

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive cancer with poor prognosis and chemotherapy with gemcitabine has limited effects and is associated with development of drug resistance. Treatment of Panc1 and MiaPaca2 pancreatic cancer cells with gemcitabine induced expression of the orphan nuclear receptor 4A2 (NURR1) and analysis of the cancer genome atlas indicated the NURR1 is overexpressed in pancreatic tumors and is a negative prognostic factor for patient survival. Results of NURR1 knockdown or treatment with the NURR1 antagonist 1,1-bis(3΄-indolyl)-1-(p-chlorophenyl)methane (C-DIM 12) demonstrated that NURR1 was pro-oncogenic in pancreatic cancer cells and regulated cancer cell and tumor growth and survival. NURR1 is induced by gemcitabine and serves as a key drug-resistance factor and is also required for gemcitabine-induced cytoprotective autophagy. NURR1 regulated genes were determined by RNA sequencing of mRNAs expressed in MiaPaCa2 cells expressing NURR1 and in CRISPR/Cas9 gene edited cells for NURR1 knockdown and KEGG enrichment analysis of the differentially expressed genes showed that autophagy was the major pathway regulated by NURR1. Moreover, NURR1 regulated expression of two major autophagic genes ATG7 and ATG12 which are also overexpressed in pancreatic tumors and like NURR1 are negative prognostic factors for patient survival. Thus, gemcitabine-induced cytoprotective autophagy is due to the NURR1 – ATG7/ATG12 axis and this can be targeted and disrupted by NURR1 antagonist C-DIM12 demonstrating the potential clinical applications for combination therapies with gemcitabine and NURR1 antagonists.
Keywords: Pancreatic cancer, NURR1, Autophagy, ATG7, ATG12, chemoresistance
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal human malignancy with a five-year survival rate of 9% and it is estimated that by 2030 pancreatic cancer will be the second leading cause of cancer deaths in United States (1). Although new therapeutic regimens against pancreatic ductal adenocarcinoma have improved treatment of this disease, the late stage detection, lack of targeted therapies and chemoresistance are still some of the major challenges for enhancing patient outcomes. The current treatments include surgical resection which is followed by adjuvant chemotherapy (2) and palliative chemotherapy [with gemcitabine or FOLFIRINOX (folinic acid, 5-flouorouracil, irinotecan and oxaliplatin)] (3–5). Gemcitabine (Gemzar; 2’,2’-difluorodeoxycytidine), an FDA approved chemotherapeutic drug is used for treatment of different cancers and overall survival and progression free survival is enhanced by gemcitabine compared to those receiving non-gemcitabine-based therapy (6). Although, these treatments are initially effective in increasing survival of pancreatic cancer patients, residual neoplastic cells usually result in relapse and subsequent appearance of more aggressive and lethal tumors (7). The success of cancer treatments for PDA patients largely depends on diagnosis during initial stages of the disease. Cellular mechanisms such as apoptosis, necroptosis and autophagy also determine the advancement and response to these cancer treatments (8). Thus, in order to find a remedy for treating this devastating disease it is important to understand the mechanism through which pancreatic cancer cells acquire resistance to current therapeutics.
NURR1 (NR4A2) is an orphan nuclear receptor and a transcription factor that activates target genes by binding as monomers or dimers to cognate cis-elements in target gene promoters (9). NURR1 has been characterized as an important regulator in neuronal development and there is also increasing evidence of a pro-oncogenic role for their receptor in solid tumors (9–11) NURR1 regulates cell growth survival and metabolism (12–14) and studies in this laboratory identified 1,1-bis (3’-indolyl)-1-(p-chlorophenyl) methane (C-DIM12) as prototypical NURR1 ligand (15, 16). A recent study showed that C-DIM12 inhibited glioblastoma cell and tumor growth and also blocked NURR1 dependent pro-oncogenic pathways in glioblastoma (15). It was previously reported that induction of NURR1 promotes 5-FU resistance in squamous cell carcinoma (17), however, the mechanism of NURR1-mediated chemoresistance and its role in pancreatic cancer has not been determined.
Autophagy is an evolutionary cellular response to diverse stress and physiological conditions during cancer progression and has been reported to induce tumor cell survival and drug resistance in cancer cells (18, 19). Autophagy is initiated with the formation of double layer membrane around the cytoplasmic components known as autophagosome which fuses with lysosomes to recycle these components for protein synthesis and energy production. It is closely regulated by a highly conserved set of genes known as ATGs (autophagy related genes) (18). A recent study demonstrates that autophagy regulates the unique properties of cancer stem cells such as differentiation and self-renewal which contributes to tumor metastasis, tumor reoccurrence and chemoresistance (20). A study, combining an autophagy inhibitor with photodynamic therapy significantly reduced the colorectal tumor size suggesting that autophagy is a cytoprotective process (21). and in another study autophagy inhibition increased the drug sensitivity (22). Thus, understanding the mechanism of autophagy and drug resistance is an unsolved problem and could lead to enhanced therapeutic efficacy.
This study demonstrates that NURR1 is essential for chemotherapeutic agent-induced cytoprotective autophagy through transcriptional regulation of autophagy-related genes. We show for first time that ATG7 and ATG12 are NURR1 regulated genes, and high expression of NURR1/ATG7/ATG12 corresponds to the poor survival and prognosis of PDA patients and the NURR1-ATG7/ATG12 axis can be target by NURR1 antagonists.
Materials and Methods
Cell lines and transfection with small-interference RNA and plasmids
MiaPaCa2 and Panc1 pancreatic cancer cell lines were obtained from ATCC (American Type Culture Collection) and were cultured in in DMEM supplemented with 10% FBS (Gibco/Invitrogen), 1% L-glutamine (Gibco/Invitrogen), and 1% penicillin-streptomycin (Invitrogen) at 37°C in 5% humidified CO2 incubators. CRISPR/Cas9-mediated knockout of NURR1 in MiaPaCa2 cells was accomplished using guide RNAs targeting NURR1, fused with CRISPR/Cas9 and GFP protein (23). CRISPR Universal Negative Control plasmid (CRISPR06–1EA) were purchased from Sigma-Aldrich (St. Louis, MO). Cells were harvested after 48 hours of transfection and GFP positive cells were single sorted by using FACS Calibur flow cytometer. The guide RNA sequences used were:
NURR1-1(gatcccgggtcgtcccacat),
NURR1-2(gggcttgtagtaaaccgacc),
For all cell cultures experiments Mycoplasma testing (MycoAlert TM Mycoplasma Detection Kit, Lonza) was performed after each thawing and at least monthly. Cells were also authenticated using short tandem repeat analysis (Biosynthesis, Lewisville, TX)
Cells were plated at 60% confluency in 6-well plates, and transient siRNA transfections (1 μM) were performed using Lipofectamine 3000 (Invitrogen) and Opti-MEM (Invitrogen) according to the manufacturer’s protocol; 48 hours after transfection, cells were treated or analyzed, as described (24, 25). Small interfering RNA (siRNA) oligos were purchased from Life Technologies (siNURR1, 4427038; siATG7, 4392420; siATG12, 4392420; siCTRL, AM4635).
Cell growth assays
Cells were plated in 96-well plates at 1×103 cells per well. After 5 days of incubation, cell growth was measured using Quant-iT™ PicoGreen™ dsDNA assay kit (Invitrogen). To estimate cell death, cells were trypsinized and counted after Trypan blue staining (Invitrogen) with a Hausser bright-line hemocytometer (Fisher Scientific)(26). Annexin V/PI staining was performed using the Dead Cell Apoptosis Kit (ThermoFisher Scientific #V13245), according to the manufacturer’s instructions. Staining was measured with a Accuri C6 flow cytometer and analyzed with FlowJo Version 10.2 software (26).
Immunoblot analysis
Cells were lysed using 1% Triton in TBS containing protease and phosphatase inhibitors. Tumors were lysed using 1X RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of total protein were separated by electrophoresis on a 4–12% Bis-Tris gel and transferred to a PVDF membrane (23). Blots were blocked in 5% BSA, and then probed with antibodies against anti- NURR1 (sc-376984, SCB), anti- ATG 7 (10088-2AP, Proteintech), anti-ATG 12(D88H11, Cell Signaling Technology), anti-LC3BI/II(3868, Cell Signaling Technology), anti-Cleaved PARP (9532S, Cell Signaling Technology) and anti-α-tubulin (Sigma). Chemiluminescent (32106, Thermo Fisher Scientific) signal was captured using a Syngene G-BOX iChemi XT imager (26).
Clonogenic assay
1000–2000 cells per well were plated in a 6-well plate. The media was not changed during experiments unless indicated. Upon completion of the experiments, colonies were fixed in reagent containing 80% methanol and stained with 0.5% crystal violet. To determine relative growth, dye was extracted from stained colonies with 10% acetic acid and the associated absorbance measured at 600 nm using a Microplate Reader/Synergy HT BioTek plate reader (23, 27).
Luciferase reporter assays
Cells were seeded at 60% confluency in six well dishes, then transfected using lipofectamine 3000 (Thermo Fisher Scientific) with 2 μg of the promoter dual reporter plasmid (pCheck2–promoter ATG7 [TSS=11272324; Upstream=1265, Downstream=295;Length=1561] or ATG12 [TSS=115841851;Upstream=1271,Downstream=228;Length=1500]), purchased from GeneCopoeia (23). The luciferase activity of the cultured supernatant was measured 48 hours after transfection using luciferase assay reporter kit (Promega) according to the manufacturer’s instructions (23).
Electron Microscopy
Cells were cultured in permanox petri dish and treated with gemcitabine (1μM) and C-DIM12 (15 μM) for 24 hours. The cells were fixed with 2.5% paraformaldehyde, 2% glutaraldehyde, 0.1M cacodylate buffer and embedded using Epon 812. The ultra-thin sections (~100nm) were cut using a Leica EM UC6 ultramicrotome and diamond knife. The sections were then placed on copper grids, post-stained with saturated Uranyl Acetate and Reynolds Lead Citrate, and imaged using an FEI Morgagni 268 transmission electron microscope equipped with a MegaView III CCD camera.
Quantitative RT-PCR
Total RNA was extracted using RNAeasy kit (Qiagen). cDNA was synthesized from 1 μg of total RNA using TagMan probes (NURR1, Hs00428691, Hs01117525; ATG7, Hs00893766, Hs04969948; ATG12, Hs00740818, Hs01047860; 18s, 99999901; Actin, Hs00157387, Life Technologies) and MultiScribe Reverse Transcriptase (Life Technologies). RT-qPCR analysis was performed using the Applied Biosystems 7500 Fast Real-Time PCR System (Life Technologies) and TagMan RT-PCR Master Mix (Life Technologies).
RNA Sequencing
RNA quality was assessed via the Agilent 2100 Bioanalyzer (Agilent Technologies). Strand-specific RNA-seq library was prepared using NEBNext Ultra II Directional RNA Library Prep Kit (NEB, Ipswich, MA) according to the manufacturer’s protocols (27). RNA-sequencing was performed using 150-bp paired-end format on a NovaSeq 6000 (Illumina) sequencer. RNA-seq quality was checked by running FastQC, and TrimGalore was used for adapter and quality trimming (27). Sequence reads were aligned to the hg19 human genome build using the STAR aligning program (28). Quantification of all genes and their isoforms was performed using FPKM normalized values using Cufflinks v2.2.1, DESeq2 analysis with an adjusted P-value <0.05 was used to get a list of differentially expressed genes (29).
Immunofluorescence Assay:
Cells were seeded in 24-well plate (50,000 cells/well). After 24 hours the media was discarded and the cells were then washed with 1X PBS. The cells were then fixed with 10% formaldehyde and permeabilized with 0.1% Triton-X 100. The cells were then incubated with primary antibody, overnight at 4°C and then with secondary antibody (Thermo Fisher Scientific #A11001 and Cell Signaling #4412S)] for 2 hours at room temperature. The images were captured using Zeiss Imager.Z1 AXIO at 40X magnification.
Chromatin Immunoprecipitation Assay
Cells were cross-linked for 10 min at room temperature by the addition of one-tenth volume of 11% formaldehyde (11% formaldehyde, 50mM HEPES pH 7.4, 100mM NaCl, 1mM EDTA pH 8.0, 0.5mM EGTA pH 8.0), followed by 5 min quenching with 1/20th volume of 2.5M glycine. Cells were washed twice with PBS, with spins after each rinse, the supernatant was aspirated and the cell pellet was flash frozen in liquid nitrogen. Frozen crosslinked cells were stored at −80 °C (30).
Crosslinked cells were lysed with lysis buffer 1 (50mM HEPES pH 7.5, 140mM NaCl, 1mM EDTA, 10% glycerol, 0.5% NP-40, and 0.25% Triton X-100), pelleted and resuspended in lysis buffer 2 (10 mM TrisHCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA), and pelleted again (30). The pellet was resuspended in sonication buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1 mM EGTA, 0.1% Na-deoxycholate, 0.1% SDS, and 1% Triton X-100), and sonicated (30) using a Branson Sonifier (power setting 5) for 10 cycles at 30 seconds each on ice (18–21 W) with 60 seconds on ice between cycles.
Dynal magnetic beads (Sigma) (50 μL) were blocked with 0.5% BSA (w/v) in PBS, and then bound with 10 μg of antibody against NURR1, Abcam (AB41917). Sonicated crosslinked lysates were incubated overnight at 4°C with magnetic beads bound with antibody. Beads were pelleted, and then washed several times: two times with sonication buffer, one time with sonication buffer with 500 mM NaCl, one time with LiCl wash buffer (10 mM TrisHCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate) and one time with TE (10 mM TrisHCl pH 8.0, 1 mM EDTA). Bound protein and cross-linked DNA was eluted in elution buffer (50 mM TrisHCl pH 8.0, 10 mM EDTA, 1% SDS), and cross-links were reversed by overnight incubation using RNase A(10mg/ml) and Proteinase K(20 mg/ml) for 1 hour, respectively and DNA was purified with phenol/chloroform extraction and ethanol precipitation and used for Q-PCR (primers summarized in Supplemental Table 1).
Mouse studies
All experiments involving mice were approved by the Texas A&M University’s Animal Care and Use Committee. Six-week-old, female, athymic nude mice (Nude-Foxn1nu) were purchased from ENVIGO. MiaPaCa2 (ctrl), CTRL.KO, NURR1.KO cells, were prepared in 100 μl solution comprised of 70% DPBS and 30% Matrigel. Suspensions of 3 × 106 cells were then injected subcutaneously into the left and right flanks of mice. Tumor volumes were measured three times per week using calipers (Volume = Length × Width2/2), along with body weight. Mice with established tumors (after 25 days, mean tumor volume of ~100 mm3) were randomly divided into 4 groups, which were then treated with vehicle (20 μl/g of 0.9%NaCl), GEM (5 mg/ml, prepared in vehicle solution) 100 mg/kg intraperitoneally (IP) twice biweekly, C-DIM12 (30 mg/kg IP Mon/Wed/Fri), LC Laboratories #R-5000) or a combination of both. Upon termination of mouse experiments, mice were euthanized using carbon dioxide inhalation followed by cervical dislocation, and tumors harvested.
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM) of at least three independent experiments. An unpaired, two-tailed student t test was used to determine the differences between groups (* p < 0.05; ** p < 0.01; *** p < 0.001). ANOVA test was used for the analysis of tumor measurements among treated groups.
Results
Prognostic significance of NURR1 in PDAC patients
Previous studies showed that NURR1 is highly expressed in glioblastoma (15) and analysis of the published TCGA (The Cancer Genome Atlas) database showed that NURR1 was also more highly expressed tumor samples from PDA patient. Kaplan-Meier analysis of NURR1 expression data showed that NURR1 overexpression PDA patients’ tumor samples was also significantly associated with their poor survival (Figure 1A). Lymph node metastasis was studied in 173 PDA patients and NURR1 expression was significantly higher in N0 and N1 PDA patients compared to normal individuals (Figure 1B). NURR1 was also overexpressed in Grade 2 tumor samples confirming that high levels of NURR1 are associated with the severity of PDAC (Figure 1C).
Figure 1. NURR1 confers to PDA chemoresistance.
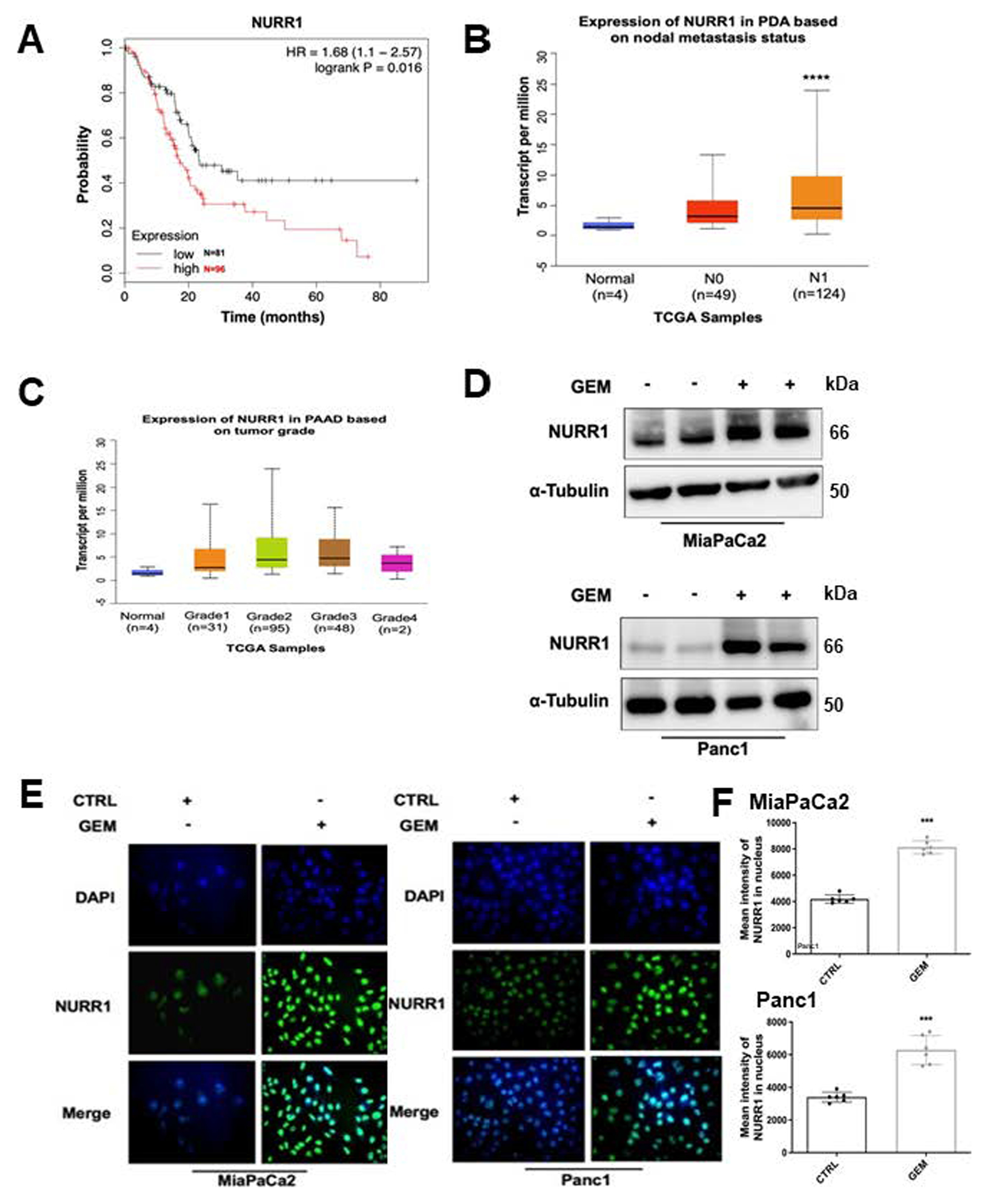
A) The Cancer Genome Atlas (TCGA) research network for PDA database was used for expression of NURR1 mRNA and correlated with patient’s disease-free survival by using Kaplan– Meier survival analysis with log-rank tests P<0.016, B) NURR1 mRNA expression levels in PDA patients based on nodal metastasis status. For each boxplot, median expression value, and 1st and 3rd quartiles are indicated, **** p < 0.00001, C) NURR1 mRNA expression levels in PDA patients based on tumor grade, D) Immunoblot analysis of NURR1 protein lysates from MiaPaCa2 and Panc1 cells after 48 h of treatment with GEM (1μM), β-Tubulin serves as a loading control (3 independent experiments performed), E) Immunofluorescence staining of MiaPaCa2 and Panc1cells after 24 h of treatment with GEM (1μM) NURR1, Alexa Fluor 488 (green); nucleus, DAPI. Merged image, nuclear localization of NURR1 (3 independent experiments performed, and at least 10 images per slide were analyzed for each condition). Magnification 40x. F. Bars represent the quantification of mean intensity of NURR1 in the nucleus. Each data point represents the mean ± SEM of three independent experiments (*** p < 0.001).
PDA is a prime example of a tumor that develops chemoresistance and in order to investigate the possible relationship between chemotherapeutic drugs and NURR1 expression pancreatic cancer cells were treated with gemcitabine and analysis by immunoblot and immunofluorescence showed that gemcitabine induced expression of NURR1 in both MiaPaCa2 and Panc1 cells (Figures 1D–1E). This demonstrates that NURR1 expression is increased during chemotherapeutic treatment and the mechanisms of GEM induction of NURR1 and GEM – C-DIM12 interactions are unknown and are currently being investigated.
NURR1 has a cytoprotective role against chemotherapeutic drug-induced cell death in pancreatic cancer cells
To study the role of NURR1 in drug resistance we treated the MiaPaCa2 cells with the NURR1 antagonist C-DIM12 (DIM-C-pPhCl) (Figure 2A) and gemcitabine and in parallel studies we also determined effects of gemcitabine alone and after NURR1 knockout (by RNA interference) in MiaPaCa cells. C-DIM12 has been characterized as an NURR1 antagonist in cancer cells and has been used as a model compound for studying the actions of NURR1 (15, 16). Crystal violet assay, phase contrast microscopy and Annexin V staining showed that gemcitabine and C-DIM12 alone were effective, however, treatment with gemcitabine in combination with C-DIM12 was a more potent inhibitor of cell proliferation and survival than the individual compounds alone (Figure 2B–2D) and similar results were observed in Panc1 cells (Suppl. Figs. 1A – 1D). Moreover, treating the cells with C-DIM12 in combination with gemcitabine increased apoptosis as determined by cleaved PARP in MiaPaCa2 (Figure 2E) and Panc1 cells (Sup Figure 2A and 2B). Results summarized in Figures 2E and 2F also show that cleaved PARP (marker of apoptosis) and inhibition of cell growth by gemcitabine were enhanced by co-treatment with C-DIM12 or by NURR1 knockout using CRISPR/Cas9 gene edited cells and this response can be attenuated by overexpression of Nurr1 (NOE) (Fig. 2G). Electron microscopy images also demonstrate that pancreatic cancer cells were more sensitive to gemcitabine when treated in combination with C-DIM12 and this is evidenced by enhanced cell shrinkage and nuclear condensation in treated compared to control cells. We also observed, progressive fragmentation and increase in number of apoptotic bodies which are indicators of apoptosis in the cells treated with gemcitabine and C-DIM12 (Sup Figure 1C).
Figure 2. NURR1 plays a cytoprotective role in pancreatic cancer.
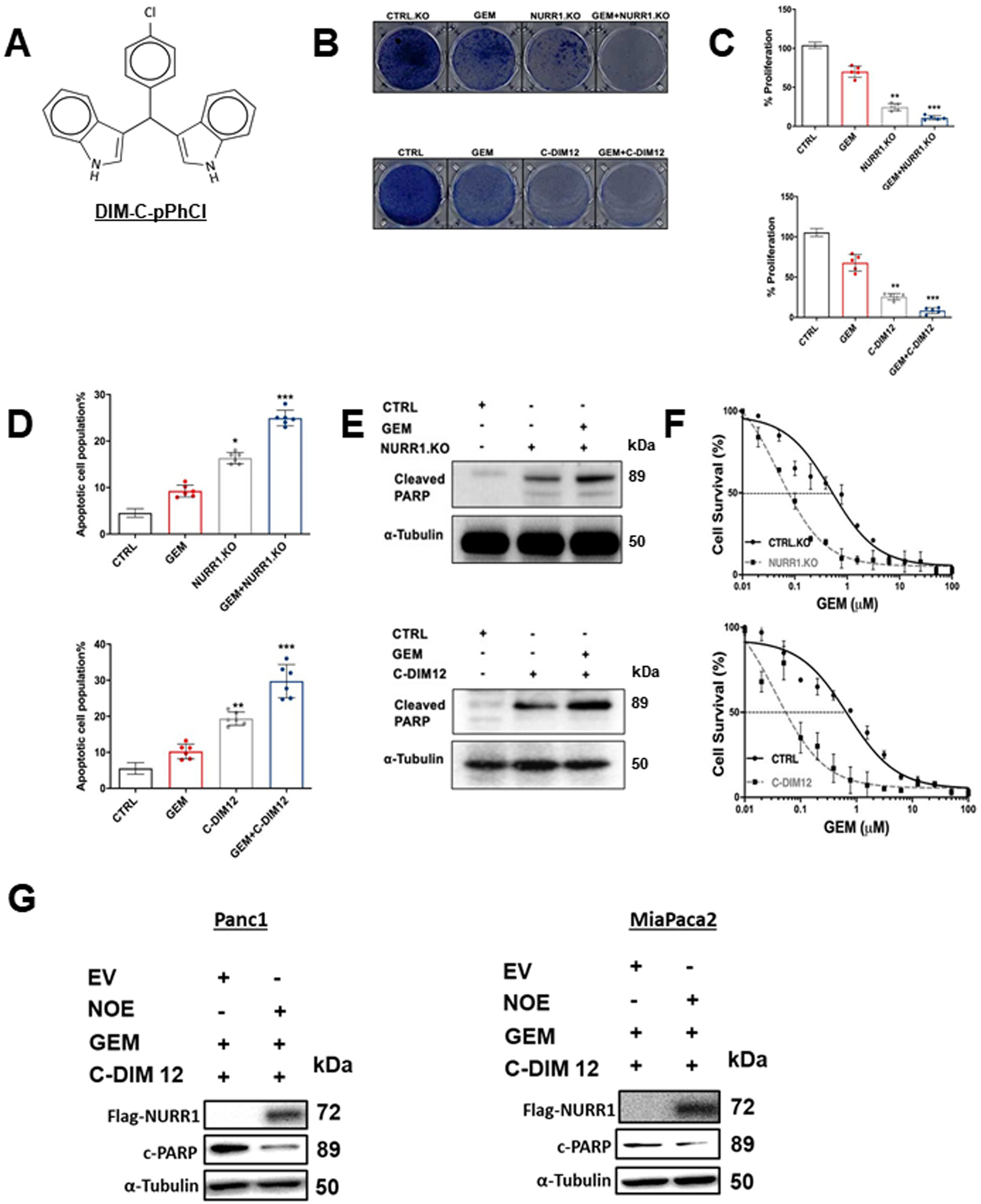
A) Chemical structure of DIM-C-pPhCl (C-DIM12), B) Images of crystal violet stained dishes that were treated with vehicle control, gemcitabine, C-DIM12 or NURR1 loss for 10 days after plating 1,000 cells per well in a 12-well plate (3 independent experiments performed). C) Quantification of crystal violet staining is shown. Error bars indicate SEM of triplicate wells from a representative experiment (3 independent experiments performed) (**p < 0.01, *** p < 0.001). D) Apoptotic cell fraction was determined after treatment control (vehicle or CTRL.KO), gemcitabine, C-DIM12 or NURR1 loss, or a combination of both (GEM+ C-DIM12/GEM+NuRR1.KO for 72 hours. Apoptotic cell death was quantified by Annexin V/ propidium iodide (PI) staining and flow cytometry and is shown as the percentage of cells that were PI positive. Each data point represents the mean ± SEM of three independent experiments. * p < 0.05; ** p < 0.01; *** p < 0.001.E) Immunoblot analysis of Cleaved PARP in control, gemcitabine, C-DIM12 or NURR1 loss treated cells with α-Tubulin as loading control in MiaPaCa2, F) Drug sensitivity measured by PicoGreen DNA quantitation, in MiaPaCa2 cells under the indicated culture conditions, and with varying doses of GEM. Each data point represents the mean of 4 independent measurements. G. Panc1 MiaPaca2 cells were treated as indicated in the presence or absence of Nurr1 overexpression (NOE) or expression of empty vector (EV) and whole cell lysates were analyzed by western blots as outlined in the Methods.
ATG7 and ATG12 are the key targets of NURR1
NURR1 regulates specific gene activity and mediates cell survival, migration, invasion and transformation (9–13, 15, 31). In order to identify NURR1-regulated genes that may be involved in drug resistance we performed RNA sequencing and KEGG enrichment pathway analysis in MiaPaCa2 cells, modified by CRISPR/Cas9 gene editing for NURR1 expression and in control cells expressing NURR1 (Figure 3A). To investigate pathways altered by the NURR1 knockout we performed KEGG enrichment pathway analysis and observed that regulation of genes associated with autophagy was enriched (Figure 3B) (Supplemental Table 2). The expression of the autophagic ATG7 and ATG12 genes (18) was downregulated in NURR1.KO cells when compared with NURR1.CTRL cells (Figure 3A, 3C) and therefore the potential roles of ATG7 and ATG12 in NURR1-mediated chemoresistance was further investigated since autophagy plays a key role in cell survival and drug resistance (32). Immunoblot analysis confirmed that NURR1 regulates the expression of ATG7 and ATG12 since knockdown of NURR1 significantly downregulated levels of ATG7 and 12 proteins (Figure 3D). Further analysis by chromatin immunoprecipitation (ChIP) revealed that NURR1 is associated with the ATG7 and 12 promoters (Figure 3E). Nurr1 was also associated with the Sp1 but not the ATG13 (negative control) promoter in a ChIP assay (Fig. 3E). We also subcloned the ATG7 and ATG12 promoter sequences into a luciferase reporter plasmid and after transfection into MiaPaCa2 cells the effects of NURR1 on luciferase activity in was determined. Knockdown of NURR1 expression significantly decreased luciferase activity in both AGT7 and AGT12 promoter-luciferase constructs (Figure 3F) confirming that ATG7 and ATG12 are key autophagic genes regulated by NURR1. Therefore, the role of NURR1, ATG7/ATG12 in gemcitabine resistance was further investigated.
Figure 3. ATG7 and ATG12 are the key targets of NURR1.
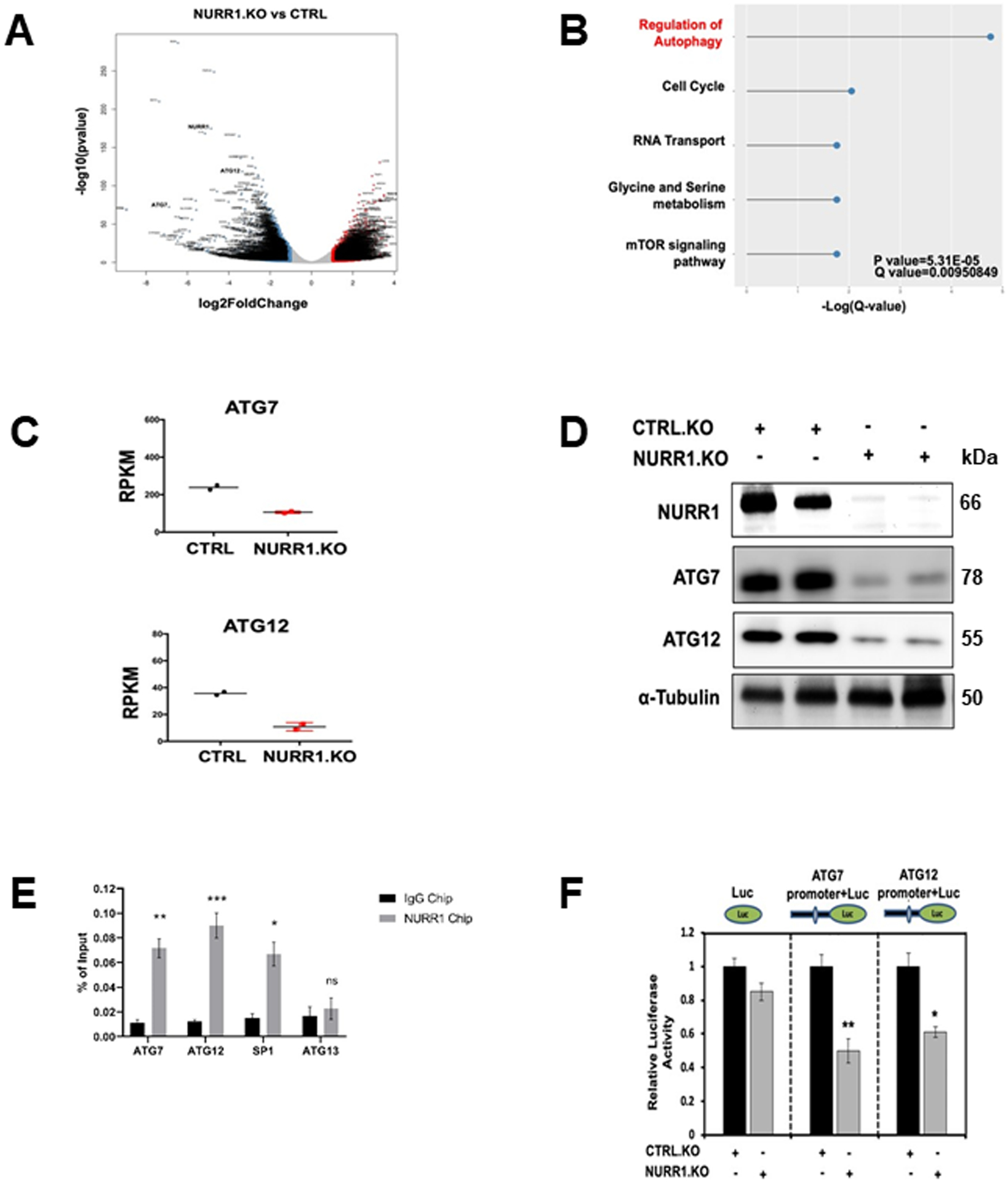
A) Volcano plots of log2fold change vs. −log10(P value) of RNA-Seq data for NURR1.KO vs. control (CTRL), for MiaPaCa2 cells subject to these manipulations. B) KEGG enrichment pathway analysis of NURR1 regulated pathways by comparing gene expressed wild-type MiaPaca2 cells and NRR1 knockout cells. C) RT-qPCR analysis of ATG7and ATG12 expression in CTRL and NURR1.KO cells. Gene expression is normalized to β-actin. Each data point represents the mean ± SEM of four independent experiments (** p < 0.01; *** p < 0.001), D) Immunoblot analysis of NURR1, ATG7 and ATG12 in NURR1.KO cells compared to CTRL MiaPaCa2 cells with α-Tubulin as loading control. E) Q-PCR analysis of a ChIP assay in MiaPaCa2 cells shows a significant increase of NURR1 interactions on the in ATG7 and ATG12 promoters relative to IgG, ** p < 0.01, *** p < 0.001. Results are expressed as the fold enrichment over input DNA. Error bars represent the mean ± SEM of three independent experiments F) Dual luciferase assay of MiaPaCa2 cells with empty vector control (EV) and NURR1.KO. Cell lines were transfected with Renilla luciferase reporter constructs fused with ATG7 or ATG12 promoter, as well as a constitutive firefly luciferase expression construct for 24 h. Renilla luciferase activity was normalized to firefly luciferase activity, and results shown are the average of 4 experiments ± SEM * p < 0.05, ** p < 0.01.
NURR1 induces autophagy, ATG7 and ATG12 in pancreatic cancer cells
Autophagy is a cellular self-degradation process that reduces cellular damage in response to stressful conditions. and the link between NURR1 expression and autophagy was further investigated in MiaPaCa2 cells treated with gemcitabine. Figures 1D and 1E show that gemcitabine induced NURR1 expression in Panc1 and MiaPaCa2 cells and this is also observed in Figure 4A where gemcitabine-induced NURR1 expression is accompanied by an increase in the autophagic marker LC3B-II whereas in NURR-KO cells ± gemcitabine low levels of LC3B-II were expressed (Figure 4A). Next, electron microscopy supports the immunoblot data showing that gemcitabine induced autophagy which is characterized by autophagosomes and autophagic compartments, however, treatment with gemcitabine in combination with the NURR1 antagonist C-DIM12 reduced evidence for autophagy (Figure 4B). Confocal microscopy also demonstrated that gemcitabine induced autophagy and that knockdown of NURR1 or treating the cells with C-DIM12 significantly reduced autophagy marked by decreased punctate staining of LC3-II (Figure 4C). The bar graph representation revealed that LC3-II punctate staining was decreased 2–3-fold when expression of NURR1 was inhibited (Figure 4D) and as a control we show that chloroquine increased LC3-II punctate staining (Fig. 4E). These data suggest that NURR1 is essential for induction of autophagy in pancreatic cancer cells. We next investigated the effects of the NURR1 antagonist C-DIM12 on expression of ATG7 and ATG12 in MiaPaCa2 cells and showed that C-DIM12 decreased expression of ATG7 and ATG12 mRNA levels (Figure 5A). Treatment with C-DIM12 in combination with gemcitabine significantly downregulated expression of ATG7 and ATG12 in immunoblot analysis in MiaPaCa2 and Panc1 cells (Figures 5B and 5C) and in the latter cell line C-DIM12 decreased Nurr1 expression. GEM alone induced PARP cleavage in MiaPac2 cells (Fig. 5D and 5E) whereas knockdown of ATG7 or ATG12 did not induce this response. In contrast, GEM alone and GEM plus knockdown of ATG 7 (Fig. 5D) or ATG12 (Fig. 5E) induced PARP cleavage and p62, and these responses were not enhanced by C-DIM12. The unexpected synergistic interactions of GEM plus knockdown of ATG7 and ATG12 on induction of p62 are being further investigated. We also observed that silencing of ATG7 or ATG12 enhanced the cytotoxicity of GEM (Figure 5F) and these results are consistent with enhanced GEM cytotoxicity after treatment with C-DIM12 or knockdown of Nurr1 (Fig. 2F).
Figure 4. NURR1 induces autophagy in pancreatic cancer.
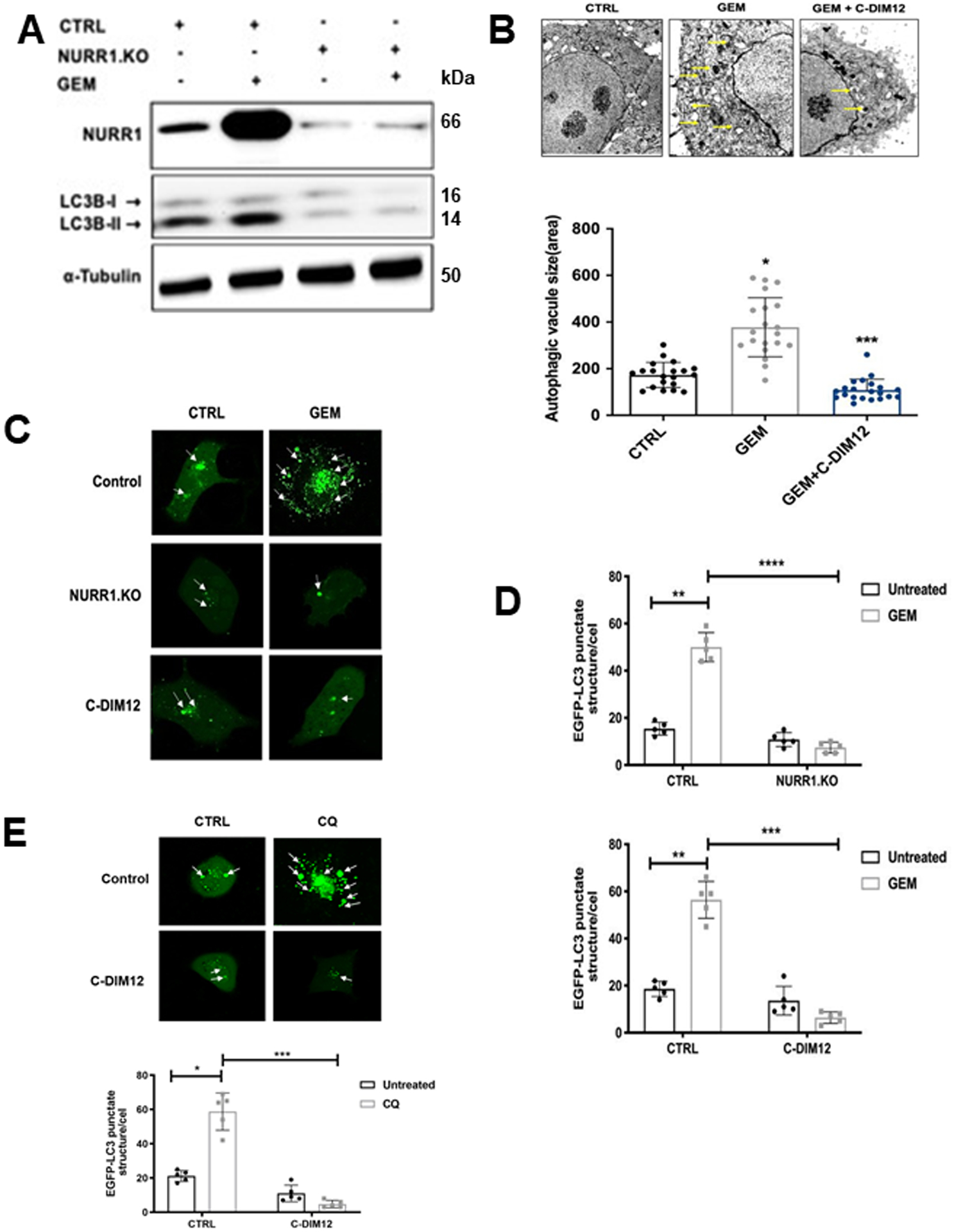
A) Immunoblot analysis of NURR1 and LC3-I and LC3-II in NURR1.KO cells compared to control (EV) MiaPaCa2 cells with α-Tubulin as loading control, B) MiaPaCa2 cells were treated with gemcitabine or C-DIM12 and autophagosomes were observed using transmission electron microscopy. The yellow arrow head indicates the autophagosomes and autolysosomes. The size of autophagosome was analyzed by measuring the area of autophagic vacuoles, C) Confocal microscopy images of MiaPaCa2 cells for EGFP-LC3 punctate staining in control (EV) and NURR1.KO cells treated with gemcitabine and C-DIM12 for 24 hours, white arrows indicate punctate EGFP-LC3 structures, D) and as a control experiment we show that chloroquine induces punctate screening E) Bar graphs representation of the average EGFP-LC3 punctate in the cells. * p < 0.05, ** p < 0.01, **** p < 0.0001. (3 independent experiments performed, and at least 10 images per slide were analyzed for each condition). Scale bar, 10 um. Magnification 40x.
Figure 5. NURR1 regulates the expression of ATG7 and ATG12.
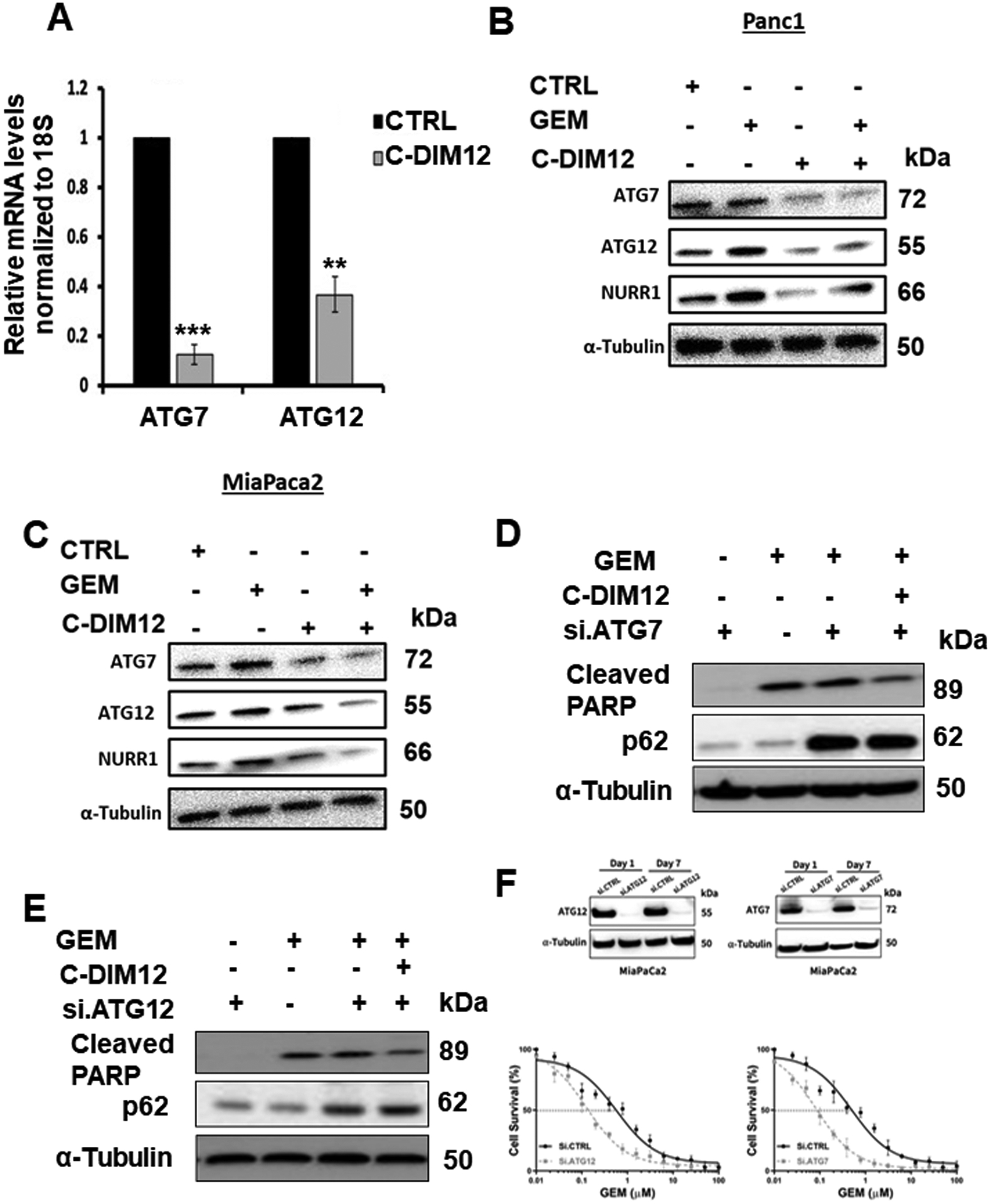
A) qPCR analysis of ATG7 and ATG12 expression after treatment with C-DIM12, ** p < 0.01, *** p < 0.001, B) Immunoblot analysis of ATG7 and ATG12 and Cleaved PARP in gemcitabine and C-DIM12 treated MiaPaCa2 cells, C) Immunoblot analysis of ATG7 and ATG12 in gemcitabine and C-DIM12 treated Panc1 cells. D/E. Immunoblot analysis of Cleaved PARP and p62 in the presence of si. ATG7 or si. ATG12 MiapaCa2 cells in the presence or absence of treatment with gemcitabine, C-DIM12, and combination of both. α-Tubulin serves as a loading control. F. Effects of ATG7 and ATG12 knockdown on GEM-induced survival of MiaPaca2 cells was determined as outlined in Figure 2F. Each data point represents the mean of 4 independent measurements.
In vivo confirmation of NURR1/ATG7/ATG12 axis
In athymic nude mice bearing MiaPaCa2 cells (Ctrl and NURR1-KO) gemcitabine alone decreased tumor volume (Figure 6A, 6B), and both tumor volumes and weights were decreased in mice bearing NURR1-KO cells alone and after treatment with gemcitabine. Compared with the vehicle treated group, the combination of gemcitabine, and NURR1 knockdown also extended the median survival of the mice from 20 to 97 days (Figure 6C). Both ATG7 and ATG12 mRNA levels were lower in tumors derived from mice bearing NURR1-KO cells and treated with gemcitabine compared to control cells (Figure 6D). A comparable set of data was obtained in tumors from mice bearing wild type MiaPaCa2 cells and treated with vehicle, gemcitabine, C-DIM12 and their combination. Both gemcitabine and C-DIM12 and their combination inhibited tumor growth (Figures 6E, 6F) to a similar extent, however, post-treatment survival (Fig. 6G) shows that GEM plus C-DIM12 was more effective than either compound alone. GEM plus C-DIM12 also inhibited ATG7 and ATG12 mRNA levels (Figure 6H) and this corresponds to results observed in the in vitro studies (Figure 5).
Figure 6. Gemcitabine in combination with C-DIM12 inhibits tumor growth in vivo.
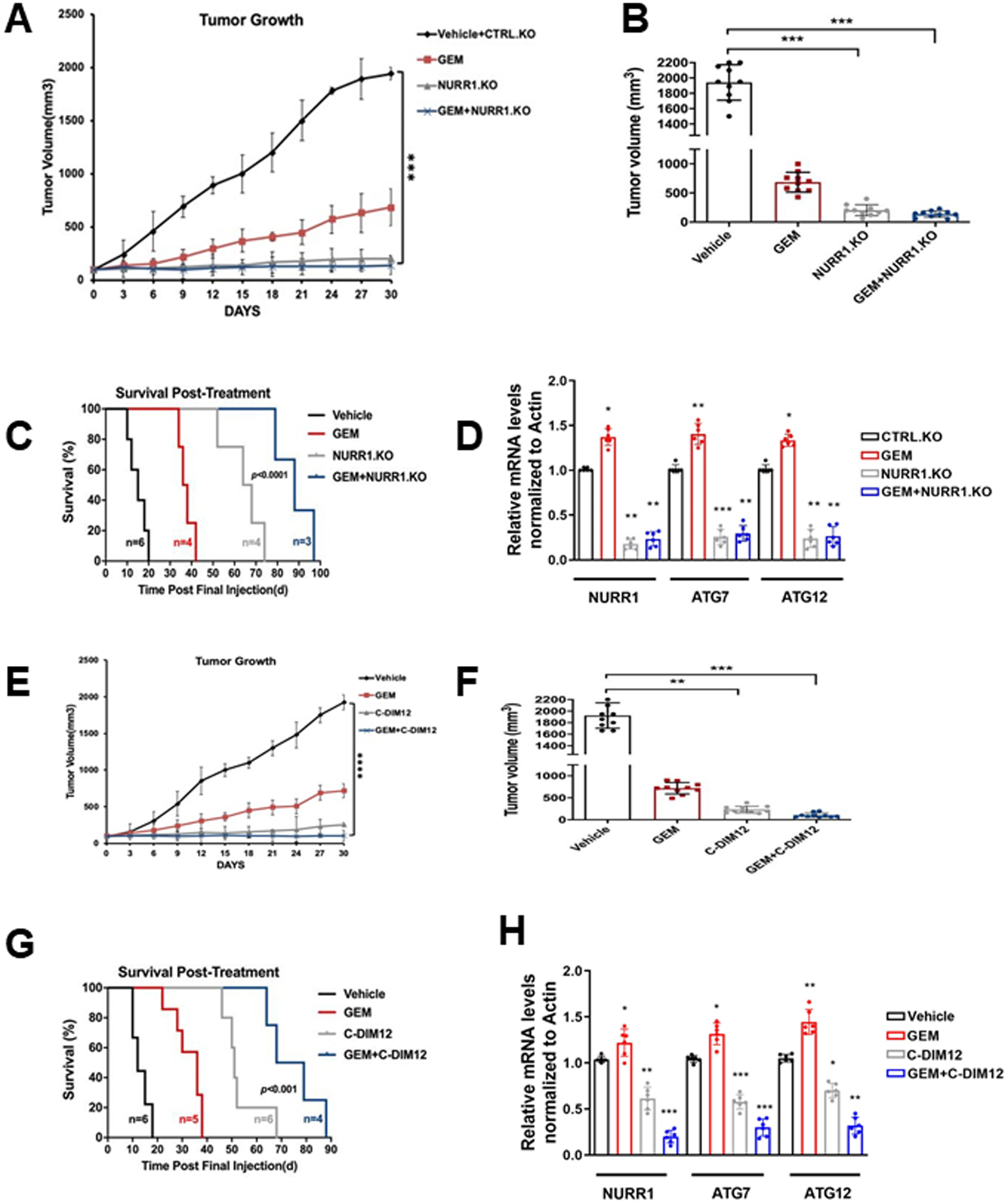
A) Tumor growth curves of MiaPca2 CTRL and NURR1.KO xenografts in nude mice treated with vehicle, GEM, or combined NURR1.KO and GEM for 30 days, when control tumors reached 100 mm3 in size. Tumor size was assessed every 3 days using digital calipers. (n=5 per group, two tumors per mouse) (***p < 0.001). Mean and standard deviation are shown, B) Average tumor volume of MiaPaca2 (vehicle, GEM, NURR1.KO and combination of both) xenografts at the end of the experiment (day 30) (n=10 tumors per group), ***p < 0.001, C) Survival was assessed after gemcitabine cessation. Mice were removed from the group when tumors achieved a volume of 1500 mm3, statistical significance was determined by Log-rank test. D) Tumor RNA was extracted from vehicle or combination of GEM and NURR1.KO xenografts and analyzed for mRNA expression of NURR1, ATG7, or ATG12. Normalized values are shown as mean ± SEM (n = 6 tumors per group; **p < 0.01), E) Tumor growth curves of MiaPca2 xenografts in nude mice treated with vehicle, GEM, C-DIM or combined for 30 days (n=5 per group, two tumors per mouse) (**** p < 0.0001), F) Average tumor volume of MiaPacA2 (vehicle, GEM, C-DIM and combination of both) xenografts at the end of the experiment (day 30) (n=10 tumors per group), ** p < 0.01, ***p < 0.001, G) Survival was assessed after treatment cessation. Mice were removed from the group when tumors achieved a volume of 1500 mm3, statistical significance was determined by Log-rank test. H) Tumor RNA was extracted from vehicle or combination of GEM and C-DIM xenografts and analyzed for mRNA expression of NURR1, ATG7, or ATG12. β-actin was used as normalization control. Normalized values are shown as mean ± SEM (n = 6 tumors per group; *p < 0.05**p < 0.01).
Prognostic significance of ATG7 and 12 and association of NURR1 and ATG7 and 12 expressions in clinical specimens
To study the prognostic significance of ATG7 and 12 in PDA we examined the Cancer Genome Atlas (TCGA) which showed a significant increase in expression of ATG7 in pancreatic tumors (T=179) compared to normal tissue samples (N=171) (Figure 7A). Kaplan-Meier Analysis survival curve showed that high expression of ATG7 correlated with poor survival rate of PDA patients (Figure 7B). This analyses also demonstrated a significant increase in the expression level of ATG12 in pancreatic tumors, (T=179) compared to normal tissue samples (N=171) (Figure 7C) and Kaplan-Meier analysis shows that high expression of ATG12 corresponds to poor patient survival rate (Figure 7D). We also investigated the correlation between the expression of NURR1 and ATGs (ATG7 and 12) in pancreatic tumor samples by performing correlation analysis. NURR1 expression was strongly correlated with ATG7 expression (R=0.2, p-value=6.2e-05) (Supplemental Figure 2C) and ATG12 expression (R=0.31, p-value=2e-05) (Supplemental Figure 2D) in PDA patient samples. Together, these data indicate prognostic significance of ATG7 and ATG12 in PDA and their correlation with NURR1 expression. These data coupled with mechanistic studies suggest a role for NURR1 in gemcitabine-induced resistance which is link to enhanced autophagy and the potential clinical applications of combination therapies using gemcitabine and NURR1 antagonists (Figure 7E). The linkage between NURR1 and ATG7/12 has been clearly demonstrated, however, the possible interactions with other important autophagic factors (e.g.: TFEB) has not been determined and is currently been investigated.
Figure 7. Prognostic significance of ATG7 and ATG12 in PDA.
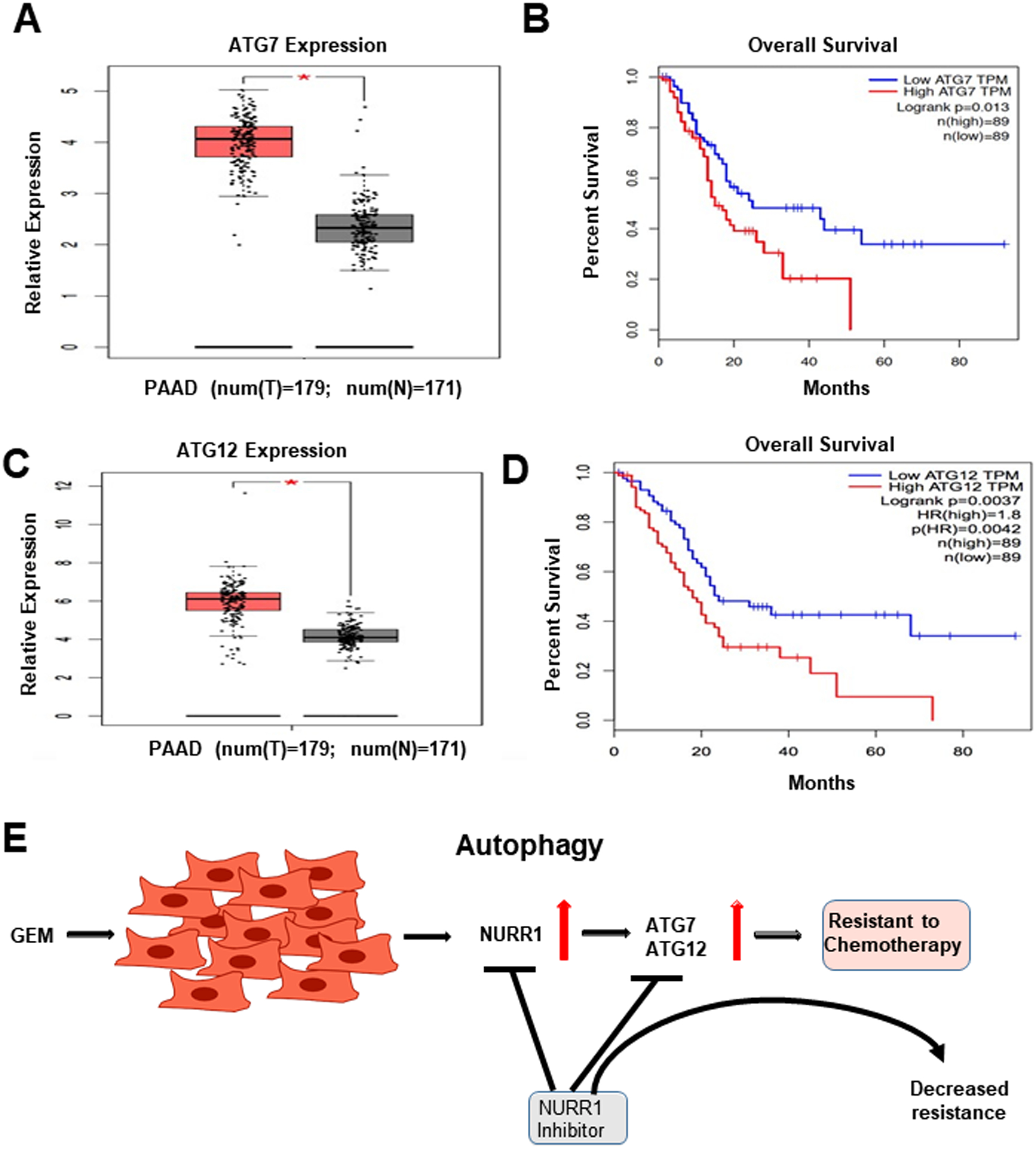
A) ATG7 expression in PDA patient tumor samples as compared to normal pancreas tissue using TCGA database, * p <0.05, B) Kaplan Meier analysis to demonstrate the prognostic significance of ATG7 in PDA patient, p=0.013, C) ATG12 expression in PDA patient tumor samples as compared to normal pancreas tissue using TCGA database, * p <0.05, D) Kaplan Meier analysis to demonstrate the prognostic significance of ATG12 in PDA patient, p=0.0042). E. Model for the role of NURR1 in gemcitabine drug resistance in PDAC and effects of NURR1 antagonists to decrease drug resistance.
Discussion
Pancreatic ductal adenocarcinoma is usually detected in later stages and PDA patients have low survival rates and current treatment options are limited in their effectiveness. Improvements in PDA patient survival will require development of validated biomarkers that appear early in the formation of these tumors and also new mechanism-based drugs and drug combinations that increase efficacy and decrease drug resistance. We initially examined the TCGA database and observed that expression of the orphan nuclear receptor NURR1 was more highly expressed in pancreatic tumors compared to the normal pancreas and pancreatic cancer patients expressing high levels of NURR1 exhibited decreased survival (Figure 1) and this was similar to our recent studies on the expression and prognostic value of NURR1 in glioblastoma (15). The functions of NURR1 have been investigated in multiple tumors (9, 33–38) and results of knockdown or overexpression studies have characterized this receptor as a pro-oncogenic factor that regulates cancer cell proliferation, survival, migration and invasion. Although an endogenous ligand for NURR1 has not been identified, a recent study shows that the bis-indole derived C-DIM12, a known NURR1 ligand, acted as a receptor antagonist in glioblastoma cells and inhibited cell growth and invasion and increased apoptosis (15). Another report showed that overexpression of NURR1 in squamous cell carcinoma cells increased resistance to 5-FU treatment (17) suggesting a possible role for NURR1 in drug resistance. Gemcitabine has replaced 5-FU for treatment of pancreatic cancer and based on the prognostic and functional characteristics of NURR1 in cancer cells this study focused on determining the functions of NURR1 in pancreatic cancer and its role in gemcitabine- resistance.
Knockout of NURR1 in pancreatic cancer cells or treatment with C-DIM12 induced apoptosis and inhibited proliferation (Figure 2). Moreover, in athymic nude mice bearing MiaPaCa2 (NURR1+/+) or NURR1-KO cells it was clear that loss of NURR1 or treatment with C-DIM12 inhibited tumor growth and enhanced survival (Figure 6). These results confirmed the pro-oncogenic activity of NURR1 in pancreatic cancer cells as previously observed in other cancer cell lines (15) and demonstrated that the NURR1 antagonist C-DIM12 was an effective anticancer agent that blocked NURR1-mediated responses.
We also investigated the role of NURR1 in drug resistance and observed that treatment of pancreatic cancer cells with gemcitabine resulted in induction of NURR1 (Figure 1). Thus, although gemcitabine alone induced apoptosis and inhibited growth of pancreatic cancer cells and tumors (Figure 1, 6) this was accompanied by induction of NURR1 which exhibits tumor promoter-like activity. This counter-intuitive effect of gemcitabine could be a component of drug resistance and this was confirmed in combination treatments of cells and mice with gemcitabine alone and in combination with NURR1 knockdown or receptor antagonist (C-DIM12) (Figures 1, 6). Results of the in vitro and in vivo effects of these combination therapies are complementary and demonstrate the NURR1 inactivation or treatment with C-DIM12 enhanced the effectiveness of gemcitabine thus demonstrating that induction of NURR1 by gemcitabine is associated with drug resistance.
The mechanism of NURR1 as a drug resistant factor was further investigated by RNAseq in wild-type and NURR1-KO cells and this resulted in identification of several genes/ pathways regulated by NURR1; however, the major pathway was associated with changes in expression of genes involved in autophagy (Figure 3) (39–43). Since autophagy has previously been linked to cytoprotection in colon cancer cells (21) we examined this gene set and identified two genes, namely ATG7 and ATG12, that were not only regulated by NURR1 (Figures 5A and 5B) but also exhibit clinical characteristics similar to that observed for NURR1 in pancreatic cancer patients (Figures 1 and 7). These results confirm that NURR1 exhibits pro-oncogenic like activity in pancreatic cancer cells and this receptor also plays a role in gemcitabine- induced drug resistance (Fig. 7E). These observations coupled with the anticarcinogenic effects of C-DIM12 suggest that the effectiveness of therapeutic regimens including gemcitabine for treatment of PDAC can be significantly enhanced using combination therapies that include a NURR1 antagonist such as C-DIM12.
Supplementary Material
Significance.
Gemcitabine induces NURR1-dependent ATG7 and ATG12 cytoprotective autophagy in PDA cells which can be reversed by NURR1 antagonists.
Funding:
This work was supported by the department of defense; DOD-W81XWH-18-0592 (M. Zarei) and the National Institutes of Health P30-ES029067 (S. Safe).
Abbreviations:
- PDA
Pancreatic ductal adenocarcinoma
- NR4A2(NURR1)
Nuclear receptor 4A2
- ATG7
Autophagy-related Protein 7
- ATG12
Autophagy-related Protein 12
- cPARP
cleaved PARP
- RT-qPCR
quantitative reverse transcription polymerase chain reaction
- TCGA
The Cancer Genome Atlas
- GEM
gemcitabine
- ChIP
Chromatin Immunoprecipitation
Footnotes
Disclosure of Conflicts of Interest: There are no other conflicts of interests to declare.
Data availability statement:
RNAseq files will be deposited in Gene Expression Omnibus (GEO).
References
- 1.McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2018;24(43):4846–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orth M, Metzger P, Gerum S, Mayerle J, Schneider G, Belka C, et al. Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol. 2019;14(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27(33):5513–8. [DOI] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie Z, Zhang Y, Jin C, Fu D. Gemcitabine-based chemotherapy as a viable option for treatment of advanced breast cancer patients: a meta-analysis and literature review. Oncotarget. 2018;9(6):7148–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capurso G, Sette C. Drug resistance in pancreatic cancer: New player caught in act. EBioMedicine. 2019;40:39–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie L, Xia L, Klaiber U, Sachsenmaier M, Hinz U, Bergmann F, et al. Effects of neoadjuvant FOLFIRONOX and gemcitabine-based chemotherapy on cancer cell survival and death in patients with pancreatic ductal adenocarcinoma. Oncotarget. 2019;10(68):7276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ke N, Claassen G, Yu DH, Albers A, Fan W, Tan P, et al. Nuclear hormone receptor NR4A2 is involved in cell transformation and apoptosis. Cancer Res. 2004;64(22):8208–12. [DOI] [PubMed] [Google Scholar]
- 10.Komiya T, Yamamoto S, Roy A, McDonald P, Perez RP. Drug screening to target nuclear orphan receptor NR4A2 for cancer therapeutics. Transl Lung Cancer Res. 2017;6(5):600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohan HM, Aherne CM, Rogers AC, Baird AW, Winter DC, Murphy EP. Molecular pathways: the role of NR4A orphan nuclear receptors in cancer. Clin Cancer Res. 2012;18(12):3223–8. [DOI] [PubMed] [Google Scholar]
- 12.Riggins RB, Mazzotta MM, Maniya OZ, Clarke R. Orphan nuclear receptors in breast cancer pathogenesis and therapeutic response. Endocr Relat Cancer. 2010;17(3):R213–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitagawa H, Ray WJ, Glantschnig H, Nantermet PV, Yu Y, Leu CT, et al. A regulatory circuit mediating convergence between Nurr1 transcriptional regulation and Wnt signaling. Mol Cell Biol. 2007;27(21):7486–96. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Nordzell M, Aarnisalo P, Benoit G, Castro DS, Perlmann T. Defining an N-terminal activation domain of the orphan nuclear receptor Nurr1. Biochem Biophys Res Commun. 2004;313(1):205–11. [DOI] [PubMed] [Google Scholar]
- 15.Karki K, Li X, Jin UH, Mohankumar K, Zarei M, Michelhaugh SK, et al. Nuclear receptor 4A2 (NR4A2) is a druggable target for glioblastomas. J Neurooncol. 2020;146(1):25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Tjalkens RB, Shrestha R, Safe S. Structure-dependent activation of gene expression by bis-indole and quinoline-derived activators of nuclear receptor 4A2. Chem Biol Drug Des. 2019;94(4):1711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shigeishi H, Higashikawa K, Hatano H, Okui G, Tanaka F, Tran TT, et al. PGE(2) targets squamous cell carcinoma cell with the activated epidermal growth factor receptor family for survival against 5-fluorouracil through NR4A2 induction. Cancer Lett. 2011;307(2):227–36. [DOI] [PubMed] [Google Scholar]
- 18.Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett. 2010;584(7):1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorente J, Velandia C, Leal JA, Garcia-Mayea Y, Lyakhovich A, Kondoh H, et al. The interplay between autophagy and tumorigenesis: exploiting autophagy as a means of anticancer therapy. Biol Rev Camb Philos Soc. 2018;93(1):152–65. [DOI] [PubMed] [Google Scholar]
- 20.Yun CW, Lee SH. The Roles of Autophagy in Cancer. Int J Mol Sci. 2018;19(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong L, Liu Z, Ouyang G, Lin L, Huang H, Kang H, et al. Autophagy inhibition enhances photocytotoxicity of Photosan-II in human colorectal cancer cells. Oncotarget. 2017;8(4):6419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng CY, Liu JC, Wang JJ, Li YH, Pan J, Zhang YR. Autophagy inhibition increased the anti-tumor effect of cisplatin on drug-resistant esophageal cancer cells. J Biol Regul Homeost Agents. 2017;31(3):645–52. [PubMed] [Google Scholar]
- 23.Zarei M, Giannikou K, Du H, Liu HJ, Duarte M, Johnson S, et al. MITF is a driver oncogene and potential therapeutic target in kidney angiomyolipoma tumors through transcriptional regulation of CYR61. Oncogene. 2021;40(1):112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Lee SO, Safe S. Structure-dependent activation of NR4A2 (Nurr1) by 1,1-bis(3’-indolyl)-1-(aromatic)methane analogs in pancreatic cancer cells. Biochemical pharmacology. 2012;83(10):1445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karki K, Harishchandra S, Safe S. Bortezomib Targets Sp Transcription Factors in Cancer Cells. Molecular pharmacology. 2018;94(4):1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA, et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer research. 2014;74(4):1128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zarei M, Lal S, Parker SJ, Nevler A, Vaziri-Gohar A, Dukleska K, et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer research. 2017;77(16):4460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zarei M, Lal S, Vaziri-Gohar A, O’Hayer K, Gunda V, Singh PK, et al. RNA-Binding Protein HuR Regulates Both Mutant and Wild-Type IDH1 in IDH1-Mutated Cancer. Molecular cancer research : MCR. 2019;17(2):508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beard JA, Tenga A, Hills J, Hoyer JD, Cherian MT, Wang YD, et al. The orphan nuclear receptor NR4A2 is part of a p53-microRNA-34 network. Sci Rep. 2016;6:25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han YF, Cao GW. Role of nuclear receptor NR4A2 in gastrointestinal inflammation and cancers. World J Gastroenterol. 2012;18(47):6865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji L, Gong C, Ge L, Song L, Chen F, Jin C, et al. Orphan nuclear receptor Nurr1 as a potential novel marker for progression in human pancreatic ductal adenocarcinoma. Exp Ther Med. 2017;13(2):551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komiya T, Coxon A, Park Y, Chen WD, Zajac-Kaye M, Meltzer P, et al. Enhanced activity of the CREB co-activator Crtc1 in LKB1 null lung cancer. Oncogene. 2010;29(11):1672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holla VR, Mann JR, Shi Q, DuBois RN. Prostaglandin E2 regulates the nuclear receptor NR4A2 in colorectal cancer. The Journal of biological chemistry. 2006;281(5):2676–82. [DOI] [PubMed] [Google Scholar]
- 37.Han Y, Cai H, Ma L, Ding Y, Tan X, Chang W, et al. Expression of orphan nuclear receptor NR4A2 in gastric cancer cells confers chemoresistance and predicts an unfavorable postoperative survival of gastric cancer patients with chemotherapy. Cancer. 2013;119(19):3436–45. [DOI] [PubMed] [Google Scholar]
- 38.Han Y, Cai H, Ma L, Ding Y, Tan X, Liu Y, et al. Nuclear orphan receptor NR4A2 confers chemoresistance and predicts unfavorable prognosis of colorectal carcinoma patients who received postoperative chemotherapy. European journal of cancer. 2013;49(16):3420–30. [DOI] [PubMed] [Google Scholar]
- 39.Sun K, Deng W, Zhang S, Cai N, Jiao S, Song J, et al. Paradoxical roles of autophagy in different stages of tumorigenesis: protector for normal or cancer cells. Cell Biosci. 2013;3(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J, Li Y, Tian Z, Hua X, Gu J, Li J, et al. ATG7 Overexpression Is Crucial for Tumorigenic Growth of Bladder Cancer In Vitro and In Vivo by Targeting the ETS2/miRNA196b/FOXO1/p27 Axis. Mol Ther Nucleic Acids. 2017;7:299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai S, Liu Z, Yao J, Patel N, Chen J, Wu Y, et al. Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). The Journal of biological chemistry. 2013;288(13):9165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun S, Wang Z, Tang F, Hu P, Yang Z, Xue C, et al. ATG7 promotes the tumorigenesis of lung cancer but might be dispensable for prognosis predication: a clinicopathologic study. Onco Targets Ther. 2016;9:4975–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu J, Tian Z, Li Y, Hua X, Zhang D, Li J, et al. ATG7 Promotes Bladder Cancer Invasion via Autophagy-Mediated Increased ARHGDIB mRNA Stability. Adv Sci (Weinh). 2019;6(8):1801927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq files will be deposited in Gene Expression Omnibus (GEO).