

SUMMARY
Aging causes stem cell dysfunction, as a result of extrinsic and intrinsic changes. Decreased function of the stem cell niche is an important contributor to this dysfunction. We use the Drosophila testis to ask what factors maintain niche cells. The testis niche comprises quiescent “hub” cells and supports two mitotic stem cell pools: germline stem cells and somatic cyst stem cells (CySCs). We identify the cell cycle-responsive Dp/E2f1 transcription factor as a crucial non-autonomous regulator required in CySCs to maintain hub cell quiescence. Dp/E2f1 inhibits local Activin ligands through production of the Activin antagonist Follistatin (Fs). Inactivation of Dp/E2f1 or Fs in CySCs or promoting Activin receptor signaling in hub cells causes transdifferentiation of hub cells into fully functional CySCs. This Activin-dependent communication between CySCs and hub regulates the physiological decay of the niche with age and demonstrates that hub cell quiescence results from signals from surrounding stem cells.
Keywords: Drosophila, testis, cyst stem cell, niche, quiescence, transdifferentiation, Activin, Follistatin, Dp/E2f, aging, fertility
INTRODUCTION
The niche creates a distinct microenvironment for stem cells and secretes short range self-renewal cues that promote ‘stemness’ in the resident population (Morrison and Spradling, 2008). Decreased stem cell function with age can at least in part be attributed to both decreased niche function and niche cell numbers (Oh et al., 2014). These observations raise the question of what factors maintain niche cells. The Drosophila testis provides an ideal system to address this important issue. The testis niche comprises approximately twelve quiescent somatic hub cells that support two mitotic stem cell pools: GSCs that ultimately produce sperm and somatic CySCs that support GSCs and produce somatic support cells. The hub and CySCs share a common lineage during development: somatic cells are initially all equivalent somatic gonadal precursors, but during embryonic stages, a subset of these are specified to become hub cells (Okegbe and DiNardo, 2011, Dinardo et al., 2011, Le Bras and Van Doren, 2006, Anllo et al., 2019, Kitadate and Kobayashi, 2010). The remaining somatic precursors become CySCs and their offspring, cyst cells. Intriguingly, despite their common origin, CySCs are the only somatic cells in the testis to proliferate, both hub and cyst cells are post-mitotic. Since they cease proliferating, hub cells need to be maintained during adulthood; work has identified several factors that act autonomously within the hub to maintain quiescence or survival (Hetie et al., 2014, Voog et al., 2014, Resende et al., 2013, Greenspan and Matunis, 2018). These include the transcription factor Escargot (Esg), as well as the cell cycle inhibitor Retinoblastoma homolog, Rbf. Most intriguingly, prior work has shown that genetic ablation of all CySCs causes hub cells to exit quiescence and transdifferentiate into CySCs, suggesting the existence of unidentified CySC-derived factors that non-autonomously maintain hub cells (Hetie et al., 2014). Finally, during normal aging, the ability of the hub to support stem cells declines. This is due both to a reduction in hub cell numbers (Sreejith et al., 2019, Wallenfang et al., 2006, Lee et al., 2016), as well as lower production of self-renewal ligands to support surrounding stem cells (Toledano et al., 2012, Boyle et al., 2007). Yet how the mechanisms maintaining hub cell quiescence are affected during aging is still unknown.
Here we identify how CySCs maintain hub cell quiescence. We find that hub cells are lost following depletion of the transcription factor that regulates S-phase gene expression, which is a complex of the activator E2f, called E2f1 in Drosophila, and the sole Dimerization Partner homolog, Dp, in CySCs. This signaling between cycling CySCs and the hub is mediated through production of Follistatin (Fs), an antagonist of Activin signaling. Finally, we show that Activin autonomously promotes hub cells to transdifferentiate into CySCs, leading to hub cell loss, and that increased activity of this pathway is responsible for age-dependent loss of hub cells.
RESULTS
Dp/E2f1 functions in CySCs to non-autonomously maintain hub cells
Our prior work focused on how cell cycle progression in CySCs, the only mitotic somatic cells in the testis, influenced cell fate (Amoyel et al., 2014). Here, we focused on the transcription factor that regulates S-phase gene expression, which is a complex of E2f1 and Dp and which we previously reported to be active in CySCs (Amoyel et al., 2014). Knockdown of Dp by RNAi using the cyst lineage driver traffic jam (tj)-GAL4 resulted in a complete loss of CySCs, as identified by Zfh1 expression, and only Eya-positive differentiated cyst cells were visible (Leatherman and Dinardo, 2008, Fabrizio et al., 2003) (Fig. 1A,B). In most cases (12/20), testes with somatic Dp depletion lacked the entire stem and early differentiated cell compartment for both somatic and germ lineages and contained only spermatocytes or spermatid fibers. This suggested that Dp was required for CySC self-renewal and that its loss resulted in ectopic and premature differentiation. To confirm this, we used mitotic recombination to generate clones mutant for Dp. Surprisingly, Dp mutant CySC clones, which we identified as Tj-positive cells adjacent to the hub, were recovered at similar rates to control clones, both 7 days post clone induction (dpci) and 14 dpci (Fig. 1C-F and Fig. S1A,B), indicating that they had no autonomous self-renewal defect. We confirmed this result using an independent Dp null allele and used an antibody against Dp to verify that the mutant clones lacked Dp protein (Fig. S1A-C). Similarly, CySC clones expressing the same Dp RNAi as in Fig. 1B were also recovered at 7 dpci (Fig. S1D). Dp null mutant clones resulting from single clonal induction events contained many cells, indicating that they had proliferated over the course of the experiment (Fig. 1D). Indeed, Dp mutant CySCs were found to incorporate the nucleotide analogue 5-ethynyl-2’-deoxyuridine (EdU), demonstrating that cells lacking Dp could undergo DNA replication (Fig. 1G,H and Fig. S1A,E, arrowheads). Consistently, we recovered clones mutant for E2f1, the sole activator E2f in Drosophila, at similar rates to control clones at 7 dpci (Fig. S1F,G). While surprising, this result concurs with recent work showing that Dp is dispensable for most larval proliferation in Drosophila and that viable adults lacking Dp in all but muscle tissues can be obtained (Zappia and Frolov, 2016). Similarly in mouse, proliferation still occurs in the absence of all activating E2fs (Chen et al., 2009).
Figure 1: Dp/E2f1 is required in CySCs to maintain hub cells.
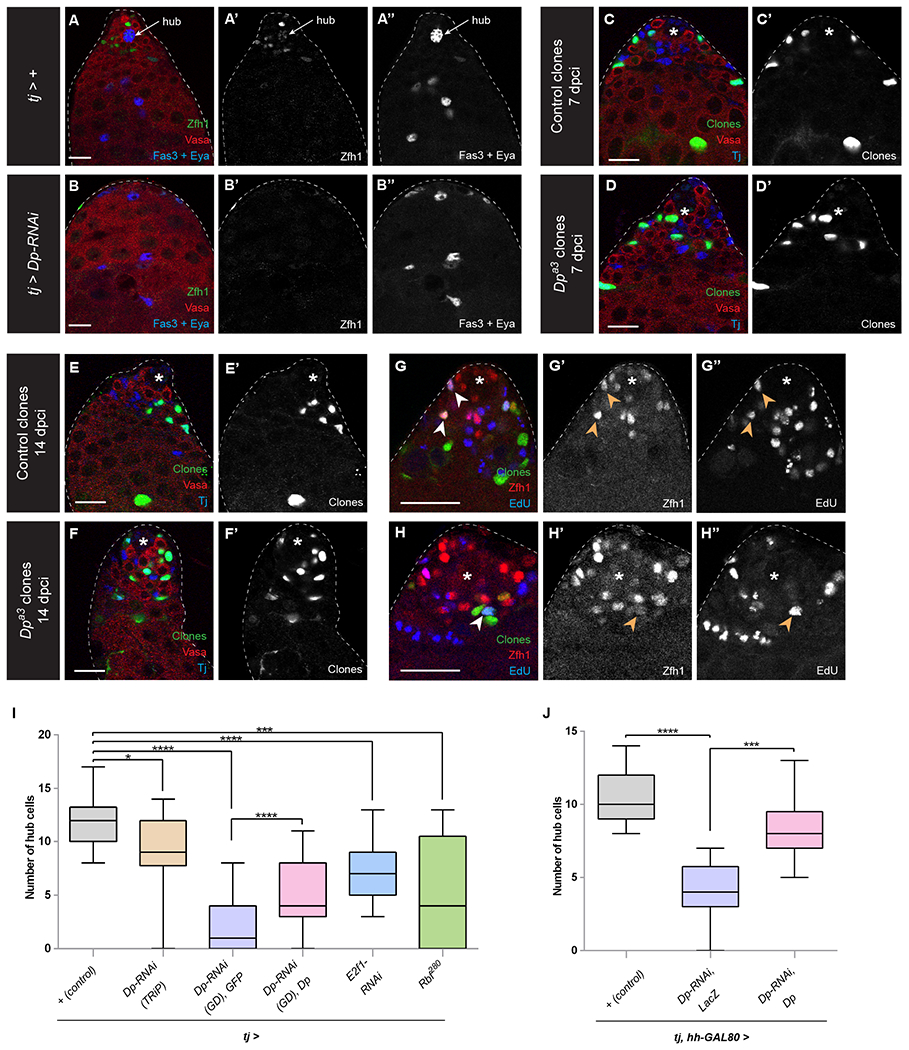
(A, B) A control tj-GAL4 (labeled tj > +) adult testis with hub cells (A, arrow) surrounded by both GSCs and CySCs. A tj > Dp RNAi (B) adult testis lacking CySCs, GSCs and hub cells. Both testes were isolated after 10 days at 29°C to induce maximal GAL4 activity. Zfh1 (green) labels CySCs; Vasa (red) marks the germline; Fas3 (blue) marks the hub cell membranes; Eya (blue) labels the nucleus of differentiating cyst cells.
(C-H) GFP-positive FRT42 control clones (C, E, G) or FRT42 Dpa3 mutant clones (D, F, H). Both types of CySC clones can be recovered at 7 days post clone induction (dpci) (C, D) and 14 dpci (E ,F) and both incorporate EdU (blue G, H), indicating that they can undergo S phase. Clones are marked by GFP (green), Vasa (red, C-F) marks the germline, and Tj (blue, C-F) marks CySCs and early cyst cells. Zfh1 (blue, G, H) marks CySCs.
(I) Graph showing the average number of hub cells after 10 days in 29°C using tj-GAL4 in control (+, gray bar, n=18), Dp-RNAi (brown and purple bars, n=14 and n=31, respectively), Dp depletion plus exogenous Dp (pink bar, n=15), E2f1 depletion (blue bar, n=17), overexpression of Rbf280 (green bar, n=12).
(I, J) Graphs showing the average number of hub cells after 10 days in 29°C using tj-GAL4 and hh-GAL80, which inhibits GAL4 activity in the hub, limiting expression of UAS-dependent constructs to CySCs in control (+, gray bar, n=27), Dp-RNAi (purple bar, n=8), Dp depletion plus exogenous Dp (pink bar, n=33).
An asterisk marks the hub.
Error bars represent the data range. **** P < 0.0001; *** P < 0.001; * P < 0.05 as assessed by Student’s t-test.
See also Tables S1 and S2, Figures S1 and S2.
Scale bar = 20 μM.
The fact that Dp was not required autonomously for CySC self-renewal, but its depletion in all CySCs led to loss of the entire stem cell population suggested that Dp may be required in a non-autonomous manner to maintain CySCs. Indeed, we noticed that in addition to loss of CySCs, hub cells were also absent in testes in which Dp was knocked down with tj-GAL4 (Fig. 1A,B,I). While in controls the hub is composed of 11.8±0.5 cells, when Dp was knocked down in the cyst lineage, there were many fewer cells (2.1±0.4). To verify this observation, we used the markers upd-LacZ and hh-LacZ to label hub cells and found a reduction of labeled cells in the Dp knockdown and in many cases no cells expressing these markers (Tulina and Matunis, 2001, Forbes et al., 1996, Amoyel et al., 2013)(Fig. S2A-D). To confirm that this phenotype was due to loss of Dp and not an off-target effect of the RNAi, we used three approaches: first, we expressed full-length Dp together with Dp RNAi and observed a partial rescue of hub cell numbers compared to co-expressing GFP as a titration control (Fig. 1I, compare purple to pink bar); second, we used an independent RNAi line targeting Dp, which gave a similar, albeit weaker loss of hub cells (Fig. 1I, compare brown to gray bar); third, we inactivated the Dp/E2f1 transcription factor by knocking down E2f1 or over-expressing a constitutively active form of the Dp/E2f1 inhibitor Rbf, called Rbf280, which recapitulated the loss of hub cells observed in Dp knockdowns in both cases (Fig. 1I, compare blue and green bars to gray bar).
We then ruled out the possibility of hub loss being due to a developmental defect of the Dp RNAi by using a temperature-sensitive form of GAL80 (GAL80TS) to only induce transgene expression in adult stages (McGuire et al., 2004). We analyzed flies at eclosion (0 days) after raising them at the permissive temperature for GAL80TS (18ºC) and observed no significant differences in hub cell numbers between control flies expressing tj-GAL4 and GAL80TS, referred to as tjTS, and flies also carrying Dp or E2f RNAi transgenes (Fig. S2E). After shifting to the restrictive temperature, we observed no significant change in hub cells in control tjTS flies over 10 days, although we see a slight but not significant decrease in 20 day-old flies. By contrast, Dp knock down, E2f1 knock down or Rbf280 overexpression in CySCs led to a progressive loss of hub cells and an almost complete loss (0.03±0.03 in the Dp knock down) by 20 days of adulthood (Fig. S2E and Table S1). Altogether, these results indicate that the hub cell loss observed in Dp knockdowns is due to a progressive defect in maintenance of hub cells, not in their establishment.
We sought to establish that Dp/E2f1 function was indeed required specifically in CySCs to maintain hub cells non-autonomously. Since tj-GAL4 is occasionally expressed in hub cells (Fairchild et al., 2016), we generated a strain expressing GAL80 under the control of the endogenous hh locus, which is expressed exclusively in hub cells (see Methods) (Amoyel et al., 2013, Michel et al., 2012). Importantly, knockdown of Dp using tj-GAL4 led to significant loss of hub cells in a hh-GAL80 background (4.0±0.8 hub cells in Dp RNAi vs 10.6±0.3 in control, Fig. 1J, purple bar). As expected, hub cell loss was suppressed by co-expressing Dp in non-hub somatic cells (8.4±0.3 hub cells vs 4.0±0.8 in controls expressing LacZ as a titration control, n=33 and 8, respectively, P < 0.001, Fig. 1J, pink bar). Finally, to rule out any possibility of ectopic hub expression of the RNAi, we used alternative GAL4 lines which drive expression in CySCs and cyst cells and which were previously shown to have sporadic or negligible expression in hub cells: eyaA3-GAL4 (Fairchild et al., 2016), fringe (fng)-GAL4 (Dinardo et al., 2011) and C587-GAL4 (Fairchild et al., 2016, Hetie et al., 2014). Knockdown of Dp using all three drivers resulted in a loss of hub cells (Fig. S2F). In the case of fng-GAL4, we used GAL80TS to show progressive hub cell loss in adult flies when Dp or E2f1 were knocked down (Fig. S2F and Table S1). In sum, our results indicate that activity of the Dp/E2f1 transcription factor is dispensable within individual CySCs for cell cycle progression but that its loss in all adult CySCs results in a progressive and non-autonomous loss of hub cells. These observations suggest a model in which Dp/E2f1 activity in CySCs causes them to signal to hub cells to promote their maintenance in adult testes.
Dp/E2f1 regulates Follistatin expression to maintain the hub non-autonomously
To determine the signals downstream of Dp/E2f1 that acted on the hub non-autonomously, we performed an RNAi screen. We used tj-GAL4 to deplete from CySCs secreted factors that are enriched in testicular stem cells and then assessed hub cell number (Terry et al., 2006, Kurusu et al., 2008) (Fig. S3A). Knockdown of Follistatin (Fs), encoding a conserved repressor of TGFβ/Activin ligands (Fig. 2A) (Pentek et al., 2009), recapitulated the phenotypes observed with Dp or E2f1 knockdown. After 14 days of RNAi expression, tj>Fs-RNAi testes contained 1.1±0.3 hub cells, whereas control tj>LacZ testes had 9.7±0.5 hub cells (Fig. 2B-D). Frequently, tj>Fs-RNAi testes contained 0 hub cells. In those testes, both stem cell populations were lost and we frequently observed only differentiated spermatids, indicating that all the more undifferentiated cell types had been lost to ectopic differentiation (Fig. S3B,C). The specificity of the Fs RNAi transgene was confirmed by RT-qPCR analysis showing a significant decrease in Fs transcripts in tj>Fs-RNAi testes (Fig. S3D,E) and by a significant rescue of hub cells when full length Fs was concomitantly overexpressed with Fs-RNAi (Fig. 2D). Overexpression of wild type Fs alone using tj-GAL4 did not affect the number of hub cells (Fig. 2D). No phenotype was observed when Fs was depleted from the germline using nanos (nos)-GAL4 (Fig. S3F).
Figure 2: Follistatin acts downstream of Dp/E2f1 in CySCs to maintain hub cells.
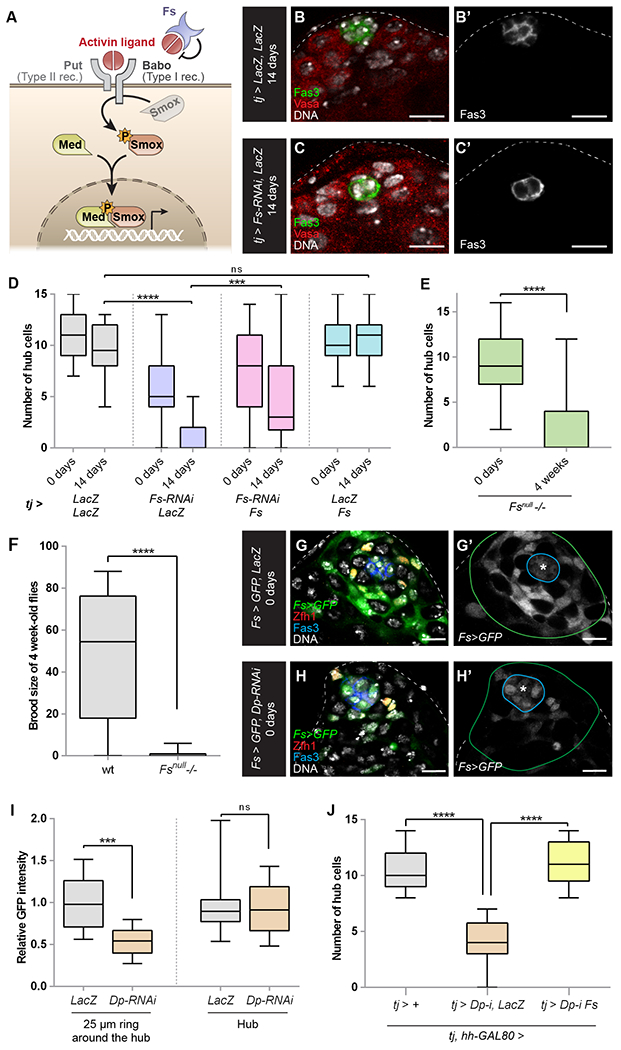
(A) Model of the Activin pathway. Fs (purple) inhibits Activin ligands (red) binding to Activin receptors, Baboon (Babo, type I receptor) and Punt (Put, type II receptor). Receptor activation causes phosphorylation (orange P star) of the SMAD3 homolog Smox (inactive Smox is gray, active Smox is brown). Active Smox associates with the Co-SMAD Medea (Med, yellow) at regulatory sites of target sites to alter transcription.
(B, C) A control tj-GAL4 (labeled tj > LacZ, LacZ) adult testis has a normal number of hub cells (B), while a tj > Fs-RNAi, LacZ (C) adult testis has only 2 hub cells. Both testes were isolated after 14 days at 29°C to induce maximal GAL4 activity. Fas3 (green) labels hub cells; Vasa (red) marks germ cells; DNA marked by DAPI is white.
(D) Graph showing the average number of hub cells at 0 and 14 days at 29°C in tj > LacZ, LacZ (gray bars, n=49 and n=22, respectively), tj > Fs-RNAi, LacZ (purple bars, n=32 and n=28, respectively), tj > Fs-RNAi, UAS-Fs (pink bars, n=31 and n=34, respectively), or tj > LacZ, UAS-Fs (blue bars, n=37 and n=31, respectively). See Table S1 for n values.
(E) Graph showing the average number of hub cells at 0 days and 4 weeks in Fsnull mutant (n=18 and n=31, respectively).
(F) Graph showing the fertility (brood size) at 4 weeks in a control and Fsnull mutant (n=50 in both cases).
(G, H) Expression at 0 days of adulthood of Fs-GAL4 in a control testis (Fs > GFP, LacZ) or a testis in which Dp was depleted throughout development (Fs > GFP, Dp-RNAi). Fs-GAL4 is expressed strongly in CySCs and early cyst cells and weakly in hub cells (G) but its expression is substantially reduced when Dp is depleted (H). GFP (green) labels Fs-GAL4 expressing cells; Zfh1 (red) marks CySCs; Fas3 (blue) marks hub cells; DNA marked by DAPI is white. Blue line surrounds hub cells and green line marks 25 uM away from the hub.
(I) Graph of relative GFP intensity in Fs > GFP, LacZ testes (labeled LacZ, gray bars, n=13) or Fs > GFP, Dp-RNAi testes (labeled Dp-RNAi, brown bars, n=13) in CySCs and early cyst cells (i.e., area between blue and green lines in (G,H) (labeled “25 μM ring around the hub”) and in hub cells (i.e., the area within the blue line, labeled “Hub”).
(J) Graph of the number of hub cells in testes from control tj-GAL4, hh-GAL80 males (gray bar, labeled “+”, n=27) or these males expressing Dp-RNAi and LacZ (brown bar, n=8) or expressing Dp-RNAi and UAS-Fs (yellow bar, n=17). UAS-Fs significantly rescues hub cell number compared to Dp-RNAi alone.
Error bars represent the data range. **** P < 0.0001; *** P < 0.001. ns = not significant as assessed by Student’s t-test.
See also Tables S1 and S2, Figures S3 and S4.
Scale bar = 10 μM.
To confirm these results, we generated a Fs null allele lacking the sequence spanning coding exons 1 and 4 (Fig. S3D, see Methods). The deletion was confirmed by sequencing and by RT-qPCR (Fig. S3E). Fsnull flies were adult viable and showed progressive hub cell loss with age: from 9.4±0.9 hub cells in 0 day-old adult flies to 2.2±0.6 hub cells in 28 day-old flies (Fig. 2E and Fig. S3G). Hub cell loss in the Fsnull mutant was significantly rescued by overexpressing wild type Fs in either somatic cells (fng-GAL4) or hub cells (updTS-GAL4) (Fig. S3H,I). Furthermore, we observed similar hub cell loss in testes from trans-heterozygous combinations of other Fs alleles (Fig. S3J). As expected, 28 day-old Fsnull males were significantly less fertile than age-matched control flies (Fig. 2F). Using a Fs-GAL4 transcriptional reporter, we found that Fs was expressed in hub cells, CySCs and early somatic cells (Fig. 2G).
Given the remarkable similarity in the non-autonomous hub cell phenotypes observed upon Dp/E2f1 or Fs loss in CySCs, we asked whether Fs functioned downstream of the Dp/E2f1 complex in CySCs. To test this, we examined expression of the Fs-GAL4 transcriptional reporter in control testes or in testes somatically depleted for Dp. Fs transcription in the somatic lineage, as assessed by UAS-GFP expression, was significantly reduced when Dp was depleted compared to controls (Fig. 2G-I). We observed a similar reduction in the levels of a Fs-GFP protein trap upon E2f1 knockdown (Fig. S4A-C). We reasoned that if Dp/E2f1 activity in CySCs non-autonomously maintained the hub by inducing Fs expression, then hub cell loss caused by Dp depletion should be prevented when Fs was concomitantly supplied. Indeed, exogenous Fs completely rescued hub cell loss compared to Dp-RNAi alone (Fig. 2J and Table S1). Thus, Fs mediates the non-autonomous effects of Dp/E2f1 on hub cell maintenance.
Activin signaling triggers hub-to-CySC transdifferentiation
Since Fs is an extracellular antagonist of Activin ligands, we hypothesized that sustained autonomous activation of the Activin pathway within hub cells would have a similar effect on hub cell maintenance. In Drosophila, three Activin ligands (Dawdle (Daw), Activinβ (Actβ), and Myoglianin (Myo)) stimulate the type I Activin receptor Baboon (Babo) (Upadhyay et al., 2017). This results in the activation of transcription factor Smox, the SMAD3 homolog, which then alters target gene transcription. Fs binds to Activin ligands and prevents them from binding to and activating Babo (Fig. 2A).
First, we determined that hub cells can indeed respond to Activin ligands, by examining the distribution of the receptor. A protein trap for Babo was present at the surface of many cells in the testis, and importantly, was found decorating the membrane of Fas3-positive hub cells (Fig. 3A). Next, we tested what effect Activin pathway signaling had within hub cells. We used upd-GAL4 and GAL80TS (termed updTS) to overexpress a constitutively active form of Babo (BaboQD) (Brummel et al., 1999) in hub cells and assessed hub cell number at 0, 7, 14, 21 and 28 days of adulthood. Freshly eclosed flies that were raised at the permissive temperature for GAL80TS had no significant differences in hub cell numbers, whether or not they carried the UAS-baboQD transgene (Fig. 3B). After shifting to the restrictive temperature, autonomous activation of the Activin pathway (updTS>baboQD) in hub cells induced their progressive and complete loss by 28 days, while controls showed a modest decrease in hub cell number over this period (Fig. 3B,C). We then asked what the fate of lost hub cells with ectopic Activin signaling could be and hypothesized that, since hub cells have the potential, under certain experimental conditions, to transdifferentiate into CySCs (Hetie et al., 2014, Voog et al., 2014, Greenspan and Matunis, 2018), Activin signaling may induce this identity switch. To test this possibility, we permanently labelled the hub cell lineage with GFP (see Methods, Fig. 3D-F). In testes where baboQD was overexpressed using updTS, 67% of CySCs and early differentiating cyst cells (labelled by Zfh1) were GFP-positive after 14 days, indicating that they originated from hub cell transdifferentiation (Fig. 3E,F). In contrast, in control testes, only 9% of Zfh1-positive CySCs and their immediate daughter cells were GFP-positive (P < 0.0001) (Fig. 3D,F).
Figure 3: Autonomous Activin signaling disrupts hub cell quiescence.
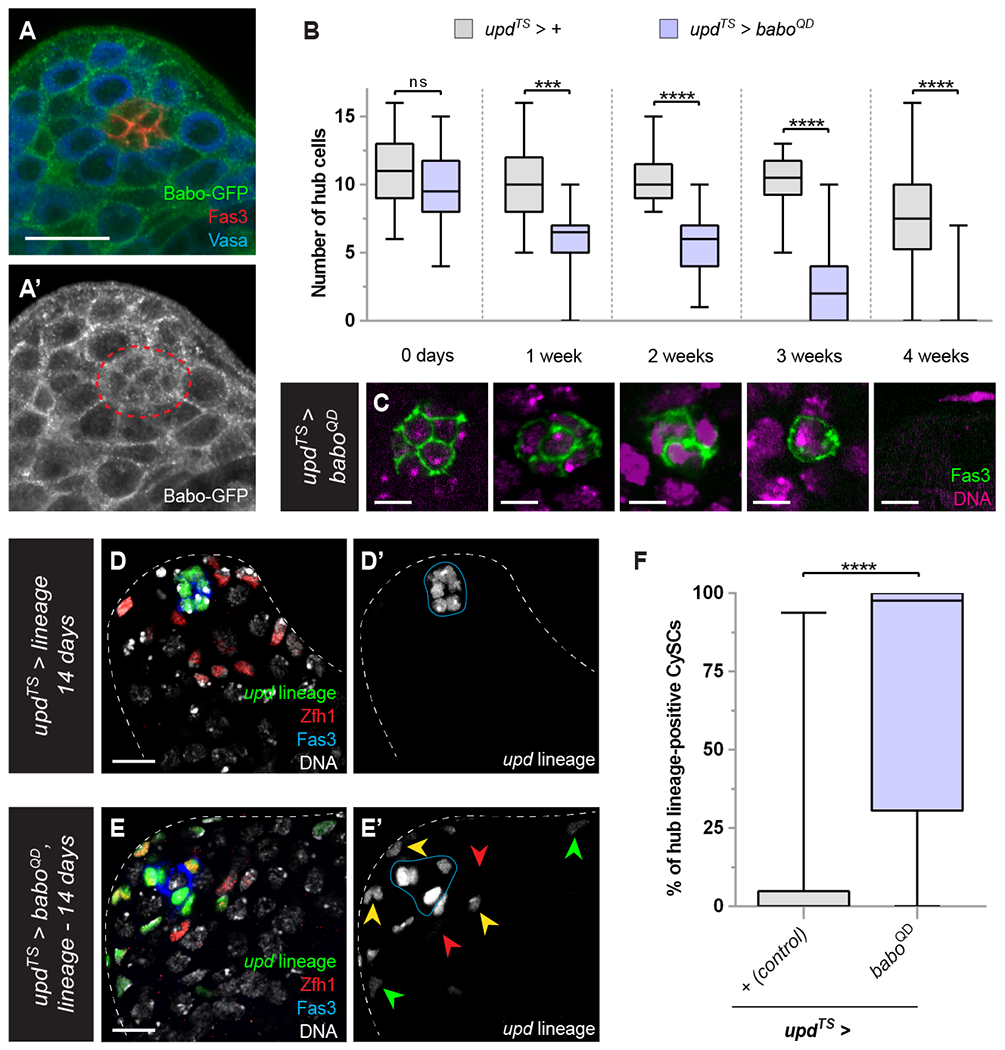
(A) Expression of Babo-GFP fusion protein (green, single channel in A’) is detected in the testis apex. In particular, Babo-GFP expression is visible in hub cells (labeled with Fas3, red), outlined with a red dashed line in A’. Vasa (blue) labels the germline.
(B) Graph showing the number of hub cells in control testes (updTS > +, gray bars, n=23 for 0 days, n=15 for 1 week, n=13 for 2 weeks, n=8 for 3 weeks, n=84 for 4 weeks) or those with sustained Activin signaling in hub cells (updTS > baboQD, purple bars, n=16 for 0 days, n=16 for 1 week, n=28 for 2 weeks, n=35 for 3 weeks, n=30 for 4 weeks) at the indicated time points. Note the progressive loss of hub cells in updTS > baboQD.
(C) Confocal sections of updTS > baboQD testes at the indicated time points. Fas3 (green) marks hub cells and TO-PRO (magenta) marks DNA.
(D, E) Lineage tracing hub cells in control updTS > + (D) and updTS > baboQD testes (E). Note in (E) the presence of hub lineage-positive (green) cells outside of the niche that express the CySC marker Zfh1 (red). Fas3 (blue) marks hub cells and TO-PRO (DNA) is white. In (E’), yellow and red arrowheads indicate hub lineage-positive CySCs and wild type CySCs, respectively, and the green arrowheads indicate differentiating cyst cell descended from a hub-lineage CySC. Blue lines in C,D indicate Fas3-positive hub cells.
(F) Graph indicating the percentage of CySCs positive for hub lineage in updTS > + (gray bar, n=34) and updTS > baboQD (purple bar, n=26) testes.
Error bars represent the data range. **** P < 0.0001; *** P < 0.001. ns = not significant as assessed by Student’s t-test.
Scale bar = 5 μM in C and 10 μM in all other panels.
Since autonomous activation of Activin signaling resulted in hub-to-CySC transdifferentiation, we asked whether knocking down Dp in CySCs also resulted in ectopic transdifferentiation of hub cells, leading to their eventual complete loss. We designed a strategy to trace the hub cell lineage while simultaneously knocking down Dp in CySCs by employing orthogonal binary expression strategies (Fig. 4A and Fig. S5A, see Methods). In control genotypes, where Dp was not depleted in CySCs, hub lineage-derived cells were detected infrequently outside the hub, in 6–14% of testes (Fig. 4B,D and Fig. S5B,C,E) consistent with prior results (Voog et al., 2014). By contrast, hub lineage-positive CySCs were detected in 87–89% of testes when Dp was depleted from all CySCs, a significant increase compared to controls (Fig. 4C,D and Fig. S5B,F). These cells lacked the hub cell marker Fas3 but expressed high levels of the CySC marker Zfh1, suggesting that they had adopted CySC identity (Leatherman and Dinardo, 2008) (Fig. 4C and Fig. S5D,F). Importantly, hub lineage-expressing cells could incorporate the S-phase marker EdU (Fig. S5E,F), a marker of CySC identity. Additionally, lineage-positive cells many cell diameters away from the niche presented long membrane extensions and expressed the differentiation marker Eya (Fig. 4C and Fig. S5G), suggesting that transdifferentiated hub cells are functional CySCs, capable of proliferating and differentiating. Taken together, our data demonstrate that autonomously increasing Activin signaling in hub cells or non-autonomously depleting Dp from CySCs lead to transdifferentiation of hub cells into CySCs, and that these new hub-derived CySCs display all the hallmarks of stem cell behavior, namely self-renewal and differentiation.
Figure 4: CySC depletion of Dp results in non-autonomous hub-to-CySC trandifferentiation.
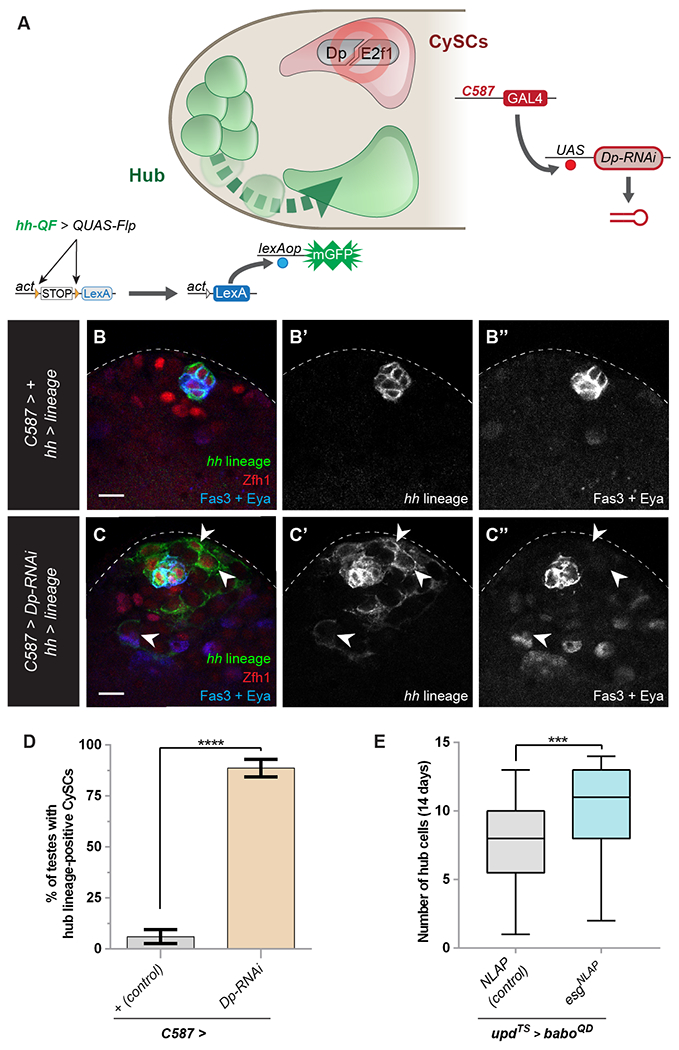
(A) Model indicating experimental design to deplete Dp from CySCs while concomitantly tracing hub cells using independent binary expression systems. To trace the lineage of hub cells, we used hh-QF, which is expressed in only hub cells, to induce QUAS-FLP. In turn, FLP recombines FRT sites in the act>STOP>LexA transgene. This leads to the production of LexA. Then LexA induces expression of lexAop-GFP, hereby exclusively labeling hub cells and their lineage with membrane GFP. In the same animal, C587-GAL4 drives expression of a Dp-RNAi transgene, which depletes Dp from CySCs but not from hub cells.
(B, C) There are GFP-positive cells expressing Zfh1 (C, arrowheads) outside the cluster of hub cells in a C587 > Dp-RNAi; hh > lineage testis but not in a control C587 > +; hh > lineage testis (B). Hub lineage is in green, Zfh1 (red) labels CySCs, Fas3 (blue) labels hub cell membranes, Eya (blue) labels the nuclei of differentiating cyst cells.
(D) Graph showing the percentage of testes in which hub lineage-positive CySCs were present in C587 > + (control) (gray bar, n=53) or C587 > Dp-RNAi (brown bar, n=49).
(E) Graph of the number of hub cells in testes in which baboQD was mis-expressed in hub cells (updTS >) with either escargot (esg-NLAP, gray bar, n=41) or the control transgene (NLAP, blue bar, n=38).
Error bars represent the data range. **** P < 0.0001; *** P < 0.001 as assessed by Fisher’s exact test (D) or Student’s t-test (E).
See also Tables S1 and S2, Figure S5.
Scale bar = 10 μM.
We sought to define the autonomous relationship between Activin signaling and Esg, a Snail family transcriptional repressor required in hub cells to prevent their transdifferentiation into CySCs (Voog et al., 2014). We reasoned that excess Esg might be able counteract the hub cell loss caused by BaboQD. To test this, we misexpressed Esg while simultaneously inducing Activin signaling and found that sustained Esg expression suppressed hub cell loss induced by BaboQD (Fig. 4E). This result indicates that Esg functions downstream of or in parallel to Activin signaling in hub cells to maintain hub cell identity.
The physiological age-dependent loss of hub cells depends on Activin signaling.
Our experiments show that loss of Dp/E2f1 or Fs from all CySCs or increased Activin signaling in all hub cells results in loss of hub cells, indicating that CySCs signal to maintain quiescence of hub cells. Since lineage-wide insults are unlikely in normal physiological conditions, we asked what the functional significance of this CySC-to-hub cell signaling could be. We hypothesized that increased Activin signaling may be responsible for the decrease in the number of hub cells shown to occur during normal aging (Wallenfang et al., 2006, Boyle et al., 2007). Consistent with this, expression of the Fs>GFP transcriptional reporter significantly declined in CySCs in testes from 4 week-old flies compared to those from 0 days-old flies (Fig. 5A-C). Importantly, overexpressing Fs in CySCs blocked the loss of hub cells in 4-week-old males (Fig. 5D). While age had a significant effect on hub cell number (P = 0.0049), it displayed a strong interaction with genotype (P = 0.0067), indicating that the number of hub cells in Fs-overexpressing testes declined significantly less with age. These data implied that in older animals, reduced Fs levels could result in increased availability of Activin ligands to induce pathway signaling in hub cells. To test this model, we knocked down the Activin receptor babo or the Activin-dependent transcription factor Smox in hub cells. In both conditions, the decline in hub cell number was significantly lower after 4 weeks than in controls (Fig. 5E). Finally, we asked which of the Activin ligands was responsible for the age-dependent loss of hub cells. After surveying transcriptional reporters and endogenously tagged Activin ligand lines (Fig. S6), we found that only daw was expressed in the testis stem cell niche, as both a Daw protein trap (Fig. 5F) and two daw-GAL4 transcriptional reporters (Fig. 5G and Fig. S6A) were expressed in hub cells and in CySCs. The transcription of daw increased with age, suggesting it could be partly responsible for the age-related decline of hub cell numbers (Fig. 5H). Indeed, knockdown of daw in hub cells significantly suppressed their loss in aged testes (Fig. 5E). In sum, our data demonstrate that the natural decline in hub cell number during aging is caused, at least in part, by increased Activin signaling in the hub leading to transdifferentiation of hub cells into CySCs.
Figure 5: Increased Activin signaling is responsible for the decline of hub cell numbers during normal aging.
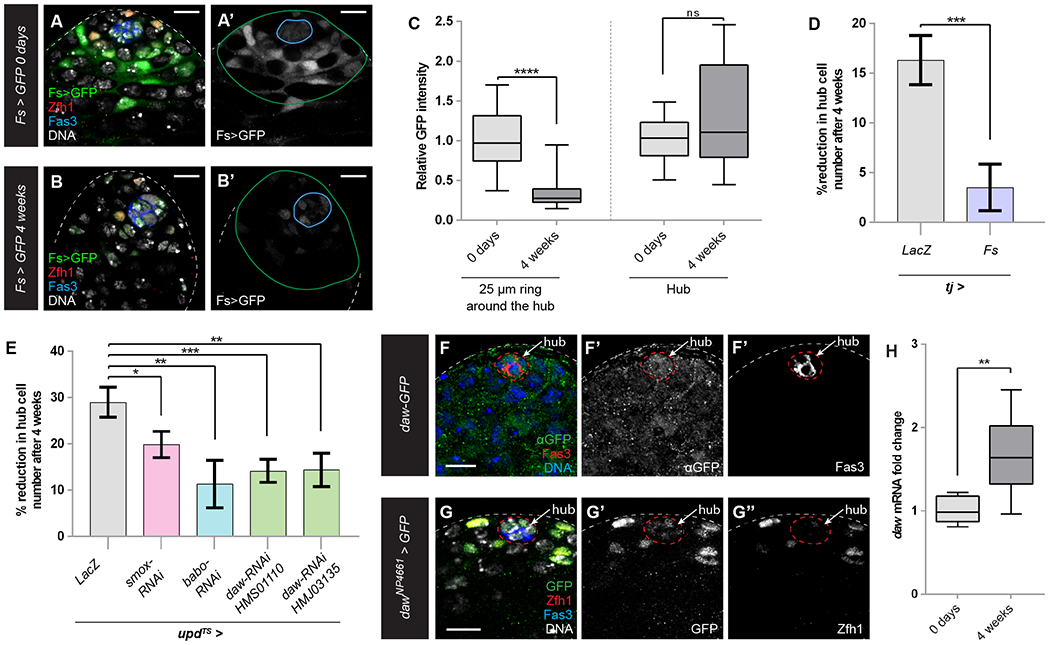
(A-C) Expression at 0 days (A) and 4 weeks (B) of adulthood of Fs-GAL4, UAS-GFP in a control testis (Fs > GFP). Fs-GAL4 is expressed strongly in CySCs and early cyst cells and weakly in hub cells (A), and its expression is substantially reduced at 4 weeks of age (B). GFP (green) labels Fs-GAL4 expressing cells; Zfh1 (red) marks CySCs; Fas3 (blue) marks hub cells; DNA labeled with DAPI is white. Blue line surrounds hub cells and green line marks a 25 μM ring from the hub. (C) Graph of relative GFP intensity in Fs > GFP testes at 0 days (light gray bars, n=15) or 4 weeks (dark gray bars, n=17) in CySCs and early cyst cells (i.e., area between blue and green lines in A,B (labeled “25 μM ring around the hub”) and in hub cells (i.e., the area within the blue line, labeled “Hub”).
(D) Graph showing the reduction of hub cells after 4 weeks relative to newly-eclosed flies in control tj > LacZ (gray bar, n=49 at 0 days and n=66 at 4 weeks; tj > Fs (purple bar, n=37 at 0 days and n=42 at 4 weeks)
(E) Graph showing the reduction of hub cells after 4 weeks relative to newly-eclosed flies in control upd > lacZ (gray bar, n=45 at 0 days and n=84 at 4 weeks); upd > smox-RNAi (pink bar, n=19 at 0 days and n=47 at 4 weeks); upd > babo-RNAi (blue bar, n=38 at 0 days and n=26 at 4 weeks); upd > daw RNAi HMS01110 (first green bar, n=33 at 0 days and n=45 at 4 weeks); upd > daw RNAi HMJ03135 (second green bar, n=29 at 0 days and n=46 at 4 weeks).
(F, G) Expression of daw in the testis tip, as detected with a protein trap (F) or enhancer trap (G). Both reporters show weak expression in the hub (marked by Fas3, red in F, blue in G, and indicated with a dotted line). Daw-GFP protein is also detected outside the hub, and the enhancer trap reveals expression in Zfh1-positive CySCs (red, G).
(H) Expression of daw mRNA increases with age in wild type testes, as measured by qPCR (n=8).
Error bars represent the data range. **** P < 0.0001; *** P < 0.001; ** P < 0.01; * P < 0.05; ns = not significant, as assessed by Student’s t-test.
See also Tables S1 and S2, Figure S6.
Scale bar = 10 μM.
DISCUSSION
We uncover a previously unknown aspect of the relationship between stem cells and their niches. While traditionally thought of as unidirectional and top-down, with niche cells secreting self-renewal factors essential for stem cells, prior work has shown that stem cells can induce their niche, both in normal development, and in malignant situations (Song et al., 2007, Ward et al., 2006, Patel et al., 2015). Here we show that signals from the stem cells are required continually throughout the lifetime of the animal to maintain their niche. We find that the hub surveils signals from resident stem cells and acts as a reserve pool of stem cells if Activin increases (Fig. 6A,B). Linking Fs production to Dp/E2f1 activity in CySCs ensures that the hub cells maintain their quiescence when surrounded with proliferating stem cells.
Figure 6: Model of the communication between CySCs and hub cells that underlies hub cell maintenance.
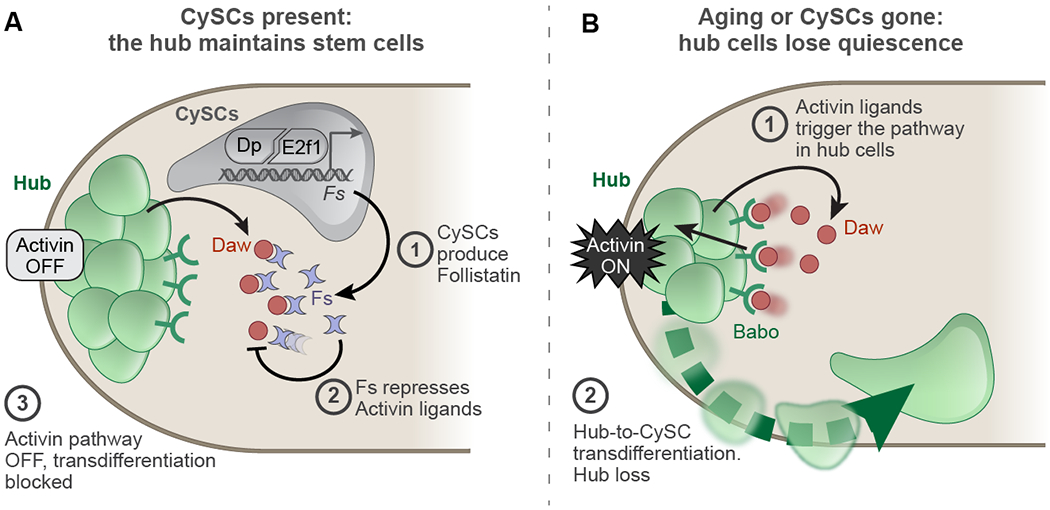
(A) In a young wild type testis, hub cells are quiescent. (1) Fs is produced by CySCs downstream of Dp/E2f1 either directly or indirectly. (2) Daw is produced by hub cells and CySCs. Daw is inactivated by Fs and does not activate its receptor Babo on hub cells. (3) In quiescent hub cells, Activin signaling is not activated.
(B) In an old testis or a testis lacking CySCs, (1) Fs expression declines while increased Daw can now activate Babo on hub cells. (2) Activin signaling disrupts hub cell quiescence, leading to hub-to-CySC transdifferentiation.
We show that expression of both Fs and daw in CySCs change with age and demonstrate that reducing Activin signaling in hub cells significantly ameliorates age-dependent hub cell loss. However, it is interesting that Fs expression in hub cells does not decrease with age. Since hub-derived Fs can rescue the Fsnull phenotype when over-expressed, suggesting it is functional, it is likely that hub-derived Fs alone is not sufficient to fully inhibit Activin signaling. Similarly, both hub cells and CySCs express daw. We show that Daw produced by the hub is relevant to physiological aging and that its knockdown produces a similar rescue to inhibiting Activin signalling in hub cells through babo or smox knockdown. Thus, it is likely that hub-produced Daw is responsible for all the effects on hub cell numbers with age; however, it would be intriguing to test what the role of Daw produced by CySCs is, both in normal homeostasis and in age-dependent dysfunction.
In mammals, the Fs homolog FST is also involved in maintaining fertility via roles in somatic cells (Fullerton et al., 2017), although it has not yet been implicated in regulating the germline stem cell niche. Additionally, similar to our result that Activin signaling can force hub cells to exit quiescence, mammalian TGFβ/Activin signaling can reverse dormancy in disseminated tumor cells in the bone marrow, leading to tumor growth and metastases (Bragado et al., 2013, Ghajar et al., 2013).
Intriguingly, loss of Retinoblastoma, the negative regulator of E2f/Dp transcription, resulted in perturbed interactions between hematopoietic cells and their niche (Walkley et al., 2007). Thus, cells may utilize the E2f/Dp transcription factor in many different contexts to transmit information about their cell cycle state and allow effective monitoring of stem cells by the niche. Gaining a better understanding of the mechanisms that maintain niches in other stem cell models will enable targeting of the interactions between cancer stem cells and their supportive niches, and provide new avenues for therapy (Plaks et al., 2015).
LIMITATIONS OF STUDY
Although our study showed no effect of Dp loss on CySC function, we cannot rule out that there are defects we failed to detect. Clones mutant for Dp persist and proliferate for up to 2 weeks after induction, but they may display defects when aged for longer periods. We showed decreased Fs expression upon knockdown of E2f1 or Dp. This could be due to direct or indirect regulation of the Fs locus by the E2f1/Dp transcription factor, which would need to be assessed by testing for occupancy of E2f1/Dp at the Fs locus. Finally, we show that ectopic Activin signaling promotes hub-to-CySC transdifferentiation and that changes in signaling with age are relevant to physiological hub cell loss. Both Fs and daw transcript levels change with age, but the mechanisms controlling these changes are unknown. It will be important in future studies to establish what directly regulates expression of these factors, and how these regulators change during aging.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erika Bach (erika.bach@nyu.edu).
Materials availability
All Drosophila stocks generated in this study are available from the Lead Contact without restriction.
Data and code availability
All data reported in this paper will be shared by the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila stocks and Maintenance
Drosophila melanogaster strains used in this study are listed in the Key Resources Tab. Drosophila were reared on food made with these ingredients: 1800mL molasses (LabScientific, Catalog no. FLY-8008-16), 266 g agar (Mooragar, Catalog no. 41004), 1800 g cornmeal (LabScientific, Catalog no. FLY-8010-20), 744g Yeast (LabScientific, Catalog no. FLY-8040-20F), 47 L water, 56 g Tegosept (Sigma no. H3647-1KG), 560mL reagent alcohol (Fisher no. A962P4), and 190mL propionic acid (Fisher no. A258500).
Key Resource Table
| Reagent or Resource | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Rabbit polyclonal anti-GFP (1:500) | Invitrogen | Cat# A-6455, RRID: AB_221570 | |
| Goat polyclonal anti-Vasa (1:200) | Santa Cruz | Cat# sc26877; RRID: AB_793877 | |
| Chicken polyclonal anti-GFP (1:500) | Abcam | Cat# ab13970;RRID: AB_300798 | |
| Rat monoclonal anti-N Cadherin (1:20) | DSHB | Cat# DN-ex #8, RRID: AB_528121 | |
| Rat monoclonal anti DE-Cadherin (1:20) | DSHB | Cat# DCad2, RRID: AB_528120 | |
| Mouse monoclonal anti-Eyes absent (Eya) (1:20) | DSHB | Cat# eya10H6; RRID: AB_528232 | |
| Rat monoclonal anti-Vasa | DSHB | Cat# anti-vasa, RRID: AB_760351 | |
| Mouse monoclonal anti- β-galactosidase (1;500) | Promega | Cat# Z3781, RRID: AB_430877 | |
| Mouse monoclonal anti-Dp (1:5) | N. Dyson (MGH Charlestown, USA | ||
| Rabbit polyclonal anti-Zfh1 (1:1000) | K. White (University of Chicago, USA) | ||
| Guinea pig polyclonal anti-Traffic jam (1:3000) | D. Godt (University of Toronto, Canada) | ||
| Cy3-AffiniPure Donkey Anti-Mouse IgG (1:400) | Jackson ImmunoResearch Labs | Cat# 715-165-150 RRID: AB_2340813 | |
| Alexa Fluor 488-AffiniPure Donkey Anti-Rabbit IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 711-545-152 RRID: AB_2313584 | |
| Cy3-AffiniPure Donkey Anti-Rabbit IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 711-165-152 RRID: AB_2307443 | |
| Cy5-AffiniPure Donkey Anti-Rabbit IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 711-175-152 RRID: AB_2340607 | |
| Alexa Fluor 488-AffiniPure Donkey Anti-Rat IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 712-545-150 RRID: AB_2340683 | |
| Cy3-AffiniPure Donkey Anti-Rat IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 712-165-150 RRID: AB_2340666 | |
| Cy5-AffiniPure Donkey Anti-Rat IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 712-175-150 RRID: AB_2340671 | |
| Alexa Fluor 488 AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 703-545-155 RRID: AB_2340375 | |
| Cy3-AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 703-165-155 RRID: AB_2340363 | |
| Cy5-AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 703-175-155 RRID: AB_2340365 | |
| Cy3-AffiniPure Donkey Anti-Guinea Pig IgG (1:400) | Jackson ImmunoResearch Labs | Cat# 706-165-148 RRID: AB_2340460 | |
| Cy5-AffiniPure Donkey Anti-Guinea Pig IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 706-175-148 RRID: AB_2340462 | |
| Cy3-AffiniPure Donkey Anti-Goat IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 705-165-003 RRID: AB_2340411 | |
| Alexa Fluor 647 AffiniPure Donkey Anti-Goat IgG (H+L) (1:400) | Jackson ImmunoResearch Labs | Cat# 705-605-003 RRID: AB_2340436 | |
| Bacterial and virus strains | N/A | ||
| Biological Samples | N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | |||
| VECTASHIELD Mounting Medium | Vector Laboratories | Cat# H-1000 RRID: AB_2336789 | |
| VECTASHIELD Mounting Medium with DAPI | Vector Laboratories | Cat# H-1200 RRID: AB_2336790 | |
| Paraformaldehyde, 16% w/v aq. soln., methanol free | Thermo Fisher Scientific | Cat# 43368-9L | |
| TO-PRO-3 iodide (1 μM) | Invitrogen | Cat# T3605 | |
| 5-ethynyl-2’-deoxyuridine (EdU) | Invitrogen | Cat# A10044 | |
| AF405 picolyl azide | Click chemistry tools | Cat# 1308-5 | |
| Tris(3-Hydroxyproyltriazolylmethyl)Amine | Sigma Aldrich | Cat# 762342 | |
| Sodium Ascorbate | Sigma Aldrich | Cat# PHR1279 | |
| Copper(II) sulfate | Fisher Scientific | Cat# 15617730 | |
| Molasses | Labscientific | Cat# FLY-8008-16 | |
| Agar | Mooragar | Cat# 41004 | |
| Cornmeal | LabScientific | Cat# FLY-8010-20 | |
| Yeast | LabScientific | Cat# FLY-8040-20F | |
| Tegosept | Sigma | Cat# H3647-1KG | |
| Reagent alcohol | Fisher | Cat# A962P4 | |
| Propionic acid | Fisher | Cat# A258500 | |
| Critical Commercial Assays | N/A | ||
| Deposited data | N/A | ||
| Experimental models: Cell lines | N/A | ||
| Experimental Models: Organisms/Strains | |||
| D. melanogaster; OregonR | Bach lab | ||
| D. melanogaster; P[GawB]NP1624/CyO (tj-GAL4) | Bach lab | Kyoto Stock Center: 104055 Flybase: FBst0302922 | |
| D. melanogaster; nos-GAL4-VP16 | Ruth Lehmann (Whitehead Institute, USA) | ||
| D. melanogaster; C587-GAL4 | Ruth Lehmann (Whitehead Institute, USA) | ||
| D. melanogaster; fng-GAL4 | Steven Dinardo (Perelman School of Medicine, University of Pennsylvania, USA) | ||
| D. melanogaster; eyaA3-GAL4 (denoted eya-GAL4) | Steven Dinardo (Perelman School of Medicine, University of Pennsylvania, USA) | ||
| D. melanogaster; upd-GAL4 | Bach lab | ||
| D. melanogaster; tub-GAL80TS | Bloomington Drosophila stock Center (BDSC) | BDSC_7017 FlyBase: FBst0007017 | |
| D. melanogaster; hh-LacZ | Bach lab | BDSC_5530 FlyBase: FBst0005530 | |
| D. melanogaster; upd-LacZ | Bach lab | ||
| D. melanogaster; hh-GAL80 | This manuscript | ||
| D. melanogaster; hh-QF | This manuscript | ||
| D. melanogaster; w1118; P[w[+mC]=UAS-lacZ.NZ]J312 Insertion on III | BDSC | BDSC_3956 FlyBase: FBst003956 | |
| D. melanogaster; w1118; P[w[+mC]=UAS-lacZ.NZ]20b Insertion on II | BDSC | BDSC_3955 FlyBase: FBst003955 | |
| D. melanogaster; UAS-GFP; Ubi-p63E(FRT.STOP)Stinger | BDSC | BDSC_32251 | |
| D. melanogaster; w1118; P[GD4444]v12722(Dp-RNAi) | Vienna Drosophila Resource Center (VDRC) | VDRC_v12722 FlyBase: FBst0450633 | |
| D. melanogaster; FRT42D, Dpa3/CyO | M. Frolov, University of Illinois at Chicago, USA | Flybase: FBgn0011763 | |
| D. melanogaster; FRT42D, Dpa4/CyO | M. Frolov, University of Illinois at Chicago, USA | Flybase: FBgn0011763 | |
| D. melanogaster; UAS-Dp | M. Frolov, University of Illinois at Chicago, USA | Flybase: FBgn0011763 | |
| D. melanogaster; FRT82B E2f1729 | M. Frolov, University of Illinois at Chicago, USA | FlyBase: FBgn0011766 | |
| D. melanogaster; y1, v1; P[TRiP.JF02519]attP2/TM3, Sb1 (Dp-RNAi) | BDSC | BDSC_30515 FlyBase: FBst0030515 | |
| D. melanogaster, w1118; P[GD4448]v15886 (E2f1-RNAi) | VDRC | VDRC: v15886 FlyBase: FBst0452055 | |
| D. melanogaster; UAS-Rbf280 Insertion on III | W. Deng (Tulane University, USA) | BDSC_50748 Flybase: FBst0050748 | |
| D. melanogaster; act>y[+]>LHV2-86Fb,13XlexAop2-myr::GFP(FLEX-AMP) | C. Desplan (New York University, USA) | ||
| D. melanogaster; UAS-FLP; QUAS-FLP | BDSC | BDSC_30126 | |
| D. melanogaster; w1118; P[GD15843]v46260(Fs-RNAi) | VDRC | VDRC v46260 FlyBase: FBst0466595 | |
| D. melanogaster; y1 v1; P[TRiP.HMJ03135]attP40(daw-RNAi) | BDSC | BDSC_50911 Flybase: FBst0050911 | |
| D. melanogaster; P[KK110248]VIE-260B(daw-RNAi) | VDRC | VDRC #v105309 Flybase: FBst0477137 | |
| D. melanogaster; y1 sc* v1 sev21; P[TRiP.HMS01110]attP2 (daw-RNAi) | BDSC | BDSC_34974 Flybase: FBst0034974 | |
| D. melanogaster; y1 v1; P[TRiP.GL01476]attP2(smox-RNAi) | BDSC | BDSC_43138 Flybase: FBst0043138 | |
| D. melanogaster; y1 w*; Mi[MIC]FsMI01433(FsMI01433) | BDSC | BDSC_33121 Flybase: FBst0033121 | |
| D. melanogaster; y1 w*; Mi[MIC]Fs MI11350 (FsMI11350) | BDSC | BDSC_56310 Flybase: FBst0056310 | |
| D. melanogaster; y1 w*;FsMI04308-TG4.1 CG8079MI04308-TG4.1-X (FsTJ4.1) | BDSC | BDSC_66838 (lost during the pandemic) | |
| D. melanogaster;y1 w*; Mi[PT-GFSTF.1]FsMI04308-GFSTF.1 CG8079MI04308-GFSTF.1-X/CyO(FsGFSTF.1 or Fs-GFP) | BDSC | BDSC_65327 Flybase: FBst0065327 | |
| D. melanogaster; Fsnull | This manuscript | ||
| D. melanogaster; UAS-Fs | O’Connor lab | ||
| D. melanogaster; Fs-GAL4 (FsMI-GAL4) | This manuscript | ||
| D. melanogaster; UAS-esgNLAP | L. Jones (UCSF, USA) | ||
| D. melanogaster; UAS-NLAP | L. Jones (UCSF, USA) | ||
| D. melanogaster; Actβ-GAL4 | O’Connor lab | ||
| D. melanogaster; myo-GAL4 | O’Connor lab | ||
| D. melanogaster; y1 w*; Mi[MIC]dawMI05383 (daw-GFP) | BDSC | BDSC_43001 Flybase: FBst0043001 | |
| D. melanogaster; UAS-babo-RNAi. | O’Connor lab | ||
| D. melanogaster; UAS-baboQD | O’Connor lab | ||
| D. melanogaster; PBac[fTRG00506.sfGFP-TVPTBF]VK00033(ActβfTRG00506.sfGFP-TVPTBF) | VDRC | VDRC #v318136 Flybase: FBst0491562 | |
| D. melanogaster;PBac[fTRG00161.sfGFP-TVPTBF]VK00033(myofTRG00161.sfGFP-TVPTBF) | VDRC | VDRC #v318065 Flybase: FBst0491390 | |
| D. melanogaster;PBac[fTRG00444.sfGFP-TVPTBF]VK00033(babofTRG00444.sfGFP-TVPTBF) | VDRC | VDRC #v318433 Flybase: FBst0491516 | |
| D. melanogaster;w*; P[GawB]dawNP4661 / CyO (dawNP4661-GAL4) | Kyoto Stock Center | Kyoto #113490 Flybase: FBst0316217 | |
| D. melanogaster;y* w*; P[GawB]dawNP6274 / CyO (dawNP6274-GAL4) | Kyoto Stock Center | Kyoto #105179 Flybase: FBst0304038 | |
| Oligonucleotides | |||
| gRNA Fs 1 | CCGGTTGCATCATGTATCTTGGC | IDTDNA | |
| gRNA Fs 2 | CCAACTGGAGGTCGCCTATCGGG | IDTDNA | |
| qPCR primer Fs-fwd: | 5’-AGTGTCATATATACTCTCCGCATGT-3’ | IDTDNA | |
| qPCR primer Fs-rev: | 5’-ACAGCAACTGCTTTTTAACTATGCC-3’ | IDTDNA | |
| qPCR primer α-tub84B-fwd: | 5’-TCGTTTTACGTTTGTCAAGCCTC-3’ | IDTDNA | |
| qPCR primer α-tub84B-rev: | 5’-GAGATACATTCACGCATATTGAGTT-3’ | IDTDNA | |
| qPCR primer daw-fwd | 5′-CCCATCTTCGACGGGATGAC-3′ | IDTDNA | |
| qPCR primer daw-rev | 5′-TTGCACTCGACCTCCTCTCT-3′ | IDTDNA | |
| Recombinant DNA | N/A | ||
| Software and Algorithms | |||
| ImageJ/Fiji | Fiji | http://fiji.sc/ | |
| Photoshop/Illustrator | Adobe | https://www.adobe.com/products/ | |
| Prism | GraphPad | https://www.graphpad.com | |
| ZEN | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html | |
| Excel | Microsoft | https://products.office.com/en-us/excel | |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/ | |
| Other | N/A | ||
Flies were kept at 25°C, except crosses with GAL80TS, which were maintained at 18°C until eclosion, and the adult flies were transferred to 29°C. Clonal experiments were analyzed at 2 dpci to assess clone induction and 7 and 14 dpci for maintenance as is standard in the field (Amoyel et al., 2014). For transgene expression in CySCs, we examined hub cell number at 0, 3, 7, 10 and 20 days of adulthood. Our reason for selecting these time points is that Dp knockdown by tj-GAL4 led to an almost complete loss of hub cells by 10 days, but since other genotypes exhibited a slower loss of hub cells, we also examined hub cells at 20 days of adulthood. We looked at earlier time points (3 and 7 days) to examine hubs before their complete loss. For the aging experiments, we examined hub cell number at 1, 2, 3, or 4 weeks (or 7, 14, 21, 28 days) of adulthood.
Males were aged separately from females and provided with fresh food every other day. For adult-onset overexpression and RNAi-depletion, we used the appropriate driver combined with the temperature-sensitive repressor tub-GAL80TS. Flies were reared at the permissive temperature (18°C) and adult males were collected and shifted to the restrictive temperature of 29°C to allow GAL4 activity. Experiments without GAL80 were raised at room temperature until eclosion and shifted to 29°C for the specified period to ensure maximum GAL4 activity.
We used the following fly stocks: Oregon-R; tj-GAL4 (Kyoto Stock Center #NP1624); nos-GAL4-VP16 (gift of Ruth Lehmann, Whitehead Institute, USA; upd-LacZ (Tsai and Sun, 2004); hh-LacZ; tub-GAL80TS (McGuire et al., 2004); hh-GAL80 (this study); hh-QF (this study); UAS-LacZ (Bloomington Drosophila Stock center (BDSC) #3955 and 3956); UAS-GFP; Ubi-p63E(FRT.STOP)Stinger (also known as GTRACE, BDSC # 32251); UAS-Dp RNAi (Vienna Drosophila Resource Center (VDRC) #v12722); FRT42D Dpa3 and FRT42D Dpa4 (both gifts of M. Frolov, University of Illinois at Chicago, USA); FRT82B E2f1729 (gift of M. Frolov); UAS-Dp RNAi (BDSC #30515); UAS-Dp (Zappia and Frolov, 2016) (gift of M. Frolov); UAS-E2f1 RNAi (VDRC #v15886); UAS-Rbf280 (BSDC #50748); act>y[+]>LHV2–86Fb,13XlexAop2-myr::GFP (also known as FLEXAMP (Bertet et al., 2014)) where LHV2–86Fb encodes an optimized version of LEXA-VP16 with reduced toxicity; UAS-FLP; QUAS-FLP (BDSC #30126); UAS-Fs-RNAi (GD15843, VDRC #v46260); UAS-daw-RNAi (HMJ03135 BDSC #50911; HMS01110 BDSC #34974; and KK110248 VDRC #v105309); UAS-smox-RNAi (GL01476 BDSC #43138); FsMI01433 (BDSC #33121); FsMI11350 (BDSC #56310); FsMI04308-TG4.1 CG8079MI04308-TG4.1-X (referred to as FsTJ4.1, BDSC #66838); FsMI04308-GFSTF.1 CG8079MI04308-GFSTF.1-X (referred to as FsGFSTF.1 or Fs-GFP) BDSC #65327); Fsnull (this study); UAS-Fs (Pentek et al., 2009); Fs-GAL4 (referred to also as FsMI-GAL4) (this study); UAS-esgNLAP and UAS-NLAP (both gifts from Leanne Jones, UCLA, USA) (Voog et al., 2014); Actβ-GAL4 (Song et al., 2017); myo-GAL4 (Awasaki et al., 2011); dawMI05383 (referred to as daw-GFP, BDSC #43001); UAS-babo-RNAi (Peterson and O’Connor, 2013); UAS-baboQD; ActβfTRG00506.sfGFP-TVPTBF (Sarov et al., 2016), VDRC #v318136); myofTRG00161.sfGFP-TVPTBF (Sarov et al., 2016), VDRC #v318065); babofTRG00444.sfGFP-TVPTBF (Sarov et al., 2016), VDRC #v318433); punt (put)fTRG00910.sfGFP-TVPTBF (Sarov et al., 2016), VDRC #v318264); dawNP4661-GAL4 and dawNP6274-GAL4,UAS-LacZ (Kyoto Stock Center #113490 and #105179, respectively).
METHOD DETAILS
Generation of transgenic Drosophila lines
The Fs-GAL4 line used to monitor Fs transcriptional activity was created by recombination mediated cassette exchange (RMCE) to insert GAL4 in the MiMIC line FsMI01433 (Venken et al., 2011). The Fsnull mutant was created by GenetiVision using CRISPR/Cas9 to delete 3 kb of the Fs locus, corresponding to the first 4 coding exons of both predicted Fs isoforms (Fig. S3D). This genomic region was replaced by a splicing acceptor site and stop codons in all the 3 frames, followed by a 3xP3-GFP cassette. We used CCGGTTGCATCATGTATCTTGGC and CCAACTGGAGGTCGCCTATCGGG as gRNAs. We validated the mutant by sequencing the PCR product using the genomic DNA from the Fsnull mutant as a template and the following primers: fwd 5’ CGGTGCATAATGCGCCAAACC 3’ rev 5’ CTTGCAGTGCACTGGATATGG 3’
hh-GAL80 and hh-QF were generated by RMCE by injection into line MI10526 (BDSC #53865), carrying a MiMIC transposon in the hh locus, using plasmids from the Drosophila Genomics Resource Center (DGRC) #1390 (Diao et al., 2015) and #1296 (Venken et al., 2011), respectively.
Drosophila genetics
To label the lineage of hub cells, we used upd-GAL4 and tub-GAL80TS to drive expression of the UAS-FLP recombinase, which excises a stop from the ubiP63E(FRT.STOP)Stinger G-TRACE cassette (Evans et al., 2009). After the excision of FRT sites following recombination, the resulting GFP serves an indelible and persistent marker to identify CySCs derived from the hub lineage. For tracing the hub lineage while manipulating expression in the cyst lineage, we used C587-GAL4 to knock down Dp in CySCs, and an orthogonal expression system, hh-QF, to drive QUAS-FLP expression in hub cells. The lineage was permanently marked upon FLP expression using either the GTRACE cassette, as above, or the FLEXAMP cassette, in which a yellow+ transgene is excised by FLP, leading to permanent expression of LexA under the actin promoter. LexA labels cells through expression of myr-GFP, driven by multimerized LexAop binding sites.
For mutant clones, crosses were raised at 25°C until eclosion. Males were collected for up to 2 days, then heat-shocked at 37°C for one hour and returned to 25°C until dissection, 2, 7 or 14 days later.
Antibodies and immunofluorescence
The following primary antibodies were used: goat anti-Vasa (1:200; Santa Cruz), mouse anti-Fasciclin3 (Fas3) (1:50; Developmental Studies Hybridoma Bank (DHSB), mouse anti-Eya (1:20, DSHB), rat anti-Vasa (1:20, DSHB), rat anti-N-Cadherin (1:20, DSHB) rabbit anti-GFP (1:500; Invitrogen), chicken anti-GFP (1:500, Aves Labs), mouse anti-β-Galactosidase (1:500, Promega), guinea pig anti-Tj (1:3000, a gift of Dorothea Godt, University of Toronto, Canada), rabbit anti-Zfh1 (1:1000), mouse anti-Dp (1:5, a gift of Nick Dyson, MGH Charlestown, USA), TO-PRO-3 iodide (1 μM; Molecular Probes); 4′,6-diamidino-2-phenylindole (DAPI) (1:500, Invitrogen). Donkey secondary antisera were used 1:400 (Jackson ImmunoResearch). Dissections and staining were carried out as previously described (Flaherty et al., 2010). Briefly, testes were dissected in 1x phosphate buffered saline (PBS), fixed for 15 minutes in 4% formaldehyde in 1x PBS, washed for 1 hour at 25°C in 1x PBS with 0.5% Triton X-100, and blocked in PBTB (1x PBS 0.2% Triton X-100 and 1% bovine serum albumin) for 1 hour at 25°C. Primary antibodies were incubated overnight at 4°C. They were washed two times for 30 minutes in PBTB and incubated 2 hours in secondary antibody in PBTB at 25°C and then washed two times for 30 minute in 1x PBS with 0.2% Triton X-100. They were mounted in Vectashield or Vectashield + DAPI (Vector Laboratories). 5-ethynyl-2′-deoxyuridine (EdU) labeling was achieved by incubating dissected samples in 10 μM EdU (Life Technologies) in Schneider’s medium prior to fixation. Following primary and secondary antibody incubations and washes, samples were incubated for 30 minutes in buffer containing 0.1M THPTA, 2mM sodium ascorbate, 1 mM CuSO4 and 2.5 μM picolyl azide conjugated to a fluorophore (Alexa 405, 488, 568 or 643, Click Chemistry Tools). Samples were then washed and mounted for imaging. Confocal images were captured using Zeiss LSM 510, LSM 700, LSM 880 and a Leica Sp8 confocal microscopes with a 63x objective.
Quantitative RT-PCR
Whole testes (n=20–25) were isolated and mRNA was extracted with PicoPure RNA Isolation system (Applied Biosystems) following the manufacturer’s instructions. Reverse transcription was performed using Maxima Reverse Transcriptase (ThermoFisher) as per manufacturer’s instructions and using 0.5 μg of RNA as template. qRT-PCR was performed using SYBR Green PCR Master Mix (ThermoFisher) and a QuantStudio 5 Real-Time PCR System machine (Applied Biosystems). 8 replicates were performed for the experiments in Fig. 5H and 4 replicates for each experiment shown in Fig. S3E. To detect Fs isoform B (FlyBase code FBtr0339996), we used the following primers and normalized expression levels to the control gene α-tub84B: Fs-fwd:
5’-AGTGTCATATATACTCTCCGCATGT-3’
Fs-rev: 5’-ACAGCAACTGCTTTTTAACTATGCC-3’
α-tub84B-fwd: 5’-TCGTTTTACGTTTGTCAAGCCTC-3’
α-tub84B-rev: 5’-GAGATACATTCACGCATATTGAGTT-3’
daw-fwd 5′-CCCATCTTCGACGGGATGAC-3′
daw-rev 5′-TTGCACTCGACCTCCTCTCT-3′
We failed to detect transcripts of Fs isoform A in the testis (FlyBase FBtr0087398), using the following primers and normalized expression levels to the control gene α-tub84B: Fs-A-fwd:
5’-GAACGGACCGCGCTAAAAAT-3’
Fs-A-rev: 5’-GGCAAACGCACTGGTTTCAT-3’
Fertility tests
Mutant males were crossed individually with two OregonR females. After 48 hours of mating and egg laying, all adults were removed from the vials. The size of the brood of each male was scored ten days later.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis and statistics
Hub cells were quantified by counting nuclei (labelled with DAPI, or low levels of Tj or Zfh1) in cells that were also Fas3-positive.
For GFP intensity quantifications, we dissected control flies and experiments on the same day and processed them simultaneously. Images were acquired with the same microscope settings. Plots show mean fluorescence intensity normalized to the average value of the controls.
The percentage of transdifferentiated CySCs was calculated as the ratio of Zfh1-positive cells simultaneously positive for the hub cell-lineage labeling.
Image processing, quantifications and figure preparation were performed with Fiji-ImageJ (Schindelin et al., 2012), Adobe Photoshop and Adobe Illustrator software. Statistical tests were performed using GraphPad Prism8 or JMP software. To test for an interaction between genotype and aging in Fig. 5D,E, we used a linear model featuring full factorial design and tested for the effect of both age and genotype on hub cell number, and their interaction. Categorical data analysis in Fig. 4D and Figs. S1B,G, S5B was performed as described in (Xu et al., 2010) using Fisher’s Exact tests. Other data were analyzed with Student’s t-tests because they were pairwise comparisons between genotypes. Data were analyzed and plotted with GraphPad Prism8. In all graphs, whiskers indicate the entire range of data, the boxes show the second and third quartiles and the line shows the median.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank members of the Bach lab, Jessica Treisman, Don Ryoo, Vilaiwan Fernandes, Will Wood, Arantza Barrios and Richard Poole for discussions, Nazif Alic for help with statistics, and Aryeh Korman for help with imaging Babo-GFP.
The authors acknowledge for antibodies the DHSB, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242, and for plasmids the DGRC at Indiana University, which is supported by a grant from the National Institutes of Health (ORIP and NIGMS). Work is the Bach lab is supported by grants from the NIH and New York State Department of Health/NYSTEM (C32584GG). Work in the Amoyel lab is supported by an Elizabeth Blackwell Institute Early Career Fellowship and an MRC Career Development Award MR/P009646/2.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- AMOYEL M, SANNY J, BUREL M & BACH EA 2013. Hedgehog is required for CySC self-renewal but does not contribute to the GSC niche in the Drosophila testis. Development, 140, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMOYEL M, SIMONS BD & BACH EA 2014. Neutral competition of stem cells is skewed by proliferative changes downstream of Hh and Hpo. EMBO J, 33, 2295–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANLLO L, PLASSCHAERT LW, SUI J & DINARDO S 2019. Live imaging reveals hub cell assembly and compaction dynamics during morphogenesis of the Drosophila testis niche. Dev Biol, 446, 102–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AWASAKI T, HUANG Y, O’CONNOR MB & LEE T 2011. Glia instruct developmental neuronal remodeling through TGF-beta signaling. Nat Neurosci, 14, 821–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERTET C, LI X, ERCLIK T, CAVEY M, WELLS B & DESPLAN C 2014. Temporal patterning of neuroblasts controls Notch-mediated cell survival through regulation of Hid or Reaper. Cell, 158, 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOYLE M, WONG C, ROCHA M & JONES DL 2007. Decline in self-renewal factors contributes to aging of the stem cell niche in the Drosophila testis. Cell Stem Cell, 1, 470–8. [DOI] [PubMed] [Google Scholar]
- BRAGADO P, ESTRADA Y, PARIKH F, KRAUSE S, CAPOBIANCO C, FARINA HG, SCHEWE DM & AGUIRRE-GHISO JA 2013. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol, 15, 1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUMMEL T, ABDOLLAH S, HAERRY TE, SHIMELL MJ, MERRIAM J, RAFTERY L, WRANA JL & O’CONNOR MB 1999. The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev, 13, 98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN D, PACAL M, WENZEL P, KNOEPFLER PS, LEONE G & BREMNER R 2009. Division and apoptosis of E2f-deficient retinal progenitors. Nature, 462, 925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIAO F, IRONFIELD H, LUAN H, DIAO F, SHROPSHIRE WC, EWER J, MARR E, POTTER CJ, LANDGRAF M & WHITE BH 2015. Plug-and-play genetic access to drosophila cell types using exchangeable exon cassettes. Cell Rep, 10, 1410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINARDO S, OKEGBE T, WINGERT L, FREILICH S & TERRY N 2011. lines and bowl affect the specification of cyst stem cells and niche cells in the Drosophila testis. Development, 138, 1687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS CJ, OLSON JM, NGO KT, KIM E, LEE NE, KUOY E, PATANANAN AN, SITZ D, TRAN P, DO MT, YACKLE K, CESPEDES A, HARTENSTEIN V, CALL GB & BANERJEE U 2009. G-TRACE: rapid Gal4-based cell lineage analysis in Drosophila. Nat Methods, 6, 603–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FABRIZIO JJ, BOYLE M & DINARDO S 2003. A somatic role for eyes absent (eya) and sine oculis (so) in Drosophila spermatocyte development. Dev Biol, 258, 117–28. [DOI] [PubMed] [Google Scholar]
- FAIRCHILD MJ, YANG L, GOODWIN K & TANENTZAPF G 2016. Occluding Junctions Maintain Stem Cell Niche Homeostasis in the Fly Testes. Curr Biol, 26, 2492–2499. [DOI] [PubMed] [Google Scholar]
- FLAHERTY MS, SALIS P, EVANS CJ, EKAS LA, MAROUF A, ZAVADIL J, BANERJEE U & BACH EA 2010. chinmo is a functional effector of the JAK/STAT pathway that regulates eye development, tumor formation, and stem cell self-renewal in Drosophila. Dev Cell, 18, 556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORBES AJ, LIN H, INGHAM PW & SPRADLING AC 1996. hedgehog is required for the proliferation and specification of ovarian somatic cells prior to egg chamber formation in Drosophila. Development, 122, 1125–35. [DOI] [PubMed] [Google Scholar]
- FULLERTON PT JR., MONSIVAIS D, KOMMAGANI R & MATZUK MM 2017. Follistatin is critical for mouse uterine receptivity and decidualization. Proc Natl Acad Sci U S A, 114, E4772–E4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHAJAR CM, PEINADO H, MORI H, MATEI IR, EVASON KJ, BRAZIER H, ALMEIDA D, KOLLER A, HAJJAR KA, STAINIER DY, CHEN EI, LYDEN D & BISSELL MJ 2013. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol, 15, 807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREENSPAN LJ & MATUNIS EL 2018. Retinoblastoma Intrinsically Regulates Niche Cell Quiescence, Identity, and Niche Number in the Adult Drosophila Testis. Cell Rep, 24, 3466–3476 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HETIE P, DE CUEVAS M & MATUNIS E 2014. Conversion of quiescent niche cells to somatic stem cells causes ectopic niche formation in the Drosophila testis. Cell Rep, 7, 715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITADATE Y & KOBAYASHI S 2010. Notch and Egfr signaling act antagonistically to regulate germ-line stem cell niche formation in Drosophila male embryonic gonads. Proc Natl Acad Sci U S A, 107, 14241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURUSU M, CORDING A, TANIGUCHI M, MENON K, SUZUKI E & ZINN K 2008. A screen of cell-surface molecules identifies leucine-rich repeat proteins as key mediators of synaptic target selection. Neuron, 59, 972–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE BRAS S & VAN DOREN M 2006. Development of the male germline stem cell niche in Drosophila. Dev Biol, 294, 92–103. [DOI] [PubMed] [Google Scholar]
- LEATHERMAN JL & DINARDO S 2008. Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell, 3, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE JY, CHEN JY, SHAW JL & CHANG KT 2016. Maintenance of Stem Cell Niche Integrity by a Novel Activator of Integrin Signaling. PLoS Genet, 12, e1006043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGUIRE SE, MAO Z & DAVIS RL 2004. Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci STKE, 2004, pl6. [DOI] [PubMed] [Google Scholar]
- MICHEL M, KUPINSKI AP, RAABE I & BOKEL C 2012. Hh signalling is essential for somatic stem cell maintenance in the Drosophila testis niche. Development, 139, 2663–9. [DOI] [PubMed] [Google Scholar]
- MORRISON SJ & SPRADLING AC 2008. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell, 132, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OH J, LEE YD & WAGERS AJ 2014. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med, 20, 870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKEGBE TC & DINARDO S 2011. The endoderm specifies the mesodermal niche for the germline in Drosophila via Delta-Notch signaling. Development, 138, 1259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATEL PH, DUTTA D & EDGAR BA 2015. Niche appropriation by Drosophila intestinal stem cell tumours. Nat Cell Biol, 17, 1182–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENTEK J, PARKER L, WU A & ARORA K 2009. Follistatin preferentially antagonizes activin rather than BMP signaling in Drosophila. Genesis, 47, 261–73. [DOI] [PubMed] [Google Scholar]
- PETERSON AJ & O’CONNOR MB 2013. Activin receptor inhibition by Smad2 regulates Drosophila wing disc patterning through BMP-response elements. Development, 140, 649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLAKS V, KONG N & WERB Z 2015. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell, 16, 225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RESENDE LP, BOYLE M, TRAN D, FELLNER T & JONES DL 2013. Headcase promotes cell survival and niche maintenance in the Drosophila testis. PLoS One, 8, e68026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAROV M, BARZ C, JAMBOR H, HEIN MY, SCHMIED C, SUCHOLD D, STENDER B, JANOSCH S, K JV, KRISHNAN RT, KRISHNAMOORTHY A, FERREIRA IR, EJSMONT RK, FINKL K, HASSE S, KAMPFER P, PLEWKA N, VINIS E, SCHLOISSNIG S, KNUST E, HARTENSTEIN V, MANN M, RAMASWAMI M, VIJAYRAGHAVAN K, TOMANCAK P & SCHNORRER F 2016. A genome-wide resource for the analysis of protein localisation in Drosophila. Elife, 5, e12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHINDELIN J, ARGANDA-CARRERAS I, FRISE E, KAYNIG V, LONGAIR M, PIETZSCH T, PREIBISCH S, RUEDEN C, SAALFELD S, SCHMID B, TINEVEZ JY, WHITE DJ, HARTENSTEIN V, ELICEIRI K, TOMANCAK P & CARDONA A 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods, 9, 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONG W, CHENG D, HONG S, SAPPE B, HU Y, WEI N, ZHU C, O’CONNOR MB, PISSIOS P & PERRIMON N 2017. Midgut-Derived Activin Regulates Glucagon-like Action in the Fat Body and Glycemic Control. Cell Metab, 25, 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONG X, CALL GB, KIRILLY D & XIE T 2007. Notch signaling controls germline stem cell niche formation in the Drosophila ovary. Development, 134, 1071–80. [DOI] [PubMed] [Google Scholar]
- SREEJITH P, JANG W, TO V, HUN JO Y, BITEAU B & KIM C 2019. Lin28 is a critical factor in the function and aging of Drosophila testis stem cell niche. Aging (Albany NY), 11, 855–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERRY NA, TULINA N, MATUNIS E & DINARDO S 2006. Novel regulators revealed by profiling Drosophila testis stem cells within their niche. Dev Biol, 294, 246–57. [DOI] [PubMed] [Google Scholar]
- TOLEDANO H, D’ALTERIO C, CZECH B, LEVINE E & JONES DL 2012. The let-7-Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature, 485, 605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSAI YC & SUN YH 2004. Long-range effect of upd, a ligand for Jak/STAT pathway, on cell cycle in Drosophila eye development. Genesis, 39, 141–53. [DOI] [PubMed] [Google Scholar]
- TULINA N & MATUNIS E 2001. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science, 294, 2546–9. [DOI] [PubMed] [Google Scholar]
- UPADHYAY A, MOSS-TAYLOR L, KIM MJ, GHOSH AC & O’CONNOR MB 2017. TGF-beta Family Signaling in Drosophila. Cold Spring Harb Perspect Biol, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENKEN KJ, SCHULZE KL, HAELTERMAN NA, PAN H, HE Y, EVANS-HOLM M, CARLSON JW, LEVIS RW, SPRADLING AC, HOSKINS RA & BELLEN HJ 2011. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods, 8, 737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOOG J, SANDALL SL, HIME GR, RESENDE LP, LOZA-COLL M, ASLANIAN A, YATES JR 3RD, HUNTER T, FULLER MT & JONES DL 2014. Escargot restricts niche cell to stem cell conversion in the Drosophila testis. Cell Rep, 7, 722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALKLEY CR, SHEA JM, SIMS NA, PURTON LE & ORKIN SH 2007. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell, 129, 1081–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLENFANG MR, NAYAK R & DINARDO S 2006. Dynamics of the male germline stem cell population during aging of Drosophila melanogaster. Aging Cell, 5, 297–304. [DOI] [PubMed] [Google Scholar]
- WARD EJ, SHCHERBATA HR, REYNOLDS SH, FISCHER KA, HATFIELD SD & RUOHOLA-BAKER H 2006. Stem cells signal to the niche through the Notch pathway in the Drosophila ovary. Curr Biol, 16, 2352–8. [DOI] [PubMed] [Google Scholar]
- XU B, FENG X & BURDINE RD 2010. Categorical data analysis in experimental biology. Dev Biol, 348, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAPPIA MP & FROLOV MV 2016. E2F function in muscle growth is necessary and sufficient for viability in Drosophila. Nat Commun, 7, 10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the Lead Contact upon request.