

Abstract
Purpose:
There is increasing recognition that progress in immuno-oncology could be accelerated by evaluating immune-based therapies in dogs with spontaneous cancers. Osteosarcoma (OS) is one tumor for which limited clinical benefit has been observed with the use of immune checkpoint inhibitors. We previously reported the angiotensin receptor blocker losartan suppressed metastasis in preclinical mouse models through blockade of CCL2-CCR2 monocyte recruitment. Here we leverage dogs with spontaneous OS to determine losartan’s safety and pharmacokinetics associated with monocyte pharmacodynamic endpoints, and assess its antitumor activity, in combination with the kinase inhibitor toceranib.
Experimental Design:
CCL2 expression, monocyte infiltration, and monocyte-recruitment by human and canine OS tumors and cell lines was assessed by gene expression, ELISA, and transwell migration assays. Safety and efficacy of losartan-toceranib therapy was evaluated in 28 dogs with lung metastatic osteosarcoma. Losartan PK and monocyte PD responses were assessed in three dose cohorts of dogs by chemotaxis, plasma CCL2 and multiplex cytokine assays, and RNAseq of losartan-treated human PMBCs.
Results:
Human and canine OS cells secrete CCL2 and elicit monocyte migration which is inhibited by losartan. Losartan PK/PD studies in dogs revealed that a ten-fold-higher dose than typical anti-hypertensive dosing was required for blockade of monocyte migration. Treatment with high-dose losartan and toceranib was well-tolerated and induced a clinical benefit rate of 50% in dogs with lung metastases.
Conclusions:
Losartan inhibits the CCL2-CCR2 axis, and in combination with toceranib, exerts significant biological activity in dogs with metastatic osteosarcoma, supporting evaluation of this drug combination in pediatric osteosarcoma patients.
Keywords: losartan, toceranib, osteosarcoma, dogs, monocytes, immunotherapy
Introduction
Tumor metastasis is a complex multi-step process governed in part by intricate overlapping interactions between tumor cells, immune cells, and their surrounding host environment (1). The chemokine receptor CCR2 and its ligand, CCL2, represent a signaling axis which is implicated as a driver of both tumor cell intrinsic and extrinsic processes of the metastatic cascade (2) (3,4). In pre-clinical mouse models of metastasis, CCR2 expressing inflammatory monocytes (IMs) are preferentially recruited early to metastatic sites via tumor and stromal cell-mediated production of CCL2 (3,5). Once present at the site of metastases, these monocytes can differentiate into metastasis-associated macrophages (MAMs), which play essential roles in metastatic colonization via promotion of tumor cell extravasation, growth, and angiogenesis (3,6). The clinical importance of CCL2-CCR2 signaling in tumor progression is underscored by numerous studies demonstrating the prognostic significance of CCL2 and/or peripheral blood monocyte counts in multiple human tumor types (7–10). Combined, these data highlight this chemotactic axis as an attractive target for metastasis-directed therapies, and small molecule therapeutics targeting CCR2 are currently being evaluated in clinical trials (ClinicalTrials.gov NCT03851237, NCT04123379, NCT03767582, and NCT03496662).
Osteosarcoma (OS) is the most common primary malignant tumor of bone and occurs more frequently in children and adolescents than any other age group (11). Overall survival rates for OS patients have remained unchanged since improvements associated with the introduction of multi-drug chemotherapy protocols in the 1980s . This clinical failure is directly attributable to our inability to treat those ~30% of individuals who remain at high risk to develop tumor recurrence, despite receiving the same first line drug combinations effective in other OS patients(11–13). Metastasis to the lung almost exclusively accounts for these cases of OS recurrence (12,13). Once pulmonary metastasis has developed, therapies with proven clinical benefit beyond surgical resection are extremely limited, and only ~20% of these patients remain alive 4 years after recurrence (12,13). A significant therapeutic advancement in human oncology over the last decade has been the FDA approval of multiple new immunotherapy approaches (14). In patients with other solid tumors such as melanoma, non-small cell lung, renal, gastric, and bladder cancers, monoclonal antibodies targeting immune checkpoint molecules such as programmed cell-death protein 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) have resulted in unprecedented responses in a subset of individuals with advanced stage/metastatic disease (15,16). However, the potential success of immune checkpoint inhibitors has fallen disappointingly short is osteosarcoma (17,18), which is surprising given the high degree of genomic instability in this tumor type (19,20). However, the prior success of the macrophage-activating agent liposomal muramyl tripeptide-phosphatidyl ethanolamine (L-MTP-PE) in this disease suggests that immunotherapeutic approaches targeting the innate immune system still hold promise for OS patients (21–24).
Spontaneously-occurring tumors in pet dogs represent a valuable intermediary animal model for evaluation of novel cancer therapeutics and validation of pre-clinical findings, a translational approach increasingly recognized by the NIH and greater cancer research community [(25) https://grants.nih.gov/grants/guide/rfa-files/rfa-ca-17-001.html]. Canine osteosarcoma (OS) is an archetype of a naturally-occurring canine cancer with the potential to inform immuno-oncology drug development (26). Dogs with OS share many clinical similarities with human pediatric OS including primary tumor location, response to conventional adjuvant therapies, the presence of microscopic lung metastases at diagnosis, and unfortunately, a lack of effective therapies for those high-risk patients with recurrent/metastatic disease (26). Our group has previously shown that elevated peripheral blood monocyte counts are independently associated with decreased survival in dogs with OS (27). More recently, we have shown that the small molecule anti-hypertensive drug losartan potently inhibits monocyte recruitment through noncompetitive inhibition of CCL2-CCR2 signaling induced ERK1/2 activation, independent of its known biological target, the angiotensin II type 1 receptor (AT1R) (28). In these studies, losartan treatment significantly reduced lung metastatic burden in mice, an effect associated with a significant decrease in CD11b+/Ly6C+-recruited monocytes in the lungs. Collectively, our results indicated that losartan could exert antimetastatic activity by inhibiting CCR2 signaling and suppressing monocyte recruitment, a previously undescribed mechanism for this drug, and provided pre-clinical rationale for repurposing losartan as a monocyte-targeted immunotherapy and potential anti-metastatic agent.
To this end, we evaluated CCL2 secretion and losartan’s effect on monocyte recruitment by human and canine OS cells in vitro, as well as the in vivo anti-metastatic activity of losartan in a clinical trial of pet dogs with lung metastatic OS. In this approach, we assessed the safety and anti-tumor activity of escalating doses of losartan in combination with toceranib phosphate (Palladia), one of the only tumor-targeted tyrosine kinase inhibitors approved in veterinary medicine. Translationally, toceranib shares significant structural and functional homology with sunitinib, a multi-kinase inhibitor approved for investigational use in human osteosarcoma. Limited clinical data in tumor-bearing dogs suggests that toceranib may also exert secondary immunomodulatory effects on Tregs, similar to that reported for sunitinib (29–32). Our data demonstrate that high dose losartan – toceranib combination therapy is associated with in vivo modulation of CCL2-CCR2 pharmacodynamic endpoints and a clinical benefit rate of 50% in dogs with advanced lung metastatic OS. Additionally, transcriptomic analysis of human peripheral blood mononuclear cells treated with losartan demonstrated similar monocyte-associated pharmacodynamic effects. Combined, these data support evaluation of this drug combination as a novel therapeutic approach for high-risk metastatic osteosarcoma patients.
Materials and Methods
Cell lines
Human osteosarcoma and THP-1 cells (RRID:CVCL_0006) were obtained from the American Type Culture Collection (Manassas, VA USA), or the University of Colorado Cancer Center Tissue Culture Shared Resource. Canine osteosarcoma cell lines were obtained from the Flint Animal Cancer Center repository, and origin details have been previously described (33). All cell lines were validated by short tandem repeat (STR) analysis and routinely tested for mycoplasma contamination using a commercially available assay (Mycoalert from Lonza Inc.). Cells were maintained in DMEM media (Gibco, Grand Island, NY USA) supplemented with 10% fetal bovine serum (FBS; Atlas Biologicals, Fort Collins, CO USA), penicillin (100 U/mL), streptomycin (100 μg/mL), L-glutamine (2 mM), and non-essential amino acids (0.1 mM) (All from Gibco). Cells were grown sterilely on standard plastic tissue culture flasks (Cell Treat, Shirley, MA), under standard conditions of 37 °C, 5% CO2, and humidified air.
Patient enrollment
Dogs enrolled in these studies were client-owned dogs presenting to the Colorado State University (CSU) Flint Animal Cancer Center (Fort Collins, CO). Trial enrollment was in compliance with the Clinical Review Board and Animal Care and Use Committee of Colorado State University recommendations, and signed informed consent was obtained from all dog owners. For study inclusion, all patients were required to have a previous histopathologically confirmed diagnosis of OS, with measurable pulmonary metastasis documented via thoracic radiographs. Prior treatment of the primary bone tumor (typically involving the limb) by surgical amputation was also required. Previous cytotoxic chemotherapy or other antineoplastic treatment was acceptable with a 2-week washout period. Prior to study entry, dogs were assessed by physical examination, and standard laboratory tests including complete blood count (CBC), serum chemistry, urinalysis, urine protein:creatinine ratio (UPCR) and systolic blood pressure to ensure that inclusion criteria were met. Dogs were included in the study if the above clinical criteria were met, were deemed to have adequate organ function (as determined by the standard laboratory tests) and had a modified Eastern Comparative Oncology Group (ECOG) score of 0 or 1. Baseline 3-view thoracic radiographs were obtained and evaluated by a board-certified radiologist. Target lesions (up to 5) were identified, and measurements (longest diameters) were recorded.
Patient monitoring and treatment evaluations
Dogs enrolled in the trial were re-assessed at weeks 1, 2, and 4 post-initiation of therapy, and every 4 weeks thereafter for the duration of the study. Adverse events (AEs) were recorded for all dogs in the study and graded according to the Veterinary Cooperative Oncology Group (VCOG) Common Terminology Criteria for Adverse Events v1.1 (34) based on owner history, physical examination, CBC, serum chemistry, urinalysis, and UPCR. Reductions in dosage and/or frequency were permitted to manage AEs which were attributed to losartan and/or toceranib. Treatment response was evaluated according to the published VCOG Response Evaluation Criteria for Solid Tumors in Dogs v1.0 (35), and were based on longest diameter measurements obtained for target lesions identified at baseline thoracic radiographs. Dogs were considered evaluable for response if they remained on trial for at least one cycle of follow-up thoracic radiographs.
Responses were monitored at week 8 and every 8 weeks thereafter in the 1 mg/kg cohort, while dogs in the 10 mg/kg losartan dose cohort were also evaluated by additional thoracic radiographs at week 2. Dogs experiencing stable disease, or a partial or complete response at week 2 (10 mg/kg cohort) or week 8 were allowed to remain on study. At each recheck examination, whole blood, serum and plasma were collected for immune endpoints. Serum and plasma were aliquoted and stored at −80 °C for batch cytokine analysis by ELISA or multiplex analysis. Blood was processed within 1 hour of collection by ACK lysis, and freshly isolated PBMCs were immediately assayed for CCL2-directed ex vivo monocyte migration (as described in the supplemental methods).
Cytokine analysis of patient samples and cell culture supernatant
Commercially available human and canine CCL2 ELISA kits (R&D Systems Inc., Minneapolis, MN USA) was used to measure the concentration of CCL2 in pre- and post-treatment patient plasma samples and in osteosarcoma tumor-conditioned cell culture media. In addition, interferon-gamma (IFN-γ) and vascular endothelial growth factor (VEGF) were also quantified in pre- and post-treatment canine patient plasma samples by commercial ELISA (R&D Systems). Multiplex cytokine analysis of patient plasma was conducted using a commercially available canine 13-plex cytokine kit run according to the manufacturer’s instructions (MilliporeSigma, Burlington, MA USA). Samples were run on a Luminex MagPix instrument. Quality control and initial processing of data was performed with Luminex xPONENT software, prior to export and further analysis with GraphPad Prism version 9.1.0 (La Jolla, CA USA).
Losartan pharmacokinetics
For assessment of losartan trough plasma concentrations in the 1 mg/kg dose cohort of treated dogs, weekly EDTA-treated blood samples were collected via jugular venipuncture approximately 6 hours following oral administration of losartan and stored at −80°C prior to batch analysis. For the losartan dose escalation cohorts, healthy dogs or dogs with OS metastasis received losartan (2.5 mg/kg or 10 mg/kg PO BID.) for 14 days. On day 14, a catheter was placed in the lateral saphenous vein for repeated sampling, and blood was collected at 0.25, 0.5, 1, 2, 4, 6, and 12 hours post dosing. Additional details regarding specific LC/MS methods and data analysis are provided in Supplementary methods.
Cancer Cell Line Encyclopedia, RNAseq data of human primary and lung metastatic osteosarcoma tumors, and canine tumor cell line microarray data.
Cancer Cell Line Encyclopedia (CCLE) RNAseq data in the form of RPKM expression values in .gct.gz file format (CCLE_RNAseq_genes_rpkm_20180929.gct.gz) was downloaded from the Broad Institute CCLE website (https://portals.broadinstitute.org/ccle) on April 14, 2020. CCL2 mRNA expression data was extracted from the gct file in RStudio and z-transformed across all cell lines for comparison between osteosarcoma cell lines versus all other tumor type categories.
CCL2 mRNA expression across the canine tumor cell line panel was evaluated using a microarray expression data set previously generated at the Flint Animal Cancer Center by the Duval Laboratory (36). Briefly, RNA samples were sent to the Genomics and Microarray Core at the University of Colorado, and were hybridized onto Affymetrix GeneChip Canine Genome 2.0 arrays, Canine Gene 1.0 ST arrays, and GeneChip miRNA 4.0 arrays. Resulting CEL files of expression data were then imported into Bioconductor (RRID:SCR_006442) (37), and intensity values were preprocessed with the Robust Multi-Array Average (RMA) algorithm (38). Expression values for CCL2 were z-transformed for comparison across cell lines. POS
FASTQ RNAseq files from de-identified human OS tumor samples previously published by Wu, Beird, et al., (19) were kindly provided by the Futreal laboratory at MD Anderson Cancer Center. Briefly, raw FASTQ files were trimmed using Trimmomatic (RRID:SCR_011848) (v0.36) and trimmed reads were mapped against the GRCh38 human genome using STAR (RRID:SCR_004463) (v2.6.1a). Genes were annotated using Ensembl (RRID:SCR_002344) (v100) gtf files. The relative expression of genes (FPKM) was generated using cufflinks (RRID:SCR_014597) (v2.2.1), and gene count data was generated using HTseq-count (v0.9.1) resources. The relative abundance of monocytes in primary tumors vs. lung metastases was determined using a combination of previously described immune cell gene expression signatures (39,40). An individual sample’s monocyte expression score was calculated as the average of FKPM expression values of the markers for that cell type.
RNA sequencing and analysis of losartan-treated human PBMCs
EDTA-treated whole blood was obtained from healthy male and female human donors following obtainment of written informed consent. The study was conducted under Institutional Review Board approval by Colorado State University and in accordance with the Declaration of Helsinki. PBMCs were obtained from whole blood via ficoll density gradient centrifugation, plated in 24 well cell culture plate at 3e6 per mL in RPMI plus 10% FBS, 1% penicillin/streptomycin and 1% L-glutamine. Losartan (TCI, cat#L0232) was reconstituted in PBS and added to treated wells to a final concentration of 10 μg/mL. Donor PBMCs were matched for use in control and losartan-treated wells. Cells were treated for 24 hours and then RNA was extracted using a Qiagen RNeasy mini kit. Extracted RNA was sent to Novogene Corp Ltd. for sequencing. Samples were tested for quality control by Agilent 2100 Bioanalyzer system and by agarose gel electrophoresis. Libraries were run on Illumina PE150 (HiSeq) for 40M raw reads/sample. FASTQ files from Novogene were analyzed using Partek Flow software, version 9.0. Briefly, raw data were filtered by removing adapters and N > 10%, and selected for Phred (RRID:SCR_001017) score >30. Filtered reads were then aligned with Bowtie2 (RRID:SCR_016368) and the number of mapped reads per gene was counted using HTseq-count (RRID:SCR_011867). The count data was read R (v3.6.1) and normalized using RUVSeq (RRID:SCR_006263) (v1.20) package, specifically using RUVr function (41). The normalized count data was used to identify differentially expressed genes between control (untreated) and losartan-treated PBMCs. Differential gene expression analysis was performed using the limma-voom (3.42.2) and EdgeR (RRID:SCR_012802) (v3.28.1) packages.
Gene Set Enrichment Analysis (GSEA; https://www.gsea-msigdb.org/gsea/downloads.jsp) was performed according to the user guide (https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html) to determine biological pathways differentially enriched between losartan-treated and control primary human donor PBMCs (42). GSEA analysis was performed using publicly available gene sets housed in the Molecular Signatures Database (MSigDB; https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) and the following parameters: the Gene Ontology Biological Process gene set (ftp.broadinstitute.org://pub/gsea/gene_sets/c5.bp.v7.1.symbols.gmt), with Signal2Noise ranking metric, permutation type set to “gene set,” and 1000 permutations used to calculate statistical significance of enrichment scores. Statistically significant pathways were defined as those with P < .05 and false discovery rate (FDR) adjusted q < 0.05.
Statistical analyses
For the comparison of mean values between three or more groups, a one-way ANOVA with Tukey’s post-test was performed. For comparison of means between two groups, or comparison of repeated measures between two groups, a two-tailed, unpaired Students’ t test or Wilcoxon matched-pairs t test was used, respectively. Progression-free survival (PFS) was calculated from the date of administration of the first dose of losartan/toceranib to the date of PD. Kaplan-Meier estimation was used to determine PFS and overall survival, and statistical comparison between groups was done by log rank test. All statistical analyses were performed using Graph Pad Prism software (RRID:SCR_002798) (La Jolla, CA, USA).
Data availability statement
The mRNA sequence data generated in this study are publicly available in the NIH Sequence Read Archive (SRA), BioProject ID: PRJNA763881.
Results
Human and canine osteosarcoma cells overexpress CCL2 and are enriched for monocytes in lung metastases
To determine whether human and canine osteosarcoma cells preferentially overexpress CCL2 as compared to other tumor types, we analyzed CCL2 gene expression z-scores obtained from microarray data of a panel of 37 canine tumor cell lines (representing 10 different histo-types) and RNAseq data from the Broad Institute Cancer Cell Line Encyclopedia (CCLE). Both human and canine OS cell lines demonstrated significant overexpression of CCL2 as compared to other tumor histo-types (Fig. 1A and B, P < 0.05). Based on this overexpression of CCL2, we also sought to assess whether there was a preferential increase in monocyte infiltration of canine patient-derived OS lung metastases as compared to primary tumors. Immunolabeling and quantitative image analysis for MAC387 demonstrated a significant increase in MAC387+ cells in canine OS lung metastases as compared to unmatched primary appendicular tumors from patients with stage II clinical disease (Fig. 1C, D, P < 0.01). Immunolabeling for CCL2, performed on these same pulmonary metastases, demonstrated that this MAC387+ infiltrated was associated with strong, peri-nuclear, cytoplasmic positive immunolabeling of CCL2 in >75% of tumor cells in all cases (Fig. 1E). Additionally, we analyzed previously published RNAseq data of human primary appendicular OS tumors and lung metastases (19) to determine whether there was enrichment for expression of previously published monocyte immune gene signatures in these tumors (see methods). In this analysis, we identified preferential monocyte immune gene signature enrichment in human OS lung metastases as compared to primary tumors (Fig. 1F, P = 0.1).
Fig. 1.
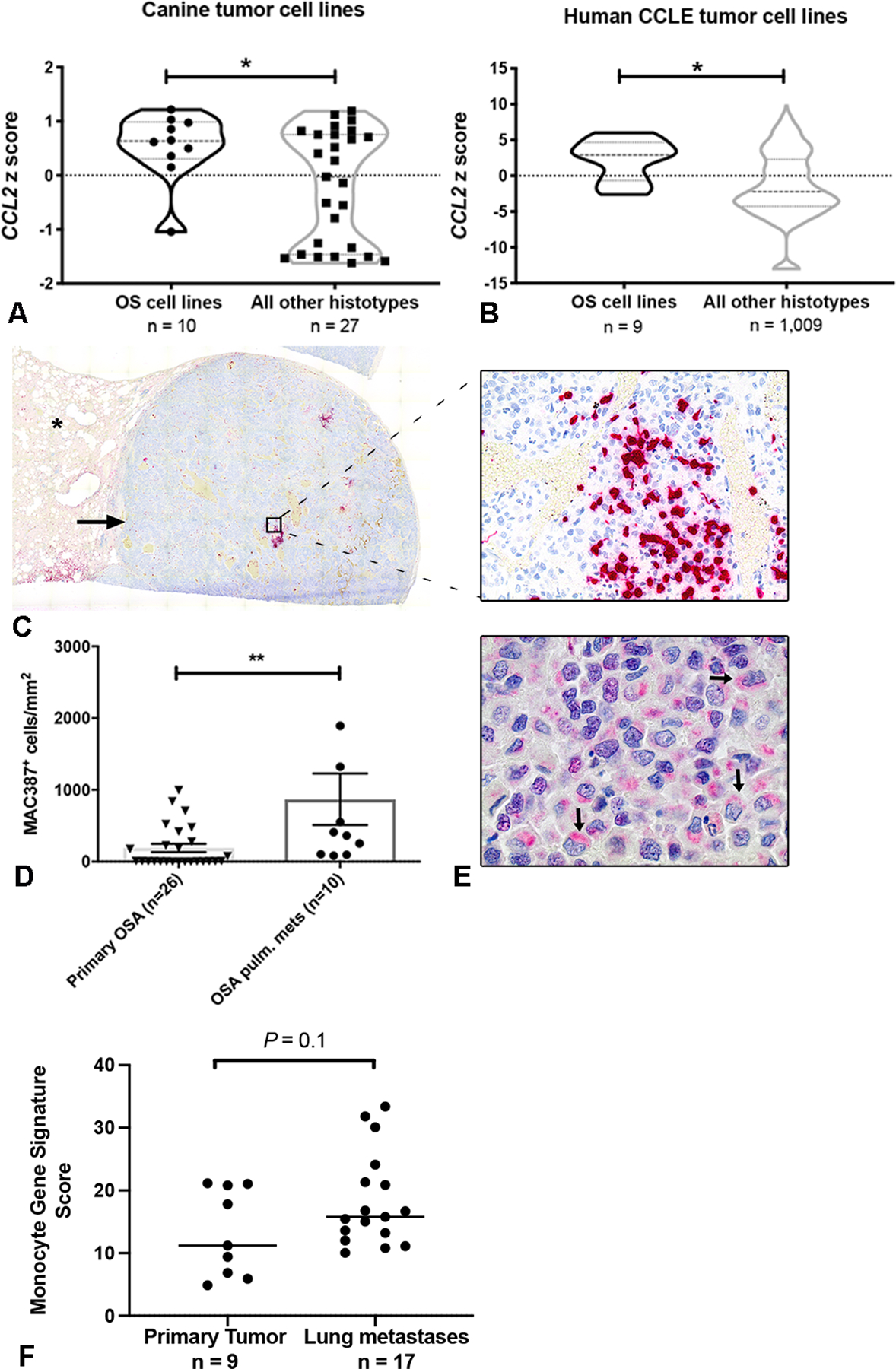
Canine and human osteosarcoma cells express CCL2 and are enriched for monocytes within pulmonary metastases. (A) CCL2 mRNA expression in canine OS cells, as compared to all cell lines of non-osteosarcoma histo-type. *P = 0.04, unpaired two-tailed Student’s t test. (B) CCL2 mRNA expression in human OS cells as compared to all cell lines of non-osteosarcoma histo-type within the Broad Institute Cancer Cell Line Encyclopedia (CCLE). *P = 0.01, unpaired two-tailed Student’s t test. (C) Sub-gross overview and corresponding 400x magnification image of a canine osteosarcoma pulmonary metastasis containing extensive intra-tumoral infiltrates of MAC387+ monocytes and macrophages (cells labeled in red; asterisk denotes normal lung parenchyma). (D) 1,000x magnification image of the same metastatic lesion shown in (D) demonstrating strong cytoplasmic, peri-nuclear positive immunolabeling of canine OS tumor cells for CCL2 (arrows). (E) Quantitative image analysis of MAC387+ myeloid cells in osteosarcoma pulmonary metastases as compared to primary tumors (n=10 and 26 animals per group, respectively). **P = 0.004, two-tailed Mann Whitney test, data plotted as mean ± SEM. (F) Mean expression of monocyte immune signature genes in human osteosarcoma pulmonary metastases vs. primary tumors using RNAseq data obtained from CC Wu et al. P = 0.1, unpaired two-tailed Student’s t test.
Losartan inhibits in vitro human and canine osteosarcoma-elicited monocyte migration
To determine whether human and canine OS cells were capable of eliciting monocyte migration, we generated OS tumor cell-conditioned media from a panel of 4 human and 4 canine OS cell lines for use in trans-well monocyte migration assays. Assessment of these supernatants by ELISA demonstrated secretion of CCL2 by 4 of 4 human and 3 of 4 canine OS cell lines evaluated (Fig. 2A and B). Significant THP-1 cell migration in response to these supernatants was observed in 2 of 4 human OS cell lines (MG63.0 and 143b, Fig. 2C), while supernatants from all 4 human OS cell lines stimulated significant migration of human PBMCs obtained from healthy male and female donors (Fig. 2D). Similarly, canine PBMCs obtained from healthy dogs demonstrated significant migration to supernatants from all 3 canine OS cell lines found to produce CCL2 by ELISA (Fig. 2E). When combined, we observed a statistically significant positive correlation between the level of CCL2 production and degree of trans-well monocyte migration across all 8 tumor cell lines and all three evaluated monocyte cell types for both humans and dogs (Fig. 2F).
Fig. 2.
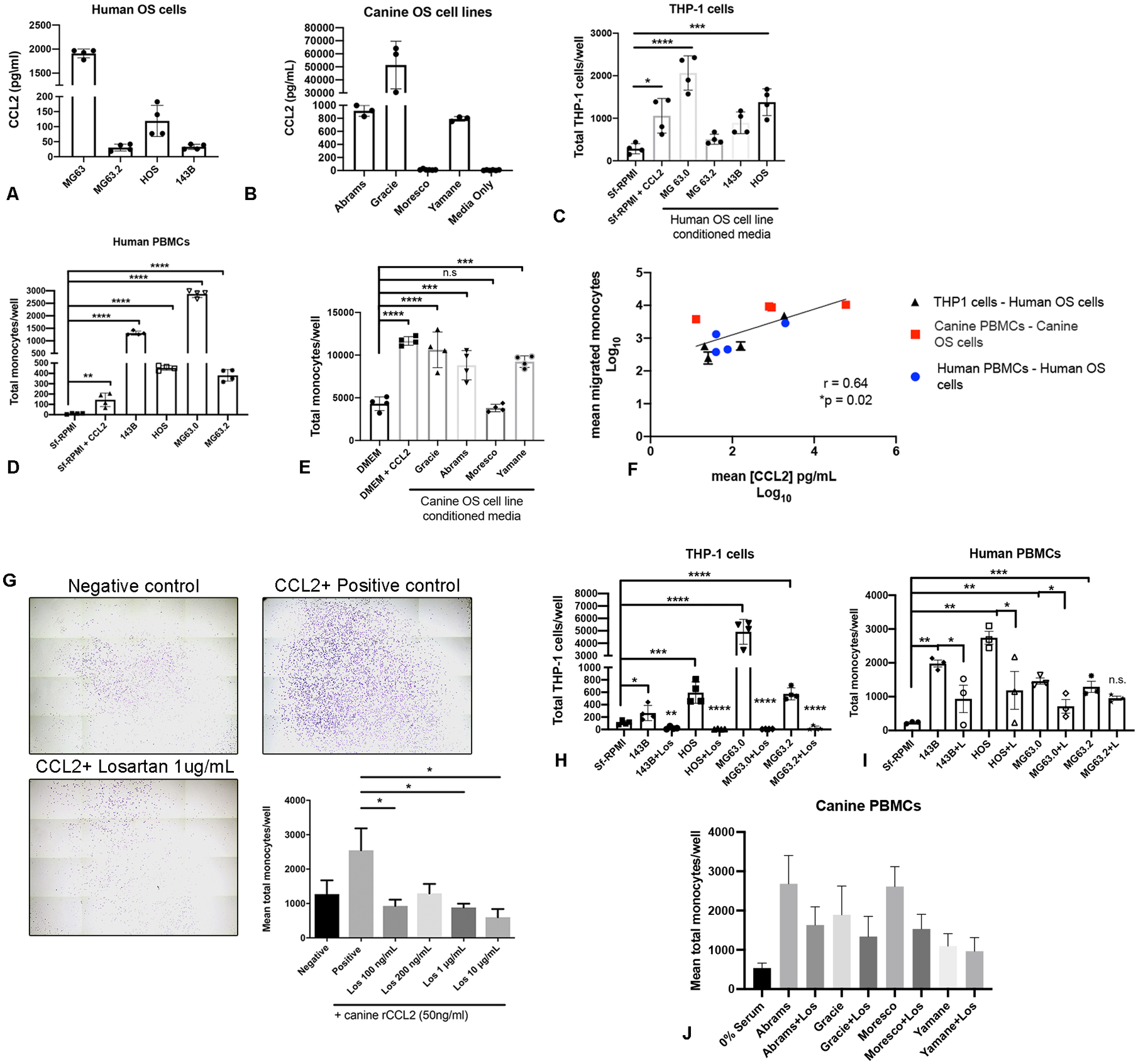
Losartan inhibits in vitro monocyte migration elicited by canine and human osteosarcoma cell secretion of CCL2. (A) CCL2 secretion by human OS cells quantified via ELISA. (B) CCL2 secretion by canine OS cells quantified via ELISA. (C) THP-1 monocyte trans-well migration to human OS cell line conditioned media. Data represents mean ± SD, analyzed by one-way ANOVA with Tukey’s post-test, *P = 0.01, ***P < 0.001, ****P < 0.0001. (D) Primary human donor peripheral blood mononuclear cell (PBMC) trans-well migration to human OS cell line conditioned media. Data represents mean ± SD, analyzed by one-way ANOVA with Tukey’s post-test, **P < 0.01, ****P < 0.0001. (E) Primary canine donor peripheral blood mononuclear cell trans-well migration to canine OS cell line conditioned media. Data represents mean ± SD, analyzed by one-way ANOVA with Tukey’s post-test, ***P < 0.001, ****P < 0.0001. (F) Correlation of log10 transformed mean CCL2 secretion (pg/mL) in both canine and human OS cell line conditioned media with log10 transformed mean human THP-1 monocyte, human PBMC and canine PBMC trans-well migration. Spearman r = 0.64. (G) Representative whole well images (10x magnification) and quantification of losartan inhibition of CCL2 directed canine peripheral blood monocyte migration (n = 4 canine donors). Crystal violet staining. Data represents mean ± SEM of each donor, analyzed by one-way ANOVA with Tukey’s post-test, *P < 0.05. (H) Losartan inhibition of THP-1 monocyte migration to human OS cell line conditioned media. Data represents mean ± SD, analyzed by one-way ANOVA with Tukey’s post-test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (I) Losartan inhibition of human PBMC migration to human OS cell line conditioned media. Data represents mean ± SD, analyzed by one-way ANOVA with Tukey’s post-test, *P < 0.05, **P < 0.01, ***P < 0.001. (J) Losartan inhibition of canine PBMC migration to canine OS cell line conditioned media, n = 5 independent canine donors. All THP-1, human PBMC, and canine PBMC trans-well migration assays were performed in technical triplicate or quadruplicate and are representative of a minimum of 2 different canine or human blood donors or 3 independent experiments (THP-1 cells).
We previously demonstrated that losartan inhibited CCL2-CCR2 mediated signaling and CCL2-directed migration in human and murine monocytes (28). However, prior to initiating clinical studies of this drug in tumor-bearing dogs, we sought to address whether 1) losartan was capable of blocking canine monocyte migration, as poor cross-species reactivity for small molecule CCR2 antagonists has been previously documented for other compounds (43), and 2) if this effect also translated to blockade of monocyte migration elicited specifically by OS tumor cells. In vitro losartan treatment of canine PBMCs significantly reduced CCL2-directed migration in a dose-dependent manner, to levels ranging from 42% (losartan [100 ng/mL]) to 12% (losartan [10 μg/mL]) of those observed for untreated positive control wells (Fig. 2G *P<0.05). Additionally, 1 hr pre-treatment of cells with 10 μg/mL losartan also resulted in significant inhibited of human THP-1 and human PBMC migration, and to a lesser extent canine PBMC migration, to OS conditioned media from all evaluated cell lines (Fig. 2H–J).
Clinical evaluation of losartan-toceranib combination therapy in dogs with spontaneous metastatic OS
Patient characteristics
Dose-escalation PK/PD studies of losartan to determine a biologically effective dose on monocyte pharmacodynamic endpoints were initiated in both healthy dogs and dogs with metastatic OS. Thirty-one dogs with metastatic OS that met eligibility criteria were enrolled in the study, with 8 dogs enrolled in the initial 1 mg/kg losartan dose cohort, 3 healthy dogs enrolled in a 2.5 mg/kg losartan dose cohort, and 20 dogs enrolled in the 10 mg/kg losartan dose cohort. All patients had prior surgical removal of their primary tumor, which included amputation for 25 cases of appendicular OS, amputation and hemi-pelvectomy for a single case of axial (right ischium) OS (10 mg/kg cohort), and hemi-mandibulectomy for two cases of mandibular OS (1 in each cohort). Prior adjuvant chemotherapy had been administered in 24 dogs and consisted of single agent carboplatin treatment (n=19), combination carboplatin-doxorubicin (n=4), and carboplatin plus rapamycin (n=1). All patients receiving post-amputation NSAID (carprofen) therapy prior to enrollment continued to receive these medications concomitantly on a daily or as needed basis and included 2 of 8 patients in the 1 mg/kg losartan cohort and 9 of 16 evaluable patients in the 10 mg/kg losartan cohort. None of the patients enrolled in the trial received prior bisphosphonate therapy. Four patients did not receive prior therapy due to the presence of pulmonary metastases at the time of diagnosis. Baseline characteristics for all patients, including information on primary tumor and prior therapies, are presented in Table S1.
Safety and toxicity
Data from 28 dogs was available for assessment of toxicity associated with combined losartan and toceranib therapy. In addition, data for single agent high dose losartan (10 mg/kg) toxicity assessment were available for 4 dogs treated with losartan only for 2 weeks prior to initiation of toceranib. Concurrent oral administration of losartan and toceranib was well tolerated by dogs in both the 1 mg/kg and 10 mg/kg losartan dose cohorts (Table S2). Importantly, no dogs experienced any significant hypotension associated with losartan treatment (repeated blood pressure measurements for dogs in the 10 mg/kg losartan cohort are shown in Fig. S1).
Toxicities directly attributable to high dose losartan therapy were assessed in a subset of 4 dogs treated with single agent losartan (10 mg/kg) for two weeks prior to initiation of toceranib therapy (Table S2). Losartan toxicities observed during this period were limited and, no significant increases in frequency, severity, or duration of toxicities were observed with concurrent administration of toceranib in the 10 mg/kg losartan dose cohort (Table S2). Observed toxicities were primarily grade 1 and 2 and most commonly included neuromuscular (fore-/hindlimb) weakness, hyporexia, and diarrhea. One dog experienced a persistently increased urine protein:creatinine (UPC) ratio above baseline. This dog had a pre-existing elevated UPC ratio of 1.6 at enrollment, but which increased to 4.2 over the 16-week course of therapy.
Losartan pharmacokinetics and monocyte pharmacodynamic responses
An initial losartan dose of 1 mg/kg was selected for the clinical trial, as this is the dose previously published and reported to be safe for the treatment of proteinuria in dogs (44). Monocyte pharmacodynamic responses to losartan therapy were evaluated via quantification of ex vivo PBMC migration to CCL2 and ELISA measurement of plasma CCL2 concentrations. Compensatory elevation in plasma CCL2 levels has been shown previously to be a valid and reliable, mechanism of action-based pharmacodynamic endpoint for CCR2 antagonists in mouse models as well as in human clinical trials [ClinicalTrials.gov NCT01215279, (45,46)]. Samples from 8 dogs in the 1 mg/kg cohort were available for evaluation of monocyte migration and losartan pharmacokinetics, while 6 of 8 dogs had plasma samples available for CCL2 measurement. In this 1 mg/kg cohort, CCL2-mediated ex vivo monocyte migration was unexpectedly increased following losartan treatment (Fig. S2 A).
Moreover, PK analysis demonstrated very low losartan plasma concentrations, with a mean plasma concentration of 6.8 ng/mL (Fig. S2 B), and at each evaluated time point (2, 4, and 8-weeks post treatment),1 to 3 dogs in this cohort had drug levels below the lower limit of quantification (<1 ng/mL). We also noted that 5 of 6 dogs had increases in plasma CCL2 at 2 weeks post treatment, ranging from 119 to 288% above pre-treatment baseline values (Fig. S2 C); though this increase was not significant.
Given the low plasma concentrations of losartan and lack of monocyte pharmacodynamic responses, an interim dose escalation study of losartan to 2.5 mg/kg BID was performed in a cohort of healthy dogs (n=3). Drug concentration curves for this cohort (Fig. 3A), revealed a mean losartan Cmax of 76.7 ng/mL, and a mean AUC0–12h of 18.2 μg • min • mL−1 (Table S3). The mean inhibition of monocyte migration in these dogs was 27%, with a monocyte chemotactic index (as % of baseline) after 2 weeks of losartan dosing of 73 % (range of 42 – 132%) (Fig. 3B). These data demonstrated a dose-dependent increased effect of losartan on CCL2-mediated monocyte pharmacodynamic responses as compared to PD effects observed in the 1 mg/kg cohort. Nonetheless, maximum losartan plasma concentrations and exposure levels were still significantly below the desired therapeutic levels identified from in vitro migration assays (Fig. 2G).
Fig. 3.
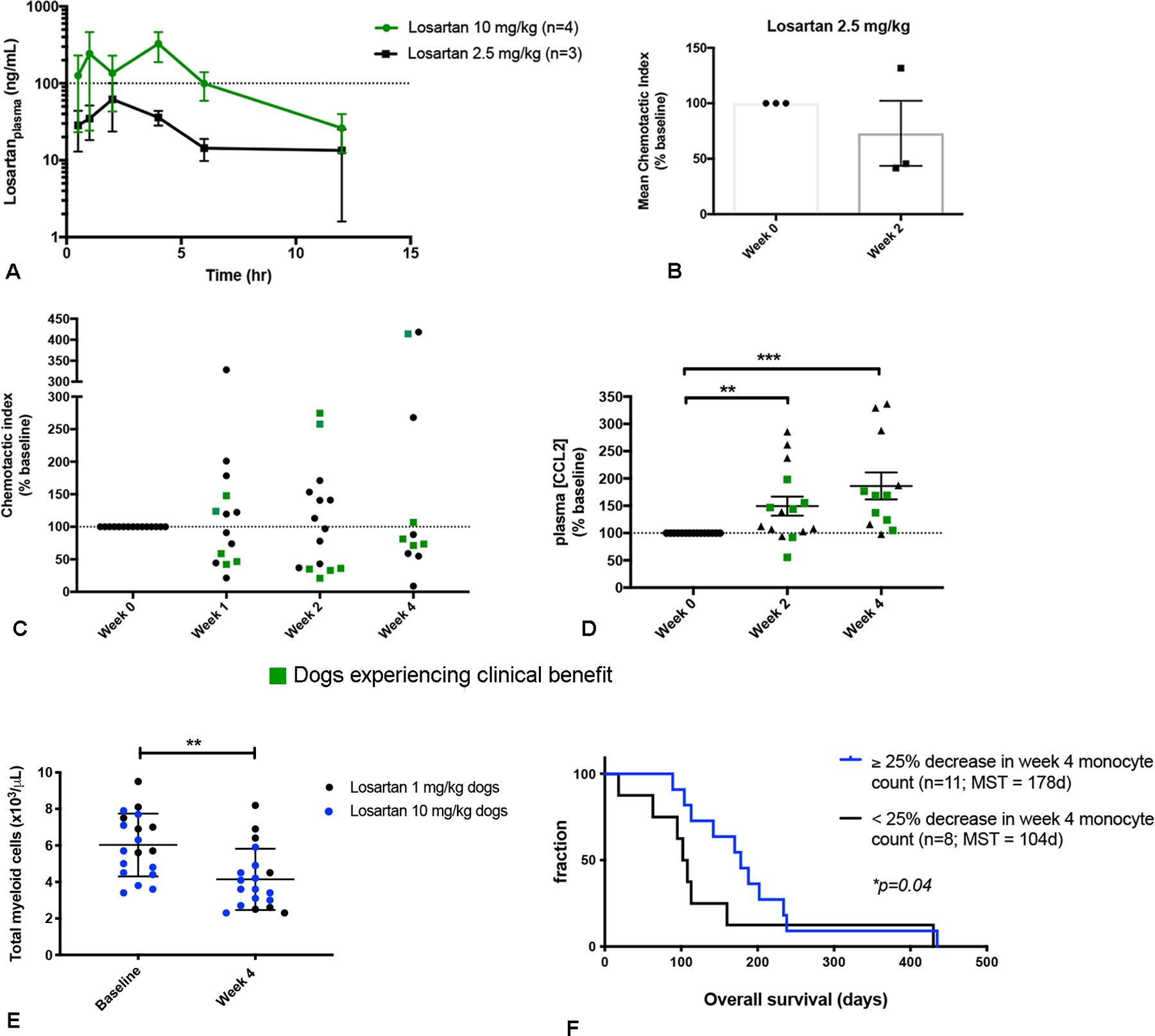
Dose-escalation of losartan in healthy and osteosarcoma metastasis-bearing dogs is associated with modulation of CCL2-CCR2 pharmacodynamic endpoints. (A) Mean (± SEM) plasma losartan concentration over time following oral administration of losartan at 2.5 mg/kg or 10 mg/kg BID for 14 consecutive days (n=3 or 4 dogs/group). (B) Pharmacodynamic assessment of ex vivo CCL2-directed monocyte migration pre- and 14 days post dosing of losartan at 2.5 mg/kg BID in healthy dogs. (C) Pharmacodynamic assessment of ex vivo CCL2-directed monocyte migration pre- and 7, 14, and 28 days post oral dosing of losartan (10 mg\/kg BID) plus toceranib (2.75 mg/kg EOD; day 28) in osteosarcoma-bearing dogs. (D) Plasma CCL2 concentration at week 0 and weeks 2 and 4 post losartan 10 mg/kg dosing, as measured by ELISA (expressed as % of week 0 baseline). (E) Peripheral blood total myeloid cell counts (combined absolute monocyte and neutrophil counts) at baseline and week 4 post losartan treatment. (F) Kaplan-Meier curve comparing overall survival in dogs experiencing a greater than 25% decrease in week 4 post-treatment total monocyte counts below baseline (n=11), vs. those that did not (n=8), for evaluable dogs in both the 1 mg/kg and 10 mg/kg cohorts. (P = 0.04, Gehan–Breslow–Wilcoxon test). Results for B & C represent the mean chemotactic index (fold-change of CCL2-directed migration over negative control wells), as percentage of week 0 baseline. Data expressed as means ± SEM and were analyzed by a two-tailed paired t test, (n=15–17 dogs/group/time-point. **p < 0.01, ***p < 0.001).
Previous pharmacokinetic evaluation of losartan demonstrated a strong linear relationship between losartan dose and plasma Cmax and systemic exposure (AUC) (47). Thus, by extrapolating from our two dose cohorts, it was predicted that a losartan dose of 10 mg/kg given twice daily would achieve the desired maximum plasma concentration and overall exposure (AUC) of losartan required for inhibition of monocyte responses in vivo. Therefore, a third dose level was initiated with dogs treated with losartan at 10 mg/kg BID, in combination with toceranib. The first 4 dogs enrolled in this third cohort received single-agent losartan therapy for an initial two-week lead-in period to eliminate the possibility of pharmacokinetic and/or toxicity interactions between losartan and toceranib. At a losartan dose of 10 mg/kg BID, the mean maximum losartan concentration and exposure were significantly above target therapeutic levels determined in our prior in vitro migration assays, with a mean losartan Cmax of 508.6 ng/mL, and a mean AUC0–12h of 94.9 μg • min • mL−1 (Fig. 3A and Table S3). Thus, the 10 mg/kg dose of losartan met the desired PK parameters.
Pharmacodynamic assessment of monocyte migration in dogs treated at the 10 mg/kg losartan dose showed a significant reduction in the mean chemotactic index (expressed as % of baseline) after 1-week of losartan therapy in all 4 dogs receiving losartan monotherapy (mean 54.4 %, range of 21.3 – 91%) (Fig. S3, *P < 0.05). For all dogs enrolled in this high dose losartan cohort with post-treatment peripheral blood samples available for evaluation, reduction of CCL2-medated ex vivo monocyte migration from baseline was observed in 7/14 dogs at week 1, 8/15 dogs at week 2, and 7/11 dogs at week 4 (Fig. 3C). Additionally, 10 mg/kg losartan therapy resulted in significant post-treatment increases in plasma CCL2 levels in 12 of 15 dogs at week 2 and 11 of 12 dogs at week 4 with plasma samples available for evaluation, with mean increases of 149.4 % and 186.3 % above baseline, respectively (Fig. 3D, **P = .0084, and ***P = .001). Furthermore, we observed changes in absolute peripheral blood total myeloid and monocyte cell counts which were associated with treatment and or overall survival. Combined assessment of losartan-treated dogs with evaluable CBC data in the low and high-dose cohorts (n=19) demonstrated a significant reduction in week 4 post-treatment total peripheral blood myeloid cells (combined monocyte and neutrophil counts; Fig. 3E). Additionally, following median dichotomization of 1-month post-treatment peripheral blood monocyte counts for all evaluable dogs in both the 1 and 10 mg/kg cohorts (n=19), overall survival was significantly longer in dogs with a ≥25% decrease in monocyte counts (median OS = 178 days), as compared to dogs experiencing a < 25% decline in monocyte count below baseline (median OS = 104 days) (Fig. 3F, Gehan-Breslow-Wilcoxon test, *P = 0.04, HR 1.7, 95% CI: 0.15 to 1.24). Together, with the observed significant elevations in plasma CCL2 levels and consistently lower ex vivo monocyte migration at 1, 2, and 4 weeks post 10 mg/kg losartan dosing, these data provide evidence for increasing modulation of the CCR2 target and inhibition of monocyte migration at escalated doses of losartan.
Clinical responses to losartan-toceranib combination therapy
Progression-free survival (PFS) was evaluable for 7 of 8 dogs in the 1 mg/kg losartan dose cohort, with 1 dog censored at time of removal from the trial due to owner perceived AE. Four dogs were censored from PFS analysis and were also not evaluable for radiographic response in the 10 mg/kg cohort due to removal of 1 dog from the trial for an AE, removal of another two dogs due to owner perceived drug safety concerns, at days 6 and 7, and euthanasia of one dog on day 4 due to hind limb paresis not attributable to disease progression. Radiographically measurable responses were evaluable in 5 of 8 dogs in the 1 mg/kg losartan trial, as 3 dogs developed progressive disease prior to repeat thoracic radiographs. Response data for all evaluable dogs are summarized in Table S4. The median progression-free survival time (PFS) was 61 days (range 17–127 days) for the 1 mg/kg cohort, and 111 days (range 14–240 days) for the 10 mg/kg cohort (Fig. 4A). In comparison, 22 dogs with metastatic OS enrolled into a contemporaneous clinical trial of single agent toceranib conducted at the Colorado State University and the University of Wisconsin Veterinary Teaching Hospitals had a median PFS of 57 days (range 7–176 days) (Fig. 4A). Best responses in the 1 mg/kg cohort included stable disease in 3 of 8 dogs (of 11-, 16-, and 17-weeks duration) for an overall clinical benefit rate of 37.5%. In the 10 mg/kg dose cohort, partial responses occurred in 4/16 dogs, with reductions in the sum diameters of metastatic target lesions of 45%, 68%, 70% and 73% (Fig. 4B), and a median duration of response of 163 days (range 119–240 days; Fig. 4C). Additionally, clinically meaningful stable disease (> 8 weeks) was noted in another 4 dogs (median response duration of 139 days, range 113–329 days; Fig. 4C), for an objective response rate of 25% and a clinical benefit rate of 50% in the 10 mg/kg cohort (Table S4). In comparison, no partial responses were observed in single agent toceranib-treated dogs, while 3 of 17 dogs experienced durable stable disease, for an overall clinical benefit rate of 18% (Table S4). Overall survival was 89 days for single agent toceranib-treated dogs, 109 days for 1 mg/kg losartan-toceranib treated dogs, and 148 days for 10 mg/kg losartan-toceranib treated dogs (Fig. 4D). Baseline and 8- and 16-week post-treatment thoracic radiographs are presented in Figs. 4E and 4F for two dogs experiencing partial responses. Of note, one of the dogs in the 10 mg/kg cohort that experienced a partial response eventually died secondary to OS metastases to the right scapula and distal ulna; however, on necropsy only microscopic evidence of pulmonary metastasis was identified (Fig. 4E H&E image inset), with no grossly visible lung metastases observed.
Fig. 4.
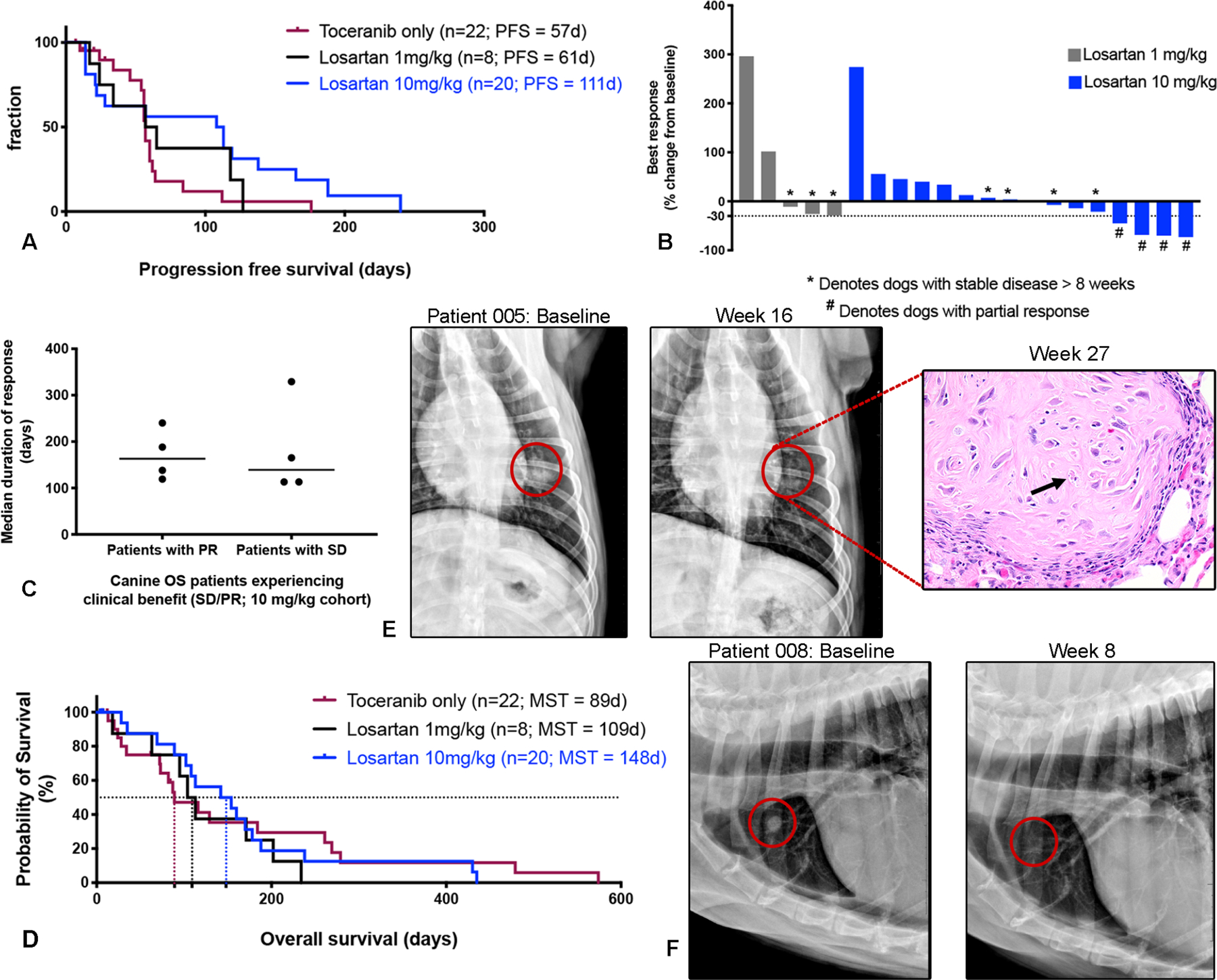
High dose losartan results in objective responses in dogs with metastatic osteosarcoma. (A) Kaplan-Meier curve comparing progression free survival in dogs treated with either single agent toceranib (n=22), low dose losartan (1 mg/kg) + toceranib (n=8), or high dose losartan (10 mg/kg) + toceranib (n=20). Log rank test for trend P =0.058. (B) Best responses to losartan-palladia combination therapy, as determined by RECIST criteria (shown as % change from baseline) for dogs which remained on study for at least one cycle of repeat thoracic radiographs. * Denotes dogs with stable disease, and # denotes dogs experiencing a partial response. (C) Median duration of response for those dogs experiencing clinical benefit (stable disease or partial response) in the high dose losartan (10 mg/kg) cohort. (D) Kaplan-Meier curve comparing overall survival in dogs treated with either single agent toceranib (n=22), low dose losartan (1 mg/kg) + toceranib (n=8), or high dose losartan (10 mg/kg) + toceranib (n=20). Log rank test for trend P =0.84. (E and F) Baseline versus 16 and 8-week post-treatment thoracic radiographs, respectively, of two patients in the 10 mg/kg high dose losartan cohort experiencing partial regression of pulmonary metastases. Both dogs experienced an ~ 70 % reduction in the sum diameter of their target lesions. For the patient shown in (E), this response was initially noted at week 8, remained stable to further reduced at weeks 16 and 24, and no grossly visible OS pulmonary metastases were evident on necropsy of this patient at 27 weeks post-treatment. Only a single residual poorly cellular, matrix-rich microscopic metastasis (shown at right), with individual tumor cell necrosis (arrows) was observed in the lung.
Evaluation of peripheral blood cytokines associated with response to losartan therapy
To determine whether baseline or post-treatment changes in peripheral blood cytokine levels, and specifically known myeloid and monocyte chemokines, were associated with clinical response to high dose losartan-toceranib therapy, we performed a bead-based multiplex ELISA analysis to measure the levels of GM-CSF, IFNγ, IL-2, IL-6, IL-7, IL-8, IL-15, CXCL10, KC-like (functional homologue to CXCL1), IL-10, IL-18, CCL2, and TNF-α in plasma samples available for pre- (n=16) and 2-week (n=14) and 4-week (n=13) post-treatment samples from dogs in the 10 mg/kg cohort. We observed statistically significant increases in the myeloid-derived suppressor cell regulating and monocyte chemoattractants IL-18 and CCL2 at both 2- and 4-weeks post treatment as compared to baseline (Fig. 5A). Additionally, significantly higher pre-treatment levels of IL-8 were present in those dogs experiencing a clinical response (stable disease or partial regression, n=8) to high dose losartan-toceranib therapy vs. non-responders (n=8; Fig. 5B), and interestingly, clinically responding dogs also demonstrated a significantly decreased fold change in IL-8 concentrations at 4-weeks post treatment as compared to non-responding dogs (Fig. 5C).
Fig. 5.
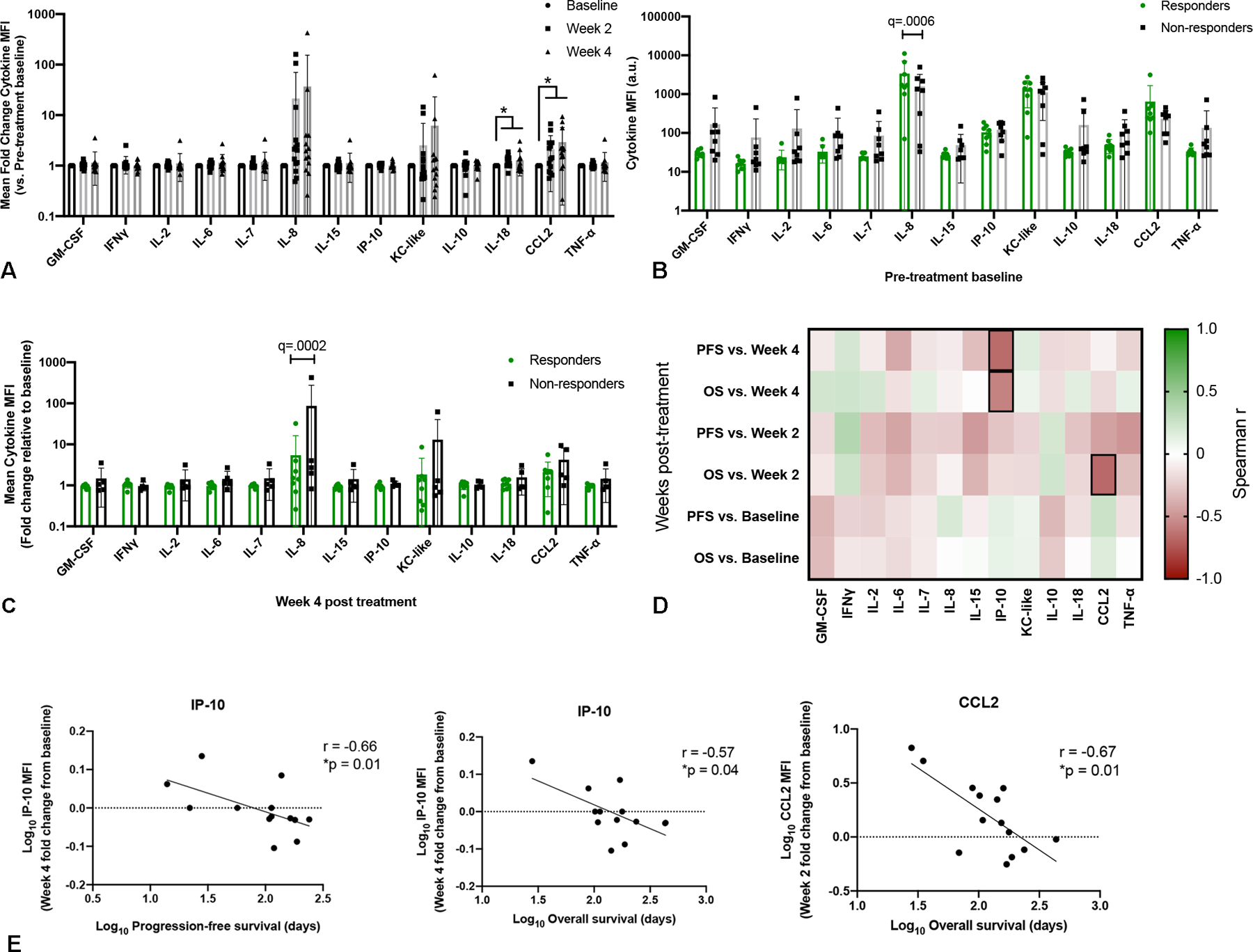
Multiplex cytokine profiling of patient peripheral blood for evaluation of immune correlates associated with response to losartan therapy. (A) Mean fold change in peripheral blood cytokine median fluorescence intensity (MFI) at 2- and 4-weeks post losartan treatment for evaluable patients in the high dose losartan (10 mg/kg) cohort (n = 13 – 16 dogs/timepoint). P value calculated by mixed-effects analysis with Benjamini, Krieger, and Yekutieli multi-comparison correction (FDR q value). * q = 0.01 (IL-18 week-2 vs. baseline), q = 0.07 (IL-18 week-4 vs baseline), q = 0.03 (CCL2 week-2 and week-4 vs. baseline). (B) Comparison of baseline pre-treatment mean peripheral blood cytokine MFI values in responders (dogs experiencing clinical benefit , n = 8) vs. non-responders (n = 8) in the high dose losartan (10 mg/kg) cohort. P value calculated by multiple unpaired t test with Benjamini, Krieger, and Yekutieli multi-comparison correction, FDR q = 0.0006 (IL-8). (C) Mean fold change, relative to pre-treatment baseline, in peripheral blood cytokine MFI values at 4-weeks post losartan treatment, in responders (dogs experiencing clinical benefit , n = 8) vs. non-responders (n = 5) in the high dose losartan (10 mg/kg) cohort. P value calculated by multiple unpaired t test with Benjamini, Krieger, and Yekutieli multi-comparison correction, FDR q = 0.0002 (IL-8). (D) Correlation matrix between peripheral blood cytokine levels at baseline, week 2 and week 4 post treatment with patient progression free and overall survival. Spearman r values, green = 1, or strong positive correlation, red = −1 or strong negative correlation, white = 0, no correlation. Outlined boxes indicate Spearman P < 0.05. (E) Individual Spearman correlation plots for those cytokines significantly correlated with clinical outcome, as outlined in (D).
When evaluating cytokines correlated with patient survival, we noted that increasing concentrations of CXCL10 (IP-10) at 4-weeks post-treatment was associated with shorter progression-free (r = −0.66, P = 0.01) and overall survival (r = −0.57, P = 0.04), while increasing concentrations of CCL2 at 2-weeks post-treatment was associated with shorter progression-free survival (r = −0.67, P = 0.01; Figs. 5D and E).
Transcriptomic responses of human peripheral blood mononuclear cells to in vitro losartan treatment
To better define the translational potential of losartan as a monocyte-targeted immunotherapy for human osteosarcoma patients, we performed RNA sequencing to identify transcriptional changes associated with losartan treatment of primary human PBMCs, with control and losartan-treated PBMC samples matched for each donor. We identified 59 significantly differentially expressed genes (Log2 FC > 1.5 or < −1.5 and adj. P < 0.05) in 24-hour in vitro losartan treated vs. control PBMCs, including 27 genes down-regulated and 32 genes up-regulated in response to losartan (Fig. 6A and B, Table S5). We noted significant downregulation of CCR2 in response to losartan, consistent with our prior published results in murine monocytes and THP-1 cells (28). We also observed significant down-regulation of the M2-associated macrophage marker CD163, a cell population associated with clinical outcome in human OS (24), and interestingly, 3 of the top-10 most down-regulated genes in response to losartan treatment are implicated as phenotypic or functional markers of tumor-associated macrophages (MS4A6A, CD163L1, and GPR34) (24,48–50). In line with the cytokine changes observed post-losartan treatment in our OS-bearing trial dogs, there was significant up-regulation of CXCL8 (IL-8) in losartan-treated human PBMCs.
Fig. 6.
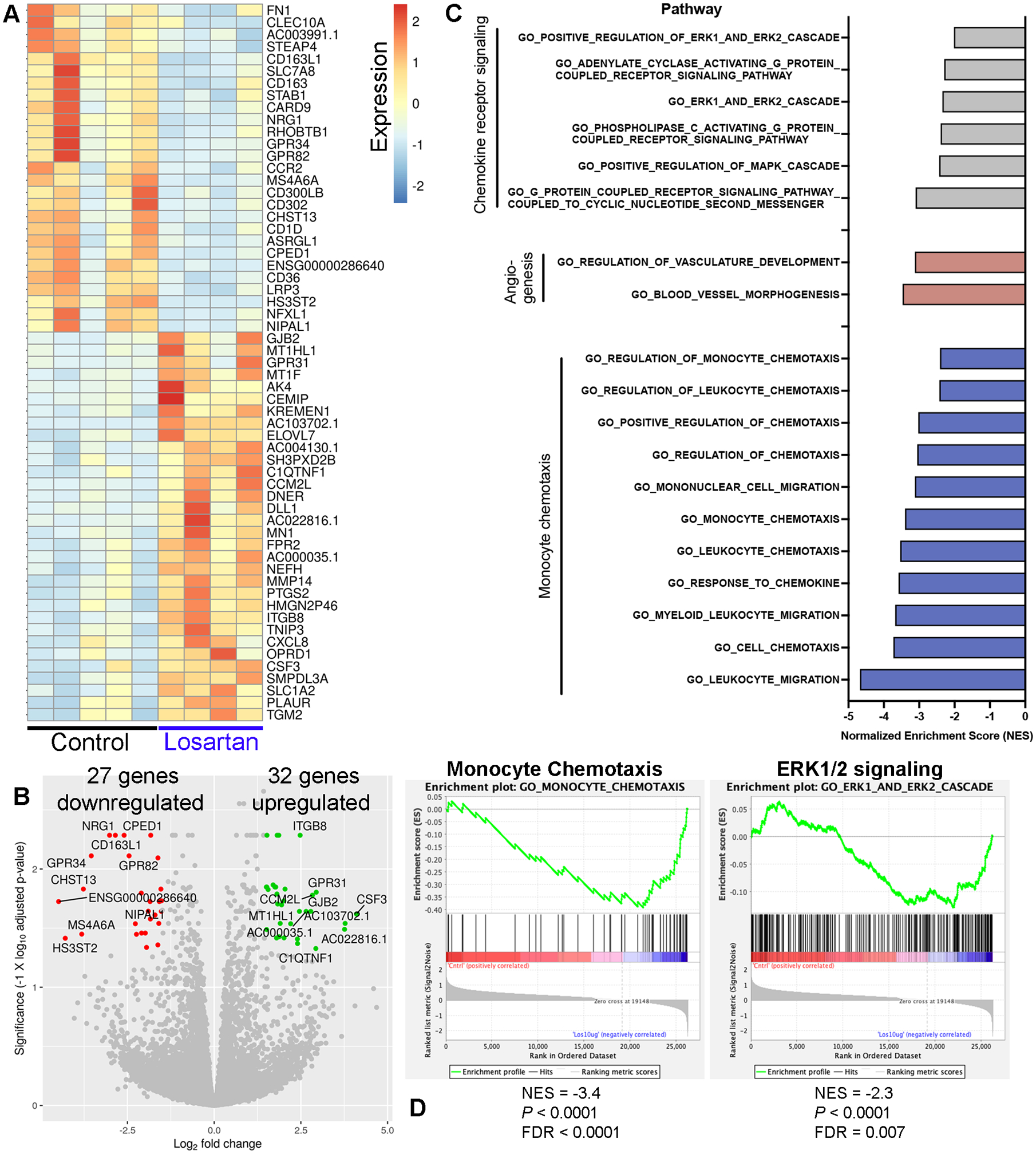
Transcriptional profiling of losartan-treated human peripheral blood mononuclear cells reveals down-regulation of genes associated with M2 macrophage polarization, monocyte migration, and chemokine receptor signaling. (A) Heatmap of the 59 differentially expressed genes (adjusted P value <0.05 and log2 fold change of +/− 1.5) between control (n = 5) and losartan-treated (n = 4; 10 μg/mL) human PBMCs. (B) Volcano plot of differentially expressed genes. The top 10 up- and down-regulated genes are highlighted in green and red, respectively. (C) Normalized Enrichment Scores (NES) for pathways related to CCL2-CCR2 signaling and monocyte chemotaxis, which were significantly down-regulated (FDR q < 0.05) in losartan-treated PBMCs vs. control, identified by Gene Set Enrichment Analysis (GSEA) using the GO-Biological Processes gene sets from the Molecular Signatures database (MSigDB). (D) GSEA negative enrichment plots for monocyte chemotaxis and ERK1/2 signaling genetic signatures in losartan-treated human PBMCs.
We then performed gene set enrichment analysis (GSEA) (42) using the gene ontology biological processes gene sets from MSigDB, in order to better characterize biological processes of human PBMCs functionally changed in response to losartan treatment. GSEA analysis identified 409 pathways which were significantly (Padj < 0.05) down-regulated in losartan treated versus control PBMCs (Table S6). Down-regulated pathways were heavily over-represented by signatures associated with immune cell processes, primarily those involving leukocyte, monocyte, myeloid cell, and granulocyte chemotaxis, chemokine and cytokine receptor signaling through G-protein coupled receptors, JAK/STAT, and the MAPK pathway, and to a lesser extent angiogenesis (Fig. 6C and D, and Table S6). Collectively, these transcriptional changes in losartan-treated human PBMCs parallel our previously described effects of losartan in murine models of inflammation and metastasis, as well as the CCL2 and monocyte-associated pharmacodynamic effects reported here in losartan-treated canine OS trial patients.
Discussion
Osteosarcoma metastasis, which exhibits an almost exclusive tropism for the lung, is present in 15–20% of OS patients at diagnosis and subsequently develops in another 30–40% of patients despite successful first-line therapy (11–13). Beyond surgical resection, we have little to offer this population of high-risk OS patients and their outcomes have not improved in four decades. To address this clinical hurdle, we leveraged the value of dogs with spontaneous OS as a biologically validated means to accelerate human OS therapeutic advancement (25,26). Our recent work demonstrated that high-dose losartan can reduce lung metastasis growth in mouse models through blockade of CCL2-CCR2 signaling and monocyte recruitment (28). Thus, we sought to test the translational potential of this preclinical data and assess the feasibility of losartan dose-escalation with respect to CCL2 and monocyte pharmacodynamics in a clinical trial of 28 dogs with lung metastatic OS. Results of this trial demonstrated that dose escalation of losartan, in combination with the multi-kinase inhibitor toceranib, is both well-tolerated and associated with significant disease stabilization and/or regression of advanced stage lung metastases in 50% of treated dogs. This observed response rate is notable, given that the discovery of intrinsic molecular drivers of OS progression has not translated to success with molecularly targeted therapies, and in spite of rapid advancements in the field of immunotherapy and the high degree of genomic instability in OS, trials of newly approved immunotherapies have failed (17,18,51). With limited therapeutic developments in the pipeline for this orphan disease, and a historical track record of therapeutic failures in the gross metastatic setting (52), the safety and clinical response data from this trial provide compelling evidence to consider clinical evaluation of this drug combination, or losartan in combination with other therapies, in human OS patients with non-resectable lung metastases.
Substantial pre-clinical and clinical data have demonstrated a pro-metastatic role for CCL2-CCR2 signaling in the tumor microenvironment and prognostic association of increased CCL2 and/or monocyte levels with poor patient outcomes for multiple types of canine and human cancers (8,27,53). Our results demonstrating enrichment of CCL2 secretion and monocyte recruitment by human and canine osteosarcoma cells and patient tumor samples suggest that in particular, this tumor type may represent an ideal target for CCL2-CCR2 targeted therapies. Therefore, one primary objective for the clinical studies reported here in tumor-bearing dogs was to determine the pharmacokinetic parameters of losartan dosing specifically with regard to CCL2 and monocyte pharmacodynamic modulation. We demonstrated in vitro that losartan was capable of inhibiting human and canine OS cell-elicited monocyte migration, and our pharmacokinetic analysis of losartan dose escalation in OS-bearing dogs demonstrated increasing maximum plasma and overall drug exposure levels, which met or exceeded the in vitro predicted therapeutic target for exposure.
In this trial, escalating doses of losartan were associated with multiple pharmacodynamic effects reflective of modulation of CCL2 signaling and monocyte migration, including post-treatment compensatory increases in plasma CCL2 levels, inhibition of ex-vivo CCL2 stimulated monocyte migration, and a reduction in total peripheral blood myeloid cells and monocytes. Specifically, significant inhibition of monocyte migration was observed in at least 50% of dogs at every post-treatment time point evaluated in the 10 mg/kg cohort. While it is possible this post-treatment inhibition of ex vivo monocyte migration could be related to variable CCR2 expression on patient PBMCs, as has previously been reported in dogs with OS (54), our additional pharmacodynamic endpoints suggests this is an unlikely mechanism. We also demonstrated statistically significant post-treatment elevations in mean plasma CCL2 concentrations in 90% of treated dogs, a previously reported robust pharmacodynamic marker of CCR2 inhibition [ClinicaTrials.gov NCT01215279, (45,46)]. It should also be noted that the greatest increases in CCL2 concentrations were observed in dogs with objective tumor responses and did not appear to be associated with tumor progression/increased tumor burden. For example, 6 of 6 dogs experiencing clinical benefit with evaluable samples demonstrated increases in plasma CCL2 levels at four weeks post-treatment, while 5 of 9 dogs experiencing significant progression of disease at two weeks post-treatment demonstrated increases in plasma CCL2 levels of only 12% or less above baseline. Together, these data suggest re-purposing this anti-hypertensive drug may represent a cost- and time-effective strategy for therapeutic targeting of the CCL2-CCR2 axis in human OS.
The repurposing of losartan as a cancer therapeutic is also being investigated by other groups. Prior retrospective studies have associated the use of losartan and other angiotensin receptor blockers (ARBs) with improved outcomes for certain tumor types (55), and a recent trial of losartan treatment for pancreatic cancer reported encouraging clinical benefit for patients, even following administration at relatively low doses (e.g., 0.5 mg/kg BID) (56). The pre-clinical rationale for this pancreatic cancer clinical trial originated from prior work which demonstrated another off-target effect of losartan via indirect inhibition of TGF-β through suppression of thrombospondin-1 expression (57). Losartan inhibition of TGF-β’s fibrogenic effects normalized the extracellular matrix to relieve tumor solid stress, resulting in blood vessel decompression and improved chemotherapeutic efficacy (57,58). Whether or not these other off-target effects of losartan are associated with clinical responses in this trial cannot be fully determined, a limitation of this study. However, our data support a mechanism whereby losartan-mediated tumor regression and/or disease stabilization in this canine trial is likely indirectly mediated by CCL2-CCR2 and monocyte modulation. Toceranib was developed primarily to suppress the proliferation of canine mast cell tumors with activating KIT mutations, and these activating KIT mutations are not known to be present in canine OS (59). More importantly, when comparing clinical responses for single agent toceranib in dogs with metastatic OS, there were no objective tumor responses observed in 22 dogs, and this ORR was not improved with the addition of low-dose 1 mg/kg losartan therapy. Instead, when the losartan dose was escalated to a monocyte-targeted therapeutic range in combination with toceranib, we observed an increased objective response rate to 25% (4/16) and associated 95% and 66% increases in median progression-free and overall survival, respectively. Additionally, at this high losartan dose, we observed no post-treatment changes in serum TGF-β or association with outcome in our patients (Fig. S4), a biomarker reported to be associated with losartan therapy in human PDAC patients (56).
Instead, pharmacodynamic endpoints from our study provide evidence suggesting that treatment responses were the result of high-dose losartan modulation of CCL2-CCR2 monocyte responses. Of the 8 dogs experiencing clinical benefit in the 10 mg/kg dose losartan cohort, 4 of 6 dogs with evaluable samples demonstrated two-week post-treatment reductions in ex vivo monocyte migration below baseline, while all 6 of these dogs demonstrated significant post-treatment increases in plasma CCL2 levels. Additionally, multiplex cytokine profiling of peripheral blood also identified unique changes in myeloid-derived suppressor cell and/or monocyte-associated biomarkers associated with response to losartan therapy. We observed significant post-treatment increases in IL-18 and CCL2. While compensatory increases in CCL2 have been reported with CCR2 targeted therapies, the observed increase in IL-18 was unexpected, but may also be related to losartan’s impact on myeloid cells. IL-18 has been reported to be a monocyte and monocytic-MDSC chemoattractant, and thus this increase may also represent a compensatory mechanism in response to losartan blockade of monocyte recruitment, similar to CCL2. Additionally, we observed that those dogs experiencing clinical benefit with high-dose losartan had increased pre-treatment plasma levels of IL-8, and also displayed significant suppression of post-treatment increases in IL-8, as compared to non-responders. Similar to CCL2 and IL-18, IL-8 is a known driver of recruitment and expansion of tumor-promoting myeloid cells in the tumor-microenvironment, and interestingly, overexpression of IL-8 in human OS lung metastases as compared to matched primary tumors has been previously reported and implicated as a promoter of OS lung metastasis in mouse models. Combined, these changes in patient circulating cytokine concentrations in response to losartan therapy suggest a unified theme of myeloid cell immunomodulation by this drug.
Based on growing evidence that the anti-tumor effects of ARB drugs are due to their immunomodulatory properties (55), a trial of losartan in combination with the PD-1 inhibitor nivolumab is currently underway (ClinicaTrials.gov Identifier: NCT03563248), and additional clinical trials are evaluating whether losartan can improve responses to various combinations of chemo-radiation (ClinicaTrials.gov Identifiers: NCT03563248, NCT01821729, NCT04106856). Our results add additional preclinical rationale for these trials, but also suggest that higher doses of losartan, to leverage its inhibitory effects on CCL2 monocyte responses, may also be an efficacious strategy for an immunotherapy combination, without inducing greater side-effects. This is supported by the transcriptomic responses we observed in response to losartan treatment of primary human peripheral blood mononuclear cells. Losartan’s primary transcriptional effects on human PBMCs were directly related to monocyte and myeloid cell chemotaxis and chemokine receptor signaling, and also demonstrated significant downregulation of known genes which regulate a pro-tumorigenic M2 macrophage phenotype. These transcriptional changes in human PBMCs are consistent with the monocyte pharmacodynamic effects we have previously described in mouse models and those reported in this present clinical trial in dogs, and highlight a second translational potential for this drug as a repurposed monocyte-targeted therapy, distinct from its current application in pancreatic cancer.
In conclusion, these studies provide clinical evidence in a translationally relevant spontaneous animal model of OS, that high-dose losartan therapy, when combined with a multi-tyrosine kinase inhibitor functionally equivalent to sunitinib, is capable of generating substantial antitumor activity with minimal toxicity. Based on these data, we are currently conducting a phase I clinical trial (NCT03900793) evaluating this two-drug combination for the treatment of lung metastatic OS in pediatric and young adult patients, designed to escalate losartan dosing to a biologically-effective dose for inhibition of CCL2-CCR2 monocyte responses in these OS patients.
Supplementary Material
Translational Relevance.
30–40% of all osteosarcoma (OS) patients develop tumor recurrence, almost exclusively in the form of lung metastasis, occurring on average only 1.6 years after diagnosis. Overall survival rates for these patients have not improved since the 1980s, due to a lack of therapeutic options for effectively treating these high-risk metastatic patients. Despite progress in our understanding of the tumor microenvironment, including how host stroma is co-opted by tumors to promote their growth and survival in distant sites and approval of immune therapies for cancer patients, these discoveries have not translated to clinical successes for OS patients. CCR2+ monocytes have been shown to play a critical role in the promotion of lung metastasis, yet to date there are currently no approved drugs to target these cells. Here we show in pre-clinical studies in dogs with metastatic osteosarcoma that the angiotensin-receptor blocker losartan, administered at high doses, can significantly interrupt the CCR2-CCL2 axis and block monocyte migration. The combination of high-dose losartan with the multi-kinase inhibitor toceranib led to significant clinical activity including tumor stabilization and/or regression in 50% of dogs with advanced relapsed metastatic osteosarcoma to the lungs.
Acknowledgements
We thank Dr. Kristen Weishaar and the entire clinical trials staff at the Colorado State University Flint Animal Cancer for all of their support with conduct of the clinical trial. Additionally, we would like to thank The Futreal Laboratory and Hannah Beird at MD Anderson for providing the human OS RNAseq data files. The work for this projected was supported by the Shipley Foundation, The Boettcher Foundation, the Flint Animal Cancer Center One Cure Fund, and NIH grants T32OD010437 (DPR), K01OD022982 (DPR), P30 CA046934 (University of Colorado Cancer Center Support Grant), and U01 CA224182 (SWD).
Footnotes
The authors declare no potential conflict of interests.
References
- 1.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011;147:275–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016;7:28697–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011;475:222–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 2012;21:488–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a Enhances Hepatic Metastases of Colon Cancer via Monocyte Chemoattractant Protein-1-mediated Inflammatory Cell Infiltration. The Journal of biological chemistry 2015;290:10667–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011;19:512–26 [DOI] [PubMed] [Google Scholar]
- 7.Ni XJ, Zhang XL, Ou-Yang QW, Qian GW, Wang L, Chen S, et al. An elevated peripheral blood lymphocyte-to-monocyte ratio predicts favorable response and prognosis in locally advanced breast cancer following neoadjuvant chemotherapy. PLoS One 2014;9:e111886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 2013;19:3404–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki A, Kai S, Endo Y, Iwaki K, Uchida H, Tominaga M, et al. Prognostic value of preoperative peripheral blood monocyte count in patients with colorectal liver metastasis after liver resection. Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract 2007;11:596–602 [DOI] [PubMed] [Google Scholar]
- 10.Svensson S, Abrahamsson A, Rodriguez GV, Olsson AK, Jensen L, Cao Y, et al. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin Cancer Res 2015;21:3794–805 [DOI] [PubMed] [Google Scholar]
- 11.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009;115:1531–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawkins DS, Arndt CA. Pattern of disease recurrence and prognostic factors in patients with osteosarcoma treated with contemporary chemotherapy. Cancer 2003;98:2447–56 [DOI] [PubMed] [Google Scholar]
- 13.Kempf-Bielack B, Bielack SS, Jurgens H, Branscheid D, Berdel WE, Exner GU, et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005;23:559–68 [DOI] [PubMed] [Google Scholar]
- 14.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nature reviews Clinical oncology 2016;13:273–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015;372:2521–32 [DOI] [PubMed] [Google Scholar]
- 16.Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. The Lancet Oncology 2015;16:257–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. The Lancet Oncology 2017;18:1493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. The Lancet Oncology 2018;19:416–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu CC, Beird HC, Andrew Livingston J, Advani S, Mitra A, Cao S, et al. Immuno-genomic landscape of osteosarcoma. Nature communications 2020;11:1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Behjati S, Tarpey PS, Haase K, Ye H, Young MD, Alexandrov LB, et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nature communications 2017;8:15936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyers PA, Schwartz CL, Krailo MD, Healey JH, Bernstein ML, Betcher D, et al. Osteosarcoma: The Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival—A Report From the Children’s Oncology Group. Journal of Clinical Oncology 2008;26:633–8 [DOI] [PubMed] [Google Scholar]
- 22.MacEwen EG, Kurzman ID, Rosenthal RC, Smith BW, Manley PA, Roush JK, et al. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J Natl Cancer Inst 1989;81:935–8 [DOI] [PubMed] [Google Scholar]
- 23.Kleinerman ES, Gano JB, Johnston DA, Benjamin RS, Jaffe N. Efficacy of liposomal muramyl tripeptide (CGP 19835A) in the treatment of relapsed osteosarcoma. Am J Clin Oncol 1995;18:93–9 [DOI] [PubMed] [Google Scholar]
- 24.Buddingh EP, Kuijjer ML, Duim RA, Bürger H, Agelopoulos K, Myklebost O, et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res 2011;17:2110–9 [DOI] [PubMed] [Google Scholar]
- 25.LeBlanc AK, Mazcko CN. Improving human cancer therapy through the evaluation of pet dogs. Nature reviews Cancer 2020;20:727–42 [DOI] [PubMed] [Google Scholar]
- 26.Khanna C, Fan TM, Gorlick R, Helman LJ, Kleinerman ES, Adamson PC, et al. Toward a drug development path that targets metastatic progression in osteosarcoma. Clin Cancer Res 2014;20:4200–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sottnik JL, Rao S, Lafferty MH, Thamm DH, Morley PS, Withrow SJ, et al. Association of blood monocyte and lymphocyte count and disease-free interval in dogs with osteosarcoma. J Vet Intern Med 2010;24:1439–44 [DOI] [PubMed] [Google Scholar]
- 28.Regan DP, Coy JW, Chahal KK, Chow L, Kurihara JN, Guth AM, et al. The Angiotensin Receptor Blocker Losartan Suppresses Growth of Pulmonary Metastases via AT1R-Independent Inhibition of CCR2 Signaling and Monocyte Recruitment. Journal of immunology (Baltimore, Md : 1950) 2019;202:3087–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009;69:2514–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guislain A, Gadiot J, Kaiser A, Jordanova ES, Broeks A, Sanders J, et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother 2015;64:1241–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res 2008;14:6674–82 [DOI] [PubMed] [Google Scholar]
- 32.London CA, Malpas PB, Wood-Follis SL, Boucher JF, Rusk AW, Rosenberg MP, et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res 2009;15:3856–65 [DOI] [PubMed] [Google Scholar]
- 33.Das S, Idate R, Cronise KE, Gustafson DL, Duval DL. Identifying Candidate Druggable Targets in Canine Cancer Cell Lines Using Whole-Exome Sequencing. Mol Cancer Ther 2019;18:1460–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veterinary cooperative oncology group – common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1. Veterinary and Comparative Oncology 2016;14:417–46 [DOI] [PubMed] [Google Scholar]
- 35.Nguyen SM, Thamm DH, Vail DM, London CA. Response evaluation criteria for solid tumours in dogs (v1.0): a Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet Comp Oncol 2015;13:176–83 [DOI] [PubMed] [Google Scholar]
- 36.Fowles JS, Dailey DD, Gustafson DL, Thamm DH, Duval DL. The Flint Animal Cancer Center (FACC) Canine Tumour Cell Line Panel: a resource for veterinary drug discovery, comparative oncology and translational medicine. Veterinary and Comparative Oncology 2017;15:481–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–64 [DOI] [PubMed] [Google Scholar]
- 39.Nirmal AJ, Regan T, Shih BB, Hume DA, Sims AH, Freeman TC. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol Res 2018;6:1388–400 [DOI] [PubMed] [Google Scholar]
- 40.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39:782–95 [DOI] [PubMed] [Google Scholar]
- 41.Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nature biotechnology 2014;32:896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brodmerkel CM, Huber R, Covington M, Diamond S, Hall L, Collins R, et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J Immunol 2005;175:5370–8 [DOI] [PubMed] [Google Scholar]
- 44.Brown S, Elliott J, Francey T, Polzin D, Vaden S. Consensus recommendations for standard therapy of glomerular disease in dogs. J Vet Intern Med 2013;27 Suppl 1:S27–43 [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Cui L, Gonsiorek W, Min SH, Anilkumar G, Rosenblum S, et al. CCR2 and CXCR4 regulate peripheral blood monocyte pharmacodynamics and link to efficacy in experimental autoimmune encephalomyelitis. Journal of inflammation (London, England) 2009;6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis and rheumatism 2008;58:1931–9 [DOI] [PubMed] [Google Scholar]
- 47.Christ DD, Wong PC, Wong YN, Hart SD, Quon CY, Lam GN. The pharmacokinetics and pharmacodynamics of the angiotensin II receptor antagonist losartan potassium (DuP 753/MK 954) in the dog. The Journal of pharmacology and experimental therapeutics 1994;268:1199–205 [PubMed] [Google Scholar]
- 48.González-Domínguez É, Samaniego R, Flores-Sevilla JL, Campos-Campos SF, Gómez-Campos G, Salas A, et al. CD163L1 and CLEC5A discriminate subsets of human resident and inflammatory macrophages in vivo. J Leukoc Biol 2015;98:453–66 [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Yan K, Lin J, Li J, Bi J. Macrophage M2 Co-expression Factors Correlate With the Immune Microenvironment and Predict Outcome of Renal Clear Cell Carcinoma. Frontiers in Genetics 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang K, Park SH, Chen J, Qiao Y, Giannopoulou E, Berg K, et al. Interferon-γ Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity 2017;47:235–50.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nature reviews Cancer 2014;14:722–35 [DOI] [PubMed] [Google Scholar]
- 52.Roberts RD, Lizardo MM, Reed DR, Hingorani P, Glover J, Allen-Rhoades W, et al. Provocative questions in osteosarcoma basic and translational biology: A report from the Children’s Oncology Group. Cancer 2019;125:3514–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biller BJ, Guth A, Burton JH, Dow SW. Decreased ratio of CD8+ T cells to regulatory T cells associated with decreased survival in dogs with osteosarcoma. J Vet Intern Med 2010;24:1118–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tuohy JL, Lascelles BD, Griffith EH, Fogle JE. Association of Canine Osteosarcoma and Monocyte Phenotype and Chemotactic Function. J Vet Intern Med 2016;30:1167–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Science translational medicine 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY, et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol 2019;5:1020–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proceedings of the National Academy of Sciences of the United States of America 2011;108:2909–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nature communications 2013;4:2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakthikumar S, Elvers I, Kim J, Arendt ML, Thomas R, Turner-Maier J, et al. SETD2 Is Recurrently Mutated in Whole-Exome Sequenced Canine Osteosarcoma. Cancer Res 2018;78:3421–31 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mRNA sequence data generated in this study are publicly available in the NIH Sequence Read Archive (SRA), BioProject ID: PRJNA763881.