

Abstract
Oxidative phosphorylation (OXPHOS) is an active metabolic pathway in many cancers. RNA from pre-treatment biopsies from patients with triple negative breast cancer (TNBC) who received neoadjuvant chemotherapy demonstrated that the top canonical pathway associated with worse outcome was higher expression of OXPHOS signature. IACS-10759, a novel inhibitor of OXPHOS, stabilized growth in multiple TNBC patient-derived xenografts (PDXs). On gene expression profiling, all of the sensitive models displayed a basal-like 1 TNBC subtype. Expression of mitochondrial genes were significantly higher in sensitive PDXs. An in vivo functional genomics screen to identify synthetic lethal targets in tumors treated with IACS-10759 found several potential targets, including CDK4. We validated the antitumor efficacy of the combination of palbociclib, a CDK4/6 inhibitor, and IACS-10759 in vitro and in vivo. In addition, the combination of IACS-10759 and multi-kinase inhibitor cabozantinib had improved antitumor efficacy. Taken together, our data suggest that OXPHOS is a metabolic vulnerability in TNBC that may be leveraged with novel therapeutics in combination regimens.
Keywords: Breast Cancer, Oxidative Phosphorylation, Chemoresistance
Introduction
Approximately 10-15% of women diagnosed with breast cancer present with triple negative breast cancer (TNBC), which lacks expression of estrogen and progesterone receptors (ER and PR) or amplification/overexpression of HER2 (1). There are limited targeted therapy options currently for TNBC patients. For early stage patients with TNBC, neoadjuvant (preoperative) cytotoxic chemotherapy can achieve complete pathologic responses (pCR) in 30-40% of patients with TNBC, resulting in improved long-term outcomes (2). However, patients with TNBC who have significant residual disease following neoadjuvant chemotherapy have substantially worse oncologic outcomes (3). There is thus a clinical need to find factors associated with residual TNBC in order to identify novel therapeutic strategies to improve TNBC outcomes.
Recent studies have suggested that some tumor cells have an enhanced dependence on OXPHOS (4–6). The increase in OXPHOS/mitochondria genes can provide/restore energy and metabolites (aspartate) needed for biosynthesis and proliferation (7). This increased reliance on OXPHOS can result from passenger genomic alterations associated with genomic deletions of neighboring tumor suppressors (7,8). These coalterations result in decreased glycolytic potential alongside the tumorigenic events (4). However, other studies have shown at least a partial reliance on OXPHOS even without these genomic alterations (9,10).
Here, we show that OXPHOS is associated with a higher risk of recurrence and death in TNBC using gene expression profiling of treatment naïve tumors. We found that TNBC can be functionally reliant on OXPHOS using a panel of molecularly diverse patient derived xenografts (PDXs) generated from chemotherapy treated, residual TNBC. IACS-10759 is a novel inhibitor of OXPHOS that is currently in clinical trials (11). IACS-10759 is clinical grade inhibitor of complex I of the mitochondrial electron transport chain. Molecular studies have shown that IACS-10759 inhibits the OXPHOS by binding Mitochondrial Respiratory Complex 1 near or in the entrance of the ubiquinone channel thereby inhibiting ubiquinone function (7). We found that targeting OXPHOS with IACS-10759 inhibits growth of TNBC PDXs across a range of molecular subtypes with greatest antitumor efficacy seen in the basal-like TNBC molecular subtype and in tumors with high mitochondrial RNA expression. We systematically assessed potential predictive and pharmacodynamic markers of response to IACS-1075, using a panel of Patient Derived Xenografts (PDXs), in order to identify potential avenues to monitor ongoing OXPHOS inhibition. We also explored potential combination therapies by targeting putative mechanisms of intrinsic resistance and by performing an in vivo synthetic lethality screen for combination partners for IACS-10759 to improve antitumor efficacy.
Materials and Methods
Patient Cohort for RNASeq
We performed RNA sequencing on pre-treatment biopsies from 43 patients with operable TNBC who received sequential taxane and anthracycline-based neoadjuvant chemotherapy. All patients gave informed consent for treatment on an open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with TNBC (NCT00499603) (12). The study did not show a difference in pCR rate between the arms so pretreatment biopsies from both arms were analyzed together for analysis of transcriptomic features associated with relapse and overall survival.
PDXs
PDXs were generated from surgical samples obtained from patients after prospective informed consent as previously described (13). Investigators obtained informed written consent from all subjects. For this study, female athymic nude mice purchased from Harlan/Envigo were used for treatment testing. All animal experiments were approved by the University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee. For AXL suppression in BCX.010, we tested 3 shRNAs in pGIPz lentiviral vector and chose one to proceed to in vivo testing. We used a firefly luciferase targeted control shRNA. We choose the cell lines with greatest knockdown of AXL by qRTPCR and injected 2.5 x 106 cells on to the mammary fat pad of each mouse.
Animal Treatments
IACS-10759 was suspended in 0.5% methycellulose and given orally for 5 days followed by 2 days off every week. Cabozantinib was dissolved in Dimethyl Sulfoxide (DMSO) and then added to 0.5% methylcellulose to a final DMSO concentration of 10%. Cabozantinib was given orally daily. BGB-324 (50 mg/kg) was dissolved in 0.5% methylcellulose and given orally 7 days a week. Palbociclib (50 mg/kg) was dissolved in 0.5% methycellulose and given 7 days a week. Talazoparib (0.3 mg/kg) was dissolved in DMAC and then added to 20% Solutol, and given orally 7 days a week. Entinostat (20 mg/kg) was suspended in PBS and given orally 7 days a week. BAY87-2243 (5 mg/kg) was dissolved in 0.5% methycellulose and given orally for 5 days followed by 2 days off every week. Tumor volume was calculated by the formula: TV (mm3) = ((width)2 x length)/2.
Molecular analysis
For RNA analysis, small fragments of frozen tumor were placed in lysis buffer and homogenized manually. RNA was isolated from the frozen tumors using Norgen BIOTEK Total RNA Purification Plus Kit. RNA was quantified using Qubit system. Methodology and gene selection for Nanostring OXPHOS panel has been described (7).
Reverse Phase Protein Arrays (RPPA) were performed at the MD Anderson Cancer Center Functional Proteomics Core Facility. Frozen tumor pieces were cut into ~3 x 3 mm size fragments and placed in bead lysis tubes for protein extraction. We chose the RPPA results originated from rabbit and goat antibodies (230 antibodies) for PDXs because validated mouse antibodies have significant background in PDX samples due to the mouse stromal tissue.
For immunohistochemistry (IHC), we used monoclonal rabbit antibodies for phosphoHistone H3 (Bethyl Cat# IHC-00061), cleaved caspase 3 (CC3) (Cell Signaling #9664), and PCNA (KLJ). Sample were collected four hours after IACS-10759 treatment and twelve days after start of treatments. The assessment was performed by a clinically trained pathologist.
In Vivo Synthetic Lethality Screen
We utilized a barcoded FDAome shRNA library correspond to targets linked to drugs approved by the Food and Drug Administration (FDA )to determine potential IACS-10759 synthetic lethal partners. Breast cancer cell line BCX.010-CL was derived from BCX.010 as previously described (14). BCX.010-CL cells were infected with our FDAome shRNA library, reinjected into mice (2 X 106 cells) and treated with either vehicle or IACS-10759. After ten days, xenografts were harvested and DNA was extracted. Deep sequencing was performed to determine shRNA abundance (barcode abundance) on the basis of shRNA performance supervised analysis. Cumulative Wilcoxon test was done to score statistical gene level significance.
In vitro Testing
Breast cancer cell lines were cultured in Dulbecco’s modified Eagle’s medium/F-12 supplemented with 10% fetal bovine serum at 37°C and in a humidified incubator containing 5% CO2. BCX.010-CL cells were isolated as previously described (14); origin validation was done by Short Tandem Repeat Fingerprinting. MDA-MB-468 cells were obtained from ATCC. Laboratory cell cultures were regularly tested for mycoplasma and passaged a maximum of ten times after thawing early passage stocks. For in vitro testing, all agents were dissolved in DMSO. For cell viability assays, cells were plated into 96-well plates at 5000 cells/well. The cells were allowed to attach overnight, and then treated with the selected drugs for 72 hours. Cell viability was evaluated by sulforhodamine B assay. IC50s from dose-response curves were determined using GraphPad Prism v.6.05 software. Combination index (CI) was calculated using the IC50-based Chou-Talalay model.
RESULTS
Oxidative phosphorylation is associated with recurrence in TNBC patients
To determine molecular features associated with higher risk of relapse in operable TNBC, we performed RNA sequencing on pre-treatment biopsies from 43 patients with TNBC who received sequential taxane-and anthracycline-based neoadjuvant chemotherapy. At >5 years median follow-up, 14 patients had recurred and of those all but two had died. At a false discovery rate of 0.05 (q value<0.05), 33 genes were differentially expressed between patients who did and did not have a subsequent recurrence (Fig 1). Twenty seven genes were differentially expressed between patients based on overall survival (Supplementary Fig. 1).
Figure 1.
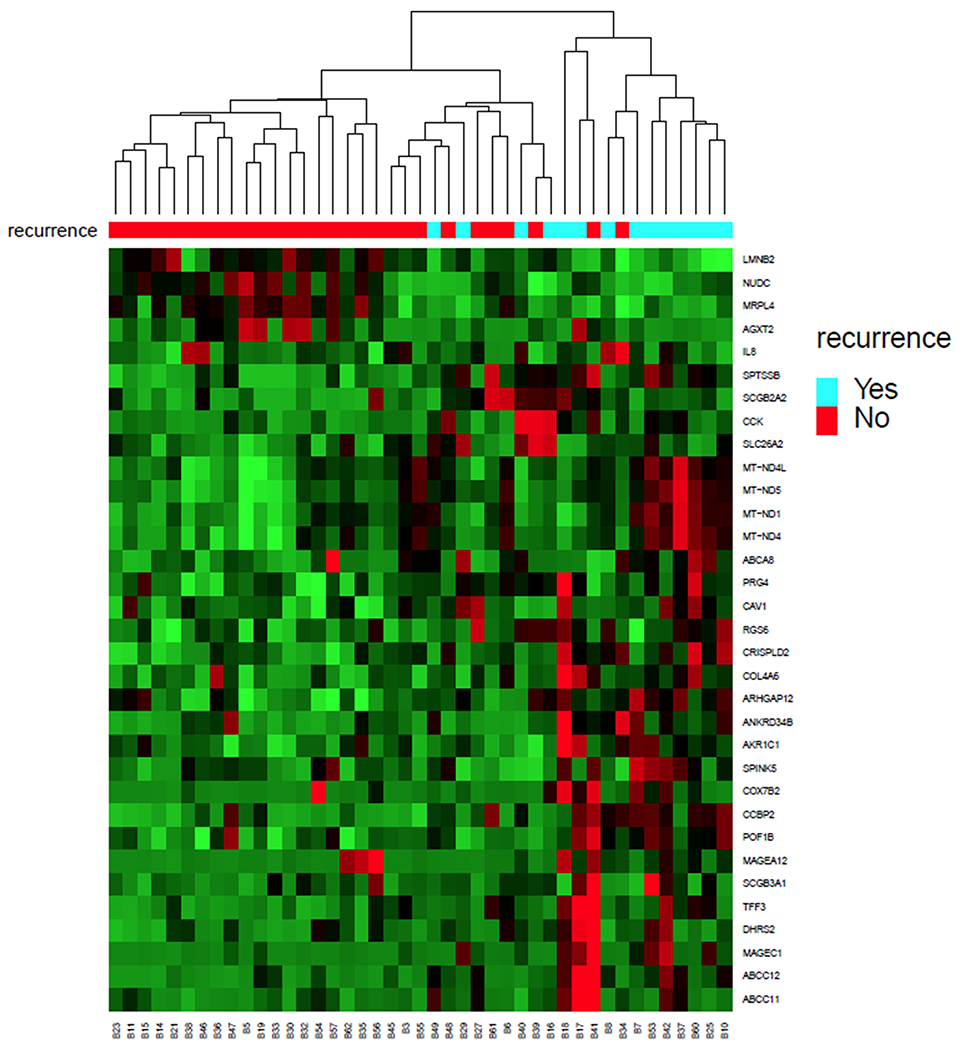
We performed RNA sequencing on pre-treatment biopsies from 43 patients with operable TNBC who received sequential taxane-and anthracycline-based neoadjuvant chemotherapy. At greater than 5 year median follow-up, 14 patients recurred and of those all but 2 patients had died. At a false discovery rate of 0.05, 33 genes were differentially expressed between patients who did and did not have a subsequent recurrence. Ingenuity pathway analysis demonstrated that one of the top canonical pathways that differed was higher expression of oxidative phosphorylation signature (p<0.001).
One of the top canonical pathways that was associated with higher risk of relapse and lower risk of survival was higher expression of OXPHOS signature (adjusted p<0.001) as determined by Ingenuity Pathway Analysis (IPA). The patients that recurred had significantly higher levels of expression of mitochondrial genes: MT-ND1 (adjusted p=0.007); MT-ND5 (p=0.03) and MT-ND4 (p=0.04). Similarly, the patients that had died had higher expression of several mitochondrial genes: MT-ND5 (p=0.001); MT-ND4 (p=0.005), MT-ND4L (p=0.015), MT-ND6 (p=0.018), and MT-ATP6 (p=0.03). All of these mitochondrial genes (excluding MT-ATP6) encode members of Complex 1. For these reasons, we hypothesized that OXPHOS pathway, and specifically mitochondrial Complex 1, may represent a potential therapeutic target in TNBC.
Inhibiting Oxidative Phosphorylation has broad antitumor effect in TNBC PDXs
To determine the functional necessity of OXPHOS in TNBC, we treated a panel of ten TNBC PDXs with OXPHOS inhibitor IACS-10759. Eight of these ten PDXs were generated from residual tumors collected after neoadjuvant chemotherapy; these patients by definition did not have a complete pathological response and thus these tumors represent relatively chemoresistant disease. Although the models differ in their chemotherapy sensitivity, most have been previously demonstrated to be resistant to several standard chemotherapy regimens (13,15). BCX.092 and BCX.024 were generated from patients that did not receive neoadjuvant therapy (13). Known clinically relevant genomic alterations in these models are listed in Supplementary Table 1. We treated the PDXs with IACS-10759 using a small cohort (n=2-4) screening approach (Fig. 2). Five of ten PDX models had either stable disease (did not increase ≥20% in size) or regressed at 28 days of treatment. One PDX (BCX.070) which is known to be resistant to several chemotherapeutic agents (13), drastically regressed with IACS-10759 treatment.
Figure 2. Inhibiting OXPHOS result in broad tumor in a range of TNBC PDXs developed from residual disease.
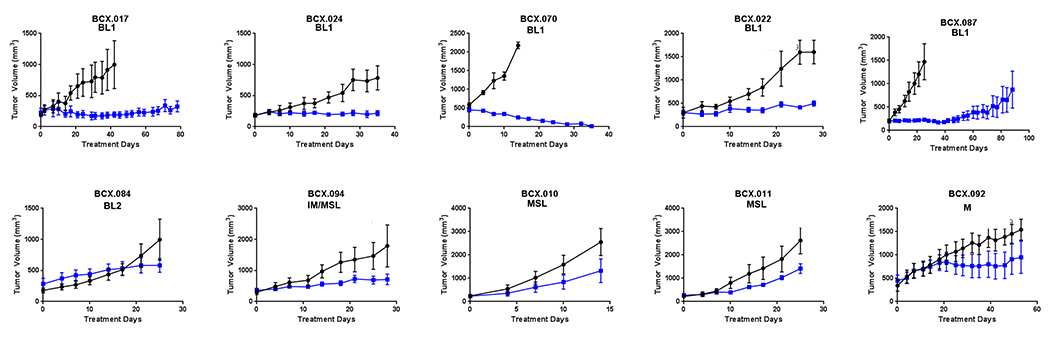
Ten TNBC PDXs were treated with IACS-10759 (5 mg/kg, po, 5 days on 2 days off). Cohorts of 2-4 mice were used for this initial screening. IACS-10759 stabilized disease (<20% median change from baseline) for at least 21 days in four PDXs and one PDX regressed to be immeasurable disease. Only one of five BL1 PDXs grew more than >20% in 21 days. Means +/− SEM are shown.
To elucidate the specificity of these effects in vivo, we treated a PDX that was less sensitive to IACS-10759 (BCX.011) and another that is more sensitive to IACS-10759 (BCX.017) with BAY87-2243, an alternative complex 1 inhibitor (16). Similar to IACS-10759, BAY87-2243 did not prevent progression of BCX.011 but did cause disease stabilization in BCX.017 (Supplementary Fig. 2). This suggests that indeed the differences we observed with IACS-10759 sensitivity may be representative of intrinsic differences in metabolism and reliance on OXPHOS for tumor growth.
Baseline biomarkers associated with intrinsic or acquired resistance and sensitivity to IACS-10759
We next performed analyses to identify potential predictive biomarkers of sensitivity using baseline RNAseq data for the PDXs. We first determined the association between IACS-10759 sensitivity and clinically relevant RNA expression-based molecular subtypes of TNBC based on the Lehman classification as previously described (TNBCtype) (13)(17). All of the IACS-10759 sensitive PDXs were TNBCtype basal-like 1 (BL1).
The baseline expression of the oxidative phosphorylation signature by IPA was not significantly different between IACS-10759 sensitive vs resistant PDX models. However, PDXs sensitive to IACS-10759 had higher expression of protein-encoding mitochondrial genes, including the specific MT genes that we found to be also differentially expressed higher in the patients more likely to develop recurrent disease (Fig. 3A; Supplementary Fig. 3).
Figure 3. Predictor of response to IACS-10759 in TNBC.
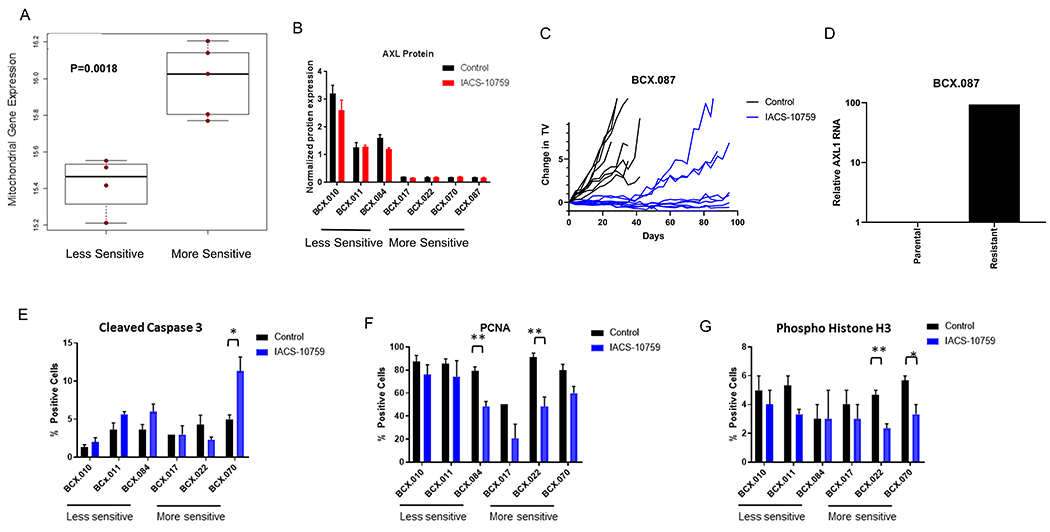
(A) Using baseline RNAseq analysis for PDX with known IACS-10759 sensitivity, we found that protein-coding mitochondrial genes are expressed significantly higher in PDXs more sensitive to IACS-10759. (B) AXL protein as determine by RPPA is higher in PDXs less sensitive to IACS-10759. (C-D) A highly sensitive PDX was treated for >90 days (individual tumors shown) and sporadic tumors began growing and a reformed tumor had increase AXL1 mRNA expression compared to the control. (E-G) We treated a select set of PDXs with ranges of responses to IACS-10759 (5 mg/kg, po, 5 days on 2 days off) and collected the tumors after 12 days of treatment. We analyzed the samples for cleaved caspase 3 (E), PCNA (F), and phosphohistone H3 (G).*= p<0.05; **=p<0.01
As loss of alpha enolase (ENO1) expression has been linked to reliance on OXPHOS (7), we assessed ENO1 expression by RNAseq and IHC. The more sensitive models trended towards lower ENO1 RNA expression, with the regressive model, BCX70, having lowest expression (Supplementary Fig. 4A). The IACS-10759 sensitive models had lower ENO1 staining intensity by IHC (p<0.001) (Supplementary Fig. 4B). Using RNASeq data, we assessed a published proliferation index (18); we found no significant difference in baseline proliferation between more and less sensitive PDXs (Supplementary Fig. 5).
Comparing more versus less IACS-10759-sensitive models, we identified 202 genes that are higher in the less sensitive models at FDR=0.01 (Supplementary Table 2). By Ingenuity Pathway analysis, epithelial mesenchymal transition (EMT) signature as well as hypoxia signatures were differentially expressed between sensitive and less-sensitive PDXs (Supplementary Table 3). PDXs that were less sensitive to IACS-10759 expressed higher levels of TWIST1, a key transcriptional factor associated with EMT, and AXL, a tyrosine kinase that has been associated with EMT in breast cancer.
AXL expression has been associated with more aggressive tumor behavior, as well as therapeutic resistance to chemotherapy and targeted therapy, and AXL inhibition has been shown to enhance sensitivity to targeted therapy (19,20). Therefore we also assessed AXL protein expression in IACS-10759 sensitive vs resistant PDXs. We found that AXL protein expression was higher in the less sensitive models by RPPA (Fig. 3B) and by western blotting (Supplementary Fig. 6). We also assessed AXL phosphorylation at Tyrosine 702 but were unable to detect baseline phosphorylation of AXL in these samples.
To determine whether AXL plays a role in acquired resistance to IACS-10759, we treated an IACS-10759 sensitive PDX (BCX.087) with IACS-10759 until tumors progressed to greater than 100% starting tumor volume (for up to 92 days) (Fig. 3C). We analyzed AXL expression in regrown BCX.087 by qRTPCR and found significant higher levels compared to the control (Fig. 3D).
These data taken together suggest that baseline molecular profiles of tumors may help separate tumors with greater reliance on OXPHOS signaling. Potential biomarkers of OXPHOS dependence include basal TNBC type and higher baseline mitochondrial gene expression while EMT signature and higher AXL expression was associated with less sensitivity to OXPHOS inhibition.
Pharmacodynamic markers of response to OXPHOS inhibition as determined by IHC
We next used a multipronged approach to determine pharmacodynamics (PD) changes induced by OXPHOS inhibition in order to find clinically relevant, early response markers for IACS-10759. We chose three models that are more sensitive to IACS-10759 and three models that are less sensitive to IACS-10759 for early PD marker assessment. We treated these PDXs with IACS-10759 for 12 days (10 treatment days) and collected the tumors four hours following final treatment. We analyzed these samples for traditional markers of apoptosis (nuclear cleaved caspase 3) (Fig. 3E) and proliferation (PCNA, pHistone H3) (Fig. 3F and 3G) by IHC. The most significant increase in cleaved caspase 3 (CC3) was seen in BCX.070 which regressed on therapy. Phosphorylated Histone H3 (pHistone mitosis) decreased with IACS-10759 treatment in two of three sensitive models. For PCNA, BCX.017 (more sensitive) and BCX.084 (less sensitive) had significantly lower expression with IACS-10759 treatment.
Pharmacodynamic markers of response to OXPHOS inhibition as determined by RNA Analysis
We next compared the gene expression profiles of seven PDXs, three less sensitive models and four more sensitive models, treated with IACS-10759 for 12 days by RNASeq. Based on global mRNA expression profiles, sensitive models clustered separately from the less sensitive models (Supplementary Fig. 7). In addition, treated and untreated models clustered as separate groups for each sensitive model. We performed Gene Set Enrichment Analysis with this data using the Reactome and Hallmark databases. We found several profiles downregulated by IACS-10759 treatment, including a lipid metabolism profile and hypoxia profile (Supplementary Fig. 8).
We assessed the role that proliferation plays in response to OXPHOS inhibition by calculating a proliferation index for each model using RNASeq data (18). We found that the proliferation index was significantly decreased upon IACS-10759 treatment in more sensitive models but not in the resistant models (Supplementary Fig. 9). We next removed the genes comprising the proliferation index and performed cluster analysis. Even without these proliferation specific genes, the more sensitive models and less sensitive models clustered separately (Supplementary Fig. 10).
We then analyzed the tumors using a Nanostring gene set that was specifically developed to assess genes found to be consistently altered by inhibition of OXPHOS in acute myeloid leukemia and pancreatic cancer (7). When this data set was subjected to unsupervised clustering, the IACS-10759 treated samples for the most part clustered together, away from their controls similar to RNASeq (Supplementary Fig. 11). The four samples that clustered away from the respective treatment groups were from the less sensitive PDXs.
When the seven PDX models were analyzed together, there were 146 genes differentially regulated with IACS-10759 treatment (Supplementary Fig. 12). Only three of four sensitive models had differentially regulated genes at FDR=0.05 (Supplementary Fig. 13–15). Ten genes were differentially expressed with IACS-10759 treatment in three of the four sensitive models (Supplementary Table 4). The differentially expressed genes included metabolic regulator ASNS that encodes asparagine synthetase, and TRIB3 (Tribbles pseudokinase 3) and DDIT3 (DNA Damage-inducible transcript 3), genes known to play a role in cellular stress response.
Pharmacodynamic markers of response to OXPHOS inhibition as determined by Reverse Phase Protein Array
We analyzed the samples based on protein expression and phosphorylation as determined by Reverse Phase Protein Array (RPPA). On unsupervised clustering, the IACS-10759 treated samples clustered together with matched untreated controls. Notably RPPA clustered IACS-10759 sensitive vs IACS-10759 PDX models separately (Supplementary. Fig. 16).
We went on to identify a set of 31 total or phospho-proteins that are differentially expressed between treated and untreated samples regardless of sensitivity (Supplementary Fig. 17). The expression of AXL protein was not significantly changed with IACS-10759 treatment versus matched controls. Phosphorylated 5′ AMP-activated protein kinase (pAMPK), well established marker of metabolic stress, was increased by OXPHOS inhibition. SRC phosphorylation on Tyr527, a phosphorylation site that renders the SRC enzyme less active, was decreased (21). MAPK and PI3K downstream signaling was repressed as demonstrated by the decrease in phospho-p44/MAPK (ERK1/2) Thr202/Tyr204) and phospho-S6 Ser235/236 and phospho-S6 Ser240/244, and phosphorylation of tumor suppressor Rb was decreased. In addition, we identified proteomic changes unique to each sensitive model (Supplementary Fig. 18–20). Three of four sensitive models had differentially expressed genes at FDR=0.05. However, the only proteomic change occurring in all three of these models was decreased phosphorylation of AKT (T308), a key mediator of PI3K signaling pathway.
Identifying Rational Combinations with IACS-10759
Building on the finding that AXL is highly expressed in PDXs that are less reliant on OXPHOS, we hypothesized that targeting AXL may enhance sensitivity to IACS-10759. We thus tested IACS-10759 in combination with cabozantinib, a multi-kinase inhibitor that targets AXL and is approved by the FDA for the treatment of renal cell carcinoma, medullary thyroid cancer and hepatocellular carcinoma. We tested the combination in two less sensitive models with high expression of AXL (BCX.010 and BCX.084) and one more sensitive model (BCX.087). In both less OXPHOS dependent PDXs, we found that the combination of cabozantinib and IACS-10759 significantly prolonged tumor inhibition compared to either single agent (Fig. 4A and 4B). We also observed tumor regression in the more OXPHOS sensitive model (BCX.087) compared to stable disease with single agent treatment (Fig. 4C). We assessed signaling in the BCX.010 treated samples by RPPA and found that S6 phosphorylation was inhibited significantly more in the combination group (Fig. 4D).
Figure 4. Targeting AXL1 expressing TNBC with combination of cabozantinib and IACS-10759.
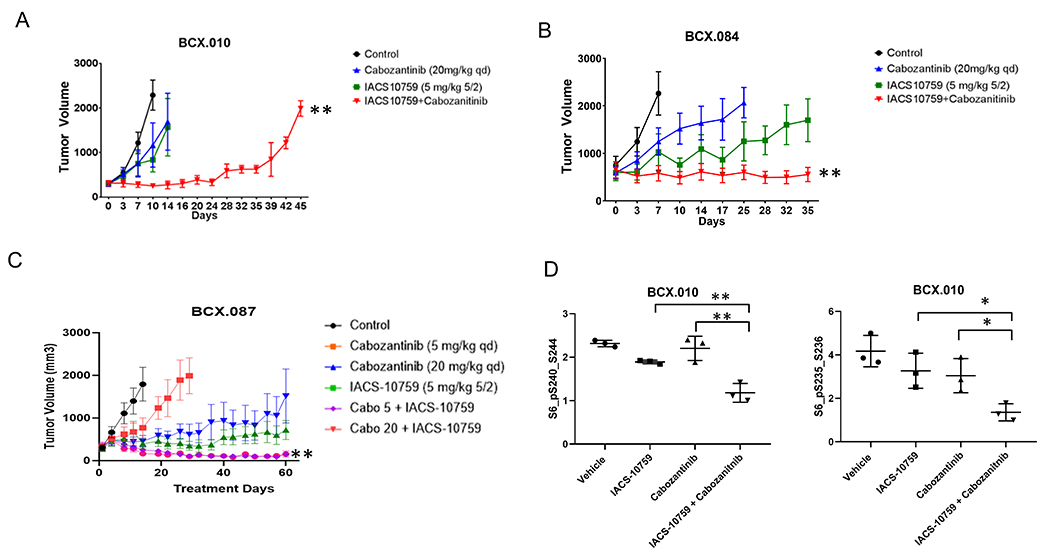
(A-B) Two AXL1 high TNBC PDXs (BCX.010; A and BCX.084, B) were treated with cabozantinib (20 mg/kg, po, daily) and IACS-10759 (5 mg/kg, po, 5 days on 2 days off) prolonged tumor stability compared to either single agent alone. (C) A low AXL1 expressing PDX that is relatively more sensitive to IACS-10759 was treated with cabozantinib (20 mg/kg or 5 mg/kg, po, daily) and IACS-10759 (5 mg/kg, po, 5 days on 2 days off) and both combinations resulted in tumor regression from baseline. Data shown mean +/−SEM. **=p<0.001. (D) In the PI3KCA mutant PDX (BCX.010), the combination of cabozantinib and IACS-10759 significantly inhibited PI3K/mTOR pathway to a greater extent that either single agent along as evidenced by decrease phosphorylation of ribosomal protein S6 on RPPA. *=p<0.05; **=p<0.01
We also tested IACS-10759 plus an AXL specific inhibitor (BGB-324) in AXL high BCX.010. BGB-324 added no benefit over IACS-10759 alone (Supplementary Fig. 21A). We also assessed effect of AXL knocked down by shRNA. AXL played an important growth regulatory role, as demonstrated by the significant growth inhibition seen by AXL shRNA in BCX.010 (Supplementary Fig. 21B–C). However, IACS-10759 did not further enhance growth inhibitory effect.
In vivo synthetic lethality to identify therapies that to sensitize to OXPHOS inhibition.
In order to identify additional combination partners for OXPHOS inhibition, we also performed an in vivo synthetic lethality screen using a set of shRNA linked to FDA-approved agents (Fig. 5A). To do so, we transduced BCX.010 cells with molecular-barcoded shRNA at a multiplicity of infection (MOI) of 0.2. We injected the cells into mice and treated the mice with vehicle or IACS-10759 for 12 days. We collected the tumors and compared remaining cells with shRNA integrations by sequencing of the barcodes. We identified several potential targets with linked FDA-approved agents with high breast cancer relevance (Fig. 5B), including CDK4, PARP1 and 2, and HDAC3.
Figure 5. Identification of combination partners for IACS-10759 using in vivo synthetic lethality screen.
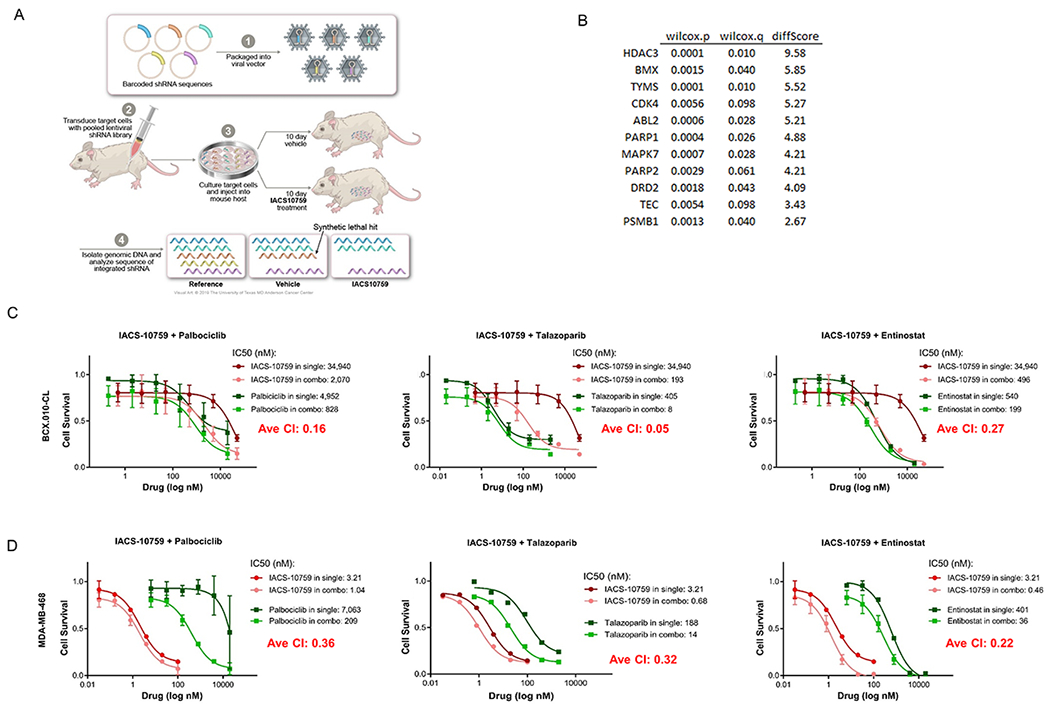
(A) Illustration of method used to identify genes (from gene panel linked to FDA-approved agents) whose suppression led to increased cell loss in present of IACS-10759 in mice. (B) List of genes identified in screen. We chose to validate CDK4, PARP1 and HDAC3 using palbociclib, talazoparib and entinostat respectively. (C-D) IACS-10759 was synergistic (CI<1) with palbociclib, talazaparib, and entinostat in BCX.010-CL and MDA-MB-468 cells as assessed by cell growth assays.
We tested the in vitro growth inhibitory efficacy of IACS-10759 in combination with palbociclib (targeting CDK4/6), talazoparib (targeting PARP), and entinostat (targeting HDAC3) in two breast cancer cell lines, BCX.010-CL, a cell line derived from BCX-10 (Fig. 5C) and MDA-MB-468 (Fig. 5D). All three combinations were synergistic, with combination indices of 0.16, 0.05, 0.27 for palbociclib, talazoparib and entinostat respectively in BCX.010-CL. Using extracellular O2 consumption assay, we noted that while IACS-10759 inhibited oxygen consumption rate (OCR) in MDA-MB-468 cells, none of the other agents directly inhibited OCR, but the combinations of agent led to a greater inhibition of OCR (Supplementary Fig. 22).
Next, we tested these three combinations on BCX.010 in vivo. We found that the combination of IACS-10759 with palbociclib significantly enhanced growth inhibition at doses tested compared to palbociclib or IACS-10759 alone in BCX.010 (Fig. 6A) and an additional Rb wild-type model, BCX.080 with low (10%) ER expression (Fig. 6B). We also developed a PDX from a patient with metastatic hormone receptor positive breast cancer who had received palbociclib and progressed (i.e. had intrinsic resistance). We treated this PDX with palbociclib and IACS-10759 and found enhanced response to the combination (Fig 6C).
Figure 6. Validation of functional shRNA screen to identify potential IACS-10759 combination partners.
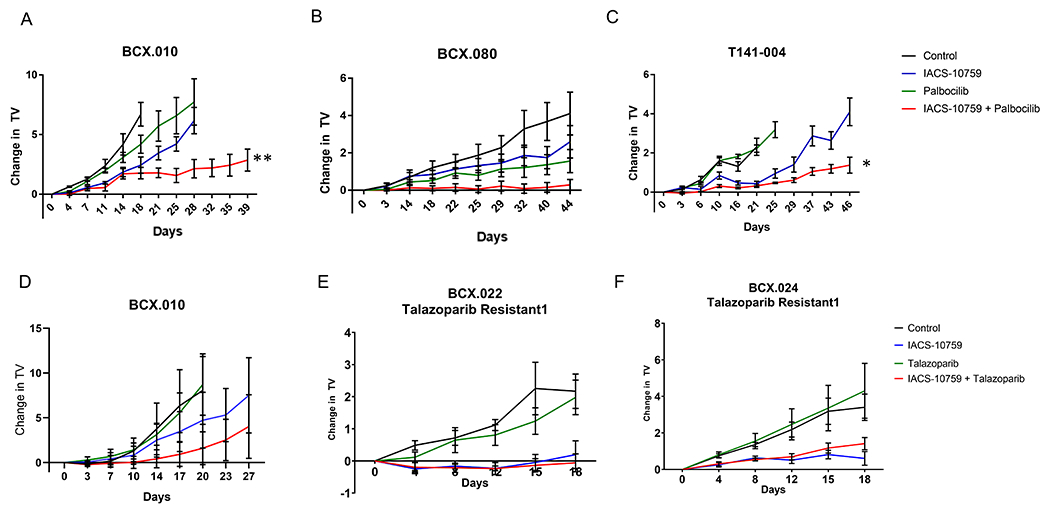
(A) We tested palbociclib (50 mg/kg daily) in BCX.010 PDX in vivo. The combination of IACS-10759 and palbociclib showed clear improvement over both single agents similar to in vitro. (B) We further validated palbociclib + IACS-10759 in an additional retinoblastoma gene positive less sensitive TNBC PDX (BCX.080). Data are shown as mean +/− SEM. **=p<0.001. (C) We also tested palbociclib + IACS-10759 using a PDX (T141-003) generated from a breast cancer patient who had received palbociclib and progressed. Data are shown as mean +/− SEM. *=p<0.01. (D) We tested talazaparib (0.3 mg/kg daily) + IACS-10759 combination in BCX.010 PDX in vivo but found with a more limited combination efficacy. (E-F) We next tested talazoparib in combination with IACS-10759 in PDXs with acquired resistance to talazoparib (BCX.022 talazoparib resistant 1 and BCX.024 talazoparib resistant 1). These models were created by treating talazoparib sensitive PDXs until tumors were not palpable and then collecting and serial passaging the grown tumors. The combination did not have improved efficacy in these models but IACS-10759 did have significantly greater inhibition versus talazoparib (BCX.022 talazoparib resistant: p<0.001; BCX.024 talazoparib resistant: p<0.01).
Although talazoparib plus IACS-10759 had greater growth inhibition in BCX.010 PDXs in vivo than with either agent alone, this difference was not statistically significant (Fig. 6D). To assess the role of IACS-10759 in modifying sensitivity to PARP inhibitors, we also tested the combination of talazoparib and IACS-10759 using two talazoparib acquired resistance models developed in our lab. These models were developed by treating two TNBC BL1 PDXs that regressed with talazoparib and viably collecting tumors that subsequently progressed (13). The combination with talazoparib had no improved efficacy in vivo using these models (Fig. 6E and 6F), but IACS-10759 alone did show statistically significant growth inhibition compared to PARP inhibition in these acquired resistance models. The combinations of entinostat and IACS-10759 did not increase efficacy over the single agent on BCX.010 in vivo (Supplementary Fig. 23).
DISCUSSION
We show here that chemoresistant TNBC can be reliant on OXPHOS to varying extents; importantly, complete regression was observed in a PDX with IACS-01759 treatment. Thus several metabolic pathways may play a role in chemoresistant breast cancer but OXPHOS may be the absolute necessary determinant in some resistant specific TNBC. Notably, progression-free survival and overall survival in TNBC may be influenced by many clinical parameters such as tumor size and nodal status as well as molecular features. Further study is needed to see how OXPHOS activity varies with other parameters. The association of OXPHOS and outcomes in this small study is hypothesis generating and would need much larger studies for validation, also taking into account to rapidly evolving treatment algorithms. However, our finding is consistent with a recent report by Echeverria et al that showed that OXPHOS was the most significantly up-regulated pathway in residual tumors after TNBC PDXs were treated with chemotherapy compared to vehicle-treated tumors (10).
We show that several basal-like PDXs comprised of cancers resistant to standard neoadjuvant therapy were sensitive to IACS-10759. Moreover, we found that basal-like TNBC, which are most often defective in DNA damage repair response pathways and often sensitive to FDA-approved PARP inhibitors, are sensitive to OXPHOS inhibition. While we found in vitro synergy with talazoparib in TNBC models, we did not observe enhanced efficacy with the combination results in vivo. However, the sensitivity of models with acquired talazoparib resistance to IACS-10759 highlights that there is no cross-resistance to PARP inhibitors.
Previous studies have shown that genomic passenger alterations can result in reliance on OXPHOS. While we found molecular features associated with sensitivity to IACS-10759 in TNBC, including high mitochondrial RNA and low ENO1 protein expression among others, further studies are required to determine if any biomarkers can reproducibly assess reliance on OXPHOS in TNBC. We show evidence that higher expression of mitochondrial protein coding RNA is correlated with functional reliance on OXPHOS in TNBC PDXs, and we believe that this demonstrates higher mitochondrial numbers in sensitive models but could also represent overcompensation due to lower mitochondrial function. We also observed an increase in phosphorylated AMPK in all models with IACS-10759 treatment. ATP, which is produced via oxidative phosphorylation, inhibits the phosphorylation of AMPK. Thus the increased level of phosphorylated AMPK may suggests that in these models ATP deficiencies induced by OXPHOS are not fully compensated for by alternative mechanisms.
Changes in traditional IHC pharmacodynamics (CC3, pHistone H3, PCNA) did not perfectly correlate with increased sensitivity to IACS-10759. However, two of three more sensitive PDXs had significantly decreased pHistone H3, but no less sensitive PDXs had changes in pHistone H3. Interesting, the PDX that regressed with IACS-10759 treatment (BCX.070) had the largest increase in cleaved caspase 3. These findings suggested that both cleaved caspase 3 and pHistone H3, along with gene expression changes observed, have potential as pharmacodynamics markers in OXPHOS inhibitor trials as potential early molecular predictors of clinical response/benefit.
RPPA analysis revealed that IACS-10759 reduces activity of several well characterized signaling pathways. In the set of PDXs tested, which includes a PIK3CA mutant PDX and a PDX with PTEN loss, the PI3K/AKT pathway was inhibited by IACS-01759 treatment as evidenced by decreased phosphorylation of downstream targets such S6 ribosomal subunit, PRAS40 (pT246) and RICTOR (pT1135). Studies have suggested that reducing energy expenditure, via reduced mTOR activity, can allow cancer cells to escape energy stress caused by OXPHOS inhibition (8). Therefore, a decrease in this important protein translation pathway may be a mechanism of adaptive response/resistance to IACS-10759.
SRC has been established as a major regulator of mitochondrial energy metabolism and OXPHOS (22). On RPPA, we also observed a decrease in SRC (pT527), suggesting increased SRC activity. Several oxidative phosphorylation components have been shown to be direct targets of SRC (23). SRC has been shown to be a preeminent inhibitor of pyruvate entering oxidative phosphorylation via inhibition of PDH (24). SRC also has been shown to interact with and phosphorylate hexokinases HK1 and HK2, the rate limiting enzymes in glycolysis (25). Thus increased SRC activity with IACS-10759 treatment may demonstrate the cells are attempting to shift reliance on OXPHOS. Further studies are needed to determine effects of SRC modulation in combination with IACS-10759.
Our PDX testing demonstrated the potential effectiveness of inhibiting OXPHOS in TNBC with IACS-10759. However, several TNBC PDXs were less sensitive to OXPHOS inhibition alone suggesting the need for future combination strategies with IACS-10759. We thus identified two potential combination partners that are FDA-approved for other indications: cabozantinib and palbociclib.
We hypothesized that cabozantinib would synergize with IACS-10759 due to high expression of AXL in the resistant PDXs. AXL is among proteins known to increase glycolytic function in cancer and be expressed in some TNBC (26,27). AXL is also known to promote breast cancer metastasis and chemoresistance (28,29). AXL has been shown to upregulate two key enzymes of aerobic glycolysis (Glucose transporter type 4 and Pyruvate Dehydrogenase Kinase 1) via Tensin 2 (TNS2) phosphorylation (30). In melanoma, AXL expression is inversely correlated with MITF expression, and low MITF expression is linked to low OXPHOS (31). Low MITF1/AXL expression ratio is linked to resistance to multiple drug targets, strengthening the hypothesis that AXL may be associated IACS-10759 resistance. AXL suppression caused strong antitumor efficacy alone and may not properly represent what could be achievable by pharmacological inhibition. However, AXL specific inhibitor and AXL suppression via shRNA were not synergistic with IACS-10759. This data suggests AXL upregulation alone may not be sufficient to drive OXPHOS resistance, and may be part of a larger reprogramming such as EMT in association with drug resistance. This is supported by upregulation of other EMT markers such as TWIST in the resistant models (Supplementary Table 3). Further work is needed to assess whether AXL expression or other EMT-associated markers could potentially serve as biomarkers of resistance to OXPHOS inhibition. Notably, cabozantinib is a multi-kinase inhibitor, and its other kinase targets such as VEGFR2 and MET and/or the resultant anti-angiogenesis effect may have mediated the enhanced efficacy of the combination of cabozantinib and IACS-10769. Cabozantinib is approved for hepatocellular cancer, medullary thyroid cancer and renal cell carcinoma and is being investigated in TNBC and multiple other tumor types. Thus these findings may have implications for many tumor types.
Palbociclib is approved for hormone receptor positive (HR+) breast cancer. In our study, we first found efficacy of the palbociclib and IACS-10759 combination in hormone receptor negative Rb1 positive breast cancer. Other studies have shown potential utility for CDK inhibition in combination with other targets in TNBC (32–34), and this could be an avenue to explore in combination with OXPHOS inhibition. However, we also showed that PDXs generated from a HR+ breast cancer patients were sensitive to this combination and thus this combination could be explored in the hormone receptor positive setting as well.
In summary, our study provided evidence for OXPHOS dependence in chemotherapy resistant TNBC. Future metabolic studies are needed to better elucidate the fuel for this increased metabolic activity. We have identified and validated biomarkers that can be analyzed as a measure of OXPHOS dependence as well as correlative studies in clinical trials of OXPHOS inhibitors for assessing target engagement as well as retrospectively accessing predictive reliance. Importantly, we illuminated potential targeted therapies where OXPHOS inhibition may represent a novel combination strategy.
Supplementary Material
Statement of Significance.
These findings suggest that TNBC is highly reliant on OXPHOS and that inhibiting OXPHOS may be a novel approach to enhance efficacy of several targeted therapies.
Acknowledgements
We appreciate Susanna Brisendine for helping with manuscript preparation. We appreciate insights from our collaboration with Charles Perou. For funding we acknowledge the following: National Cancer Institute PDX Development and Trial Center U54 #CA224065, National Institutes of Health T32 CA009599, The MD Anderson Cancer Center Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer, MD Anderson Breast Cancer Moonshot Program, NIH Clinical Translational Science Award 1UL1TR003167, the Nellie B. Connally Breast Cancer Research Endowment, the Barr funds, Cancer Prevention & Research Institute of Texas grant RP180248, and the MD Anderson Cancer Center Support Grant (P30 CA016672).
Disclosure of Potential Conflicts of interest:
The University of Texas MD Anderson Cancer Center holds Intellectual Property on IACS-010759.
K. Evans, S. Moulder-Thompson, E. Yuca, S. S. Scott, M. Zhao, N. Paez Arango, C. Cruz-Pico, T. Saridogan, M. Shariati, C. P. Vellano, X. Zheng, A. M. Gonzalez-Angulo, X. Su, K. Chen, A. Akcakanat, B. Lim, M. E. Di Francesco, G. Draetta, J. R. Marszalek declare no potential conflicts.
C. Tapia is an employee of Epizyme Inc. and performed contract work for Armo Bioscience.
D. Tripathy reports receiving commercial research grants from Novartis, Polyphor. He also served as a consultant for Pfizer, AstraZeneca, GlaxoSmithKline, Genomic Heath, OncoPep.
T. Yap reports receiving commercial research grants from Artios, AstraZeneca, Bayer, Clovis, Constellation, Cyteir , Eli Lilly, EMD Serono, Forbius, F-Star, GlaxoSmithKline, Genentech, ImmuneSensor, Ipsen, Jounce, Karyopharm, Kyowa, Merck, Novartis, Pfizer, Ribon Therapeutics, Regeneron, Repare, Sanofi, Scholar Rock, Seattle Genetics, Tesaro, and Vertex Pharmaceuticals. He has also served as a consultant for Almac, Aduro, AstraZeneca, Atrin, Axiom, Bayer, Bristol Myers Squibb, Calithera, Clovis, Cybrexa, EMD Serono, F-Star, Guidepoint, Ignyta, I-Mab, Jansen, Merck, Pfizer, Repare, Roche, Rubius, Schrodinger, Seattle Genetics, Varian and Zai Labs.
P. Jones reports serving as a consultant for Tvardi Therapeutics.
T.P. Heffernan reports personal fees and stock ownership from Cullgen Inc.
F. Meric-Bernstam reports receiving commercial research grants from Aileron Therapeutics Inc., AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Millennium Pharmaceuticals Inc., Novartis, Puma Biotechnology Inc., and Taiho Pharmaceutical Co. She also served as a consultant for Aduro BioTech Inc., DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech Inc., IBM Watson, Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics Inc., Tyra Biosciences, Xencor, and Zymeworks. Additionally, she serves on the advisory committee for Immunomedics, Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology Inc., Seattle Genetics, Silverback Therapeutics, and Spectrum Pharmaceuticals.
References
- 1.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer 2009;9 Suppl 2:S73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 2014;384:164–72 [DOI] [PubMed] [Google Scholar]
- 3.Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res 2013;19:5533–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013;23:302–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013;23:811–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toulza E, Mattiuzzo NR, Galliano MF, Jonca N, Dossat C, Jacob D, et al. Large-scale identification of human genes implicated in epidermal barrier function. Genome Biol 2007;8:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036–46 [DOI] [PubMed] [Google Scholar]
- 8.Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med 2018;24:1047–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med 2019;11. [DOI] [PubMed] [Google Scholar]
- 10.Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap TA, Ahnert JR, Piha-Paul SA, Fu S, Janku F, Karp DD, et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. Journal of Clinical Oncology 2019;37:3014- [Google Scholar]
- 12.Gonzalez-Angulo AM, Akcakanat A, Liu S, Green MC, Murray JL, Chen H, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann Oncol 2014;25:1122–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans KW, Yuca E, Akcakanat A, Scott SM, Arango NP, Zheng X, et al. A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin Cancer Res 2017;23:6468–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H, et al. Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. PLoS One 2015;10:e0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arango NP, Yuca E, Zhao M, Evans KW, Scott S, Kim C, et al. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res 2017;19:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schockel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G, et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab 2015;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121:2750–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol 2011;7:e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balaji K, Vijayaraghavan S, Diao L, Tong P, Fan Y, Carey JP, et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol Cancer Res 2017;15:45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies AXL as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19:279–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas JE, Soriano P, Brugge JS. Phosphorylation of c-SRC on tyrosine 527 by another protein tyrosine kinase. Science 1991;254:568–71 [DOI] [PubMed] [Google Scholar]
- 22.Hunter CA, Koc H, Koc EC. c-SRC kinase impairs the expression of mitochondrial OXPHOS complexes in liver cancer. Cell Signal 2020;72:109651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hebert-Chatelain E SRC kinases are important regulators of mitochondrial functions. Int J Biochem Cell Biol 2013;45:90–8 [DOI] [PubMed] [Google Scholar]
- 24.Jin Y, Cai Q, Shenoy AK, Lim S, Zhang Y, Charles S, et al. SRC drives the Warburg effect and therapy resistance by inactivating pyruvate dehydrogenase through tyrosine-289 phosphorylation. Oncotarget 2016;7:25113–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Wang S, Jiang B, Huang L, Ji Z, Li X, et al. c-SRC phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat Commun 2017;8:13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian M, Chen XS, Li LY, Wu HZ, Zeng D, Wang XL, et al. Inhibition of AXL enhances chemosensitivity of human ovarian cancer cells to cisplatin via decreasing glycolysis. Acta Pharmacol Sin 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alfonso TM, Hannah J, Chen Z, Liu Y, Zhou P, Shin SJ. AXL receptor tyrosine kinase expression in breast cancer. J Clin Pathol 2014;67:690–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, et al. AXL is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A 2010;107:1124–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y, et al. Gas6/AXL Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016;6:1205–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng LC, Chen YL, Cheng AN, Lee AY, Cho CY, Huang JS, et al. AXL phosphorylates and up-regulates TNS2 and its implications in IRS-1-associated metabolism in cancer cells. J Biomed Sci 2018;25:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014;5:5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foidart P, Yip C, Radermacher J, Blacher S, Lienard M, Montero-Ruiz L, et al. Expression of MT4-MMP, EGFR, and RB in Triple-Negative Breast Cancer Strongly Sensitizes Tumors to Erlotinib and Palbociclib Combination Therapy. Clin Cancer Res 2019;25:1838–50 [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto T, Kanaya N, Somlo G, Chen S. Synergistic anti-cancer activity of CDK4/6 inhibitor palbociclib and dual mTOR kinase inhibitor MLN0128 in pRb-expressing ER-negative breast cancer. Breast Cancer Res Treat 2019;174:615–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cretella D, Fumarola C, Bonelli M, Alfieri R, La Monica S, Digiacomo G, et al. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci Rep 2019;9:13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.