

Abstract
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is an aggressive leukemia of plasmacytoid dendritic cells (pDCs). BPDCN occurs at least three times more frequently in men than women, but the reasons for this sex bias are unknown. Here, studying genomics of primary BPDCN and modeling disease-associated mutations, we link acquired alterations in RNA splicing to abnormal pDC development and inflammatory response through Toll-like receptors. Loss-of-function mutations in ZRSR2, an X chromosome gene encoding a splicing factor, are enriched in BPDCN and nearly all mutations occur in males. ZRSR2 mutation impairs pDC activation and apoptosis after inflammatory stimuli, associated with intron retention and inability to upregulate the transcription factor IRF7. In vivo, BPDCN-associated mutations promote pDC expansion and signatures of decreased activation. These data support a model in which male-biased mutations in hematopoietic progenitors alter pDC function and confer protection from apoptosis, which may impair immunity and predispose to leukemic transformation.
INTRODUCTION
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a hematologic malignancy in which patients can have leukemic involvement of the blood and bone marrow, and also tumor formation in the skin (in ~90% of cases), lymphoid organs, and other tissues (1). Among the unique epidemiologic features of the disease is the extreme male predominance, with a male:female incidence ratio of at least 3:1 in adults (2,3). There is no explanation to date for why males are predisposed to the disease, or alternatively, why females are relatively protected. Outcomes for patients with BPDCN are poor, with median survival of 12–24 months from diagnosis (3,4), demonstrating the unmet need for additional biological insight.
DNA sequencing of bone marrow or skin involved with BPDCN has identified mutations in genes often affected in other blood cancers, particularly myelodysplastic syndrome (MDS) and related myeloid malignancies. These include point mutations or indel/frameshift mutations in ASXL1, TET2, and TP53, and copy number alterations affecting one or more cell cycle regulators (5–9). However, BPDCN frequently arises in the context of a pre-existing or concurrent myeloid malignancy, such as MDS or chronic myelomonocytic leukemia (CMML) (10–12), but most of the prior genetic landscape studies of BPDCN analyzed unsorted bone marrow or skin biopsies with heterogeneous composition. Those biopsies likely included hematopoietic cells, possibly neoplastic, of other lineages, and non-hematopoietic cells. Therefore, one goal of this project was to define the genetic alterations and transcriptional changes present in highly purified BPDCN cells separated from any background clonal hematopoietic disorder and to link these to patient sex. We found that loss-of-function mutations in the RNA splicing factor ZRSR2, encoded on chromosome X, are enriched in BPDCN and are associated with a significant fraction of the male predominance of the disease.
BPDCN is thought to develop from plasmacytoid dendritic cells (pDCs) or their precursors, based primarily on similarities in gene expression and cellular function (13–15). However, mechanisms of how genetic alterations promote leukemic transformation in the dendritic cell lineage are unclear. Furthermore, studies have not linked the myeloid neoplasm-associated mutations described in BPDCN to consequences on pDC development, growth, or function. Thus, a second goal of this work was to connect genes mutated in BPDCN to dendritic cell transformation via interrogation of their effects specifically in pDCs, myeloid/dendritic progenitors, and BPDCN cells. We found that ZRSR2 mutations impair pDC activation and apoptosis in the setting of inflammatory stimuli, at least in part via misregulation of the interferon regulatory factor, IRF7, downstream of Toll-like receptor (TLR) signaling.
RESULTS
BPDCN has male-biased ZRSR2 mutations and a UV-associated signature
We performed whole exome sequencing (WES) on sorted BPDCN (CD45+ CD4+ CD56+ CD123+ BDCA4+ CD3−; n=11 patients) from blood or bone marrow and on paired CD3+ cells to identify acquired mutations in the malignant cells. We also performed targeted sequencing using a 95-gene panel in an extended set of bone marrows from patients with BPDCN (n=27) (16). The most frequently mutated genes across all BPDCNs were TET2, ASXL1, and genes involved in RNA splicing, including ZRSR2, SRSF2, U2AF1, and SF3B1 (Figure 1a, Supplementary Table 1). Additional mutations included genes recurrently altered in other myeloid malignancies, such as TP53, NRAS, KRAS, and GNB1. Of potential clinical relevance, we found an oncogenic IDH2 mutation in 4 of 38 cases (~11%), which we previously reported was associated with sensitivity to the IDH2 inhibitor enasidenib in a patient with BPDCN (3). TET2 and IDH2 mutations were mutually exclusive, as has been reported in acute myeloid leukemia (AML) (17). In contrast, other common myeloid malignancy-associated mutations were absent or rare, such as in DNMT3A, NPM1, or FLT3-ITD (fms-like tyrosine kinase 3, internal tandem duplication). One BPDCN harbored a single nucleotide variant that encodes an IRF8 R404W missense mutation. While the functional consequence of this specific variant is unknown, IRF8 is important for pDC development and function (18,19). WES also identified a small number of recurrently mutated genes in this set of BPDCNs that have not been previously identified as mutated in other cancers or in prior BPDCN sequencing (Supplementary Table 2). Some of these variants are in homopolymer tracts that are at risk for sequencing errors or are in genes that are not expressed in BPDCN or pDCs, and thus, their contribution to the BPDCN phenotype is not clear.
Figure 1. Recurrently mutated genes and ultraviolet light-induced global mutation signature in BPDCN.
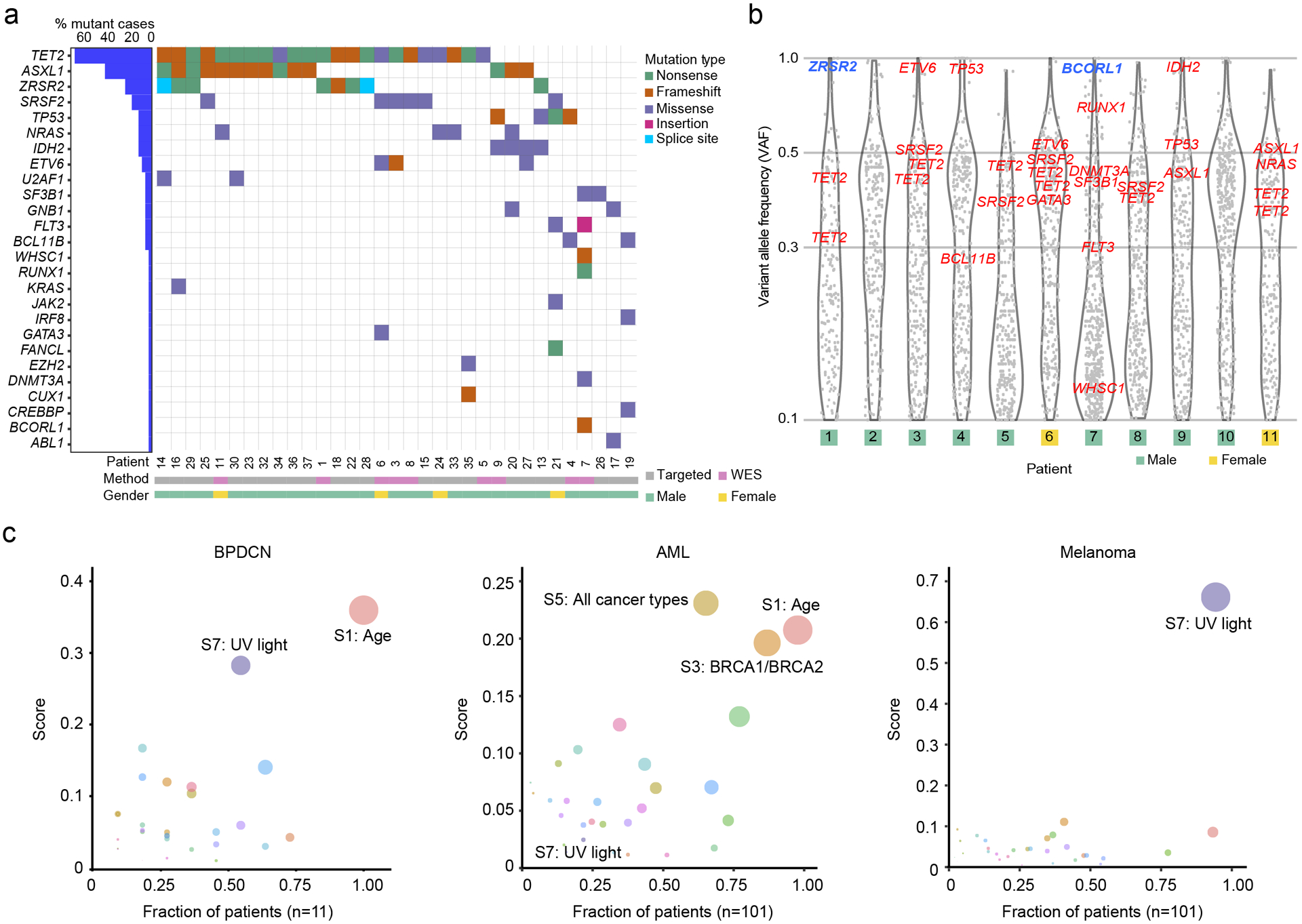
a. Co-mutation plot of single nucleotide variants (SNVs) and insertions/deletions in BPDCN samples among genes recurrently mutated in other hematologic malignancies. Each column represents an individual patient, genes are in rows and mutation types are annotated by color. Percentage of patients with a given gene altered are plotted to the left. Samples are annotated by gender and sequencing method (WES, whole exome sequencing). b. Variant allele frequency (VAF) of all somatic mutations (including synonymous) with VAF ≥ 0.1 detected by WES in eleven BPDCNs plotted as gray dots. VAFs of known hematologic malignancy-related genes from panel (a) are annotated in red (autosomes) or blue (X chromosome). c. Global somatic mutational signatures in BPDCN, AML, and melanoma plotted as fraction of samples (x-axis) having a specific signature with the mean signature score in those patients (y-axis). The color of the circle represents a specific signature, and the size of the circle represents the strength of association as a combined measure of the fraction of patients having a signature and the contribution score.
In the BPDCNs studied by WES, the distribution of variant allele frequency (VAF) for all somatic mutations allowed us to define their clonal structure (Figure 1b). Most cases harbored known hematologic malignancy associated genes in a single dominant clone (clustered around VAF ~50% for presumed heterozygous mutations), which included all of the variants in the most frequently mutated genes: TET2, ASXL1, and spliceosome components. Some mutations were detected at nearly 100% VAF, including in ZRSR2 and BCORL1, which reside on the X chromosome and were mutated nearly exclusively in males in this cohort, and in autosomal genes ETV6, IDH2, and TP53, which likely represent loss-of-heterozygosity events.
Given that many genes mutated in BPDCN were shared with other myeloid malignancies, we asked if the global mutation pattern might indicate unique BPDCN-specific features. We performed a mutation analysis on all somatic alterations and their surrounding nucleotide context using signatures previously defined in the Catalog of Somatic Mutations in Cancer (COSMIC) (20). We found that BPDCNs harbored an age-associated pattern (“Signature 1”), which is detected in most cancers in COSMIC (Figure 1c, Supplementary Figure 1). However, we also detected a strong association with ultraviolet (UV)-induced mutation signatures in the majority of BPDCNs (signature score >0.25 in 6/11 or 55%; Figure 1c). In contrast, there was no UV signature associated with AML (zero of 101 cases, P<0.0001 by Fisher’s exact test), even though AML shares many recurrently mutated genes with BPDCN and may arise from a similar myeloid progenitor. For comparison, melanoma harbors a uniformly high UV signature in most tumors (Figure 1c). The BPDCN cells we analyzed were harvested from bone marrow or blood and not directly from UV-exposed tissue (i.e., skin), suggesting that at least some leukemic BPDCN cells maintain a UV signature presumably acquired during a prior skin phase of malignant evolution.
Sequencing purified BPDCN cells rather than bulk marrow or skin samples also allowed us to determine DNA copy number variants (CNVs) that were more confidently BPDCN-associated. Some of the chromosome and arm level CNVs were similar to those described previously in BPDCN (Figure 2a) (7,21). These included loss of 7p (which harbors IKZF1), 9p (CDKN2A, CDKN2B), 12p (CDKN1B, ETV6), 13q (RB1), and 17p (TP53) (Supplementary Figure 2). Copy number analysis also clarified the disease genetics for patients 2 and 10, who did not have any single nucleotide variants in known blood cancer-associated genes from WES (Figure 1a). In those patients, we detected deletions in IKZF1 and RB1 (patient 2) and TET2, RB1, and ZRSR2 (patient 10), which supports that analysis of CNVs in addition to point mutations/indels can detect additional relevant driver events. Integrated analysis of single nucleotide variants/indels and copy loss highlighted four putative BPDCN tumor suppressors targeted by both types of alteration: ZRSR2, TET2, TP53, and SETD2 (Figure 2b). PCR confirmed the WES finding that chromosome Xp22.2 copy loss in patient 10, a male, resulted in complete absence of ZRSR2 DNA in tumor but not in germline cells (Figure 2c).
Figure 2. ZRSR2 mutations and their association with BPDCN sex bias.
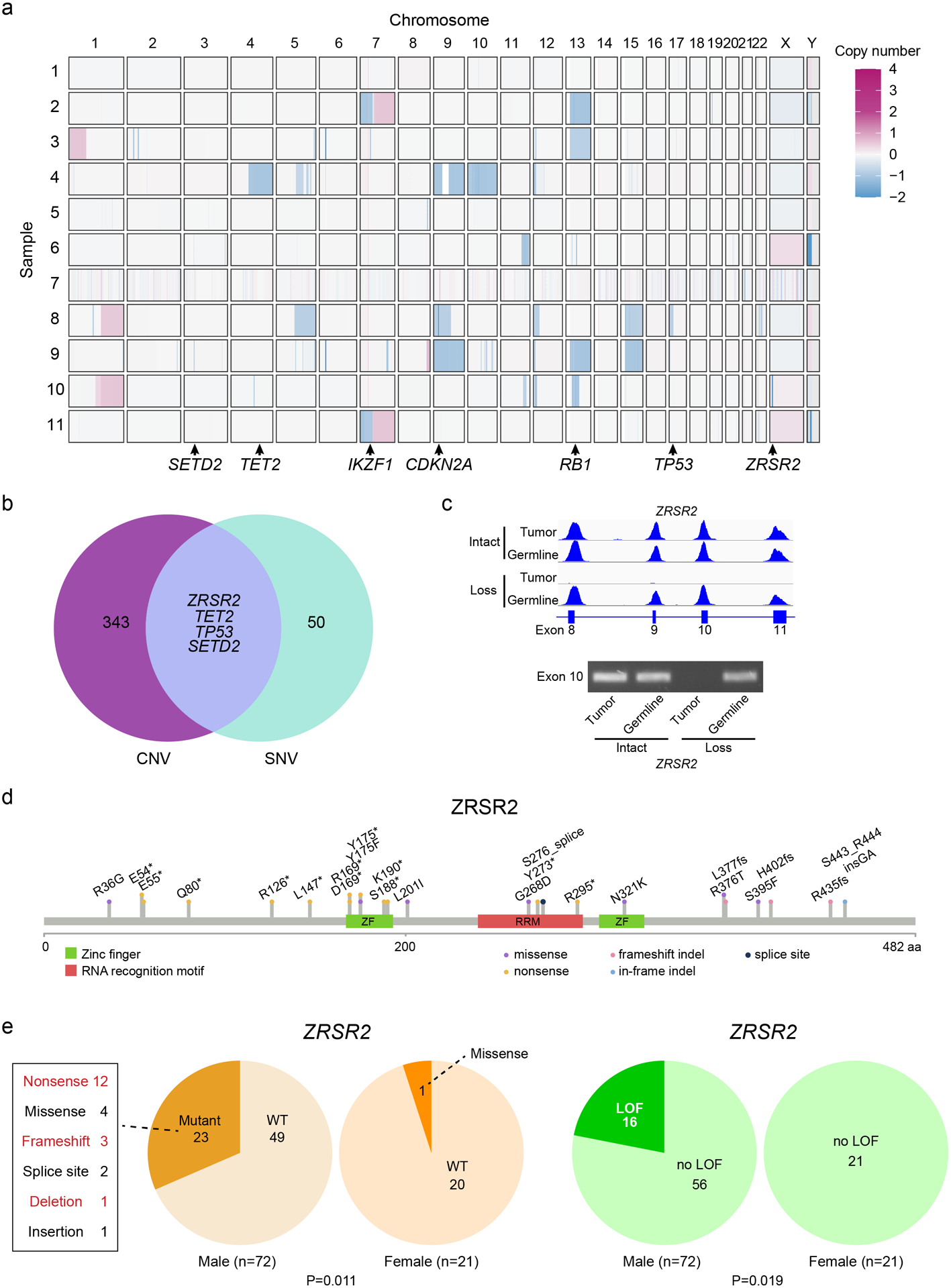
a. Somatic DNA copy number changes in 11 BPDCNs identified by WES shown from blue (copy loss) to red (gain). b. Venn diagram showing the four genes with overlap of copy number variations (among 347 protein coding genes observed in at least two patients with VAF ≥ 0.2) and single nucleotide variations (among 54 targeted panel genes or protein coding gene mutations observed in at least two patients by WES). c. Sequencing read traces in the ZRSR2 locus from WES and PCR of exon 10 DNA in tumor/germline pairs from representative BPDCNs in males with intact or somatic copy number loss of ZRSR2. d. Schematic of the ZRSR2 protein with amino acid locations and specific mutations (n=24) detected in BPDCN (n=93). e. ZRSR2 mutations are male-biased. 23 of 72 male BPDCNs had ZRSR2 mutations vs. 1 of 21 female (P=0.011 by Fisher’s exact test). When restricted to obvious loss-of-function (LOF) mutations (nonsense, frame shift, deletion; marked in red), 16 of 72 male BPDCNs had ZRSR2 LOF mutations vs. 0 of 21 female (P=0.019).
ZRSR2 mutations in BPDCN are of particular interest for several reasons. First, while other splicing factors are mutated across many solid and blood cancers, ZRSR2 is nearly exclusively associated with myeloid disorders, such as MDS, CMML, and AML. However, in contrast to MDS, where SRSF2, SF3B1, and U2AF1 mutations are more common, ZRSR2 was the most frequently mutated spliceosome gene in this BPDCN cohort. To confirm this finding in a larger and more geographically diverse cohort, we examined BPDCN DNA from three separate groups (from France, Italy, and MD Anderson Cancer Center), and found ZRSR2 mutations in 24 of 93 (26%) (Figure 2d). This is significantly higher than the incidence of ZRSR2 mutations in MDS, where the gene was mutated in only 13 of 288 patients in one large cohort (4.5%; P<0.0001 by Fisher’s exact test) (22).
Second, nearly all ZRSR2 mutations in our cohort were in males (mutated in 23 of 72 males vs. 1 of 21 females, P=0.011 by Fisher’s exact test) (Figure 2e). The mutational pattern is consistent with loss-of-function, with most variants predicting inactivating events (nonsense, frame shift, deletion), which implicates ZRSR2 as a male-biased X chromosome tumor suppressor gene. In fact, strictly defined inactivating mutations in our cohort were exclusively in males (16 of 72 males vs. 0 of 21 females, P=0.019) (Figure 2e). This is similar to MDS, where loss-of-function mutations in ZRSR2 are almost always restricted to males (22). ZRSR2 belongs to the minor subset of X chromosome genes that escape X inactivation silencing and is biallelically expressed in female cells (23). The finding that ZRSR2 is also preferentially mutated in BPDCN and MDS from males strongly suggests that ZRSR2 is an Escape from X Inactivation Tumor Suppressor (EXITS) gene (24). As we previously described for other EXITS genes across a range of cancers, females are relatively protected from BPDCN because they have two active ZRSR2 alleles and therefore require two mutations to eliminate function, whereas males require only one. The degree of sex bias in BPDCN incidence linked to ZRSR2 can be expressed as the number of excess ZRSR2 mutations in males per excess case of BPDCN in males (24). Using that calculation, in this cohort 43% of the excess male risk of BPDCN is associated with a ZRSR2 mutation.
Unique transcriptomic features of spliceosome mutated BPDCN
Next, we asked how BPDCN transcriptomes relate to their genetics. We performed RNA-sequencing on the same sorted BPDCN samples that we had profiled by whole exome sequencing. Peripheral blood pDCs (CD45+ CD123+ BDCA2+ CD3−) from healthy donors were analyzed in parallel for comparison (Supplementary Table 3). Differentially expressed genes between BPDCN and pDCs were similar to those we previously defined that distinguished BPDCNs from normal dendritic cells using single cell RNA-sequencing (Figure 3a) (14). We also observed differences in individual oncogenes and tumor suppressors (BCL2, MYB, IRF4, and MAP3K1) known to be dysregulated in BPDCN (Figure 3b). Furthermore, gene set enrichment analysis (GSEA) identified oncogene-associated signatures in BPDCN, including in both splicing factor mutated or wild-type compared to normal pDCs, such as overexpression of MYC and E2F targets, and of PI3K/AKT/MTORC1 signaling pathway genes (Figure 3c). In addition to these expected gene expression changes, we also found that BPDCNs had upregulation of RNA splicing machinery and associated genes when compared to normal pDCs (Figure 3d). Furthermore, within BPDCN, splicing factor mutant cases showed upregulation of RNA splicing genes and were enriched for markers of active nonsense mediated decay (NMD) RNA catabolism compared to BPDCNs without ZRSR2, SRSF2, SF3B1, or U2AF1 mutations (Figure 3d), consistent with what has been observed in other splicesosome-mutant cancers (25,26). This was of particular interest given that ZRSR2 loss promotes intron retention (27), which is known to trigger NMD activation and cause altered myelopoiesis (28).
Figure 3. BPDCN transcriptomes have alterations in oncogene, dendritic cell development, and RNA processing genes.
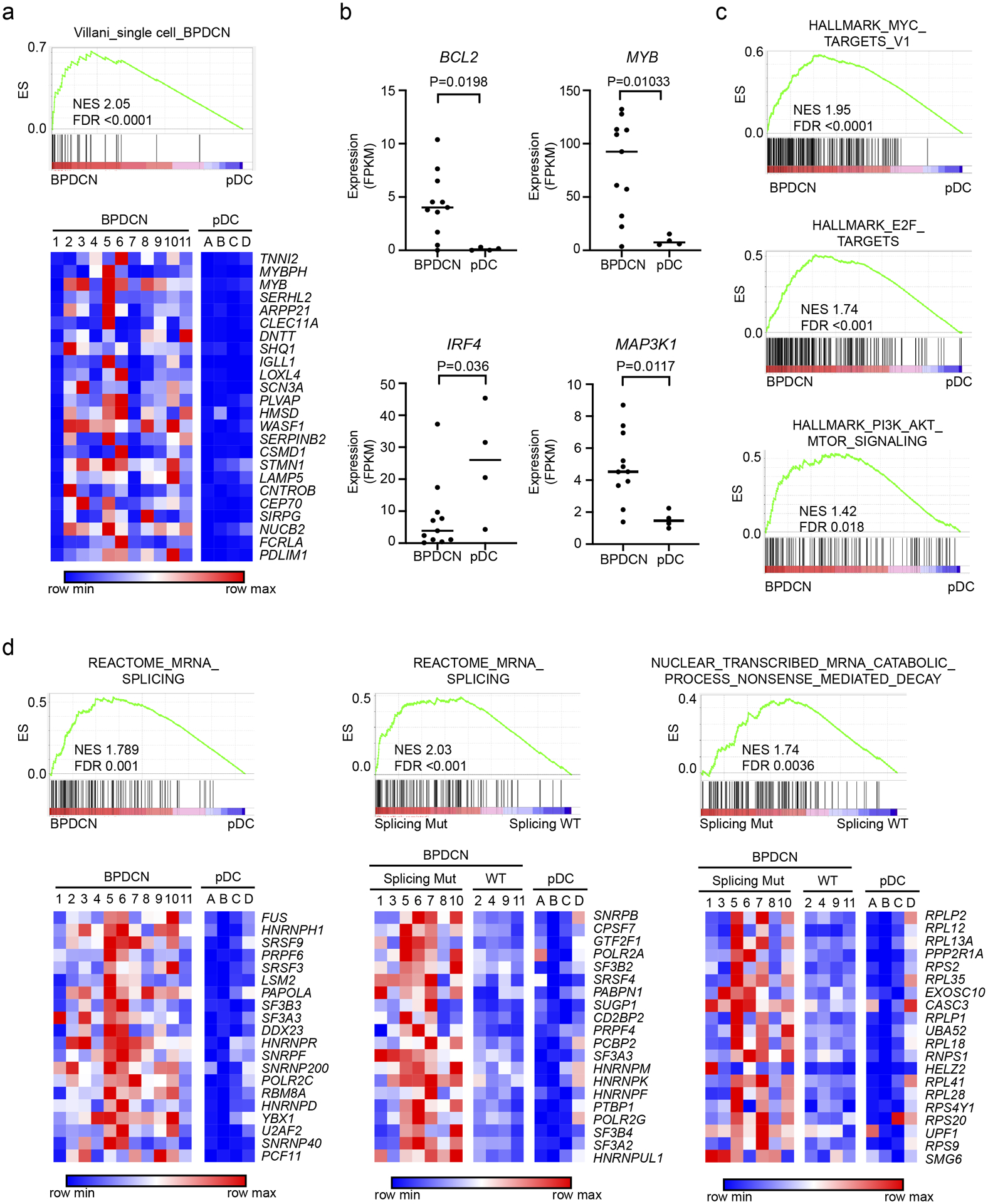
a. Gene set enrichment analysis (GSEA) showing association of a previously defined gene signature from single cell RNA-sequencing that differentiated BPDCN from normal human DC subtypes (14) in BPDCN (n=11) compared to normal pDCs (n=4) from the current cohort. ES, enrichment score; NES, normalized enrichment score; FDR, false discovery rate. Heatmaps of the same genes plotted as low (blue) to high (red) relative expression. b. RNA expression of the indicated genes in BPDCN (n=11) compared to normal pDCs (n=4), groups compared by t test. c. GSEA comparing BPDCN to normal pDCs showing enrichment of the indicated hallmarks of cancer gene sets (MSigDB collection “H”) in BPDCN. d. GSEA as in (a) for the indicated RNA splicing and nonsense mediated decay gene sets with heatmaps of the top 20 leading edge genes plotted as low (blue) to red (high) relative expression. In the indicated plots, BPDCNs are separated by whether (Splicing Mut) or not (Splicing WT) they harbor a mutation in a splicing factor (SF3B1, SRSF2, U2AF1, or ZRSR2).
Next, we analyzed RNA splicing in splicing factor mutated BPDCN compared to normal pDCs and to BPDCN without any splicing factor mutation. BPDCNs with splicing factor mutations showed several types of abnormal splicing, including intron retention (IR), cryptic 3’ splice site usage, and exon skipping (Supplementary Table 4). ZRSR2, SRSF2, SF3B1, and U2AF1 are all involved in U2 intron splicing (99.7% of introns in the human genome). In contrast, ZRSR2 is uniquely necessary for proper splicing of U12 introns (0.3% of human introns), an evolutionarily conserved genomic feature with distinct branch and splice-site sequences (29,30). In MDS, ZRSR2 mutations promote aberrant IR with a bias toward retention of U12-type introns (31). Similarly, we found that BPDCNs with ZRSR2 mutation or loss had increased IR compared to those without spliceosome mutations (Figure 4a). Both U2- and U12-type introns were affected, but IR was markedly weighted towards U12-type introns in BPDCNs with ZRSR2 mutation (Figure 4b). Consistent with possible activation of NMD, genes with an IR event in ZRSR2-mutant BPDCNs had decreased expression compared to splicing factor wild-type BPDCNs, albeit modestly across all IR events, versus genes without IR events (Supplementary Figure 3a). We also detected some of the stereotypical mis-splicing patterns associated with specific splicing factor mutations in other cancers (31). These included preferential exon inclusion or exclusion related to CCNG/GGNG exonic splicing enhancer motifs in BPDCN with SRSF2 mutation (32) and alternative 3’ splice sites usage with SF3B1 mutation (Supplementary Figure 3b–c) (33). To confirm ZRSR2-associated splicing changes in a separate set of BPDCNs, we analyzed patient-derived xenografts (PDXs) (34). We performed RNA-seq on six BPDCN PDXs, two with ZRSR2 mutations and four without. Similar to what we observed in patient cells, PDXs with ZRSR2 mutations had increased IR events that were biased toward aberrant retention of U12-type introns (Figure 4b–c).
Figure 4. ZRSR2 mutations promote specific mis-splicing events.
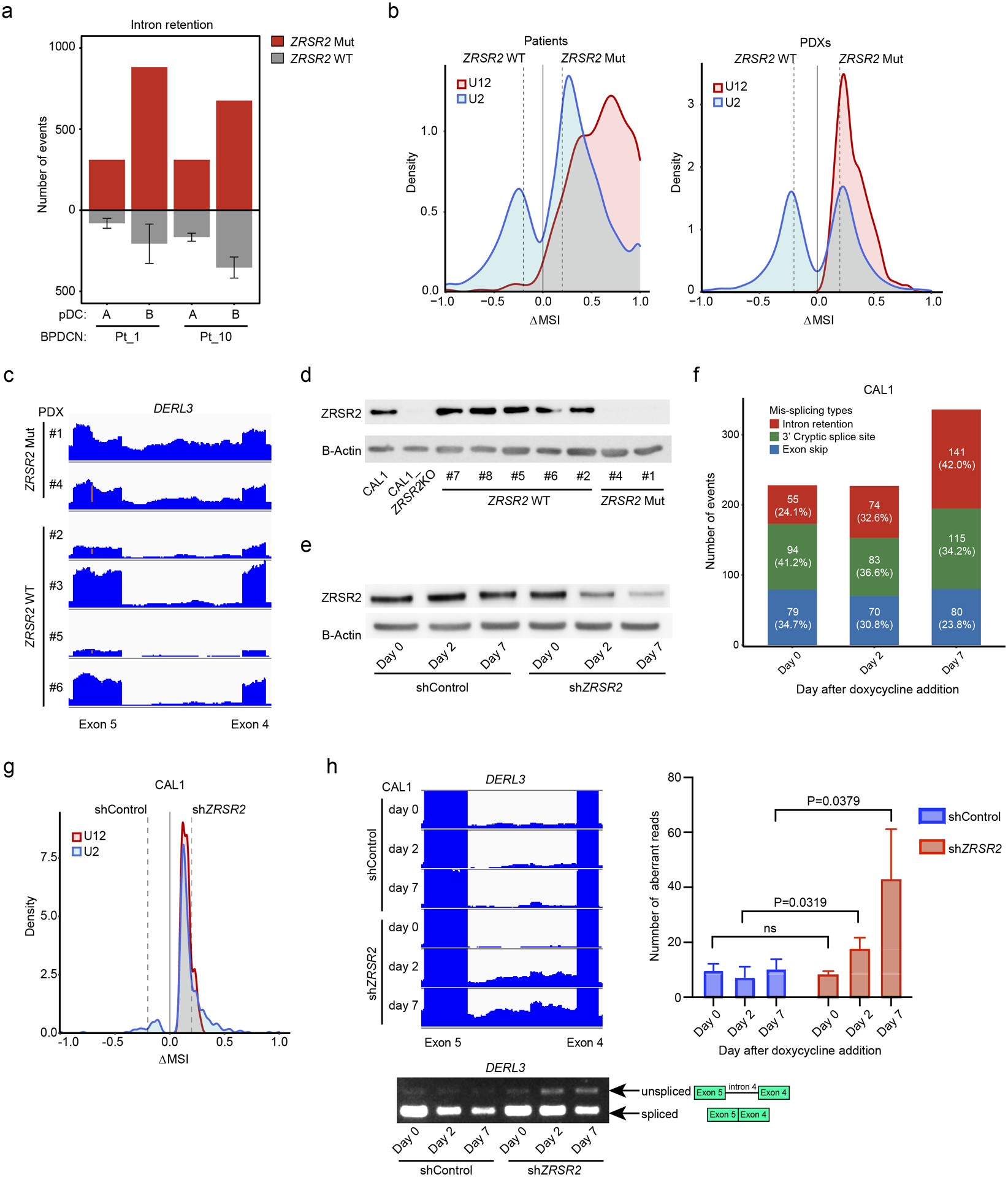
a. Intron retention events in BPDCNs with a ZRSR2 mutation (n=2, red) compared pairwise to normal pDCs (n=2, noted as samples A and B) and to those events in BPDCNs with no known splicing factor mutation (n=4, gray). b. Density plots of frequencies of U2-type (blue) and U12-type (red) introns among aberrantly retained introns (Fisher’s exact test p-value ≤ 0.05) in pairwise analyses of ZRSR2 mutant versus wild-type BPDCNs and patient-derived xenografts (PDXs). N=2 ZRSR2 mutant and n=4 splicing factor wild-type in each. Dotted lines represent ΔMSI −0.2 and +0.2. c. RNA-seq reads of an aberrantly retained U12 intron (intron 4) in human DERL3 from BPDCN PDXs with or without ZRSR2 mutation (equivalent scale for all samples). d. Western blot for ZRSR2 and beta-actin in CAL1 cells (parental or ZRSR2 knockout) and in BPDCN PDXs with wild-type or mutant ZRSR2. e. Western blot from CAL1 cells at baseline (day 0) and at 2 and 7 days after doxycycline induction of non-targeting control or ZRSR2-targeted shRNAs. f. Mis-splicing events after knockdown of ZRSR2 from cells shown in panel (e), calculated by pairwise comparison between ZRSR2 knockdown and controls (n=3 biological replicates each) on days 0, 2, and 7 of doxycycline induction. The number (and percentage) of events on each day is depicted in a single bar colored by different types of mis-splicing events: intron retention (red), 3’ cryptic splice site (green), and exon skip (blue). g. Density plots of frequencies of U2-type (blue) and U12-type (red) introns in pairwise analysis of CAL1 with ZRSR2 knockdown versus control on day 7 after the doxycycline induction (Fisher’s exact test p-value ≤ 0.05), plotted as in (b). h. (left) RNA-seq reads and RT-PCR from CAL1 cells after the induction of control or ZRSR2-targeted shRNA showing retention of DERL3 intron 4. (right) Number of DERL3 intron 4 RNA-seq reads plotted for control and ZRSR2 knockdown cells at days 0, 2, and 7 after doxycycline shRNA induction (n=3 biological replicates each, groups compared by t test).
Next, we asked if the splicing abnormalities observed were directly related to loss of ZRSR2. The predominant nonsense and frameshift ZRSR2 mutations we detected in BPDCN (Figure 2d–e) are predicted to result in absence of a mature protein, which is what we observed by western blotting in mutated compared to non-mutated BPDCNs (Figure 4d). Therefore, we used engineered knockdown or knockout cell line models to study ZRSR2 mutations in BPDCN cells. First, we generated CAL1 BPDCN cell lines harboring doxycycline-inducible shRNAs targeting ZRSR2 to model protein loss and facilitate time-dependent analysis of ZRSR2 depletion effects on splicing (Figure 4e). Seven days after induction of knockdown, intron retention was the most prominent acquired aberrant splicing event in shZRSR2 compared to shControl cells (Figure 4f). Both U2- and U12-type intron retention events were increased in ZRSR2 knockdown cells (Figure 4g) and intron retention events detected by RNA-seq could be confirmed by RT-PCR (Figure 4h). Knockdown of the NMD factor UPF1 in ZRSR2 deficient cells led to increased DERL3 RNA, one of the U12 genes consistently retained across BPDCNs and cell lines with ZRSR2 alterations, suggesting misspliced DERL3 is subject to NMD (Supplementary Figure 3d). Together, these experiments showed that U2- and U12-type intron retention is a consequence of ZRSR2 depletion on RNA splicing in human BPDCN cells.
ZRSR2 mutation impairs pDC activation and apoptosis induced by inflammatory stimuli
Genes that were misspliced in primary BPDCNs with ZRSR2, SRSF2, or SF3B1 mutations included several that are important in dendritic cell development and/or function, including CSF2RB, IRF4, IRF7, IRF8, and LILRB4 (Supplementary Table 4). Therefore, to connect missplicing with cellular phenotype we asked how ZRSR2 mutations affect pDC function. ZRSR2 knockdown was inversely correlated with expression of genes normally upregulated in Toll-like receptor 7 (TLR7) agonist (R848, resiquimod) treated dendritic cells (Figure 5a). This was of particular interest because studies have reported that primary BPDCN cells are less responsive to TLR/infectious stimulation than normal pDCs or have signatures of decreased activation, but the mechanisms are unknown (13,35). To test if complete loss of ZRSR2, as we saw in patient samples, was sufficient to impair pDC activation, we stimulated CAL1 cells harboring CRISPR/Cas9 knockout of ZRSR2 with TLR agonists (Figure 5b). Upregulation of the activation marker CD80 after stimulation with lipopolysaccharide (LPS, TLR4 agonist) or R848 (TLR7) was markedly reduced in cells with knockout of ZRSR2 (Figure 5c). Next, we assessed the same phenotype in primary human BPDCN and pDCs. BPDCN, across genotypes, had decreased induction of CD80 compared to pDCs after R848 stimulation (Figure 5d). Knockdown of ZRSR2 in otherwise normal primary pDCs also impaired the R848 response (Figure 5e). These data suggest that loss of ZRSR2 is one, but not necessarily the only, mechanism by which BPDCNs are rendered hyporesponsive to TLR activation.
Figure 5. pDC activation by Toll-like receptor stimulation is impaired by loss of ZRSR2 and in BPDCN.
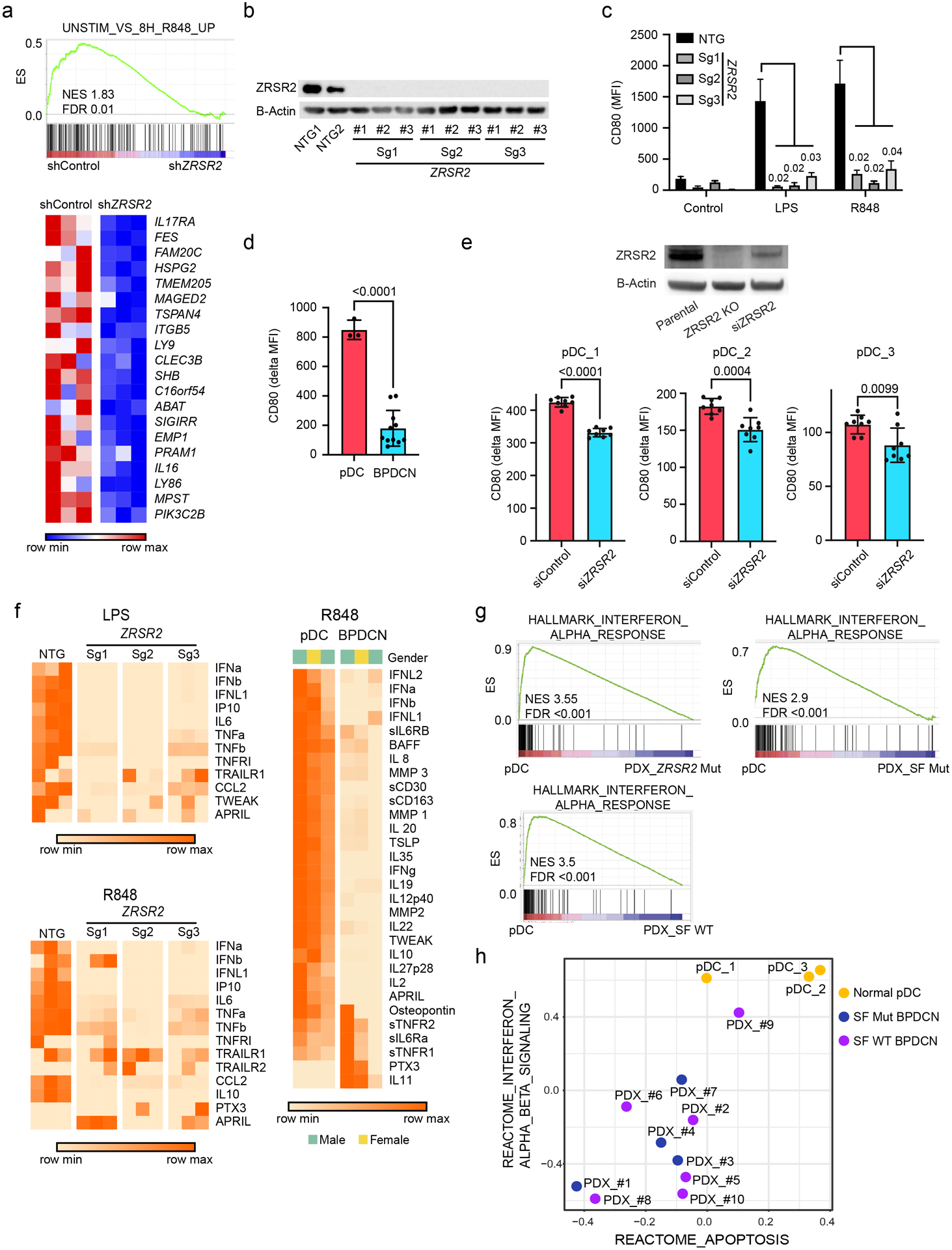
a. GSEA of RNA-seq showing decreased enrichment of TLR7 (R848) stimulated genes in ZRSR2 knockdown cells (shZRSR2 vs shControl CAL1 cells, each after 7 days of doxycycline shRNA induction; n=3 per condition). Heatmap shows expression levels of the leading edge genes in GSEA from blue (low) to red (high). ES, enrichment score; NES, normalized enrichment score; FDR, false discovery rate. b. Western blot for ZRSR2 and beta-actin in control or ZRSR2 knockout CAL1 cells. NTG1 and NTG2 are independent non-targeting sgRNAs and Sg1, Sg2, and Sg3 are independent ZRSR2-targeted sgRNAs, each assessed in biological triplicate samples. c. Mean fluorescence intensity of cell surface CD80 on control or ZRSR2 knockout CAL1 cells is shown after stimulation with either LPS or R848 (n=3 biological replicates of each sgRNA, groups compared by t test). d. CD80 upregulation (delta MFI; MFI stimulated-unstimulated) 24 hours after R848 stimulation in 3 sets of independent normal donor purified pDCs and in 11 primary BPDCN PDXs, groups compared by t test. e. (Top) Western blot for ZRSR2 and beta-actin in parental CAL1 cells and in CAL1 cells with CRISPR knockout (ZRSR2 KO) or transient knockdown (siZRSR2) of ZRSR2. (Bottom) CD80 upregulation after R848 treatment in primary pDCs from 3 independent healthy donors (pDC_1–3) each with 7–9 biological replicates each transfected with control or ZRSR2 siRNAs, groups compared by t test. f. Heatmaps showing protein quantitation of the indicated cytokines in supernatants of control or ZRSR2 knockout CAL1 cells (n=3 independent biological replicates of each sgRNA, non-targeting or ZRSR2-targeting), and normal pDCs or BPDCN PDXs (each column is from an independent individual donor or PDX) after stimulation with LPS or R848. BPDCN genotypes left-right: splicing factor wild-type, splicing factor wild-type, ZRSR2-mutant. g. GSEA of RNA-seq from R848 stimulated normal human pDCs or primary BPDCN PDXs that were ZRSR2 mutated (ZRSR2 Mut), any splicing factor mutated (SF Mut), or without known splicing factor mutation (SF WT). h. Bubble plot of enrichment scores from gene set variation analysis (GSVA) for the indicated type 1 interferon and apoptosis signatures from RNA-seq in normal pDCs (yellow) and BPDCN PDXs (blue, splicing factor mutated; purple, splicing factor wild-type) after treatment with R848.
TLR stimulation of normal pDCs induces secretion of numerous inflammatory cytokines, including type 1 interferons (IFN-α and IFN-β). In contrast, CAL1 cells with ZRSR2 knockout had defective secretion of several cytokines (e.g., IFN-α, IFN-β, IL-6, TNF-α) after LPS or R848 stimulation compared to controls (Figure 5f). We also found similar defective cytokine production in primary BPDCN cells compared to normal pDCs upon stimulation with R848 (Figure 5f). Of note, ZRSR2 loss did not simply cause a complete block in TLR signaling or protein secretion, because other cytokines were produced equivalently in ZRSR2-mutant cells and BPDCN cultures after stimulation (e.g., TRAILR1). Together, these data suggest that ZRSR2 mutations in BPDCN cells cause defective activation and impaired secretion of specific cytokines, including type 1 interferons, in the setting of TLR stimulation.
After activation, normal pDCs undergo apoptotic cell death as part of a negative feedback process that limits the inflammatory response (36). We hypothesized that BPDCN-associated alterations might protect pDCs from apoptosis in the setting of TLR stimulation and this could be a mechanistic link between spliceosome mutations and malignant transformation. Consistent with this model, we observed that gene expression changes in R848-stimulated primary BPDCNs compared to normal pDCs were enriched for decreased type 1 interferon and decreased apoptosis signatures (Figure 5g–h, Supplementary Figure 4a).
Next, we asked if loss of ZRSR2 conferred protection from apoptosis in the setting of inflammation. We stimulated CAL1 cells with LPS or R848 and found that both initiated apoptosis as measured by activation of caspase 3 and 7 (Figure 6a). Knockout of ZRSR2 conferred relative protection from LPS or R848-induced apoptosis (Figure 6b). The growth rate of CAL1 cells at steady state was not changed by ZRSR2 mutation. In contrast, while growth of wild-type cells was inhibited by LPS, ZRSR2 knockout cells were protected from LPS-induced growth arrest (Figure 6c).
Figure 6. ZRSR2 mutation impairs pDC apoptosis following TLR stimulation, associated with blunted IRF7 and TRAIL induction.
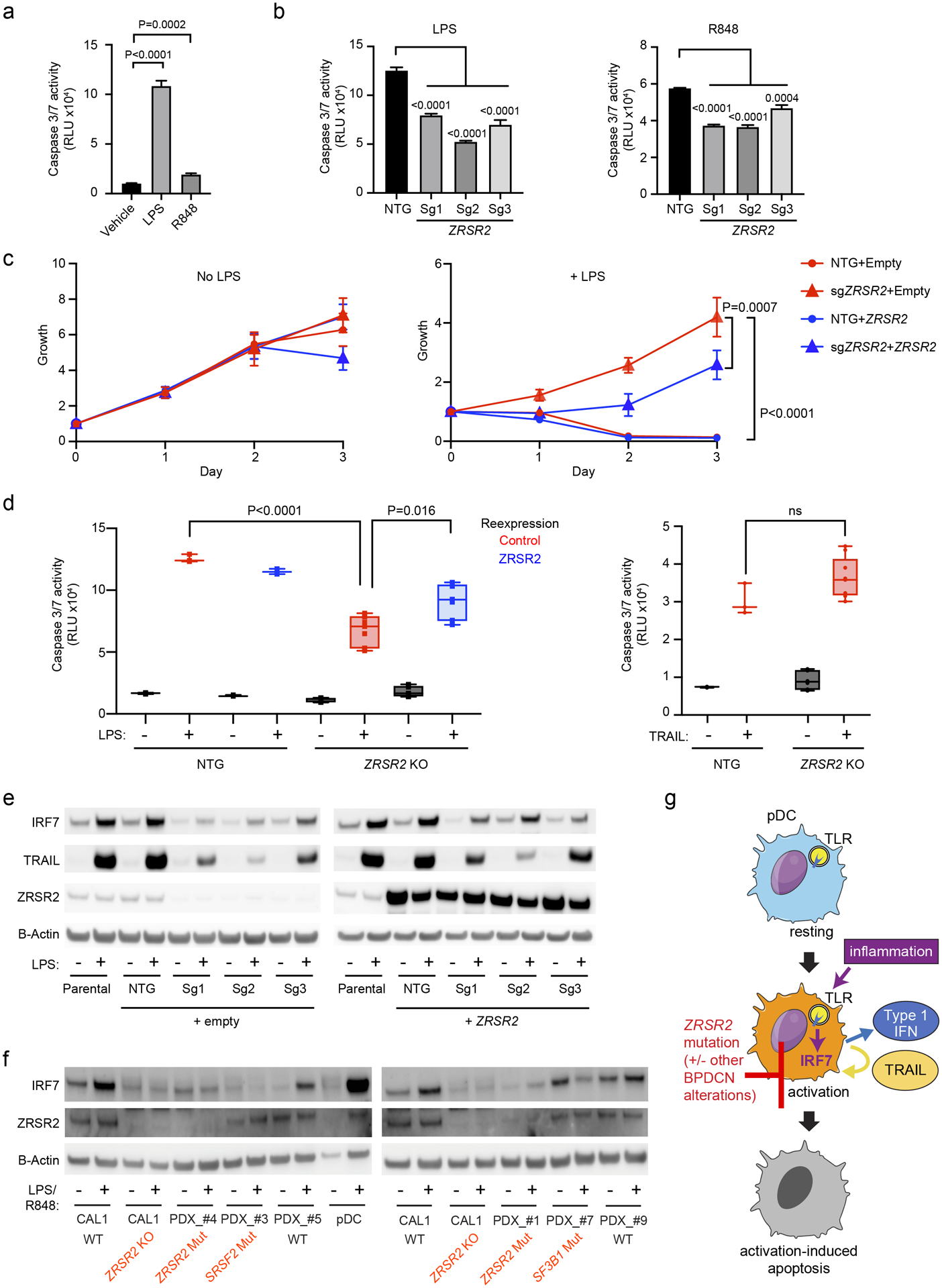
a. Caspase 3/7 activity in parental CAL1 cells after treatment with LPS or R848 compared to vehicle control. b. Caspase 3/7 activity in control or ZRSR2 knockout cells after stimulation with LPS or R848 compared to control sgRNA-expressing cells. c. Relative growth of control and ZRSR2 knockout cells, with wild-type ZRSR2 reexpression or empty vector control, are shown in normal medium (left) or in medium containing LPS (right). d. Caspase 3/7 activity in control and ZRSR2 knockout cells, with wild-type ZRSR2 reexpression or empty vector control, is shown after treatment with LPS or TRAIL. In panels a-d, n=3 biologically independent replicates, groups compared by t test. e. Western blot for IRF7, TRAIL, ZRSR2 and beta-actin in parental, non-targeting control, and ZRSR2 knockout CAL1 cells, with or without ZRSR2 reexpression, 24 hours after stimulation with LPS or vehicle. f. Western blot for IRF7, ZRSR2, and beta-actin in wild-type or ZRSR2 knockout (KO) CAL1 cells, BPDCN PDXs of the indicated genotypes, or normal pDCs, with and without LPS (CAL1) or R848 (BPDCN and normal pDCs) treatment. g. Model for BPDCN-associated mutations’ contribution to disease pathogenesis. Normal pDCs respond to inflammation via TLR signaling to IRF proteins, such as IRF7, which causes production of inflammatory mediators, such as type 1 interferons and TRAIL, and promotion of a feedback loop that leads to activation-induced apoptosis. pDCs with ZRSR2 mutations, and likely also in the presence of other BPDCN-associated alterations, are relatively protected from activation-induced apoptosis because they have impaired upregulation of IRF7 and downstream inflammatory mediators.
We confirmed the specificity of CRISPR knockout and that loss of ZRSR2 was necessary for protection from apoptosis by demonstrating that reexpression of wild-type ZRSR2 in mutant cells partially rescued the ability of LPS to induce apoptosis and impair growth (Figure 6c–d). Finally, we confirmed that ZRSR2-mutant CAL1 cells had not simply lost the ability to undergo cell death. A downstream consequence of pDC stimulation is production of TRAIL, a TNF-family pro-apoptotic cytokine, which promotes autocrine and paracrine apoptosis (37,38). Treatment with exogenous TRAIL promoted apoptosis equivalently in control and ZRSR2-mutant CAL1 cells, and in normal pDCs and BPDCNs, demonstrating that the cell death response downstream of TRAIL was intact (Figure 6d, Supplementary Figure 4b). Furthermore, while ZRSR2-mutant CAL1 cells had decreased surface expression of the TRAIL receptor DR5 after exogenous TRAIL treatment, a known consequence of receptor internalization after ligand binding, they did not after LPS or R848 treatment, consistent with a lack of autocrine TRAIL production and stimulation (Supplementary Figure 4c).
Collectively, these data suggested that ZRSR2 mutation perturbs specific pathways downstream of TLRs that result in impaired activation-induced cell death. While it is likely that multiple effects of splicing mutations contribute to BPDCN, we asked if any common targets across BPDCNs might be involved in the hypo-activation phenotype. When we analyzed aberrant splicing across BPDCNs harboring any splicing factor mutation, IRF7 was among the overlapping candidates. The IRF7 gene encodes a transcription factor, interferon regulatory factor 7, that is activated by TLR signaling and is important for induction of downstream genes, including type 1 interferons and TRAIL (39,40). Interestingly, the IRF7 mRNA transcript is not classified as having U12-type introns but it does contain a similarly behaving so-called “weak intron” (intron 4) that is known to be subject to intron retention and NMD in normal dendritic cells during activation (41). Rate-limiting splicing is also a feature of U12-type introns and more generally of a group of genes (with either U2 or U12 introns) that participate in “programmed delayed splicing” associated with inflammatory response regulation at the level of RNA processing (42,43). IRF7 intron 4 was aberrantly retained in ZRSR2 mutant BPDCNs (2 of 2) and the same intron-exon region was also misspliced in SRSF2 (4 of 4) and SF3B1-mutated (1 of 1) cases but not in splicing factor wild-type BPDCNs or in normal pDCs (Supplementary Table 4).
Therefore, we hypothesized that TLR stimulation of BPDCN with a ZRSR2 mutation would result in impairment of IRF7 induction and downstream gene activation including pro-apoptotic pathways. Indeed, whereas the baseline IRF7 protein levels were minimally affected by ZRSR2 mutation, mutant CAL1 cells had impaired ability to increase IRF7 protein after LPS stimulation (Figure 6e). Spliced IRF7 RNA increased, as expected, after stimulation in both control and mutant cells, whereas IRF7 intron 4 expression was selectively increased in mutant cells (Supplementary Figure 5a–b), which supports that the impairment of IRF7 protein induction by LPS is associated with intron retention. Induction of TRAIL was also impaired in ZRSR2-mutant cells, as expected for an IRF7 target gene (Figure 6e). IRF7 and TRAIL induction by LPS were partially rescued in ZRSR2-mutant cells by overexpression of wild-type ZRSR2 (Figure 6e). Furthermore, expression of an intronless, constitutively active IRF7 (44) impaired growth of both wild-type and ZRSR2-mutant cells (Supplementary Figure 5c). This suggested that loss of ZRSR2 affected the induction of IRF7 protein after LPS stimulation, but function downstream of activated IRF7 remained intact.
We also generated ZRSR2-deficient CAL1 cells with an intronless wild-type or transcriptionally inactive IRF7 cassette knocked into the IRF7 endogenous locus. Wild-type intronless IRF7, which would not require ZRSR2 for splicing, restored LPS-induced growth arrest in ZRSR2-mutant cells, whereas inactive IRF7 did not (Supplementary Figure 5d). Finally, we asked if impaired IRF7 upregulation after TLR stimulation was seen in primary BPDCN cells. In contrast to normal pDCs, ZRSR2-, SRSF2-, and SF3B1-mutant BPDCNs failed to upregulate IRF7 protein after stimulation (Figure 6f). Two splicing factor wild-type BPDCNs had normal IRF7 upregulation after TLR stimulation, which is consistent with the RNA splicing analysis where IRF7 was affected in all splicing factor mutant compared to non-mutant BPDCNs. Together, these data support a model in which ZRSR2 mutations provide a growth advantage in the setting of TLR activation, at least in part by impairing IRF7 induction and activation-induced apoptosis (Figure 6g). Given that impaired activation is observed in malignant cells across several genotypes, other disease-associated alterations may also contribute to this general feature of BPDCN.
BPDCN-associated Zrsr2 and Tet2 mutations promote aberrant pDC phenotypes in vivo
To evaluate the contribution of BPDCN-associated mutations to pDC development and function in vivo, we generated bone marrow chimeras with hematopoietic cells harboring mutations in Zrsr2, Tet2, or both. Tet2 was chosen because it is the most frequently mutated gene in BPDCN and is the most commonly co-occurring mutated gene in BPDCNs with mutated ZRSR2 (Figure 1a). We harvested c-kit+ bone marrow cells from Rosa-Cas9 knock-in animals, transduced with lentiviruses encoding sgRNAs targeting Zrsr2, Tet2, or control non-targeting guides in pairwise fashion, each co-expressing GFP or tagRFP. Eight weeks after transplantation of transduced marrow into lethally irradiated wild-type recipients, we observed approximately equivalent single- and double-positive GFP/tagRFP cells in the bone marrow across genotypes (Figure 7a). We confirmed CRISPR insertion-deletion events at sgRNA target sites qualitatively and quantitatively using a T7 endonuclease assay and by barcoded pooled sequencing (Supplementary Figure 6a–b).
Figure 7. BPDCN-associated mutations in hematopoietic progenitors affect dendritic cell differentiation, RNA splicing, and activation signatures in vivo.
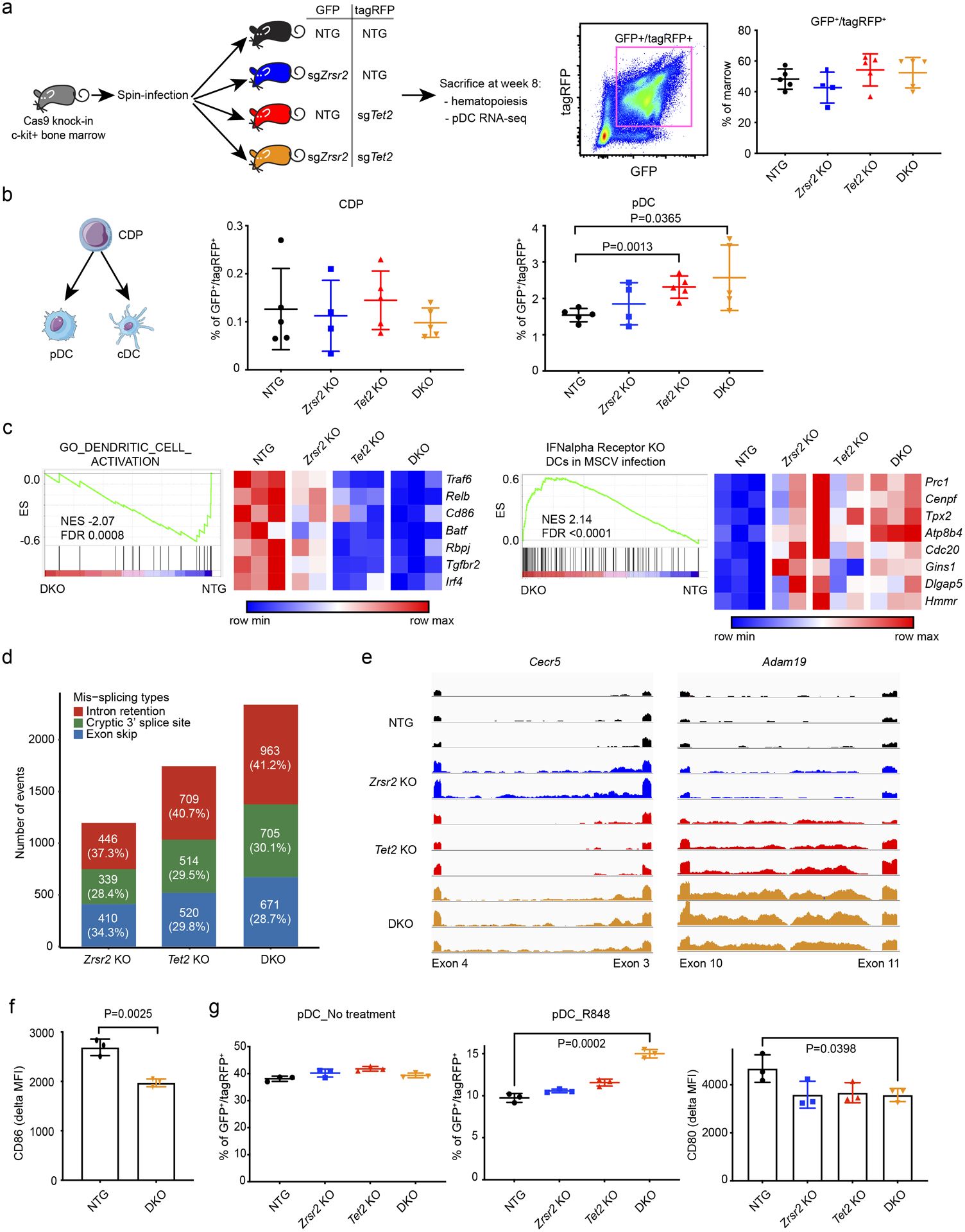
a. Schematic of the in vivo experiment. sgRNA-transduced Cas9 knock-in bone marrow c-kit+ progenitors were injected into lethally irradiated wild-type recipient mice. Each guide was marked with either GFP or tagRFP. The percentage of double positive cells in recipient bone marrow 8 weeks after transplantation is shown. b. Flow cytometry analysis of dendritic progenitor and mature pDCs in sgRNA positive marrow cells, groups compared by t test. CDP, common dendritic progenitor; pDC, plasmacytoid dendritic cell; cDC, conventional dendritic cell. c. GSEA of RNA-seq in control (NTG) and Zrsr2/Tet2-targeted (DKO) pDCs showing changes in signatures related to DC activation and IFN alpha receptor-dependent gene expression in the setting of viral infection. Heatmap shows expression of the leading edge genes in GSEA from low (blue) to high (red). d. Mis-splicing events in Zrsr2, Tet2, and Zrsr2/Tet2-targeted (DKO) pDCs. Events in each condition were calculated by pairwise comparisons between control knockout (n=3) and Zrsr2 (n=2), Tet2 (n=3), and Zrsr2/Tet2-targeted (DKO, n=3) biologically independent replicates. The number (and percentage) of events in each condition is shown in a single bar. The colors indicate different event types, intron retention (red), cryptic splice site (green), and exon skip (blue). e. Representative RNA-seq reads in Zrsr2, Tet2, and Zrsr2/Tet2-targeted (DKO) pDCs visualized on the same scale for Cecr5 (intron retention associated with Zrsr2 loss) and Adam19 (intron retention in Zrsr2 and Tet2-targeted pDCs with additive retention in Zrsr2/Tet2-targeted DKO pDCs). f. CD86 upregulation (delta MFI) in vivo after systemic treatment with R848 compared to vehicle in control (NTG) or Zrsr2/Tet2-targeted DKO bone marrow pDCs (n=3 independent animals/group, compared by t test). g. (Left 2 panels) Percentage of GFP+/tagRFP+ cells representing the indicated genotypes from in vitro cultures of Cas9 transgenic bone marrow expressing the indicated sgRNAs after vehicle or R848 treatment. (Right panel) CD80 upregulation (delta MFI) on pDCs of the indicated genotypes from R848 compared to vehicle-treated cultures. N=3 independent cultures per genotype, groups compared by t test.
We compared the GFP/tagRFP double positive populations in each of the four recipient groups (sgControl/sgControl, sgZrsr2/sgControl, sgControl/sgTet2, and sgZrsr2/sgTet2). We focused on dendritic cells and their precursors, but we also analyzed other hematopoietic stem and progenitor (HSPC) populations. Zrsr2 and Tet2 targeting was associated with relative expansion of lineage-negative, Sca1+, cKit+ (LSK) HSPCs, common myeloid progenitors (CMP), and megakaryocyte-erythroid progenitors (MEPs) in the bone marrow (Supplementary Figures 7, 8a–b). In dendritic cell development, common dendritic progenitors (CDP) differentiate into conventional DCs (cDCs) and pDCs. In recipients of transduced and transplanted bone marrow, CDP and cDC percentage and absolute number were not different in Zrsr2, Tet2, or Zrsr2/Tet2 double mutants compared to controls. In contrast, pDCs were modestly expanded, particularly in Tet2 and Zrsr2/Tet2 double mutant bone marrow (Figure 7b, Supplementary Figure 8a–b).
We assessed the transcriptome of mutant cells by sorting pDCs from bone marrow and performing RNA-seq. GSEA in mutant pDCs was negatively correlated with gene sets associated with DC activation and positively correlated with signatures of interferon alpha receptor knockout DCs exposed to viral infection (Figure 7c). This suggests that mutant mouse pDCs, similar to ZRSR2-mutant BPDCN cells, harbored signatures of decreased pDC activation and impaired type 1 interferon-dependent signaling. We also detected a higher frequency of mis-splicing events in the presence of Zrsr2, Tet2, and Zrsr2/Tet2 mutation. Similar to what we observed in BPDCN, pDCs harboring these mutations accumulated several types of aberrant splicing (Figure 7d). Comparison of each group revealed genotype-specific patterns, such as Zrsr2 mutation associated with a bias toward U12-type intron retention events, as we observed in human BPDCN cells with ZRSR2 mutations (Supplementary Figure 8c). Mutation of Tet2 alone was associated with splicing abnormalities including both U2 and U12 IR, and in some genes, we observed additivity between Tet2 and Zrsr2 mutations to promote increased intron retention (Figure 7e). These data are consistent with existing evidence that mutations in TET2 and other epigenetic modifiers can themselves promote splicing abnormalities, and in concert with splicing factor mutations produce cooperative effects on splicing and hematopoiesis (33,45–47). In vivo, Zrsr2/Tet2 mutant pDCs had impaired activation after systemic exposure to R848 (Figure 7f). In vitro differentiation of dendritic cells from gene-edited multipotent HSPCs yielded Zrsr2, Tet2, or Zrsr2/Tet2 mutant pDCs that had hypoactivation and a relative growth advantage, particularly in double mutant cells after R848 treatment (Figure 7g). Together, these data support our findings from human cells that ZRSR2 and TET2 loss (genes co-mutated in nearly all ZRSR2-mutant BPDCNs) promote specific transcriptome changes and provide a clonal advantage to pDCs associated with impaired activation.
DISCUSSION
Our findings contribute to understanding how acquired mutations in hematopoietic cells drive dendritic cell transformation. By sorting tumor cells, we are confident that the somatic mutations detected are not simply passengers of concomitant myeloid malignancies, but indeed are present in the BPDCN cells. Furthermore, these data provide evidence that specific mutations, particularly in RNA splicing factors and TET2, may have cell-type specific effects on pDCs, impairing their activation and apoptosis in response to TLR stimulation. We propose that innate immune signaling via the TLR-IRF pathway functions as a tumor suppressor in the pDC lineage to prevent malignant transformation. Further work will be required to determine if and how other mutations in BPDCN affect this pathway and contribute to similar phenotypes.
The high frequency of ZRSR2 loss-of-function mutations in BPDCN, exclusively in male patients, nominate it as a lineage-specific Escape from X Inactivation Tumor Suppressor (EXITS) gene. By conservative estimation including only unambiguously inactivating alterations, ZRSR2 mutations are associated with nearly half the male bias of BPDCN. We did not identify ZRSR2 as an EXITS gene in our prior study (24) because it is most often mutated in BPDCN, MDS, and other myeloid neoplasms such as CMML, cancers that were not included in large-scale sequencing efforts like The Cancer Genome Atlas (TCGA). Therefore, this suggests that additional sex-biased cancer genes remain discoverable in understudied diseases. These data also illustrate how identifying functional consequences of a sex-biased mutation may uncover more generalized tumor biology that is relevant even in cases without the specific mutation. For example, sex bias in non-malignant pDC function has been reported previously, specifically that females have more robust pDC activation and type 1 interferon production than males. This has been attributed to higher female expression of X chromosome (e.g., TLR7) (48) or autosomal (e.g., IRF5) (49) genes. Our data nominate ZRSR2 as another candidate gene that, particularly because of its X inactivation escape and higher basal expression in female cells, might render male pDCs less susceptible to activation-induced cell death and could contribute to transformation even in the absence of a mutation.
The effect of ZRSR2 loss was much more dramatic in TLR-stimulated cells than at baseline. The fact that IRF7 is regulated by delayed splicing during inflammation (42) again points to the TLR-IRF pathway as a common node that may contribute to BPDCN. We previously found that IRF7 was among the key pDC genes downregulated in BPDCN using single cell RNA-seq, suggesting that suppression of this pathway might be a common feature of pDC transformation even in the absence of a splicing factor mutation (14). Distinct acquired alterations in pDCs may converge on TLR signaling to confer a clonal advantage by evasion of activation-induced apoptosis, which could explain why we observed decreased activation after TLR stimulation across BPDCN genotypes. This also suggests that optimal modeling of transformation by BPDCN-associated mutations may need to be performed in the presence of inflammation.
Genetically engineered mouse models that develop spontaneous BPDCN in vivo are lacking. Clonal hematopoiesis (CH)-type mutations, such as in TET2 or ZRSR2, on their own are unlikely to be sufficient to cause BPDCN. We saw no BPDCN or obvious clonal evolution, albeit in relatively short-term in vivo experiments with Zrsr2 and/or Tet2 loss. Similarly, no overt myeloid or dendritic leukemia was seen in two other animal models of Zrsr2 knockout, with or without Tet2 co-mutation, that were followed at least one year (50,51). The variant allele frequencies of ZRSR2 and TET2 from patients harboring both mutations do not clearly indicate the order of acquisition (Supplementary Table 1), but this could be important for disease modeling and warrants additional investigation (52). Generating a BPDCN phenotype in mouse models may require other cooperating mutations, targeting alterations to a specific pDC maturation state or developmental stage, the addition of chronic inflammation, or skin ultraviolet light exposure, all of which will be important future experiments to pursue. Another priority is to develop improved methods to propagate and study primary human pDCs in vitro and/or to differentiate human pDCs from gene-edited HSPCs or from donor HSPCs that harbor CH mutations. These types of experiments are currently limited by normal pDCs being post-mitotic and sensitive to activation-induced cell death caused by lentiviral infection and other transfection methods (53).
TET2, ASXL1, and RNA splicing factor mutations are associated with clonal hematopoiesis and myeloid malignancies, such as MDS and CMML, that can pre-date BPDCN (54–56). The pathogenesis model proposed here suggests that specific mutations in CH/MDS/CMML contribute to a pDC pool that is poised for transformation. A testable hypothesis for future clinical research is that patients with these precursor conditions who develop BPDCN are enriched for having a history of abnormal inflammation. These findings also suggest that mutations in patients with myeloid malignancies or in individuals with age-related clonal hematopoiesis (ARCH or clonal hematopoiesis of indeterminate potential, CHIP) may affect pDC response to TLR stimulation and thereby contribute to impaired immunity. In support of this hypothesis, TET2 regulates Irf7 expression via DNA methylation and is important for pDC type 1 interferon production and survival during viral infection in a mouse model (57). Similarly, defective type 1 interferon production by pDCs is linked to deleterious IRF7 variants in patients with inferior outcomes during severe COVID-19 / SARS-CoV-2 infection, a subgroup that is also male-biased and older than the general population (58). Our data suggest that age-associated clonal hematopoiesis mutations, such as in TET2 or ZRSR2, might also contribute to these hypoactive pDC phenotypes.
Finally, there may be therapeutic implications of these data. Splicing modulator drugs are in development and malignancies with splicing factor mutations are more sensitive to these agents (59). Patients with BPDCN, stratified by presence of a splicing factor mutation or RNA mis-splicing pattern, could be included in splicing modulator clinical trials. Also, the UV mutational signature enrichment we observed may indicate BPDCN as a candidate for immune checkpoint blockade (ICB). ICB is only modestly active in other myeloid malignancies (60) but the cancers tested (AML, MDS) do not have a UV signature. In contrast, UV signatures predict response to checkpoint blockade in solid tumors, possibly due to qualitative differences in tumor neoantigens (61). PD-L1 protein is expressed in approximately half of BPDCNs (62), which also supports clinical evaluation of ICB via PD-1/PD-L1 in the disease. Lastly, we previously found that BPDCN is highly dependent on BCL2 and sensitive to the BCL2 inhibitor venetoclax (34). We do not yet know if this is directly related to splicing mutations and impaired activation-induced apoptosis, but the data presented here provide additional rationale for evaluating BCL2 inhibition in BPDCN.
Genes mutated in BPDCN are similar to those in MDS and AML. However, many clinical characteristics of BPDCN and AML are distinct, including epidemiology, clinical presentation, pathology, and response to certain therapies. These data suggest a mechanism by which the consequences of the same gene mutation (the ‘seed’; e.g., ZRSR2), could have lineage-specific effects based on cell context (the dendritic cell ‘soil’). The TLR-IRF7-type 1 interferon-apoptosis axis that is dysfunctional in BPDCN may not be the target of the same splicing mutations in other, even closely related, malignancies. This highlights the need to study BPDCN and other rare cancers as unique entities and to consider the importance of cellular milieu when evaluating the function of cancer genes.
METHODS
Patient samples
All studies were approved by local institutional review boards and conformed to guidelines for ethical research conduct in the Declaration of Helsinki, and patients provided written informed consent.
Cell lines
293T packaging cells were obtained from ATCC. CAL1 cells, a male BPDCN cell line that has a TET2 p.Q481fs* nonsense mutation at 0.97 VAF, were provided by T. Maeda (Nagasaki University, Nagasaki, Japan). Cell line identity was verified by short tandem repeat (STR) profiling in the DFCI Molecular Diagnostics Core and cells were verified to be mycoplasma-free by regular testing at least every 6 months. CAL1 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin (Gibco, 15140122), and 1% GlutaMAX (Gibco, 35050061). 293T packaging cells were cultured in DMEM supplemented with 10% FBS, penicillin/streptomycin. Proliferation and apoptosis were measured at the indicated times starting with a concentration of 2×105 cells/ml. Proliferation was measured by CellTiter-Glo (Promega, G7572) and caspase activity was measured by Caspase-Glo 3/7 (Promega, G8092).
Primary pDC/BPDCN cell isolation and stimulation
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation (Ficoll®-Paque PLUS, GE Healthcare, 17-1440-03) from healthy donors. Normal pDCs were enriched using magnetic beads (Miltenyi Biotec, 130-097-415) for transcriptomes. CD123+BDCA2+ cells were sorted for normal pDC and CD45dimCD123+ cells were sorted for BPDCN PDX tumor cells using a FACSAria. For stimulation experiments, pDCs were collected by negative selection using the EasySep Human Plasmacytoid DC Isolation Kit (StemCell Technologies, #17977) to avoid activation by sorting. In all cases, pDCs and BPDCNs (from patient and PDX) were >95% pure by flow cytometry. CAL1 cells were seeded at 2×105/ml, BPDCN tumor cells and normal pDCs were seeded at 1×105/ml. Cells were stimulated with either 1 μg/ml LPS (SigmaAldrich, L3129), 1 μg/ml R848 (Invitrogen, tlrl-r848), or 1 μg/ml TRAIL (BioLegend, #752906, 100ng/ml) for 24 hours. Supernatants were collected and cytokines were measured by either Bio-Plex Pro (BIO RAD, #17AL001M) or ProcartaPlex (Thermo Fisher, PPX-24). Cells were stained with 7-AAD, Annexin V, anti-CD80, and anti-DR5 for analysis of viability and activation.
Lentiviral infections
293T cells were transfected with 10.8 μg psPAX2, 2.4 μg pVSV-G, and 10.8 μg of a lentiviral expression vector with Lipofectamine 2000 (Thermo Fisher, 11668500). Viral supernatant was harvested 48 and 72 hours after transfection and concentrated by ultracentrifugation at 23,000g for 2 hours at 4°C. 2×105 cells were infected in the presence of 1.5 ml of viral supernatant and 4 μg/ml polybrene (Santa Cruz Biotechnology, SC-134220).
Animal experiments including mouse bone marrow derived pDCs
All animal experiments were performed with approval from the Dana-Farber Cancer Institute (DFCI) Animal Care and Use Committee. C57BL/6J and Rosa-Cas9 knock-in mice (B6J.129(Cg)-Gt (ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J) were purchased from Jackson Laboratory (026179). Cas9 knock-in mice were sacrificed and bone marrow cells were harvested from both legs, iliac bones, and spine for transplantation. CD117/c-kit+ cells were sorted by using magnetic beads (Miltenyi Biotec #130-094-224) after red cell lysis. After culture in StemSpan SFEM with 50 ng/ml SCF (Gold Bio Technology, 1320–01) and 50 ng/ml TPO (Gold Bio Technology, 1320–06) overnight, CD117/c-kit+ cells were infected with virus particles in the presence of 4 μg/ml polybrene. Transduced cells were injected into the tail vein of lethally irradiated (5.5 Gy x2 split doses) C57BL/6J mice with rescue marrow. The experiment was performed twice with 5 mice/group with similar results; in the first experiment donors and recipients were female and in the second experiment donors and recipients were male. Animals were sacrificed 8 weeks after the transplantation. Bone marrow and splenocytes were harvested for analysis. For generation of bone marrow derived pDCs, transduced cells were cultured in RPMI 1640 supplemented with 10% FBS, mouse Flt3-ligand (Peprotech, #250–31L, 100ng/ml) and 2-mercaptoethanol (Sigma-Aldrich, M3138, 50 μM) for 7 days. For in vivo treatment with TLR ligand, mice were injected with R848 (Invitrogen, tlrl-r848, 5 μg in 200 μl of PBS) via the tail vein.
Plasmids for cDNA and shRNA expression and CRISPR/Cas9 gene targeting
shRNAs were subcloned into a Tet-on pLKO-puro (Addgene, #21915) via AgeI and EcoRI restriction sites. shRNAs were induced with 1 μg/ml doxycycline after selecting transduced cells in 1 μg/ml puromycin. sgRNAs were subcloned into a lentiviral expression vector that coexpresses GFP (pLKO5.sgRNA.EFS.GFP; Addgene, #57822) or tagRFP (pLKO5.sgRNA.EFS.tagRFP; Addgene, #57823) via the BsmBI restriction site. Cell lines stably expressing the Cas9 nuclease were generated by infection with the empty lentiCRISPRv2 lentivirus (Addgene, #52961, not containing an sgRNA guide) using standard methods (https://www.addgene.org/viral-vectors/lentivirus/lenti-guide/). Cells were selected in puromycin and FLAG-Cas9 expression was confirmed by Western blot. Cas9-expressing cell lines were infected at a density of 2×105 cells in 1.5 mL media in the presence of 4 μg/mL polybrene (Santa Cruz Biotechnology, SC-134220). sgRNA resistant human ZRSR2 DNA fragment was synthesized by Twist Bioscience and cloned into pRRL.idTomato. sgRNA, shRNA, and sgRNA resistant ZRSR2 cDNA sequences are in Supplementary Methods.
Primary pDC and cell line siRNA transfection
siZRSR2 (Dharmacon, L-006596–02), siUPF1(Dharmacon, L-011763–00), Non-targeting Control Pool siRNA (Dharmacon, D-001810–10), siGLO Cyclopholin B control siRNA (Dharmacon, D-001610–01) were transfected using N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP; Roche Diagnostics, #11202375001) as in (53). Equal volumes of siRNA (final concentration 160 nM) and DOTAP were mixed and incubated for 15 minutes at room temperature. The mixture was added on top of cells. pDCs were seeded at 1×105 cells/100 μl in 96 well plates and CAL1 cells were seeded 2×105 cells/ml in 6 well plates. Cells were incubated at 37°C for 24 hours then washed before performing experiments.
Flow cytometry
Cells were washed with PBS containing 2% FBS before staining and were then incubated with the indicated antibodies indicated for 30 minutes in the dark at 4° followed by a final wash in PBS containing 2% FBS and analyzed. In the case of murine samples, we lysed red blood cells prior to staining. Antibodies for staining are indicated in the following table. Gating strategy for bone marrow cells are defined as indicated below. Flow cytometry was performed using a BD LSR Fortessa X-20 and analyzed using FlowJo software, version 10. Antibodies for flow cytometry are in Supplementary Table 5. Cell surface markers for HSPC populations are in Supplementary Table 6.
Western blotting
Samples were prepared by lysing in RIPA buffer (Boston BioProducts, BP-115) with protease inhibitor cocktail (Thermo Fisher Scientific, 862209) and sonicated before quantification by BCA assay (Thermo Fisher Scientific, 23225). Samples were prepared with SDS sample buffer (Boston BioProducts, BP-110R), boiled for 10 minutes at 98°C, and recovered by spinning at 12,000g for 5 minutes at 4°C before loading onto the gel. The gel was run for 80 minutes at 120V in SDS running buffer (Boston BioProducts, BP-177) before being transferred to a PVDF membrane (Thermo Fisher Scientific, 7462) for 7 minutes at 20V using iBlot 2 (Thermo Fisher Scientific). Blots were blocked in 5% dried milk (AppliChem, A0830) in tris-buffered saline (TBS) with 0.1% Tween-20 for 1 hour before being incubated overnight in antibodies recognizing ZRSR2 (kindly provided by Dr. Michael Green, University of Massachusetts Medical School, Massachusetts, 1:2000), IRF7 (Cell Signaling technology, #4920, 1:1000), TRAIL (Cell Signaling technology, #3219, 1:1000), UPF1 (Abcam, #ab109363, 1:10000), or β-actin (Sigma-Aldrich, A5441, 1:10,000). The blots were washed 3 times in TBS with 0.1% Tween-20 before being incubated with either rabbit (Santa Cruz Biotechnology, SC-2004) or mouse (Santa Cruz Biotechnology, SC-2005) secondary HRP-conjugated antibodies before being imaged using ECL substrate (Bio-Rad, 170–5061) on an ImageQuant LAS-4000 (GE Healthcare, 28-9607-59AB).
T7 endonuclease (T7E1) assay
Genomic DNA from mouse bone marrow cells was extracted by the DNeasy Blood and Tissue Kit (Qiagen, #69504). The DNA region containing sgRNA target sites was amplified by PCR using specific primers (Zrsr2 Forward, CCCATGGCATCTTTGTCTATAATCT-; Zrsr2 Reverse, GCTCAGCTAAGCACTTACTCAATG; Tet2_Forward, ACATACTCCTCAGACGCAGG; Tet2_Reverse, CTGGCATGTACCTGGATTGC). PCR products were purified with NucleoSpin Gel and PCR Clean-Up (Takara Bio, #740609). 400 ng of PCR products mixed with NEB buffer 2 were denatured at 95°C for 5 minutes and ramped down to 25°C. T7 Endonuclease 1 (NEB, M0302) was added to the mixture and incubated at 37°C for 15 minutes. 0.25 M EDTA was added to stop the reaction and samples were analyzed on 2% agarose gel.
CRISPR/Cas9 editing site sequencing
Genomic DNA was extracted from GFP+/tagRFP+ mouse Lineage−c-kit+ bone marrow cells using TRIzol (Life Technologies, 15596018). PCR was performed spanning the predicted Cas9 cut sites, using the primers as below. Illumina compatible adapters with unique barcodes were ligated onto each sample during library construction. Libraries were pooled in equimolar concentrations for multiplexed sequencing on the Illumina MiSeq platform with 2×150 run parameters. Upon completion of the sequencing run, data were demultiplexed and subsequently entered into an automated de novo assembly pipeline. Sequencing and analysis were performed by the CCIB DNA Core Facility at Massachusetts General Hospital (Cambridge, MA). Tet2 For, GGGGGTTGGAGCAAGTACAAA; Tet2 Rev, GCCCCTGTAGAGACGAAGC; Zrsr2 For, CACACTCCCATCTTCACTGCT; Zrsr2 Rev, CAGCTAAGCACTTACTCAATGAACT
RT-PCR
RNA was extracted from cell lines using TRIzol (Life Technologies, 15596018) and quantified via NanoDrop. The High-Capacity cDNA Reverse Transcriptase Kit (Thermo Fisher Scientific, 4368814) was used to generate cDNA. qRT-PCR was performed for human IRF7, using GAPDH as the internal control, using the primers as below, and SYBR Green PCR master mix per the manufacturer’s instructions (Thermo Fisher Scientific, 4367659). Relative quantification was calculated using the ΔΔCt method. IRF7 For, TACCATCTACCTGGGCTTCG; IRF7 Rev, GAAGA-CACACCCTCACGCTG; IRF7 intron4 For, TGCACCTGGACGGACACTTTAG; IRF7 intron4 Rev, CCTCCTAATTCTCCAGCTCC; GAPDH For, GCACCGTCAAGGCTGAGAAC; GAPDH Rev, TGGTGA-AGACGCCAGTGGA. For RT-PCR, the following primers were used: DERL3 For, GGCCGACTTCGTCTTCATGTTTC; DERL3 Rev, CAGGTCCACGAGGATGGAGT.
IRF7 knock-in cell line generation
The FLAG tag of pFETCh_Donor (Addgene, #63934) was replaced with a V5 tag via the KasI and the BsrGI restriction sites. Homology arm 1 (HOM1) and Homology arm 2 (HOM2) were designed for intronless wild-type IRF7 and inactive form IRF7 (9A) with Gibson Assembly tails and synthesized by Twist Bioscience. Homology arms were subcloned into pFETCh_Donor with Gibson assembly (New England Biolabs, #E2611). sgRNAs were subcloned into pXPR_BRD202 via the BsmBI restriction site. 12 μg of pFETCh, 6 μg of each sgRNA, 60 μl of Lipofectamine, and 3 ml of Opti-MEM (Life Technologies, #11058021) were mixed and incubated for 20 minutes at room temperature, then added to 1 million Cas9-expressing CAL1 cells in 10 ml media. Cells were selected in G418 (Fisher Scientific, #MT61234RG) and V5 tag expression was confirmed by Western blot. IRF7 sgRNA and homology arm sequences are in Supplementary Methods.
Whole exome sequencing and analysis
Tumor and paired germline DNA samples were collected and prepared for whole-exome sequencing using the Agilent SureSelect Human All Exon V5 (50M) capture. Sequencing was performed on Hiseq4000 platform with a 150 base pair paired-end protocol targeted to 100x mean coverage. The sequences were generated as 90/100 base paired-end reads using Illumina base calling Software (ver. 1.7). The adapter sequences and low-quality reads were filtered from the raw sequencing data and the “clean data” was aligned by Burrows-Wheeler Aligner (BWA) (63) with the reference of human genome build37 (hg19). The BAM files were validated by steps of fixing mate information of the alignment, adding read group information, removing duplicate reads, and were then applied to variant calling analysis using GATK toolkit (https://gatk.broadinstitute.org/hc/en-us). The reference (hg19) and germline variant sites were used to detect somatic or tumor-specific mutations and copy number alterations. The details of the methods in GATK toolkit are described in (64,65). The identified BPDCN somatic mutations were analyzed with those reported for acute myeloid leukemia (AML) and skin melanoma (66) for presence of previously defined global DNA mutational signatures in the Catalog of Somatic Mutations in Cancer (COSMIC) database (20) using the R package, “MutationalPatterns” (67).
RNA sequencing and analysis
Samples from pDCs, BPDCN patients, PDXs, CAL1, and mouse models were collected for total RNA sequencing using the Arcturus PicoPure RNA Isolation Kit (Life Technologies). Libraries were prepared using the Ovation kit (Nugen, #0340) using 50 ng of input total RNA and 20 cycles of amplification. The Illumina Hi-Seq platform was used to generate single or paired-end sequencing results. The raw FASTQ data were analyzed by the VIPER pipeline (68) which combines STAR (69) alignment (mapped to hg19 or mm10) and gene expression analysis such as unsupervised clustering, PCA based on Cufflinks (70) and differential expression with DESeq2 (71). The expression data were used for pathway enrichment analysis with GSEA and GSVA (72,73).
Differential splicing analysis
Splicing events were identified as described in (27). The method calculates a mis-splicing index (MSI) for each splicing event in comparison to the reference genome (human hg19 or mouse mm10) and classifies them as intron retention, exon skip, and incorrect splice site usage. The difference in MSI between two samples was used for direct comparisons by defining the delta MSI (ΔMSI) as the difference in MSI at a given site between two samples. The statistical significance of differences in splicing events was evaluated by Fishers’ exact test with adjusted P-value for multiple hypothesis testing. Differences with ΔMSI>0.2 and P≤0.05 were considered significant.
Data visualization and availability
Analysis results were visualized using R 3.5.2 (R Foundation for Statistical Computing) (74) and Python (https://www.python.org). A co-mutation plot was created using the “GenVisR” package in R (75). RNA sequencing data are deposited in Gene Expression Omnibus (GEO), accession number GSE184656.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Sex bias in cancer is well recognized but the underlying mechanisms are incompletely defined. We connect X chromosome mutations in ZRSR2 to an extremely male-predominant leukemia. Aberrant RNA splicing induced by ZRSR2 mutation impairs dendritic cell inflammatory signaling, interferon production, and apoptosis, revealing a sex- and lineage-related tumor suppressor pathway.
ACKNOWLEDGEMENTS
This work was supported by the Sumitomo Life Welfare and Culture Foundation (KT), the National Cancer Institute R37 CA225191 (AAL) and R35 CA231958 (DMW), the Department of Defense CA190171 (MK, NP, OAW, and AAL), the Damon Runyon Cancer Research Foundation (GKG), Mark Foundation for Cancer Research (AAL), Doris Duke Charitable Foundation (AAL), Bertarelli Rare Cancers Fund (AAL), Ludwig Center at Harvard (AAL), and the American Society of Hematology (AAL).
Conflict of Interest Statement:
AAL has received research funding from AbbVie and Stemline Therapeutics and consulting fees from N-of-One and Qiagen. GKG has received research funding from Calico Life Sciences and consulting fees from Moderna Therapeutics. MS and SB were employees of H3 Biomedicine at the time of this study. MS is currently an employee of Remix Therapeutics (Cambridge, MA).
REFERENCES
- 1.Deconinck E, Petrella T, Garnache Ottou F. Blastic Plasmacytoid Dendritic Cell Neoplasm: Clinical Presentation and Diagnosis. Hematol Oncol Clin North Am 2020;34(3):491–500 doi 10.1016/j.hoc.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Garnache-Ottou F, Vidal C, Biichle S, Renosi F, Poret E, Pagadoy M, et al. How should we diagnose and treat blastic plasmacytoid dendritic cell neoplasm patients? Blood Adv 2019;3(24):4238–51 doi 10.1182/bloodadvances.2019000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor J, Haddadin M, Upadhyay VA, Grussie E, Mehta-Shah N, Brunner AM, et al. Multicenter analysis of outcomes in blastic plasmacytoid dendritic cell neoplasm offers a pretargeted therapy benchmark. Blood 2019;134(8):678–87 doi 10.1182/blood.2019001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagano L, Valentini CG, Pulsoni A, Fisogni S, Carluccio P, Mannelli F, et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica 2013;98(2):239–46 doi 10.3324/haematol.2012.072645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bastidas Torres AN, Cats D, Mei H, Fanoni D, Gliozzo J, Corti L, et al. Whole-genome analysis uncovers recurrent IKZF1 inactivation and aberrant cell adhesion in blastic plasmacytoid dendritic cell neoplasm. Genes Chromosomes Cancer 2020;59(5):295–308 doi 10.1002/gcc.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jardin F, Ruminy P, Parmentier F, Troussard X, Vaida I, Stamatoullas A, et al. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm. Br J Haematol 2011;153(3):413–6 doi 10.1111/j.1365-2141.2010.08556.x. [DOI] [PubMed] [Google Scholar]
- 7.Lucioni M, Novara F, Fiandrino G, Riboni R, Fanoni D, Arra M, et al. Twenty-one cases of blastic plasmacytoid dendritic cell neoplasm: focus on biallelic locus 9p21.3 deletion. Blood 2011;118(17):4591–4 doi 10.1182/blood-2011-03-337501. [DOI] [PubMed] [Google Scholar]
- 8.Menezes J, Acquadro F, Wiseman M, Gomez-Lopez G, Salgado RN, Talavera-Casanas JG, et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2014;28(4):823–9 doi 10.1038/leu.2013.283. [DOI] [PubMed] [Google Scholar]
- 9.Sapienza MR, Abate F, Melle F, Orecchioni S, Fuligni F, Etebari M, et al. Blastic plasmacytoid dendritic cell neoplasm: genomics mark epigenetic dysregulation as a primary therapeutic target. Haematologica 2019;104(4):729–37 doi 10.3324/haematol.2018.202093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alayed K, Patel KP, Konoplev S, Singh RR, Routbort MJ, Reddy N, et al. TET2 mutations, myelodysplastic features, and a distinct immunoprofile characterize blastic plasmacytoid dendritic cell neoplasm in the bone marrow. Am J Hematol 2013;88(12):1055–61 doi 10.1002/ajh.23567. [DOI] [PubMed] [Google Scholar]
- 11.Brunetti L, Di Battista V, Venanzi A, Schiavoni G, Martelli MP, Ascani S, et al. Blastic plasmacytoid dendritic cell neoplasm and chronic myelomonocytic leukemia: a shared clonal origin. Leukemia 2017;31(5):1238–40 doi 10.1038/leu.2017.38. [DOI] [PubMed] [Google Scholar]
- 12.Luskin MR, Kim AS, Patel SS, Wright K, LeBoeuf NR, Lane AA. Evidence for separate transformation to acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm from a shared ancestral hematopoietic clone. Leuk Lymphoma 2020:1–4 doi 10.1080/10428194.2020.1755856. [DOI] [PubMed] [Google Scholar]
- 13.Chaperot L, Bendriss N, Manches O, Gressin R, Maynadie M, Trimoreau F, et al. Identification of a leukemic counterpart of the plasmacytoid dendritic cells. Blood 2001;97(10):3210–7 doi 10.1182/blood.v97.10.3210. [DOI] [PubMed] [Google Scholar]
- 14.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017;356(6335) doi 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sapienza MR, Fuligni F, Agostinelli C, Tripodo C, Righi S, Laginestra MA, et al. Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia 2014;28(8):1606–16 doi 10.1038/leu.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kluk MJ, Lindsley RC, Aster JC, Lindeman NI, Szeto D, Hall D, et al. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J Mol Diagn 2016;18(4):507–15 doi 10.1016/j.jmoldx.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18(6):553–67 doi 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 2011;365(2):127–38 doi 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 2016;45(3):626–40 doi 10.1016/j.immuni.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 20.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jardin F, Callanan M, Penther D, Ruminy P, Troussard X, Kerckaert JP, et al. Recurrent genomic aberrations combined with deletions of various tumour suppressor genes may deregulate the G1/S transition in CD4+CD56+ haematodermic neoplasms and contribute to the aggressiveness of the disease. Leukemia 2009;23(4):698–707 doi 10.1038/leu.2008.359. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478(7367):64–9 doi 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 23.Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, et al. Landscape of X chromosome inactivation across human tissues. Nature 2017;550(7675):244–8 doi 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunford A, Weinstock DM, Savova V, Schumacher SE, Cleary JP, Yoda A, et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat Genet 2017;49(1):10–6 doi 10.1038/ng.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep 2018;23(1):282–96 e4 doi 10.1016/j.celrep.2018.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Yoshimi A, Wang J, Cho H, Chun-Wei Lee S, Ki M, et al. Mutations in the RNA Splicing Factor SF3B1 Promote Tumorigenesis through MYC Stabilization. Cancer Discov 2020;10(6):806–21 doi 10.1158/2159-8290.CD-19-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 2015;6:6042 doi 10.1038/ncomms7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013;154(3):583–95 doi 10.1016/j.cell.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 29.Shen H, Zheng X, Luecke S, Green MR. The U2AF35-related protein Urp contacts the 3’ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev 2010;24(21):2389–94 doi 10.1101/gad.1974810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turunen JJ, Niemela EH, Verma B, Frilander MJ. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA 2013;4(1):61–76 doi 10.1002/wrna.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madan V, Li J, Zhou S, Teoh WW, Han L, Meggendorfer M, et al. Distinct and convergent consequences of splice factor mutations in myelodysplastic syndromes. Am J Hematol 2020;95(2):133–43 doi 10.1002/ajh.25673. [DOI] [PubMed] [Google Scholar]
- 32.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015;27(5):617–30 doi 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016;30(3):404–17 doi 10.1016/j.ccell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montero J, Stephansky J, Cai T, Griffin GK, Cabal-Hierro L, Togami K, et al. Blastic Plasmacytoid Dendritic Cell Neoplasm Is Dependent on BCL2 and Sensitive to Venetoclax. Cancer Discov 2017;7(2):156–64 doi 10.1158/2159-8290.CD-16-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beird HC, Khan M, Wang F, Alfayez M, Cai T, Zhao L, et al. Features of non-activation dendritic state and immune deficiency in blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood Cancer J 2019;9(12):99 doi 10.1038/s41408-019-0262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med 2011;208(12):2367–74 doi 10.1084/jem.20110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blum A, Chaperot L, Molens JP, Foissaud V, Plantaz D, Plumas J. Mechanisms of TRAIL-induced apoptosis in leukemic plasmacytoid dendritic cells. Exp Hematol 2006;34(12):1655–62 doi 10.1016/j.exphem.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 38.Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens JP, et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol 2006;176(1):248–55 doi 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- 39.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005;434(7034):772–7 doi 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 40.Romieu-Mourez R, Solis M, Nardin A, Goubau D, Baron-Bodo V, Lin R, et al. Distinct roles for IFN regulatory factor (IRF)-3 and IRF-7 in the activation of antitumor properties of human macrophages. Cancer Res 2006;66(21):10576–85 doi 10.1158/0008-5472.CAN-06-1279. [DOI] [PubMed] [Google Scholar]
- 41.Frankiw L, Majumdar D, Burns C, Vlach L, Moradian A, Sweredoski MJ, et al. BUD13 Promotes a Type I Interferon Response by Countering Intron Retention in Irf7. Mol Cell 2019;73(4):803–14 e6 doi 10.1016/j.molcel.2018.11.038. [DOI] [PubMed] [Google Scholar]
- 42.Majumdar DS, Frankiw L, Burns CH, Garcia-Flores Y, Baltimore D. Programmed Delayed Splicing: A Mechanism for Timed Inflammatory Gene Expression. bioRxiv 2018. doi 10.1101/443796. [DOI] [Google Scholar]
- 43.Patel AA, McCarthy M, Steitz JA. The splicing of U12-type introns can be a rate-limiting step in gene expression. EMBO J 2002;21(14):3804–15 doi 10.1093/emboj/cdf297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG, Siggers T. DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer-specific gene regulation. Nucleic Acids Res 2018;46(5):2509–20 doi 10.1093/nar/gky002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marina RJ, Sturgill D, Bailly MA, Thenoz M, Varma G, Prigge MF, et al. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. EMBO J 2016;35(3):335–55 doi 10.15252/embj.201593235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Valiente C, Garcia-Ruiz C, Roson B, Liquori A, Gonzalez-Romero E, Fernandez-Gonzalez R, et al. Aberrant Alternative Splicing in U2af1/Tet2 Double Mutant Mice Contributes to Major Hematological Phenotypes. Int J Mol Sci 2021;22(13) doi 10.3390/ijms22136963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019;574(7777):273–7 doi 10.1038/s41586-019-1618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Webb K, Peckham H, Radziszewska A, Menon M, Oliveri P, Simpson F, et al. Sex and Pubertal Differences in the Type 1 Interferon Pathway Associate With Both X Chromosome Number and Serum Sex Hormone Concentration. Front Immunol 2018;9:3167 doi 10.3389/fimmu.2018.03167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-alpha Production in Women. J Immunol 2015;195(11):5327–36 doi 10.4049/jimmunol.1501684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madan V, Cao Z, Teoh WW, Dakle P, Han L, Shyamsunder P, et al. ZRSR1 cooperates with ZRSR2 in regulating splicing of U12-type introns in murine hematopoietic cells. Haematologica 2021. doi 10.3324/haematol.2020.260562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inoue D, Polaski JT, Taylor J, Castel P, Chen S, Kobayashi S, et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet 2021;53(5):707–18 doi 10.1038/s41588-021-00828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 2015;372(7):601–12 doi 10.1056/NEJMoa1412098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith N, Vidalain PO, Nisole S, Herbeuval JP. An efficient method for gene silencing in human primary plasmacytoid dendritic cells: silencing of the TLR7/IRF-7 pathway as a proof of concept. Sci Rep 2016;6:29891 doi 10.1038/srep29891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–98 doi 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014;28(2):241–7 doi 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mason CC, Khorashad JS, Tantravahi SK, Kelley TW, Zabriskie MS, Yan D, et al. Age-related mutations and chronic myelomonocytic leukemia. Leukemia 2016;30(4):906–13 doi 10.1038/leu.2015.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma S, Wan X, Deng Z, Shi L, Hao C, Zhou Z, et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J Exp Med 2017;214(5):1471–91 doi 10.1084/jem.20161149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020;370(6515) doi 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 2016;22(6):672–8 doi 10.1038/nm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bewersdorf JP, Stahl M, Zeidan AM. Immune checkpoint-based therapy in myeloid malignancies: a promise yet to be fulfilled. Expert Rev Anticancer Ther 2019;19(5):393–404 doi 10.1080/14737140.2019.1589374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pham TV, Boichard A, Goodman A, Riviere P, Yeerna H, Tamayo P, et al. Role of ultraviolet mutational signature versus tumor mutation burden in predicting response to immunotherapy. Mol Oncol 2020;14(8):1680–94 doi 10.1002/1878-0261.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aung PP, Sukswai N, Nejati R, Loghavi S, Chen W, Torres-Cabala CA, et al. PD1/PD-L1 Expression in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers (Basel) 2019;11(5) doi 10.3390/cancers11050695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature 2011;470(7333):214–20 doi 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011;471(7339):467–72 doi 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med 2018;10(1):33 doi 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cornwell M, Vangala M, Taing L, Herbert Z, Koster J, Li B, et al. VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics 2018;19(1):135 doi 10.1186/s12859-018-2139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28(5):511–5 doi 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. [Google Scholar]
- 75.Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, et al. GenVisR: Genomic Visualizations in R. Bioinformatics 2016;32(19):3012–4 doi 10.1093/bioinformatics/btw325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.