

Abstract
Purpose.
In metastatic castration resistant prostate cancer (mCRPC) low serum androgens prior to starting Abiraterone Acetate (AA) is associated with more rapid progression. We evaluated the effect of AA on androgens in CRPC metastases and associations of intratumoral androgens with response.
Experimental Design.
We performed a phase II study of AA plus prednisone in mCRPC. The primary outcome was tissue testosterone at 4 weeks. Exploratory outcomes were association of steroid levels and genomic alterations with response, and escalating AA to 2000mg at progression.
Results.
29 of 30 men were evaluable. Testosterone in metastatic biopsies became undetectable at 4 weeks (p<0.001). Serum and tissue DHEAS remained detectable in many patients and was not increased at progression. Serum and tissue DHEAS in the lowest quartile (pre-treatment), serum DHEAS in the lowest quartile (4 weeks), and undetectable tissue DHEAS (on therapy) associated with rapid progression (20 vs 48 weeks p=0.0018; 20 vs 52 weeks p=0.0003; 14 vs 40 weeks p=0.0001; 20 vs 56 weeks p=0.02, respectively). One of 16 men escalating to 2000mg had a 30% PSA decline; 13 developed radiographic progression by 12 weeks. Among patients with high serum DHEAS at baseline, wild type PTEN status associated with longer response (61 vs 33 weeks; p=0.02).
Conclusion.
Low circulating adrenal androgens levels are strongly associated with an androgen-poor tumor microenvironment and with poor response to AA. CRPC patients with higher serum DHEAS levels may benefit from dual-AR pathway inhibition, while those in the lowest quartile may require combinations with non-AR directed therapy.
Trial registration
INTRODUCTION
While initially effective, treatment of prostate cancer with androgen deprivation therapy (ADT) is uniformly characterized by progression to castration resistant prostate cancer (CRPC). Despite castrate serum testosterone (T) levels, levels of T and dihydrotestosterone (DHT) in CRPC metastases and patient derived xenografts (PDX) are sufficient for AR activation (1, 2). Residual androgens in CRPC tumors may reflect de novo androgen synthesis or intratumoral uptake and conversion of adrenally-derived steroid precursors to T and DHT (3, 4). In particular, serum levels of dehydroepiandrosterone-sulfate (DHEA-S), the primary circulating form of the adrenal androgen DHEA, are extremely high and are not suppressed by ADT (5, 6).
CYP17A, expressed in the adrenal gland, testes and ovary, catalyzes sequential reactions converting pregnenolone and progesterone to the adrenal androgens DHEA and androstenedione (AED). The role of adrenal androgens in promoting CRPC is supported by the efficacy of the CYP17A inhibitor abiraterone acetate (AA) in decreasing circulating adrenal androgens and improving overall survival (OS) in CRPC (7–10). The proposed mechanism is suppression of androgen levels in tumor tissue as a result of suppressing testicular, adrenal and tumoral CYP17A activity. This regimen markedly decreases prostate androgens below that achieved by standard ADT in the neoadjuvant setting (5) and studies using CRPC xenografts similarly demonstrate suppression of tissue androgens below castration alone (11). However, the efficacy of AA and prednisone in reducing intratumoral androgens in CRPC metastases has not been reported. Moreover, lower serum androgen levels prior to starting this regimen have been associated with worse outcomes in CRPC patients (12, 13). Whether a similar association exists with intratumoral androgens levels prior to starting therapy, or with on-treatment androgen levels in serum or tissue is unknown.
We carried out a phase II study to evaluate the efficacy of AA plus prednisone in reducing androgen levels in CRPC metastases. The primary outcome was tissue T levels at 4 weeks. Exploratory outcomes were the association of steroid levels and genomic alterations with response, and the impact of escalating AA to 2000mg at progression.
PATIENTS AND METHODS
Study Design and Patient Population
This was an investigator-initiated open-label single center three-arm study of AA (1000mg daily) and prednisone (5mg twice daily) followed by escalation of AA to 2000mg daily at disease progression in men with mCRPC. Eligible patients had metastatic prostate cancer resistant to ADT comprised of medical or surgical castration ± standard antiandrogen (bicalutamide, flutamide, or nilutamide) and were not previously treated with AA or enzalutamide. Prior ketoconazole and docetaxel were allowed.
Study Procedures and Treatment
Patients underwent pretreatment biopsy of a bone, node or soft tissue metastasis and were then alternately enrolled in cohorts undergoing repeat biopsy at 4 weeks (cohort 1) or 12 weeks (cohort 2) until 10 patients were enrolled in each cohort (Study Schema provided in Supplementary Figure 1). Patients with radiographic or clinical progression prior to planned biopsy were reassigned to cohort 3 (biopsy at progression) and another patient placed in cohort 1 or 2. After cohorts 1 and 2 were filled, patients were sequentially assigned to cohort 3 with biopsy at progression, until a total of 10 patients were enrolled in cohort 3 (of these, 2 had been reassigned from cohort 1 or 2 for clinical progression at 4 and 8 weeks). Patients remained on ADT and received AA 1000mg orally daily with prednisone 5mg orally twice daily until development of radiographic or clinical progression. Computed tomography of the chest, abdomen and pelvis, and bone scan were obtained before enrollment and every 12 weeks while on study treatment. Radiographic progression was determined based on Prostate Cancer Working Group 2 (PCWG2) criteria. Serum for PSA was drawn monthly. At radiographic progression asymptomatic patients were offered dose escalation of AA to 2000 mg daily. Therapy with standard or dose-escalated AA was continued until radiographic or clinical progression or for a maximum of 2 years (104 weeks).
Steroid and Abiraterone Measurements
Methods for determination of steroids and abiraterone in serum and prostate tissue by mass spectrometry were as previously reported (14). Similar methods were used for detection of the abiraterone metabolites D4-abiraterone, and 3-keto-5α-abiraterone. Additional information on assay methodology and limits of detection and quantitation is provided in Supplementary Figure 2. For purposes of calculation, analyte values below the lower limit of detection were set at the lower limit of quantitation specific for that analyte.
Genomic Analysis
Genomic DNA was prepared from clinical samples (buffy coat, metastatic CRPC tumor tissue) using DNeasy Blood and Tissue Kit (QIAGEN, Germantown, MD). Next-generation sequencing was performed on CRPC tissue biopsies using the clinically validated UW-OncoPlex platform (15). We determined HSD3B1 genotype in DNA extracted from buffy coat using a melting assay with an unlabeled, locked, nucleic acid oligonucleotide probe in an asymmetric PCR as previously described (16).
Statistical Analysis
A sample size of 6 patients per arm provided 94% power to detect the anticipated 0.660 pg/mg difference in tissue T levels relative to baseline, based on a 2-sided paired t-test with alpha 5%. Ten patients per cohort were enrolled to account for potentially unproductive biopsies. Demographic and clinical characteristics were summarized using descriptive statistics. Comparisons of continuous variables among groups were assessed using the non-parametric Wilcoxon rank sum test (Mann Whitney test). Comparison of continuous variables between baseline and subsequent timepoints were assessed using the Wilcoxon matched-pairs test. Progression-free survival was estimated using Kaplan-Meier methods and compared using the Gehan-Wilcoxan test. The statistical significance was set at p < 0.05. Due to the small sample size, all findings were considered hypothesis generating and no multiple testing adjustments were performed. All statistical analyses were done using GraphPad Prism (version 8.3.0).
Study Approval
All procedures were carried out in accordance with the U.S. Common Rule and approved by the institutional review board of the University of Washington. All subjects signed written informed consent. The trial was registered with the clinicaltrials.gov identifier NCT01503229.
RESULTS
Patient Characteristics
Thirty patients enrolled and 29 were evaluable for analysis (Figure 1). Baseline characteristics are shown in Table 1. Median age was 71 years (range 44–83). The median PSA was 78 ng/ml (range 2–908). In the 18 patients with soft tissue +/− bone disease, the biopsy site was equally divided between bone and lymph node based solely on tumor location most accessible to biopsy. Eleven patients had received previous definitive therapy for localized disease while 19 were metastatic at diagnosis. Eleven patients had received one or more treatments beyond bicalutamide for CRPC.
Figure 1. Flow diagram of patient recruitment, enrollment, and participation.
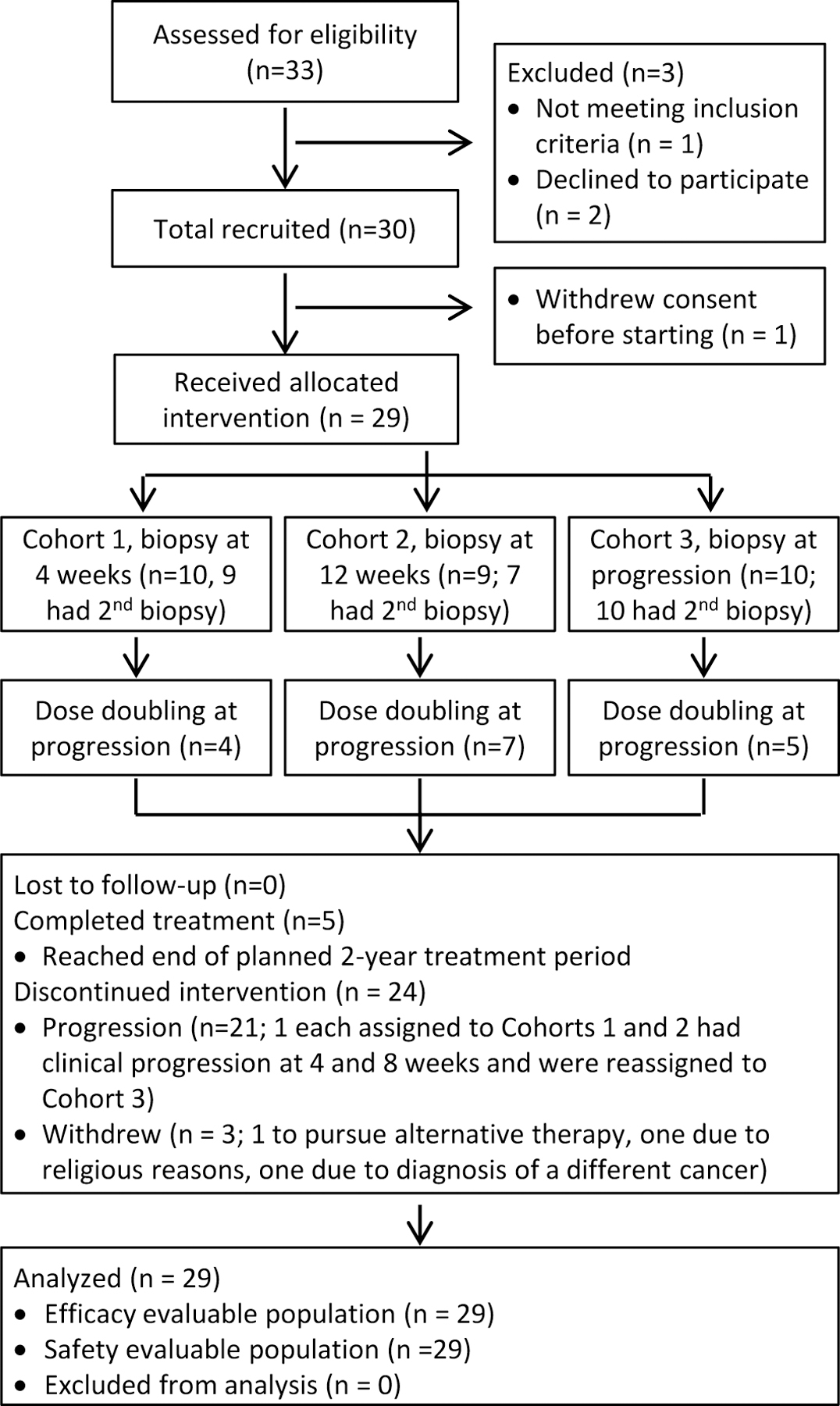
Diagram depicts participant flow through the study process from patient screening to data analysis.
Table 1.
Baseline Clinical Characteristics at Time of Study Enrollment
| Patients, n | 29 |
| Median Age (Range), years | 71 (44–83) |
| Median PSA (Range), ng/ml | 78 (2–908) |
| Extent of disease | |
| Bone only | 10 (33%) |
| Bone and LAD | 15 (50%) |
| Bone and LAD/Visceral disease | 3 (10%) |
| LAD only | 2 (7%) |
| Median tumor fraction in pre-study biopsy | 55% (4–88%) |
| Previous Therapies, n | |
| Radical prostatectomy | 5 |
| Definitive radiation therapy | 6 |
| Neither (metastatic at diagnosis) | 19 |
| Systemic Therapy | |
| Combined androgen blockade | 8 |
| Bicalutamide | 9 |
| Nilutamide/Flutamide | 3 |
| High dose ketoconazole | 5 |
| Diethylstilbestrol | 2 |
| Sipuleucel-T | 4 |
| Docetaxel | 4 (1 CSPC) |
| Lines of therapy for CRPC | |
| 0 | 7 |
| 1 | 14 |
| 2 | 3 |
| 3–4 | 5 |
PSA – prostate specific antigen; CRPC – Castration resistant prostate cancer; CSPC – Castration sensitive prostate cancer; LAD - lymphadenopathy
Clinical Response to Standard and Dose Escalated AA and Prednisone
The median time to radiographic/clinical progression in the entire cohort was 36 weeks (range 4–104; Fig 2A). Twenty of 29 (69%) achieved a PSA decline of ≥30% (PSA30) after 12 weeks of standard-dose AA plus prednisone (Fig 2B), with 22 (75%) eventually doing so. The median time to progression was longer in those achieving PSA30 decline at 12 weeks (40 vs 34 weeks; p=0.05; with a similar but nonsignificant trend in those achieving a PSA50 decline (Supplementary Fig 3A, 3B). (17) Notably, 4 of the 9 patients without PSA30 at 12 weeks had responses lasting 36, 40, 41 and 104 months, underscoring the importance of radiographic progression vs PSA decline as a primary endpoint. Three patients withdrew while responding to therapy (one each to pursue alternative therapy, for religious reasons, and due to diagnosis of an unrelated malignancy) and were censored at the time of study withdrawal (at 28, 24 and 20 months). Three patients did not progress on standard dose therapy by the end of the two-year treatment period and were censored at 104 months.
Figure 2. Clinical Response and Steroid Levels in Serum and Metastatic Tissue on Abiraterone Acetate plus Prednisone.
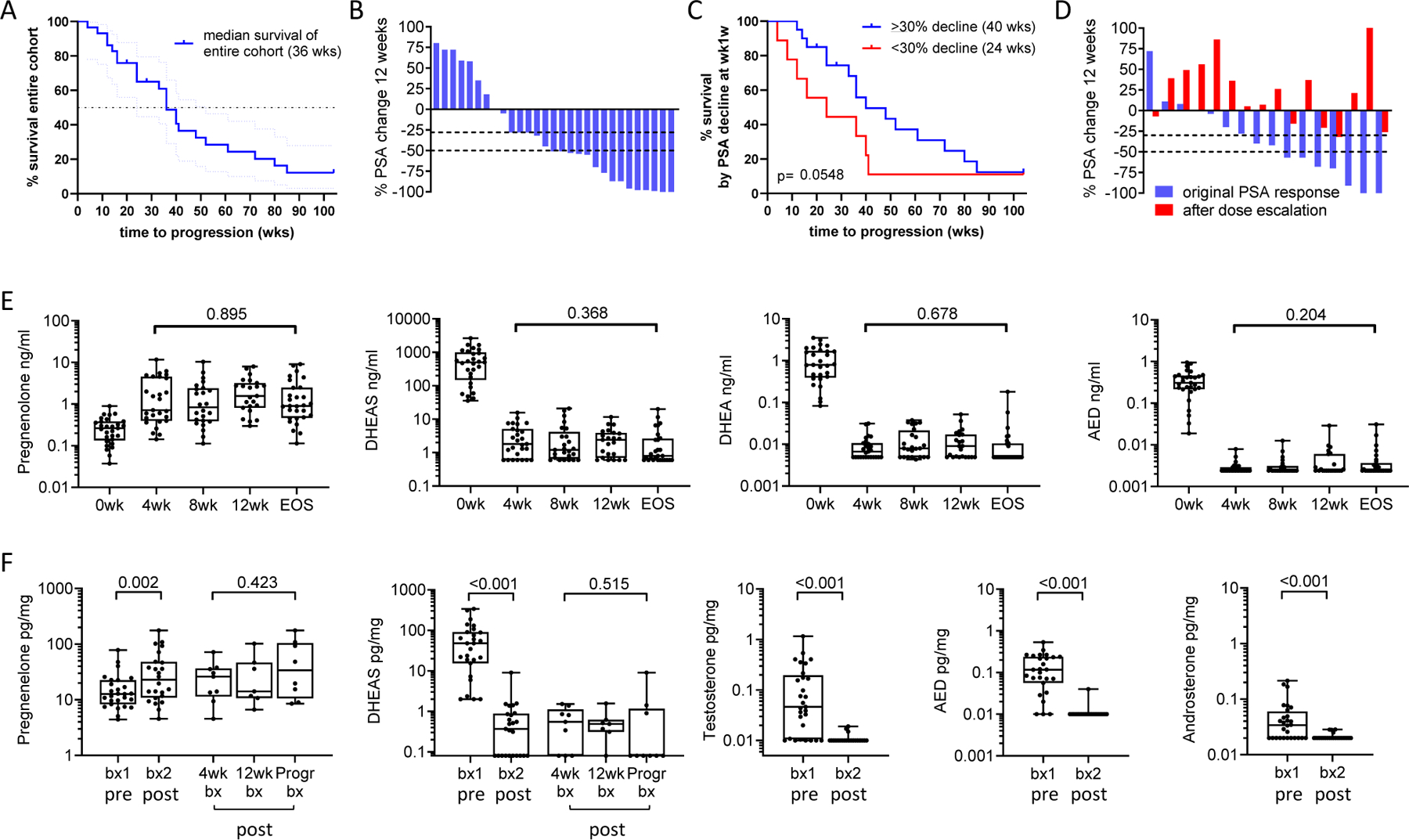
A. Kaplan Meier plot of time to radiographic progression on standard dose abiraterone acetate. B. Waterfall plot showing percent change in PSA at 12-weeks. C. Kaplan Meier plot of time to radiographic progression comparing patients with or without a 30% PSA decline at 12 weeks. D. Waterfall plot comparing the original change in PSA (blue bars) to the percent change in PSA after dose escalation of abiraterone acetate to 2000mg per day (red bars). E. Change in serum steroid levels after standard dose abiraterone acetate plus prednisone at baseline (0 weeks), at 4, 8 and 12 weeks (4wk, 8wk, 12wk) and at end of study (EOS) at the time of radiographic progression. F. Change in steroid levels in metastatic tissue (tx) biopsies prior to therapy (biopsy 1, bx1) and while on therapy (biopsy 2, bx2). Biopsy 2 was taken at either 4 weeks (4wk bx), 12 weeks (12wk bx), or at progression (Progr bx). P values for the indicated comparison calculated via the Wilcoxon matched-pairs test. Data are shown as box and-whisker plots, where horizontal lines indicate median values; white boxes denote the 75th (upper margin) and 25th percentiles (lower margin), and upper and lower bars indicate the minimum and maximum values, respectively. Dehydroepiandrosterone sulfate (DHEAS), dehydroepiandrosterone (DHEA), androstenedione (AED).
At radiographic progression, 16 of 29 patients underwent dose escalation of AA to 2000mg per day (with continuation of prednisone). There was no decrease in the median PSA (Supplementary Fig 3C), with only one patient achieving a PSA30 decline (Fig 2C). Thirteen patients had clinical or radiographic progression and discontinued therapy by week 12 after dose escalation. Two patients remained on dose escalated therapy for 56 weeks and 36 weeks, respectively, prior to second radiographic progression, while one patient remained on dose escalated therapy for 20 weeks at which time he reached the end of the two-year treatment period and was censored at 104 months. No consistent findings with regards to PSA, steroid, or abiraterone levels, or tumor genomic alterations were observed in these 3 patients.
Steroid and Abiraterone Levels During Standard and Dose-Escalated AA Therapy
Steroid levels in serum and tissue prior to and during AA and prednisone therapy are summarized in Supplementary Tables 1 and 2. In serum levels showed a significant increase in steroids upstream of CYP17A (pregnenolone) and decreases in downstream steroids (DHEAS, DHEA, AED, T, and DHT) at all timepoints (Fig 2D; see steroid metabolism schema, Supplementary Fig 4), consistent with prior observations in serum and urine. (7) While median levels of DHEAS and DHEA decreased by over two orders of magnitude in response to therapy in all patients, there appeared to be two modes of response. In one, levels became undetectable by week 4 and remained undetectable, while in the other subset, levels were still detectable at week 4 and remained detectable at all time points of treatment. AED, T and DHT in serum were largely undetectable at all time points after starting therapy. Serum steroids were not increased at progression compared to earlier time points.
In tissue as in serum, steroids upstream of CYP17A (pregnenolone) were increased in the on-treatment tissue biopsies, while downstream steroids (DHEAS, AED, T, androsterone) were significantly decreased (Fig 2E). Levels of AED, T and androsterone in tissue fell below the limit of detection by week 4 and remained essentially undetectable. DHEAS in tissue remained detectable in the majority of biopsies obtained at 4 and 12 weeks (6 of 9 and 6 of 7, respectively), but was undetectable in 6 of 9 samples progression. DHT levels were below the limit of detection in 24 of 29 tissue samples at baseline and undetectable in all tissues after therapy. These observations suggest that an increase in tissue levels of androgens is not responsible for resistance to AA and prednisone and explains the lack of response to AA dose escalation.
AA is a prodrug that rapidly dissociates to abiraterone once ingested, therefore, all serum and tissue measurements are of abiraterone, not AA (summarized in Supplementary Table 3). Abiraterone is converted by 3βHSD1 to D4-abiraterone, and then by SRD5A to 3-keto-5α-abiraterone, metabolites with AR antagonist and AR agonist activity, respectively (18). Levels of abiraterone metabolites in serum and tissue were similar at all time points and were not decreased at progression compared to earlier time points (Supplementary Figure 5A-B). These observations suggest that resistance to AA is not associated with an increase in serum steroid levels or decrease in serum or tissue drug levels at the time of progression.
Dose escalation of AA from 1000mg to 2000mg resulted in the expected pharmacokinetic changes with a nonsignificant increase in the median level of abiraterone (22.4 to 78.2 ng/ml), and a significant increase in D4 abiraterone (1.48 to 4.0; p=0.012) and 3-keto-5α-abiraterone (10.6 to 14.3 ng/ml; p=0.005) at four weeks (Supplementary Figure 5C). Accordingly, serum levels of DHEAS and DHEA decreased in 9 of 16 patients, and AED levels remained or became undetectable in 15 of 16 (Supplementary Figure 5D), although the overall decrease in median levels was nonsignificant for DHEAS, DHEA and of borderline significance for AED (p=0.21, p=0.36, p=0.06, respectively). These observations suggest that the lack of clinical response to AA dose escalation is not accounted for by a failure of pharmacokinetic or pharmacodynamic responses in serum. Moreover, in the 3 patients that experienced prolonged responses, serum abiraterone and androgen levels after dose escalation were variably altered (decreased in 2 and increased in one), although changes in tissue levels before and after dose escalation were not assayed.
Correlation of Steroid Levels in Serum and Metastatic Tissue Before and After Therapy with AA and Prednisone
Serum levels of DHEAS, DHEA, AED, ASD, T and DHT, were highly correlated with each other and with levels of DHEAS, AED and androsterone in pre-treatment biopsies (Fig 3A). Tissue levels of DHEAS and AED, DHEAS and ASD, and AED and ASD correlated with each other (r=0.51, 0.56 and 0.46, respectively; p<0.05 for all), while tissue T levels were not significantly correlated with steroid levels in either serum or tissue. After starting AA plus prednisone, levels of DHEAS, DHEA and AED remained significantly correlated with each other and showed the expected inverse correlations with abiraterone and its metabolites (Fig 3B), while progesterone showed the expected positive correlation with abiraterone and its metabolites. These treatment-related correlations were present by 4 weeks and become notably more pronounced at 8 and 12 weeks. Abiraterone and metabolites levels were strongly correlated with each other in serum and tissue all time points (Supplementary Fig 6A).
Figure 3. Correlation of Steroid Levels in Serum and Metastatic Tissue Before and After Treatment with Abiraterone Acetate plus Prednisone.
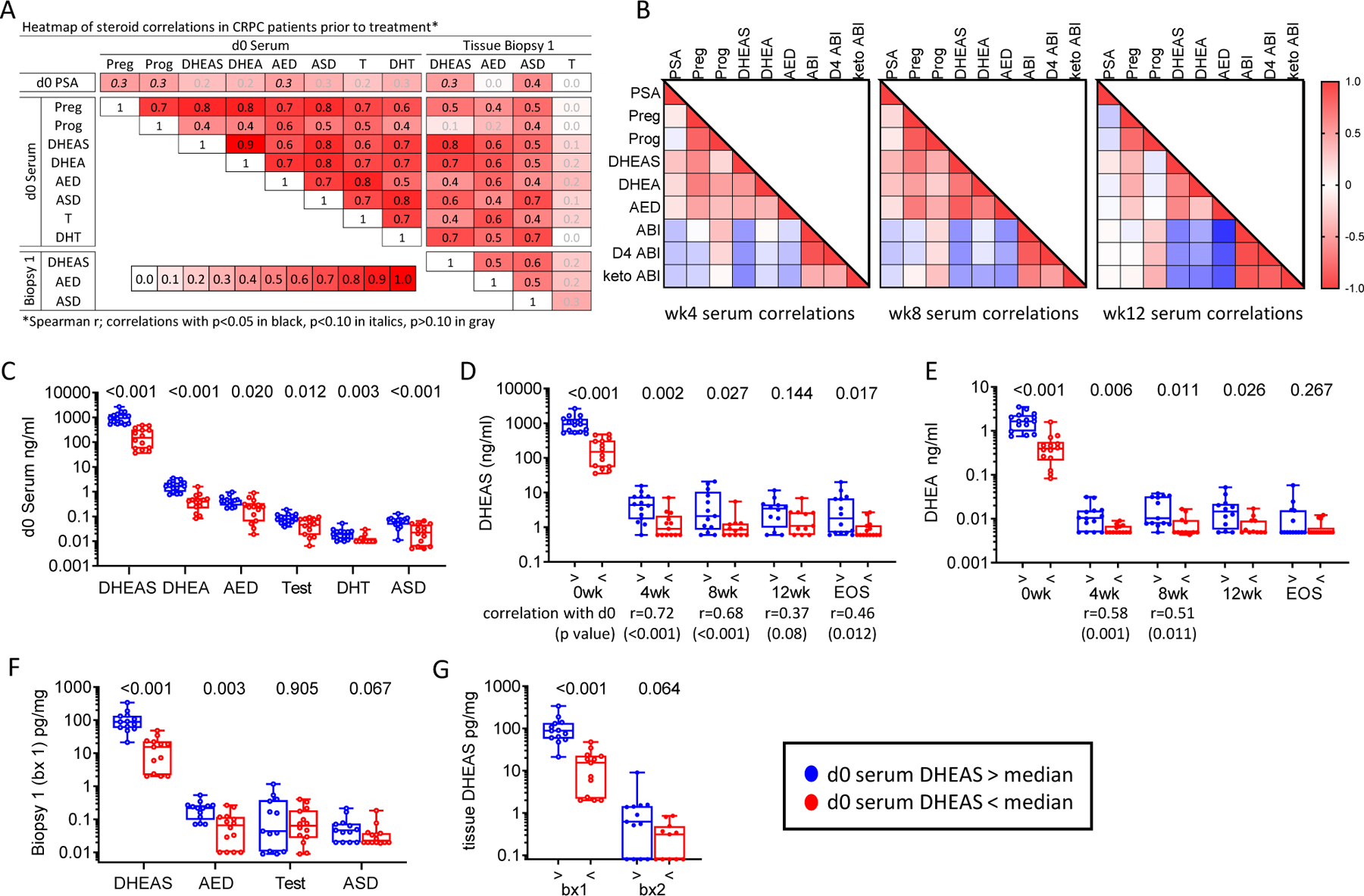
A. Heatmap of steroid correlations in pre-treatment serum (day 0) and metastatic tissue biopsies (Biopsy 1). The Spearman r value for each correlation is shown in the box. Correlations with p<0.05 in black, p<0.10 in italics, p>0.10 in gray. B. Heatmap of Spearman correlations for steroids in serum at week 4 (wk4), week 8 (wk8), and week 12 (wk12). C. Comparison of baseline serum steroid levels stratified by serum DHEAS levels above (blue) vs below (red) the median at baseline (d0). D. Comparison of DHEAS levels and E. DHEA levels in serum stratified by baseline serum DHEAS levels at baseline (0 weeks), at 4, 8 and 12 weeks (4wk, 8wk, 12wk) and at end of study (EOS). Significant Spearmen correlations between on-treatment and baseline values at each time point is indicated below the timepoint. F. Comparison of steroid levels in pre-treatment tissue biopsies (bx 1) stratified by serum DHEAS levels above (blue) vs below (red) the median at baseline. G. Comparison of tissue (tx) DHEAS levels stratified by baseline serum DHEAS levels in pre-treatment and on-treatment tissue biopsies (bx1 and bx2). P values for the indicated comparison calculated via non-parametric Mann Whitney t tests. Pregnenolone (Preg), progesterone (Prog), dehydroepiandrosterone sulfate (DHEAS), dehydroepiandrosterone (DHEA), androstenedione (AED), testosterone (test), dihydrotestosterone (DHT), androsterone (ASD), abiraterone (ABI), D4 abiraterone (D4 Abi), 3-keto-5a-abiraterone (Keto ABI).
These baseline and on-treatment steroid correlations are further illustrated by the observation that subjects with baseline serum DHEAS levels above vs below the median also had higher baseline levels of DHEA, AED, T, DHT and androsterone (Fig 3C) and maintained significantly higher serum levels of DHEAS (Fig 3D) and DHEA (Fig 3E) at all time points (including, for DHEAS, at the end of study (EOS) measurement taken at progression). Moreover, strong correlations between baseline and on-treatment serum levels were observed at all time points for DHEAS (Fig 3D), and at 4 and 8 weeks for DHEA (Fig 3E). Likewise, subjects with baseline serum DHEAS above vs below the median had higher tissue levels of DHEAS, AED and androsterone at baseline (Fig 3F), as well as higher DHEAS in on-treatment tissue biopsies (Fig 3G). These observations demonstrate that serum DHEAS levels prior to AA therapy can identify CRPC patients with concomitantly higher levels of all androgens in serum and tissue, and that this subset of patients with high baseline androgen levels maintains persistently higher serum and tissue DHEAS levels after treatment with AA and prednisone.
Association of Steroid and Abiraterone Levels with PSA Decline and Progression Free Survival
Compared to patients who achieved a PSA30 decline at 12 weeks, those with less than a PSA30 decline had numerically lower pre-treatment levels of serum and tissue androgens (Fig 4A). These differences were strongest for T (p=0.055) and DHT (p=0.069) in serum, and for AED (p=0.063), T (p=0.034) and androsterone (p=0.088) in tissue. Accordingly, the rate of achieving a PSA30 decline at 12 weeks was 37% (3/8) in patients in the lowest quartile of serum DHEAS, but 81% (17/21) in patients in the top 3 quartiles (Fig 4C; PSA levels at baseline were not associated with serum DHEAS levels). These observations suggest that being in the lowest quartile of serum androgens prior to starting AA therapy is associated with a decreased incidence of PSA30 decline at 12 weeks.
Figure 4. Association of Steroid and Abiraterone Levels with PSA Decline and Radiographic Progression on Abiraterone Acetate plus Prednisone.
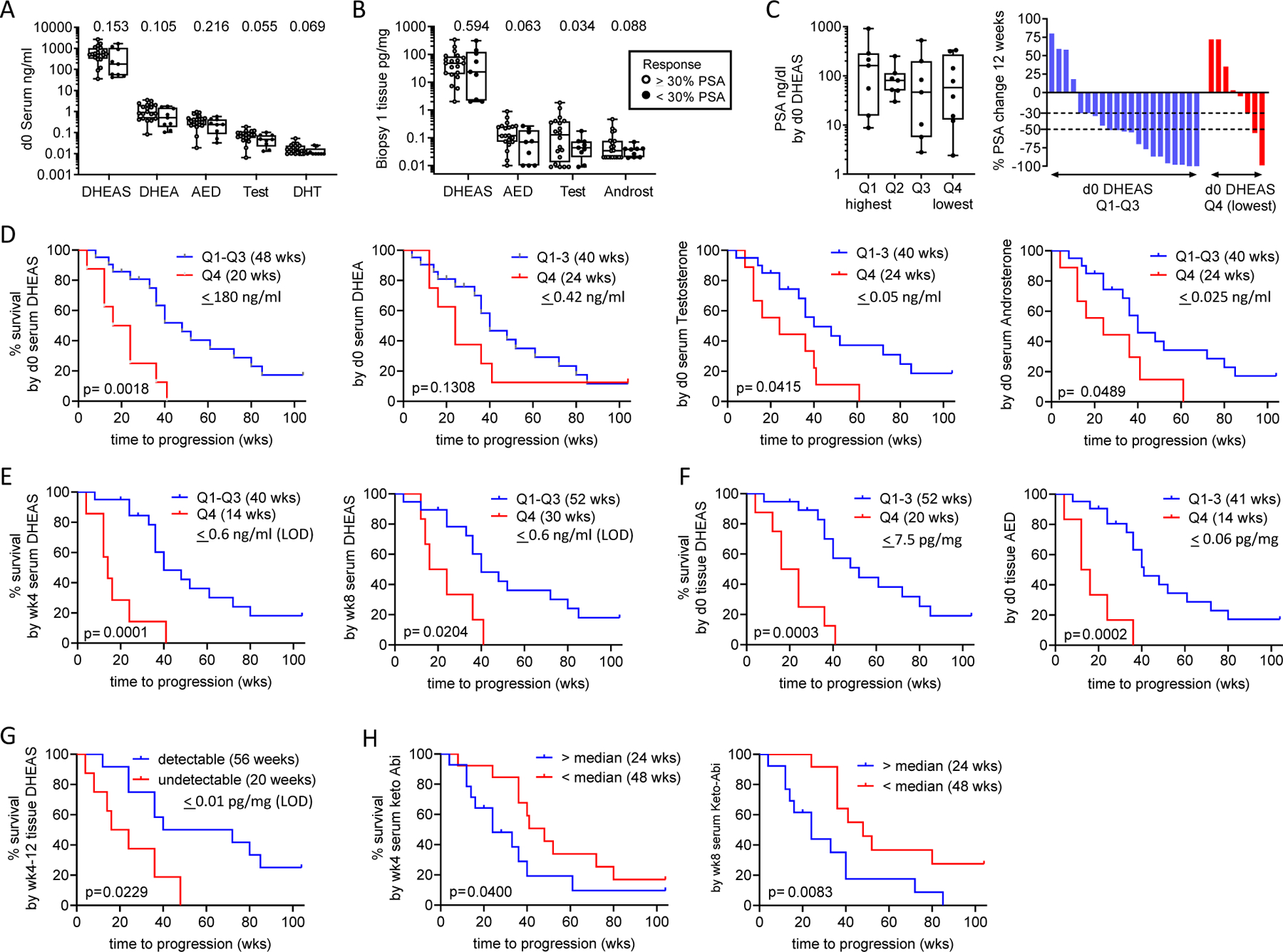
Comparison of steroid levels in A. serum and B. metastatic tissue biopsies, based on achieving a 30% percent PSA decline at 12 weeks. C. Distribution of pre-treatment PSA levels and waterfall plot showing percent change in PSA at 12-weeks by quartile (Q1-Q4) of pre-treatment serum DHEAS levels. P values for the indicated comparison calculated via non-parametric Wilcoxon rank sum test (Mann Whitney test). D. Radiographic progression free survival (rPFS) as a function of baseline serum androgen levels comparing subjects in the lowest vs highest three quartiles (Q4 vs Q1–3). E. rPFS as a function of on-treatment serum DHEAS levels at week 4 (wk4) and week 8 (wk8) comparing subjects in the lowest vs highest three quartiles (Q4 vs Q1–3). F. rPFS as a function of pre-treatment tissue DHEAS and AED levels comparing subjects in the lowest vs highest three quartiles (Q4 vs Q1–3). In each case, the cut-off value reflects the highest number of the bottom 1/4th of the values. The quartiles were separately assessed in the pre and on-treatment populations. G. rPFS comparing subjects with detectable vs undetectable levels of DHEAS in tissue biopsies taken at 4 and 12 weeks of therapy. H. rPFS as a function of serum 3-keto-5a-abiraterone levels (keto-Abi) at week 4 (wk4) and week 8 (wk8) comparing subjects above vs below the median. Progression-free survival was estimated using Kaplan-Meier methods and compared using the Gehan-Wilcoxan test. Dehydroepi-androsterone sulfate (DHEAS), dehydroepiandrosterone (DHEA), androstenedione (AED), testosterone (test), dihydrotestosterone (DHT), androsterone (androst), quartile (Q).
Consistent with these observations, serum steroid levels in the lowest quartile prior to therapy were associated with more rapid progression. This was highly significant for DHEAS (20 vs 48 weeks, p=0.0018) with less significant trends noted for DHEA, T and ASD (p=0.13, p=0.041, and p=0.049 respectively; Fig 4D). Notably, on-treatment levels of DHEAS in the lowest quartile remained associated with more rapid progression (14 vs 40 weeks, p=0.0001 at 4 weeks; 20 vs 40 weeks, p=0.02 at 8 weeks; Fig 4E). Low adrenal androgens in pre-treatment tissue biopsies were even more strongly associated with worse outcomes (20 vs 52 weeks, p=0.0003 for DHEAS), and (14 vs 41 weeks; p=0.0002 for AED; Fig 4F). Likewise, undetectable vs detectable DHEAS levels in biopsies taken at 4 and 12 weeks were associated with shorter time to progression (20 vs 56 weeks, p=0.023; Fig 4G). These observations demonstrate that the association of lower serum adrenal androgen levels with more rapid progression on AA plus prednisone holds true for lower adrenal androgen levels in pre-treatment metastasis biopsies, and for lower on-treatment levels of DHEAS in serum and tissue.
Levels of abiraterone and D4 abiraterone in serum were not associated with differences in time to progression, nor were levels of abiraterone and its metabolites in tissue (Supplementary Fig 6B, 6C). However, serum levels of 3-keto-5α-abiraterone above the median at week 4 and 8 were associated with more rapid progression (24 vs 48 weeks, p=0.04, and 24 vs 48 weeks, p=0.008, respectively; Fig 4H), consistent with the ability of this metabolite to act as an AR agonist.
Association of Genomic Alterations with Radiographic PFS on AA and Prednisone
All patients had pre-treatment tumor biopsy tissue that was adequate for sequencing. The frequency and distribution of pathogenic alterations were consistent with metastatic CRPC (Supplementary Table 4). Wild type (WT) PTEN was weakly associated with longer time to progression (52 vs 33 weeks, p=0.06; Fig 5A). Patients in the lowest quartile of serum DHEAS (3 PTEN WT and 5 PTEN loss) were previously shown to have a poor response to AA (Fig 4D) and this was not influenced by PTEN status. However, in the top 3 quartiles of serum DHEAS (with median survival 48 weeks, Fig 4D), wild type PTEN vs PTEN loss distinguished patients with a significantly more prolonged vs intermediate response (61 vs 33 weeks; p=0.02) vs those in the lowest quartile of DHEAS (20 weeks; p <0.001 for trend; Fig 5A). These findings demonstrate that patients in the lowest quartile of serum DHEAS levels have the most rapid progression on AA, regardless of PTEN status, while those with DHEAS levels in the top 3 quartiles can be meaningfully stratified by PTEN status. In this small study WT MYC was also weakly correlated with longer survival (40 vs 16 weeks, p=0.08) while TP53 status or the presence of AR mutation or amplification were not associated with progression (Fig 5B).
Figure 5. Association of PTEN status with Radiographic Progression on Abiraterone Acetate plus Prednisone.
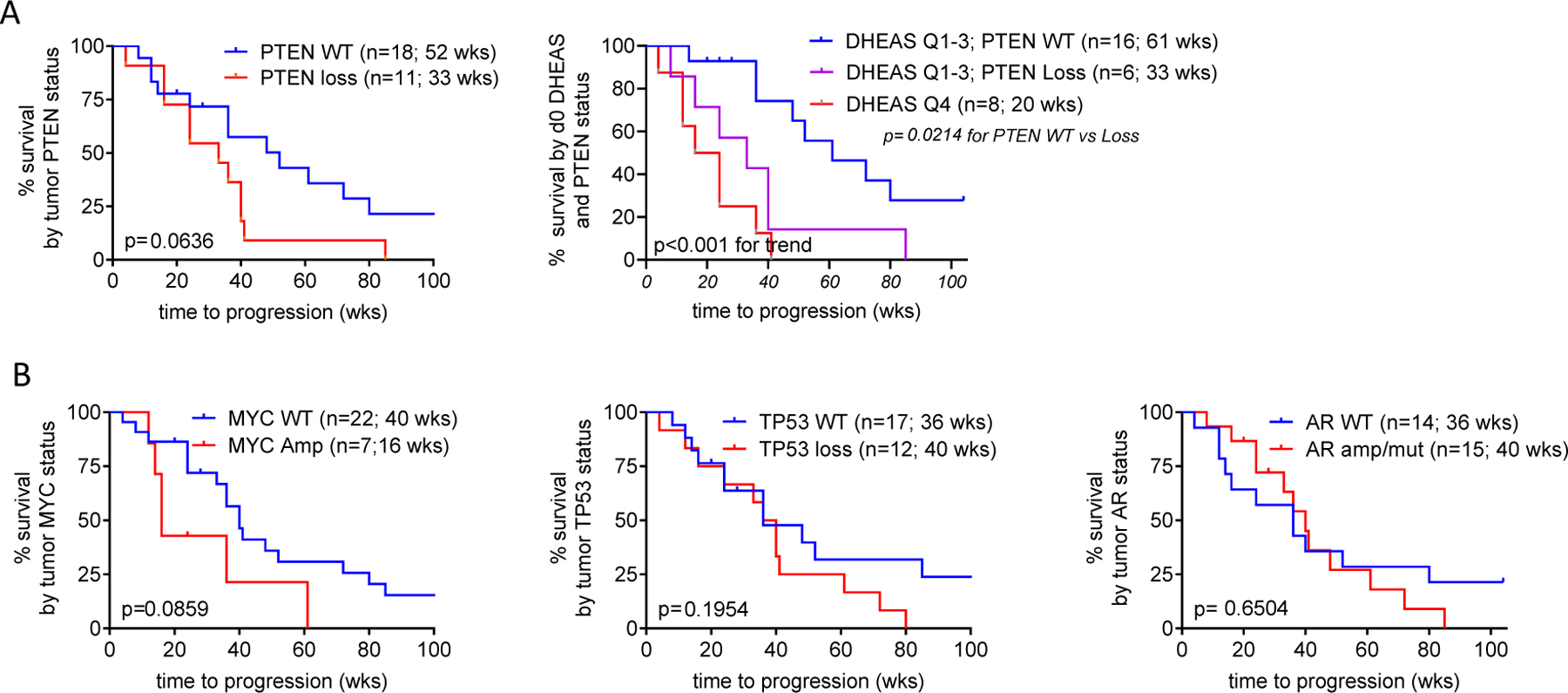
A. Radiographic progression free survival (rPFS) as a function of PTEN status alone or as a function of baseline serum DHEAS levels stratified by PTEN status, comparing subjects in the lowest vs highest three quartiles (Q4 vs Q1–3). B. rPFS as a function of MYC, TP53 or AR status.
As inhibition of AR signaling has been associated with reciprocal induction of PTEN signaling, (19, 20) we determined if lower androgens were associated with PTEN loss. Median serum levels of T and DHT at baseline were numerically but not statistically lower in patients with PTEN loss vs WT PTEN (p=0.09 and 0.084, respectively; Supplementary Fig 7B, as were on-treatment levels of DHEAS in tissue (p=0.09). Median serum levels of DHEAS and DHEA at baseline were also numerically lower in patients with TP53 loss vs WT TP53 (p=0.053 and 0.047, respectively; Supplementary Fig 7C). These observations suggest that while specific AR independent tumor drivers such as PTEN and TP53 may be selected to emerge in a low androgen environment, a low androgen environment is likely to promotes a more aggressive tumor biology by multiple mechanisms not limited to specific pathway alterations.
Association of HSD3B1 Genotype with Steroid and Abiraterone Levels and Radiographic PFS
The 1245C variant of HSD3B1 produces a more stable enzyme, increasing conversion of DHEA to AED, a precursor of T and DHT (Supplementary Fig 4), as well as increasing conversion of abiraterone to D4-abiraterone (21). Germline DNA testing identified 14 men who were homozygous wild type (48%), 10 men who were heterozygous (34%), and 6 who were homozygous for the allelic variant (21%). Consistent with the impact of the variant on enhancing conversion of abiraterone to D4-abiraterone, serum levels of this metabolite were highest in subjects with two variant alleles (median 2.7 ng/ml) vs one variant allele (2.1 ng/ml) vs two wild-type alleles (1.2 ng/ml; ANOVA p=0.02; Supplementary Fig 8A). Serum levels of 3-keto-5α-abiraterone did not differ by HSDB1 genotype, nor did levels of abiraterone metabolite in tissue (Supplementary Fig 8A). There was no difference in serum or tissue androgen levels based on HSD3B1 status (including the 5 tissue samples with detectable levels of DHT; Supplementary Fig 8B), nor in time to progression (Supplementary Fig 8C).
DISCUSSION
In this phase 2 study we demonstrate that in metastatic CRPC AA plus prednisone decreases T levels in metastatic tissue biopsies to undetectable by 4 and 12 weeks of therapy, consistent with the predicted mechanism of activity. Steroid and abiraterone levels in serum and tissue remained stable at progression compared to earlier time points, consistent with prior reports regarding serum adrenal androgens (8), but demonstrating for the first time that tissue androgen levels also remain unchanged at progression. Overall, these findings suggest that the development of resistance to AA plus prednisone is not accounted for by a decrease in serum or tissue drug levels below the levels originally achieved in response to therapy. Nor is it accounted for by an increase in serum or intra-tumoral androgens above the levels originally achieved in response to AA plus prednisone. The observations that tumor steroid levels do not become re-elevated at progression and that persistent serum DHEAS levels do not become undetectable after dose escalation of AA to 2000mg likely accounts for the lack of clinical response to high dose AA at progression, now reported in two studies (22).
At baseline, serum levels of DHEAS and DHEA varied by over an order of magnitude (range 36–2659 ng/ml) and demonstrated two patterns of response to treatment. While median levels of steroids downstream of CYP17A decreased by a similar order of magnitude in all patients, the subset of patients defined by higher serum DHEAS levels prior to therapy also had higher serum levels of other steroids at baseline, higher levels of DHEAS, AED and androsterone in pre-treatment tissues biopsies, and maintained higher DHEAS levels in on-treatment serum samples and tissue biopsies. These observations demonstrate that serum DHEAS levels prior to therapy can distinguish patients with correspondingly lower or higher serum and tissue levels of all androgens and can identify those likely to maintain persistent serum and tissue DHEAS levels while on treatment with AA plus prednisone.
The correlation of low baseline with low on-treatment adrenal androgen levels is of particular interest as lower adrenal androgens prior to therapy have been consistently associated with worse response to AA plus prednisone (13, 23–25). We show for the first time that this association with worse outcomes extends to low on-treatment level of DHEAS in serum and tissue, and with low adrenal androgens in pre-treatment metastasis biopsies. At baseline, the association of worse response with lower levels was highly significant for DHEAS in serum (p=0.0018) and for DHEAS and AED in pre-treatment tissue biopsies (p=0.0003 and p=0.0002, respectively). On-therapy, serum DHEAS in the lowest quartile at 4 and 8 weeks (p=0.0001 and 0.024, respectively), as well as undetectable vs detectable DHEAS levels in metastatic biopsies taken at 4 and 12 weeks (p=0.023 and p=0.04, respectively) all associated with more rapid progression on AA therapy. These specific associations with DHEAS are consistent with fact that is DHEAS the predominant steroid in serum and tissue prior to therapy, and remains the only steroid consistently present at measurable levels in metastatic tissue after AA therapy.
While response to AA is clearly linked with suppression of circulating and intra-tumor androgens, the paradoxical association of better outcomes with higher on-treatment androgen levels suggests that response to AA is not dictated solely by a pharmacodynamic effect on serum or tissue androgens, but may reflect an intrinsic resistance to AR-pathway therapy associated with tumor outgrowth in a low androgen microenvironment (26). These observations suggest that baseline serum androgen levels, by identifying patients with a more aggressive tumor biology that is less androgen-dependent, may be prognostic as well as predictive of response to AR-pathway directed therapy. As such, patients with higher androgens may have better survival independent of an effect of AA. This is consistent with the association of lower serum androgen levels with decreased survival in both the placebo and AA arms of the phase III study of AA plus prednisone in men with metastatic CRPC (13), and with a recent meta-analysis demonstrating that PFS and OS were lower in CRPC patients with lower vs higher T levels (27).
Importantly, the observation that lower androgen levels in CRPC associate with worse outcomes is not incompatible with data linking the achievement of lower T levels with improved time to progression in men with CSPC treated with ADT (28, 29). While time to development of CRPC may be delayed, CRPC tumors that emerge n patients with lower T levels (due to low adrenal contribution and/or optimally suppressed T levels while on ADT) are likely to represent a more aggressive, less androgen-dependent phenotype vs tumors that emerge with more rapid kinetics in context of higher androgen levels (due to more robust adrenal contribution and/or sub-optimally suppressed T levels). While this may not influence response to subsequent non-AR specific agents such as docetaxel, it is likely to adversely influence response to next-generation AR targeted therapy. This is consistent with findings in the meta-analysis discussed above, in which lower T levels were a consistently poor prognostic factor for OS in CRPC patients treated with AR targeting agents, but not in those treated with chemotherapy (27).
The significant variation in adrenal androgen levels is not well understood. Functional polymorphisms in genes encoding critical steroidogenic enzymes such as CYP17A, HSD3B1, SULT2A1, AKR1C3, SRD5A, and UGT2B17 influence enzyme activity and/or production of downstream steroids (21, 30–32). Population-based studies will be required to determine whether a composite haplotype of genes involved in adrenal androgen production exists that may account for the observed spectrum of serum androgen levels. Alternatively, the variability might reflect genetic variation in regulatory factors such as CYB5A1, NR5A1 (also known as SF-1 or steroidogenic factor 1) or MC2R (the ACTH receptor) that are upstream of adrenal steroid synthesis (33–35).
We found no association of abiraterone levels with PSA decline or with time to radiographic progression, with nearly all patients having serum levels above the 8.4 ng/ml value previously proposed as a cut-off for PSA response (36). This contrasts with the observation of Friedlander et al who found in a study of 41 men with mCRPC that patients who failed to show a PSA response to AA had lower abiraterone levels after 4 weeks and at progression (22). This difference may reflect variability in timing of the blood draws, which, while mandated before the daily dose, were not specifically recorded precluding correction for differences based on pharmacokinetics (18). However, our findings are consistent with the work of Smulewitz et al, who also found no correlation between abiraterone levels and change in PSA in a study of 72 men with mCRPC randomized to standard or low dose AA (37), as well as with an analysis performed by the US Food and Drug Administration in which no association was found between trough abiraterone levels and OS (38).
The adrenal permissive variant of HSD3B1 (promotes conversion of AED and T to downstream androgens, and of abiraterone to D4 abiraterone and 3-keto-5α-abiraterone) has been consistently associated with more rapid progression on ADT (39), and potentially with worse outcomes in response to AA and enzalutamide in some but not all studies (40–43). Although we did find an association of HSD3B1 status with D4 abiraterone (the immediate downstream product of HSD3B1), we did not observe an association of HSD3B1 genotype with response to abiraterone, nor the previously reported association with 3-keto-5α-abiraterone, potentially reflecting that serum levels were not corrected for differences based on pharmacokinetics. (18) However, we did observe that serum levels of 3-keto-5α-abiraterone above the median at week 4 and 8 were associated with a more rapid time to progression. These observations are consistent with the ability of this metabolite to act as an AR agonist, and suggest that dose escalation of AA may be in fact be detrimental to patient outcomes. These findings provide support for the low dose AA strategy proposed by Smulewitz (37), and provide impetus for clinical trials combining AA therapy with an SRD5A inhibitor to prevent conversion of D4-abiraterone to 5a-keto metabolite (44, 45).
Next generation sequencing of CRPC tumors has identified aberrations in multiple genes including AR, TP53, PTEN and SPOP which have been explored as predictive biomarkers of response to AR pathway inhibition. In this small study we found no association of AR, TP53 or SPOP status with response to AA (46–48). In our cohort, WT PTEN was weakly associated with longer time to progression on AA plus prednisone, consistent with Ferraldeschi et al who reported a retrospective study of 144 patients in which PTEN loss was associated with shorter OS (14 vs 21 months; p = 0.004) and shorter duration of abiraterone treatment (24 vs 28 weeks; p = 0.009) (49). Moreover, whereas poor response to AA in men with the lowest serum DHEAS levels was not additionally influenced by PTEN status, in those with higher androgen levels WT PTEN vs PTEN loss conferred a significantly longer time to progression (61 vs 33 weeks; p=0.02). While our study is too small to meaningfully predict genomic alterations associated with response to AR pathway inhibition, these data suggest that interpretation of tumor sequencing data may be more informative if analyzed in context of the risk conferred by low vs high serum androgen levels.
This study has several important limitations, including small sample size, that abiraterone and metabolite levels were not corrected for pharmacokinetic differences based on timing of trough blood draws, and that analyses were exploratory with no attempt to correct for multiple testing. As such, our observations must be considered hypothesis generating and require validation in larger data sets.
Our data show that higher circulating adrenal androgens in CRPC patients are strongly associated with an androgen-rich tumor microenvironment before and during therapy with AA and prednisone, and suggest that ambient androgen levels in the castrate tumor microenvironment are an important determinant of prostate tumor biology and response to therapy (13). Baseline and on-treatment androgen levels are likely to be important in the stratification and interpretation of trials evaluating AR pathway directed therapy. In particular, while addition of AA to enzalutamide did not prolong survival in metastatic CRPC (50), higher baseline DHEAS levels may distinguish a subset of patients who do benefit from dual AR therapy. Conversely, CRPC patients in the lowest quartile of serum DHEAS levels may warrant stratification to regimens that include non-AR directed therapy. Whether adrenal androgens associate with response to AA in metastatic castration sensitive prostate cancer is unknown but requires exploration. Prospective studies evaluating baseline and on-treatment androgen levels as predictive biomarkers of response to AR-directed therapy are required to test these hypotheses.
Supplementary Material
Statement of Translational Relevance.
In men with metastatic castration resistant prostate cancer (CRPC), low circulating adrenal androgens levels are strongly associated with an androgen-poor tumor microenvironment, with lower androgen levels while on-therapy, and with poor response to abiraterone acetate (AA) plus prednisone. While response to AA is clearly linked with its ability to suppress circulating and intra-tumor androgen levels, the paradoxical association of lower adrenal androgen levels while on-therapy with more rapid radiographic progression suggests an intrinsic resistance to AR-pathway therapy related to tumor outgrowth in a low androgen microenvironment. These data suggest that clinical benefit from dual-AR pathway inhibition is likely to be observed in patients with higher serum adrenal androgen levels, but will be limited in those with low/undetectable levels in whom treatment combinations with non-AR directed therapy are likely to be required. Assessment of baseline and on-treatment androgen levels can inform the stratification and interpretation of trials evaluating the efficacy of AR pathway directed therapy.
ACKNOWLEDGEMENTS
DOD W81XWH-14–1–0004 (E Mostaghel); Pacific Northwest Prostate Cancer SPORE P50CA097186 (E Mostaghel, P Nelson, R Montgomery); P01 CA163227 (E Mostaghel, P Nelson, S Balk); R01 CA234715 (P Nelson); R01 CA172382 and R01 CA236780 (both to N Sharifi). Department of Veterans Affairs Puget Sound Health Care System (E Mostaghel, A Matsumoto); Funding of the clinical trial was provided by Janssen.
Footnotes
Conflict of interest:
EA Mostaghel reports research support from ESSA Pharma.
EY Yu, MT Schwiezer, HH Cheng report research support
PW Kantoff reports investment interest in Congent Biosciences, Context Therapeutics LLC, DRGT, Mirati, Placon, PrognomIQ, Seer Biosciences, SnyDevRx and XLink, he is a company board member for Context Therapeutics LLC, he is a company founder for XLink, and is/was a consultant/scientific advisory board member for Anji, Bavarian Nordic Immunotherapeutics, Candel, DRGT, Immunis, AI (previously OncoCellMDX), Janssen, Progenity, PrognomIQ, Seer Biosciences, SynDevRX, Tarveda Therapeutics, Veru, and serves on data safety monitoring boards for Genentech/Roche and Merck. He reports spousal association with Bayer.
S Balk reports pending patents related to induction of NKT cells for treatment of metabolic disorders and to use of novel aryl sulfonamide compounds for treatment of conditions related to the androgen receptor.
ME Taplin reports income support from Arcus Bioscience
N Sharifi reports pending patents related to HSD3B1.
PS Nelson, RB Montgomery report research support.
BT Marck, O Kolokythas, F Chew and A Matsumoto report no disclosures.
Presented in part at the 2019 American Society of Clinical Oncology GenitoUrinary Cancers Meeting, Feb 11–13, 2019, San Francisco, CA (Abstract #208).
REFERENCES
- 1.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68(11):4447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohler JL, Gregory CW, Ford OH 3rd, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10(2):440–8. [DOI] [PubMed] [Google Scholar]
- 3.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–25. [DOI] [PubMed] [Google Scholar]
- 4.Sharifi N. The 5alpha-androstanedione pathway to dihydrotestosterone in castration-resistant prostate cancer. J Investig Med. 2012;60(2):504–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taplin ME, Montgomery B, Logothetis CJ, Bubley GJ, Richie JP, Dalkin BL, et al. Intense Androgen-Deprivation Therapy With Abiraterone Acetate Plus Leuprolide Acetate in Patients With Localized High-Risk Prostate Cancer: Results of a Randomized Phase II Neoadjuvant Study. J Clin Oncol. 2014. [DOI] [PMC free article] [PubMed]
- 6.Tamae D, Mostaghel E, Montgomery B, Nelson PS, Balk SP, Kantoff PW, et al. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Interact. 2015;234:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attard G, Reid AH, Auchus RJ, Hughes BA, Cassidy AM, Thompson E, et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab. 2012;97(2):507–16. [DOI] [PubMed] [Google Scholar]
- 8.Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28(9):1481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13(10):983–92. [DOI] [PubMed] [Google Scholar]
- 10.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mostaghel EA, Marck B, Plymate S, Vessella RL, Balk SP, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011. [DOI] [PMC free article] [PubMed]
- 12.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan CJ, Molina A, Li J, Kheoh T, Small EJ, Haqq CM, et al. Serum Androgens As Prognostic Biomarkers in Castration-Resistant Prostate Cancer: Results From an Analysis of a Randomized Phase III Trial. J Clin Oncol. 2013;31(22):2791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mostaghel EA, Cho E, Zhang A, Alyamani M, Kaipainen A, Green S, et al. Association of Tissue Abiraterone Levels and SLCO Genotype with Intraprostatic Steroids and Pathologic Response in Men with High-Risk Localized Prostate Cancer. Clin Cancer Res. 2017;23(16):4592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hearn JWD, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang KH, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. Lancet Oncol. 2016;17(10):1435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrylak DP, Ankerst DP, Jiang CS, Tangen CM, Hussain MH, Lara PN Jr., et al. Evaluation of prostate-specific antigen declines for surrogacy in patients treated on SWOG 99–16. J Natl Cancer Inst. 2006;98(8):516–21. [DOI] [PubMed] [Google Scholar]
- 18.Alyamani M, Emamekhoo H, Park S, Taylor J, Almassi N, Upadhyay S, et al. HSD3B1(1245A>C) variant regulates dueling abiraterone metabolite effects in prostate cancer. J Clin Invest. 2018;128(8):3333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19(6):792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang K, Li R, B K, Lotan Y, Roehrborn C, Liu J, et al. A Gain-of-Function Mutation in DHTSynthesis in Castration Resistant Prostate Cancer. Cell. 2013;154:1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedlander TW, Graff JN, Zejnullahu K, Anantharaman A, Zhang L, Paz R, et al. High-Dose Abiraterone Acetate in Men With Castration Resistant Prostate Cancer. Clin Genitourin Cancer. 2017;15(6):733–41 e1. [DOI] [PubMed] [Google Scholar]
- 23.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27(23):3742–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim W, Zhang L, Wilton JH, Fetterly G, Mohler JL, Weinberg V, et al. Sequential use of the androgen synthesis inhibitors ketoconazole and abiraterone acetate in castration-resistant prostate cancer and the predictive value of circulating androgens. Clin Cancer Res. 2014;20(24):6269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki K, Sakamoto M, Terakawa T, Furukawa J, Harada K, Hinata N, et al. Serum DHEA-S Is a Predictive Parameter of Abiraterone Acetate in Patients with Castration-resistant Prostate Cancer. Anticancer Res. 2018;38(10):5929–35. [DOI] [PubMed] [Google Scholar]
- 26.Valcamonico F, Ferrari L, Consoli F, Amoroso V, Berruti A. Testosterone serum levels and prostate cancer prognosis: the double face of Janus. Future Oncol. 2014;10(7):1113–5. [DOI] [PubMed] [Google Scholar]
- 27.Miura N, Mori K, Mostafaei H, Quhal F, Sari Motlagh R, Abufaraj M, et al. Prognostic value of testosterone for the castration-resistant prostate cancer patients: a systematic review and meta-analysis. Int J Clin Oncol. 2020;25(11):1881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toren P, Hoffman A, Ding K, Joncas FH, Turcotte V, Caron P, et al. Serum Sex Steroids as Prognostic Biomarkers in Patients Receiving Androgen Deprivation Therapy for Recurrent Prostate Cancer: A Post Hoc Analysis of the PR.7 Trial. Clin Cancer Res. 2018;24(21):5305–12. [DOI] [PubMed] [Google Scholar]
- 29.Klotz L, O’Callaghan C, Ding K, Toren P, Dearnaley D, Higano CS, et al. Nadir testosterone within first year of androgen-deprivation therapy (ADT) predicts for time to castration-resistant progression: a secondary analysis of the PR-7 trial of intermittent versus continuous ADT. J Clin Oncol. 2015;33(10):1151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilborn TW, Lang NP, Smith M, Meleth S, Falany CN. Association of SULT2A1 allelic variants with plasma adrenal androgens and prostate cancer in African American men. J Steroid Biochem Mol Biol. 2006;99(4–5):209–14. [DOI] [PubMed] [Google Scholar]
- 31.Shiota M, Fujimoto N, Yokomizo A, Takeuchi A, Kashiwagi E, Dejima T, et al. The prognostic impact of serum testosterone during androgen-deprivation therapy in patients with metastatic prostate cancer and the SRD5A2 polymorphism. Prostate Cancer Prostatic Dis. 2016;19(2):191–6. [DOI] [PubMed] [Google Scholar]
- 32.Nadeau G, Bellemare J, Audet-Walsh E, Flageole C, Huang SP, Bao BY, et al. Deletions of the Androgen-Metabolizing UGT2B Genes Have an Effect on Circulating Steroid Levels and Biochemical Recurrence after Radical Prostatectomy in Localized Prostate Cancer. J Clin Endocrinol Metab. 2011. [DOI] [PubMed]
- 33.Stark K, Straub RH, Rovenský J, Blažičková S, Eiselt G, Schmidt M. CYB5A polymorphism increases androgens and reduces risk of rheumatoid arthritis in women. Arthritis Res Ther. 2015;17(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schimmer BP, Cordova M, Tsao J, Frigeri C. SF1 polymorphisms in the mouse and steroidogenic potential. Endocr Res. 2002;28(4):519–25. [DOI] [PubMed] [Google Scholar]
- 35.Liu ZL, He B, Fang F, Tang CY, Zou LP. Genetic polymorphisms of MC2R gene associated with responsiveness to adrenocorticotropic hormone therapy in infantile spasms. Chin Med J (Engl). 2008;121(17):1627–32. [PubMed] [Google Scholar]
- 36.Carton E, Noe G, Huillard O, Golmard L, Giroux J, Cessot A, et al. Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur J Cancer. 2017;72:54–61. [DOI] [PubMed] [Google Scholar]
- 37.Szmulewitz RZ, Peer CJ, Ibraheem A, Martinez E, Kozloff MF, Carthon B, et al. Prospective International Randomized Phase II Study of Low-Dose Abiraterone With Food Versus Standard Dose Abiraterone In Castration-Resistant Prostate Cancer. J Clin Oncol. 2018;36(14):1389–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.US Food and Drug Administration CfDEaR. Clinical Pharmacology and Biopharmaceutics Review(s) Abiraterone Acetate. APPLICATION NUMBER: 202379Orig1s000. 2011;https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000PharmR.pdf.
- 39.Thomas L, Sharifi N. Germline HSD3B1 Genetics and Prostate Cancer Outcomes. Urology. 2020;145:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahn AW, Gill DM, Nussenzveig RH, Poole A, Farnham J, Cannon-Albright L, et al. Germline Variant in HSD3B1 (1245 A > C) and Response to Abiraterone Acetate Plus Prednisone in Men With New-Onset Metastatic Castration-Resistant Prostate Cancer. Clin Genitourin Cancer. 2018;16(4):288–92. [DOI] [PubMed] [Google Scholar]
- 41.Lu C, Terbuch A, Dolling D, Yu J, Wang H, Chen Y, et al. Treatment with abiraterone and enzalutamide does not overcome poor outcome from metastatic castration-resistant prostate cancer in men with the germline homozygous HSD3B1 c.1245C genotype. Ann Oncol. 2020;31(9):1178–85. [DOI] [PubMed] [Google Scholar]
- 42.Khalaf DJ, Aragón IM, Annala M, Lozano R, Taavitsainen S, Lorente D, et al. HSD3B1 (1245A>C) germline variant and clinical outcomes in metastatic castration-resistant prostate cancer patients treated with abiraterone and enzalutamide: results from two prospective studies. Ann Oncol. 2020;31(9):1186–97. [DOI] [PubMed] [Google Scholar]
- 43.Shiota M, Narita S, Akamatsu S, Fujimoto N, Sumiyoshi T, Fujiwara M, et al. Association of Missense Polymorphism in HSD3B1 With Outcomes Among Men With Prostate Cancer Treated With Androgen-Deprivation Therapy or Abiraterone. JAMA Netw Open. 2019;2(2):e190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKay RR, Werner L, Mostaghel EA, Lis R, Voznesensky O, Zhang Z, et al. A Phase II Trial of Abiraterone Combined with Dutasteride for Men with Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2017;23(4):935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, et al. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533(7604):547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol. 2017;28(7):1508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boysen G, Rodrigues DN, Rescigno P, Seed G, Dolling D, Riisnaes R, et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin Cancer Res. 2018;24(22):5585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Laere B, Oeyen S, Mayrhofer M, Whitington T, van Dam PJ, Van Oyen P, et al. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2019;25(6):1766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferraldeschi R, Nava Rodrigues D, Riisnaes R, Miranda S, Figueiredo I, Rescigno P, et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur Urol. 2015;67(4):795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris MJ, Heller G, Bryce AH, Armstrong AJ, Beltran H, Hahn OM, et al. Alliance A031201: A phase III trial of enzalutamide (ENZ) versus enzalutamide, abiraterone, and prednisone (ENZ/AAP) for metastatic castration resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2019;37(15_suppl):5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.