

Abstract
Genomic analysis has recently identified multiple ESR1 gene translocations in estrogen receptor-alpha positive (ERα+) metastatic breast cancer (MBC) that encode chimeric proteins whereby the ESR1 ligand binding domain (LBD) is replaced by C-terminal sequences from many different gene partners. Here we functionally screened 15 ESR1 fusions and identified 10 that promoted estradiol-independent cell growth, motility, invasion, EMT and resistance to fulvestrant. RNA sequencing identified a gene expression pattern specific to functionally active ESR1 gene fusions that was subsequently reduced to a diagnostic 24-gene signature. This signature was further examined in 20 ERα+ patient-derived xenografts (PDXs) and in 55 ERα+ MBC samples. The 24-gene signature successfully identified cases harboring ESR1 gene fusions and also accurately diagnosed the presence of activating ESR1 LBD point mutations. Therefore, the 24-gene signature represents an efficient approach to screening samples for the presence of diverse somatic ESR1 mutations and translocations that drive endocrine treatment failure in MBC.
Introduction
The majority of breast cancers (~70%) are initially diagnosed as estrogen receptor-alpha positive (ERα+) and are dependent on 17-β estradiol (E2) for growth (1). Thus, endocrine therapies (ET) either induce estrogen deprivation, for example through aromatase inhibition (AI), or directly target the ERα ligand binding domain (LBD) with selective ER modulation (e.g., tamoxifen) or degradation (e.g., fulvestrant) (1). However acquired ET resistance is common, and is often associated with somatic mutations in the gene encoding ERα, ESR1. The most extensively studied examples are point mutations in the LBD that result in ERα proteins with ligand-independent activity. Common examples include Y537S and D538G (2,3). These mutations typically arise after patients have undergone extensive endocrine treatment and can be present in up to 40% of patients with ERα+ metastatic breast cancer (MBC) (4,5).
Emerging evidence indicates that chromosomal translocations involving the ESR1 gene can also drive ET-resistance through the formation of chimeric transcription factors with constitutive activity (TF) (6,7). The first described example was an ESR1-e6>YAP1 fusion detected by whole genome sequencing and RNA sequencing (RNA-Seq) in samples from a patient with rapid onset ET resistance (6). The fusion protein was encoded by an inter-chromosomal translocation event that brought ESR1 exons 1 to 6 (ESR1-e6) on chromosome (chr) 6q into the YAP1 locus on chr11q thereby replacing the entire LBD with transactivation domain (TAD) sequences from this Hippo pathway transcriptional coactivator (CoA). A PDX established from the patient’s tumor (WHIM18) also exhibited ET resistance. We subsequently identified another in-frame exon 6 fusion, ESR1-e6>PCDH11X in a male patient with ER+ MBC as a result of a chr6q>Xq translocation. This second example was a harbinger of complexity to come, since PCDH11X encodes a protocadherin without known transcriptional functions. Both fusions not only induced ET-resistant tumor growth but also increased lung metastasis in xenograft mice models (7). Another in-frame ESR1-e6 fusion, ESR1-e6>NOP2, was detected in a primary tumor but was found to be transcriptionally inactive (7). This example suggested that the mere presence of an ESR1-e6 fusion that generates a stable chimeric protein is insufficient evidence that an ET-resistance driver has been identified.
Multiple additional ESR1-e6 fusions have now been identified from ER+ MBC patients. In a study by Lee et al., three ESR1-e6 fusions (ESR1-e6>DAB2, ESR1-e6>GYG1, and ESR1-e6>SOX9) were shown to activate an estrogen response element (ERE)-driven luciferase reporter construct in transfected HEK293 cells (8). In the MET500 study (9), three in-frame fusions (ESR1-e6>ARNT2-e18, ESR1-e6>PCMT1, and ESR1-e6>ARID1B) were identified in samples from MBC patients and were provided to us for functional studies in a pre-publication personal communication. To further investigate the significance of in-frame ESR1-e6 fusion genes, each example was screened for ET-resistance induction in vitro, defined as E2-independent and fulvestrant-resistant growth and increased motility, in two ER+ breast cancer cell lines (T47D and MCF7). RNA-Seq was undertaken to understand the transcriptional reprogramming induced by active ESR1 fusions. We subsequently trained a 24-gene signature that was characteristic of the presence of an active ESR1 fusion as compared to an inactive fusion or wild-type (WT) ESR1. In the effort to validate this signature in ER+ PDXs and in clinical samples, activating LBD point mutations were discovered to also induce this gene expression signature. These data suggest that despite the remarkable diversity of mutations in the ESR1 gene, these somatic events converge on a common pathogenic transcriptional reprogramming mechanism to drive poor outcome and ET-resistance in MBC.
Materials and Methods
Cell Culture
Growth conditions for T47D (ATCC Cat# HTB-133, RRID:CVCL_0553) and MCF7 (ATCC Cat# HTB-22, RRID:CVCL_0031) cells are described in Lei et al. (7) and detailed in Supplementary information. ERα ligands (E2 and fulvestrant) were purchased from Sigma (E4389) and Selleckchem (S1191), respectively.
Subcloning of ESR1 mutants into a lentiviral expression vector
Lentiviral vectors expressing C-terminal HA-tagged YFP, truncated ESR1-e6, ESR1-WT, ESR1-Y537S, ESR1-D538G, ESR1-e6>YAP1, and ESR1-e6>PCDH11X were previously described (6,7). HA-tagged cDNAs encoding the new ESR1-e6 fusions in this study were constructed in a similar fashion as detailed in Supplementary information.
Generation of lentiviral stable ESR1 mutant expressing cell lines
Lentivirus were produced as described (6) by co-transfecting the above ESR1 cDNA lentiviral vectors with the packaging plasmids pMD2.G (RRID:Addgene_12259) and psPAX2 (RRID:Addgene_12260) into HEK293T (ATCC Cat# CRL-3216, RRID:CVCL_0063) cells using lipofectamine 2000 (Invitrogen, cat#11668–027). Transduced breast cancer cells were selected with 2 μg/mL puromycin (Sigma, cat# P8833) for 7 days. Expression of various ESR1 proteins was validated using immunoblotting.
Immunoblotting and immunoprecipitation
Cells were harvested and whole cell lysates were prepared in RIPA lysis buffer as described (7) or in MIB lysis buffer (10) supplemented with 1x protease inhibitors and 1x phosphatase inhibitors (Roche) by sonication for 2 minutes. To make ER+ PDX tumor lysates, frozen PDX tumors were cryopulverized with a Covaris CP02 Pulverizer and then protein was extracted in MIB lysis buffer with sonication. Protein concentration determination and SDS-PAGE (20 μg protein per lane) were performed as described (7). Immunoblotting of nitrocellulose membranes was performed as described (7). Primary and HRP-conjugated secondary antibodies employed are listed in the Supplementary information.
Immunoprecipitation was performed as described (7), using 2 mg of lysates from hormone-deprived T47D cells with or without E2 treatment (100 nM for 45 minutes). Lysates were incubated with 2 μg anti-HA tag antibody (Santa Cruz Biotechnology Cat# sc-7392, RRID:AB_627809) or mouse IgG (Cell Signaling Technology Cat# 61656, RRID:AB_2799613) control, followed by capture of antibody-antigen complexes with protein A magnetic beads (Bio-Rad, cat# 1614013) as described (7). Immunoprecipitated proteins, as well as 20 μg of whole cell lysates (1% inputs), were analyzed by immunoblotting.
Cell growth, motility and invasion assays
Cell growth assays of different ESR1 fusion protein expressing breast cancer cells that were first hormone-deprived and then subsequently treated either with 100 nM fulvestrant in the presence or absence of 10 nM E2 for 7–10 days were performed in 96-well plates using an alamarBlue assay as described (7). Cell growth reading values were normalized to that of control YFP cells, –E2.
Cell motility was detected using a scratch wound assay of hormone-deprived stable cells in a 96-well ImageLock plate (Essen BioScience) that were pre-treated for 2h with mitomycin C (50 ng/mL for T47D and 200 ng/mL for MCF7; Sigma, M4287) before wounding as described (7). Wound images were acquired every 6 hours for 72 hours by an IncuCyte camera (Essen Bioscience) in a cell culture incubator. Relative wound densities (RWD) were calculated as density in the wound area relative to that outside the wound area to account for confounding proliferation.
The cell invasion assay was performed and analyzed in a similar manner to the scratch wound assay except that cells were plated on Matrigel-coated plate. After the scratch was generated on cell monolayer, 50 μL Matrigel solution was added to the wells thus filling the scratch region and 100 μl of additional culture media containing mitomycin C.
RNA-Seq and analysis
Different ESR1 cDNA stably expressing T47D cell lines were cultured in CSS media for 5 days followed by treatment with or without 10 nM E2 for 2 days. RNA was isolated using RNeasy Mini Kit (QIAGEN, cat#74106) and treated with DNase (QIAGEN, cat#79254) to remove genomic DNA. The Genomic and RNA Profiling (GARP) Core at BCM confirmed concentration (using a NanoDrop spectrophotometer) and integrity (using an Agilent Bioanalyzer). The GARP core then made mRNA libraries and performed sequencing on an Illumina NovaSeq 6000 sequencing instrument as described in detail in Supplementary information. For RNA-Seq on isolated ER+ PDX tumors, frozen PDX tumors were cryopulverized as above and total RNA was isolated using the RNeasy kit. RNA-Seq was performed at the Human Genome Sequencing Center at BCM as described in detail in Supplementary information.
For RNA-seq analysis, paired-end 150 bp reads were aligned to the hg19 (GRCh37) reference genome using RSEM v1.2.31 (11) (RSEM, RRID:SCR_013027) and Bowtie 2 (12). Transcripts per million values calculated by RSEM were log2 transformed and subjected to heatmap generation using Morpheus (https://software.broadinstitute.org/morpheus) (Morpheus, RRID:SCR_014975). Unsupervised hierarchical clustering and identification of differentially expressed genes in active ESR1 fusion protein expressing cells to cells expressing inactive fusions and controls are described in Supplementary information.
Whole exome sequencing (WES) and analysis
DNA was isolated from the ER+ PDX tumors using a QIAamp DNA Mini Kit (QIAGEN, cat#51304). WES data was generated by the Human Genome Sequencing Center at BCM using the Illumina platform as described in detail in Supplementary information. Tools used for somatic ESR1 LBD gene variant calling were Strelka2, Mutect2, and CARNAC (v 0.2.5b9) as described in Supplementary information. ESR1-e6>YAP1 fusion was detected in WHIM18 previously (6).
Reverse transcription - quantitative PCR (RT-qPCR)
RNA was isolated from hormone-deprived stable T47D cells as above with concentration determined using a NanoDrop spectrophotometer. One step RT-qPCR was conducted using 50 ng RNA incubated with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, cat#1725274), iScript reverse transcriptase (Bio-Rad, cat#170–8891) and 0.5 μM primers (Sigma) as described (7). All samples were run in triplicate on a CFX96 thermal cycler (Bio-Rad).
Immunofluorescence
Immunofluorescence was performed of different HA-tagged ESR1 fusion proteins expressed in hormone-deprived T47D cells as described (7). These proteins were detected using an anti-HA antibody (Cell Signaling Technology Cat# 2367, RRID:AB_10691311, 1:50) and goat anti-mouse IgG secondary antibody (Alexa Fluor 568, Molecular Probes Cat# A-11004, RRID:AB_2534072, 1:1000) as described in Supplementary information. Nuclei were detected by DAPI staining as described (7).
PDX models
The PDX models were previously described (6,13,14). All animal procedures were approved by the Institutional Animal Care and Use Committee at BCM (protocol# AN-6934). Two-three mm pieces from PDX tumors were engrafted into cleared mammary fat pads of 3 to 4-week-old ovariectomized SCID/beige mice (Charles River). Mice were randomized to receive sterile drinking water with or without 8 μg/ml E2 supplementation (n=7–16 per PDX line per arm). Tumor volumes were measured by caliper every 3–4 days, and were calculated by V = 4/3 × π × (width/2)2 × (length/2). Mice were sacrificed when tumors reached 1.5 cm3 or at the study end point. Tumors were harvested and frozen in liquid nitrogen for storage. Additional information on BCM and HCI PDX models is available at pdxportal.research.bcm.edu/.
Gene signature and ROC curve analysis
The signature performance was calculated as follows: Accuracy= (TP+TN)/(TP+TN+FP+FN), Sensitivity=TP/(TP+FN), Specificity=TN/(TN+FP), in which TP, true positive; TN, true negative; FP, false positive; FN, false negative. ROC curve analysis was performed using “pROC” package in R (15).
ERE DNA pulldown assays
These assays were modified from the established protocol of HeLa cell nuclear extract (NE) supplemented with recombinant estrogen receptors (16,17). Briefly, nuclear extracts were made from T47D cell lines expressing YFP or different ESR1 fusion proteins (15–25 15 cm dishes employed) exactly as published (18). Pulldown assays employed 1 mg of T47D cell NE to resuspend 60 μl Dynabeads M-280 Streptavidin that was pre-bound to 3 μg biotinylated 4xERE-E4 921 bp DNA. Incubation occurred at 4°C with gentle rotation for roughly 2 hours, followed by pelleting beads with a magnetic rack and quick washes as described (16,17). Final beads were resuspended in 30 μl 2xSDS-sample buffer, boiled, and 30% of the final supernatants were loaded onto 4–15% gradient SDS-PAGE gels. After transfer to nitrocellulose, immunoblots were probed with N-terminal ERα (Millipore Cat# 04–820, RRID:AB_1587018) or DNA-PKcs (Santa Cruz Biotechnology Cat# sc-5282, RRID:AB_2172848) antibodies with ECL-based signal detection on a Bio-Rad Imaging System.
Statistical tests and analyses of publically-available data
Unpaired two-tailed Student’s t-test and ANOVA were performed with GraphPad Prism 9 (GraphPad Prism, RRID:SCR_002798), as indicated in the figure legends. P-values less than 0.05 were considered statistically significant. The protein domains with functional information in Figures 1B and 5A were extracted from UniProt Knowledgebase (19). RNA-Seq data derived from ER+ MBC along with ESR1 mutation status (n=55) were downloaded from the MET500 web portal (https://met500.path.med.umich.edu/). Cases with an ESR1 mRNA expression >1 FPKM were considered as ER+, using a previously described criteria (20).
Figure 1. In-frame ESR1-e6 fusions identified in ER+ MBC patients.
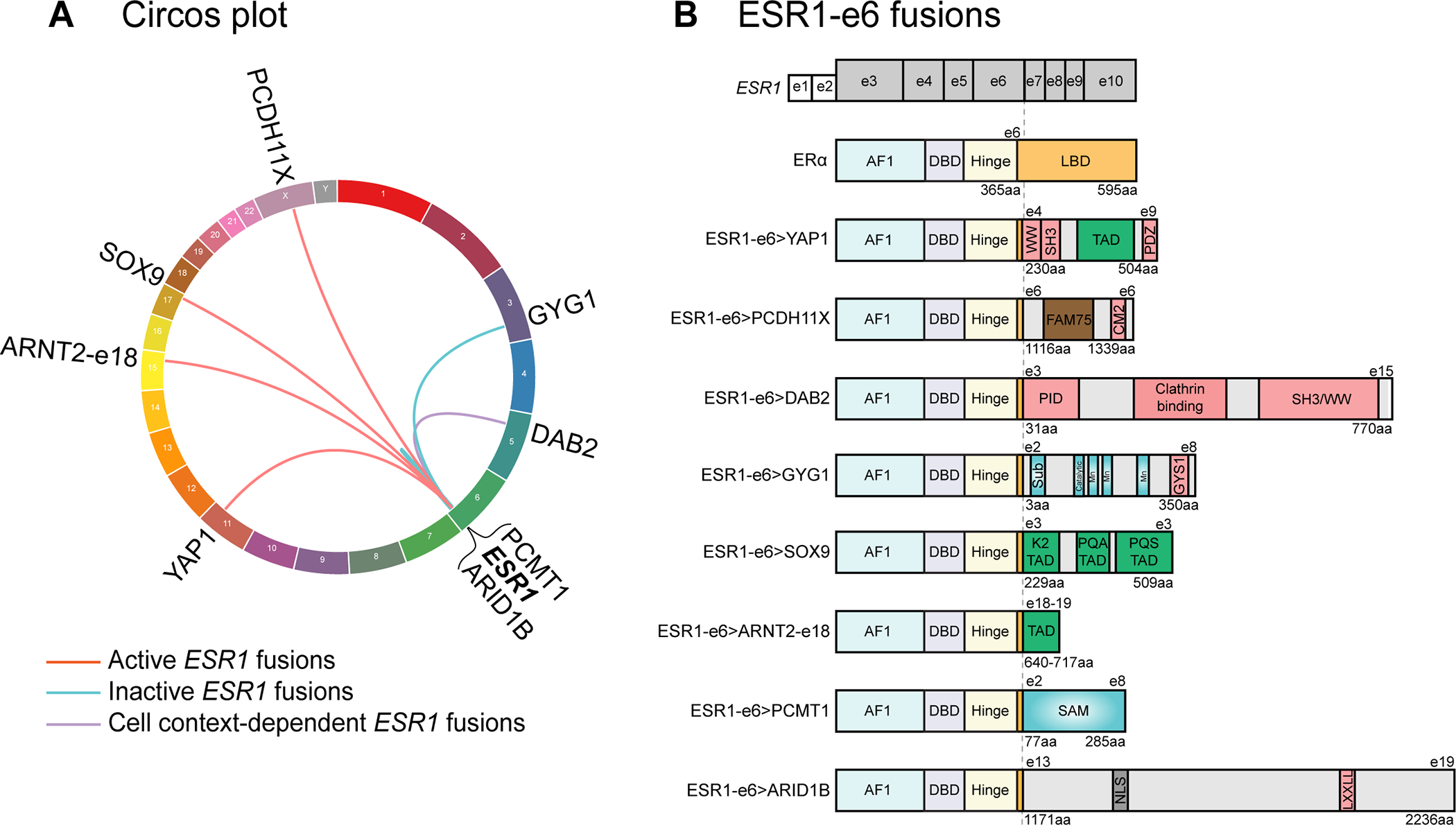
(A) Circos plot depicting ESR1 fusion events identified from ER+ MBC patients. The ESR1 gene is connected to its 3’ partner genes with lines. (B) In-frame ESR1 fusions in ER+ MBC possess a common structure whereby the first 6 exons (two untranslated exons and four coding exons in grey, exons 3–6) of ESR1 fuse in-frame to C-terminal sequences from partner genes. Key for domains in the WT ERα protein: AF1: activation function 1 domain; DBD: DNA-binding domain; Hinge: domain connecting DBD and LBD; and LBD: ligand-binding domain. Pink boxes in partner proteins mediate protein-protein interactions, including WW binding motifs, SH3 binding motifs, a PDZ domain, a conserved motif 2 (CM2), a phospho-tyrosine interaction domain (PID), an interaction with Glycogen Synthase 1 region (GYS1), and a LXXLL motif. Green boxes represent known transcriptional activation domains (TADs). The brown box represents the FAM75 domain of unknown function. Blue domains have enzymatic activities, including substrate binding site (Sub), catalytic site, three manganese binding sites (Mn) and an S-adenosylmethionine-dependent methyltransferase domain (SAM). The grey box labeled NLS represents a nuclear localization signal.
Figure 5. The MOTERA signature predicts activity of additional ESR1 fusions identified in ER+ MBC patients.
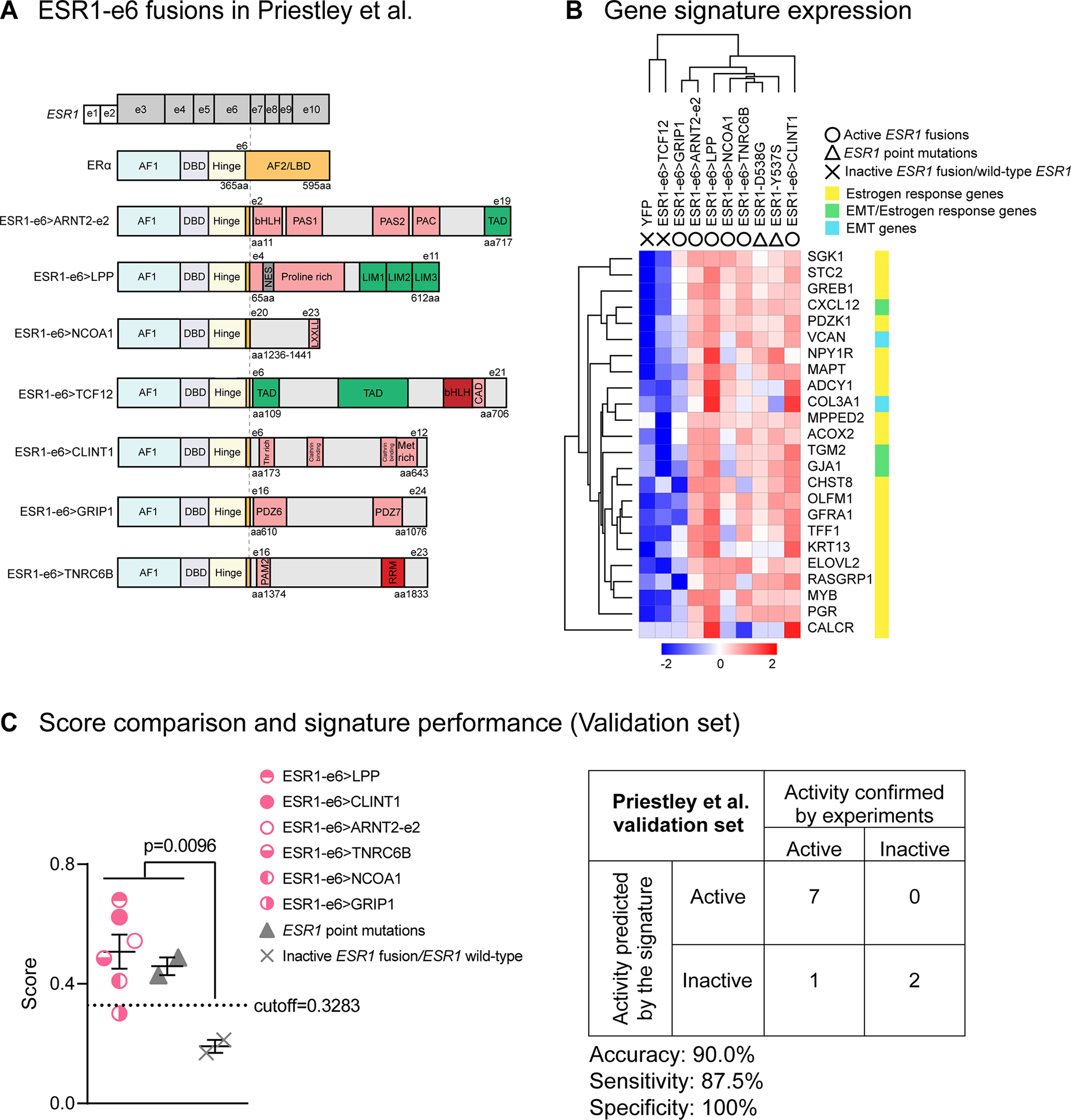
(A) Seven additional ESR1-e6 fusions identified in Priestley et al.(27) are illustrated. These in-frame fusions possess a common structure as shown in Figure 1B. Pink boxes represent protein–protein interactions, including the Per-Arnt-Sim (PAS) domain, PAC motif, LXXLL motif, Class A specific domain (CAD), Threonine-rich domain (Thr rich), Methionine-rich domain (Met rich), PDZ domain and PABPC1-interacting motif-2 (PAM2). Green boxes either represent transcriptional activation domains (TADs) or LIM zinc-binding (LIM) domains that provide coactivator function for LPP. The grey box represents a nuclear export signal (NES) in LPP. Red boxes represent the bHLH DNA binding domain and the RNA Recognition Motif (RRM). (B) Heatmap showing the expression of the 24-gene signature in T47D cells expressing additional ESR1 fusions and LBD point mutations (Y537S and D538G). Scale bar indicates row Z scores. (C) Left panel: Scatter plot showing signature scores of ESR1 mutations (including fusions and LBD point mutations) and YFP control cells expressing endogenous ERα. Two-tailed t test was used to compare scores. The ESR1-GRIP1 fusion was the only active fusion that did not reach score significance. Right panel: Confusion matrix to measure the performance of the signature to predict the activities of ESR1 fusions. Accuracy is the proportion of correctly predicted events in all cases. Sensitivity is the ability of the signature to predict an active fusion event to be active. Specificity is the ability of the signature to predict an inactive fusion event to be inactive.
Data Availability
RNA-Seq data from T47D cells and WES and RNA-Seq data from ER+ PDX tumors in this study are being submitted to Gene Expression Omnibus (GEO). Until an accession number is provided, these raw data are available upon request from the corresponding author.
A full description of all methods and reagents can be found in Supplementary information.
Results
A subset of in-frame ESR1-e6 fusions identified in ER+ MBC patients drive ET-resistant growth and promote hormone-independent motility and invasion of ER+ breast cancer cells.
We initially studied six newly identified in-frame ESR1 fusions detected in samples from MBC patients and compared them to the ESR1-e6>YAP1 and ESR1-e6>PCDH11X examples we described previously (7). Some fusion examples arose from inter-chromosomal translocations, such as ESR1-e6>DAB2, ESR1-e6>GYG1, ESR1-e6>SOX9 (Lee laboratory (8)) and ESR1-e6>ARNT2-e18 (Robinson, D. personal communication). Two other fusions were formed by rearrangements within chromosome 6, ESR1-e6>PCMT1 and ESR1-e6>ARID1B (Robinson, D. personal communication) (Figure 1A). All six examples followed a structure established by the original ESR1-e6>YAP1 fusion whereby the first six exons of ESR1 were fused in-frame to C-terminal partner genes, completely replacing the ERα LBD with an alternative C-terminus. We noted two classes functionally, 1) transcription factor (TF) and transcription coactivator (CoA) fusions or 2) fusions with genes without previously established (direct) functions in gene transcription (Figure 1B).
To characterize each chimeric ESR1 fusion protein, HA-tagged cDNA constructs were expressed in two ER+ breast cancer cell lines (T47D and MCF7) by lentiviral transduction. Stable cell lines expressing yellow fluorescent protein (YFP) were generated as negative controls. Truncated ESR1 (ESR1-e6 protein) and wild-type ESR1 (ESR1-WT protein) were also stably expressed to provide over-expression controls (Figure 2A, Supplementary figure 1A). When cells were treated with 100 nM fulvestrant, a selective ERα degrader that inhibits endogenous ERα (21), the level of ESR1 fusion protein was predictably unaffected. In comparison, the WT ERα protein was reliably degraded, providing an endogenous control for fulvestrant activity (Figure 2A, Supplementary figure 1A). To investigate whether ESR1 fusion proteins drove ET resistance, cell lines expressing ESR1 fusion cDNAs were hormone-deprived for 7 days in charcoal stripped serum-containing phenol red-free, RPMI media (CSS media) and then treated for 7–10 days with or without 10 nM E2 and with or without 100 nM fulvestrant. Cell growth was measured using an alamarBlue assay. Similar to ESR1-e6>YAP1 and ESR1-e6>PCDH11X (7), ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18 conferred E2-independent growth of T47D cells compared to the YFP controls (–E2, +DMSO) (Figure 2B, all four conditions are shown in Supplementary figure 1B) in a manner that was uniformly resistant to fulvestrant (Figure 2B). Although the four other ESR1-e6 fusions studied (ESR1-e6>DAB2, ESR1-e6>GYG1, ESR1-e6>PCMT1, and ESR1-e6>ARID1B) produced stable proteins, they did not promote ET-resistant growth of T47D cells with inactivity resembling the controls (truncated ESR1-e6 protein alone, ESR1-WT and YFP). The GYG1 example is an important exception, since this is an in-frame, inter-chromosomal translocation that might have been expected to be active. While cell growth was induced by E2 treatment regardless of the presence of an ESR1 fusion protein, only ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18 drove significantly higher growth than YFP control cells in presence of fulvestrant (+E2, +Fulvestrant) (Supplementary figure 1B). The elevated E2-independent, fulvestrant-resistant growth phenotypes were further validated in MCF7 cells (Supplementary figure 1C). Interestingly, ESR1-e6>DAB2 demonstrated E2-independent, fulvestrant-resistant growth in MCF7 cells, but not in T47D cells, suggesting the activity of this fusion was cell line selective.
Figure 2. ESR1 fusion proteins drive ET-resistant growth and promote hormone-independent motility of ER+ breast cancer cells.
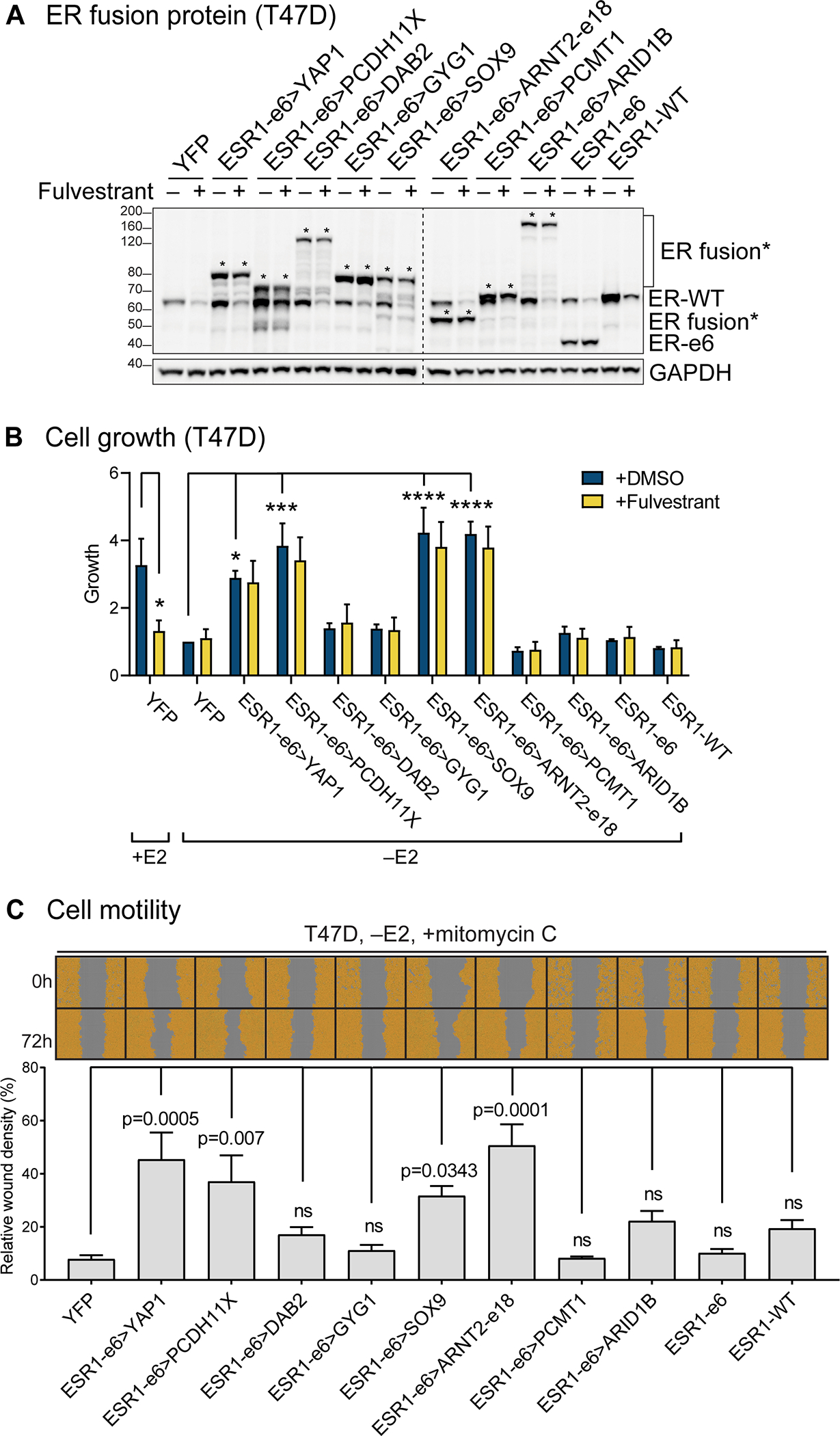
(A) Immunoblotting of ERα and ESR1 fusion proteins with an N-terminal ERα antibody in lysates made from hormone-deprived stable T47D cells. Asterisks indicate ER fusion proteins. GAPDH serves as a loading control. The dashed line indicates two separate blots that were conducted at the same time. The representative image is from three independent experiments. (B) Cell growth was assayed in hormone-deprived stable cells (mean ± SEM, n=3). One-way ANOVA followed by Dunnett’s multiple comparisons test was used to compare data of hormone-deprived ESR1 fusion expressing cells to YFP control cells in the vehicle (+DMSO) group. Two-way ANOVA followed by Bonferroni’s test was used for multiple comparisons for each stable cell line after 100 nM fulvestrant treatment in the presence or absence of 10 nM estradiol (E2). *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. See Supplementary figure 1B for the complete data. (C) Cell motility was detected using scratch wound assays in hormone-deprived stable T47D cells, treated with mitomycin C to block proliferation (mean ± SEM, n=3). Cells are pseudo-colored orange to aid visualization. One-way ANOVA followed by Dunnett’s multiple comparisons test was used to compare each stable T47D cell line to YFP control cells (ns: not significant).
To determine whether each fusion protein promoted cell motility, as an initial measure of metastasis-driving potential, stable T47D or MCF7 cells were hormone-deprived and pre-treated with mitomycin-C to inhibit cellular proliferation. Cell monolayers were scratched and wound images were monitored for 72 hours. Relative wound densities (RWD) were measured as density in the wound area relative to that outside the wound area. All four growth-promoting ESR1 fusion proteins, ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18, induced higher cell migration than controls in a hormone-independent manner (–E2) (Figure 2C, Supplementary figure 1D). Consistent with the proliferation data, ESR1-e6>DAB2 also promoted cell motility in MCF7, but not in T47D cells. Importantly, the expression levels of the functionally active ESR1 fusion proteins were similar to the inactive examples (Figure 2A, Supplementary figure 1A), suggesting that the inactivity of individual ESR1 fusion proteins was not due to differential expression or stability. ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18 also promoted more invasion through matrigel than either the controls (YFP, ESR1-e6, and ESR1-WT) or the inactive fusions (Supplementary figure 2).
Active ESR1 fusion proteins upregulate expression of estrogen response genes and EMT genes.
To define the transcriptional profile driven by active ESR1 fusion proteins, RNA-Seq was performed on T47D cells expressing these ESR1 fusion cDNAs as well as control (YFP, ESR1-e6, and ESR1-WT) cells in the presence and absence of E2. Hierarchical clustering showed that T47D cells expressing ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18 fusions clustered distinctly from other ESR1-e6 fusions and control cells under E2-deprived conditions (–E2) (Figure 3A). All four active fusions demonstrated an expression pattern similar to control cells treated with E2, consistent with potent hormone-independent transcriptional activation of “estrogen response” genes (Figure 3B). Interestingly, active ESR1 fusion proteins also upregulated a cluster of genes that were not observed in the control cells stimulated by E2 (Figure 3C). Over-representation analysis revealed a significant enrichment of “estrogen response” pathways and an epithelial-to-mesenchymal transition (EMT) signature specific to active ESR1 fusion proteins and thus consistent with data presented above on invasion and motility (Figure 3B and C). The expression of three canonical estrogen response genes in stably transfected T47D cells were validated using reverse transcription-quantitative PCR (RT-qPCR). ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18 significantly induced the expression of GREB1, TFF1, and PGR mRNA, all three known as direct ERα targets (22) (Figure 3D), in a hormone-independent, fulvestrant-resistant manner compared to YFP controls. These transcriptionally active ESR1 fusion proteins also upregulated two EMT-related genes, SNAI1 (Snail), encoding a master TF that induces EMT (23) by transcriptional repression of epithelial genes such as E-cadherin (24), and VCAN (versican) (Figure 3E). The elevated expression of Snail protein and a corresponding decrease of E-cadherin (E-cad) were confirmed by immunoblotting (Figure 3F). As expected, the expression of these genes were unaffected by fulvestrant treatment. The other ESR1-e6 fusion examples, ESR1-e6>DAB2, ESR1-e6>GYG1, ESR1-e6>PCMT1, and ESR1-e6>ARID1B did not induce E2-independent activation of ERα target genes and EMT-related genes in T47D cells. The induction of Snail protein was also reproduced in MCF7 cells, however ESR1-e6>PCDH11X displayed a minor upregulation compared to other transcriptionally active fusions (Supplementary figure 1A). Consistent with the observed MCF7 cell line-selective increase in cell growth and migration, ESR1-e6>DAB2 also upregulated Snail expression compared to YFP control and the inactive fusions. Consistent with above T47D cell line data, E-cadherin protein was reduced in MCF7 cells expressing active ESR1 fusion proteins (Supplementary figure 1A).
Figure 3. Active ESR1 fusion proteins upregulate expression of estrogen response and EMT genes.
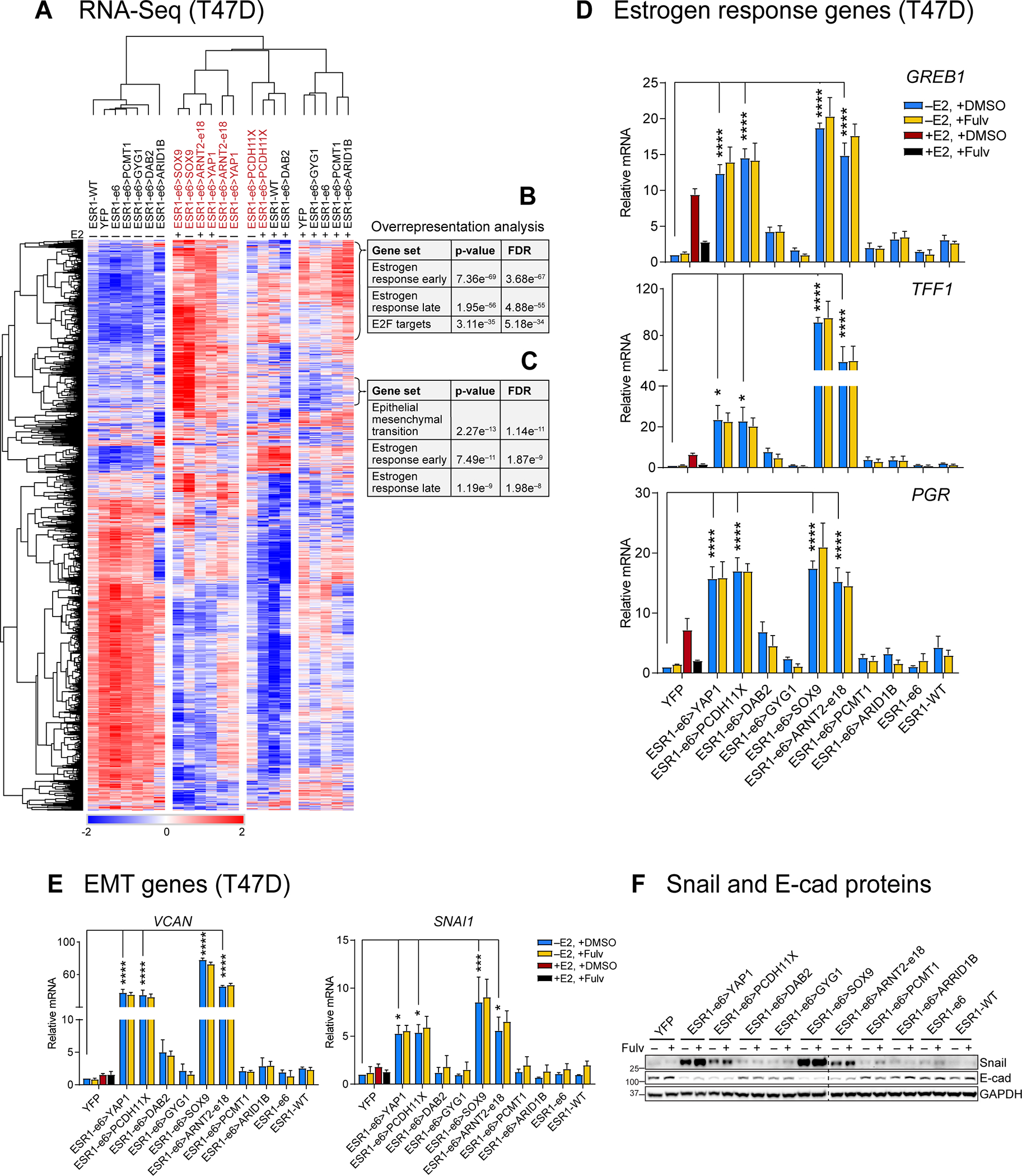
(A) Heatmap showing the differently expressed genes in the T47D RNA-Seq data. (B and C) Active ESR1 fusions upregulate expression of two clusters of estrogen response (early and late) and EMT-related genes as indicated. (D and E) Expression of estrogen response genes (GREB1, TFF1 and PGR) and EMT-related genes (VCAN and SNAI1) were measured by RT-qPCR in E2-deprived T47D cells treated with vehicle (+DMSO) or 100 nM fulvestrant (+Fulv) in the absence (–E2) or presence (+E2) of 10 nM E2 for 48 hours. Values were normalized to GAPDH mRNA and relative expression was calculated as fold change to YFP, –E2 (mean ± SEM, n=3). One-way ANOVA followed by Dunnett’s multiple comparisons test was used to compare each E2-deprived T47D cell line to YFP control cells (*p<0.05, **p<0.01, ***p<0.001, and ****p< 0.0001). Two-way ANOVA followed by Bonferroni’s test was used for multiple comparisons for each stable cell after 100 nM fulvestrant treatment. (F) Snail and E-cadherin proteins were measured by immunoblotting in E2-deprived cells treated with or without 100 nM fulvestrant (Fulv). GAPDH protein served as a loading control. The dashed line indicates two separate blots that were conducted at the same time. The representative image is from three independent experiments.
Additional experiments were conducted to demonstrate that inactive ESR1 fusion proteins enter the nucleus as the nuclear translocation signal is preserved. Also there is no biochemical evidence for heterodimer formation with WT ERα (Supplementary Figure 3). These data imply that the inactive ESR1 fusion genes are not dominant negative, a result consistent with the normal E2-induced growth in inactive fusion expressing cells.
Active ESR1 fusion proteins induce a characteristic, hormone-independent transcriptional signature.
The 3’ partners of ESR1 fusion genes are highly diverse, consequently their presence is only revealed by unbiased genomic techniques such as whole genome sequencing or RNA-Seq. These techniques are not routinely used clinically, and it is currently unknown how sensitive unbiased techniques are as screens for an ESR1 gene fusion event, because an orthogonal assay is requited to determine sensitivity. Adding to diagnostic complexity, some ESR1 fusion proteins are inactive and therefore not clinically actionable. An in vitro assay such as the ones described above are feasible but difficult to conduct within a clinically useful time-frame. We therefore sought to develop a gene expression signature that is diagnostic for the presence of a transcriptionally active ESR1 fusion protein. RNA-Seq was applied to T47D cells expressing ESR1 fusion cDNAs to identify genes that were selectively upregulated by the four transcriptionally active ESR1 fusion proteins as compared to: 1) three inactive ESR1 fusions and 2) three controls (Figure 4A). These two comparisons yielded an overlapping group of 66 candidate genes with a fold change (FC) greater than 4 and a false discovery rate (FDR) less than 0.05. Over-representation analysis using Hallmark pathways from MSigDB (25,26) identified candidate genes that were overrepresented in the estrogen response (early and late) and EMT gene sets (Figure 4B). An active ESR1 fusion signature was then devised based on estrogen response and EMT genes, as these were the top two pathways modulated by expression of an active ESR1 fusion protein. Specifically, we identified 24 Hallmark genes, including 19 genes in the estrogen response set (CHST8, MAPT, OLFM1, PDZK1, RASGRP1, MPPED2, GREB1, MYB, GFRA1, PGR, ELOVL2, ADCY1, NPY1R, TFF1, ACOX2, SGK1, STC2, CALCR and KRT13), two genes in the EMT gene set (VCAN and COL3A1), and three genes in both gene sets (CXCL12, GJA1 and TGM2). The expression of each gene was ranked by percentile within each sample and scores were computed as the mean percentile of the signature gene sets. ESR1 fusions were predicted as encoding active or inactive proteins according to the cutoff obtained by the receiver operating characteristic (ROC) curve analysis (cutoff, 0.3283) (Supplementary figure 4). In this training set, transcriptionally active ESR1 fusion proteins showed significantly higher scores as compared to inactive fusions and controls, as expected (Figure 4C).
Figure 4. Active ESR1 fusions program a unique, 24-gene transcriptional signature.
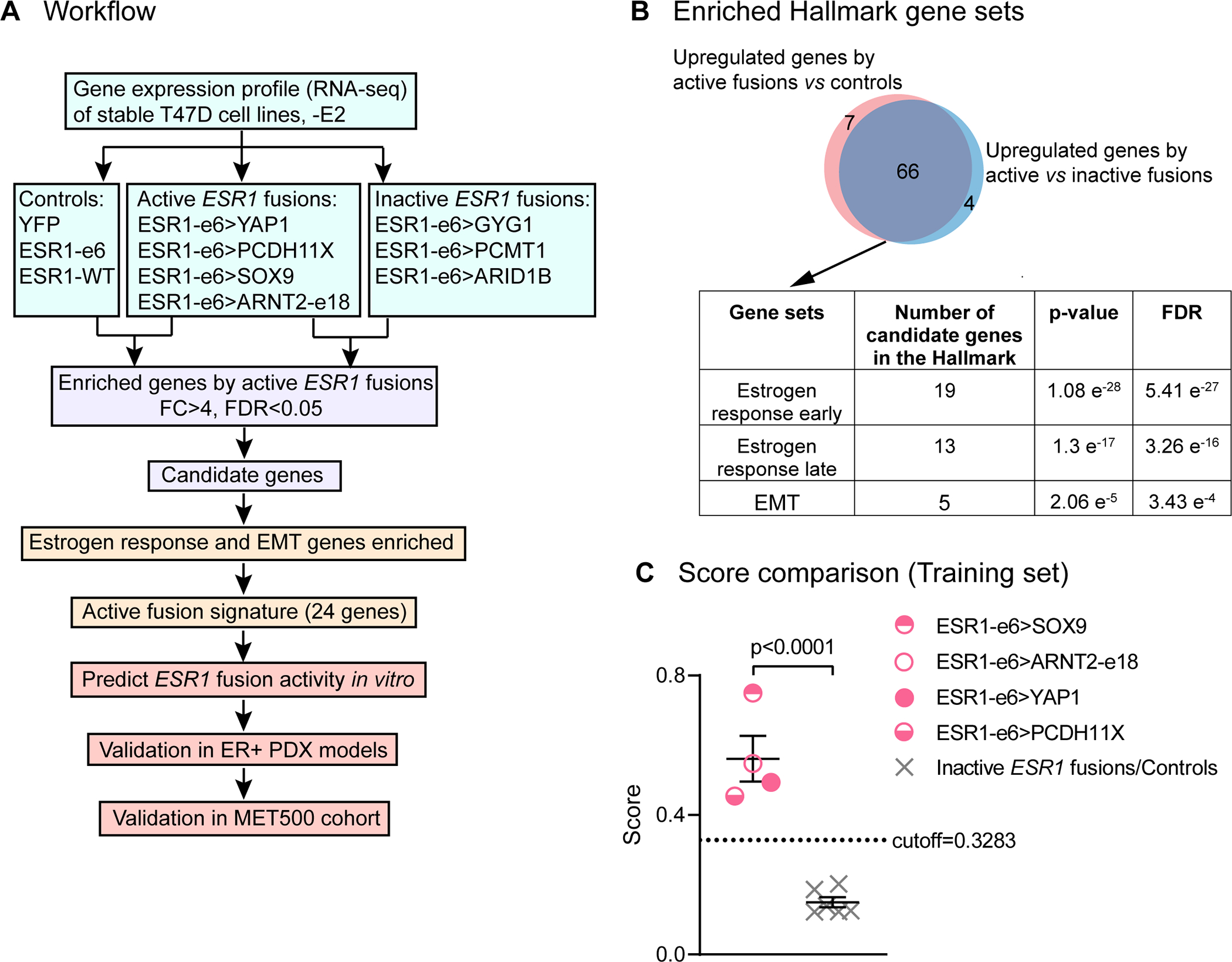
(A) Workflow to identify the gene signature to predict active fusions (FC: fold change. FDR: false discovery rate). (B) Venn diagram showing overlapping upregulated genes by active ESR1 fusions compared to inactive fusions or control cells. Table below shows the top three Hallmark gene sets enriched in the candidate genes. (C) Scatter plot showing signature scores of active ESR1 fusions (ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9, and ESR1-e6>ARNT2-e18) compared to inactive fusions (ESR1-e6>GYG1, ESR1-e6>PCMT1 and ESR1-e6>ARID1B) and control cells (YFP, ESR1-e6 and ESR1-WT) all minus E2. A two-tailed t test was used to calculate statistical significance.
The 24-gene transcriptional signature predicts the in vitro activity of additional ESR1-e6 fusion genes
To validate the 24-gene ESR1 fusion activity signature, we studied seven additional ESR1 gene fusions published by Priestly et al. (27). These in-frame ESR1-e6 fusions were identified in ER+ MBC patients by whole-genome sequencing, including four fusions with TF or CoA partners, ESR1-e6>ARNT2-e2, ESR1-e6>LPP, ESR1-e6>NCOA1 and ESR1-e6>TCF12 (Figure 5A). Another three fusions, analogous to PCDH11X, involved genes encoding protein-protein interaction motifs that serve non-transcriptional cellular functions, including ESR1-e6>CLINT1, ESR1-e6>GRIP1 and ESR1-e6>TNRC6B. The same approach as in Figure 2 was taken to assess the function of these new fusions in vitro. All of the seven ESR1 fusion cDNAs expressed stable chimeric proteins in T47D and MCF7 cells (Supplementary figure 5A and B). Three fusions that involve TF/CoA partners, ESR1-e6>ARNT2-e2, ESR1-e6>LPP, and ESR1-e6>NCOA1, drove E2-independent and fulvestrant-resistant growth, as well as increased motility of T47D cells, when compared to the YFP controls (–E2, +DMSO) (Supplementary figure 5C–E). Surprisingly the fourth fusion, ESR1-e6>TCF12, which involves a TF in the basic helix-loop-helix (bHLH) E-box family, expressed a stable chimeric protein, but was inactive in both T47D and MCF7 cells (Supplementary figure 5C–E). The ESR1-e6>TCF12 fusion was able to bind to concatenated EREs in a pulldown assay similar to active fusion examples (ESR1-e6>YAP1, ESR1-e6>SOX9 and ESR1-e6>CLINT1) (Supplementary figure 6), thus suggesting that the transcriptional inactivity of ESR1-e6>TCF12 was not due to lack of an ability to bind DNA.
Three gene fusions that did not involve a known TF/CoA partner, ESR1-e6>CLINT1, ESR1-e6>GRIP1 and ESR1-e6>TNRC6B, but all demonstrated ET-resistant cell growth and enhanced E2-independent motility, although the effect of ESR1-e6>GRIP1 on proliferation was statistically marginal (Supplementary Figure 5C–E). RNA-Seq was then performed on RNA extracted from T47D cells that expressed each new ESR1 fusion protein, as well on RNA from YFP control cells. In this experiment we also included two common ESR1 LBD point mutations (Y537S and D538G) to compare the active ESR1 fusion signature with the transcriptional profile associated with known activating ESR1 point mutants. Five out of six active ESR1 fusions (ESR1-e6>ARNT2-e2, ESR1-e6>LPP, ESR1-e6>NCOA1, ESR1-e6>CLINT1 and ESR1-e6>TNRC6B) demonstrated similar elevated expression of the 24-gene signature in sum, although there was some variability at the level of individual genes (Figure 5B). ESR1-e6>GRIP1 induced lower expression of the 24-gene signature than other active fusion examples, consistent with its weaker activity in proliferation assays compared to the five other fusions studied. Interestingly, the two ESR1 LBD point mutant proteins expressed in T47D cells induced similar levels of gene expression from the 24-gene signature as active ESR1 fusion proteins, suggesting that despite different mutational mechanisms for ESR1 protein activation, LBD point mutants and translocated ERs activate a similar pathogenic transcriptional pattern (Figure 5B). The mean signature scores of active ESR1 fusions and LBD point mutants were significantly increased compared to those of the inactive ESR1-e6>TCF12 fusion and YFP (endogenous ERα) control (Figure 5C). As expected, the mean score of the weakly active ESR1-e6>GRIP1 fusion fell below the cutoff value. The validation statistics of the independent Priestley et al. (27) set showed an accuracy of 90.0% (specificity, 100%; sensitivity, 87.5%) (Figure 5C). Since the 24-gene signature was similarly induced by ESR1 LBD point mutants and active ESR1 fusion proteins, it was given the moniker “MOTERA” for Mutant or Translocated Estrogen Receptor Alpha.
The MOTERA signature accurately predicts the presence and functional status of ESR1 mutations and gene fusions in ER+ PDX tumors and clinical samples.
To test the properties of the MOTERA signature in human tumors that naturally express either ESR1 gene fusions or mutations, we examined performance for ESR1 fusion or point mutation detection in a panel of 20 ER+ PDX tumors. The E2 dependence of each PDX tumor was evaluated in ovariectomized SCID/beige mice with or without 8 μg/ml E2 in the drinking water (Figure 6). ESR1 mutation status was determined by whole exome sequencing (WES) and gene expression was determined by RNA-Seq under both plus estradiol (+E2) and minus estradiol (–E2) conditions. When tumors were completely E2 dependent, the –E2 transcriptome was established by replacing the +E2 water with control (–E2) water for one week and then harvesting the tumors. As expected, the MOTERA signature was highly expressed in the E2-independent WHIM18 PDX naturally expressing the ESR1-YAP1 fusion protein (6) (Figure 7A), thus demonstrating a high degree of similarity between the experimental context of the ESR1-e6>YAP1 cDNA in T47D cells and the natural context in a PDX where this fusion was first identified. Consistent with T47D-based gene expression findings displayed in Figure 5B, ET-resistant PDXs bearing a variety of ESR1 LBD point mutations also induced the MOTERA signature, confirming an overlap between the transcriptional properties of active ESR1 fusion proteins and LBD point mutants noted in T47D cell experiments (Figure 7A). For example, the MOTERA signature score was enriched over the cutoff derived from the T47D training set in the cases of BCM15100, WHIM20, WHIM40, and HCI013 (all expressing ESR1-Y537S), WHIM37 and WHIM43 (expressing ESR1-D538G), WHIM24 (expressing ESR1-E380Q), WHIM27 (expressing ESR1-Y537N), and HCI005 and HCI007 (expressing ESR1-L536P) mutants (Figure 7B). PDX tumors expressing ESR1-WT (HCI003, HCI011, BCM15057, BCM4888, BCM15034, BCM3277, BCM7441, WHIM9 and WHIM16) had MOTERA scores below the cutoff in low estradiol (–E2) conditions in each case (Figure 7B). We note that the mean signature scores for ESR1-WT tumors increased with E2, consistent with some genes in the signature being E2-induced (Figure 7A and B). Thus, as a screening tool, the MOTERA signature is likely to be more specific if the biopsy sample is taken while the patient is taking an AI or an anti-estrogen. Paradoxically, the HCI013 PDX example harbors the Y537S ESR1 mutation but remained E2-dependent as previously reported by Welm et al. (28) (Figure 6). Similarly, HCI007 harbors an ESR1 L536P mutation, but also grew in an E2-dependent manner. These tumors have lower MOTERA scores but still above the training set defined cutoff. Presumably in these examples, ESR1-WT is functionally dominant over the LBD mutant ERα, although the mechanism remains obscure. Under –E2 conditions, the MOTERA signature successfully distinguished between ET-resistant tumors driven by mutant or translocated ESR1 proteins from ESR1-WT PDXs, with an accuracy of 95.0% (specificity, 88.9%; sensitivity, 100%) (Figure 7B). Although the MOTERA transcriptional signature was largely composed of estrogen response genes, expression levels were not affected by E2 supplementation to the WHIM18 ESR1-YAP1 expressing PDX or other PDXs expressing ESR1 LBD point mutations, underscoring sensitivity for the activated ESR1 mutant/translocated protein state (Figure 7A and B). Upon E2 treatment, the MOTERA scores of ESR1-WT bearing PDX lines still remained significantly lower than those of ESR1 mutated tumors, although in several cases expression levels rose above the cutoff established in E2-deprived conditions (Figure 7B).
Figure 6. The growth of 20 ER+ PDX tumors in xenografted mice in the absence and presence of E2.
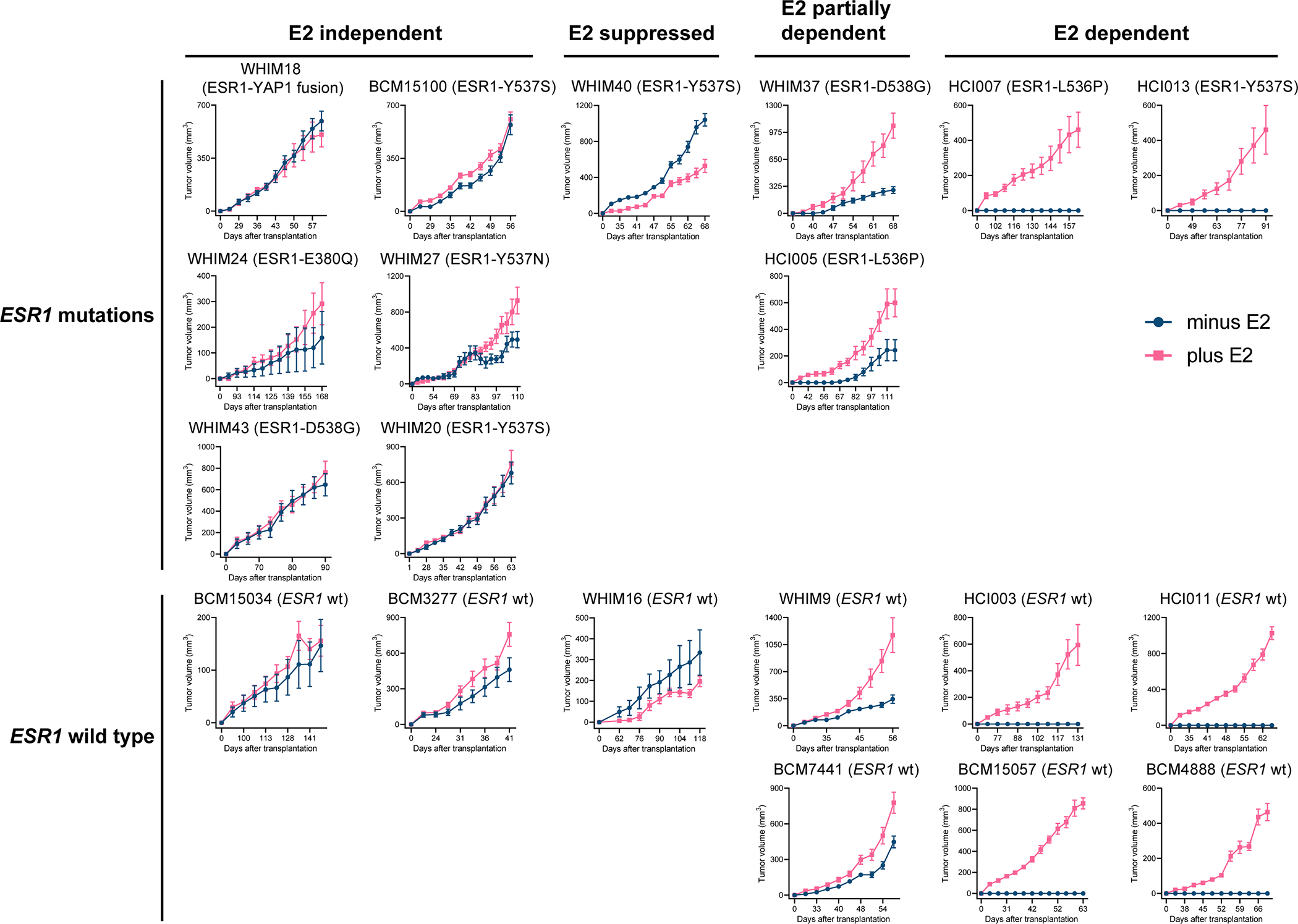
Volumes of 20 ER+ PDX tumors were measured in ovariectomized SCID/beige mice supplemented with or without 8 μg/ml E2 in the drinking water (mean ± SEM, n=7–16 per PDX line per arm). PDX tumors were categorized based on ESR1 status (mutations listed or wild-type, wt) and E2 dependency for growth (E2-independent, E2-suppressed, E2-partially dependent and E2-dependent).
Figure 7. The MOTERA signature predicts activity of ESR1 fusions/point mutations in ER+ PDX tumors and in MBC patients.
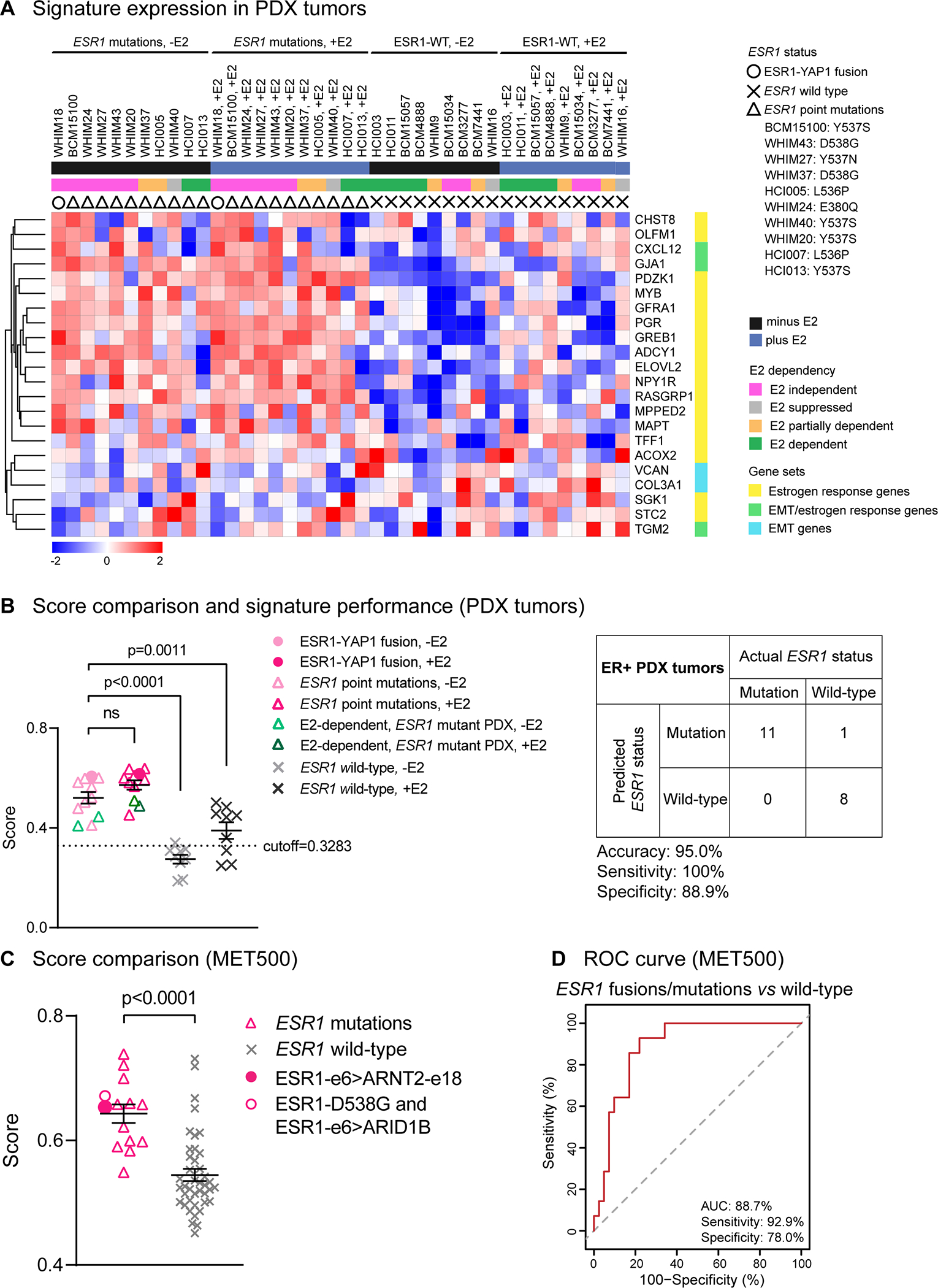
(A) Heatmap showing the expression of the 24-gene signature in 20 ER+ PDX tumors. Scale bar indicates row Z scores. CALCR and KRT13 in the signature were missing in the PDX RNA-Seq data, so they were not included in the heatmap. (B) Left panel: Scatter plot showing mean signature scores of ESR1 mutations (including the ESR1-YAP1 fusion and LBD point mutations) and ESR1-WT expressing tumors. One-way ANOVA with Dunnett’s multiple comparisons test was used to calculate statistical significance. Right panel: Confusion matrix to measure the performance of the signature to predict the presence of ESR1 mutations. Accuracy is the proportion of correctly predicted events in all cases. Sensitivity is the ability of the signature to predict an ESR1 mutation to be a mutant. Specificity is the ability of the signature to predict an ESR1-WT to be wild-type. (C) Scatter plot showing mean signature scores of MBC patient tumors expressing ESR1 mutations versus ESR1-WT in the MET500 cohort(9). Two-tailed t test was used to compare scores. (D) ROC curve for the 24-gene signature performance to differentiate ESR1 mutations from ESR1-WT in the MET500 cohort. The AUC is the probability that the signature ranks a randomly chosen ESR1 mutation higher than a randomly chosen WT ESR1 (100% is the best test and the dashed diagonal line illustrates the performance of a random signature).
An independent RNA-Seq data set of 55 ER+ mRNA positive MBC cases from the MET500 study (9) was used to further evaluate the performance of the MOTERA gene signature in tumor samples. Signature scores were significantly elevated in tumors expressing ESR1 LBD point mutations, such as Y537S and D538G, versus ESR1-WT samples (Figure 7C). Two ESR1 fusions that were functionally studied in Figure 2 (ESR1-e6>ARNT2-e18 and ESR1-e6>ARID1B) were both originally identified from the MET500 study. As expected, the ESR1-e6>ARNT2-e18 fusion drove a high MOTERA score in the sample in which it was identified. Against predictions, the functionally inactive ESR1-e6>ARID1B fusion also had a positive MOTERA signature score. However, this patient sample also harbored an ESR1-D538G LBD mutation, likely explaining the discordance. In terms of performance, the MOTERA signature score significantly distinguished active ESR1 mutations (Y537S, D538G, and Y537C point mutations and the ESR1-e6>ARNT2-e18 fusion) from WT ESR1, with a sensitivity of 92.9% and a specificity of 78.0% for an AUC of 88.7% (95% confidence interval, 80.0%−97.3%; Figure 7D).
Discussion
The data presented herein clearly demonstrate that most in-frame ESR1-e6 fusion proteins derived from inter-chromosomal translocations are drivers of ET resistance. Hitherto the clinical importance of ESR1 gene translocation has been underappreciated because the diversity of C-terminal partner genes creates a considerable diagnostic challenge. Fluorescence In Situ Hybridization (FISH) and PCR approaches that require the identification of both partners in a gene fusion event are not applicable. Even in the case of ESR1-e6>ARNT2, where we identified two examples, the ARNT2 exons present at the fusion junctions were different. Break-apart FISH probes can be considered in a setting where only one partner in the fusion is known. However, this approach does not identify the unknown 3’ partner gene or the reading frame, which is critical because inactive out-of-frame fusions are common (Supplementary table 1). RNA-Seq is clearly an applicable unbiased discovery approach, but sensitive detection requires the identification of a sufficient number of fusion junction reads to confidently diagnose the presence of an in-frame translocation. When RNA-Seq coverage is low, or the RNA is of low quality, fusion junction sequences could easily remain undetected.
Adding to the difficulty of understanding the clinical significance of ESR1 gene fusions is the fact that only a subset of ESR1 fusion proteins are active, and therefore clinically actionable. Consistent rules to diagnose whether a fusion is active based on the known functions of the C-terminal fusion partners proved hard to define. While ESR1-e6 fusions with YAP1, SOX9, ARNT2, LPP, and NCOA1 are all known positive regulators of transcription and produce active fusion proteins, our analysis of the ESR1-e6>TCF12 fusion protein produced an interesting exception. TCF12 encodes a bHLH E-box TF and its two TADs (29) are present in the fusion. Nonetheless the synthetic ESR1-e6>TCF12 cDNA was inactive in both T47D and MCF7 cells. We cannot exclude the possibility that this particular fusion is only active in the context of the cancer in which it evolved, i.e. the indicator cell lines we used lack the requisite coactivators. If truly inactive, however, the ESR1-e6>TCF12 fusion event raises the question of how this example could have been selected during clonal evolution. A potential explanation is provided by the ESR1-e6>ARID1B fusion protein, which is transcriptionally inactive with a 3’ partner gene related to the established tumor suppressor ARID1A (30,31). It has been proposed that TCF12 encodes a tumor suppressor (29,32). Thus, selection of transcriptionally inactive ESR1 fusions could be explained if these fusions inactivate tumor suppressor functions encoded by the 3’ partner gene. One could even speculate that these putative ESR1 tumor suppressor fusion proteins act in a dominant negative fashion, thereby interrupting the function of the remaining intact TCF12 or the ARID1A activity. Multiple active non-TF/CoA fusions (PCDH11X, DAB2, CLINT1, GRIP1 and TNRC6B) dramatically add to the complex landscape of ESR1 fusion genes. The activity of these fusions cannot, by definition, be predicted from an understanding of the normal function of each 3’ partner gene involved since none are known to be a TF or CoA and the wild-type protein is not nuclear-localized. Presumably the fusion partners have diverse protein-protein interaction domains that are subverted for the purposes of activating gene transcription in the context of a pathological fusion with ESR1. These questions must be addressed in follow up mechanistic studies.
One diagnostic approach after the detection of an in-frame ESR1 fusion gene would be to test the newly identified example in vitro. However, this is inefficient for clinical care, and may not always produce an accurate result. These concerns stimulated the development of the MOTERA gene signature to screen for tumors driven by the diverse somatic events that activate ESR1 through the presence of a diagnostic gene signature. In a setting where an ESR1 activating mutation has already been identified, the MOTERA signature could be used to confirm the mutant ESR1 gene is indeed driving ET-resistance. However, the MOTERA signature is likely to be of most value in the setting where a canonical ESR1 LBD point mutation has not been detected. Here, a high MOTERA score would warrant further investigation to detect a functional in-frame ESR1-e6 fusion. Reflex diagnostic approaches for these cases could include unbiased RNA-Seq, ESR1-specific 3’ Rapid Amplification of cDNA Ends (3’-RACE) or break-apart ESR1 FISH. While break-apart FISH would not identify the C-terminal partner, its presence has already been signaled by a positive MOTERA score implying the unknown partner in the chimera is transcriptionally active.
Analysis of the MET500 data indicates that MBC with high MOTERA scores but without an ESR1 point mutation detected by genome sequencing or translocation detected by RNA-Seq are not infrequent (Figure 7C). Possibilities for these cases include: 1) the RNA-Seq result was false-negative for the presence of an active ESR1 fusion; 2) the exome sequencing was a false-negative for the presence of an ESR1 mutation; 3) the MOTERA score was a false-positive that reflects wild-type ERα activity because the sample was taken when the patient was not taking ET and the tumor was still E2-dependent; and 4) some wild-type ERα MBC persist by expressing a similar MOTERA signature that might be driven by other mechanisms, like transcription factors other than ERα. The prospective evaluation of the MOTERA signature is therefore the next phase of our investigation.
An important focus for future studies will be to determine the clinical characteristics of ESR1 fusion-driven tumors. Of particular interest is an examination of the metastatic spread associated with tumors expressing ESR1 gene fusions, as in mouse xenograft systems active ESR1 fusions drive lung metastasis (7). Distinct from WT ERα, active ESR1 fusions strongly induce EMT-related genes, which we functionally annotated using motility and invasion assays. This property differentiates the MOTERA signature from other gene sets that measure activity of the ERα pathway, such as the Hallmark early/late estrogen response gene set. Consistent with this, the MOTERA scores of ESR1 mutated PDX tumors were still significantly higher than those of ESR1-WT bearing lines that received E2 treatment. Interestingly, EMT-related gene expression is elevated during mammary gland development as the nascent ducts invade the mammary fat pad, and then EMT gene expression is reduced after puberty (33,34). Thus, active ESR1-e6 fusion proteins may be reactivating a developmental EMT program that is usually silenced in mature breast epithelial cells. Specific examples of ESR1 fusion-induced genes in the MOTERA signature that are related to metastasis include SGK1, which encodes serum- and glucocorticoid-inducible kinase 1 and promotes breast cancer bone metastasis (35). VCAN encodes versican, whose expression is significantly correlated with metastasis and poor overall survival (36). GJA1 encodes connexin-43, a gap junction protein that mediates tumor cell migration and invasion (37,38). GFRA1 encodes GFRα that acts as a co-receptor in conjunction with the RET receptor, and activation of GFRα-RET signaling by binding the glial derived neurotrophic factor (GDNF) ligand leads to ERα serine phosphorylation and enhanced transcriptional activity (39).
At least one ESR1 fusion partner gene described herein has been observed in other settings. Gene fusions involving LPP, the gene encoding the Lipoma Preferred Partner protein, such as a recurrent HMGA2-LPP fusion have been found in multiple tumors, including lipoma (40), pulmonary chondroid hamartomas (41), and chondromas (42). In leukemia, an MLL-LPP fusion has been identified (43). Similar to the ESR1-e6>LPP fusion, these fusions preserve the three C-terminal LIM domains encoded by the LPP gene, which serve as the binding site for the ETS domain transcription factor PEA3 and contain coactivator activity (44). It is therefore likely that in larger studies, some ESR1 gene fusions will be observed to be recurrent, making the diagnosis of some ESR1 translocations easier.
In conclusion, ESR1-e6 gene fusions are part of the spectrum of the somatic mutations that constitutively activate ESR1 proteins in advanced ER+ breast cancer to drive poor outcomes. The MOTERA signature should be useful to answer the question how common these events are, because it will focus sensitive fusion detection approaches on cases where there is transcriptional evidence for an activating ESR1 fusion (or mutation) that has not been diagnosed yet. Once the clinical significance of ESR1 gene fusions becomes more widely recognized and the diagnostic approach becomes more efficient, specific treatment approaches for tumors expressing active ESR1 fusion proteins can be developed.
Supplementary Material
Significance.
This study identifies a gene signature diagnostic for functional ESR1 fusions that drive poor outcome in advanced breast cancer, which could also help guide precision medicine approaches in patients harboring ESR1 mutations.
Acknowledgments
We gratefully acknowledge Drs. Bing Zhang and Eric C. Chang at BCM for their scientific input.
This work was supported by the following grants to M.J. Ellis from the Susan G. Komen Foundation (SAC190059, PG12220321, SAC130059, BCTR0707808), the DOD Breast Cancer Research Program Grant (W81XWH-21-1-0119), and National Cancer Institute Grants P50CA186784 and U54CA233223 (to N. Mitsiades and M.J. Ellis) and U54CA224076 to M.T. Lewis and A.L. Welm. M.J. Ellis is also a Susan G. Komen Foundation Scholar, a McNair Scholar supported by the McNair Medical Institute at The Robert and Janice McNair Foundation, and a recipient of a CPRIT Established Investigator Award (RR140027). X. Gou was supported by CPRIT RP210027 - Baylor College of Medicine Comprehensive Cancer Training Program. J.T. Lei was supported by T32CA203690 and T32GM088129. This project was also supported in part by the GARP Core at BCM with funding from the NIH NCI grant (P30CA125123) and the NIH S10 grant (1S10OD023469). We also acknowledge the Adrienne Helis Malvin Medical Research Foundation through direct engagement with the continuous active conduct of medical research in conjunction with BCM (M-2017 to C.E. Foulds; M-2020 to M.J. Ellis). The BCM ER+ PDX were generated with a CPRIT Core Facility Support Grant to M.T. Lewis (RP170691) and P30 Cancer Center Support Grant (P30CA125123).
Footnotes
Disclosure of Potential Conflicts of Interest
M.J. Ellis received consulting fees from Abbvie, Sermonix, Pfizer, AstraZeneca, Celgene, NanoString, Puma, Veracyte, Eli Lilly and Novartis, and he is an equity stockholder and Board Director member of BioClassifier. M.J. Ellis is an inventor on a patent for the Breast Cancer PAM50-based assay, Prosigna, which is marketed by Veracyte. M.J Ellis also receives royalties from Washington University in St Louis when the WHIM PDX lines are licensed to for-profit companies. C.E. Foulds holds an equity position in Coactigon, Inc. M.T. Lewis is a Manager in StemMed Holdings L.L.C., a limited partner in StemMed Ltd., and holds an equity stake in Tvardi Therapeutics. L.E. Dobrolecki is a compensated employee of StemMed, Ltd. A.L. Welm may receive tangible property royalties if the University of Utah chooses to license HCI PDX models to for-profit companies. A.V. Lee is a stock holder and receives compensation from Ocean Genomics. All other authors declare no conflict of interests.
References
- 1.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011;62:233–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer 2015;15:261–75 [DOI] [PubMed] [Google Scholar]
- 3.Dustin D, Gu G, Fuqua SAW. ESR1 mutations in breast cancer. Cancer 2019;125:3714–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol 2016;34:2961–8 [DOI] [PubMed] [Google Scholar]
- 5.Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 2016;7:11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 2013;4:1116–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, et al. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep 2018;24:1434–44 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, Vanden Borre P, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann Oncol 2018;29:872–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012;149:307–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res 2013;73:4885–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 2011;17:1514–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foulds CE, Feng Q, Ding C, Bailey S, Hunsaker TL, Malovannaya A, et al. Proteomic analysis of coregulators bound to ERalpha on DNA and nucleosomes reveals coregulator dynamics. Mol Cell 2013;51:185–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, Hamilton RA, et al. Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene 2018;37:4581–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagchi MK, Tsai SY, Tsai MJ, O’Malley BW. Identification of a functional intermediate in receptor activation in progesterone-dependent cell-free transcription. Nature 1990;345:547–50 [DOI] [PubMed] [Google Scholar]
- 19.UniProt C UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2021;49:D480–D9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaRese TP, Rheaume BA, Abraham R, Eipper BA, Mains RE. Sex-Specific Gene Expression in the Mouse Nucleus Accumbens Before and After Cocaine Exposure. J Endocr Soc 2019;3:468–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex): development of a novel, “pure” antiestrogen. Cancer 2000;89:817–25 [DOI] [PubMed] [Google Scholar]
- 22.Carroll JS, Brown M. Estrogen receptor target gene: an evolving concept. Mol Endocrinol 2006;20:1707–14 [DOI] [PubMed] [Google Scholar]
- 23.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015;525:256–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000;2:76–83 [DOI] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27:1739–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019;575:210–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sikora MJ, Cooper KL, Bahreini A, Luthra S, Wang G, Chandran UR, et al. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res 2014;74:1463–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labreche K, Simeonova I, Kamoun A, Gleize V, Chubb D, Letouze E, et al. TCF12 is mutated in anaplastic oligodendroglioma. Nat Commun 2015;6:7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagl NG Jr., Patsialou A, Haines DS, Dallas PB, Beck GR Jr., Moran E The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res 2005;65:9236–44 [DOI] [PubMed] [Google Scholar]
- 31.Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A 2012;109:E252–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen QB, Liang YK, Zhang YQ, Jiang MY, Han ZD, Liang YX, et al. Decreased expression of TCF12 contributes to progression and predicts biochemical recurrence in patients with prostate cancer. Tumour Biol 2017;39:1010428317703924. [DOI] [PubMed] [Google Scholar]
- 33.Sikandar SS, Kuo AH, Kalisky T, Cai S, Zabala M, Hsieh RW, et al. Role of epithelial to mesenchymal transition associated genes in mammary gland regeneration and breast tumorigenesis. Nat Commun 2017;8:1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foubert E, De Craene B, Berx G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res 2010;12:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Xu Q, Song C, Mi B, Zhang H, Kang H, et al. Serum- and Glucocorticoid-inducible Kinase 1 is Essential for Osteoclastogenesis and Promotes Breast Cancer Bone Metastasis. Mol Cancer Ther 2020;19:650–60 [DOI] [PubMed] [Google Scholar]
- 36.Mitsui Y, Shiina H, Kato T, Maekawa S, Hashimoto Y, Shiina M, et al. Versican Promotes Tumor Progression, Metastasis and Predicts Poor Prognosis in Renal Carcinoma. Mol Cancer Res 2017;15:884–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollmann MA, Shao Q, Laird DW, Sandig M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res 2005;7:R522–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotini M, Mayor R. Connexins in migration during development and cancer. Dev Biol 2015;401:143–51 [DOI] [PubMed] [Google Scholar]
- 39.Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, et al. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene 2010;29:4648–57 [DOI] [PubMed] [Google Scholar]
- 40.Crombez KR, Vanoirbeek EM, Van de Ven WJ, Petit MM. Transactivation functions of the tumor-specific HMGA2/LPP fusion protein are augmented by wild-type HMGA2. Mol Cancer Res 2005;3:63–70 [DOI] [PubMed] [Google Scholar]
- 41.Rogalla P, Lemke I, Kazmierczak B, Bullerdiek J. An identical HMGIC-LPP fusion transcript is consistently expressed in pulmonary chondroid hamartomas with t(3;12)(q27–28;q14–15). Genes Chromosomes Cancer 2000;29:363–6 [PubMed] [Google Scholar]
- 42.Dahlen A, Mertens F, Rydholm A, Brosjo O, Wejde J, Mandahl N, et al. Fusion, disruption, and expression of HMGA2 in bone and soft tissue chondromas. Mod Pathol 2003;16:1132–40 [DOI] [PubMed] [Google Scholar]
- 43.Daheron L, Veinstein A, Brizard F, Drabkin H, Lacotte L, Guilhot F, et al. Human LPP gene is fused to MLL in a secondary acute leukemia with a t(3;11) (q28;q23). Genes Chromosomes Cancer 2001;31:382–9 [DOI] [PubMed] [Google Scholar]
- 44.Guo B, Sallis RE, Greenall A, Petit MM, Jansen E, Young L, et al. The LIM domain protein LPP is a coactivator for the ETS domain transcription factor PEA3. Mol Cell Biol 2006;26:4529–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data from T47D cells and WES and RNA-Seq data from ER+ PDX tumors in this study are being submitted to Gene Expression Omnibus (GEO). Until an accession number is provided, these raw data are available upon request from the corresponding author.
A full description of all methods and reagents can be found in Supplementary information.