

Abstract
Rationale:
The initial hypertrophy response to cardiac pressure overload is considered compensatory, but with sustained stress, it eventually leads to heart failure. Recently, a role for recruited macrophages (mφs) in determining the transition from compensated to decompensated hypertrophy has been established. However, whether cardiac-resident immune cells influence the early phase of hypertrophy development has not been established.
Objective:
To assess the role of cardiac immune cells in the early hypertrophy response to cardiac pressure overload-induced by transverse aortic constriction (TAC).
Methods and Results:
We performed cytometry-by-time-of-flight to determine the identity and abundance of immune cells in the heart at 1 and 4 weeks after TAC. We observed a substantial increase in cardiac mφs 1 week after TAC. We then conducted Cite-Seq single-cell RNA sequencing of cardiac immune cells isolated from 4 sham and 6 TAC hearts. We identified 12 clusters of monocytes and mφs, categorized as either resident or recruited mφs, that showed remarkable changes in their abundance between sham and TAC conditions. To determine the role of cardiac-resident mφs early in the response to a hypertrophic stimulus, we used a blocking antibody against macrophage colony-stimulating factor 1 receptor (CD115). As blocking CD115 initially depletes all macrophages, we allowed the replenishment of recruited mφs by monocytes before performing TAC. This preferential depletion of resident mφs resulted in enhanced fibrosis and a blunted angiogenesis response to TAC. Mφ-depletion in CCR2 knockout mice showed that aggravated fibrosis was primarily caused by the recruitment of monocyte-derived mφs. Finally, 6 weeks after TAC these early events lead to depressed cardiac function and enhanced fibrosis, despite complete restoration of cardiac immune cells.
Conclusions:
Cardiac resident mφs are a heterogeneous population of immune cells with key roles in stimulating angiogenesis and inhibiting fibrosis in response to cardiac pressure overload.
Keywords: Basic Science Research, Fibrosis, Hypertrophy, Inflammation
Graphical Abstract
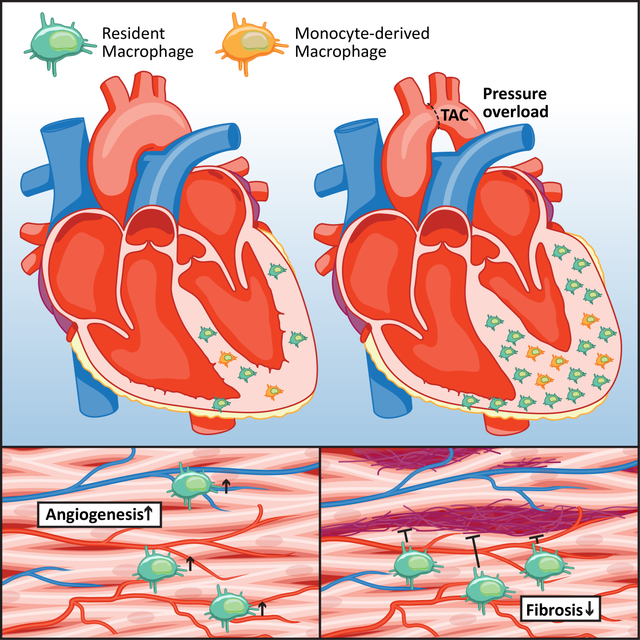
INTRODUCTION
The heart responds to stress such as pressure overload or myocardial infarction with an increase in cardiac muscle mass1, 2. This hypertrophic response is initially considered adaptive and involves an increase in microvascular density3. With sustained stress, the heart eventually transitions into a phase of maladaptive growth, resulting in fibrosis, microvascular rarefaction, and reduced cardiac function4, 5. Most of the research into the mechanisms that drive cardiac hypertrophy and failure focused on cardiomyocyte-specific signaling. More recently, a functional role for monocyte-derived mφs and T cells has been established in the transition from compensated hypertrophy to heart failure (HF)6, 7. In humans and mice, cardiac mφs can be primarily discriminated according to their expression of C-C chemokine receptor type 2 (CCR2) into resident CCR2− and recruited CCR2+ subsets derived from embryonic and hematopoietic origin, respectively8, 9. However, a subset of CCR2+ resident macrophages that promote monocyte recruitment after injury has been recently identified, highlighting the heterogeneity of cardiac mφs10. Cardiac resident CCR2− mφs mediate cardiac electrical conduction and metabolic stability under homeostatic conditions11, 12 and promote cardiac recovery in the early postnatal period through stimulation of cardiomyocyte proliferation and angiogenesis13. In contrast, recruited CCR2+ mφs become activated during myocardial injury and promote inflammation8, 10, 14. Following TAC, the number of cardiac macrophages increases due to CCR2+ monocyte recruitment and expansion of resident CCR2− mφs6. The notion that cardiac resident mφs facilitate reparative processes after injury, while recruited monocytes and monocyte-derived mφs generate inflammation and oxidative stress, is emerging13. However, the precise role of cardiac resident mφs in the early adaptation to cardiac pressure overload and the transition to HF is unclear.
Here, we used innovative tools such as cytometry by time-of-flight (CyTOF) and Cite-seq single-cell RNA sequencing to better define the functional heterogeneity of resident and recruited cardiac immune cells. We then depleted cardiac mφs and identified an important role for cardiac resident mφs in regulating fibrosis and angiogenesis during the early compensatory response to pressure overload. Depletion of cardiac resident mφs prior to cardiac pressure overload leads to accelerated decline in cardiac function and exaggerated fibrosis development.
METHODS
Data Availability.
A detailed description of all experimental procedures and statistical tests can be found in the Expanded Materials & Methods section in the Online Data Supplement. RNA sequencing datasets are available at Geo Datasets GSE179276.
RESULTS
Pressure overload-induced cardiac remodeling profoundly alters the cardiac immune cell composition.
To better understand the role of cardiac immune cells in the regulation of cardiac hypertrophy, we performed flow cytometry on cardiac non-cardiomyocytes isolated from sham or TAC-operated mice 1 week after surgery15, 16. Compared with sham controls, TAC mice showed a substantial increase in the frequency and number of CD45+ immune cells, one week following pressure overload surgery (Online Fig. IA). We observed an accumulation of mφs, B and T cells, as well as NK cells and dendritic cells in TAC-operated mice compared with sham controls (Online Fig. IB). Changes in immune cells after TAC were specific to the pressure overload and not caused to the surgical intervention in the mediastinum (Online Fig. IC–E). Given the large increase in mφ abundance following TAC, we next used CyTOF17 to characterize these cells. Similar to our flow cytometry results, we observed a substantial accumulation of immune cells 1 week after TAC surgery (Fig. 1A). However, by week four post-surgery, the number of cardiac immune cells receded to levels equivalent to those of sham controls (Fig. 1A). Using CyTOF, we identified all major immune cells based on well-established cell surface markers (Online Fig IF). ViSNE analysis of CyTOF data was used to visualize the changes in immune cell clusters in response to pressure overload (Fig. 1B). The majority of immune cells that accumulated in the heart after TAC were CD64+F4/80+ mφs, followed by monocytes, dendritic cells, polymorphonuclear leukocytes (PMNs) and B cells as shown by ViSNE and quantification using manual gating (Fig. 1B, Online Fig. IIA).
Figure 1. Cytometry by time-of-flight of cardiac immune cells shows an expansion of macrophages early after cardiac pressure overload.
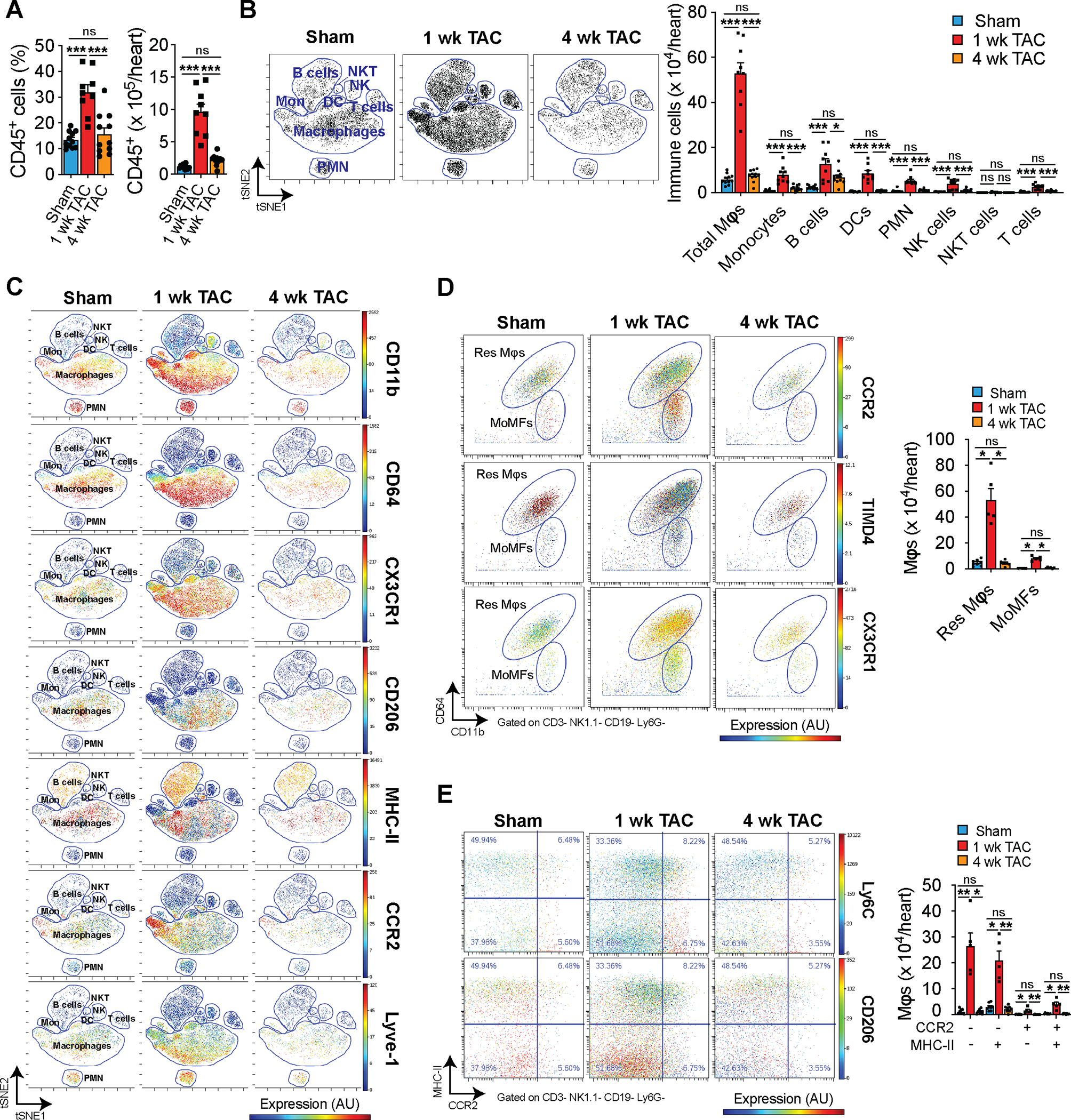
A. Relative abundance (left) and total number (right) of cardiac CD45+ immune cells in response to sham or TAC surgery at 1 or 4 weeks (n=11, 9, 11) determined by cytometry by time of flight (CyTOF). Statistical significance was evaluated by a one-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. B. Representative viSNE plot showing unsupervised clustering of cardiac immune cells (left) and quantification of their abundance (right) in sham and TAC conditions (n=11, 9, 11). Each dot represents a single cell in the viSNE plot. Statistical significance by cell type was evaluated by a one-way ANOVA (normally distributed data) or a Kruskal-Wallis test (non-normally distributed data). All pairwise comparisons were made. Tukey’s (normally distributed data) or Dunn’s (non-normally distributed data) tests were used to correct for multiple comparisons. C. Representative viSNE plots from CyTOF data showing colored expression in arbitrary units (AU) of CD11b, CD64, CX3CR1, CD206, MHC-II, CCR2, and Lyve-1 in cardiac immune cells in sham and TAC conditions (n=11, 9, 11). D. Representative CyTOF plots (left) showing cardiac resident (Res Mφs) and monocyte-derived (MoMFs) macrophages gated based on CD11b and CD64. The dot color represents the level of CCR2, TIMD4, and CX3CR1 expression in AU. Quantification (right) of Res Mφs and MoMFs in sham and TAC conditions (n=8, 5, 6). Statistical significance was evaluated by a Kruskal-Wallis test. All pairwise comparisons were made. Dunn’s tests were used to correct for multiple comparisons. E. Representative CyTOF plots (left) showing an alternative gating strategy of cardiac macrophages based on CCR2 and MHC-II expression. The dot color represents the level of Ly6C and CD206 expression in AU. Quantification (right) of CCR2 and MHC-II macrophage subsets in sham and TAC conditions (n=8, 5, 6). Statistical significance was evaluated by a Kruskal-Wallis test. All pairwise comparisons were made. Dunn’s tests were used to correct for multiple comparisons. Data are presented as mean±SEM or median±95% CI. Statistical significance is summarized as *(p<0.05), **(p<0.01), and ***(p<0.001).
We further analyzed the expression and distribution of mφ markers in cardiac immune cells and found that CD11b+ CD64+ mφ express varying levels of CX3CR1, CD206, MHC-II, CCR2, and Lyve-1 (Fig. 1C). We then gated resident (Res) and monocyte-derived (MoMF) mφs based on CD11b and CD6418 and verified their expression of Timd4 and CXC3CR1 or CCR2, respectively (Fig. 1D, left). The number of res mφs and MoMFs were increased 1 week after TAC. However, by 4 weeks after TAC, these were mostly restored to sham levels (Fig. 1D, right). We also used an alternative gating strategy based on the expression of CCR2 and MHCII and, consistent with our res and MoMF strategy, we found an expansion of all mφs at 1 week after TAC with a larger increase in CCR2− subsets (Fig. 1E). Similarly, mφ activation markers showed enhanced levels at 1 week with return to sham levels by 4 weeks after TAC (Online Fig. IIB). Furthermore, we observed a positive correlation between the number of mφs present in the heart and the level of cardiac hypertrophy following TAC (Online Fig. IIC). Overall, these findings suggest an important role for mφs early in the hypertrophic remodeling process.
Single-cell RNA sequencing identifies 12 different monocyte/macrophage clusters.
To explore the role of cardiac mφs during the compensatory hypertrophy phase in response to cardiac pressure overload, we performed multiplexed single-cell RNA sequencing of cardiac immune cells (CD45+) from sham and TAC-operated mice 1 week after surgery (Fig. 2A). After labeling the immune cells with barcoded antibodies, we combined the cells from 4 sham and 6 TAC hearts and performed droplet-based RNA sequencing. We initially detected 60,981 cells that were captured and sequenced at an average depth of 40,000 reads, based on Unique Molecular Identifier (UMI) codes. After quality control filtering and deconvolution of barcodes (Fig. 2B), we identified 33,566 unique cells that had a single barcode, including 5,854 cells from sham samples, and 27,712 cells from TAC samples (Fig. 2C). We performed unsupervised graph-based clustering of both sham and TAC cells using uniform manifold approximation and projection (UMAP), which revealed 25 clusters of CD45+ immune cells present in sham and TAC conditions (Fig. 2D). The identity of each cluster was determined based on the differential expression of established immune subset marker genes (Fig. 2E, Online Fig. IID, Online Data Set). In response to cardiac pressure overload, mφs and monocytes were the most abundant and diverse cell populations in the heart (12 clusters), followed by T cells (5 clusters), and B cells (3 clusters) (Fig. 2D–E). Importantly, based on the relative presence of sequenced cells from each mouse within each cluster, we detected an altered abundance of 8 different mφ clusters in response to TAC (Fig. 2F), indicating a critical role for cardiac mφs in the response to cardiac pressure overload.
Figure 2. Single-cell RNA sequencing of cardiac immune cells after pressure overload.

A. Design of multiplexed single-cell sequencing experiment using barcoded antibodies to identify mouse of origin of each cell. B. Overview of identified barcodes used to select singlets for further analysis. C. Pie chart indicating the number of single cells sequenced for each condition (upper) and per mouse (lower). D. UMAP projection of single cells clustered in 25 unique clusters with identification of immune cell identity. Sham- and TAC-derived cells are plotted separately to visualize abundance differences. E. Heatmap of top 5 identified genes that are specifically expressed within clusters using unsupervised clustering. F. Bar graph of relative abundance of each cluster of cells in sham vs TAC conditions (n=4, 6). Statistical significance between the sham and 1-week TAC groups by cluster was determined by two-tailed Mann-Whitney U tests. Data are presented as mean±SEM. Statistical significance is summarized as *(p<0.05). Single-cell data is shown as scaled, variance-stabilized unique molecular identifiers (UMI) counts.
Gene expression analysis distinguishes resident and monocyte-derived macrophages.
To probe the spectrum of gene expression profiles among mφs, we analyzed their gene expression within each cluster and identified both resident and recruited mφ populations based on established marker gene expression patterns. We focused on the expression of Ccr2 and Timd4 to identify CCR2+ recruited mφs and Timd4+CCR2− resident mφs (Fig. 3A). Res mφs specifically expressed Lyve1, Cd163 and Ccl24, while recruited mφs were characterized by the expression of antigen processing/presentation genes (H2-Aa, H2-Ab1, H2-Eb1, H2-DMa, H2-DMb1, and Cd74), and monocytes expressed Ly6c2 and Plac8 (Fig. 3A). To determine gene expression differences between Res and recruited mφs, we grouped mφ clusters into Res mφs and MoMF (Online Fig. IIIA) and analyzed their differentially expressed genes (Fig. 3B, Online Table I). Res mφs were characterized by high expression levels of Lyve1, Cd163, Timd4, as well as various CC Chemokine ligands, including Ccl2, 6, 7, 8, 9 and 24, Mrc1, and Lgals1. MoMFs were characterized by high expression levels of Ccr2, various MHC-II genes (H2-DMa, H2-DMb1, H2-Ab), Lgals3, Ccl5, Cxcl10 and 16, Ifit1, Il1b and fibrosis mediating genes Spp1, Thbs1 and Fn1 (Fig. 3B). Visualization of several of these genes shows the distinct expression patterns in mφ subsets (Fig. 3C). Ingenuity Pathway Analysis (IPA) of differentially regulated genes between Res mφs and MoMF identified pathways such as “Cardiac hypertrophy signaling” to be activated in Res mφs, which included genes encoding adrenergic receptors involved in cardiac remodeling19 (Online Fig. IIIB, Online Table II). Notably, IPA predicted the activation of upstream regulators IL10RA, IL4, STAT3 and IRF2 pathways, while IFNG, IRF3 and 7 regulated genes were predicted to be inhibited in Res mφs, compared with MoMFs (Fig. 3D). IPA analysis of individual clusters of Res mφs and MoMFs confirmed their heterogeneity by predicting the top activated upstream regulators IL10RA (clusters 1 and 11), MYD88 (cluster 3), and PTGER4 (cluster 4) in Res mφs, and NFAT5 (cluster 0), PPARG (cluster 5), IFNG (cluster 10), and ITGB2 (cluster 14) in MoMFs (Online Fig. IIIC). These analyses indicate distinct cell-intrinsic regulatory mechanisms in Res mφs and MoMFs that likely reflect their function in tissue homeostasis. Next, we analyzed the differential gene expression between mφs isolated from sham- and TAC-operated mice. In response to TAC, we detected a downregulation of Res mφ genes, such as Lyve1, Cd163, as well as the transcription factors Klf2 and Klf4, and the transcription factor AP-1 genes Jun, JunB, JunD, Fos, and FosB, and NfκB genes Nfkbiz and Nfkbia (Fig. 3E, Online Table III). Upregulated genes in response to TAC included Ccr2, Trem2, Cxcl10, and Lgals3 (Fig. 3E). IPA analysis of upstream regulators of differentially expressed genes predicted activation of the key mφ regulators NR1H2 and NR1H3, as well as STAT3 and STAT6 signaling (Fig. 3F). Predicted inhibited regulators included IRF3 and IRF7, as well as NFATC2 and MYD88 (Fig. 3F). These results highlight key changes that occur within Res mφs and MoMFs as the heart transitions from an adaptive hypertrophic state towards HF.
Figure 3. Analysis of cardiac macrophage gene expression patterns.
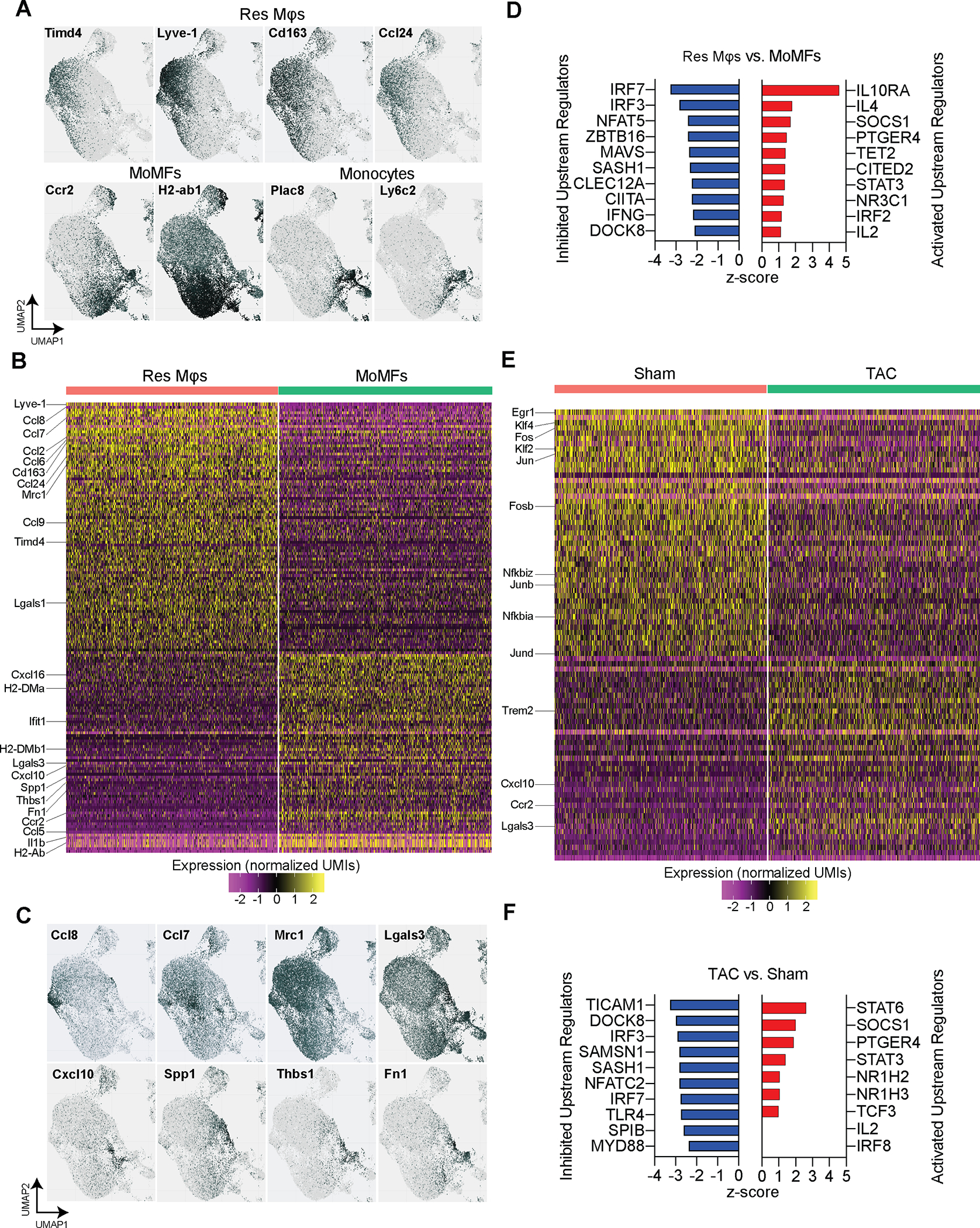
A. Identification of Res Mφs and MoMFs based on the expression of marker genes Lyve1, Timd4, Ccr2, and H2-Ab1. Additional genes that correspond with Res Mφs (Cd163 and Ccl24) or monocytes (Ly6c2 and Plac8) are shown. B. Heatmap of Differentially Expressed Genes between Res Mφs and MoMFs. Selected genes are indicated. C. Gene expression patterns of selected genes that were differentially expressed between Res Mφs and MoMFs show distinct expression patterns within mφ clusters. D. List of inhibited and activated upstream regulators identified with Ingenuity Pathway analysis from DEGs between Res Mφs and MoMFs. E. Heatmap of Differentially Expressed Genes between sham and TAC-derived cardiac mφs. Selected genes are indicated. F. List of inhibited and activated upstream regulators identified with Ingenuity Pathway analysis from DEGs between sham and TAC-derived mφs. Single-cell data is shown as scaled, variance-stabilized unique molecular identifiers (UMI) counts.
α-CD115-mediated macrophage depletion preferentially affects cardiac resident macrophages.
To determine if mφs play a causal role during the initial adaptive hypertrophic response to cardiac pressure overload, we depleted mφs with a monoclonal α-CD115 antibody that blocks CSF-1 binding to CD115-expressing cells, leading to the depletion of myeloid cells. We first examined the extent to which the α-CD115 antibody depletes cardiac mφs. We administered 2 doses of either α-CD115 or isotype control antibodies, followed by flow cytometry of cardiac non-cardiomyocytes starting at 1 week after the first injection at the timepoint when we would perform sham or TAC surgery (Fig. 4A). Indeed, α-CD115 antibody treatment resulted in a preferential reduction of Res mφs, while MoMFs were relatively spared (Fig. 4B). Although the TAC-induced increase in MoMFs was blunted 3 days after the surgery in α-CD115-treated mice, the number of MoMFs was comparable between α-CD115 and isotype controls 7 days after TAC (Fig. 4B). Peripheral blood monocytes fully recovered as early as 4 days following the second injection of α-CD115 antibody (Online Fig. IVA), suggesting that the resilience of MoMFs in the heart is due to a quick replenishment from circulating monocytes. More importantly, the depletion of Res mφ was sustained throughout the first week after TAC (Fig. 4B, Online Fig. IVB). Having validated our cardiac mφ depletion strategy, we performed TAC surgeries after α-CD115 or isotype control administration, followed by CyTOF analysis one week later. Quantification of the total number of CD45+ immune cells present in TAC hearts showed a three-fold reduction in α-CD115 treated hearts compared with isotype controls (Fig. 4C, left). To determine the potential effects of mφ depletion on other immune cell types, we quantified all major immune cell types in the heart in response to TAC and found a substantial reduction in cardiac mφs without an effect on other immune cell types (Fig. 4C, middle and right). While α-CD115 treatment depleted CCR2− res mφs, we detected a proportional increase in MHC-II+CCR2+ MoMFs after TAC in α-CD115 treated mice (Online Fig. IVC). Using flow cytometry, we noted a consistent reduction in MHC-II+ total mφs following α-CD115 treatment (Online Fig. IVD). We next used immunohistochemistry to assess the expression of the Res mφ marker CD163 (Fig. 3A) and found an increase in the abundance of CD163+ mφs in response to TAC in isotype-treated mice, with a near-complete depletion of CD163+ mφs in response to α-CD115 antibody treatment (Fig. 4D). Despite the successful depletion of Res mφs, we did not detect changes in cardiac hypertrophy, ejection fraction, and fractional shortening within the first week after cardiac pressure overload (Fig. 4E, Online Fig IVE–F). These results show that our antibody-mediated depletion strategy preferentially attenuates Res mφs while MoMFs have recovered at the time of the surgeries. Furthermore, these results show that depletion of Res mφs did not affect cardiac hypertrophy or function during the first week after TAC, although depletion might result in cardiac dysfunction at a later stage following pressure overload.
Figure 4. Cd115 antibody-mediated depletion of cardiac macrophages.
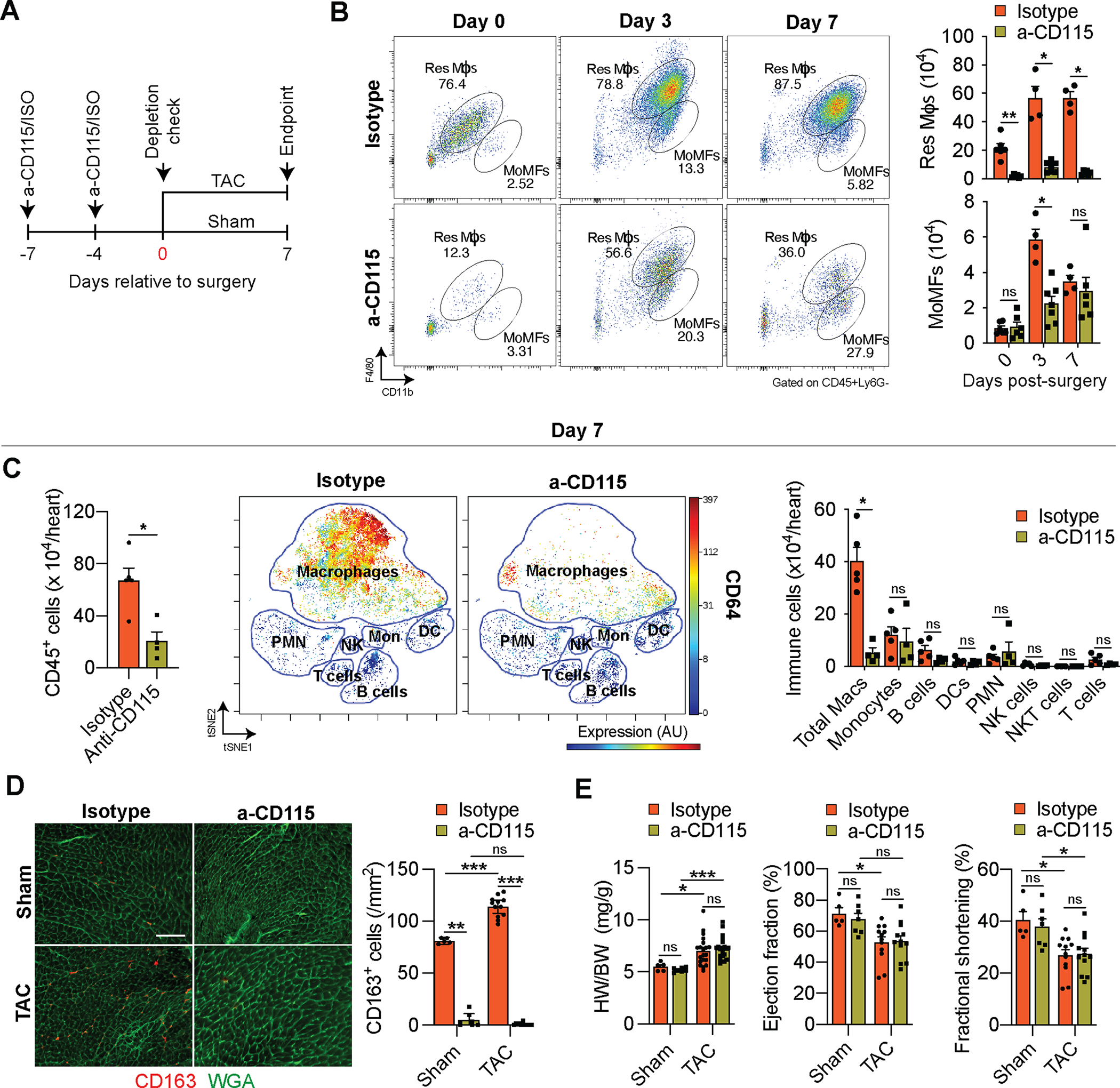
A. Schematic showing the timing of isotype or α-CD115 antibody administration relative to sham or TAC surgery. B. Representative flow cytometry plots (left) and quantification (right) of Res Mφs and MoMFs 0, 3, and 7 days after TAC surgery in isotype or α-CD115 antibody-treated mice (n=4, 6). Statistical significance between the isotype and α-CD115 antibody-treated groups by day was determined by two-tailed Mann-Whitney U tests. C. CyTOF-based quantification of cardiac immune cells (left), viSNE plot showing the abundance of different immune cells and colored expression of CD64 in arbitrary units (AU, middle), and quantification of immune cell abundance (right) after TAC surgery in isotype or α-CD115 antibody administration (n=5, 4). Statistical significance between the isotype and α-CD115 antibody-treated groups by cell type was determined by two-tailed Mann-Whitney U tests. D. Representative histological images (left, bar=100μm) of CD163 staining (red) with Wheat Germ Agglutinin (green) and quantification (right) of CD163+ cells (n=5, 7, 12, 12). Statistical significance was evaluated by a Kruskal-Wallis test. All pairwise comparisons were made. Dunn’s tests were used to correct for multiple comparisons. E. Cardiac hypertrophy [left, heart weight (HW) to body weight (BW) ratio], ejection fraction (middle), and fractional Shortening (right, n=5, 7, 12, 12). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. Data are presented as mean±SEM or median±95% CI. Statistical significance is summarized as *(p<0.05), **(p<0.01), and ***(p<0.001).
Resident macrophages inhibit fibrosis.
Although mφs appeared to be dispensable for the initial hypertrophy response to pressure overload, it is possible that they regulate initial adaptive processes such as the increase in microvascular density and the development of fibrosis20–22. To determine if cardiac mφs regulate angiogenesis and fibrosis during the early response to cardiac pressure overload, we first isolated mφs from sham or TAC operated hearts and measured their production of TNFα, IL6, and IL10 ex vivo. Using flow cytometry detection of intracellular proteins, we noticed an increase in the production of the pro-inflammatory cytokine IL6 and the anti-inflammatory cytokine IL10, suggesting an overall immune activation (Fig. 5A). To assess the consequences of the activation of mφ in response to TAC we isolated mRNA from cardiac mφs after sham and TAC surgeries and measured their expression of the pro-fibrotic Transforming Growth Factor β1 (Tgfβ1), which increased in response to TAC (Fig. 5B). Next, we determined whether depletion of Res mφs affected the development of fibrosis in response to TAC. Indeed, TAC promoted fibrosis, which was further aggravated in α-CD115 treated mice (Fig. 5C–D, Online Fig. VA–B). To identify mφ-derived factors whose production is stimulated by TAC and could promote fibrosis, we isolated cardiac mφs after sham or TAC surgery and measured their production of 10 analytes ex vivo using a bead-based immunoassay. Compared with sham, total cardiac mφs from TAC mice released increased amounts of most cytokines and growth factors including TGFβ1 and IL10, previously implicated in the development of cardiac fibrosis. Indeed, both TGFβ1 and IL10 showed a dose-dependent stimulation of myofibroblast differentiation, myofibroblast growth and collagen production, at concentrations produced by cardiac mφs in our ex vivo experiments (Fig. 5F). Together, these results show that cardiac mφs play a critical role in ameliorating fibrosis in response to pressure overload.
Figure 5. Cardiac macrophages regulate fibrosis.
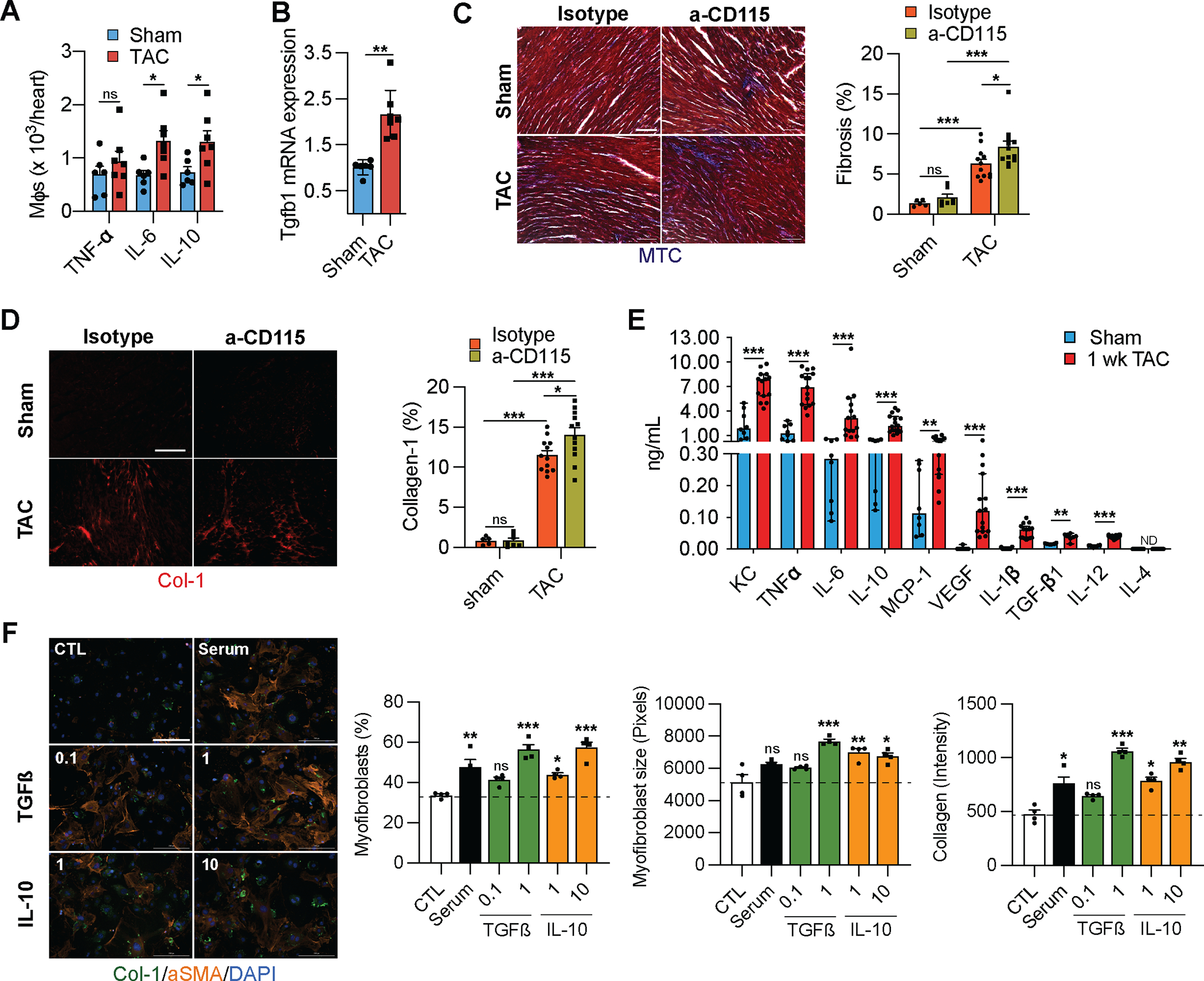
A. Quantification of cytokine-producing mφs isolated from sham or TAC operated mice. (n=6, 7). Statistical significance by cytokine was determined by two-tailed unpaired Student’s t test. B. Taqman qPCR for Tgfb1 on cDNA from freshly isolated mφs from sham or TAC operated mice. (n=6, 7). Statistical significance was evaluated by a Kruskal-Wallis test. C. Representative images (left) of Masson’s Trichrome-stained sections derived from sham or TAC operated mice after isotype or α-CD115 antibody administration. Bar graph (right) shows quantification of fibrosis (blue staining). (n=5, 7, 12, 12). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. D. Representative images (left) of collagen-1-stained sections derived from sham or TAC operated mice after isotype or α-CD115 antibody administration. Bar graph (right) shows the percent of collagen-1 staining (n=5, 7, 12, 12). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. E. Quantification of cytokines in the supernatants from mφs isolated from sham or TAC operated mice (n=8, 15) using a Legendplex™ assay following 16 hours of culture with 10 ng/mL of LPS. Statistical significance between the sham and TAC groups by cytokine was determined by two-tailed Mann-Whitney U tests. F. Representative images of fibroblasts stained for collagen-1 (green), αSMA (orange), and DAPI (blue) following treatment with 0.1 and 10 ng/mL of TGFß or IL-10 (n=4). Bar graphs show quantification of myofibroblast percentage, myofibroblast size, and collagen-1 intensity. Statistical significance was evaluated by a Kruskal-Wallis test. All comparisons were made against the control (CTL). Dunn’s tests were used to correct for multiple comparisons. for C, D and 200μm for F.
Resident macrophages stimulate angiogenesis.
To assess if cardiac mφs regulate angiogenesis, we first determined the expression of Vascular endothelial growth factor a (Vegfa) in isolated mφs from sham and TAC-operated hearts, and noted an increased expression in the mφs from TAC hearts (Fig. 6A). To determine if cardiac mφs stimulate angiogenesis, we measured the microvascular density (MVD) in isotype and α-CD115 treated hearts and found an increase in the MVD in response to TAC, which was blunted in α-CD115 treated hearts (Fig. 6B). Finally, to establish a direct role for mφs in regulating MVD, we isolated cardiac mφs and co-cultured them with HUVECs. Importantly, mφs stimulated tube formation with increases in mesh area, and number of nodes, junctions and meshes compared with the negative control (Fig. 6C). As we detected increased VEGF-A production by mφs from TAC-operated mice (Fig. 6A), we treated HUVECs with VEGF-A and confirmed a dose-dependent stimulation of HUVEC tube formation (Online Fig. VC). These results show that cardiac mφs promote vascular growth in pressure-overloaded hearts.
Figure 6. Cardiac macrophages regulate angiogenesis.
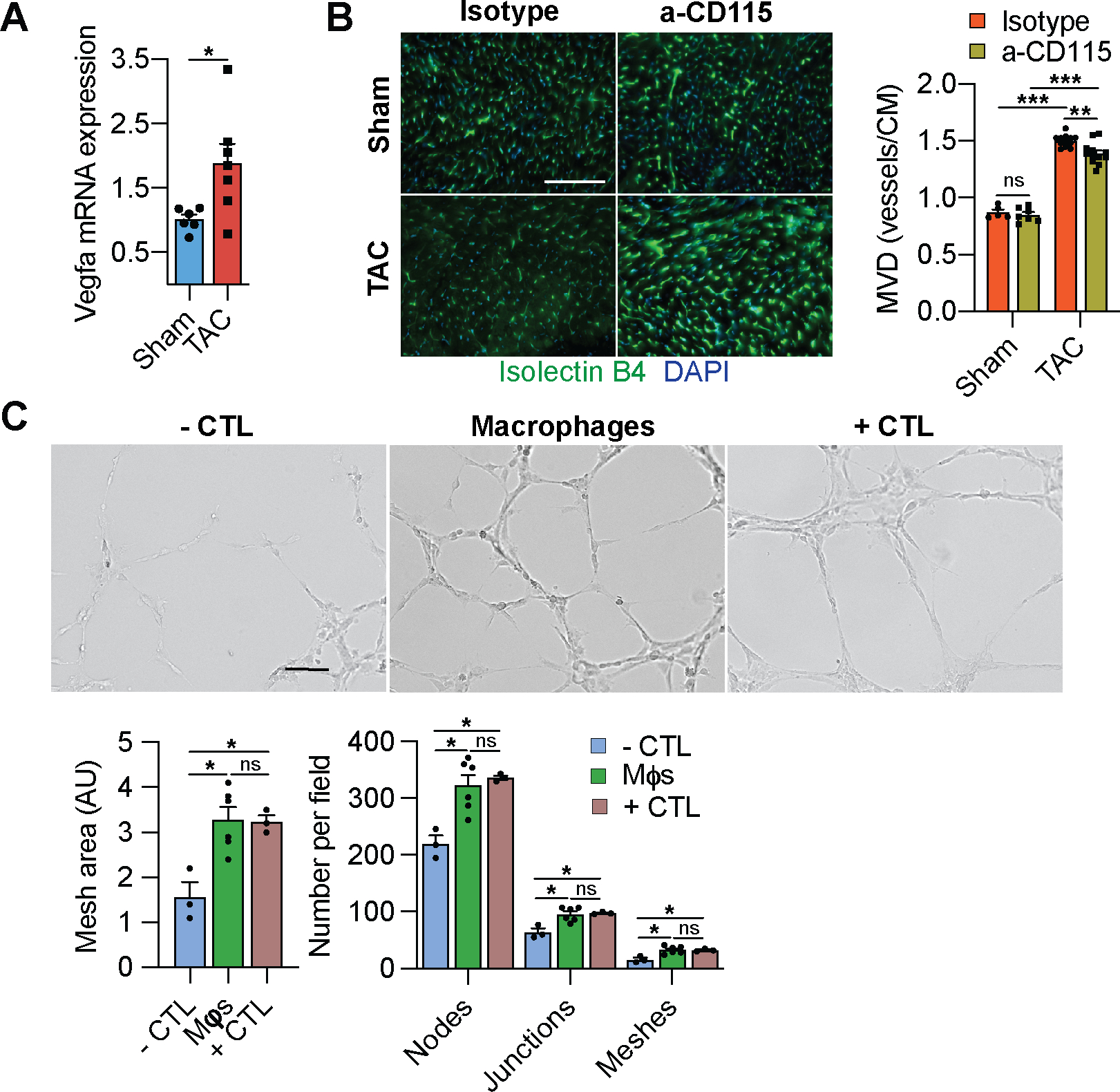
A. Taqman qPCR for Vegfa on cDNA from freshly isolated mφs from sham or TAC operated mice (n=6, 7). Statistical significance by cytokine was determined by two-tailed unpaired Student’s t test. B. Representative images (left) of isolectin B4-stained sections (green) derived from sham or TAC operated mice after isotype or α-CD115 antibody administration. Bar graph (right) shows quantification of microvascular density (vessels/cardiomyocyte) (n=5, 7, 12, 12). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. C. Representative images of tube formation assay using HUVECs co-cultured with negative control (CTL) or with cardiac mφs compared to positive control. Bar graphs show the quantification of various aspects of tube formation (mesh area, number of nodes, junctions, and meshes) (n=3, 6, 3). Statistical significance was evaluated by a Kruskal-Wallis test. All comparisons were made against the negative CTL. Dunn’s tests were used to correct for multiple comparisons. Data are presented as mean±SEM or median±95% CI. Statistical significance is summarized as *(p<0.05), **(p<0.01), and ***(p<0.001). Scale bar=100μm for C and 200μm for B.
Recruited monocyte-derived macrophages stimulate fibrosis.
Since we noticed a preferential depletion of Res mφs that aggravated fibrosis in α-CD115-treated TAC mice, while MoMFs were relatively spared, we hypothesized that recruited mφs might be partially responsible for stimulating cardiac fibrosis. To test this hypothesis, we administered α-CD115 or isotype control antibodies to CCR2 KO and C57Bl/6j mice prior to TAC (Fig. 7A). Seven days after TAC, Res mφs were depleted in C57Bl/6j and CCR2 KO mice while MoMFs were relatively spared in C57Bl/6j mice but barely detectable in CCR2 KO mice by flow cytometry (Fig. 7B) and immunohistochemistry (Online Fig. VIA). Similar to our previous results, we did not detect any effect of α-CD115-treatment on cardiac hypertrophy or function at 1-week post-TAC (Fig. 7C), although we observed increased cardiomyocyte size on histological sections (Online Fig VIE). Importantly, depletion of Res mφs alone aggravated fibrosis in C57Bl/6j in response to TAC, similar to our previous result. Furthermore, isotype-treated CCR2 KO mice showed the lowest fibrosis, consistent with a pro-fibrotic role of MoMFs. However, α-CD115-treated CCR2KO mice, that lack both Res mφs and MoMFs, were protected from the increased fibrosis (Fig. 7D–E and Online Fig. VIB–C). Surprisingly, CCR2KO mice did not show a blunted angiogenesis response to TAC when Res mφs were depleted (Online Fig. VID). These findings indicate that recruited MoMFs are strong promoters of cardiac fibrosis in a process that ultimately depends on the counteracting role of Res mφs.
Figure 7. Recruitment of monocytes promotes fibrosis after pressure overload.
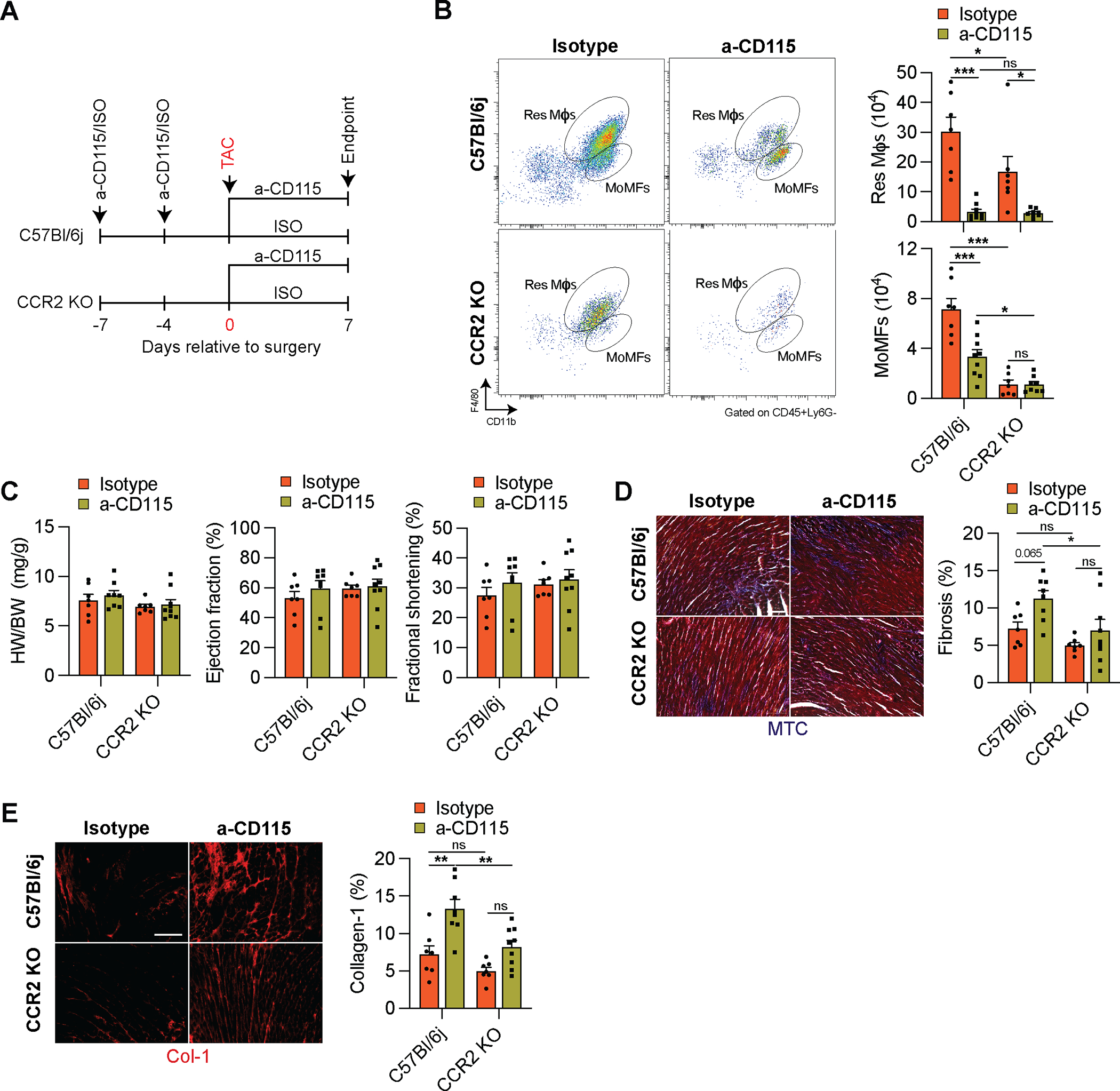
A. Experimental design of isotype or α-CD115 antibody administration to either C57Bl/6j or CCR2 KO mice relative to the time of sham or TAC surgery. B. Representative flow cytometry plots (left) and quantification (right) of Res Mφs and MoMFs in C57Bl/6j or CCR2 KO mice 1 week after isotype or α-CD115 antibody treatment (n=7, 9, 7, 8). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. C. Cardiac hypertrophy; heart weight (HW) to body weight (BW) ratio (left), ejection fraction (middle), and fractional shortening (right) of isotype or α-CD115-treated C57Bl/6j and CCR2 KO mice (n=7, 9, 7, 8). Statistical significance was evaluated by a two-way ANOVA. D. Representative images (left) and quantification (right) of Masson’s Trichrome-stained sections from isotype or α-CD115-treated C57Bl/6j and CCR2 KO mice (n=7, 9, 7, 8). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. E. Representative images (left) of collagen-1-stained sections derived from sham or TAC operated mice after isotype or α-CD115 antibody administration. Bar graph (right) shows the percent of collagen-1 staining (n=7, 9, 7, 8). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. Data are presented as mean±SEM. Statistical significance is summarized as *(p<0.05), **(p<0.01), and ***(p<0.001). Scale bar=100μm.
Depletion of resident macrophages leads to accelerated heart failure development.
As depletion of Res mφs decreased angiogenesis and aggravated fibrosis without detectable changes in cardiac function 1-week post-TAC, we assessed the long-term consequences of α-CD115-treatment. We administered α-CD115 or isotype control antibodies to C57Bl/6j mice prior to performing sham or TAC surgery and assessed cardiac function 6 weeks after surgeries (Online Fig. VIIA). We noticed that Res mφs started to partially recover as early as 2 weeks after performing TAC (Online Fig. VIIB–D). At 6 weeks post-TAC, there were no differences in the number of total immune cells, mφs, and other immune cell subsets between α-CD115 and isotype treated TAC mice (Fig. 8B). Similarly, there were no differences in the percent and number of Res mφs or MoMFs (Fig. 8C), suggesting that Res mφs have been replenished. Although the heart weight to body weight ratio was similar, we detected depressed cardiac function in α-CD115 antibody-treated TAC mice, relative to isotype controls (Fig. 8D). At the cardiomyocyte level, we detected enhanced cardiomyocyte cross-sectional areas (Fig. 8E). Importantly, the aggravated fibrosis observed in earlier timepoints was starker 6 weeks after TAC, particularly in the α-CD115 antibody-treated TAC mice (Fig. 8F–G). These results demonstrate an important role for Res mφs early in the remodeling process in preventing the deterioration of cardiac function and ameliorating the development of fibrosis following pressure overload.
Figure 8. Depletion of cardiac macrophages leads to decreased cardiac function.
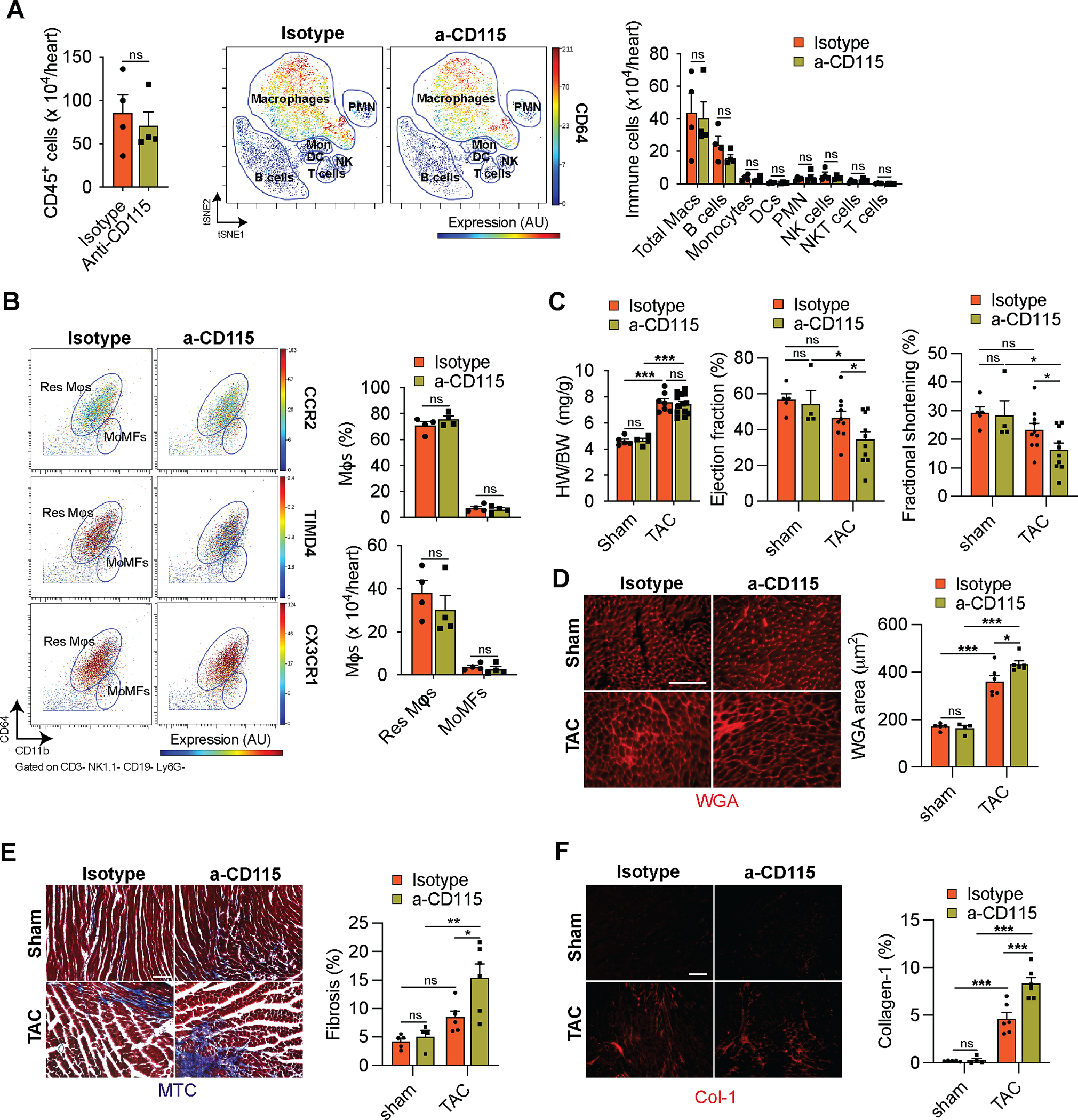
A. CyTOF-based quantification of cardiac immune cells (left), viSNE plot showing the abundance of different immune cells and colored expression of CD64 in arbitrary units (AU, middle), and quantification of immune cell abundance (right) 6 weeks after TAC surgery in isotype or α-CD115 antibody administration (n=4). Statistical significance between the isotype and α-CD115 antibody-treated groups by cell type was determined by two-tailed Mann-Whitney U tests. B. Representative CyTOF plots (left) showing Res Mφs and MoMFs gated based on CD11b and CD64. The dot color represents the level of CCR2, TIMD4, and CX3CR1 expression. Quantification (right) of Res Mφs and MoMFs 6 weeks after TAC in mice which received isotype or α-CD115 antibody administration (n=4). Statistical significance between the isotype and α-CD115 antibody-treated groups was determined by two-tailed Mann-Whitney U tests. C. Cardiac hypertrophy (left, heart weight (HW) to body weight (BW) ratio), ejection fraction (middle), and fractional shortening (right) in sham and TAC mice after isotype or α-CD115 antibody administration (n=5, 4, 6, 6). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. D. Representative images (left) and area quantification (right) of Wheat Germ Agglutinin-stained sections from isotype or α-CD115 antibody administered mice 6 weeks after sham and TAC (n=5, 4, 6, 6). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. E. Representative images (left) and quantification (right) of Masson’s Trichrome-stained sections from isotype or α-CD115 antibody administered mice 6 weeks after sham and TAC (n=5, 4, 6, 6). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. F. Representative images (left) and quantification (right) of collagen-1-stained sections from isotype or α-CD115 antibody administered mice 6 weeks after sham and TAC (n=5, 4, 6, 6). Statistical significance was evaluated by a two-way ANOVA. All pairwise comparisons were made. Tukey’s tests were used to correct for multiple comparisons. Statistical significance is summarized as *(p<0.05), **(p<0.01), and ***(p<0.001). Scale bar=100μm
DISCUSSION
Our findings, integrating CyTOF17 and multiplexed single-cell RNA sequencing23, underscore an important role for cardiac immune cells early in the response to cardiac pressure overload. Our results show a substantial increase in the abundance of cardiac mφs within 1 week after cardiac pressure overload. These findings are consistent with previous research showing that an expansion of CCR2− cardiac mφs occurs during the first week followed by a detrimental infiltration of CCR2+ monocytes in the later phase of pressure overload hypertrophy7. Our single-cell RNA sequencing experiments identified 12 distinct mφ and monocyte populations that were classified as Res mφs or MoMFs, based on previously published marker genes, including Timd4, Lyve1, Ccr2, and MHC-II genes7–10, 18. We identified dynamic changes in 8 of the 12 mφ clusters suggesting an important function for cardiac mφs early in the response to cardiac pressure overload. To assess their role in orchestrating a compensatory response to a hypertrophic stimulus, we used an antibody-based strategy to deplete Res mφs24. Depletion of Res mφs had no effect on cardiac hypertrophy or function at 1 week after cardiac pressure overload, suggesting that cardiac mφs are dispensable for a normal hypertrophy response. However, we noticed reduced MVD and enhanced fibrosis, indicating that Res mφs are critical for stimulating angiogenesis and inhibiting fibrosis. These early changes appeared to be maladaptive as we detected aggravated fibrosis and worsening of cardiac function at the 6-week timepoint. Our findings are consistent with previous work showing that Res CCR2− mφs promote angiogenesis in neonatal injury models13. In contrast, CCR2+ MoMFs have been shown to promote cardiac inflammation through IL-1ß release8 and neutrophil recruitment14 early after cardiac injury. Recent single-cell genomics studies have identified different populations of immune cells, including the recognition of CCR2+ pro-inflammatory mφs as key players in the later phase of pressure overload hypertrophy25, 26.
Cardiac mφs are continually replenished from different pools, and we assessed mφ populations in the heart to distinguish Res mφs from MoMFs8. When we assessed the role of recruited mφs using CCR2-deficient mice, we found that recruited mφs stimulate cardiac fibrosis after pressure overload. Recruited mφs have been implicated in the development of cardiac fibrosis in a CCR2-dependent process6, 10, 14. One caveat of using CCR2 expression to distinguish recruited from resident mφs is that a population of CCR2+ resident cardiac mφs from hematopoietic origins has been recently identified10. These CCR2+ resident mφs promote leukocyte recruitment and myocardial injury, but unlike CCR2+ recruited mφs, they are recruited to the healthy myocardium early in life10. Future work should clarify the population dynamics and functional heterogeneity of CCR2+ resident mφs following cardiac pressure overload. Nevertheless, our results suggest that while recruited mφs are pro-fibrotic, resident mφs are reparative as they are needed for tissue homeostasis and beneficial remodeling7. In a model of myocardial infarction (MI), selective depletion of CX3CR1+ mφs prior to MI impaired cardiac healing, reduced cardiac function, and increased mortality, suggesting that cardiac resident mφs support a regenerative response after injury18. Similarly, we found that depletion of resident mφs reduced the angiogenic response, with a concomitant lower MVD, and an enhanced development of fibrosis. Mechanistically, it is possible that the effects of CD115 blocking on fibrosis are mediated by depletion of IL-10-producing macrophages, which we found to increase in response to TAC. In humans and mice, macrophage-derived IL-10 activates fibroblasts, and promotes collagen deposition leading to diastolic dysfunction27, 28. Here, we show that IL-10 and TGFβ are produced by cardiac mφs in response to pressure overload and that they stimulate myofibroblast differentiation and collagen production.
Recently, several scRNA sequencing studies have assessed the response of cardiac cells to stress, including cardiac pressure overload25, 26, 29, 30. For example, scRNA sequencing of more than 17,000 cardiac immune cells from sham and TAC hearts at 1 and 4 weeks after TAC (one mouse each condition) revealed 1 cluster of reparative/resident, 2 clusters of proinflammatory, and 1 cluster of phagocytic mφs25. In another recent study, a comparison between all cardiac cells at 2 and 5 weeks post-TAC revealed that activation of mφ inflammatory function was the most notable change in this period when the heart loses cardiac function26. Similarly, we identified multiple types of immune cells residing within and recruited to the heart. The use of cells from 10 mice allowed us to assess the contribution of each mouse within each cluster and test the dynamic changes in clusters of cells in response to TAC. We identified gene expression patterns in specific clusters of mφs, such as the expression of Thbs1 and Fn1 in cluster 14, as well as Spp1 in cluster 5, suggesting that these cells could mediate cardiac fibrosis. Furthermore, Cxcl10 showed an expression pattern restricted to cluster 10, suggesting that this cluster of mφs may orchestrate the recruitment of monocytes and Th1 cells, which drive adverse cardiac remodeling31, 32. As infiltration of inflammatory monocytes occurs during the transition to cardiac decompensation following TAC7, our findings highlight specific subpopulations of cardiac-resident mφ that could mediate this event. Similar to previous studies18, 33, we identified resident mφs that resemble an alternatively-activated state and express Lyve1, CD163, and MRC1 (CD206).
Here, we used a blocking antibody against colony-stimulating factor 1 receptor (CSF1R or CD115) to block CSF1 from maintaining a pool of resident mφs34. Since mφs depend on the continuous input of CSF1R for their maintenance, blocking this receptor has been used in prior studies to deplete mφs in other organs24, 35. Previous work has shown that resident mφs are exclusively replenished by local proliferation, while CCR2+ MoMFs are maintained through both monocyte recruitment and proliferation9. Therefore, we took advantage of the rapid replenishment of MoMFs from circulatory monocytes following CD115 antibody treatment to preferentially deplete resident mφs. It is important to note that this strategy resulted in distinct functional outcomes, compared with liposomal clodronate depletion of mφs. One important difference between our approach to deplete mφs and liposomal clodronate, is the severe mortality that is observed after TAC in clodronate-treated mice7. As we did not observe excessive mortality in response to CD115 blocking, it is unclear whether the high mortality in response to clodronate treatment is entirely driven by mφ-mediated mechanisms or through other off-target effects. A limitation of macrophage depletion strategies, including our own, is the targeting of myeloid populations in other tissues such as the spleen or the lung that could influence a cardiac phenotype36. Future experiments should employ more precise and tissue-specific strategies to deplete cardiac mφs to discern specific functions of various populations of mφs.
In conclusion, we used a combination of CyTOF, scRNA sequencing and functional assays to identify the role of cardiac mφs early in the process of cardiac remodeling in response to pressure overload. We identified different mφ populations that show dynamic gene expression and abundance differences in the heart after pressure overload. These changes are important for regulating cardiac remodeling, as we identified an important role for resident mφs to stimulate angiogenesis, and limit cardiac fibrosis. Ultimately, a better understanding of the regulatory mechanisms of cardiac mφ stimulation and recruitment could lead to targeted therapies for HF.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Cardiac pressure overload initially leads to hypertrophy and angiogenesis, followed by the development of fibrosis and vascular rarefaction as the heart transitions to failure.
Immune cells such as macrophages and T-cells reside and accumulate in the heart where they regulate the transition to heart failure.
Cardiac macrophages can be classified broadly as resident or recruited macrophages, with recent fate-mapping and single-cell RNA sequencing experiments showing that cardiac macrophages are composed of multiple subsets of different origins and functions.
What New Information Does This Article Contribute?
Cardiac macrophages, particularly resident cells, accumulate in the heart within 1 week of cardiac pressure overload.
Multiplexed single-cell RNA sequencing of cardiac immune cells revealed 12 transcriptionally distinct clusters of macrophages and monocytes, where cardiac pressure overload altered the abundance of 8 of these macrophage clusters 1 week following injury.
Antibody-mediated depletion of resident macrophages prior to cardiac pressure overload decreased angiogenesis and aggravated cardiac fibrosis, suggesting that cardiac resident macrophages regulate these processes.
Macrophage-mediated stimulation of angiogenesis and inhibition of fibrosis early after cardiac pressure overload ameliorate the subsequent development of heart failure.
Cardiac pressure overload results in hypertrophy of the myocardium, which is considered adaptive at first. Histopathological changes like vascular rarefaction and development of fibrosis are important contributors to the development of heart failure. Previous research has shown important roles for recruited macrophages and T cells in mediating the transition to heart failure at later stages after cardiac pressure overload. The precise role of cardiac immune cells in regulating the early response of the heart to cardiac pressure overload is unclear. Our results show a substantial increase in resident macrophage abundance in the first week after cardiac pressure overload. Based on antibody-mediated depletion of resident macrophages we show their importance in stimulating angiogenesis and inhibiting cardiac fibrosis in the first week after cardiac pressure overload. Our results furthermore show that these early adaptations are important to postpone the development of heart failure. These findings suggest that resident macrophages regulate early adaptations, which are critical for the ultimate risk of development of heart failure.
Acknowledgments
SOURCES OF FUNDING
XR and JHvB are supported by grants from the NIH (DK122056, HL130072 and HL155993), the University of Minnesota Informatics Institute, and MnDRIVE. XR is the recipient of a Careers in Immunology Award by the American Association of Immunologists. JHvB is supported by an Individual Biomedical Research Award of The Hartwell Foundation. The Mass Cytometry Shared Resource at the University of Minnesota is supported by the Minnesota Partnership for Biotechnology and Medical Genomics.
Nonstandard Abbreviations and Acronyms:
- CyTOF
Cytometry by time-of-flight
- HF
Heart Failure
- mφ
Macrophage
- Res mφ
Cardiac resident macrophage
- MoMF
Monocyte-derived macrophage
- TAC
Transverse Aortic Constriction
Footnotes
DISCLOSURES
None
SUPPLEMENTAL MATERIALS
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.van Berlo JH, Maillet M and Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. The Journal of clinical investigation. 2013;123:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamura M and Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. [DOI] [PubMed] [Google Scholar]
- 3.Sano M, Minamino T, Toko H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–8. [DOI] [PubMed] [Google Scholar]
- 4.Schiattarella GG and Hill JA. Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation. 2015;131:1435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O and Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. The Journal of clinical investigation. 2006;116:1547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M and Prabhu SD. CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci. 2018;3:230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao X, Shen Y, Zhang R, et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc Natl Acad Sci U S A. 2018;115:E4661–E4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bajpai G, Schneider C, Wong N, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nature medicine. 2018;24:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bajpai G, Bredemeyer A, Li W, et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res. 2019;124:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulsmans M, Clauss S, Xiao L, et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell. 2017;169:510–522 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell. 2020. [DOI] [PubMed] [Google Scholar]
- 13.Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ and Mann DL. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:16029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Hsiao HM, Higashikubo R, et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fliegner D, Schubert C, Penkalla A, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1597–606. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, Zhu W, Bender I, et al. Pathologic Stimulus Determines Lineage Commitment of Cardiac C-kit. Circulation. 2017;136:2359–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bendall SC, Simonds EF, Qiu P, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dick SA, Macklin JA, Nejat S, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grisanti LA, Gumpert AM, Traynham CJ, et al. Leukocyte-Expressed beta2-Adrenergic Receptors Are Essential for Survival After Acute Myocardial Injury. Circulation. 2016;134:153–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okyere AD and Tilley DG. Leukocyte-Dependent Regulation of Cardiac Fibrosis. Front Physiol. 2020;11:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deniset JF, Belke D, Lee WY, et al. Gata6(+) Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity. 2019;51:131–140 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlson S, Helterline D, Asbe L, Dupras S, Minami E, Farris S and Stempien-Otero A. Cardiac macrophages adopt profibrotic/M2 phenotype in infarcted hearts: Role of urokinase plasminogen activator. Journal of molecular and cellular cardiology. 2017;108:42–49. [DOI] [PubMed] [Google Scholar]
- 23.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R and Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martini E, Kunderfranco P, Peano C, et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation. 2019;140:2089–2107. [DOI] [PubMed] [Google Scholar]
- 26.Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, Zhou B and Wang L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation. 2020;141:1704–1719. [DOI] [PubMed] [Google Scholar]
- 27.Verma SK, Garikipati VNS, Krishnamurthy P, et al. Interleukin-10 Inhibits Bone Marrow Fibroblast Progenitor Cell-Mediated Cardiac Fibrosis in Pressure-Overloaded Myocardium. Circulation. 2017;136:940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hulsmans M, Sager HB, Roh JD, et al. Cardiac macrophages promote diastolic dysfunction. The Journal of experimental medicine. 2018;215:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tucker NR, Chaffin M, Fleming SJ, et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gladka MM, Molenaar B, de Ruiter H, et al. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation. 2018;138:166–180. [DOI] [PubMed] [Google Scholar]
- 31.Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM and Alcaide P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. The Journal of experimental medicine. 2017;214:3311–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ngwenyama N, Salvador AM, Velazquez F, Nevers T, Levy A, Aronovitz M, Luster AD, Huggins GS and Alcaide P. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW and Rosenthal NA. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One. 2012;7:e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guilliams M, Thierry GR, Bonnardel J and Bajenoff M. Establishment and Maintenance of the Macrophage Niche. Immunity. 2020;52:434–451. [DOI] [PubMed] [Google Scholar]
- 35.MacDonald KP, Palmer JS, Cronau S, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–63. [DOI] [PubMed] [Google Scholar]
- 36.Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yellamilli A, Ren Y, McElmurry RT, et al. Abcg2-expressing side population cells contribute to cardiomyocyte renewal through fusion. Faseb j. 2020;34:5642–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafemeister C and Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F and Newell EW. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of all experimental procedures and statistical tests can be found in the Expanded Materials & Methods section in the Online Data Supplement. RNA sequencing datasets are available at Geo Datasets GSE179276.