

Abstract
BCMA-specific CAR T-cells have substantial therapeutic potential in multiple myeloma (MM), but most patients eventually relapse. Determinants of response and mechanisms of resistance are most likely multifactorial and include MM-related factors, premanufacturing T-cell characteristics, CAR T-cell-related features, and several components of the immunosuppressive microenvironment. Efforts to improve the potency and safety of CAR T-cell therapy include optimizing CAR design, combinatorial approaches to enhance persistence and activity, treatment of less heavily pretreated patients, and dual-antigen targeting to prevent antigen escape. We expect that these rationally designed strategies will contribute to further improvement in the clinical outcome of MM patients.
Introduction
Although the introduction of new anti-MM agents markedly improved the survival of multiple myeloma (MM) patients, there is an unmet need for new drugs for patients, who develop resistance to immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and CD38-targeting antibodies (triple-class refractory disease), which carries a poor prognosis(1). Also newly diagnosed patients with high-risk disease (e.g. presence of del(17p), t(4;14), or t(14;16)) or suboptimal response have an impaired outcome, and these patients may benefit from the incorporation of new drugs with novel mechanisms of action in first-line regimens.
A promising new strategy is the reprogramming of T-cells to target MM cells by introducing genes encoding chimeric antigen receptors (CARs). CARs are fusion proteins, combining an antigen-recognition moiety (commonly a monoclonal antibody-derived single-chain variable fragment (scFv), but other formats such as natural ligands are also possible(2)) with a T-cell activation domain, typically CD3ζ. These 2 parts are connected via an extracellular spacer region (hinge) and a transmembrane-spanning element. Second generation CARs incorporating a costimulatory domain, such as CD28, 4-1BB, OX40, or ICOS, into the CAR endodomain, result in enhanced anti-tumor activity of the modified T-cells, when compared to first-generation CARs without such domain (Figure 1)(3). Importantly, CAR T-cells eliminate tumor cells in a non-major histocompatibility complex-restricted manner.
Figure 1. Evolution of CAR design.
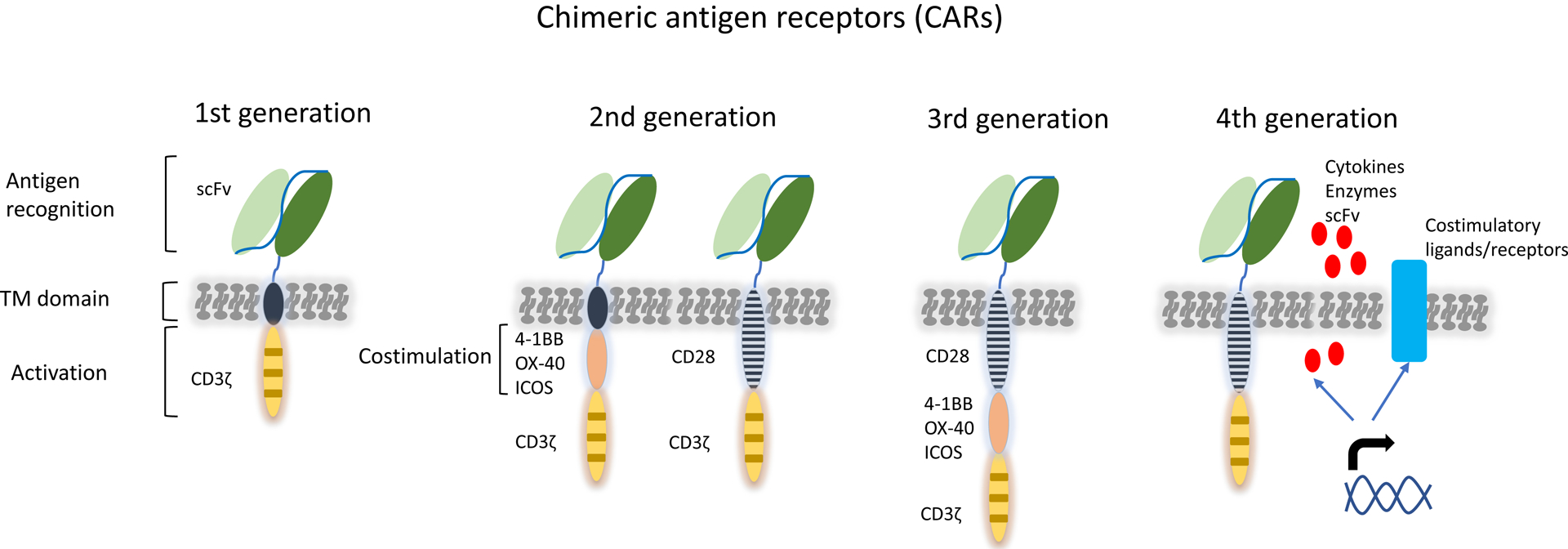
The 1st generation CARs mediate antigen recognition and T-cell activation through the fusion of an extracellular antigen-binding single-chain-variable region (scFv) with an intracellular signaling domain from the CD3ζ chain. In this way surface antigens can be recognized by CAR T cells independent of major-histocompatibility-complex (HLA)-mediated presentation. Second generation CARs provide combined activation and costimulatory signals through the addition of the intracellular domain of costimulatory receptors. The 3rd generation CARs consist of two costimulatory domains. In the latest 4th generation design, CARs are co-expressed with enzymes, cytokines and costimulatory ligands or receptors transferred with the same vector construct.
Abbreviations: scFv, single-chain fragment variable; TM: transmembrane.
Most of the CAR T-cell products, currently evaluated in clinical trials for MM patients, target B-cell maturation antigen (BCMA), which is uniformly expressed on the cell surface of MM cells, normal plasma cells, and a subset of mature B-cells. Characteristics, as well as key efficacy and safety data from several studies, evaluating BCMA-targeted CAR T-cells, are provided in Tables 1–2. CAR T-cells specific for other MM-associated antigens, such as CD19, SLAMF7, CD38, GPRC5D, are also investigated in MM. BCMA-specific CAR T-cells have significant therapeutic potential in MM, as evidenced by the high quality responses with a substantial rate of complete response (CR) and minimal-residual disease (MRD) negativity, obtained in heavily pretreated, often triple-class refractory, patients(4–11). Similar to what is observed with other therapies, depth of response is associated with improved PFS in patients treated with CAR T-cell therapy, with best outcomes in patients achieving CR or MRD-negativity(10,12). Most advanced in clinical development are the BCMA-targeting CAR T-cell products idecabtagene vicleucel (ide-cel, bb2121) and ciltacabtagene autoleucel (cilta-cel, JNJ4528), both of which have received FDA breakthrough designation based on promising results in heavily pretreated patients(6,10,11). However, not all patients achieve a remission after CAR T-cell therapy. Furthermore, there is, until now, no indication of a plateau in the survival curves, which contrasts with results obtained with CD19 CAR T-cells in acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL). In this review, we will provide an overview of the determinants of response and the mechanisms that contribute to the development of treatment failure after initial remission (acquired resistance). A better understanding of these mechanisms underlying lack of disease response and acquired resistance, may lead to new strategies to improve the effectiveness of CAR T-cell therapy.
Table 1.
Selected studies evaluating BCMA-specific CAR T-cells: CAR T-cell features and patient characteristics.
| NCI | UPenn (Novartis) | Ide-cel (BMS) | bb21217 (BMS) | LCAR-B38M | Cilta-cel (Janssen) | Orva-cel (JCARH125; BMS) | ||
|---|---|---|---|---|---|---|---|---|
| Stage of trial; Clinicaltrials.gov Identifier; reference | Phase 1; NCT02215967(5) | Phase 1; NCT02546167(9) | Phase 1; NCT02658929 (CRB-401)(12) | Phase 2; NCT03361748 (KarMMa)(10) | Phase 1; NCT03274219 (CRB-402)(82) | Phase1; NCT03090659 (LEGEND-2)(14) | Phase 1b/2; NCT03548207 (CARTITUDE-1)(11) | Phase 1/2; NCT0340011 (EVOLVE)(23) |
| Antigen-binding domain | scFv (murine) | scFv (human) | scFv (murine) | scFv (murine) | scFv (murine) | Bispecific variable fragments of llama heavy-chain antibodies; two distinct BCMA epitopes are targeted | Bispecific variable fragments of llama heavy-chain antibodies; two distinct BCMA epitopes are targeted | scFv (human) |
| Signaling domains | CD3ζ/CD28 | CD3ζ/4-1BB | CD3ζ/4-1BB | CD3ζ/4-1BB | CD3ζ/4-1BB | CD3ζ/4-1BB | CD3ζ/4-1BB | CD3ζ/4-1BB |
| Lymphodepletion | Flu/Cy | +/− Cy | Flu/Cy | Flu/Cy | Flu/Cy | Cy | Flu/Cy | Flu/Cy |
| Bridging therapy | 0% | 84% | 52% | 88% | NR | Not allowed | 65% | 63% |
| BCMA expression required | yes | no | In dose escalation phase BCMA expression on ≥50% MM cells required, not in dose expansion cohort | No | In dose escalation phase BCMA expression on ≥50% MM cells required, not in dose expansion cohort | yes | no | no |
| Number of patients included in analysis | 24 (16 patients received 9×106 CAR T cells/kg (highest dose-level)) | 25 | 62 | 128 | 69 | 57 | 97 | 62 |
| Number of prior therapies (median) | 10 in highest dose-level | 7 | 6 | 6 | 6 | 3 | 6 | 6 |
| IMID and PI-refractory | NR | 96% | 81% | 89% | 80% | NR (60% exposed to prior PI and IMiD) | NR; 88% triple-class refractory | NR; 94% triple-class refractory |
| CD38 antibody refractory | NR | 72% | ≥69% | 94% | 74% | NR, probably 0% | 99% | ≥94% |
| CAR T-cell dose | 0.3–9×106/kg | Cohort 1: 1–5×108 Cohort 2: Cy+1–5×107 Cohort 3: Cy+1–5×108 |
50–800×106 | 150–450×106 | 150–450×106 | Median dose: 0.5×106/kg | Median dose: 0.71×106/kg | 300–600×106 |
Abbreviations: NR, not reported.
Table 2.
Selected studies evaluating BCMA-specific CAR T-cells: efficacy and safety data.
| NCI | UPenn (Novartis) | Ide-cel (BMS) | bb21217 (BMS) | LCAR-B38M | Cilta-cel (Janssen) | Orva-cel (JCARH125; BMS) | ||
|---|---|---|---|---|---|---|---|---|
| Stage of trial; Clinicaltrials.gov Identifier; reference | Phase 1; NCT02215967(5) | Phase 1; NCT02546167(9) | Phase 1; NCT02658929 (CRB-401)(12) | Phase 2; NCT03361748 (KarMMa)(10) | Phase 1; NCT03274219 (CRB-402)(82) | Phase1; NCT03090659 (LEGEND-2)(14) | Phase 1b/2; NCT03548207 (CARTITUDE-1)(11) | Phase 1/2; NCT0340011 (EVOLVE)(23) |
| ≥PR | 0.3–3×106/kg: 20% 9×106/kg: 81% |
48% | 50–800×106: 76% 450×106 (n=38): 90% |
150–450×106: 73% 150×106 : 50% 300×106 : 69% 450×106 : 81% |
150–450×106: 68% 150×106 : 83% 300×106 : 43% 450×106 : 73% |
88% | 97% | 92% |
| ≥CR | 0.3–3×106/kg: 0% 9×106/kg: 13% |
8% | 50–800×106: 39% 450×106 (n=38): 37% |
150–450×106: 33% 150×106 : 25% 300×106 : 29% 450×106 : 39% |
150–450×106: 29% 150×106 : 42% 300×106 : 14% 450×106 : 30% |
74% | 67% | 36% |
| MRD-negativity (assay used to assess MRD is also provided) |
|
|
|
|
|
|
|
|
| Median PFS | 9×106/kg: 31 weeks (EFS) | Cohort 1: 65 days Cohort 2: 57 days Cohort 3: 125 days |
50–800×106 cells: 8.8 months 450×106 cells: 9.0 months |
150–450×106: 8.8 months 150×106 : 2.8 months 300×106 : 5.8 months 450×106 : 12.1 months |
PFS not reported; median duration of response: 17.0 months | 19.9 months | Not reached; 12-month PFS rate: 77% | 300×106 : 9.3 months 450×106 :not reached 600×106 : not reached |
| Cytokine release syndrome (any grade) |
|
88% | 76% | 84% | 65% | 90% | 95% | 89% |
| Grade ≥3 cytokine release syndrome |
|
32% | 7% | 5% | 4% | 7% | 4% | 3% |
| Neurotoxicity (any grade) | NR | 32% | 36% | 18% | 16% | 2% | 21%* | 13% |
| Grade ≥3 neurotoxicity | NR | 12% | 2% | 3% | 4% | 0% | 10%* | 3% |
Twelve out of 97 patients treated with cilta-cel in the CARTITUDE-1 study experienced neurotoxicity which occurred after resolution of cytokine release syndrome and/or immune effector cell-associated neurotoxicity syndrome (ICANS). Five patients experienced movement and/or neurocognitive changes, and 7 had adverse events including nerve palsy and peripheral motor neuropathy.
Abbreviations: NR, not reported; NGS, next-generation sequencing; NGF, next-generation flow cytometry; PR, partial response; VGPR, very good partial response; CR, complete response; sCR, stringent complete response.
Determinants of response and mechanisms of resistance to CAR T-cells
Mechanisms that influence CAR T-cell efficacy are multifactorial and include tumor-, host- (tumor microenvironment and T-cells), and product-related factors (Figure 2). However, the precise impact of these characteristics on primary and acquired resistance is difficult to assess, because of the limited number of patients enrolled in individual studies.
Figure 2. Determinants of response to CAR T-cell therapy.
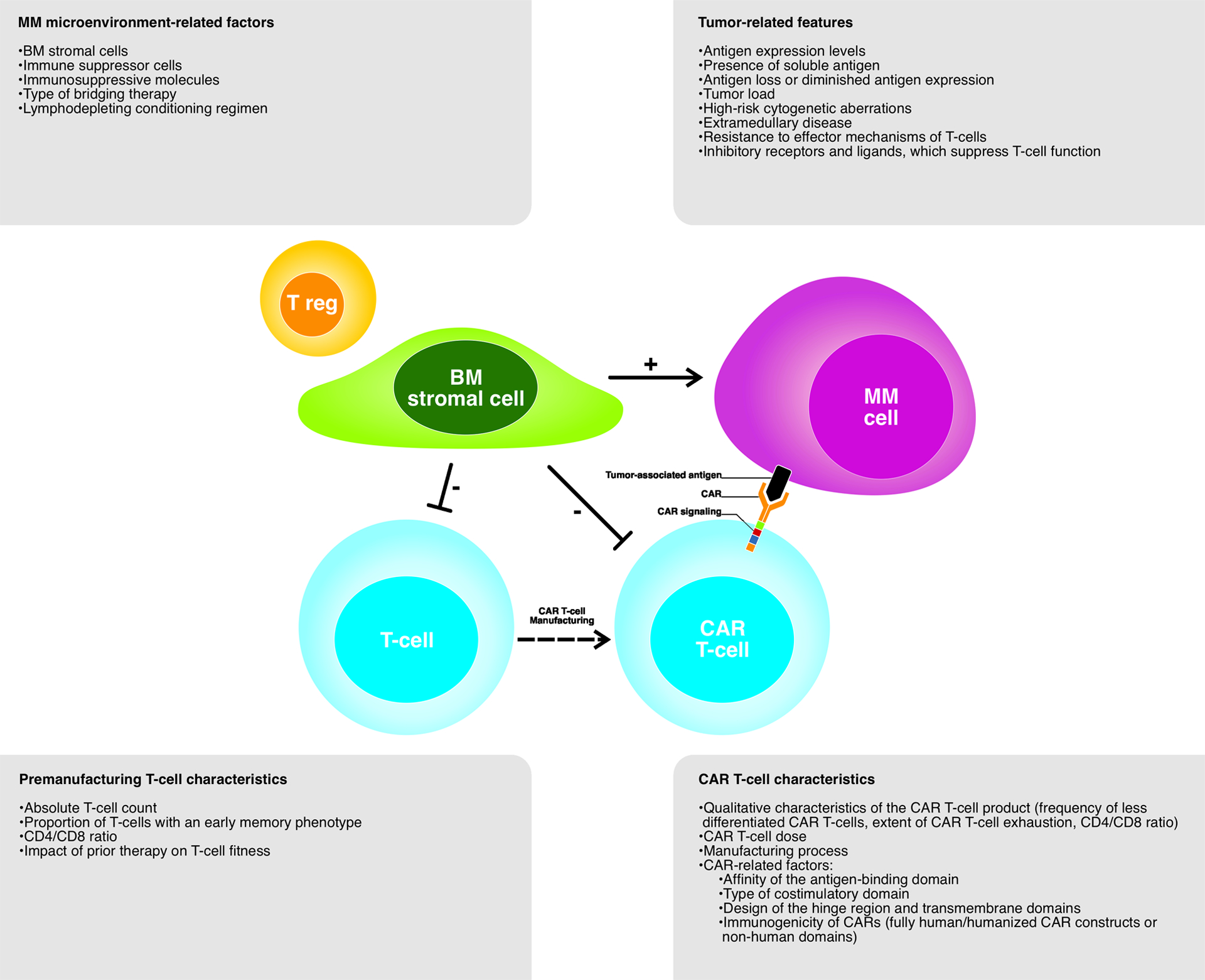
Various factors, including tumor-related features, MM microenvironment-related factors, premanufacturing T-cell characteristics and CAR T-cell characteristics, have an impact on response to CAR T-cell therapy.
Tumor-related resistance mechanisms
(Soluble) BCMA
The impact of the marked heterogeneity in BCMA antigen density among MM patients on clinical outcomes following CAR T-cell therapy is not completely clear(2). Several studies show that response, progression-free survival (PFS) and overall survival (OS) after BCMA CAR T-cell therapy are not associated with baseline BCMA expression levels on tumor cells(6,7,9,10,13,14), while in other trials pre-treatment BCMA levels have an impact on depth or durability of response(5,15,16). Discrepancies between these studies may be explained in part by differences in assays used to quantify BCMA expression with flow cytometry being more sensitive than immunohistochemistry(17). Membrane-bound BCMA can also be shed from the tumor cell surface, leading to circulation of soluble BCMA (sBCMA). The effect of sBCMA on binding of CAR T-cells to BCMA on the tumor cell surface, is controversial, with some preclinical studies showing that high levels of sBCMA impair cytolytic activity of BCMA-directed CAR T-cells(2,18,19), while in other preclinical studies sBCMA did not affect CAR T-cell activity(20–22). More importantly, in clinical trials, baseline sBCMA levels had no effect on response(4,6,9,23).
Changes in antigen expression over time may also affect the effectiveness of CAR T-cell treatment. Cohen et al. showed that following BCMA CAR T-cell infusions BCMA expression levels decreased on residual MM cells in 67% of patients, both in responding and in non-responding patients(9). In some of these patients BCMA levels were restored back to baseline levels at later time points(9). Several other studies also demonstrated reduced BCMA expression at the time of progression(4,5,15,24–26). The mechanism whereby BCMA CAR T-cells reduce BCMA cell surface expression levels probably includes selection of cells with lower BCMA levels, while tumor cells with higher BCMA expression are eliminated. Furthermore, biallelic BCMA deletions, resulting in lack of BCMA expression, have also recently been found to trigger resistance to BCMA CAR T-cells(27–29). Although BCMA antigen loss seems to be an uncommon mechanism of escape from BCMA-directed CAR T-cell therapy (4% in the KarMMa study(10)), it may have major therapeutic implications because biallelic loss of BCMA will also confer resistance to retreatment with similar or other BCMA-targeted therapies(28). This highlights the need to examine for BCMA gene alterations or to assess BCMA expression, when treatment with another BCMA-directed immunotherapy is considered. Deletion of 16p, including the BCMA locus, is present in approximately 6–7% of newly diagnosed or relapsed/refractory patients, frequently co-occurring with del(17p)(28,29). It is currently unknown whether these patients are at an increased risk for BCMA loss after immunotherapy. Loss of BCMA expression was also associated with the absence of an increase in sBCMA levels at the time of progression(28). CAR T-cell studies should incorporate serial sBCMA assessments to investigate the potential value of sBCMA as an indicator of BCMA loss at relapse(28). In addition, preclinical MM models showed that transfer of BCMA from the tumor cell surface to CAR T-cells (trogocytosis) may also contribute to antigen loss and antigen-low tumor relapse(30). At the same time, trogocytosis also leads to CAR T-cell fratricide with a negative effect on CAR T-cell activity(30).
The use of CAR T-cells with high BCMA binding affinity may prevent the outgrowth of BCMAlow tumor clones and may be more effective in patients with low target antigen density at baseline or with substantial heterogeneity in BCMA expression. Such patients, with potential resistant clones, may also benefit from CAR T-cells targeting other MM-associated antigens. Based on preclinical studies showing substantial anti-MM activity of CAR T-cells targeting SLAMF7(31), CD38(32), CD138(33), GPRC5D(34), integrinβ(7)(35), and CD44v6(36), various clinical studies are currently ongoing to evaluate the efficacy and safety of CAR T-cell products targeting these alternative MM-associated antigens. However, because some of these targets, such as CD38 and CD138, are also expressed in critical normal tissues, there is a potential risk of on-target, off-tumor toxicity. CD19 CAR T-cells are also evaluated in MM, based on the identification of a small subset of CD19+ MM cells with a less differentiated phenotype and possibly disease-propagating properties(37,38). In addition, super-resolution microscopy revealed that a substantial fraction of MM cells has low or ultra-low levels of CD19, that triggers elimination by CD19 CAR T-cells(39). CD19 CAR T-cells were clinically evaluated directly after treatment with a second course of melphalan and autologous stem cell transplantation (auto-SCT) in MM patients who previously received auto-SCT with a PFS of less than 1 year(37,38). Two out of 10 patients exhibited more durable responses, compared with the first transplantation(37,38).
However, probably most effective will be the use of combinatorial approaches to prevent antigen-loss relapses and to address antigenic heterogeneity. This includes the use of pooled CAR T-cells (co-infusion of 2 CAR T-cell products, each expressing a different CAR). Disadvantages of this strategy include the possibility of selective expansion of one of these CAR T-cell products and the requirement of manufacturing 2 clinical products(40). Growth competition can be avoided by using dual-targeted CAR-T cells (single CAR T-cells expressing 2 distinct CARs with different binding domains, or CAR T-cells expressing a single CAR molecule with two separate binding domains in tandem [tandem CAR])(40–42). Dual-targeted CAR T-cells can be more effective than pooled CAR T-cells, possibly because of enhanced bivalent immune synapse formation, resulting in improved activation and expansion(41–43). Several preclinical studies demonstrated superior tumor control and prevention of BCMA escape-mediated relapse by simultaneous targeting of BCMA and SLAMF7, BCMA and GPRC5D, or BCMA and TACI(2,19,43–45). Similarly, ex vivo treatment of MM cells with a mixture of both CD19 CAR T-cells and BCMA CAR T-cells was more effective in reducing colony formation capability, than either CD19 CAR T-cells or BCMA CAR T-cells alone(38,46). Several clinical studies in RRMM patients demonstrate a high response rate with the combination of CD19- and BCMA-targeting CAR T-cells(47–49). On-target/off-tumor toxicity consisted of B-cell aplasia and hypogammaglobulinemia(47). Also dual BCMA and CD19-targeted CAR T-cells show promising activity and a favorable safety profile in RRMM(46). A limitation of these studies is the single-arm design, which makes it difficult to assess the added value of CD19 targeting. Randomized studies are needed to answer this question.(47,48) Also a tandem CAR T-cell product, targeting CD38 and BCMA, shows promising activity with acceptable toxicity in RRMM(50). However, the phase 1 study (AUTO2) evaluating APRIL-based CAR T-cells (dual targeting of BCMA and TACI) in RRMM was stopped because of insufficient activity(51).
Combination approaches that increase antigen density may also enhance CAR T-cell efficacy. Small molecule inhibitors of γ-secretase (GSI) reduce shedding of BCMA and increase BCMA expression, resulting in enhanced CAR T-cell activity in MM mouse models(18). A limitation of GSI is their possible negative impact on CAR T-cell function because of Notch pathway inhibition(18). Preliminary results from a clinical study (NCT03502577) show high activity of the combination of BCMA CAR T-cells and GSI, also in patients that previously failed BCMA-targeted therapy, which may be related to the GSI-mediated increase in BCMA expression and reduction of sBCMA(52). However, there was also a high rate of CRS and neurotoxicity(52). Inhibitors of HDAC7 or the Sec61 complex also increase BCMA cell surface expression(53).
Tumor load
Although all studies clearly demonstrate that CAR T-cell therapy is effective in patients with high tumor load, there was a trend towards a moderately lower CR rate with ide-cel in patients with high disease burden (≥50% bone marrow (BM)-localized MM cells), when compared to patients with a relatively low tumor load (29 versus 37% in the KarMMa study)(6,10,54). A high tumor burden with chronic antigen exposure may result in CAR T-cell exhaustion, and thereby impaired antitumor activity(55). Immune checkpoint blockade may reverse the hyporesponsiveness of exhausted T-cells. A better understanding of the impact of tumor burden in studies evaluating other CAR T-cell products is essential, because this may translate into more effective treatment strategies (e.g. reinduction therapy prior to cell therapy to debulk the disease).
High-risk cytogenetics and extramedullary disease
The precise impact of cytogenetic abnormalities on the clinical outcome of heavily pretreated patients is currently unknown because of small numbers of patients and limited data on response duration in high-risk subgroups. Nevertheless, across all studies deep and durable responses were also achieved in RRMM patients with high-risk cytogenetics.(5,6,9–11,54). Extramedullary disease seems to confer a poor outcome in some studies(8,16,26,56,57), while response was similar in others(10,54). The strongest predictor for clinical activity of ide-cel was the revised ISS staging system (reflecting a combination of risk factors including high tumor load, high-risk cytogenetic abnormalities and/or elevated LDH [≥PR: 48 versus 80%; ≥CR: 10 versus 39%; median PFS: 4.9 versus 11.3 months for R-ISS stage 3 versus R-ISS stage 1 or 2 in the KarMMa study])(10,54).
Inhibitory receptors and ligands
MM cells have several properties that enable immune evasion. This includes the expression of inhibitory ligands on the MM cell surface (PD-L1 and PD-L2: ligands for PD-1; galectin-9: ligand for TIM-3; and MHC class II: ligand for LAG-3), which contribute to suppression of T-cell responses. There is increasing evidence that these ligands also impair CAR T-cell activity (for details see section “Nature of CAR T-cells infused in patients”).
In addition, a CRISPR-based screen in a MM cell line identified several novel mechanisms that control the response to BCMA CAR T-cells(53). Knockdown of genes in the sialic acid biosynthesis pathway sensitized MM cells to BCMA CAR T-cells(53). This is in line with prior studies showing that sialic acids, present on the tumor cell surface, impair T-cell mediated tumor immunity(58). This study also showed that ICAM-1 expression is important for BCMA CAR T-cell-mediated tumor cell lysis, while knockdown of genes belonging to the family of diacylglycerol kinases (DGK) increased sensitivity to BCMA CAR T-cells(53).
Resistance to the effector mechanisms of T-cells
T-cells kill their targets through exocytosis of cytotoxic granules that contain pore-forming perforin, as well as serine proteases such as granzyme B. Also induction of apoptosis via cross-linking of death receptors (such as Fas, TRAIL-R1, and TRAIL-R2) results in target cell lysis. Tumor cells can be resistant to T-cell-mediated killing by increased expression of several anti-apoptotic molecules, including serine protease inhibitors (serpins), which inactivate granzyme B(59). Furthermore, death receptor-mediated apoptosis can be prevented by overexpression of the anti-apoptotic protein c-FLIP, death receptor downregulation, cleavage of death receptors, or increased expression of decoy receptors(60,61). It is currently unknown whether resistance of tumor cells to the effector mechanisms of T-cells contributes to escape from CAR T-cell therapy in MM. However, a recent report showed that baseline death receptor expression on leukemic cells, correlates with response after CD19 CAR T-cell therapy in ALL(62). Other defense mechanisms against T-cell-mediated lysis, such as downregulation of MHC class 1 or 2 molecules, or defects in the antigen-processing machinery, will not impair CAR T-cell-mediated tumor cell killing.
Characteristics of the T-cells collected from the patient
Mechanisms of relapses with retained target expression, include decreased persistence and/or decreased function of CAR T-cells. However, the optimal duration of CAR T-cell persistence is unknown, and may also differ between CAR T-cell products. In this section we will discuss several baseline characteristics of the pre-manufacturing T-cells, which have an impact on CAR T-cell persistence and activity, as well as strategies to improve CAR T-cell fitness.
Baseline T-cell characteristics
MM is characterized by a broad range of active immune evasion strategies, that result in qualitative and quantitative abnormalities in immune cells, including T-cells. In addition, there is marked variability between MM patients in T-cell subset composition, including frequencies of CD4+ and CD8+ T-cells, and proportions of the different T-cell differentiation subsets, which can be explained by differences in age, pathogen exposure, and extent of treatment with immunosuppressive (alkylating drugs, proteasome inhibitors and dexamethasone) or immunostimulating anti-MM therapies (IMiDs) (Figure 3)(63,64). There is increasing evidence that the heterogeneity of T-cell subsets in the apheresis product explains part of the variability of the activity of the CAR T-cells infused to the patients in clinical trials. First, several BCMA CAR T-cell studies show that MM patients with a high frequency of early memory T-cells in the leukapheresis product experience a higher response rate and superior peak CAR T-cell expansion, when compared to patients with a low frequency of these cells(9,27,65,66). Similarly, the presence of early memory T-cells in the leukapheresis product was correlated with response in chronic lymphocytic leukemia (CLL), ALL, and lymphoma patients treated with CD19 CAR T-cells(65,67,68). These findings can be explained by the ability of T-cells with memory properties to undergo self-renewal and by their superior proliferative response, compared to more differentiated T-cells(69). In addition, a higher CD4/CD8 ratio in the leucapheresis product was associated with greater in vivo BCMA CAR T-cell expansion and response in MM(9,27). CD4+ T-cells promote the proliferation, survival and activity of CD8+ T-cells by providing a variety of cytokines including IL-2, which explains the synergy between CD4+ and CD8+ CAR T-cells in mediating antitumor responses(70,71). Furthermore, T-cells from BCMA CAR T-cell resistant patients, were enriched with terminally exhausted and senescent cells with high expression of checkpoint inhibitors, such as LAG-3, TIGIT, and PD-1(27). Altogether, this indicates that premanufacturing T-cell characteristics are important determinants of response to CAR T-cell therapy.
Figure 3. Impact of therapy on CAR T-cell activity.
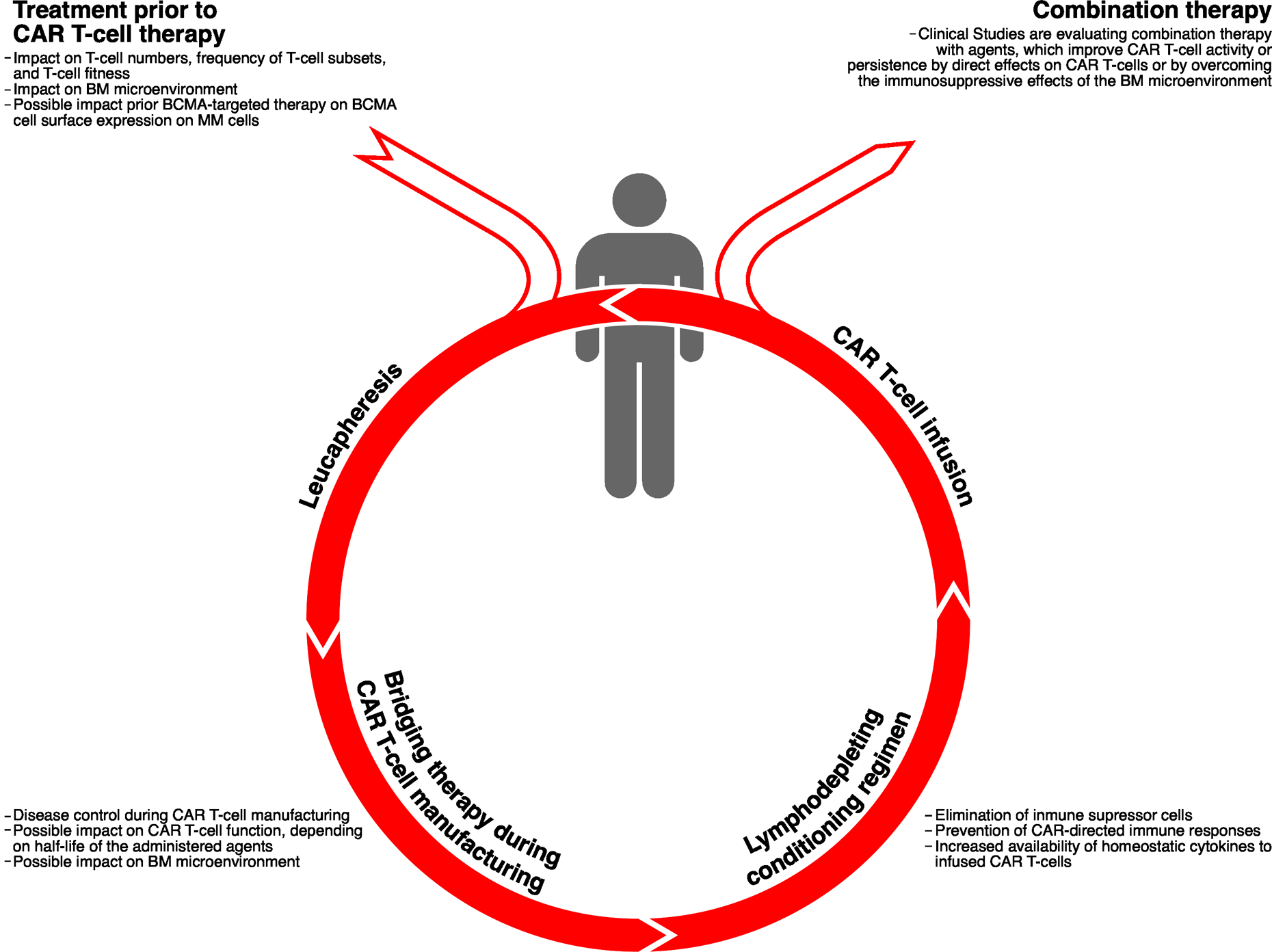
Treatment prior to leucapheresis, as well as bridging therapy administered to the patient during the production of CAR T-cells, and lymphodepleting chemotherapy prior to CAR T-cell infusion, can have an impact on the anti-tumor effect of CAR T-cells. In addition, in the setting of clinical trials, several agents are administered post CAR T-cell infusion (e.g., IMiDs and CD38-targeting antibodies) to enhance CAR T-cell efficacy or improve CAR T-cell persistence.
Effect of prior therapy on the nature of T-cells collected from the patient
Cumulative exposure to several anti-MM drugs, will reduce T-cell numbers or induce functional T-cell defects (Figure 3)(72,73). Interestingly, the frequency of early memory T-cells and CD4/CD8 ratio were higher in apheresis samples from MM patients who were early in their disease course, compared to heavily pretreated patients(74). This translated in significantly higher capacity for ex vivo proliferation during manufacturing(74). This is similar to what is observed in other malignancies where chemotherapy leads to depletion of naïve and early memory T-cells over time, and thereby poor in vitro T-cell expansion(75).
Also type of treatment administered prior to leucapheresis may affect the quality and phenotype of the harvested T cells. In ALL it has been shown that clofarabine treatment directly before leukapheresis contributes to inadequate T-cell function and probably suboptimal response to CD19 CAR T-cell therapy(76). Conversely, CLL patients treated with ibrutinib before T-cell collection had improved CD19 CAR T-cell expansion(77). Furthermore, early memory T-cells are depleted by cyclophosphamide and cytarabine in ALL and NHL patients(68). There is currently only limited data available in MM. One study showed that type of therapy prior to apheresis was not associated with response or CAR T-cell expansion(9), while another study demonstrated that patients with daratumumab as part of last line or as bridging therapy had a modestly higher response rate following ide-cel infusion when compared to patients without daratumumab as part of last line treatment (≥PR: 91% versus 75%)(6). The impact of prior therapy on T-cell fitness should be studied in larger cohorts of patients, with a focus on the potential beneficial effects of early collection of T-cells and of immunostimulatory drugs directly prior to T-cell collection. Alternatively, allogeneic T-cells obtained from healthy donors can be used to improve CAR T-cell fitness. Because of the “off-the-shelf” availability, allogeneic therapy may also overcome the logistical challenges of autologous CAR T-cell therapy. Preliminary results from the UNIVERSAL study show promising activity and a manageable safety profile (no graft-versus-host disease or neurotoxicity) of the allogeneic BCMA CAR T-cell product, ALLO-715, in patients with heavily pretreated MM(78). Approximately 90% of patients started treatment within 5 days of study enrollment(78).
BCMA also forms the target for antibody-drug conjugates (e.g. belantamab mafodotin) and bispecific antibodies (e.g. teclistamab, AMG-701, and CC-93269). A small case series showed that serial treatment with different BCMA-targeting agents is feasible(79). Ongoing studies are evaluating in a larger number of patients the efficacy of BCMA CAR T-cell therapy after prior BCMA-directed therapy.
CAR T-cell-related factors
Nature of CAR T-cells infused in patients
Extent of CAR T-cell expansion is dependent on the number of CAR T-cells administered to patients(5,6,10,12,80). In addition, several studies have demonstrated that peak expansion as well as CAR T-cell persistence are important determinants of response to BCMA CAR T-cell therapy(5,6,9,10,80–83). However, CAR T-cell expansion and persistence were not correlated with best response to cilta-cel, which may be explained by the high affinity binding of these CAR T-cells resulting in rapid elimination of disease(11,13).
Next to quantitative aspects, several qualitative characteristics of the CAR T-cell product, including T-cell functionality, extent of T-cell exhaustion, frequency of less differentiated T-cells, and CD4/CD8 ratio, may influence the efficacy of CAR T-cell therapy. In the bb21217, orva-cel (JCARH125), and P-BCMA-101 studies, patients with a higher proportion of memory-like T-cells in the infused BCMA CAR T-cell product experienced superior expansion, and had a higher probability of response and reduced risk of progression(66,82–84), which was also observed in a CLL trial with CD19 CAR T-cells(67). Preliminary evidence from clinical studies demonstrates that qualitative characteristics of the expanding CAR T-cells are also predictive for response. For example, cell expansion in patients responding to ide-cel was characterized by an increased proportion of CAR T-cells with an effector memory phenotype for both CD4+ and CD8+ subsets.(81) In addition, preclinical studies show that upon repeated antigen encounter, CAR T-cells upregulate inhibitory receptors such as PD-1, TIM-3, and LAG-3(45,85,86). Similarly, BCMA-targeted CAR T-cells acquire higher PD-1 expression after infusion in patients, which may lead to immune exhaustion and contribute to progression(4,5). Indeed, it was recently shown that expanding CAR T-cells from patients with sustained response following bb21217 treatment, expressed lower levels of PD-1 and LAG-3, compared to patients who experienced disease progression(66).
Importantly, PD-1 checkpoint blockade with antibodies has the ability to improve CAR T-cell activity and promote tumor cell death(45,85–87). CAR T-cells can also be engineered to secrete PD-1 or PD-L1 antibodies at the tumor site(88,89). Moreover, interference with signaling through the endogenous PD-1 receptor by co-transducing a PD-1 dominant negative receptor or a PD-1/CD28 chimeric receptor, enhanced CAR T cell function(85,90). Similarly, knockdown or knockout of PD-1 in CAR T-cells improved their anti-tumor efficacy(86,91). Preliminary results show that PD-1 inhibitor-based combination therapy may result in CAR T-cell expansion and anti-MM activity in a subset of patients progressing after BCMA CAR T-cell therapy(92). Other strategies to revert CAR T-cell exhaustion are also explored, including inhibition of different inhibitory immune checkpoints (e.g. LAG-3 or TIM-3) or use of co-stimulatory receptor agonists (e.g., utomilumab).
Manufacturing process
The manufacturing process includes different procedures such as T-cell activation, T-cell expansion, transduction, and storage, which may all affect the characteristics of the CAR T-cell product. Several strategies are explored to improve CAR T-cell fitness by optimizing the manufacturing process(93).
Manufacturing can be adapted to generate cell products enriched for specific subsets of T-cells with superior intrinsic abilities for survival and proliferation after infusion in patients (e.g. early memory cells). One strategy is the transduction and expansion of CAR T-cells in the presence of PI3K inhibitors (e.g. idelalisib or bb007), which results in an increased frequency of less differentiated CAR T-cells, decreased expression of PD-1 and TIM-3, improved in vivo persistence, and enhanced activity in preclinical leukemia and MM models(82,94,95). The CAR T-cell product bb21217 uses the same CAR molecule as ide-cel, but cells are cultured in the presence of bb007, resulting in enrichment for T-cells displaying a memory-like phenotype(82). In the first-in-human study, prolonged CAR T-cell persistence was observed in patients treated with escalating doses of bb21217. However, longer follow-up is required to determine whether this will translate into improved PFS(82). The manufacturing process for orva-cel and P-BCMA-101 is also designed to produce CAR T-cells enriched for central memory T-cell phenotype, but details are not disclosed(23).
Increased understanding of molecular, epigenetic, and metabolic factors that are critical for the formation and maintenance of stem cell-like memory T-cells, may also lead to novel strategies to improve CAR T-cell therapy(96–98). For example, disruption of TET2, depletion of REGNASE-1, or increasing c-Myb levels may also promote the development of memory CAR T-cells and improve CAR T-cell persistence(96–98). Generation of CAR T-cells with optimal differentiation potential and effector activity may also be achieved by using alternative cytokines during manufacturing(67,75). Furthermore, application of modified antigen presenting cells, that provide optimal signals to the CAR T-cells during manufacturing, may enhance overall CAR T-cell expansion or enable the preferential expansion of CAR T-cells with memory phenotype(99).
In addition, the variability of CD4+ and CD8+ T-cells in the apheresis product results in the production of heterogeneous CAR T-cell products with a large variation in CD4/CD8 ratio, which may contribute to differences in toxicity and activity among patients. Because CD4+ T-cell help is essential for durable T-cell immunity, several studies administer CAR T-cells with a consistent CD4/CD8 ratio after separate production of CD4+ and CD8+ CAR T-cells(24,52). However, it remains an important open research question whether generation of products with homogeneous characteristics will lead to more consistent results in clinical trials. Furthermore, “off-the-shelf” healthy donor-derived CAR T-cells with defined release criteria and minimal inter-donor variability may also lead to more consistent outcomes.
The starting material used to manufacture CAR T-cells may also contribute to manufacturing outcome. For example, high levels of myeloid cells in the starting material results in lower yields of CAR T-cells and increases the risk of product failure(100). This issue can be addressed by applying a T-cell separation strategy.
CAR structure
CAR structure affects CAR T-cell fitness, highlighting the importance of improving CAR engineering (Figure 1). CAR T-cell function may be enhanced by changing the antigen-binding domain or costimulatory domains(101). In addition, the design of the hinge region and transmembrane domains of the CAR construct may contribute to the efficiency of immune synapse formation(40).
T-cells expressing a first-generation CAR with the CD3ζ intracellular signaling domain alone have limited activity due to suboptimal activation leading to development of anergy and failure to persist. These limitations can be overcome by the incorporation of additional signaling domains from either CD28, 4-1BB, or OX40, which results in improved CAR T-cell expansion, activation, persistence and anti-tumor activity. Type of costimulatory signaling, has an impact on activity and persistence of CAR T-cells. CD28 costimulatory domains are associated with more rapid expansion and effector cell differentiation and cytotoxic ability of CAR T-cells, while 4-1BB domains may lead to superior persistence with better maintenance of a memory phenotype and reduced exhaustion(55,102–106). Distinct activation of signaling pathways and differential effects on cellular metabolism (with CD28 leading to increased glycolysis and 4-1BB to enhanced mitochondrial oxidative phosphorylation) conferred by these co-receptors, can explain these differences in CAR T-cell function(105). Most BCMA CAR T-cell products, including ide-cel and cilta-cel, use a CAR construct with 4-1BB as costimulatory molecule. Application of third generation CARs containing two costimulatory domains may further contribute to improved persistence and enhanced antitumor effects (Figure 1)(101,102).
Excessive CAR signaling as a result of high antigen burden or persistent antigen-independent (tonic) CAR signaling can induce CAR T-cell differentiation and exhaustion, resulting in poor activity(55,95,101). The incorporation of a 4-1BB endodomain instead of CD28 reduced T-cell exhaustion induced by antigen-independent signaling or by persistent antigen exposure, which may explain better persistence of CAR T-cells incorporating 4-1BB in clinical trials(22,55). Tonic signaling can also be reduced by optimizing the length of the spacer, which links the antigen-binding and transmembrane domains, or by targeting of the CAR to the T-cell receptor α constant (TRAC) locus as opposed to random insertion during conventional CAR T-cell manufacturing(107,108). The targeting of CARs to the TRAC locus with CRISPR/Cas9, places the CAR under the control of endogenous regulatory elements, leading to optimal basal and dynamic CAR expression, which improves T-cell potency by preventing tonic CAR signaling, reducing exhaustion, and delaying effector T-cell differentiation.(108) Transient rest from CAR signaling has also been shown to protect against T-cell exhaustion.(109) In this respect, several innovative strategies are explored to rapidly and reversibly control CAR expression at the cell surface. Both transcriptional(110) and post-transcriptional(111) approaches are currently evaluated in preclinical models. Beyond preventing CAR T-cell exhaustion, controlling CAR expression also has the potential to improve the safety profile of CAR T-cell therapy.
Immune-mediated rejection
Immune-mediated rejection may contribute to limited CAR T-cell persistence. In solid tumors and B-cell malignancies non-human antigen-recognition domains or suicide domains can induce humoral or cellular immune responses directed against CAR T-cells, which may result in reduced CAR T-cell counts and loss of activity(76,112,113). Similarly, in the Chinese study with LCAR-B38M, progression was associated with reduced BCMA CAR T-cell numbers and emergence of anti-CAR antibodies(8). Immune-mediated rejection may also limit the ability to treat patients with repeat CAR T-cell infusions. Indeed, development of CAR-specific immune responses explained the limited efficacy of a second infusion with CD19 CAR T-cells containing a murine scFv(114). Similarly, effectiveness of retreatment with ide-cel is limited (≥PR: 21%; median PFS: 1.0 month), which may in part be related to immune-mediated CAR T-cell rejection(10). All 6 patients, who had a response to retreatment with ide-cel, were antidrug antibody (ADA)-negative, while 73% of the 22 non-responders were ADA-positive(10).
The lymphodepleting conditioning regimen is important to suppress the development of anti-CAR immune responses(113,115). In addition, the immunogenicity of CARs may be reduced by using fully human or humanized CAR constructs(57,112,115). Orva-cel has a fully human BCMA-binding domain. In a phase 1/2 study orva-cel induced a high response rate (≥PR: 92% and ≥CR in 36%) and had an acceptable toxicity profile in 62 patients (94% triple-class refractory), who were treated with 300–600 ×106 CAR T-cells(23). Several other BCMA CAR T-cell products with fully human antigen-binding domains are currently evaluated in clinical studies(24,52,56,116), including MCARH171(80,117), FCARH143 (same CAR construct as used for orva-cel)(117), P-BCMA-101(83), CT103A(26), and the CAR T-cell product developed by UPenn(9). Interestingly, deep and durable responses were observed in 4 patients, who received CT103A after failure of a murine BCMA CAR T-cell product(26). Furthermore, less complex binding domains, such as heavy-chain-only domains, have the potential to decrease immunogenicity(22,25).
CAR T-cells are usually generated by retro- or lentiviral transduction. Non-viral vectors are also explored as a mode of gene transfer, which may decrease immunogenicity and reduce the cost of CAR T-cell production. In this context, transposon vectors (e.g. Sleeping Beauty and PiggyBac DNA transposons) have been shown to mediate stable integration and expression of CAR genes(83). In addition, CAR T-cells can be engineered by mRNA transfection, which eliminates the risk of transgene-mediated mutagenesis(118). However, the transient CAR expression with this method, may require repetitive CAR T-cell dosing(118).
Bridging therapy
Bridging therapy is administered to the majority of patients to control disease during the manufacturing process (Figure 3). Ideally, bridging therapy should not interfere with subsequent CAR T-cell expansion and persistence. Therefore, the half-life of the anti-MM agents should be taken into account. In addition, a better understanding is needed to what extent certain bridging therapies can reshape the immune suppressive BM microenvironment into a more permissive microenvironment for CAR T-cell therapy.
Lymphodepleting conditioning regimen
The lymphodepleting conditioning regimen (typically fludarabine/cyclophosphamide) prior to CAR T-cell infusion, is important for CAR T-cell expansion and persistence as a result of elimination of immune suppressor cells, prevention of CAR-directed immune responses, and through increased availability of homeostatic cytokines to newly infused cells (Figure 3)(9,114). Although lymphodepletion with fludarabine/cyclophosphamide is effective in patients with MM and other hematological malignancies, this lymphodepleting regimen is also associated with toxicity, such as long-lasting cytopenias and infections(119). Therefore further investigations are warranted to define the most optimal lymphodepleting conditioning regimen prior to CAR T-cell immunotherapy in MM.
Immune resistance conferred by the tumor microenvironment
The MM microenvironment, which consists of several components, including BM stromal cells (BMSCs), immune suppressor cells and immunosuppressive molecules, promotes tumor growth and impairs immune responses. Importantly, BMSCs also protect MM cells against CAR T-cells through various mechanisms including secretion of TGF-β and induction of anti-apoptotic proteins in MM cells(120,121). BMSC-mediated resistance can be overcome by increasing the avidity of CAR T-cells or through combination of immunotherapy with inhibitors of anti-apoptotic mediators(120).
Immune suppressor cells impair CAR T-cell activity in different types of cancer(122–125). Although Treg expansion has been described in MM patients without response to BCMA CAR T-cell therapy(27), the precise role of Tregs in mediating CAR T-cell resistance remains unclear. The impact of other immune suppressor cells, such as myeloid-derived suppressor cells (MDSCs) and immunosuppressive macrophages, on CAR T-cell activity is currently unknown in MM, and therefore all ongoing CAR T-cell trials should be accompanied by immune monitoring studies to increase our understanding of the potential ability of immune suppressor cells to impair both CAR T-cell function and persistence. The MM microenvironment is also rich in immunosuppressive cytokines and molecules. Interestingly, pretreatment levels of IL-10 are elevated in MM patients with suboptimal response following ide-cel treatment(81). Other immunosuppressive molecules (e.g. TGF-β, indoleamine 2,3-dioxygenase (IDO), arginase and adenosine) have been shown to confer resistance to CAR T-cells in various malignancies(126), but their role in MM is unclear.
Inhibitory effects from the tumor microenvironment can be partly reversed by engineering “armored” CAR T-cells that have improved ability to withstand the tumor milieu (Figure 1). Such genetic modification strategies include 1) CAR T-cells engineered to release immune stimulatory cytokines upon CAR engagement, 2) neutralization of immune suppressive signals (e.g. incorporation of dominant negative TGF-β receptor), 3) transforming an immunosuppressive signal into an immunostimulatory one by introducing a hybrid receptor, or 4) by removing genes encoding inhibitory immune checkpoints (e.g. PD-1)(87,107,127–129). Furthermore, strategies aiming at depleting, deactivating, or inducing the differentiation of immune suppressor cells, may improve the efficacy of CAR T-cells to eliminate tumor cells(125). The fludarabine/cyclophosphamide lymphodepletion regimen has the ability to induce non-selective Treg depletion. In contrast, low-dose continuous cyclophosphamide has been shown to selectively deplete Tregs in MM and solid tumors, while sparing conventional T-cells, resulting in enhanced conventional T-cell and NK cell functions(130,131). This suggests that low-dose cyclophosphamide may improve the activity of cellular therapy. Furthermore, all-trans retinoic acid is capable of reducing MDSCs numbers as well as their suppressive capacity(125). Combination therapy with inhibitors of IDO, adenosine, or arginase may also be a promising strategy to overcome the immunosuppression conferred by the tumor microenvironment. In addition, CAR T-cells simultaneously targeting tumor cells as well as components of the supportive microenvironment, may lead to CAR T-cells that are resistant to microenvironment-induced immunosuppression. For example, CD38-specific CAR T-cells or BCMA/CD38 dual-targeted CAR T-cells have the ability to eliminate CD38+ immune suppressor cells, such as Bregs, in MM patients(50,132). Bregs were also eradicated by CD19-specific CAR T-cells(49). Furthermore, in the face of Treg-mediated inhibition, superior functionality of CD28 over 4-1BB signaling was reported, which is possibly explained through enhanced secretion of proinflammatory cytokines in the presence of Tregs by CD28-based CAR T-cells(123).
Additionally, more advanced and already evaluated in clinical trials, are combination strategies with approved anti-MM agents to improve CAR T-cell function and overcome the immunosuppressive effects of the BM microenvironment. First, CD38-targeting antibodies, such as daratumumab, have the ability to eliminate CD38+ immune suppressor cells such as CD38+ Tregs, Bregs, and MDSCs, which makes this class of anti-MM agents, potential combination partners for CAR T-cell therapy, or an important component of bridging therapy to reshape the tumor microenvironment(133). CD38 is also contributing to T-cell immunosuppression through the generation of adenosine. Reducing adenosine production with CD38-targeting antibodies may further improve CAR T-cell function(134). On the other hand, CD38-targeting antibodies may also have a negative effect on CAR T-cell therapy, because activated T-cells have increased CD38 expression. However, we recently demonstrated that CD38 expression on the T-cell surface is rapidly reduced following daratumumab exposure, which prevents their elimination(135). Second, IMiDs may also be a valuable adjunct to CAR T-cells, because of their broad immunomodulatory effects, including the inhibition of Treg development in MM. Furthermore, IMiDs enhance T-cell function through the cereblon-dependent degradation of the T-cell repressors Ikaros and Aiolos(136). There is substantial preclinical evidence that the T-cell stimulatory effects of IMiDs can be used in concert with CAR T-cell therapy. Indeed, lenalidomide enhances T-helper (Th) 1-associated cytokine production, decreases secretion of Th2-associated cytokines, and improves immune synapse formation between CAR T-cells and tumor cells, resulting in enhanced cytotoxic activity of CAR T-cells(31,137,138). In MM mouse models, lenalidomide also enhanced the activity and persistence of SLAMF7- and BCMA-targeting CAR T-cells(31,137). Based on these preclinical data, several ongoing clinical studies are evaluating the combination of lenalidomide and CAR T-cells.
Conclusions
Approximately 30 years after the first reports describing engineered T-cells with chimeric scFv receptors(139), CAR T-cell therapy holds great promise in MM with regulatory approval of the first BCMA CAR T-cell products expected in the nearby future. Despite promising results, new strategies are needed to further improve the outcome of CAR T-cell therapy. A better understanding of tumor, host, and product-related features has already resulted into the design of next-generation CAR T-cell products with enhanced cytotoxic ability and improved persistence, as well as better protection against the immunosuppressive microenvironment. Also the application of immunostimulatory anti-MM agents, as opposed to drugs with immunosuppressive effects, prior to T-cell collection, may contribute to improved CAR T-cell activity. Furthermore, several clinical studies are currently evaluating the combination of CAR T-cells with therapies that are able to reduce the impact of the immunosuppressive microenvironment. However, attention should also be paid to increased toxicities such as cytokine release syndrome, that may occur in combination therapies. Finally, the introduction of new targets for CAR T-cell therapy will allow for combinatorial treatment to prevent antigen escape.
Other strategies to redirect T-cells to MM cells are also explored in MM, with promising activity of “off-the-shelf available” bispecific antibodies in patients with triple-class refractory MM(140–145). As opposed to a single infusion of CAR T-cells, bispecific antibodies are typically administered until disease progression. BCMA is also the target for antibody-drug conjugates, such as belantamab mafodotin(146–148). While cross-trial comparisons are challenging because of differences in patient characteristics, design and follow-up duration, single agent activity of CAR T-cells and bispecific antibodies is substantially higher than that of antibody-drug conjugates(146). On the other hand, depth of response with several CAR T-cell products is superior to what has been achieved with bispecific antibodies(11,14). However, studies evaluating bispecific antibodies have relatively short follow-up duration, and therefore depth of response may still improve over time. In addition, the CAR T-cell manufacturing period delays administration, which may be problematic for patients with rapidly progressive disease. Such patients may therefore be underrepresented in CAR T-cell studies, which should be taken into account when CAR T-cell therapy is compared with other types of immunotherapy. Besides efficacy, choice of modality is also dependent on other factors including patient characteristics, disease features, safety profile, availability (approval status and costs), and practical considerations (see Table 3). The safety profile of bispecific antibodies compares favorably with CAR T-cell therapy, with a lower frequency of grade ≥3 neurotoxicity and CRS, and therefore elderly patients may also benefit from treatment with bispecific antibodies(141–145). Although anti-MM activity of antibody-drug conjugates is modest in triple-class refractory MM, CRS and neurotoxicity are not observed(146,147). Hence, these agents may be applied to a larger and more diverse patient population. A limitation of belantamab mafodotin is the frequent development of keratopathy, which may substantially impair quality of life(146). Other cell types, with different killing mechanisms, such as NK cells, invariant NKT cells, γδ T-cells, or myeloid cells, can also be engineered to express a CAR(149,150). However, at this moment, in the absence of clinical data, it is unknown whether adoptive therapy with alternative cell types will be able to overcome resistance conferred by the tumor microenvironment. Altogether, we expect that these strategies will contribute to further improvement in survival of MM patients with preserved quality of life.
Table 3.
Comparison of BCMA-directed immunotherapies for patients with advanced MM (mostly triple-class refractory MM).
| Autologous CAR T-cell therapy(10,11,23,82) | Bispecific antibodies(140–145) | Antibody-drug conjugates(146–148) | ||
|---|---|---|---|---|
| Efficacy | ≥PR |
|
|
|
| ≥CR |
|
|
|
|
| Safety | CRS |
|
|
|
| Neurotoxicity |
|
|
|
|
| Ocular toxicity |
|
|
|
|
| Practical considerations | Availability |
|
|
|
| Number of administrations |
|
|
|
|
| Hospitalization |
|
|
|
|
| Route of administration |
|
|
|
|
| Infrastructure |
|
|
|
|
| Off-the-shelf available |
|
|
|
|
| Other features |
|
|
|
|
Statement of significance.
Although BCMA-specific CAR T-cell therapies are highly effective in heavily pretreated MM patients, there is until now no indication of a plateau in the survival curves. In this review, we will provide an overview of the determinants of response and the mechanisms that contribute to the development of treatment failure after initial remission (acquired resistance). A better understanding of these mechanisms, underlying lack of disease response and acquired resistance, may lead to further improvements in the effectiveness of CAR T-cell therapy.
Acknowledgements
The authors thank Victor Muñoz Sanz (Sanz Serif Research + Design Agency) for creating Figures 2 and 3.
Conflicts of interest:
NvdD: Research support from Janssen Pharmaceutical, Amgen, Celgene, Cellectis, and Bristol-Myers Squibb; Advisory boards for Janssen Pharmaceuticals, Amgen, Adaptive, Celgene, Bristol-Myers Squibb, Novartis, Roche, Takeda, Bayer and Servier.
MT holds licensed patent on CAR T cells.
SU: Research funding: Amgen, Array Biopharma, BMS, Celgene, GSK, Janssen, Merck, Pharmacyclics, Sanofi, Seattle Genetics, SkylineDX, Takeda; Consulting Fees: Abbvie, Amgen, BMS, Celgene, GSK, Genentech/Roche, Janssen, Karyopharm, Merck, Oncopeptides, Sanofi, Seattle Genetics, SkylineDx, Takeda; Speaking Fees: Celgene, Janssen, Sanofi, Takeda.
REFERENCES
- 1.Gandhi UH, Cornell RF, Lakshman A, Gahvari ZJ, McGehee E, Jagosky MH, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019;33(9):2266–75 doi 10.1038/s41375-019-0435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 2018;131(7):746–58 doi 10.1182/blood-2017-05-781351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nature biotechnology 2002;20(1):70–5 doi 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 4.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016;128(13):1688–700 doi 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol 2018;36(22):2267–80 doi 10.1200/jco.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. The New England journal of medicine 2019;380(18):1726–37 doi 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. Journal of hematology & oncology 2018;11(1):141 doi 10.1186/s13045-018-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America 2019;116(19):9543–51 doi 10.1073/pnas.1819745116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. The Journal of clinical investigation 2019;129(6):2210–21 doi 10.1172/jci126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. The New England journal of medicine 2021;384(8):705–16 doi 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 11.Madduri D, Berdeja J, Usmani S, Jakubowiak A, Agha M, Cohen A, et al. CARTITUDE-1: phase 1b/2 study of ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy, in relapsed/refractory multiple myeloma. Blood 2020;136:177. [Google Scholar]
- 12.Lin Y, Raje N, Berdeja J, Siegel D, Jagannath S, Madduri D, et al. Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed CAR T cell therapy, in patients with relapsed and refractory multipe myeloma: updated results from phase 1 CRB-401 study. Blood 2020;136:131. [Google Scholar]
- 13.Zudaire E, Madduri D, Usmani S, Jakubowiak A, Berdeja J, Geng D, et al. Translational Analysis from CARTITUDE-1, an Ongoing Phase 1b/2 Study of JNJ-4528 BCMA-targeted CAR-T Cell Therapy in Relapsed and/or Refractory Multiple Myeloma (R/R MM), Indicates Preferential Expansion of CD8+ T Cell Central Memory Cell Subset. Blood 2019;134:928. [Google Scholar]
- 14.Wang B, Zhao W, Liu J, Chen Y, Cao X, Yang Y, et al. Long-term follow-up of a phase 1, first-in-human open-label study of LCAR-B38M, a structurally differentiated chimeric antigen receptor T (CAR-T) cell therapy targeting B-cell maturation antigen (BCMA), in patients (pts) with relapsed/refractory multiple myeloma (RRMM). Blood 2019;134:579.31076443 [Google Scholar]
- 15.Green D, Pont M, Cowan A, Cole G, Sather B, Nagengast A, et al. Response to Bcma CAR-T Cells Correlates with Pretreatment Target Antigen Density and Is Improved By Small Molecule Inhibition of Gamma Secretase. Blood 2019;134:1856. [Google Scholar]
- 16.Li C, Wang Q, Zhu H, Mao X, Wang Y, Zhang Y. T Cells Expressing Anti B-Cell Maturation Antigen Chimeric Antigen Receptors for Plasma Cell Malignancies. Blood 2018;132:1013.30049811 [Google Scholar]
- 17.Salem DA, Maric I, Yuan CM, Liewehr DJ, Venzon DJ, Kochenderfer J, et al. Quantification of B-cell maturation antigen, a target for novel chimeric antigen receptor T-cell therapy in Myeloma. Leukemia research 2018;71:106–11 doi 10.1016/j.leukres.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019;134(19):1585–97 doi 10.1182/blood.2019000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidts A, Ormhoj M, Choi BD, Taylor AO, Bouffard AA, Scarfo I, et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv 2019;3(21):3248–60 doi 10.1182/bloodadvances.2019000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res 2013;19(8):2048–60 doi 10.1158/1078-0432.ccr-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bu DX, Singh R, Choi EE, Ruella M, Nunez-Cruz S, Mansfield KG, et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget 2018;9(40):25764–80 doi 10.18632/oncotarget.25359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam N, Trinklein ND, Buelow B, Patterson GH, Ojha N, Kochenderfer JN. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nature communications 2020;11(1):283 doi 10.1038/s41467-019-14119-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mailankody S, Jakubowiak A, Htut M, Costa L, Lee K, Ganguly S, et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with relapsed/refractory multiple myeloma (RRMM): update of the phase 1/2 EVOLVE study (NCT03430011). J clin Oncol 2020;38:8504. [Google Scholar]
- 24.Green D, Pont M, Sather B, Cowan A, Turtle CJ, Till B, et al. Fully human BCMA targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma Blood 2018;132:1011. [Google Scholar]
- 25.Mikkilineni L, Manasanch EE, Vanasse V, Brudno JN, Mann J, Sherry R, et al. Deep and Durable Remissions of Relapsed Multiple Myeloma on a First-in-Humans Clinical Trial of T Cells Expressing an Anti-B-Cell Maturation Antigen (BCMA) Chimeric Antigen Receptor (CAR) with a Fully-Human Heavy-Chain-Only Antigen Recognition Domain. Blood 2020;136:498. [Google Scholar]
- 26.Wang D, Wang J, Hu G, Wang W, Xiao Y, Cai H, et al. A Phase I Study of a Novel Fully Human BCMA-Targeting CAR (CT103A) in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2021. doi 10.1182/blood.2020008936. [DOI] [PubMed] [Google Scholar]
- 27.Leblay N, Maity R, Barakat E, McCulloch S, Duggan P, Jimenez-Zepeda VH, et al. Cite-seq profiling of T-cells in multiple myeloma patients undergoing BCMA targeting CAR T or Bites immunotherapy. Blood 2020;136:719. [Google Scholar]
- 28.Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai YT, Prabhala R, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nature communications 2021;12(1):868 doi 10.1038/s41467-021-21177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Da Vià MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nature medicine 2021. doi 10.1038/s41591-021-01245-5. [DOI] [PubMed] [Google Scholar]
- 30.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019;568(7750):112–6 doi 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Walter M, Urak R, Weng L, Huynh C, Lim L, et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin Cancer Res 2018;24(1):106–19 doi 10.1158/1078-0432.Ccr-17-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PW, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016;101(5):616–25 doi 10.3324/haematol.2015.137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Globerson Levin A, Rawet Slobodkin M, Waks T, Horn G, Ninio-Many L, Deshet Unger N, et al. Treatment of Multiple Myeloma using Chimeric Antigen Receptor T Cells with Dual Specificity. Cancer immunology research 2020:canimm.0118.2020 doi 10.1158/2326-6066.Cir-20-0118. [DOI] [PubMed] [Google Scholar]
- 34.Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Science translational medicine 2019;11(485) doi 10.1126/scitranslmed.aau7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosen N, Matsunaga Y, Hasegawa K, Matsuno H, Nakamura Y, Makita M, et al. The activated conformation of integrin β(7) is a novel multiple myeloma-specific target for CAR T cell therapy. Nature medicine 2017;23(12):1436–43 doi 10.1038/nm.4431. [DOI] [PubMed] [Google Scholar]
- 36.Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013;122(20):3461–72 doi 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 37.Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. The New England journal of medicine 2015;373(11):1040–7 doi 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2018;3(8) doi 10.1172/jci.insight.120505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nerreter T, Letschert S, Götz R, Doose S, Danhof S, Einsele H, et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nature communications 2019;10(1):3137 doi 10.1038/s41467-019-10948-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer immunology research 2016;4(6):498–508 doi 10.1158/2326-6066.Cir-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest 2016;126(10):3814–26 doi 10.1172/jci87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest 2016;126(8):3036–52 doi 10.1172/jci83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernández de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discovery 2020;1(2):146–54 doi 10.1158/2643-3230.Bcd-20-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen KH, Wada M, Pinz KG, Liu H, Shuai X, Chen X, et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2018;32(2):402–12 doi 10.1038/leu.2017.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zah E, Nam E, Bhuvan V, Tran U, Ji BY, Gosliner SB, et al. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nature communications 2020;11(1):2283 doi 10.1038/s41467-020-16160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang H, Dong B, Gao L, Liu L, Ge J, He A, et al. Clinical Results of a Multicenter Study of the First-in-Human Dual BCMA and CD19 Targeted Novel Platform Fast CAR-T Cell Therapy for Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020;136:178. [Google Scholar]
- 47.Yan Z, Cao J, Cheng H, Qiao J, Zhang H, Wang Y, et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol 2019;6(10):e521–e9 doi 10.1016/s2352-3026(19)30115-2. [DOI] [PubMed] [Google Scholar]
- 48.Yan L, Yan Z, Shang J, Shi X, Jin S, Kang L, et al. Sequential CD19- and Bcma-Specific Chimeric Antigen Receptor T Cell Treatment for RRMM: Report from a Single Center Study Blood 2019;134:578. [Google Scholar]
- 49.Yan L, Qu S, Shang J, Shi X, Kang L, Xu N, et al. Sequential CD19 and BCMA-specific CAR T-cell treatment elicits sustained remission of relapsed and/or refractory myeloma. Cancer medicine 2021;10(2):563–74 doi 10.1002/cam4.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C, Mei H, Hu Y, Guo T, L. L, Jiang H, et al. A bispecific CAR T-cell therapy targeting BCMA and CD38 for relapsed/refractory multiple myeloma: updated results from a phase 1dose-climbing trial Blood 2019;134:930. [Google Scholar]
- 51.Popat R, Zweegman S, Cavet J, Yong K, Lee L, Faulkner J, et al. Phase 1 First-in-Human Study of AUTO2, the First Chimeric Antigen Receptor (CAR) T Cell Targeting APRIL for Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019;134:3112. [Google Scholar]
- 52.Cowan A, Pont M, Sather B, Turtle CJ, Till B, Nagengast A, et al. Efficacy and safety of fully human Bcma CAR T cells in combination with a gamma secretase inhibitor to increase Bcma surface expression in patients with relapsed or refractory multiple myeloma. Blood 2019;134:204. [Google Scholar]
- 53.Ramkumar P, Abarientos AB, Tian R, Seyler M, Leong JT, Chen M, et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood advances 2020;4(13):2899–911 doi 10.1182/bloodadvances.2019001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raje N, Siegel D, Jagannath S, Lonial S, Munshi N, Moreau P, et al. Idecabtagene vicleucel (ide-cel, bb2121) in relapsed and refractory multiple myeloma: analyses of high-risk subgroups in the KarMMa study Blood 2020;136:3234. [Google Scholar]
- 55.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21(6):581–90 doi 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jie J, Hao S, Jiang S, Li Z, Yang M, Zhang W, et al. Phase 1 trial of the safety and efficacy of fully human anti-BCMA CAR T-cells in relapsed/refractory multiple myeloma. Blood 2019;134:4435. [Google Scholar]
- 57.Hao S, Jin J, Jiang S, Li Z, Zhang W, Yang M, et al. Two-Year Follow-up of Investigator-Initiated Phase 1 Trials of the Safety and Efficacy of Fully Human Anti-Bcma CAR T Cells (CT053) in Relapsed/Refractory Multiple Myeloma. Blood 2020;136:132. [Google Scholar]
- 58.Büll C, Boltje TJ, Balneger N, Weischer SM, Wassink M, van Gemst JJ, et al. Sialic Acid Blockade Suppresses Tumor Growth by Enhancing T-cell-Mediated Tumor Immunity. Cancer Res 2018;78(13):3574–88 doi 10.1158/0008-5472.Can-17-3376. [DOI] [PubMed] [Google Scholar]
- 59.Medema JP, de Jong J, Peltenburg LT, Verdegaal EM, Gorter A, Bres SA, et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proceedings of the National Academy of Sciences of the United States of America 2001;98(20):11515–20 doi 10.1073/pnas.201398198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pitti RM, Marsters SA, Lawrence DA, Roy M, Kischkel FC, Dowd P, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 1998;396(6712):699–703 doi 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 61.Medema JP, de Jong J, van Hall T, Melief CJ, Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. The Journal of experimental medicine 1999;190(7):1033–8 doi 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov 2020;10(4):552–67 doi 10.1158/2159-8290.Cd-19-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kay NE, Leong TL, Bone N, Vesole DH, Greipp PR, Van Ness B, et al. Blood levels of immune cells predict survival in myeloma patients: results of an Eastern Cooperative Oncology Group phase 3 trial for newly diagnosed multiple myeloma patients. Blood 2001;98(1):23–8 doi 10.1182/blood.v98.1.23. [DOI] [PubMed] [Google Scholar]
- 64.Rytlewski J, Madduri D, Fuller J, Campbell T, Mashadi-Hossein A, Thompson E, et al. Effects of prior alkylating therapies on preinfusion patient characteristics and starting material for CAR T-cell product manufacturing in late-line multiple myeloma. Blood 2020;136:1405. [Google Scholar]
- 65.Wang M, Pruteneau-Malinici I, Cohen A, Garfall AL, Milone MC, Tian L, et al. Identification and Validation of Predictive Biomarkers to CD19- and BCMA-Specific CAR T-Cell Responses in CAR T-Cell Precursors Blood 2019;134:622. [Google Scholar]
- 66.Finney O, Yeri A, Mao P, Pandya C, Alonzo E, Hopkins G, et al. Molecular and Phenotypic Profiling of Drug Product and Post-Infusion Samples from CRB-402, an Ongoing: Phase I Clinical Study of bb21217 a BCMA-Directed CAR T Cell Therapy. Blood 2020;136:1401. [Google Scholar]
- 67.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24(5):563–71 doi 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Science translational medicine 2016;8(320):320ra3 doi 10.1126/scitranslmed.aad5222. [DOI] [PubMed] [Google Scholar]
- 69.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the United States of America 2005;102(27):9571–6 doi 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016;30(2):492–500 doi 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Science translational medicine 2014;6(261):261ra151 doi 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heider U, Rademacher J, Kaiser M, Kleeberg L, von Metzler I, Sezer O. Decrease in CD4+ T-cell counts in patients with multiple myeloma treated with bortezomib. Clinical lymphoma, myeloma & leukemia 2010;10(2):134–7 doi 10.3816/CLML.2010.n.019. [DOI] [PubMed] [Google Scholar]
- 73.Chung DJ, Pronschinske KB, Shyer JA, Sharma S, Leung S, Curran SA, et al. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer immunology research 2016;4(1):61–71 doi 10.1158/2326-6066.Cir-15-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garfall AL, Dancy EK, Cohen AD, Hwang WT, Fraietta JA, Davis MM, et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood advances 2019;3(19):2812–5 doi 10.1182/bloodadvances.2019000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Das RK, Vernau L, Grupp SA, Barrett DM. Naïve T-cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov 2019;9(4):492–9 doi 10.1158/2159-8290.Cd-18-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385(9967):517–28 doi 10.1016/s0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016;127(9):1117–27 doi 10.1182/blood-2015-11-679134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mailankody S, Matous J, Liedtke M, Sidana S, Malik S, Nath R, et al. First-in-human study of the allogeneic anti-BCMA ALLO-715 CAR T-cell therapy and the anti-CD52 mab ALLO-647 in relapsed/refractory multiple myelloma (UNIVERSAL Study). Blood 2020;136:129. [Google Scholar]
- 79.Cohen AD, Garfall AL, Dogan A, Lacey SF, Martin C, Lendvai N, et al. Serial treatment of relapsed/refractory multiple myeloma with different BCMA-targeting therapies. Blood advances 2019;3(16):2487–90 doi 10.1182/bloodadvances.2019000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mailankody S, Ghosh A, Staehr M, Purdon TJ, Roshal M, Halton E, et al. Clinical responses and pharmacokinetics of MCARH171, a human-derived BCMA targeted CAR T cell therapy in relapsed/refractory multiple myeloma: final results of a phase 1 clinical trial. Blood 2018;132:959. [Google Scholar]
- 81.Martin M, Thompson E, Brown W, Finney O, Rytlewski J, Jiang Y, et al. Idecabtagene vicleucel (ide-cel, bb2121) responders are characterized by early and temporally consistent activation and expansion of CAR T cells with a T effector phenotype Blood 2020136:2315. [Google Scholar]
- 82.Alsina M, Shah N, Raje N, Jagannath S, Madduri D, Kaufman JL, et al. Updated results from the phase 1 CRB-402 study of anti-BCMA CAR T-cell therapy bb21217 in patients with relpased and refractory multiple myeloma: correlation of expansion and duration of response with T-cell phenotypes BLood 2020(136):130.32430495 [Google Scholar]
- 83.Costello C, Cohen A, Patel K, Ali S, Berdeja J, Shah N, et al. Phase 1/2 Study of the Safety and Response of P-BCMA-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) Blood 2020;136:134. [Google Scholar]
- 84.Colonna M, Navarro G, DeVries T, Beckett V, Amsberry A, Radhakrishnan A, et al. Orvacabtagene Autoleucel (orva-cel; JCARH125): A Fully Human BCMA-Targeted Second-Generation CAR T Cell Product Characterized By a Predominant Central Memory Phenotype with High in Vitro and In Vivo Proliferative Potential and Sustained In Vivo Persistence. Blood 2020;136:2358. [Google Scholar]
- 85.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016;126(8):3130–44 doi 10.1172/jci83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Molecular therapy : the journal of the American Society of Gene Therapy 2016;24(6):1135–49 doi 10.1038/mt.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep 2017;7(1):10541 doi 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016;7(23):34341–55 doi 10.18632/oncotarget.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(22):6982–92 doi 10.1158/1078-0432.Ccr-17-0867. [DOI] [PubMed] [Google Scholar]
- 90.Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res 2016;76(6):1578–90 doi 10.1158/0008-5472.Can-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific reports 2017;7(1):737 doi 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bernabei L, Garfall AL, Melenhorst JJ, Lacey SF, Stadtmauer E, Vogl DT, et al. PD-1 Inhibitor Combinations As Salvage Therapy for Relapsed/Refractory Multiple Myeloma (MM) Patients Progressing after Bcma-Directed CAR T Cells. Blood 2018;132:1973. [Google Scholar]
- 93.Gargett T, Truong N, Ebert LM, Yu W, Brown MP. Optimization of manufacturing conditions for chimeric antigen receptor T cells to favor cells with a central memory phenotype. Cytotherapy 2019;21(6):593–602 doi 10.1016/j.jcyt.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 94.Stock S, Ubelhart R, Schubert ML, Fan F, He B, Hoffmann JM, et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. International journal of cancer 2019;145(5):1312–24 doi 10.1002/ijc.32201. [DOI] [PubMed] [Google Scholar]
- 95.Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2018;32(5):1157–67 doi 10.1038/s41375-017-0008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018;558(7709):307–12 doi 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nature immunology 2019;20(3):337–49 doi 10.1038/s41590-018-0311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2019;576(7787):471–6 doi 10.1038/s41586-019-1821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hasan AN, Selvakumar A, O’Reilly RJ. Artificial Antigen Presenting Cells: An Off the Shelf Approach for Generation of Desirable T-Cell Populations for Broad Application of Adoptive Immunotherapy. Advancements in genetic engineering 2015;4(3). [PMC free article] [PubMed] [Google Scholar]
- 100.Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy 2016;18(7):893–901 doi 10.1016/j.jcyt.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI insight 2018;3(1) doi 10.1172/jci.insight.96976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2019;25(13):4014–25 doi 10.1158/1078-0432.Ccr-18-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer cell 2015;28(4):415–28 doi 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular therapy : the journal of the American Society of Gene Therapy 2009;17(8):1453–64 doi 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016;44(2):380–90 doi 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 106.Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 2004;172(1):104–13 doi 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 107.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020;367(6481) doi 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543(7643):113–7 doi 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weber E, Lynn R, Parker K, Anbunathan H, Lattin J, Sotillo E, et al. Transient “rest” induces functional reinvigoration and epigenetic remodeling in exhausted CAR-T cells. bioRxiv 202001.26.920496. [Google Scholar]
- 110.Drent E, Poels R, Mulders MJ, van de Donk N, Themeli M, Lokhorst HM, et al. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PloS one 2018;13(5):e0197349 doi 10.1371/journal.pone.0197349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Science translational medicine 2021;13(575) doi 10.1126/scitranslmed.abb6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011;117(1):72–82 doi 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 113.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Science translational medicine 2016;8(355):355ra116 doi 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. The Journal of clinical investigation 2016;126(6):2123–38 doi 10.1172/jci85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gorovits B, Koren E. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy 2019;33(3):275–84 doi 10.1007/s40259-019-00354-5. [DOI] [PubMed] [Google Scholar]
- 116.Li C, Wang J, Wang D, Hu G, Yang Y, Zhou X, et al. Efficacy and safety of fully human BCMA targeting CAR T-cell therapy in relapsed/refractory MM. Blood 2019;134:929. [Google Scholar]
- 117.Smith EL, Staehr M, Masakayan R, Tatake IJ, Purdon TJ, Wang X, et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Molecular therapy : the journal of the American Society of Gene Therapy 2018;26(6):1447–56 doi 10.1016/j.ymthe.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lin L, Cho SF, Xing L, Wen K, Li Y, Yu T, et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia 2020. doi 10.1038/s41375-020-0951-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bechman N, Maher J. Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy - what are we doing; where are we going? Expert opinion on biological therapy 2020:1–11 doi 10.1080/14712598.2021.1857361. [DOI] [PubMed] [Google Scholar]
- 120.Holthof L, van der Horst H, Poels R, van der Schans J, Gelderloos A, Fenghzi L, et al. The Impact and Modulation of Microenvironment-Induced Immune Resistance Against CAR T Cell and Antibody Treatments in Multiple Myeloma. Blood 2019;134:137. [Google Scholar]
- 121.Sakemura R, Cox M, Hansen M, Hefazi M, Roman C, Schick K, et al. Targeting Cancer Associated Fibroblasts in the Bone Marrow Prevents Resistance to Chimeric Antigen Receptor T Cell Therapy in Multiple Myeloma. Blood 2019;134:865. [Google Scholar]
- 122.Reiss D, Do T, Kuo D, Gray V, Olson N, Lee C, et al. Multiplexed Immunofluorescence (IF) Analysis and Gene Expression Profiling of Biopsies from Patients with Relapsed/Refractory (R/R) Diffuse Large B Cell Lymphoma (DLBCL) Treated with Lisocabtagene Maraleucel (liso-cel) in Transcend NHL 001 Reveal Patterns of Immune Infiltration Associated with Durable Response Blood 2019;134:202. [Google Scholar]
- 123.Kegler A, Koristka S, Bergmann R, Berndt N, Arndt C, Feldmann A, et al. T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. Oncoimmunology 2019;8(9):e1621676 doi 10.1080/2162402x.2019.1621676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov 2017;7(10):1154–67 doi 10.1158/2159-8290.Cd-16-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer immunology research 2016;4(10):869–80 doi 10.1158/2326-6066.Cir-15-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015;125(25):3905–16 doi 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.DeRenzo C, Gottschalk S. Genetic Modification Strategies to Enhance CAR T Cell Persistence for Patients With Solid Tumors. Front Immunol 2019;10:218 doi 10.3389/fimmu.2019.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell reports 2018;23(7):2130–41 doi 10.1016/j.celrep.2018.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-zeta CAR T Cells Resist TGF-beta Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Molecular therapy : the journal of the American Society of Gene Therapy 2018;26(9):2218–30 doi 10.1016/j.ymthe.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer immunology, immunotherapy : CII 2007;56(5):641–8 doi 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Franssen LE, Nijhof IS, Bjorklund CC, Chiu H, Doorn R, van Velzen J, et al. Lenalidomide combined with low-dose cyclophosphamide and prednisone modulates Ikaros and Aiolos in lymphocytes, resulting in immunostimulatory effects in lenalidomide-refractory multiple myeloma patients. Oncotarget 2018;9(74):34009–21 doi 10.18632/oncotarget.26131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther 2017;25(8):1946–58 doi 10.1016/j.ymthe.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016;128(3):384–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov 2018;8(9):1156–75 doi 10.1158/2159-8290.Cd-17-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Krejcik J, Frerichs KA, Nijhof IS, van Kessel B, van Velzen JF, Bloem AC, et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin Cancer Res 2017;23(24):7498–511 doi 10.1158/1078-0432.Ccr-17-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol 2014;164(6):811–21 doi 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Works M, Soni N, Hauskins C, Sierra C, Baturevych A, Jones JC, et al. Anti-B-cell maturation antigen chimeric antigen receptor T cell function against multiple myeloma is enhanced in the presence of lenalidomide. Molecular cancer therapeutics 2019. doi 10.1158/1535-7163.Mct-18-1146. [DOI] [PubMed] [Google Scholar]
- 138.Kuramitsu S, Ohno M, Ohka F, Shiina S, Yamamichi A, Kato A, et al. Lenalidomide enhances the function of chimeric antigen receptor T cells against the epidermal growth factor receptor variant III by enhancing immune synapses. Cancer gene therapy 2015;22(10):487–95 doi 10.1038/cgt.2015.47. [DOI] [PubMed] [Google Scholar]
- 139.Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplantation proceedings 1989;21(1 Pt 1):127–30. [PubMed] [Google Scholar]
- 140.Harrison B, Minnema M, Lee HC, Spencer A, Kapoor P, Madduri D, et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (bispecific T-cell engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020;136:28. [Google Scholar]
- 141.Garfall AL, Usmani S, Mateos MV, Nahi H, Van de Donk N, San-Miguel J, et al. Updated phase 1 results of teclistamab, a B-cell maturation antigen (BCMA) × CD3 bispecific antibody, in relapsed and/or refractory multiple myeloma (RRMM). Blood 2020;136:27. [Google Scholar]
- 142.Costa L, Wong S, Bermudez A, de la Rubia J, Mateos MV, Ocio E, et al. Interim results from the first phase 1 clinical study of the B-cell maturation antigen (BCMA) 2+1 T-cell engager (TCE) CC-93269 in patients (pts) with relapsed/refractory multiple myeloma (RRMM) EHA 2020:S205. [Google Scholar]
- 143.Madduri D, Rosko A, Brayer J, Zonder J, Bensinger W, Li J, et al. REGN5458, a BCMA × CD3 Bispecific Monoclonal Antibody, Induces Deep and Durable Responses in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020;136:291. [Google Scholar]
- 144.Lesokhin A, Levy M, Dalovisio A, Bahlis N, Solh M, Sebag M, et al. Preliminary Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of Subcutaneously (SC) Administered PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020;136:3206. [Google Scholar]
- 145.Rodriguez C, D’Souza A, Shah N, Voorhees P, Buelow B, Vij R, et al. Initial Results of a Phase I Study of TNB-383B, a BCMA × CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020;136:293. [Google Scholar]
- 146.Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol 2020;21(2):207–21 doi 10.1016/s1470-2045(19)30788-0. [DOI] [PubMed] [Google Scholar]
- 147.Kumar S, Migkou M, Bhutani M, Spencer A, Ailawadhi S, Kalff A, et al. Phase 1 First-in-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020;136:26. [Google Scholar]
- 148.Lee HC, Raje NS, Landgren O, Upreti VV, Wang J, Avilion AA, et al. Phase 1 study of the anti-BCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia 2021;35(1):255–8 doi 10.1038/s41375-020-0834-9. [DOI] [PubMed] [Google Scholar]
- 149.Poels R, Drent E, Lameris R, Katsarou A, Themeli M, van der Vliet HJ, et al. Preclinical Evaluation of Invariant Natural Killer T Cells Modified with CD38 or BCMA Chimeric Antigen Receptors for Multiple Myeloma. International journal of molecular sciences 2021;22(3) doi 10.3390/ijms22031096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014;28(4):917–27 doi 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]