

Abstract
Sepsis is a life-threatening process related to a dysregulated host response to an underlying infection which results in organ dysfunction and poor outcomes. Therapeutic strategies using mesenchymal stromal cells (MSCs) are under investigation for sepsis, with efforts to improve cellular utility. Syndecan (SDC) proteins are transmembrane proteoglycans involved with cellular signaling events including tissue repair and modulating inflammation. Bone marrow-derived human MSCs express syndecan-2 (SDC2) at a level higher than other SDC family members, thus we explored SDC2 in MSC function. Administration of human MSCs silenced for SDC2 in experimental sepsis resulted in decreased bacterial clearance, and increased tissue injury and mortality compared with wild-type MSCs. These findings were associated with a loss of resolution of inflammation in the peritoneal cavity, and higher levels of pro-inflammatory mediators in organs. MSCs silenced for SDC2 had a decreased ability to promote phagocytosis of apoptotic neutrophils by macrophages in the peritoneum, and also a diminished capability to convert macrophages from a pro-inflammatory to a pro-resolution phenotype via cellular or paracrine actions. Extracellular vesicles are a paracrine effector of MSCs that may contribute to resolution of inflammation, and their production was dramatically reduced in SDC2 silenced human MSCs. Collectively, these data demonstrate the importance of SDC2 for cellular and paracrine function of human MSCs during sepsis.
Keywords: mesenchymal stromal cells, sepsis, macrophage polarization, efferocytosis, resolution of inflammation, extracellular vesicles
INTRODUCTION
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection [1]. While comorbidities of the host, genetic determinants, and environmental or other factors contribute to this dysregulated response resulting in sepsis, the ability of the immune system to clear the inciting pathogen is critical. Without control of the underlying infection, and the appropriate resolution of the inflammatory response, collateral organ injury occurs contributing to organ dysfunction and a poor outcome. The interaction of innate immune cells, including neutrophils and macrophages, allow the host to efficiently clear pathogens and to return to homeostasis [2, 3].
Due to the challenges of therapy for the sepsis syndrome, and the fact that management remains predominantly supportive, new advances are being explored including the use of cell-based therapies [4–8]. Mesenchymal stromal cells (MSCs) [9] have shown promise in experimental models of sepsis [8]. The immune evasive properties of MSCs allow the use of allogeneic cells in humans, and also permits the use of human cells in mouse models of disease for preclinical investigation [10]. A critical property of MSCs, or their paracrine components [11], is modulation of the immune response that allows clearance of the invading organism(s), and limits tissue injury during sepsis [12]. While our laboratory and others have demonstrated that MSCs improve outcomes in experimental models of sepsis in mice [13–19], we seek to further understand how MSCs control the immune response during sepsis, with a special interest in the interaction of macrophages and neutrophils.
In an effort to identify a more homogeneous population of human MSCs, that does not require plastic adherence in culture and in vitro cell surface marker profiles and differentiation assays, investigators have recently evaluated the use of Syndecan-2 (SDC2 or CD362) as a cell surface marker of a subpopulation of MSCs. SDC2+ bone marrow-derived human MSCs were shown to decrease the severity of Escherichia (E.) coli-induced pneumonia and improved recovery from ventilator-induced lung injury in rats, and this response was superior to SDC2− cells and comparable to the heterogeneous MSC population in vivo [20]. Similarly, human umbilical cord-derived SDC2+ MSCs were shown to be as effective as the total heterogeneous MSC population in reducing E. coli-induced acute lung injury in rats [21]. While these studies used SDC2 as a marker for isolation of a homogeneous population of MSCs, the purpose of our study was to determine whether SDC2 plays an important role in MSC biology and function during experimental sepsis.
SDCs are heparan sulfate transmembrane proteoglycans that interact with a large number of ligands, and these molecules play a role in many cellular signaling events related to cell adhesion, tissue repair, and inflammation [22]. Interestingly, while involved in tissue repair, the expression of SDC2 is increased in alveolar macrophages in patients with pulmonary fibrosis, and exerts antifibrotic effects in experimental models of lung fibrosis [23, 24]. In regard to inflammation, SDCs have been shown to regulate leukocyte extravasation and cytokine/chemokine function [25], and SDCs are involved in many aspects of the inflammatory response, from leukocyte recruitment to resolution of inflammation [26]. Since MSCs are also known to modulate inflammation and tissue repair, we propose that SDC2 plays an important role in MSC function during sepsis to alleviate organ dysfunction.
RESULTS
SDC2 expression is higher in human MSCs compared with fibroblasts, and silencing of SDC2 alters cell growth
We analyzed the level of SDC2 in human bone marrow-derived MSCs (hMSCs) and human dermal fibroblasts, a control mesenchymal cell. Using quantitative real-time PCR (qRT-PCR), the level of SDC2 mRNA in hMSCs was 6.8 fold higher than human fibroblasts (Figure 1A). Furthermore, when assessing the expression of SDC2 compared with other family members, SDC2 was expressed significantly higher than SDC1, 3, and 4 in bone marrow-derived hMSCs (Figure 1A). To explore the impact of SDC2 on hMSCs function, we silenced SDC2 using a short hairpin RNA lentiviral construct (shSDC2) compared with a scrambled control construct (shSCR). Figures 1B and 1C demonstrate that silencing of SDC2 resulted in decreased mRNA levels (more than 96% reduction) and protein expression (~67% decrease) in hMSCs. shSDC2 and shSCR hMSCs were next phenotyped using flow cytometry and exhibited comparable expression of mesenchymal markers (Figure 1D), including CD90, CD73, and CD105. In both lines of hMSCs, they showed a very low expression of MHCII. Silencing of SDC2 in hMSCs demonstrated a significant reduction in cell growth at days 3, 4, and 5, compared with shSCR hMSCs (Figure 1E). Furthermore, while shSCR hMSCs continued to grow over the 5 days of evaluation, the growth of shSDC2 cells was not statistically different between any of the days 1 – 5.
Figure 1.
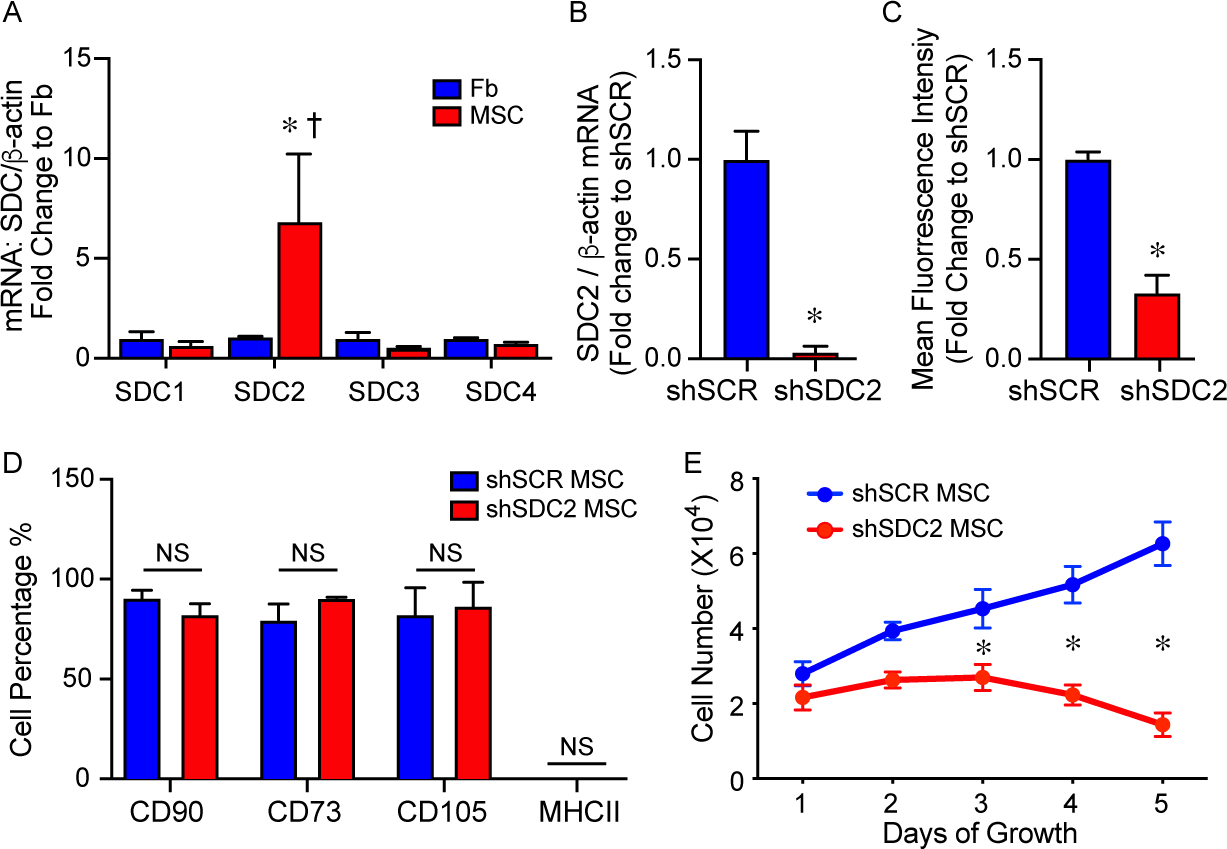
Syndecan-2 (SDC2) is highly expressed in bone marrow-derived hMSCs, and silencing results in no change in mesenchymal markers but decreased cell growth. A) RNA was extracted from human dermal fibroblasts (Fb, blue bars) and bone-marrow derived hMSCs (MSC, red bars), and quantitative real-time polymerase chain reaction (qRT- PCR) was performed for SDC 1, 2, 3, 4. Data are presented as mRNA levels of SDCs normalized for β-actin, as a fold change to Fb, mean±SEM, n=3 in each group. *p=0.032 versus Fb, and † p=0.03 SDC2 versus other SDCs. B) RNA was extracted from scrambled control short hairpin construct (shSCR) hMSCs (blue bar) and from silencing of SDC2 (shSDC2, red bar), and qRT-PCR was performed for SDC2. Data are presented as mRNA levels of SDC2 normalized for β-actin, as a fold change to shSCR hMSCs, mean±SEM, n=3 in each group, *p=0.0027 versus shSCR. C) Flow cytometry was also performed for SDC2 in shSCR (blue bar) and shSDC2 (red bar) hMSCs. Data are presented as mean fluorescent intensity, fold change to shSCR hMSCs, mean ±SEM, n=3 in each group, *p=0.002 versus shSCR. D) Flow cytometry was performed for CD90, CD73, CD105, and MHCII in shSCR (blue bar) and shSDC2 (red bar) hMSCs. Data are presented as percentage of cells expressing the markers in each group, mean±SEM, n=3 in each group, NS = not significant between groups. E) shSCR hMSCs (blue dots/line) and shSDC2 (red dots/line) hMSCs were seeded on day 0, and counted daily through day 5. Data are presented as cell number ×104, mean±SEM, n=3 experiments in each group, *p=0.011 shSDC2 versus shSCR hMSC.
Silencing SDC2 leads to a loss of hMSC survival benefit, failure to protect from tissue injury, and ineffective bacterial clearance in experimental sepsis
We next assessed the therapeutic impact of hMSC-derived SDC2 function in vivo. After the induction of sepsis by cecal ligation and puncture (CLP), mice received vehicle (phosphate buffered saline [PBS]), shSCR hMSCs, or shSDC2 hMSCs. hMSCs (5×105 cells/200 μL PBS) or vehicle (PBS 200 μL) were administered intravenously 2 hours, and then again 24 hours after CLP, and survival was assessed over 7 days. Mice treated with PBS alone had a survival rate of ~23% (Figure 2A). Injection of shSCR hMSCs led to a marked increase in mouse survival (~67%), whereas the survival of mice receiving shSDC2 hMSCs was significantly diminished (~31%). Assessment of organ injury at 48 hours after CLP or sham surgery revealed mice receiving shSDC2 hMSCs or PBS after CLP had a similar increase in apoptosis of spleen and bowel (distal small intestine), whereas mice receiving shSCR hMSCs after CLP had a blunted apoptotic response, comparable to sham surgery (Figure 2B). Lung injury is another consequence of sepsis, and hMSCs have been shown to restore fluid clearance and have antimicrobial activity in human lungs ex vivo exposed to live bacterial in a pneumonia model [27]. The mice undergoing CLP showed evidence of thickening of the alveolar walls, with some alveolar collapse and edema at 48 hours (Figure 2C). While these changes were more evident in mice that received PBS or shSCD2 MSCs after CLP, overall the histological findings in the lungs were modest in this model of peritoneal sepsis.
Figure 2.

Silencing of SDC2 in hMSCs results in decreased survival and increased tissue injury. and a lack of bacterial clearance when administered during sepsis. A) Septic mice were randomly separated into three groups: phosphate buffered saline (PBS) control (gray line, n=13), shSCR hMSCs (blue line, n=15), and shSDC2 hMSCs (red line, n=16). All mice were subjected to cecal ligation and puncture (CLP). 2 hours after CLP, the mice received PBS (200 μL) or hMSCs (5×105 cells in 200 μL PBS)) via tail vein injection. This treatment was also repeated at 24 hours after CLP. Survival of mice was monitored over 7 days, and data are presented as Kaplan-Meier survival curves, *p=0.025 versus shSCR hMSC. B) Tissue injury was assessed by terminal deoxynucleotidetransferase–mediated dUTP nick end-labeling (TUNEL) staining of spleen (left panel) and bowel (right panel) tissues 48 hours after Sham (−) or CLP (+) surgery. Data are presented as quantification of apoptotic cells per mm2, mean±SEM, from random images of fluorescent microscope (×20 objective) from random images in sham (white bars, n=10 and 13 images respectively from left and right panels), CLP+PBS (gray bars, n=24 and 18 respectively), CLP+shSCR hMSCs (blue bars, n=27 and 18 images respectively), and CLP+shSDC2 hMSCs (red bars, n=27 and 20 images respectively). p<0.0001 for spleen and bowel, with significant comparisons * versus sham, † versus PBS, ‡ versus shSCR hMSCs. C) Lung architecture was assessed by hematoxylin and eosin staining of representative tissue sections from Sham (left upper panel), CLP+PBS (right upper panel), CLP+shSCR hMSCs (left lower panel), and CLP+shSDC2 hMSCs (right lower panel). Arrows point to areas of injury. Scale bar represents 100 μm.
Bacterial clearance was also assessed at 48 hours after CLP. Mice in all sepsis groups demonstrated bacteria in the peritoneum and blood. The administration of shSCR hMSCs after the onset of sepsis resulted in a significant decrease in bacteria in both the peritoneum and the blood compared with the PBS group (Figures 3A and 3B). In contrast, mice receiving shSDC2 hMSCs had significantly higher numbers of bacteria in the peritoneum and blood compared with mice receiving shSCR hMSCs, analogous to mice receiving PBS alone. To further understand bacterial clearance, we assessed the influence of hMSCs on neutrophil and macrophage phagocytosis. Compared with no hMSCs, shSCR hMSCs increased the percentage of neutrophils phagocytizing bacteria (E. coli), and also the total amount of bacteria engulfed (Figure 3C). In the presence of shSDC2 hMSCs, the percentage and amount of E. coli phagocytized by neutrophils was not different than neutrophils not exposed to hMSCs. While macrophages phagocytized E. coli, the effect of shSCR and shSDC2 hMSCs was not different from macrophages not exposed to hMSCs (data not shown).
Figure 3.
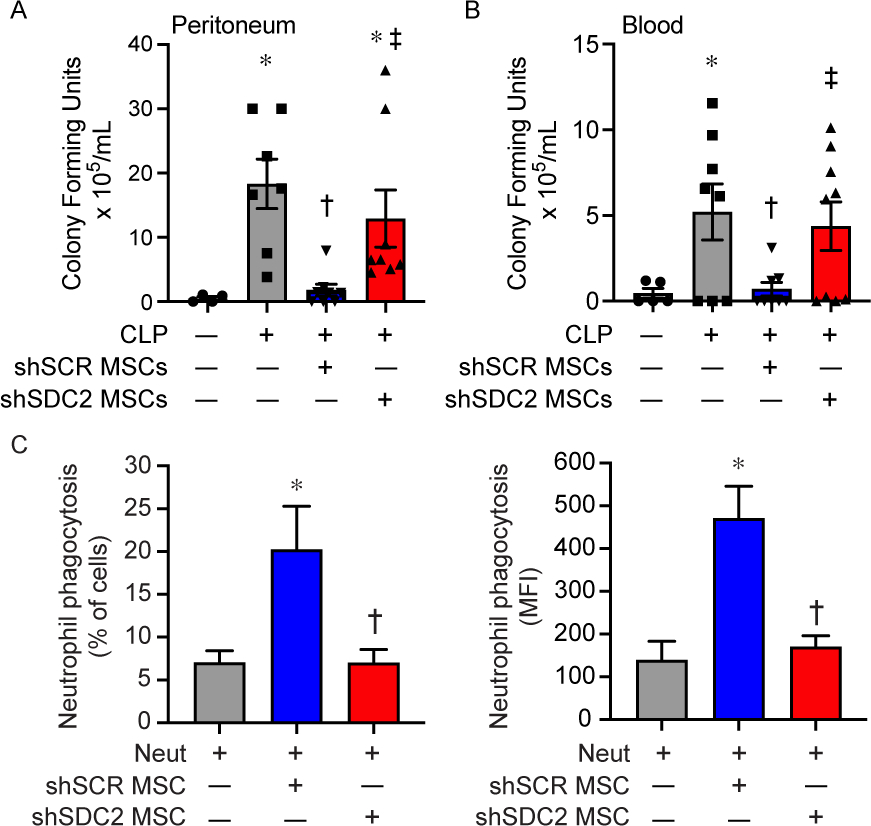
Enhanced bacterial clearance and neutrophil phagocytosis by hMSC is lost after SDC2 silencing. Assessment of bacteria colony forming units was assessed in the peritoneum (A) and blood (B) 48 hours after sham (circles, n=4 in peritoneum, n=5 in blood), PBS+CLP (squares, n=7 in peritoneum, n=8 in blood), shSCR hMSCs+CLP (downward triangle, n=8 in peritoneum and blood), and shSDC2 hMSCs+CLP (upward triangle, n=8 in peritoneum, n=9 in blood). Data are depicted graphically as a mean±SEM. p=0.003 (A) and p=0.0004 (B) with significant comparisons * versus sham, † versus PBS, ‡ versus shSCR hMSCs. C) Isolated neutrophils were incubated with GFP-labeled E. coli in the presence of no hMSCs (gray bars, n=4 in each group), shSCR hMSCs (blue bars, n=3 and 4 respectively), or shSDC2 hMSCs (red bars, n=3 and 4 respectively). Data are presented as percent of neutrophils phagocytizing bacteria (left panel) or as mean fluorescent intensity (MFI) of bacteria taken up by neutrophils (right panel). Data are presented as mean±SEM. p=0.023 for % of neutrophils phagocytizing bacteria, and p=0.0048 for neutrophil MFI of FITC, with significant comparisons * versus no hMSCs, and † versus shSCR hMSCs.
SDC2 contributes to the ability of hMSCs to modulate inflammation in sepsis
The spleen is an organ of functional importance during sepsis, including to help clear bacteria, and it is also susceptible to injury due to the immune response during systemic infection [28]. Thus, we assessed the infiltration of innate immune cells into splenic tissue after CLP, using immunostaining for neutrophils (Ly6G+) and macrophages (CD68+). There was increased infiltration of Ly6G+ neutrophils and CD68+ macrophages in the PBS and the shSDC2 hMSC groups after CLP, compared with the shSCR hMSC group (Figures 4A and 4B). In addition, we measured the mRNA levels of the inflammatory cytokine IL-6, which is important in the pathobiology of sepsis [29, 30], TNFα, and the chemokine MCP-1, at 24 hours after CLP. These mediators are biomarkers of the inflammatory response in sepsis [31]. The level of IL-6 mRNA was significantly higher in the shSDC2 hMSC group compared with the shSCR hMSC group, a level comparable to PBS in spleen, liver, and lung tissues (Figure 5A). A similar pattern of MCP-1 (Figure 5B) and TNFα (data not shown) mRNA levels was seen in the spleen, with elevated levels in the shSDC2 hMSC group compared with the shSCR hMSC group. The levels of MCP-1 and TNFα mRNA had a similar trend in liver and lung tissues, but the changes were not statistically significant.
Figure 4.
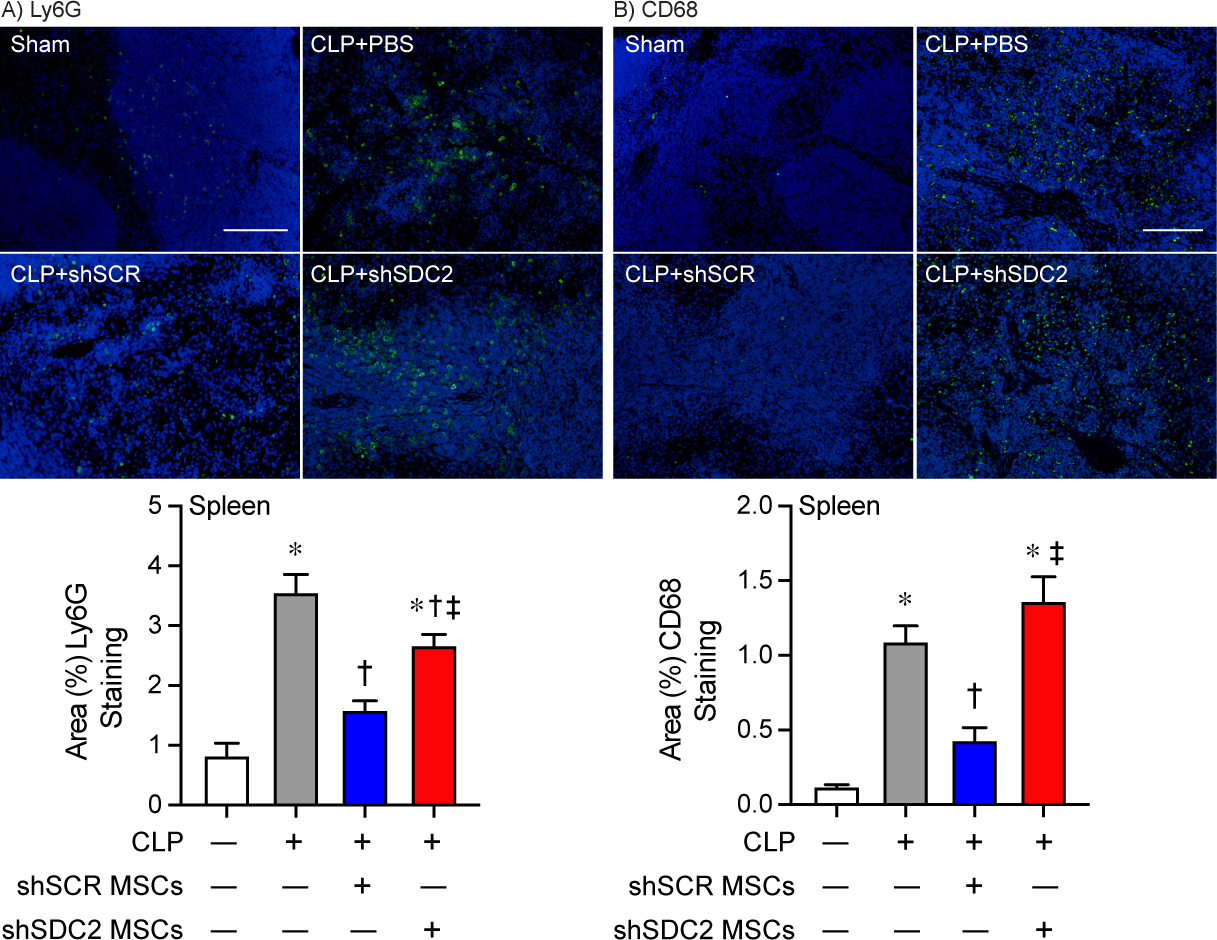
Silencing of SDC2 in hMSCs results in worse tissue Inflammation when administered during sepsis. Mice were subjected to sham or CLP surgery, and spleens were harvested at 48 hours. Immunostaining (green) was performed for neutrophils (Ly6G+, A) and macrophages (CD68+, B). Representative images of the groups are provided in the upper panels. Scale bars represents 100 μm. Mice either underwent Sham (white bars, images n=7 and 11 respectively) or CLP surgery, and septic mice were randomly assigned to receive PBS control (gray bars, images n=11 and 25 respectively), shSCR hMSCs (blue bars, images n=16 and 28 respectively), or shSDC2 hMSCs (red bars, images n=15 and 31 respectively). Quantitative data are presented as % area of staining for Ly6G+ and CD68+ cells, mean±SEM. p<0.0001 for Ly6G+ and CD68+, with significant comparisons * versus sham, † versus PBS, ‡ versus shSCR hMSCs.
Figure 5.
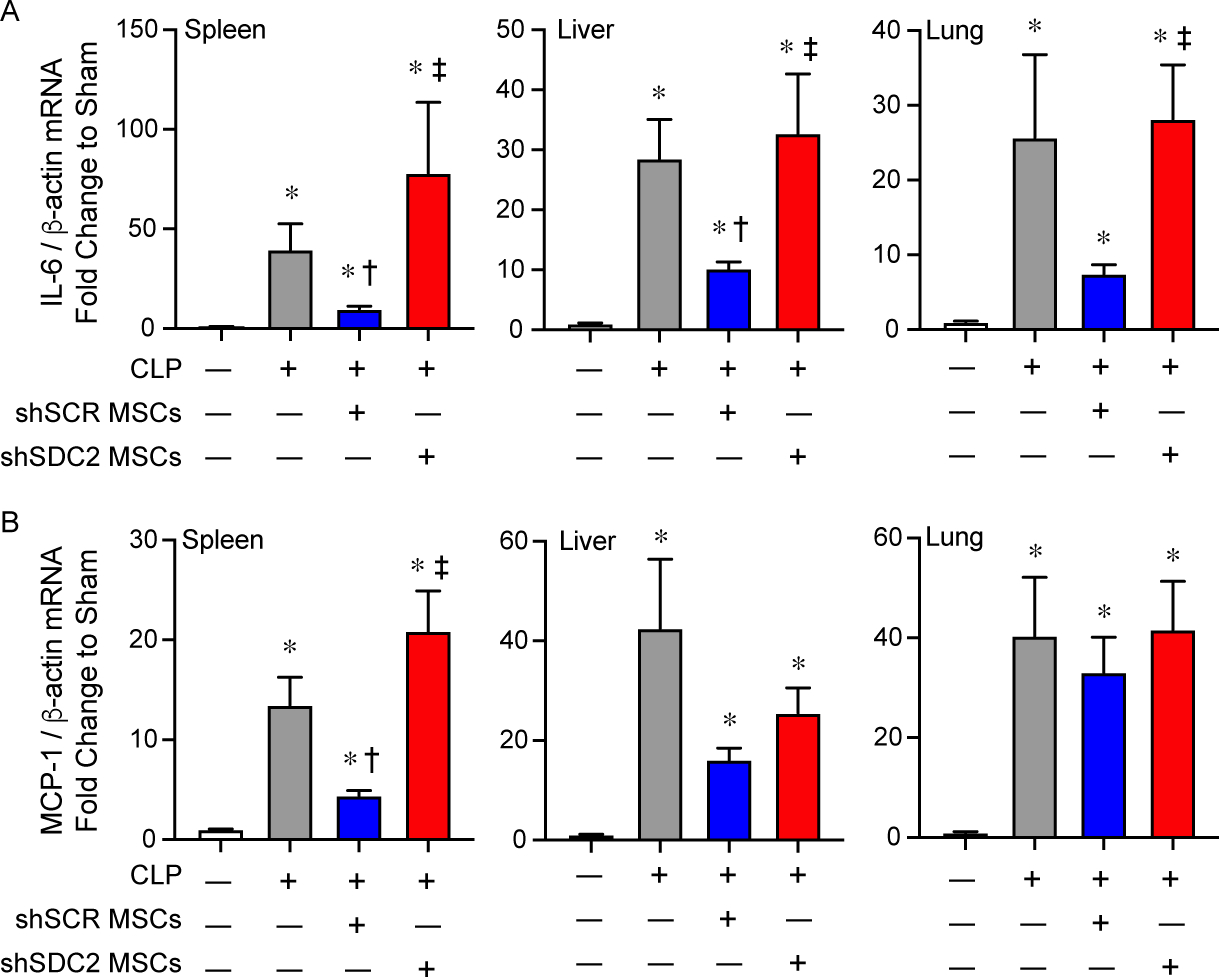
Silencing of SDC2 in hMSCs results in worse tissue expression of pro-inflammatory mediators. RNA was also harvested from spleen, liver, and lung tissue, and qRT-PCR was performed for IL-6 (A) and MCP-1 (B). Mice undergoing CLP were randomly assigned to receive PBS control (gray bars, n=20 in each group), shSCR hMSCs (blue bars, n=20 in each group), or shSDC2 hMSCs (red bars, n=16 for IL-6, n=18 for MCP-1). Data are presented as mRNA levels of IL-6 or MCP-1 normalized for β-actin, as a fold change to sham (white bars), mean±SEM. p<0.0001 with significant comparisons * versus sham, † versus PBS, ‡ versus shSCR hMSCs.
We next assessed the inflammatory response in the peritoneum, the site of initial injury in CLP-induced sepsis. At 48 hours after CLP, the total number of cells in the peritoneal fluid was significantly decreased in the shSCR hMSC group, compared with the shSDC2 hMSC and PBS groups (Figure 6A). Quantification of neutrophils demonstrated an analogous response, with more neutrophils in the shSDC2 hMSC group compared with the shSCR hMSC group (Figure 6B). Finally, we assessed total macrophages (Figure 6C) in the peritoneal fluid. In contrast to the spleen, the peritoneal fluid demonstrated no significant difference in the number of macrophages in mice receiving shSCR hMSC, shSDC2 hMSC, or PBS during CLP. We next further assessed the effect of hMSCs on macrophage subtypes and function.
Figure 6.

Silencing SDC2 in hMSCs results in less efficient resolution of neutrophilic inflammation in the peritoneum when given during CLP-induced sepsis. Total cell counts (A), innate immune neutrophils (B) and macrophages (C) were assessed in peritoneal fluid (PF) of mice undergoing sham or CLP surgery after 48 hours. Mice were randomly assigned to sham (circles/white bars, n=5 in A, n=4 in B and C) or CLP surgery, and received PBS control (− MSCs, squares/gray bars, n=7 in each group), shSCR hMSCs (+, downward triangles/blue bars, n=8 in A and C, n=7 in B), or shSDC2 hMSCs (+, upward triangles/red bars, n=9 in A and C, n=8 in B). Data are presented as cells × 106, mean±SEM. For total cells and neutrophils, p<0.0001 and p=0.0005 respectively, with significant comparisons * versus sham, † versus PBS, ‡ versus shSCR hMSCs. For macrophages, NS=not significant between groups.
SDC2 is important for hMSCs to promote efferocytosis and macrophage polarization
Macrophages and neutrophils work in concert to eliminate pathogens, and after bacterial clearance is initiated, macrophages phagocytize apoptotic neutrophils and cellular debris (efferocytosis), while concomitantly shifting from an M1-like pro-inflammatory phenotype to an M2-like pro-resolution phenotype [2, 3, 32, 33]. To further assess the impact of hMSC-derived SDC2 on macrophage function, we assessed efferocytosis, an important process during the resolution of inflammation [34]. Administration of shSCR hMSCs at 2 and 24 hours after CLP increased the clearance of apoptotic neutrophils by macrophages in the peritoneum at 48 hours (Figure 7A). However, mice receiving shSDC2 hMSCs had a level of efferocytosis similar to mice receiving PBS. We also explored this concept in vitro, utilizing the conditioned medium (CM) of hMSCs and their ability to promote macrophage phagocytosis of apoptotic neutrophils in culture. CM from shSCR hMSCs was able to increase macrophage phagocytosis of apoptotic neutrophils, while shSDC2 CM had significantly reduced efferocytosis (Figure 7B). These data suggest that silencing of SDC2 in hMSCs promotes a loss of efferocytosis, and this process relates to its paracrine actions.
Figure 7.
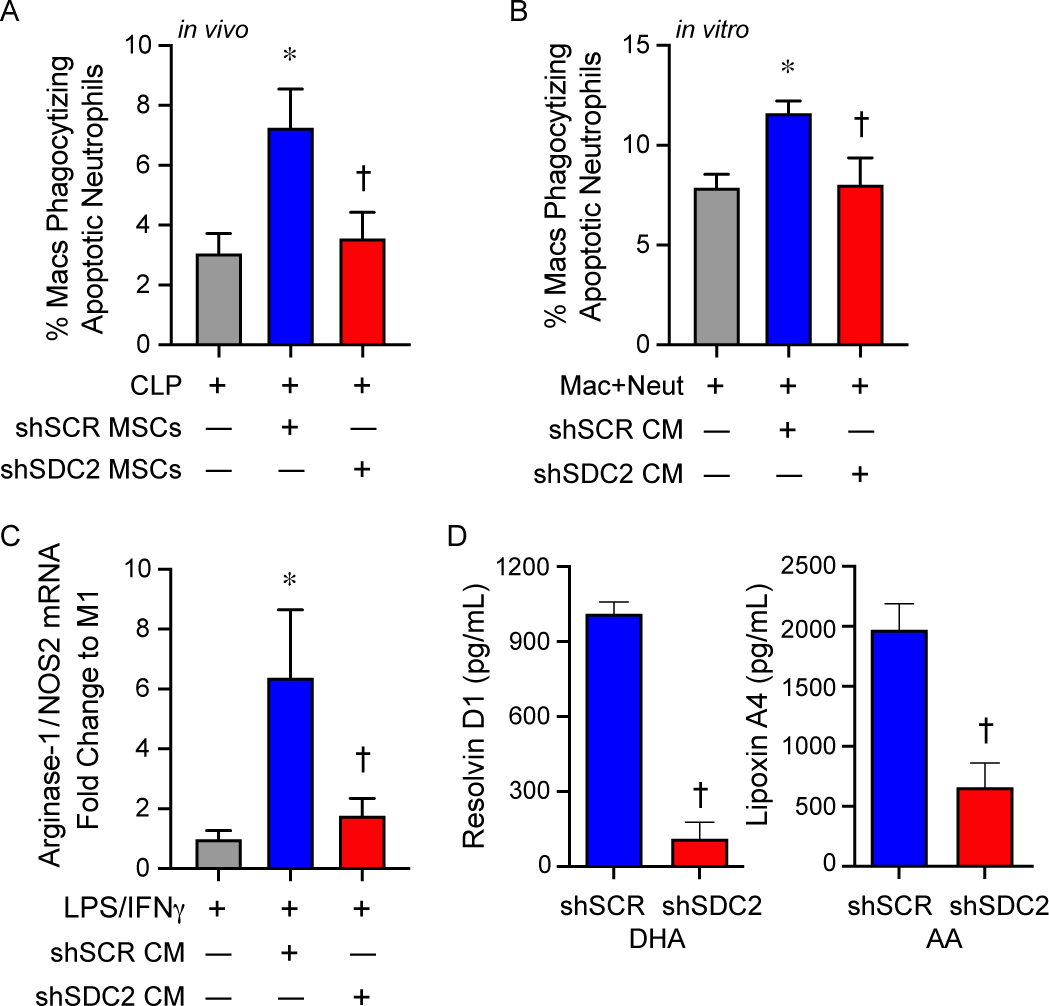
SDC2 is important for the paracrine functions of hMSCs to promote efferocytosis and macrophage polarization to an M2-like phenotype. A) Mice were subjected to CLP surgery, and randomly assigned to receive PBS control (gray bar, n=6), shSCR hMSCs (blue bar, n=7), or shSDC2 hMSCs (red bar, n=6). Efferocytosis was assessed in the peritoneal fluid of septic mice by flow cytometry. Data are presented as the percentage of macrophages phagocytizing apoptotic neutrophils, mean±SEM. p=0.015, with significant comparisons * versus PBS and † versus shSCR hMSC. B) Next, we performed the efferocytosis assay in vitro. Macrophages were exposed to apoptotic neutrophils in the presence of PBS (gray bar, n=5), conditioned medium (CM) from shSCR hMSCs (blue bar, n=5), or CM form shSDC2 hMSCs (red bar, n=5). Data are presented as the percentage of macrophages phagocytizing apoptotic neutrophils, mean ±SEM. p=0.021, with significant comparisons * versus PBS and † versus shSCR hMSC. C) M0 macrophages were stimulated with IFNγ (10 ng/mL) and LPS (10 ng/mL) to induce an M1 phenotype. At the time of LPS/IFNγ stimulation, the cells were exposed to PBS (gray bar, n=4), shSCR CM (blue bar, n=4), or shSDC2 CM (red bar, n=4). Macrophage RNA was harvested, and qRT-PCR performed to assess arginase-1/NOS2 ratio as a marker of macrophage polarization, with an increase consistent with M2-like phenotype. Data are presented as mean±SEM. p=0.0048, with significant comparisons * versus PBS and † versus shSCR hMSC. D) shSCR hMSCs (blue bars, n=5) and shSDC2 hMSCs (red bars, n=5) were exposed to SPM substrates DHA or AA for 24 hours, and then the CM from the cells were analyzed by ELISAs for resolvin D1 (left) or lipoxin A4 (right), respectively. Data are presented as mean±SEM. p<0.0001, with significance comparison † versus CM of shSCR hMSCs.
Next, we assessed whether this effect on efferocytosis by the CM of hMSCs translated into a change in macrophage subtype, from an M1-like to an M2-like phenotype. We took non-activated murine macrophages (M0) and stimulated them with LPS and IFNγ to induce an M1-like pro-inflammatory phenotype, and at the same time added CM from shSCR or shSDC2 hMSC. To identify macrophage subtypes, we used markers of macrophage arginine metabolism to characterize M1-like macrophages (expressing more NOS2) and M2-like macrophages (expressing more arginase-1) [35]. A ratio of arginase-1/NOS2 was used as a biomarker of macrophage subtype. After 48 hours of stimulation with LPS and IFNγ, in the presence or absence of hMSC CM, macrophage RNA was harvested and qRT-PCR performed to assess arginase-1/NOS2 ratio. Figure 7C demonstrated that CM from shSCR hMSCs promoted an M2-like phenotype, with an increased ratio of arginase-1/NOS2, while in the presence of CM from shSDC2 hMSC the arginase-1/NOS2 ratio was comparable to M1-like macrophages (no CM). Specialized pro-resolving lipid mediators (SPMs) are known to orchestrate the resolution of inflammation, involving efferocytosis and polarization to an M2-like pro-resolution macrophage phenotype, and SPMs also have anti-inflammatory properties [36–40]. Resolvin D1 and lipoxin A4, SPMs expressed in MSCs, have been shown to regulate the inflammatory response during models of sepsis and acute lung injury [18, 41]. Interestingly, when we assessed these SPMs in the CM of hMSCs, we found that the production of both resolvin D1 and lipoxin A4 were significantly reduced in shSDC2 MSCs compared with shSCR MSCs (Figure 7D).
SDC2 is important for the ability of hMSC-derived extracellular vesicles (EVs) to promote macrophage polarization and efferocytosis
An important component of the paracrine actions of MSCs is EVs [42]. Thus, we next explored the capacity of EVs derived from shSCR and shSDC2 hMSC to modulate macrophage polarization and efferocytosis to initiate the resolution of inflammation. We harvested EVs from hMSCs as described [43]. The isolation and characterization of EVs were in accordance with the 2018 Minimal Information for Studies of Extracellular Vesicles (MISEV) [44], as outlined by the International Society for Extracellular Vesicles. In the present studies, EVs represented a heterogeneous vesicle population that occupy a diameter of <200 nm, express established EV-associated markers including CD9 and CD81, and adhere to the typical biconcave features of EVs (Figure 8A). Exosomes comprised a subpopulation of EVs. Beyond establishing the presence of known markers for EVs from both shSCR and shSDC2 hMSCs (Figure 8B), we importantly demonstrated that Histone H3 (not released in EVs [45]) was only evident in the cells and not the EVs. As a whole, these data confirm the isolation and purification of EVs from hMSCs. Moreover, SDC2 was present in shSCR EVs (Figure 8C), and was reduced in shSDC2 EVs. We exposed macrophages (stimulated with IFNγ and LPS) to full CM, EVs, or the soluble fraction of CM (EV deficient) from shSCR hMSCs. In the experiments using EVs, a cell equivalent number of EVs from shSCR or shSDC2 hMSCs was administered, unless stated otherwise. Notably, EVs from shSCR hMSCs promoted polarization of macrophages from M1-like to M2-like phenotype (Figure 8D). Although less dramatically, the soluble fraction also induced polarization to an M2-like phenotype. We next assessed the impact of EVs from shSDC2 hMSCs, and shSCR hMSCs, on macrophage polarization and efferocytosis. EVs from shSDC2 hMSCs lost the ability to polarize M1-like to M2-like macrophages, in comparison to EVs from shSCR hMSCs (Figure 8E). Moreover, EVs from shSCR hMSCs were able to increase efferocytosis of apoptotic neutrophils by macrophages (Figure 8F), but after silencing SDC2, this function of EVs from shSDC2 hMSCs was lost.
Figure 8.
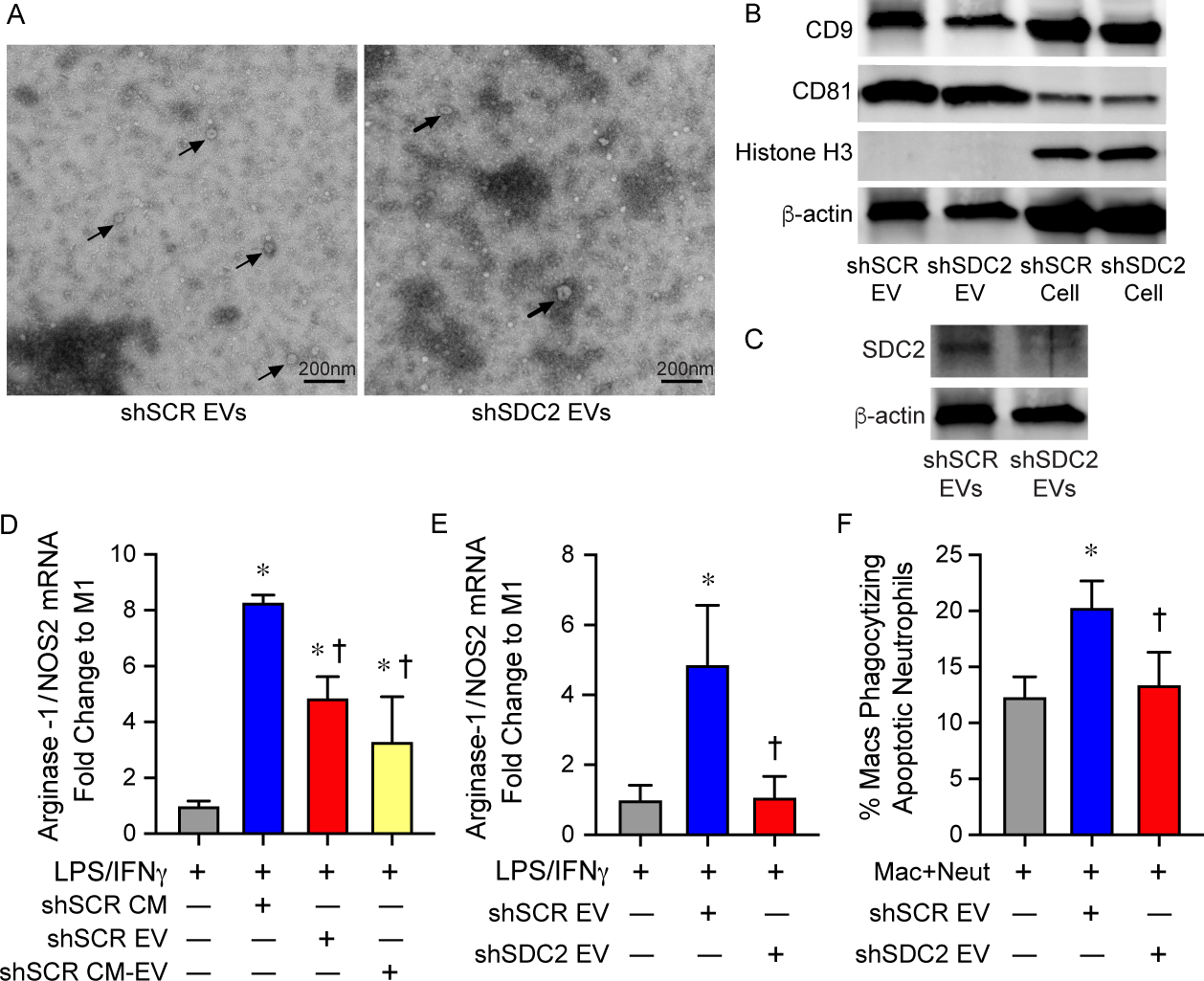
Extracellular vesicles (EVs) secreted by shSDC2 hMSCs lose the ability to promote efferocytosis and macrophage polarization to an M2-like phenotype. A) TEM was performed on EVs harvested from shSCR hMSCs (left panel) and shSDC2 hMSCs (right panel). Arrows point to representative EVs. Scale bar represents 200 nm. B) EVs isolated from shSCR hMSCs and shSDC2 hMSCs were characterized by Western blot analyses, using antibodies to CD9 and CD81 as markers of EVs, and Histone H3 as a negative control of EVs (upper panel). Total cell protein lysate was loaded as a control, and β-actin was used to signify total protein content. C) EVs isolated from shSCR hMSCs and shSDC2 hMSCs were also characterized by Western blot analyses, using an antibody to SDC2 (lower panel). Total cell protein lysate was loaded as a control, and β-actin was used to signify total protein content. D) M0 macrophages were stimulated with IFNγ (10 ng/mL) and LPS (10 ng/mL) to induce an M1 phenotype. At the time of LPS/IFNγ stimulation, the cells were exposed to PBS (gray bar, n=4), shSCR CM (blue bar, n=4), shSCR EV (red bar, n=5) or shSCR CM deplete of EVs (shSCR CM-EV, yellow bar, n=3). Macrophage RNA was harvested, and qRT-PCR was performed to assess the arginase-1/NOS2 ratio as a marker of macrophage polarization. Data are presented as mean±SEM. p<0.0001, with significant comparisons * versus PBS and † versus shSCR hMSC. E) M0 macrophages were stimulated with IFNγ (10 ng/mL) and LPS (10 ng/mL) to induce an M1 phenotype. At the time of LPS/IFNγ stimulation, the cells were exposed to PBS (gray bar, n=5), shSCR EV (blue bar, n=4), or shSDC2 EV (red bar, n=5). Macrophage RNA was harvested, and qRT-PCR was performed to assess the arginase-1/NOS2 ratio as a marker of macrophage polarization. Data are presented as mean±SEM. p=0.005, with significant comparisons * versus PBS and † versus shSCR hMSC. F) Macrophages were exposed to apoptotic neutrophils in the presence of PBS (gray bar, n=7), shSCR EV (blue bar, n=7), or shSDC2 EV (red bar, n=7). Data are presented as the percentage of macrophages phagocytizing apoptotic neutrophils, mean ±SEM. p=0.0027, with significant comparisons * versus PBS and † versus shSCR hMSC.
Silencing of SDC2 in hMSCs results in decreased EV production
To assess the impact of SDC2 on EV production, we next performed nanoparticle tracking analysis (NTA). EV production was dramatically reduced in hMSCs silenced for SDC2 (Figure 9A). Specifically, these data demonstrated that the EVs released from shSCR hMSCs were >2.5-fold greater than that of shSDC2 hMSCs. To determine whether fewer EVs are contributing to the alteration in shSDC2 paracrine function, we repeated the experiment shown in Figure 8E, but instead of using EVs from an equivalent number of shSCR and shSDC2 hMSCs, we normalized to the number of EVs. As demonstrated in Figure 9B, when we used an analogous number of EVs, both shSDC2 (1.5×106 cell equivalents) and shSCR (0.5×106 cell equivalents) hMSC EVs were able to polarize cells from an M1-like to an M2-like phenotype to a comparable degree. To further investigate EVs in shSDC2 hMSCs, we performed immunoblotting of EVs to assess key EV-associated markers, using β-actin expression for normalization. The expression of Syntenin, Tsg101, CD63, and ALIX were all decreased in EVs of shSDC2 hMSCs compared with shSCR hMSCs (Figure 9C). Taken together, these data suggest that decreased EV production, and fewer EVs in the CM of shSDC2 hMSCs, compared with shSCR hMSCs, contributed to the aberrant paracrine function.
Figure 9.

Silencing SDC2 in hMSC reduces EV production. A) EVs were counted by nanoparticle tracking analysis after harvesting EVs from an equivalent number of shSCR hMSCs (blue line, n=3) and shSDC2 hMSCs (red line, n=3). Data are presented as the mean of the experiments, particles / mL over the size distribution / nm, assessed by the area under the curve. p=0.0005, with significant difference between shSCR and shSDC2 EVs. B) M0 macrophages were stimulated with IFNγ (10 ng/mL) and LPS (10 ng/mL) to induce an M1 phenotype. At the time of LPS/IFNγ stimulation, the cells were exposed to PBS (gray bar, n=7), shSCR EV (blue bar, n=8), or shSDC2 EV (number normalized to shSCR EV, red bar, n=7). Macrophage RNA was harvested, and qRT-PCR was performed to assess the arginase-1/NOS2 ratio as a marker of macrophage polarization. Data are presented as mean±SEM. p=0.0008, with significant comparisons * versus PBS. NS=not significant. C) EVs isolated from shSCR hMSCs and shSDC2 hMSCs were characterized by Western blot analyses (left panels), using antibodies to Syntenin (n=4), Tsg101 (n=4), CD63 (n=4), and ALIX (n=3), markers of exosome biogenesis. β-actin was used to normalize for total protein content. The Western blot data were quantified protein/β-actin, fold change to shSCR (right panels), and presented as mean ±SEM. p=0.0017 Syntenin, p=0.0078 Tsg101, p=0.016 CD63, and p=0.031 ALIX. Significant comparison † versus shSCR.
DISCUSSION
SDC family members are transmembrane proteoglycans, with heparan sulfate extracellular domains that can be released from the cell membrane by sheddase enzymes [26, 46]. Both the expression levels and the proteolytic cleavage of extracellular domains of SDCs are known to be upregulated during inflammatory responses [26]. Circulating levels of soluble SDC1 and SDC3 have been reported to be increased during critical illnesses (including sepsis) compared with control patients [47]. By regulating leukocyte extravasation and cytokine/chemokine function, SDCs have a role in the regulation of the inflammatory response, from leukocyte recruitment to the resolution of inflammation [26]. Recently, using SDC2 as a marker on the surface of hMSCs, investigators identified a population of cells that was beneficial against bacterial pneumonia, and ventilator-induced lung injury [20, 21]. This population of SDC2+ cells was as effective as standard heterogeneous hMSCs to decrease severity, and improve recovery, from these acute lung injury models. Administration of SDC2+ hMSCs during experimental sepsis was also reported to be advantageous compared with a vehicle control [48]. Given that human MSCs expressing SDC2 on their cell surface were comparable to, but not more effective than the heterogeneous MSC population, we sought to further elucidate the importance of endogenous SDC2 on hMSC function.
SDCs in mammals are expressed in cell and tissue specific patterns, and SDC2 is known to be expressed in mesenchymal cells [49]. Interestingly, we demonstrated that the expression of SDC2 is more than 6-fold higher in bone marrow-derived hMSCs than in control mesenchymal fibroblasts, and SDC2 is much more highly expressed than SDC1, SDC3, or SDC4 in hMSCs. Silencing of SDC2 resulted in a loss of survival benefit, more tissue cell death, and less bacterial clearance when administered after the onset of polymicrobial sepsis compared with shSCR hMSCs. While the administration of shSCR hMSCs resulted in a decrease in the infiltration of neutrophils (Ly6G+) and macrophages (CD68+) into splenic tissue during sepsis, this decrease in innate immune cell infiltration was not evident in mice receiving shSDC2 hMSCs. Evaluation of the inflammatory response in the peritoneum (site of injury) revealed a similar decrease in neutrophils in mice receiving shSCR hMSCs after the onset of sepsis, and this response was lost in mice receiving shSDC2 hMSCs.
With evidence that the inflammatory response was not resolving as efficiently after receiving shSDC2 hMSCs, compared with shSCR hMSCs, we further assessed the phagocytosis of apoptotic neutrophils by macrophages (efferocytosis), a critical process during the resolution of inflammation [34, 50]. Here we found that efferocytosis was significantly greater in mice receiving shSCR hMSCs than mice receiving shSDC2 hMSCs. The clearance of apoptotic neutrophils by macrophages is associated with a shift from an M1-like pro-inflammatory to an M2-like pro-resolution phenotype [2, 3, 33]. Thus, even though the overall number of peritoneal macrophages was not different between the groups, we hypothesized a shift in macrophage phenotype. The CM from shSCR hMSCs was able to promote the conversion of M1-like macrophages to M2-like macrophages, and this effect was not present when using the CM of shSDC2 hMSCs. Furthermore, we demonstrate for the first time that the production of resolvin D1 and lipoxin A4, SPMs known to promote efferocytosis and polarization of macrophages to a pro-resolution phenotype, were decreased in the CM of shSDC2 compared with shSCR hMSCs. SPMs are dependent on lipoxygenase (LOX) enzymes for their biosynthesis [50], and exposure of human and mouse MSCs to a LOX inhibitor (baicalein), or silencing of the 5-LOX and 12/15-LOX enzymes in mouse MSCs resulted in a blunted effect on neutrophil phagocytosis of bacteria and a loss of survival benefit during peritoneal sepsis [18]. Thus, the defect in SPM production has important consequences on the function of shSDC2 hMSCs during sepsis.
The ability of MSCs to modulate an inflammatory response and to protect tissue from injury, or to promote tissue repair, is largely via their paracrine actions [6, 51, 52]. An important component of the paracrine actions of MSCs occurs through EVs, and MSC-derived EVs have been shown to be beneficial in sepsis [52]. Uptake of EVs by macrophages is able to induce a switch from the M1-like to M2-like phenotype [53, 54], and this modulation of macrophages has been shown to be important in experimental models of infection, inflammatory organ injury, acute lung injury, and pulmonary hypertension [52, 54, 55]. Interestingly, biodistribution of EVs accumulate mainly in organs such as the spleen, liver, and lung [56], organs in which the inflammatory mediators IL-6 and MCP-1 were decreased after administration of shSCR hMSCs, compared with shSCD2 hMSC. It is uncertain whether there is a potential for EV homing to specific organs during disease, although the route of administration influences the biodistribution [56]. Moreover, Mansouri and colleagues also demonstrated that MSC-derived exosomes/EVs were able to reprogram myeloid cells in the bone marrow, leading to lower proinflammatory monocytes in the lung after administration of bleomycin [57]. Thus, the effect of EVs on bone marrow-derived myeloid cells may also provide an immunomodulatory mechanism by which EVs have a systemic response.
In the present study, the paracrine actions of shSCR hMSCs to induce the conversion of M1-like to M2-like macrophages was in part related to EVs in the CM, along with soluble factors. However, EVs harvested from shSDC2 hMSCs (from an equivalent number of cells as shSCR hMSCs) failed to induce a change in macrophage phenotype. Additionally, the EVs from shSDC2 hMSCs were not able to increase the phagocytosis of apoptotic neutrophils, as seen with shSCR EVs.
In an effort to understand the mechanism behind this abnormal response of EVs from shSDC2 hMSCs, we assessed the number of EVs present in the CM of an equivalent number of cells in each groups. We noted there was more than ~2.5-fold fewer EVs in the CM of shSDC2 hMSCs compared with shSCR hMSCs, and when we normalized for the number of EVs, the paracrine actions of shSDC2 EVs was analogous to shSCR EVs in converting M1-like to M2-like macrophages. Previously it has been shown that the cytosolic adaptor syntenin connects to ALIX via the heparan sulfate proteoglycans of SDC1 and SDC4, and then interacts with other endosomal-sorting complex required for transport (ESCRT) machinery to support membrane budding and biogenesis of exosomes (subpopulation of EVs) [58, 59]. Interestingly, we found that silencing of SDC2 in hMSCs resulted in decreased expression of syntenin, ALIX, Tsg101, and CD63, established EV-associated markers. Taken together, these data support the concept that SDC2 is critical for the production of EVs in hMSCs, and we hypothesize that decreased numbers of EVs from shSDC2 hMSCs may contribute to decreased efferocytosis, loss of macrophage polarization to the M2-like phenotype, and altered resolution of inflammation. Since SDCs 1 and 4 are known to influence the cargo of EVs [60–62], we cannot exclude the impact of SDC2 on the composition of MSC-derived EVs and this influence on sepsis pathobiology.
In conclusion, this study demonstrates an important role for endogenous SDC2 on bone marrow-derived hMSC function (both cellular and paracrine actions) during experimental sepsis. Beyond the ability of hMSC to promote bacterial clearance by neutrophils, SDC2 is important to allow prompt resolution of inflammation resulting in less tissue injury and improved survival. The ability of hMSCs to enhance the clearance of apoptotic neutrophils from the peritoneum, and transition of macrophages from an M1-like pro-inflammatory phenotype to an M2-like pro-resolution phenotype, is lost after silencing of SDC2 in the cells. In addition, the paracrine actions of hMSC-derived EVs contributes to efferocytosis and M2-like polarization of macrophages in vitro, and these actions are related in part to the impact of SDC2 on cellular EV production. Collectively, these data advance our understanding of how SDC2 promotes hMSC function during experimental polymicrobial sepsis.
MATERIALS AND METHODS
Cells.
Primary human bone marrow-derived mesenchymal stromal cells (hMSC) were obtained from the Institute for Regenerative Medicine, Texas A&M Health Science Center. hMSCs were cultured in MEMα (Gibco, Gaithersburg, MD) supplemented with 20% fetal bovine serum (FBS), and used at passage 5–6. Control mesenchymal cells were human dermal fibroblasts.
Lentivirus silencing of SDC2 in hMSC.
The vector for SDC2 (shSDC2), target sequence 5’-GTCATTGCTGGTGGAGTTATT-3’ (TRCN0000298635), and scrambled control (shSCR) construct (SHC016) were purchased from Sigma-Aldrich (St. Louis, MO). For production of lentiviral particles, a second-generation packaging mix and LentiFectin™ Transfection reagent (Applied Biological Materials, Richmond, BC) was used. shSDC2 and shSCR lentiviral particles were added to hMSCs for 24 hours, followed by selection using puromycin (10 ug/ml) as described [18].
Assessment of SDC2 silencing by quantitative real-time PCR (qRT-PCR) and flow cytometry.
Total RNA was extracted from shSDC2 and shSCR infected hMSCs, and qRT-PCR was performed as described [63, 64] using the human primers of SDC2 forward 5’-CAACATCTCGACCACTTCCA-3’ and reverse 5’-TGGGTCCATTTTCCTTTCTG-3’. qRT-PCR of β-actin was used for normalization of SDC2 expression by the comparative Ct method using primers of human β-actin forward 5’-AGGCACCAGGGCGTGAT-3’ and reverse 5’- GCCCACATAGGAATCCTTCTGAC-3’. Cells were harvested after silencing and flow cytometry was performed, using the CD362(SDC2)-PE antibody (Table 1). The cells were then assessed using a BD FACS Canto II, and analyzed by FlowJo software.
Table 1:
Antibodies used for flow cytometry, Western blot analyses, and immunofluorescent staining.
| Flow cytometry | ||||
| Target | Company | Catalog number | Clone | Fluorophore |
| Syndecan-2 | MACS | 130–107–480 | REA468 | PE |
| CD90 | Biolegend | 328115 | 5E10 | AF647 |
| CD73 | Biolegend | 344015 | AD2 | FITC |
| CD105 | Biolegend | 323207 | 43A3 | APC |
| HLA DR | Biolegend | 307609 | L243 | APC |
| Ly6G | Biolegend | 127613 | 1A8 | APC |
| Ly6G | Biolegend | 127606 | 1A8 | FITC |
| CD11b | BD bioscience | 557397 | M1/70 | PE |
| F4/80 | Biolegend | 123116 | BM8 | APC |
| Western blot analyses | ||||
| Target | Company | Catalog number | Isotype | Host species |
| Syndecan-2 | Abcam | ab191062 | IgG | Rabbit |
| Alix | Abcam | ab117600 | IgG1 | Mouse |
| Tsg101 | Abcam | ab30871 | IgG | Rabbit |
| Syntenin-1 | Santa Cruz | sc-515538 | IgG2a | Mouse |
| CD63 | Santa Cruz | sc-5275 | IgG1 | Mouse |
| CD81 | Santa Cruz | sc-166029 | IgG2b | Mouse |
| CD9 | Santa Cruz | sc-13118 | IgG1 | Mouse |
| β-actin | Santa Cruz | sc-47778 | IgG1 | Mouse |
| Histone H3 | Abcam | ab1791 | IgG | Rabbit |
| Immunofluorescent staining | ||||
| Target | Company | Catalog number | Isotype | Host species |
| Ly6G | Biolegend | 127602 | IgG2a, K | Rat |
| CD68 | Abcam | ab125212 | IgG | Rabbit |
Growth curve.
72 hours after puromycin selection, shSCR and shSDC2 hMSCs were plated in 35 mm dishes at a density of 2×104 cells/35mm dish (day 0). The medium was changed every other day. The cell number was counted daily, from day 1 to day 5.
Cecal ligation and puncture (CLP).
C57BL/6 male mice, 6–8 weeks of age, underwent CLP as described [17–19, 65], with two-thirds of the cecum ligated and punctured with two 21-gauge holes. In sham experiments, surgery was performed, without CLP. The mice received hMSCs (5 × 105 cells/200 μL PBS) or vehicle (PBS 200 μL) via intravenous administration at 2 hours after CLP, and then again at 24 hours after CLP (5 × 105 cells/200 μL PBS or PBS 200 μL only). The mice were sacrificed at 24–48 hours after CLP, or they were monitored over 7 days to determine survival.
Bacteria clearance.
Peritoneal fluid and blood were drawn 48 hours after CLP. Serial dilutions of whole blood and peritoneal fluid were performed and then incubated overnight at 37°C on LB agar plates. CFUs of bacteria were counted and calculated as described [17].
Flow cytometry and efferocytosis of peritoneal cells.
Peritoneal lavage was performed 48 hours after CLP or sham surgery. Cells from the recovered fluid were stained with antibodies targeting Ly6G-APC and CD11b-PE to identify neutrophils, and F4/80-APC to identify macrophages [19]. For in vivo efferocytosis [18], peritoneal cells were stained with F4/80-APC antibody. After washing with 1x PBS, the cells were permeabilized with Cytofix/Cytoperm (BD Biosciences, Billerica, MA) and stained intracellularly with Ly6G-FITC antibody. The cell population positive for both F4/80-APC and Ly6G-FITC by flow cytometry was identified as macrophages phagocytizing apoptotic neutrophils (efferocytosis). The antibodies used for flow cytometry are detailed in Table 1.
Histology and immunohistochemistry.
Mice were sacrificed 48 hours following CLP or sham surgery, and organs were harvested and fixed in 10% formalin, processed, embedded in paraffin, and sectioned (5 μm). Tissue sections were assessed by hematoxylin and eosin stain, or stained for apoptotic cells using ApoAlert DNA Fragmentation Assay Kit (Clontech, Mountain View, CA). Tissues were also immunostained with Ly6G and CD68 antibodies (Table 1) for assessment of neutrophil and macrophage infiltration. The area of positively stained cells was calculated per 20X objective using ImageJ Software or Adobe Photoshop respectively, and numerous random fields were assessed per tissue section.
Assessment of organ inflammatory mediators by qRT-PCR.
The liver, spleen, and lung were harvest 24 hours after CLP or sham surgery. Total RNA was extracted and qRT-PCR performed [63, 64]. The primers used to assess inflammation were mouse IL-6 forward 5’-ACAAGTCGGAGGCTTAATTACACA T-3’ and reverse 5’-TTGCCATTGCACAACTCTTTT C-3’, and mouse MCP-1 forward 5’-ACTGAAGCCAGCTCTCTCTTCCTC-3’ and reverse 5’-TTCCTTGGGGTCAGCACAGAC-3’. Mouse β-actin was used to normalize gene expression, using primers for mouse β-actin forward 5’-ACCAACTGGGACGATATGGAGAAGA-3’ and reverse 5’-TACGACCAGAGGCATACAGGGACAA-3’.
Preparation of hMSC conditioned medium (CM).
hMSCs were cultured to 80–90% confluence, washed with PBS, and replenished with supplement-free MEMα medium. After 24 hours, the CM was collected, and centrifuged at 2000 rpm for 5 minutes to remove cell debris. The CM was then concentrated using an Amicon Ultra-4 centrifugal filter units with a 3-kDa cut-off (Millipore, Billerica, MA). Aliquots of the concentrated hMSC CM were then kept at −80°C until they were used.
Isolation of hMSC extracellular vesicles (EVs).
Growth medium of hMSCs (MEMα plus 20% FBS) was subjected to ultracentrifugation (100,000 × g for 2 hours at 4°C) to deplete EVs. The EV-depleted medium was then added to hMSCs for 36 hours, collected, and HEPES solution (1M Sigma, pH 7.4) was added as 1:40 dilution for a final concentration of 25 mM. The supernatant was then spun at 300 × g for 10 minutes at 4°C. The supernatant was again collected, filtered (0.22 μm), and spun at 2,000 g for 10 minutes at 4°C. Next, the supernatant was subjected to ultracentrifugation at 100,000 × g for 90 minutes at 4°C, and the pellet was collected. Finally, this pellet was resuspended in cold PBS and subjected to a final ultracentrifugation step (100,000 g for 90 minutes at 4°C) to isolate EVs for use in the experiments [43].
Transmission electron microscopy (TEM).
EVs were assessed morphologically by TEM. An aliquot of EVs were absorbed to a formvar/carbon grid, stained with 2% uranyl acetate, and visualized on a JEOL 1200EX TEM as previously described [66].
Nanoparticle tracking analysis (NTA).
EVs from 1×106 hMSCs were harvested as previously described, and then diluted in 100 μl of PBS. The size and concentration of the EVs were then determined using NTA (NanoSight LM10 system, Malvern Instruments, Westborough, MA) as described previously [66, 67].
Western blotting of EVs.
EVs from shSCR and shSDC2 hMSCs were lysed in 1X RIPA buffer (Cell Signaling, Danvers, MA) and 1X mini protease inhibitor cocktail (cOmplete™). After adding Laemmli’s SDS-Sample Buffer (6X, Boston BioProducts, Ashland, MA), the lysed EVs were boiled at 100°C for 5 minutes and then equal protein concentration was electrophoresed on 4–20% Mini-PROTEIN TGX Gels (Bio-Rad Laboratories, Hercules, CA). Antibodies for blotting (SDC2, ALIX, Tsg101, Syntenin-1, Histone H3, CD63, CD81, CD9, and β-actin) are detailed in Table 1. Protein expression was assessed using Image J software.
Isolation of murine macrophages and neutrophils.
Mice were given an intraperitoneal injection of Bio-Gel P100 polyacrylamide beads (2% solution, Bio-Rad Laboratories, Hercules, CA) [17]. For harvesting neutrophils, after 16–17 hours, the mice were anesthetized and 10 ml of sterile PBS was used to lavage the peritoneal cavity, and cells were washed and filtered through a 40 μm nylon mesh. Macrophages were harvested 5 days after injection of the Bio-Gel P100 polyacrylamide beads in a similar manner [18].
Macrophage polarization assay.
Murine macrophages were seeded at 2×106 cells/60mm dish in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% FBS, and after 2 hours the attached cells were M0 macrophages. Interferon (IFN)γ 10 ng/mL and E. coli LPS 10ng/mL was added to each dish to induce M1 macrophage polarization. CM or EVs equivalents from 5×105 hMSCs, PBS, or recombinant human syndecan-2 (R&D system, Minneapolis, MN – 500 ng/mL [24]) was added to each dish. The cells were cultured for 48 hours, and RNA was then extracted from the macrophages, and the expression of arginase-1 and nitric oxide synthase (NOS)2 was assessed by qRT-PCR. The primers used for mouse arginase-1 were forward 5’-ATGGAAGAGACCTTCAGCTAC-3’ and reverse 5’-GCTGTCTTCCCAAGAGTTGGG-3’. The primers used for mouse NOS2 were forward 5’-GCCACCAACAATGGCAACA-3’ and reverse 5’-CGTACCGGATGAGCTGTGAATT-3’.
Efferocytosis assay in vitro.
Murine macrophages were harvested as described [18] and seeded at 2×106cells per 60 mm dish for 2 hours. The medium and unattached cells were removed, and CM or EVs from 5×105 hMSC equivalents was added to the macrophages for another 2 hours. Finally, 4×106 apoptotic neutrophils (induced by overnight culture) were added to each dish, incubated for 1 hour, and then harvested for flow cytometry. F4/80 and Ly6G antibodies were used to label macrophages and neutrophils as described [18].
Enzyme linked immunosorbent assay (ELISA) for human resolvin D1 and lipoxin A4.
shSCR and shSDC2 hMSCs were plated on a 24 well dish (50,000 cells per well). The cells were placed in Hanks Balanced Salt Solution (HBSS) supplemented with 0.1% FBS, plus substrate docosahexaenoic acid (DHA, 10 μM)) or arachidonic acid (AA, 10 μM). After 24 hours, human resolvin D1 and lipoxin A4 were assessed by ELISA kits from Cayman Chemicals (Ann Arbor, MI), and performed as suggested by the manufacturer.
Animals.
Studies using mice were carried out in accordance with the Public Health Service policy on the humane care and use of laboratory animals, and approved by the Institutional Animal Care and Use Committee (IACUC) of Brigham and Women’s Hospital.
Statistical Analysis.
For comparisons between two groups, we used Student’s unpaired t test. For EV particles / ml, the area under the curve was assess by Student’s unpaired t test. For analysis of more than two groups, one-way or two-way analysis of variance (ANOVA) was performed. When data were not normally distributed, non-parametric analyses were performed using Kruskal-Wallis testing. Comparisons of mortality were made by analyzing Kaplan-Meier survival curves, and then log-rank test to assess for differences in survival. Statistical significance was accepted at p<0.05.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01GM118456 and R01GM136804 (to M. A. Perrella), U01AI38318 (to M. A. Perrella and J. A. Lederer), K08GM126313 (to S. Ghanta), R01HL146128 and R21AI134025 (to S. Kourembanas), 1K99HL146986 (to G.R. Willis), and T32HL007633 (to S. Ghanta and J. Ng).
Abbreviations:
- MSCs
mesenchymal stromal cells
- hMSCs
human mesenchymal stromal cells
- SDC
syndecan
- SDC2
syndecan-2
- shSDC2
silenced SDC2 short hairpin construct
- shSCR
scrambled control short hairpin construct
- qRT-PCR
quantitative real-time PCR
- CLP
cecal ligation and puncture
- PBS
phosphate buffered saline
- IL-6
interleukin-6
- MCP-1
monocyte chemoattractant protein-1
- TNFα
tumor necrosis factor alpha
- IFNγ
interferon gamma
- LPS
lipopolysaccharide
- CM
conditioned medium
- SPMs
specialized pro-resolving lipid mediators
- LOX
lipoxygenase
- DHA
docosahexaenoic acid
- AA
arachidonic acid
- EVs
extracellular vesicles
- TEM
transmission electron microscopy
- NTA
nanoparticle tracking analysis
- EMSA
enzyme linked immunosorbent assay
- TUNEL
terminal deoxynucleotidetransferase–mediated dUTP nick end-labeling
Footnotes
Conflicts of Interest: The authors declare no conflicting interests.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL & Angus DC (2016) The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3), JAMA. 315, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prame Kumar K, Nicholls AJ & Wong CHY (2018) Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease, Cell Tissue Res. 371, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silva MT (2011) Macrophage phagocytosis of neutrophils at inflammatory/infectious foci: a cooperative mechanism in the control of infection and infectious inflammation, J Leukoc Biol. 89, 675–683. [DOI] [PubMed] [Google Scholar]
- 4.McIntyre LA, Stewart DJ, Mei SHJ, Courtman D, Watpool I, Granton J, Marshall J, Dos Santos C, Walley KR, Winston BW, Schlosser K & Fergusson DA (2017) Cellular Immunotherapy for Septic Shock (CISS): A Phase I Clinical Trial, Am J Respir Crit Care Med. 197, 337–347. [DOI] [PubMed] [Google Scholar]
- 5.Kusadasi N & Groeneveld AB (2013) A perspective on mesenchymal stromal cell transplantation in the treatment of sepsis, Shock. 40, 352–357. [DOI] [PubMed] [Google Scholar]
- 6.Walter J, Ware LB & Matthay MA (2014) Mesenchymal stem cells: mechanisms of potential therapeutic benefit in ARDS and sepsis, Lancet Respir Med. 2, 1016–1026. [DOI] [PubMed] [Google Scholar]
- 7.Wannemuehler TJ, Manukyan MC, Brewster BD, Rouch J, Poynter JA, Wang Y & Meldrum DR (2011) Advances in Mesenchymal Stem Cell Research in Sepsis, J Surg Res. 173, 113–126. [DOI] [PubMed] [Google Scholar]
- 8.Keane C, Jerkic M & Laffey JG (2017) Stem Cell-based Therapies for Sepsis, Anesthesiology. 127, 1017–1034. [DOI] [PubMed] [Google Scholar]
- 9.Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF & Keiliss-Borok IV (1974) Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo, Transplantation. 17, 331–340. [DOI] [PubMed] [Google Scholar]
- 10.Ankrum JA, Ong JF & Karp JM (2014) Mesenchymal stem cells: immune evasive, not immune privileged, Nat Biotechnol. 32, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang CL, Sung PH, Chen KH, Shao PL, Yang CC, Cheng BC, Lin KC, Chen CH, Chai HT, Chang HW, Yip HK & Chen HH (2018) Adipose-derived mesenchymal stem cell-derived exosomes alleviate overwhelming systemic inflammatory reaction and organ damage and improve outcome in rat sepsis syndrome, Am J Transl Res. 10, 1053–1070. [PMC free article] [PubMed] [Google Scholar]
- 12.Keating A (2012) Mesenchymal stromal cells: new directions, Cell Stem Cell. 10, 709–716. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Rey E, Anderson P, Gonzalez MA, Rico L, Buscher D & Delgado M (2009) Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis, Gut. 58, 929–939. [DOI] [PubMed] [Google Scholar]
- 14.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA & Mezey E (2009) Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production, Nat Med. 15, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC & Stewart DJ (2010) Mesenchymal Stem Cells Reduce Inflammation while Enhancing Bacterial Clearance and Improving Survival in Sepsis, Am J Respir Crit Care Med. 182, 1047–1057. [DOI] [PubMed] [Google Scholar]
- 16.Krasnodembskaya A, Samarani G, Song Y, Zhuo H, Su X, Lee JW, Gupta N, Petrini M & Matthay MA (2012) Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes, Am J Physiol Lung Cell Mol Physiol. 302, L1003–L1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall SR, Tsoyi K, Ith B, Padera RF Jr., Lederer JA, Wang Z, Liu X & Perrella MA (2012) Mesenchymal Stromal Cells Improve Survival During Sepsis in the Absence of Heme Oxygenase-1: The Importance of Neutrophils, Stem Cells. 31, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsoyi K, Hall SR, Dalli J, Colas RA, Ghanta S, Ith B, Coronata A, Fredenburgh LE, Baron RM, Choi AM, Serhan CN, Liu X & Perrella MA (2016) Carbon Monoxide Improves Efficacy of Mesenchymal Stromal Cells During Sepsis by Production of Specialized Proresolving Lipid Mediators, Crit Care Med. 44, e1236–e1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon MY, Ghanta S, Ng J, Tsoyi K, Lederer JA, Bronson RT, El-Chemaly S, Chung SW, Liu X & Perrella MA (2020) Expression of Stromal Cell-Derived Factor-1 by Mesenchymal Stromal Cells Impacts Neutrophil Function During Sepsis, Crit Care Med. 48, e409–e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masterson C, Devaney J, Horie S, O’Flynn L, Deedigan L, Elliman S, Barry F, O’Brien T, O’Toole D & Laffey JG (2018) Syndecan-2-positive, Bone Marrow-derived Human Mesenchymal Stromal Cells Attenuate Bacterial-induced Acute Lung Injury and Enhance Resolution of Ventilator-induced Lung Injury in Rats, Anesthesiology. 129, 502–516. [DOI] [PubMed] [Google Scholar]
- 21.Horie S, Masterson C, Brady J, Loftus P, Horan E, O’Flynn L, Elliman S, Barry F, O’Brien T, Laffey JG & O’Toole D (2020) Umbilical cord-derived CD362(+) mesenchymal stromal cells for E. coli pneumonia: impact of dose regimen, passage, cryopreservation, and antibiotic therapy, Stem Cell Res Ther. 11, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manon-Jensen T, Itoh Y & Couchman JR (2010) Proteoglycans in health and disease: the multiple roles of syndecan shedding, FEBS J. 277, 3876–3889. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y, Gochuico BR, Yu G, Tang X, Osorio JC, Fernandez IE, Risquez CF, Patel AS, Shi Y, Wathelet MG, Goodwin AJ, Haspel JA, Ryter SW, Billings EM, Kaminski N, Morse D & Rosas IO (2013) Syndecan-2 exerts antifibrotic effects by promoting caveolin-1-mediated transforming growth factor-beta receptor I internalization and inhibiting transforming growth factor-beta1 signaling, Am J Respir Crit Care Med. 188, 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsoyi K, Chu SG, Patino-Jaramillo NG, Wilder J, Villalba J, Doyle-Eisele M, McDonald J, Liu X, El-Chemaly S, Perrella MA & Rosas IO (2018) Syndecan-2 Attenuates Radiation-induced Pulmonary Fibrosis and Inhibits Fibroblast Activation by Regulating PI3K/Akt/ROCK Pathway via CD148, Am J Respir Cell Mol Biol. 58, 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashida K, Parks WC & Park PW (2009) Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines, Blood. 114, 3033–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopal S (2020) Syndecans in Inflammation at a Glance, Front Immunol. 11, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J & Matthay MA (2013) Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria, Am J Respir Crit Care Med. 187, 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altamura M, Caradonna L, Amati L, Pellegrino NM, Urgesi G & Miniello S (2001) Splenectomy and sepsis: the role of the spleen in the immune-mediated bacterial clearance, Immunopharmacol Immunotoxicol. 23, 153–161. [DOI] [PubMed] [Google Scholar]
- 29.Pettila V, Hynninen M, Takkunen O, Kuusela P & Valtonen M (2002) Predictive value of procalcitonin and interleukin 6 in critically ill patients with suspected sepsis, Intensive Care Med. 28, 1220–1225. [DOI] [PubMed] [Google Scholar]
- 30.Wu HP, Chen CK, Chung K, Tseng JC, Hua CC, Liu YC, Chuang DY & Yang CH (2009) Serial cytokine levels in patients with severe sepsis, Inflamm Res. 58, 385–393. [DOI] [PubMed] [Google Scholar]
- 31.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR & Vincent JL (2016) Sepsis and septic shock, Nat Rev Dis Primers. 2, 16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ariel A & Serhan CN (2012) New Lives Given by Cell Death: Macrophage Differentiation Following Their Encounter with Apoptotic Leukocytes during the Resolution of Inflammation, Front Immunol. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva MT (2010) Neutrophils and macrophages work in concert as inducers and effectors of adaptive immunity against extracellular and intracellular microbial pathogens, J Leukoc Biol. 87, 805–813. [DOI] [PubMed] [Google Scholar]
- 34.Serhan CN & Savill J (2005) Resolution of inflammation: the beginning programs the end, Nat Immunol. 6, 1191–1197. [DOI] [PubMed] [Google Scholar]
- 35.Rath M, Muller I, Kropf P, Closs EI & Munder M (2014) Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages, Front Immunol. 5, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang JW & Lee SM (2016) Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis, Biochim Biophys Acta. 1861, 1025–1035. [DOI] [PubMed] [Google Scholar]
- 37.Schmid M, Gemperle C, Rimann N & Hersberger M (2016) Resolvin D1 Polarizes Primary Human Macrophages toward a Proresolution Phenotype through GPR32, J Immunol. 196, 3429–3437. [DOI] [PubMed] [Google Scholar]
- 38.Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology, Nature. 510, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serhan CN, Chiang N & Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators, Nat Rev Immunol. 8, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasconcelos DP, Costa M, Amaral IF, Barbosa MA, Aguas AP & Barbosa JN (2015) Modulation of the inflammatory response to chitosan through M2 macrophage polarization using pro-resolution mediators, Biomaterials. 37, 116–123. [DOI] [PubMed] [Google Scholar]
- 41.Fang X, Abbott J, Cheng L, Colby JK, Lee JW, Levy BD & Matthay MA (2015) Human Mesenchymal Stem (Stromal) Cells Promote the Resolution of Acute Lung Injury in Part through Lipoxin A4, J Immunol. 195, 875–881. [DOI] [PubMed] [Google Scholar]
- 42.Rani S, Ryan AE, Griffin MD & Ritter T (2015) Mesenchymal Stem Cell-derived Extracellular Vesicles: Toward Cell-free Therapeutic Applications, Mol Ther. 23, 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang YT, Huang YY, Zheng L, Qin SH, Xu XP, An TX, Xu Y, Wu YS, Hu XM, Ping BH & Wang Q (2017) Comparison of isolation methods of exosomes and exosomal RNA from cell culture medium and serum, Int J Mol Med. 40, 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thery C.Witwer, Aikawa KW, Alcaraz E, Anderson MJ, Andriantsitohaina JD, Antoniou R, Arab A, Archer T, Atkin-Smith F, Ayre GK, Bach DC, Bachurski JM, Baharvand D, Balaj H, Baldacchino L, Bauer S, Baxter NN, Bebawy AA, Beckham M, Bedina Zavec C, Benmoussa A, Berardi A, Bergese AC, Bielska P, Blenkiron E, Bobis-Wozowicz C, Boilard S, Boireau E, Bongiovanni W, Borras A, Bosch FE, Boulanger S, Breakefield CM, Breglio X, Brennan AM, Brigstock MA, Brisson DR, Broekman A, Bromberg ML, Bryl-Gorecka JF, Buch P, Buck S, Burger AH, Busatto D, Buschmann S, Bussolati D, Buzas B, Byrd EI, Camussi JB, Carter G, Caruso DR, Chamley S, Chang LW, Chen YT, Chen C, Cheng S, Chin L, Clayton AR, Clerici A, Cocks SP, Cocucci A, Coffey E, Cordeiro-da-Silva RJ, Couch A, Coumans Y, Coyle FA, Crescitelli B, Criado R, D’Souza-Schorey MF, Das C, Datta Chaudhuri S, de Candia A, De Santana P, De Wever EF, Del Portillo O, Demaret HA, Deville T, Devitt S, Dhondt A, Di Vizio B, Dieterich D, Dolo LC, Dominguez Rubio V, Dominici AP, Dourado M, Driedonks MR, Duarte TA, Duncan FV, Eichenberger HM, Ekstrom RM, El Andaloussi K, Elie-Caille S, Erdbrugger C, Falcon-Perez U, Fatima JM, Fish F, Flores-Bellver JE, Forsonits M, Frelet-Barrand A, A., et al. (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines, J Extracell Vesicles. 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu Q, Evans R, Fissell WH, Patton JG, Rome LH, Burnette DT & Coffey RJ (2019) Reassessment of Exosome Composition, Cell. 177, 428–445 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xian X, Gopal S & Couchman JR (2010) Syndecans as receptors and organizers of the extracellular matrix, Cell Tissue Res. 339, 31–46. [DOI] [PubMed] [Google Scholar]
- 47.Nelson A, Johansson J, Tyden J & Bodelsson M (2017) Circulating syndecans during critical illness, APMIS. 125, 468–475. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez H, Keane C, Masterson CH, Horie S, Elliman SJ, Higgins BD, Scully M, Laffey JG & O’Toole D (2020) Umbilical Cord-Derived CD362(+) Mesenchymal Stromal Cells Attenuate Polymicrobial Sepsis Induced by Caecal Ligation and Puncture, Int J Mol Sci. 21, 8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung H, Multhaupt HA, Oh ES & Couchman JR (2016) Minireview: Syndecans and their crucial roles during tissue regeneration, FEBS Lett. 590, 2408–2417. [DOI] [PubMed] [Google Scholar]
- 50.Levy BD & Serhan CN (2014) Resolution of acute inflammation in the lung, Annu Rev Physiol. 76, 467–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho MS, Mei SH & Stewart DJ (2015) The Immunomodulatory and Therapeutic Effects of Mesenchymal Stromal Cells for Acute Lung Injury and Sepsis, J Cell Physiol. 230, 2606–2617. [DOI] [PubMed] [Google Scholar]
- 52.Zheng G, Huang R, Qiu G, Ge M, Wang J, Shu Q & Xu J (2018) Mesenchymal stromal cell-derived extracellular vesicles: regenerative and immunomodulatory effects and potential applications in sepsis, Cell Tissue Res. 374, 1–15. [DOI] [PubMed] [Google Scholar]
- 53.Lo Sicco C, Reverberi D, Balbi C, Ulivi V, Principi E, Pascucci L, Becherini P, Bosco MC, Varesio L, Franzin C, Pozzobon M, Cancedda R & Tasso R (2017) Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization, Stem Cells Transl Med. 6, 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM & Krasnodembskaya AD (2017) Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer, Am J Respir Crit Care Med. 196, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willis GR, Fernandez-Gonzalez A, Reis M, Mitsialis SA & Kourembanas S (2018) Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension?, Int J Mol Sci. 19, 2534. doi: 10.3390/ijms19092534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wiklander OP, Nordin JZ, O’Loughlin A, Gustafsson Y, Corso G, Mager I, Vader P, Lee Y, Sork H, Seow Y, Heldring N, Alvarez-Erviti L, Smith CI, Le Blanc K, Macchiarini P, Jungebluth P, Wood MJ & Andaloussi SE (2015) Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting, J Extracell Vesicles. 4, 26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mansouri N, Willis GR, Fernandez-Gonzalez A, Reis M, Nassiri S, Mitsialis SA & Kourembanas S (2019) Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes, JCI Insight. 4, e128060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C, Vermeiren E, Zimmermann P & David G (2012) Syndecan-syntenin-ALIX regulates the biogenesis of exosomes, Nat Cell Biol. 14, 677–685. [DOI] [PubMed] [Google Scholar]
- 59.Friand V, David G & Zimmermann P (2015) Syntenin and syndecan in the biogenesis of exosomes, Biol Cell. 107, 331–341. [DOI] [PubMed] [Google Scholar]
- 60.Ju R, Zhuang ZW, Zhang J, Lanahan AA, Kyriakides T, Sessa WC & Simons M (2014) Angiopoietin-2 secretion by endothelial cell exosomes: regulation by the phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) and syndecan-4/syntenin pathways, J Biol Chem. 289, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monteforte A, Lam B, Sherman MB, Henderson K, Sligar AD, Spencer A, Tang B, Dunn AK & Baker AB (2017) (*) Glioblastoma Exosomes for Therapeutic Angiogenesis in Peripheral Ischemia, Tissue Eng Part A. 23, 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parimon T, Brauer R, Schlesinger SY, Xie T, Jiang D, Ge L, Huang Y, Birkland TP, Parks WC, Habiel DM, Hogaboam CM, Gharib SA, Deng N, Liu Z & Chen P (2018) Syndecan-1 Controls Lung Tumorigenesis by Regulating miRNAs Packaged in Exosomes, Am J Pathol. 188, 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baron RM, Kwon MY, Castano AP, Ghanta S, Riascos-Bernal DF, Lopez-Guzman S, Macias AA, Ith B, Schissel SL, Lederer JA, Reeves R, Yet SF, Layne MD, Liu X & Perrella MA (2018) Frontline Science: Targeted expression of a dominant-negative high mobility group A1 transgene improves outcome in sepsis, J Leukoc Biol. 104, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsoyi K, Geldart AM, Christou H, Liu X, Chung SW & Perrella MA (2015) Elk-3 is a KLF4-regulated gene that modulates the phagocytosis of bacteria by macrophages, J Leukoc Biol. 97, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fredenburgh LE, Velandia MM, Ma J, Olszak T, Cernadas M, Englert JA, Chung SW, Liu X, Begay C, Padera RF, Blumberg RS, Walsh SR, Baron RM & Perrella MA (2011) Cyclooxygenase-2 deficiency leads to intestinal barrier dysfunction and increased mortality during polymicrobial sepsis., J Immunol. 187, 5255–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willis GR, Fernandez-Gonzalez A, Anastas J, Vitali SH, Liu X, Ericsson M, Kwong A, Mitsialis SA & Kourembanas S (2018) Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation, Am J Respir Crit Care Med. 197, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA & Kourembanas S (2009) Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease, Am J Respir Crit Care Med. 180, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]