

Abstract
Diabetic retinopathy (DR) is the leading cause of vision loss in working adults in developed countries. The disease traditionally classified as a microvascular complication of diabetes is now widely recognized as a neurovascular disorder resulting from disruption of the retinal neurovascular unit (NVU). The NVU comprising retinal neurons, glia and vascular cells coordinately regulates blood flow, vascular density and permeability to maintain homeostasis. Disturbance of the NVU during DR can lead to vision-threatening clinical manifestations. A limited number of signaling pathways have been identified for intercellular communication within the NVU, including vascular endothelial growth factor (VEGF), the master switch for angiogenesis. VEGF inhibitors are now widely used to treat DR, but their limited efficacy implies that other signaling molecules are involved in the pathogenesis of DR. By applying a novel screening technology called comparative ligandomics, we recently discovered secretogranin III (Scg3) as a unique DR-selective angiogenic and vascular leakage factor with therapeutic potential for DR. This review proposes neuron-derived Scg3 as the first diabetes-selective neurovascular regulator and discusses important features of Scg3 inhibition for next-generation disease-targeted anti-angiogenic therapies of DR.
Keywords: Diabetic retinopathy, neurovascular unit, neurovascular crosstalk, VEGF, Scg3, ligandomics
Introduction
Diabetic retinopathy (DR) is the leading cause of vision impairment in working adults in developed countries with a prevalence that parallels the incidence of diabetes [1]. About one third of diabetic patients develop DR, and the diabetic population presently estimated at 463 million worldwide is rapidly expanding [2]. DR is traditionally considered to be the most common microvascular complication of diabetes. The vascular manifestations of DR, including capillary loss, microaneurysms, venous beading, retinal vascular leakage, diabetic macular edema (DME), proliferative DR with pathological retinal neovascularization (RNV), retinal and vitreous hemorrhage, can be conveniently detected by ophthalmic examinations through the transparent cornea (Fig. 1) [3]. If untreated, these microvascular complications typically lead to progressive vision impairment. However, as early as the 1960s, post-mortem histopathological examinations found that neuronal degeneration, in addition to microvascular issues, played an important role in DR pathogenesis [4, 5]. Subsequently, irrefutable evidence supports neurovascular disease with both microvascular and neurodegenerative complications as central components of DR etiology [6].
Fig. 1.

Stages of diabetic retinopathy (DR) progression. a. No apparent DR at the early stage of diabetes. No abnormalities are present in the retina and macula. b. Non-proliferative diabetic retinopathy (NPDR) in the early stage of symptomatic DR. Microaneurysms, small retinal hemorrhage spots and cotton wool spots are detectable by fundoscopic examination. According to the severity, it can be further divided into mild, moderate and severe NPDR. c. Proliferative diabetic retinopathy (PDR) in the advanced stage of DR. Severe NPDR may progress toward PDR with the hallmark of retinal neovascularization that could lead to retinal and/or vitreous hemorrhage. Diabetic macular edema (DME) can occur during either NPDR or PDR.
Retinal Structure and Vasculature
Retinal cells are organized and structured into multiple layers and vascular plexuses with distinct demarcation and functions to sense light, transduce visual signals and maintain homeostasis and a robust metabolism (Fig. 2) [7]. At the inner most layer are the nerve fibers that derive from the retinal ganglion cells (RGCs) below. Beneath the RGCs is the inner plexiform layer (IPL), followed by the inner nuclear layer (INL) that comprises bipolar, horizontal and amacrine cells. Beneath the INL are the outer plexiform layer (OPL), the outer nuclear layer (ONL) of the photoreceptors, and photoreceptor inner segments (PIS) and outer segments (POS). The outermost layer is the retinal pigment epithelium (RPE) and Bruch’s membrane. The functional roles of these layers with different cell types are discussed extensively in recent reviews [7].
Fig. 2.
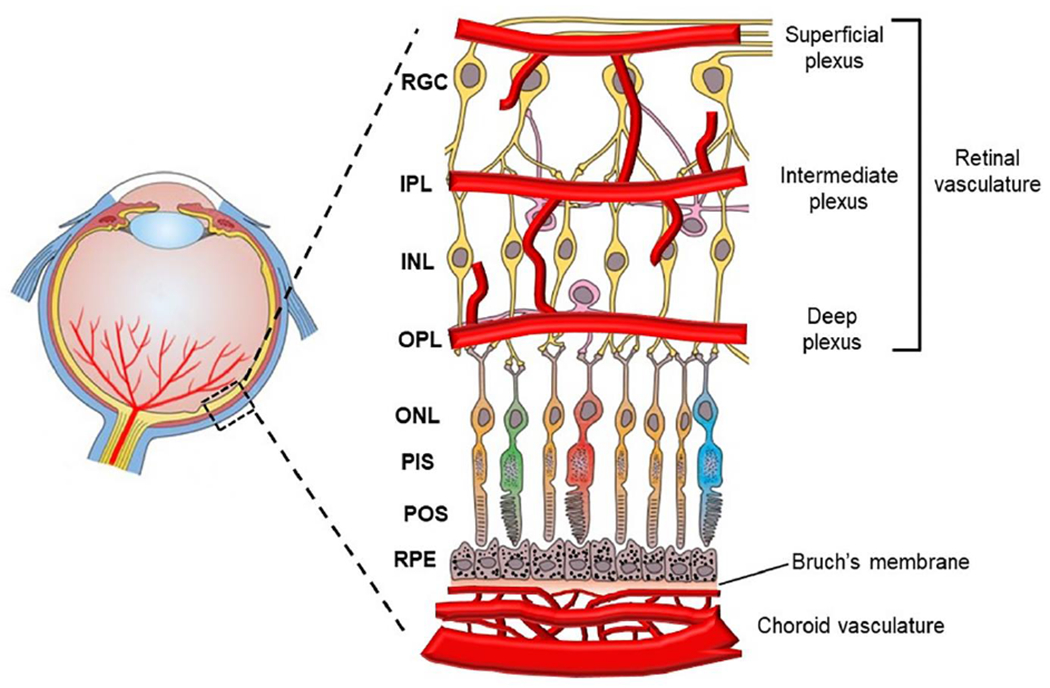
Retinal structure and vasculature. The retina is composed of multiple distinct layers, including the ganglion cells (RGC), inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), outer nuclear layer (ONL), photoreceptor inner segments (PIS), photoreceptor outer segments (POS), retinal pigment epithelial (RPE) cells and Bruch’s membrane. The retina is supplied with two sets of vessels, including retinal and choroidal vasculatures. The retinal vasculature comprises superficial, intermediate and deep plexuses that are interconnected to vascularize the upper half of the retina. The bottom half of the retina is avascular.
The retina is supplied by two vascular systems: the central retinal artery supports the inner retinal layers and the choriocapillaris supplies the RPE and outer retina (Fig. 2) [8]. The central retinal artery enters the retina from the back of the eye through the center of the optical nerve head and branches out toward the peripheral retina on the retinal surface. This superficial vascular plexus further penetrates into the upper half of the retina to form the intermediate and deep plexuses. These three layers of plexuses vascularize the RGC, IPL and INL layers. Because the bottom half of the retina is not vascularized, the photoreceptors obtain oxygen and nutrients through diffusion from the retinal and choroidal vasculatures.
Neurovascular Unit
The NVU is composed of a structural and functional complex made up of neurons that include the RGCs, bipolar, amacrine and horizontal cells; non-neuronal glia, consisting of Müller cells and astrocytes and microglia; and vascular cells, comprising endothelial cells (ECs) and pericytes [6, 9, 10]. Within the NVU, cells, including pericytes, ECs and glia, can communicate directly through physical interactions, while cells without physical connections communicate at a distance via secreted soluble ligands and/or exosomes (Fig. 3a). Such intercellular signaling and communications pathways between NVU cells support complex and dynamically interacting networks that critically regulate blood flow, vascular outgrowth and permeability in response to the similarly dynamic requirements of retinal neurons. By supplying oxygen and nutrients, recycling neurotransmitters and removing metabolic wastes, such elaborate fine-tuning of vascular function is critical to maintain retinal homeostasis.
Fig. 3.

The neurovascular units (NVU) and Scg3 in healthy and diabetic retina. The NVU is composed of retinal neurons (e.g., retinal ganglion, bipolar, horizontal amacrine cells), glial cells (e.g. Müller cells, astrocytes and microglia) and vascular cells (e.g., endothelial cells and pericytes). a. The NVU in healthy retina. Scg3 is secreted via neurotransmitter vesicles, but its receptor is not expressed on healthy endothelium. b. The NVU in diabetic retina. Both Scg3 and VEGF stimulates retinal leakage and retinal neovascularization but by different mechanisms. Secreted Scg3 is expressed at the similar levels in the diabetic and healthy retina and binds to its receptor, which is present only on diabetic endothelia, to selectively promote angiogenesis and vascular leakage. In contrast, VEGF is markedly induced in the diabetic retina and binds its receptors that are minimally upregulated on diabetic endothelia.
Arterioles and capillaries are primarily responsible for the dynamic autoregulation of blood flow through the inner retina. Microcapillaries also play important roles in regulating local circulation and permeability, and the tight junctions of ECs in the inner lining of vessels form the blood-retina barrier (BRB) that is critical to prevent potentially toxic solutes from the blood to access the NVU compartment. Pericytes with cytoplasmic processes wrap around blood microvessels and modulate local blood flow [8, 11]. Pericytes and ECs jointly create a basement membrane as part of the extracellular matrix and respond in concert to signaling cues from neurons and glia, adjusting blood flow according to changing metabolic demands.
The photoreceptors and retinal neurons that support phototransduction and neurotransmission for vision have extremely high energy demands, and blood flow through the retinal vasculature is determined directly by neuronal activity [12]. Increased metabolic demands of retinal neurons must be met by correspondingly increased and rapidly adjustable supplies of oxygen and nutrients as well as removal of metabolic wastes and recycling of neurotransmitters. Therefore, blood flow through the NVU is subjected to elaborate regulation at multiple levels and is highly responsive to neuronal activity and metabolic demands.
Müller cells are principal glial cells of the retina, form a pan-retinal network and primarily reside in the retinal inner nuclear layer [13]. Müller cells provide elaborate support and protection for neurons, are typically in close contact with blood vessels and extend processes that envelope neurons, synapses and microvessels from the inner to the outer limiting membranes, providing additional avenues for intimate neurovascular communication [14]. Glial astrocytes also wrap around blood vessels of the retina, make contacts with synapses and play important roles in maintaining the BRB [15]. Microglia, the resident macrophages of the retina, normally function as phagocytes to clear metabolic products and tissue debris downstream of synaptic remodeling and apoptosis. During DR, activated microglia release excess pro-inflammatory mediators, including cytokines, chemokines, caspases and glutamate [16]. These mediators induce additional apoptosis of retinal neurons and ECs and also compromise the function of the BRB.
Diabetic Retinopathy
Based on clinical manifestations, DR is typically classified into two categories: an early stage of non-proliferative DR (NPDR) and an advanced stage of proliferative DR (PDR) [17]. NPDR is characterized by microaneurysms, increased vascular permeability, thickening of the basement membrane, loss of pericytes, capillary occlusion, microcapillary loss, cotton wool spots, retinal hemorrhages and exudates. Vascular leakage with a compromised BRB may lead to DME, the most common cause of vision loss in patients with DR. DME causes serous retinal detachment, distortion of vision images and decreased visual acuity and can occur at any stage during DR progression. NPDR frequently progresses to PDR with pathological RNV, the hallmark feature of PDR. The abnormal neovessels generated in the PDR condition are leaky and fragile and may bleed into the vitreous (vitreous hemorrhage), leading to vision loss and tractional retinal detachment (Fig. 1).
In contrast to clinically detectable microvascular complications, neurodegeneration during DR is more subtle. Diabetes-induced neuroglial degeneration includes reactive gliosis (glial activation), diminished function of retinal neurons and increased neuronal apoptosis that can be observed in diabetic animal models and in the retina of diabetic donors before overt microvascular complications [18, 19]. In DR with glial activation, retinal neurodegeneration share some common pathways with brain neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases [20]. Stressed neurons release molecular cues that promote local circulation and may confer early vascular remodeling. Astrocytes and Müller cells are essential to maintain retinal homeostasis by regulating retinal blood flow, water balance in the neural parenchyma and vascular permeability [21]. Gliosis is typically associated with increased expression of VEGF and pro-inflammatory cytokines and induces BRB dysfunction. Thus, reactive gliosis may play a pivotal role in damage to retinal neurons and link the neurodegenerative process with microvascular complications.
Regulation of DR Vessels by Neural Cues
Retinal neurons and glial cells secrete molecular cues that regulate blood flow, vascular permeability and revascularization in response to physiological, metabolic and stress-related stimuli. Whereas microcapillaries normally support the function and survival of retinal neurons by providing precisely calibrated oxygen and nutrient delivery and waste removal, such a balance can be disturbed during the progress of DR. Adaptive molecular cues that increase blood and nutrient supplies and promote neuron survival in the early stages of the disease can switch to become maladaptive and exacerbate vision impairment as DR severity progresses. Such molecular cues, including VEGF, placental growth factor, semaphoring 3A, platelet-derived growth factor (PDGF), pigment epithelium-derived factor, norrin, prostanoids, hypoxia and insufficient nutrients, contribute importantly to DR pathogenesis, as discussed in previous reviews [6, 9, 22, 23]. Other mediators, such as extracellular glutamate, oxidative stress, pigment epithelial-derived factor (PEDF), somatostatin, neuroprotectin D1, brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF) and nerve growth factor (NGF), may also involve in neurodegenerative process in DR [24]. Here we focus on the roles of VEGF and Scg3 in DR pathogenesis.
VEGF
In mammals, the VEGF family is composed of five related growth factors: VEGFA (the prototype), VEGFB, VEGFC, VEGFD and placental growth factor (PlGF) [25]. VEGFA is also known as vascular permeability factor (VPF) [26]. The complexity of the VEGF family is further increased by alternative splicing. For example, human VEGFA is alternatively spliced into VEGF111, VEGF121, VEGF145, VEGF165, VEGF189 and VEGF206 with the numbers indicating amino acid residues [25]. These isoforms display different interactions with VEGF coreceptor neurophilin-1 and heparin sulphate, affecting VEGF activity and bioavailability. The prototypical VEGF165 with balanced receptor binding activity and bioavailability is the most active isoform to induce angiogenesis. Three structurally related mammalian VEGF receptor tyrosine kinases (VEGFRs) have been identified, including VEGFR1, VEGFR2 and VEGFR3 that are encoded by Flt1, Kdr and Flt4 genes, respectively [27]. VEGFRs form homo- or heterodimers that have different binding specificity for VEGFs. VEGFR2 is the dominant receptor and master regulator of VEGF-induced angiogenesis. VEGFs and their receptors have been extensively reviewed by others [25, 27].
In the retina, VEGF is expressed by multiple retinal cells, including Müller cells, ECs, pericytes, astrocytes, RPE cells, and RGCs [28–31]. VEGF is markedly upregulated in the diabetic retina by complex molecular cues, including hypoxia [17]. Photoreceptor inner segments have abundant mitochondria and the highest oxygen demands of the retina [32, 33]. Vasoregression and occlusion of microcapillaries during the progression of DR result in local hypoxia and induction of hypoxia-inducible factors (HIFs) that are transcription factors positively regulating the expression of VEGF and numerous other adaptive responses to oxygen deprivation [34]. Regulation of HIFs by hypoxia is primarily post-translational involving direct oxygen-dependent stabilization of the protein [34]. The prototypic HIF-1 α subunit (HIF-1α) is hydroxylated, ubiquitinated, and rapidly degraded under normoxia. In hypoxic conditions, HIF-1α is stabilized to enhance the expression of target genes, including VEGF. HIFs are considered to be central to both physiological and pathological angiogenesis [35].
VEGF is a growth factor not only for ECs but also a growth and survival factor for retinal neurons and RPE [36, 37]. Upregulated VEGF may initially promote survival of stressed neurons by mechanisms that include: i) VEGF-mediated neuroprotection via activation of survival signal pathways, such as PI3K/Akt that directly enhance neuronal survival [36, 38]; ii) VEGF-induced vascular permeability that facilitates nutrient delivery and metabolite recycling in poorly perfused retinal regions resulting from capillary loss, thereby improving local neuronal function and survival; and iii) VEGF-induced local angiogenesis that improves neuronal survival by revascularizing poorly-perfused areas. However, excessive secretion of VEGF during prolonged DR can lead directly to DME with uncontrollable vascular leakage and ultimately serous retinal detachment. Excessive VEGF may also trigger PDR with the associated production of abnormal neovessels and retinal or vitreous hemorrhage. Under such extreme conditions, the neurotrophic and neuroprotective functions of VEGF are overridden, and the VEGF-VEGFR pathway becomes the driving force for DME and PDR with consequent impairment and loss of vision. The apparently opposing neuronal protective versus pathological angiogenic actions of the VEGF-VEGFR pathways in the setting of DR creates a dichotomy wherein the positive actions of VEGF inhibitors to alleviate DME and PDR are compromised by a parallel loss of VEGF-mediated neuroprotection. The adverse effects can be especially detrimental when high doses of VEGF inhibitors are administered for long-term outcomes [39, 40].
Scg3
Scg3 belongs to the nine-member chromogranin/secretogranin family that regulate the biogenesis of secretory granules [41, 42]. Scg3 shares minimal protein sequence homology with its family members or any other proteins. Instead, the granin family is defined by their common molecular properties, such as location to secretory vesicles, acidic isoelectric point (pI), calcium-binding activity, propensity to form aggregates and presence of multiple proteolytic cleavage sites. Scg3 is predominantly expressed in endocrine and neuroendocrine cells as well as neurons with secretory and neurotransmitter vesicles. Single-nucleotide polymorphisms (SNPs) in the SCG3 gene are associated with obesity in humans [43]. Although maladaptation of Scg3−/− mice to an inadequate diet and stress involving impaired processing of prohormones has been reported [44], Scg3-null mice present with a normal gross phenotype, including fertility, body weight, blood glucose, basic functional activities and behavior [45]. This finding suggests that putative roles for Scg3 in the processing of neurotransmitters and vital prohormones, such as proinsulin, can be at least partially compensated by other granins.
By applying our novel comparative ligandomics technique to live mice, we recently discovered that Scg3 functions as a unique diabetes-selective pro-angiogenic endothelial ligand with preferential binding to diabetic vs. healthy vessels [46]. Our results showed >1,700-fold increase in apparent binding of Scg3 to diabetic mouse retinal vessels, indicating that the Scg3 receptor(s) (Scg3R), as yet to be identified, is markedly upregulated on diabetic vessels, similar to the induction of the receptor of advanced glycation end products (RAGE) on diabetic endothelium [47]. Subsequent in vivo functional analyses independently confirmed that Scg3 selectively promotes angiogenesis and vascular leakage of diabetic but not healthy mouse vessels [46, 48]. In contrast, VEGF binds to and induces angiogenesis and vascular leakage of both diabetic and healthy vessels.
Angiogenic factors initiate neovascularization by binding their cognate EC receptors. Such activation may occur via upregulation of the angiogenic factors and/or increased expression or availability of their receptors. In DR, the levels of conventional angiogenic factors, such as VEGF and PDGF, increase while the receptor abundances change only minimally [49]. For example, VEGF was upregulated 36 – 110-fold in the vitreous of PDR patients, whereas soluble VEGFR1 increased only moderately by 2.5-fold [50–52]. In contrast, Scg3 binding to retinal vessels of diabetic mice increased by 1,731-fold but with only 1.38-fold increase in Scg3 expression [46]. These findings suggest marked upregulation of Scg3R and only minimal change in Scg3 ligand abundance in DR, whereas the VEGF induction is coupled only with moderate increase in VEGFR1 [52]. A similar upregulation of the CCR3 angiogenic receptor was reported in choroidal neovascularization (CNV) [53]. To our knowledge, retinal Scg3 is a unique neuron-derived diabetes-selective angiogenic and vascular leakage factor, representing a new type of neurovascular crosstalk because of its high disease selectivity in DR (Fig. 3) [46]. However, its receptor in the DR retina is yet to be identified.
An advantage of upregulation of receptors over ligands in a disease state involves the opportunity to exploit a disease-selective therapy. Secreted angiogenic factors, such as VEGF induced in a disease-associated manner, diffuse extracellularly and activate pro-angiogenic and/or-survival pathways in both diseased and healthy cells. Therapies targeting these factors may alleviate pathological angiogenesis but risk adverse side effects because of the simultaneous inhibitory actions in diseased and healthy retinal vessels. In contrast, angiogenic receptors, such as Scg3R upregulated selectively on diabetic endothelium, are stationary, and their inhibition by ligand interference is selective for the disease state with minimal side effects because of low or absent receptor expression and activity on healthy, non-DR endothelia (Fig. 3). A caveat is that during DR, all retinal vessels are exposed to hyperglycemia, and therefore, in contrast to CNV or cancer, there may not be distinct separation of diabetic and healthy vessels. However, DR is well recognized for local pathogenesis, including regional events of capillary loss, microaneurysms, venous beading, DME and PDR. Anti-Scg3 therapy is predicted to preferentially target local retinal regions with severe DR pathogenesis relative to the less severely affected area. Another issue is potential neurotoxicity related to inhibition of Scg3 versus VEGF. Whereas we do not yet know whether Scg3/Scg3R signaling is neuroprotective or whether Scg3 even binds neurons, the neuroprotective actions of VEGF are well established (Fig. 3) [36], and neurotoxicity caused by the loss of such protection is a potential side effect of long-term anti-VEGF therapy. While we have no evidence that Scg3 antagonism is similarly neurotoxic, Scg3 does not activate the PI3K/Akt survival pathway [46]. Even if present, such toxicity may be limited to regions with severe DR pathogenesis where Scg3 binding is predicted to be higher, sparing healthy neurons.
Therapeutic Interventions
DR was traditionally treated with laser therapy with limited efficacy and potential side effects [54]. Over the past decade, therapeutic strategies for DR have evolved rapidly with the emergence of two drug categories, including anti-inflammatory corticosteroids and anti-angiogenic VEGF inhibitors. Corticosteroid implants with long-lasting duration and reduced treatment burden are approved only as second-line therapies for DME due to high risks of adverse side effects, including increased intraocular pressure and cataract, as summarized in a recent review [55]. The anti-VEGF drugs ranibizumab and aflibercept are approved for all forms of DR, with or without DME. A number of investigational drugs directed at different targets are currently under development and will further shape the landscape of the disease management [56, 57]. Instead of covering all drugs in the pipeline, we focus on the comparison of anti-Scg3 vs. anti-VEGF here.
Anti-VEGF Therapy
The advent of anti-VEGF drugs represented a major breakthrough for DR therapy with well-defined mechanism of action and improved efficacy and safety over conventional laser therapy [58]. In contrast to ranibizumab that is a humanized VEGF-neutralizing antibody Fab fragment, aflibercept is an engineered chimeric decoy receptor derived from the extracellular Ig-like domain 2 of VEGFR1 and Ig-like domain 3 of VEGFR2 covalently fused to a human IgG1 Fc domain [59]. Because of the lack of the transmembrane and intracellular domain, aflibercept acts as a soluble decoy receptor that sequesters and neutralizes VEGF. Whereas ranibizumab inhibits only VEGFA, aflibercept neutralizes VEGFA, P1GF and VEGFB [60].
Although anti-VEGF drugs provide superior efficacy relative to laser therapy, about 30 – 40 % of DR patients respond poorly and require alternative therapies, including switching to other VEGF inhibitors or corticosteroids [55, 61, 62]. Side effects of anti-VEGF therapy represent additional concerns. Systemic administration of the anti-VEGF drug bevacizumab or zivaflibercept for cancer therapy triggers severe side effects, including gastrointestinal perforation, delayed wound healing, hypertension, proteinuria, heart failure, hemorrhage and thromboembolic events [63]. In general, VEGF inhibitors administered through intravitreal injection for the therapy of DR and CNV have excellent short-term safety profiles. However, long-term anti-VEGF therapy for aged patients with CNV may increase the risk of geographic atrophy [64]. Because VEGF is a well-characterized neurotrophic factor, another concern is whether repetitive treatments with anti-VEGF agents have long-term negative consequences on the function of retinal neurons. Such a possibility is implicated by clinical trials, in which high dose of ranibizumab inversely reduced long-term visual acuity despite improvement of anatomical retinal structure, including retinal thickness [39, 40].
Anti-Scg3 as an Emerging Therapy
Disease-selective anti-angiogenic therapy against Scg3 is currently under development by our group [46]. This novel therapy has the following features over anti-VEGF therapy: i) Scg3 secreted by retinal neurons is a prototypical neurovascular regulator. Successful development of anti-Scg3 therapy will highlight the importance of neurovascular crosstalk, not only for mechanistic research but also disease therapy. ii) Scg3 is the first drug target discovered by ligandomics. Successful development of this new therapy will support this innovative omics approach as a valuable tool for drug target discovery. iii) Scg3 is a unique disease-selective angiogenic factor that regulates diseased but not healthy vessels. Our recent studies showed that Scg3-neutralizing mAbs have significant safety advantages over anti-VEGF agents [65]. We further predict that anti-Scg3 antibodies selectively inhibit DME and PDR by blocking angiogenesis within severely affected areas with minimal side effects on the healthy vasculature. iv) It will be important to determine whether neurotoxicity is a side effect of anti-Scg3 therapy, as it is for high-dose ranibizumab [39, 40]. Given the normal phenotype of Scg3-null mice with no reported defects in neurogenesis, we predict that Scg3-neutralizing antibodies will not adversely affect retinal neurons. v) Anti-Scg3 therapy targets VEGF-independent angiogenic pathways and may improve treatment efficacy of DR as sole or combination therapy with VEGF inhibitors. Despite these important features, anti-Scg3 therapy is still only at preclinical stage and is yet to be tested in clinical trials.
Conclusion
DR is a leading cause of vision impairment and blindness involving the NVU. Although two VEGF inhibitors have been approved for DR therapy, an unmet clinical need still remains because of limited efficacy and/or potential adverse side effects. Multiple investigational drugs are currently under development and will provide additional options to combat the DR epidemic. The NVU plays an important role in the progression of the disease through different molecular signaling pathways of intercellular crosstalk, many of which are yet to be delineated and will likely present additional therapeutic opportunities. To our knowledge, Scg3 is the first diabetes-selective neurovascular regulator with therapeutic potential. In contrast to conventional case-by-case approaches, the emerging technologies of comparative ligandomics may help systematically uncover new neurovascular crosstalk pathways, disease-selective targets and novel targeted DR therapies. The successful development of anti-Scg3 therapy will support ligandomics as a valuable technology for drug target discovery.
Acknowledgement.
We would like to thank Dr. Yingbin Fu for scientific discussion. This work was supported by NIH R01EY027749 (WL), R24EY028764 (WL and KAW), R24EY028764-01A1S1 (WL and KAW), R43EY031238 (HT, KAW and WL), R43EY031643 (HT), R41EY027665 (WL and HT), American Diabetes Association 1-18-IBS-172 (WL), NIH P30EY002520, Knights Templar Eye Foundation Endowment in Ophthalmology (WL) and an unrestricted institutional grant from Research to Prevent Blindness (RPB) to Department of Ophthalmology, Baylor College of Medicine.
Footnotes
Conflict of interest. HT and WL are shareholders of Everglades Biopharma, LLC and LigandomicsRx, LLC. WL is the inventor of pending patents related to ligandomics and anti-Scg3 therapy.
Ethics approval and consent to participate: Not applicable.
Consent for publication: Not applicable.
Data availability.
All data are included in this article.
References
- 1.Yau JWY, Rogers SL, Kawasaki R, et al. (2012) Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35:556–564. 10.2337/dcll-1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Worldwide toll of diabetes. https://diabetesatlas.org/en/sections/worldwide-toll-of-diabetes.html. Accessed 18 Jan 2021
- 3.Gangaputra S, Lovato JF, Hubbard L, et al. (2013) Comparison of standardized clinical classification with fundus photograph grading for the assessment of diabetic retinopathy and diabetic macular edema severity. Retina 33:1393–1399. 10.1097/IAE.0b013e318286c952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloodworth JM (1962) Diabetic retinopathy. Diabetes 11:1–22 [PubMed] [Google Scholar]
- 5.Wolter JR (1961) Diabetic retinopathy. Am J Ophthalmol 51:1123–1141. 10.1016/0002-9394(61)91802-5 [DOI] [PubMed] [Google Scholar]
- 6.Simó R, Stitt AW, Gardner TW (2018) Neurodegeneration in diabetic retinopathy: does it really matter? Diabetologia 61:1902–1912. 10.1007/s00125-018-4692-l [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoon M, Okawa H, Della Santina L, Wong ROL (2014) Functional architecture of the retina: development and disease. Prog Retin Eye Res 42:44–84. 10.1016/j.preteyeres.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kur J, Newman EA, Chan-Ling T (2012) Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog Retin Eye Res 31:377–406. 10.1016/j.preteyeres.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran EP, Wang Z, Chen J, et al. (2016) Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am J Physiol Heart Circ Physiol 311:H738–749. 10.1152/ajpheart.00005.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duh EJ, Sun JK, Stitt AW (2017) Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2:. 10.1172/jci.insight.93751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geevarghese A, Herman IM (2014) Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res 163:296–306. 10.1016/j.trsl.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noonan JE, Lamoureux EL, Sarossy M (2015) Neuronal activity-dependent regulation of retinal blood flow. Clin Exp Ophthalmol 43:673–682. 10.llll/ceo.12530 [DOI] [PubMed] [Google Scholar]
- 13.Wang J, O’Sullivan ML, Mukherjee D, et al. (2017) Anatomy and spatial organization of Müller glia in mouse retina. J Comp Neurol 525:1759–1777. 10.1002/cne.24153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reichenbach A, Bringmann A (2013) New functions of Müller cells. Glia 61:651–678. 10.1002/glia.22477 [DOI] [PubMed] [Google Scholar]
- 15.Allen NJ, Lyons DA (2018) Glia as architects of central nervous system formation and function. Science 362:181–185. 10.1126/science.aat0473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grigsby JG, Cardona SM, Pouw CE, et al. (2014) The role of microglia in diabetic retinopathy. J Ophthalmol 2014:705783. 10.1155/2014/705783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong TY, Cheung CMG, Larsen M, et al. (2016) Diabetic retinopathy. Nat Rev Dis Primers 2:16012. 10.1038/nrdp.2016.12 [DOI] [PubMed] [Google Scholar]
- 18.Barber AJ, Lieth E, Khin SA, et al. (1998) Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 102:783–791. 10.1172/JCI2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrasco E, Hernández C, Miralles A, et al. (2007) Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care 30:2902–2908. 10.2337/dc07-0332 [DOI] [PubMed] [Google Scholar]
- 20.Sundstrom JM, Hernández C, Weber SR, et al. (2018) Proteomic Analysis of Early Diabetic Retinopathy Reveals Mediators of Neurodegenerative Brain Diseases. Invest Ophthalmol Vis Sci 59:2264–2274. 10.1167/iovs.17-23678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bringmann A, Wiedemann P (2012) Müller glial cells in retinal disease. Ophthalmologica 227:1–19. 10.1159/000328979 [DOI] [PubMed] [Google Scholar]
- 22.Fu Z, Sun Y, Cakir B, et al. (2020) Targeting Neurovascular Interaction in Retinal Disorders. Int J Mol Sci 21:. 10.3390/ijms21041503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerani A, Tetreault N, Menard C, et al. (2013) Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab 18:505–518. 10.1016/j.cmet.2013.09.003 [DOI] [PubMed] [Google Scholar]
- 24.Simó R, Hernández C, European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR) (2014) Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab 25:23–33. 10.1016/j.tem.2013.09.005 [DOI] [PubMed] [Google Scholar]
- 25.Peach CJ, Mignone VW, Arruda MA, et al. (2018) Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int J Mol Sci 19:. 10.3390/ijmsl9041264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senger DR, Van de Water L, Brown LF, et al. (1993) Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev 12:303–324. 10.1007/BF00665960 [DOI] [PubMed] [Google Scholar]
- 27.Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17:611–625. 10.1038/nrm.2016.87 [DOI] [PubMed] [Google Scholar]
- 28.Aiello LP, Northrup JM, Keyt BA, et al. (1995) Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol 113:1538–1544. 10.1001/archopht.1995.01100120068012 [DOI] [PubMed] [Google Scholar]
- 29.Froger N, Matonti F, Roubeix C, et al. (2020) VEGF is an autocrine/paracrine neuroprotective factor for injured retinal ganglion neurons. Sci Rep 10:12409. 10.1038/s41598-020-68488-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierce EA, Avery RL, Foley ED, et al. (1995) Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A 92:905–909. 10.1073/pnas.92.3.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stone J, Itin A, Alon T, et al. (1995) Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 15:4738–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoang QV, Linsenmeier RA, Chung CK, Curcio CA (2002) Photoreceptor inner segments in monkey and human retina: mitochondrial density, optics, and regional variation. Vis Neurosci 19:395–407. 10.1017/s0952523802194028 [DOI] [PubMed] [Google Scholar]
- 33.Yu DY, Cringle SJ (2001) Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res 20:175–208. 10.1016/sl350-9462(00)00027-6 [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez FJ, Xie C, Jiang C (2018) The role of hypoxia-inducible factors in metabolic diseases. Nat Rev Endocrinol 15:21–32. 10.1038/s41574-018-0096-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krock BL, Skuli N, Simon MC (2011) Hypoxia-induced angiogenesis: good and evil. Genes Cancer 2:1117–1133. 10.1177/1947601911423654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calvo PM, Pastor AM, de la Cruz RR (2018) Vascular endothelial growth factor: an essential neurotrophic factor for motoneurons? Neural Regen Res 13:1181–1182. 10.4103/1673-5374.235024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Byeon SH, Lee SC, Choi SH, et al. (2010) Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol Vis Sci 51:1190–1197. 10.1167/iovs.09-4144 [DOI] [PubMed] [Google Scholar]
- 38.Ruan G-X, Kazlauskas A (2012) VEGF-A engages at least three tyrosine kinases to activate PI3K/Akt. Cell Cycle 11:2047–2048. 10.4161/cc.20535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sepah YJ, Sadiq MA, Boyer D, et al. (2016) Twenty-four-Month Outcomes of the Ranibizumab for Edema of the Macula in Diabetes - Protocol 3 with High Dose (READ-3) Study. Ophthalmology 123:2581–2587. 10.1016/j.ophtha.2016.08.040 [DOI] [PubMed] [Google Scholar]
- 40.Ho AC, Busbee BG, Regillo CD, et al. (2014) Twenty-four-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology 121:2181–2192. 10.1016/j.ophtha.2014.05.009 [DOI] [PubMed] [Google Scholar]
- 41.Li W, Webster KA, LeBlanc ME, Tian H (2018) Secretogranin III: a diabetic retinopathy-selective angiogenic factor. Cell Mol Life Sci 75:635–647. 10.1007/s00018-`017-2635-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosaka M, Watanabe T (2010) Secretogranin III: a bridge between core hormone aggregates and the secretory granule membrane. Endocr J 57:275–286. 10.1507/endocrj.kl0e-038 [DOI] [PubMed] [Google Scholar]
- 43.Tanabe A, Yanagiya T, Iida A, et al. (2007) Functional single-nucleotide polymorphisms in the secretogranin III (SCG3) gene that form secretory granules with appetite-related neuropeptides are associated with obesity. J Clin Endocrinol Metab 92:1145–1154. 10.1210/jc.2006-1808 [DOI] [PubMed] [Google Scholar]
- 44.Maeda Y, Kudo S, Tsushima K, et al. (2018) Impaired Processing of Prohormones in Secretogranin III-Null Mice Causes Maladaptation to an Inadequate Diet and Stress. Endocrinology 159:1213–1227. 10.1210/en.2017-00636 [DOI] [PubMed] [Google Scholar]
- 45.Kingsley DM, Rinchik EM, Russell LB, et al. (1990) Genetic ablation of a mouse gene expressed specifically in brain. EMBO J 9:395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LeBlanc ME, Wang W, Chen X, et al. (2017) Secretogranin III as a disease-associated ligand for anti angiogenic therapy of diabetic retinopathy. J Exp Med 214:1029–1047. 10.1084/jem.20161802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramasamy R, Yan SF, Schmidt AM (2011) Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci 1243:88–102. 10.llll/j.1749-6632.2011.06320.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rong X, Tian H, Yang L, Li W (2019) Function-first ligandomics for ocular vascular research and drug target discovery. Exp Eye Res 182:57–64. 10.1016/j.exer.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freyberger H, Bröcker M, Yakut H, et al. (2000) Increased levels of platelet-derived growth factor in vitreous fluid of patients with proliferative diabetic retinopathy. Exp Clin Endocrinol Diabetes 108:106–109. 10.1055/s-2000-5803 [DOI] [PubMed] [Google Scholar]
- 50.Watanabe D, Suzuma K, Matsui S, et al. (2005) Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med 353:782–792. 10.1056/NEJMoa041773 [DOI] [PubMed] [Google Scholar]
- 51.Aiello LP, Avery RL, Arrigg PG, et al. (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 331:1480–1487. 10.1056/NEJM199412013312203 [DOI] [PubMed] [Google Scholar]
- 52.Matsunaga N, Chikaraishi Y, Izuta H, et al. (2008) Role of soluble vascular endothelial growth factor receptor-1 in the vitreous in proliferative diabetic retinopathy. Ophthalmology 115:1916–1922. 10.1016/j.ophtha.2008.06.025 [DOI] [PubMed] [Google Scholar]
- 53.Takeda A, Baffi JZ, Kleinman ME, et al. (2009) CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature 460:225–230. 10.1038/nature08151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dowler JGF (2003) Laser management of diabetic retinopathy. J R Soc Med 96:277–279. 10.1258/jrsm.96.6.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whitcup SM, Cidlowski JA, Csaky KG, Ambati J (2018) Pharmacology of Corticosteroids for Diabetic Macular Edema. Invest Ophthalmol Vis Sci 59:1–12. 10.1167/iovs.17-22259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mansour SE, Browning DJ, Wong K, et al. (2020) The Evolving Treatment of Diabetic Retinopathy. Clin Ophthalmol 14:653–678. 10.2147/OPTH.S236637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Honasoge A, Nudleman E, Smith M, Rajagopal R (2019) Emerging Insights and Interventions for Diabetic Retinopathy. Curr Diab Rep 19:100. 10.1007/sll892-019-1218-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stewart MW (2016) Treatment of diabetic retinopathy: Recent advances and unresolved challenges. World J Diabetes 7:333–341. 10.4239/wjd.v7.il6.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holash J, Davis S, Papadopoulos N, et al. (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 99:11393–11398. 10.1073/pnas.172398299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papadopoulos N, Martin J, Ruan Q, et al. (2012) Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 15:171–185. 10.1007/sl0456-011-9249-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ellis MP, Lent-Schochet D, Lo T, Yiu G (2019) Emerging Concepts in the Treatment of Diabetic Retinopathy. Curr Diab Rep 19:137. 10.1007/sll892-019-1276-5 [DOI] [PubMed] [Google Scholar]
- 62.Mira F, Paulo M, Henriques F, Figueira J (2017) Switch to Aflibercept in Diabetic Macular Edema Patients Unresponsive to Previous Anti-VEGF Therapy. J Ophthalmol 2017:5632634. 10.1155/2017/5632634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kazazi-Hyseni F, Beijnen JH, Schellens JHM (2010) Bevacizumab. Oncologist 15:819–825. 10.1634/theoncologist.2009-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grunwald JE, Pistilli M, Daniel E, et al. (2017) Incidence and Growth of Geographic Atrophy during 5 Years of Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 124:97–104. 10.1016/j.ophtha.2016.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang F, LeBlanc ME, Wang W, et al. (2019) Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis 22:369–382. 10.1007/s10456-019-09662-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in this article.