were aligned during the docking process. Rotations along the symmetry axes were sampled with 3° angular spacing, and translations, in 0.5Å intervals along the aligned axis. Among the sampled conformations, the top 50 samples having the best centroid level energy combined with Motif Dock Score were subjected to full-atom relaxation, and five models having the lowest energy were selected after clustering.
Results
Overall performance
The modeling strategies we used for 29 CASP14 assembly targets are summarized in Table 1. 15 out of 29 targets were modeled using the template-based approach, 8 using the gradient-based fold-and-dock approach, and 6 with the ab initio docking approach. The quality of the predicted multimeric structures was assessed in terms of Interface Patch Similarity (IPS) score, Interface Contact Similarity (ICS) score10, oligomer lDDT34, and oligomer TM-score measured by MM-align35. The modified Z-score was calculated based on CASP conventions (recalculating Z-score without outliers having Z-score < −2.0) for each of the evaluation metrics. The average Z-score is reported in Table 1 as well as raw ICS and oligomer TM-score. For 16 targets, we failed to submit the best model as model 1. For H1045, H1060v3, T1048, and T1078, the differences in ICS score between model 1 and the best model are larger than 0.05 points. This scoring failure might be overcome by a better model accuracy estimation method for complex structures in the future.
The best relative performance was with the gradient-based fold-and-dock protocol (Figure 4A) with an average Z-score > 2.0; there were no oligomeric templates for most of these targets. We also generated relatively good models by 1) generating complex structures with ab initio docking for two medium difficulty targets and 2) finding distant homologs based on structural template search for one hard and two medium difficulty targets. Those examples will be discussed in the following sections.
Figure 4.
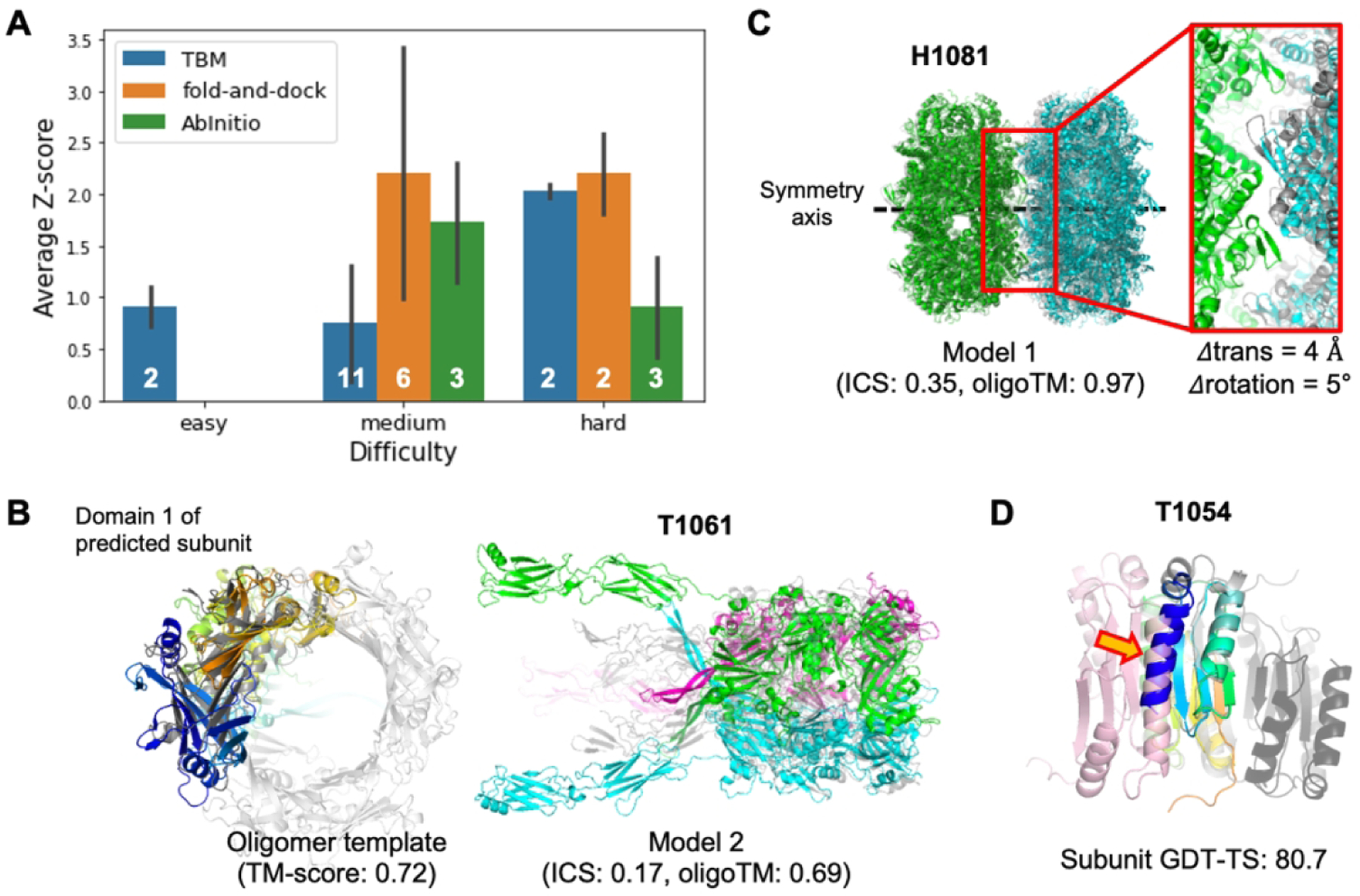
Oligomer modeling performance of BAKER-experimental group. A) The relative performance in terms of average Z-score for the best out of five submissions for each target difficulty and modeling strategy we used. B) A successful example (T1061) of template-based approach by detecting a distant oligomer template based on structural similarity. Left; The subunit structure (colored in rainbow) used to search oligomer templates and the detected template (colored in gray, PDB ID: 3CDD) are shown. Right; The predicted structure (submitted as model 2) is shown with the native structure colored in gray. C) A successful example (H1081) of ab initio docking with a constraint to match symmetry axes of two subunits. The native structure is colored in gray. D) A failed example to generate a correct binding pose by ab initio docking with the subunit structure (colored in rainbow colors from the N-terminus in blue to the C-terminus in red) having high GDT-TS. The problematic N-terminal helix is highlighted by an orange arrow. The correct binding pose is colored in pink while the predicted one is colored in dark gray.
Improvements in subunit modeling led to better template detection for oligomer modeling
Improvements in our tertiary structure prediction method combined with structure-based template search made it easier to find distant oligomer templates hard to detect using sequence-based search. For example, for T1061 (Figure 4B), our tertiary structure modeling protocol with metagenome sequence database (BAKER group) predicted reasonable subunit structures (subunit TM-score to native: 0.67). Using these structures, we were able to find distant oligomer templates (PDB ID: 3CDD) with TM-align and generated complex structures using the template-based approach. The resulting model submitted as model 2 showed a reasonable global arrangement of each subunit (complex TM-score: 0.69) but failed to recapitulate accurate interfaces (ICS score: 0.17) because it was too large to refine (2,847 residues in total) starting from the medium quality of initial subunit structure (Figure S1). In addition, C-terminal domains were not covered by detected oligomer templates resulting in huge errors (interface RMSD: 20.76 Å). For H1060v2, H1060v3, H1060v4 and H1060v5, we were able to find oligomer templates through either sequence-based or structure-based search (PDB ID: 5NGJ for H1060v2 and H1060v3, 6V8I for H1060v4 and 4V96 for H1060v5). Based on these templates, we generated oligomer structures having reasonable global subunit arrangements (oligomer TM-score: 0.88, 0.84, 0.73 and 0.95, respectively) but again failed to accurately model the interfaces (ICS score: 0.10, 0.12, 0.21 and 0.50, respectively) in part due to the large sizes of the proteins (1,392 residues, 894 residues, 1,680 residues, and 1,224 residues for each target).
Inter-chain contact predictions enabled to generate oligomer structures from scratch
As shown in Figure 4A, the predictions that most stood out from those of other groups were made primarily with the gradient-based fold-and-dock protocol that models oligomer structures starting from scratch based on inter-chain contacts predicted by deep learning-based methods or derived from partial templates. For the eight targets for which we used the gradient-based fold-and-dock approach, the resulting models were better than those produced using traditional template-based or ab initio docking approaches, with summed Z-scores 5.4 units higher than the next best group. For H1065, T1048, T1083, T1084, and T1087, reasonable inter-chain contacts were predicted using our deep learning-based methods resulting in better oligomer models with Z-score > 1.5 in all cases. Four cases (T1048, T1083, T1084, and T1087) are helical bundles; this simplicity in topology likely makes it easier to predict inter-chain contacts and to generate accurate models based on the gradient-based fold-and-dock method.
The quality of the predicted oligomer structures is correlated with the predicted inter-chain contact quality measured by F1-score as shown in Figure 3B. When inter-chain contacts were predicted accurately (T1048 and T1084, both having F1-score > 30.0), we were able to predict high accuracy oligomer structures not only having good global arrangements (oligomer TM-score: 0.83 and 0.91, respectively) but also having accurate interface structures (ICS: 0.58 and 0.84, respectively). For T1048, we were the only group predicting the correct oligomer structure, exceeding the next best group by 0.4 in ICS score and by 0.5 in oligomer TM-score; accurate oligomer structure modeling was made possible by accurate prediction of both intra-chain and inter-chain contacts (Figure 3C). For T1080, which forms a highly intertwined homo-trimer structure (Figure 3D), we generated a relatively better model (Z-score: 2.6) based on accurate predicted inter-chain contacts for the intertwined interactions at the C-terminal part of the target, but we failed to capture intertwining patterns at the N-terminal resulting in a less accurate overall oligomer structure (ICS: 0.13, oligomer TM-score: 0.61).
For H1097 (Figure 3E), we identified 121 quite divergent oligomer templates from the PDB100 database. These contained many insertions and deletions, and it was expected to form highly intertwined oligomer structures from the templates. To generate intertwined models, we used the gradient-based fold-and-dock protocol guided by inter-chain pairwise distance and orientation distributions from 121 detected oligomer templates. With inter-chain restraints derived from templates together with intra-chain restraints derived from trRosetta outputs, the gradient-based fold-and-dock protocol built a reasonable quality model (ICS: 0.44, oligomer TM-score: 0.75) that ranked first. The oligomer model of the next best group (likely using a template-based approach) has an ICS score of 0.31 and oligomer TM-score of 0.68.
Ab initio docking approach was successful only for a few cases
With ab initio docking, we were able to predict structures having oligomer TM-score higher than 0.6 only for two targets: H1081 and T1078. For H1081 (Figure 4C), we were asked to build a homo 20-mer structure by combining two homo-decamer subunits. The homo-decamer subunit structure was first predicted by RosettaCM28 based on two close templates (PDB ID: 2VYC and 5XX1) having sequence identity over 70%. Homo-20-mer structures were generated by sampling the rigid body degrees of freedom (rotation and translation along the common symmetry axis) as described in the method section. Because decamer subunits were quite accurate and the system symmetry reduces six rigid-body degrees of freedom to just two, a reasonable quality complex structure (ICS: 0.35, oligomer TM-score: 0.97) was generated. The errors in translational and rotational degrees of freedom are 4Å and 5°, respectively. For T1078, we modeled a complex structure by symmetric docking with the subunit structure submitted as model 1 for the BAKER group in the TS category. As the N-terminal of the subunit was predicted to have low accuracy by DeepAccNet36 (Figure S2A), our accuracy prediction method, we trimmed the N-terminal part (residue 1–13) for docking and reconstructed it after selecting the final five models to submit. We generated a roughly correct oligomer structure (ICS: 0.25, oligomer TM-score: 0.67, Figure S2B) reflecting the quality of the subunit structure used for docking (GDT-TS: 66.7).
For H1060v1 and T1054, we failed to predict correct binding poses despite having subunit models with the right fold (subunit TM-score > 0.7), primarily due to local inaccuracies at the interface. For example, for T1054 the subunit has 80.7 GDT-TS to the experimental structure, but the N-terminal helix (which is missing in the crystal structure) was mislocated to the interface region as shown in Figure 4D. It hindered generating correct binding pose during docking, resulting in a complex structure having the wrong interface.
Assembly modeling can improve subunit quality when it provides correct interface information
To see the effects of considering binding partners on the subunit modeling for oligomer targets, we compared the GDT-TS values of model 1 structures from BAKER-experimental to those of the subunit structures modeled as monomers (Figure 5A). To eliminate differences coming from the quality of MSAs used to model the structures, we re-modeled subunit structures using our CASP14 tertiary structure modeling method (BAKER-ROSETTASERVER) with the same MSA used for oligomer modeling. The evaluation unit definition posted on the CASP14 web page (https://predictioncenter.org/casp14/domains_summary.cgi) was used for analysis.
Figure 5.
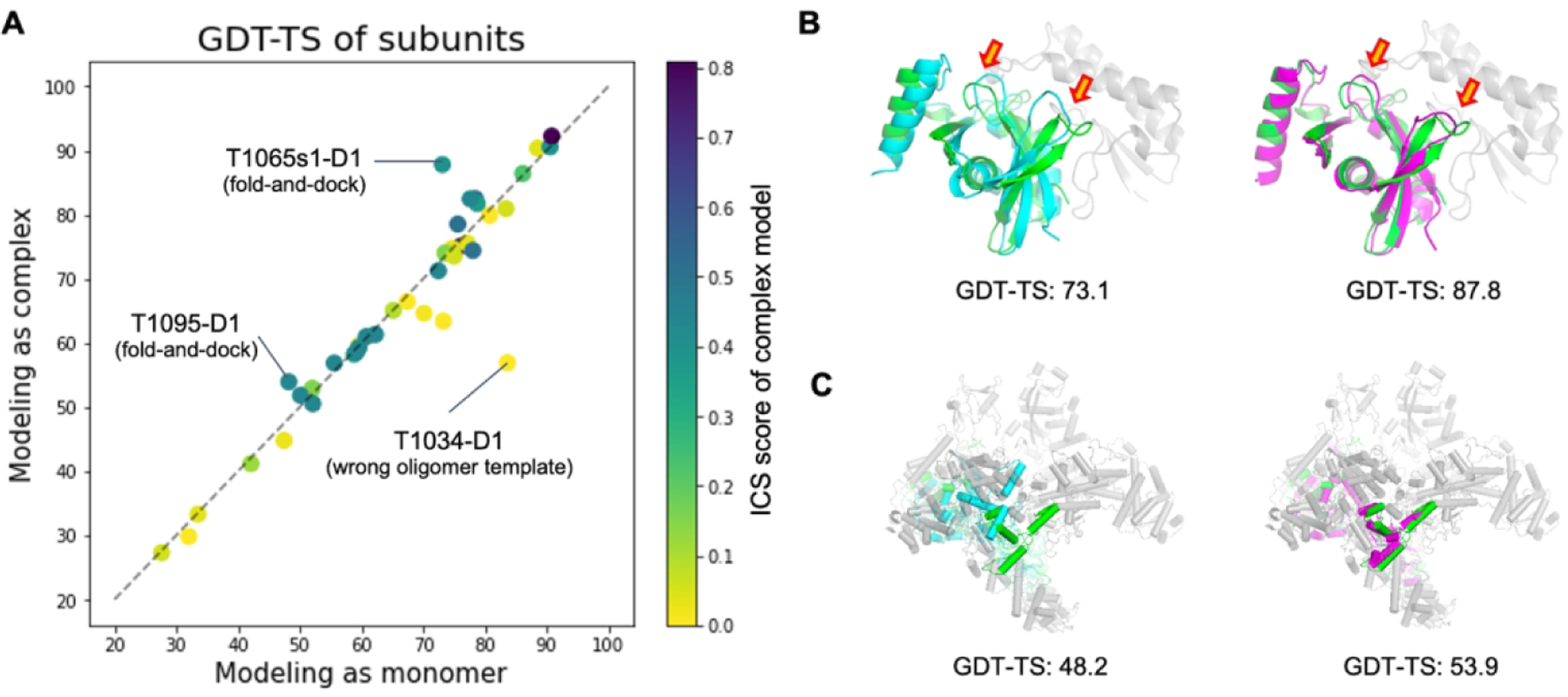
Comparison of subunit structures modeled as complexes to those modeled as monomers. A) Head-to-head comparison of the subunit qualities in terms of the evaluation unit-wise GDT-TS score. Dots are colored by the ICS score of predicted complex structures. Panel B) and C) show two successful examples (T1065s1-D1 and T1095-D1, respectively). The native structure of the target subunit and its binding partners are shown in green and gray, respectively. The subunit structures predicted as a monomer are shown in cyan (left), while those predicted in oligomer contexts are colored in magenta (right).
The cases where modeling in oligomer contexts generated better subunit structures tended to have flexible regions at the interface (52% of interface residues didn’t have regular secondary structures), and we were able to predict interface contacts correctly using deep learning-based methods or templates. For T1065s1-D1 (Figure 5B), two beta hairpins (residues 87–92 and 111–118) interact with an adjacent subunit. By modeling the entire complex together using the gradient-based fold-and-dock method, those hairpins moved to more correct positions to have better interactions with the binding partner resulting in overall rearrangement of secondary structure components with a 14.7 GDT-TS improvement. For T1095-D1 (one of the subunits for H1097, Figure 5C), the orientations of C-terminal helices are stabilized by interactions with neighboring subunits, making models without considering binding partners less accurate than models generated for the holo-complex. In some cases like T1034-D1, the subunit quality modeled as complex was worse than that modeled as monomer because our oligomer models were generated based on the wrong oligomer template (Figure S3).
Conclusion
We used a new gradient-based fold-and-dock approach incorporating predicted intra- and inter-chain contacts to build reasonably accurate models of protein assemblies in CASP14. This new gradient-based fold-and-dock approach outperformed the other more traditional template-based or ab initio docking approaches. Moreover, the inclusion of binding partners during the folding/docking process led to improvements in subunit modeling in regions at oligomer interfaces. We also obtained good results with a template-based approach, using subunit structures generated by deep learning-based structure prediction methods to find distant templates based on structural similarity search.
There is still considerable room for improvement in the modeling of higher-order assemblies. The performance of the fold-and-dock approach highly depended on the quality of predicted inter-chain contacts, and advances in deep learning-based inter-chain contact or distance prediction methods could considerably improve this approach. Predicting high accuracy complex structures based on distant templates remains challenging, as they only provide clues to the overall structure but not detailed interaction information on the interface. Moving forward, deep learning methods that utilize both MSA and template information, either to predict residue pairwise interactions for use in fold-and-dock protocols or to predict complex structure coordinates directly, are likely to become increasingly powerful.
Supplementary Material
Acknowledgements
This work is supported by NIAID Federal Contract # HHSN272201700059C (M.B.), a gift from Microsoft (M.B.), National Science Foundation Award # DBI 1937533 (I.A.), Eric and Wendy Schmidt by recommendation of the Schmidt Futures program (H.P.), a gift from Amgen (I.H.), the Howard Hughes Medical Institute (D.B.), the Open Philanthropy Project Improving Protein Design Fund (D.B.), and the Audacious Project at the Institute for Protein Design (D.B.).
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Availability
Deep learning models (trRosetta-homo, trRosetta-discont) and a pyRosetta37 script for gradient-based fold-and-dock are available at https://github.com/RosettaCommons/trRosetta2 under the MIT license.
References
- 1.Berggård T, Linse S, James P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 2007;7(16):2833–2842. [DOI] [PubMed] [Google Scholar]
- 2.Keskin O, Tuncbag N, Gursoy A. Predicting Protein-Protein Interactions from the Molecular to the Proteome Level. Chem Rev. 2016;116(8):4884–4909. [DOI] [PubMed] [Google Scholar]
- 3.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. [DOI] [PubMed] [Google Scholar]
- 4.Baek M, Park T, Heo L, Park C, Seok C. GalaxyHomomer: a web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res. 2017;45(W1):W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep. 2017;7(1):10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alekseenko A, Ignatov M, Jones G, Sabitova M, Kozakov D. Protein-Protein and Protein-Peptide Docking with ClusPro Server. Methods Mol Biol. 2020;2165:157–174. [DOI] [PubMed] [Google Scholar]
- 7.Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy Burman SS, Yovanno RA, Gray JJ. Flexible Backbone Assembly and Refinement of Symmetrical Homomeric Complexes. Structure. 2019;27(6):1041–1051.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marze NA, Roy Burman SS, Sheffler W, Gray JJ. Efficient flexible backbone protein-protein docking for challenging targets. Bioinformatics. 2018;34(20):3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lafita A, Bliven S, Kryshtafovych A, et al. Assessment of protein assembly prediction in CASP12. Proteins. 2018;86 Suppl 1:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guzenko D, Lafita A, Monastyrskyy B, Kryshtafovych A, Duarte JM. Assessment of protein assembly prediction in CASP13. Proteins. 2019;87(12):1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife. 2014;3:e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopf TA, Schärfe CPI, Rodrigues JPGLM, et al. Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife. 2014;3. doi: 10.7554/eLife.03430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng H, Wang S, Zhou T, et al. ComplexContact: a web server for inter-protein contact prediction using deep learning. Nucleic Acids Res. 2018;46(W1):W432–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577(7792):706–710. [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci USA. 2020;117(3):1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J Distance-based protein folding powered by deep learning. Proc Natl Acad Sci USA. 2019;116(34):16856–16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greener JG, Kandathil SM, Jones DT. Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat Commun. 2019;10(1):3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirdita M, von den Driesch L, Galiez C, Martin MJ, Söding J, Steinegger M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017;45(D1):D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen I-MA, Markowitz VM, Chu K, et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017;45(D1):D507–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balakrishnan S, Kamisetty H, Carbonell JG, Lee S-I, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79(4):1061–1078. [DOI] [PubMed] [Google Scholar]
- 23.Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci USA. 2013;110(39):15674–15679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ovchinnikov S, Kim DE, Wang RY-R, Liu Y, DiMaio F, Baker D. Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta. Proteins. 2016;84 Suppl 1:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Söding J Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–960. [DOI] [PubMed] [Google Scholar]
- 28.Song Y, DiMaio F, Wang RY-R, et al. High-resolution comparative modeling with RosettaCM. Structure. 2013;21(10):1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park H, Bradley P, Greisen P Jr, et al. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J Chem Theory Comput. 2016;12(12):6201–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baek M, Park T, Woo H, Seok C. Prediction of protein oligomer structures using GALAXY in CASP13. Proteins. 2019;87(12):1233–1240. [DOI] [PubMed] [Google Scholar]
- 32.Das R, André I, Shen Y, et al. Simultaneous prediction of protein folding and docking at high resolution. Proc Natl Acad Sci USA. 2009;106(45):18978–18983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rohl CA, Strauss CEM, Misura KMS, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. [DOI] [PubMed] [Google Scholar]
- 34.Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013;29(21):2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee S, Zhang Y. MM-align: a quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic Acids Res. 2009;37(11):e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiranuma N, Park H, Baek M, Anishchenko I, Dauparas J, Baker D. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat Commun. 2021;12(1):1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaudhury S, Lyskov S, Gray JJ. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26(5):689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Deep learning models (trRosetta-homo, trRosetta-discont) and a pyRosetta37 script for gradient-based fold-and-dock are available at https://github.com/RosettaCommons/trRosetta2 under the MIT license.