

Abstract
Purpose:
Selinexor is an oral selective inhibitor of exportin-1 (XPO1) with efficacy in various solid and hematological tumors. We assessed intra-tumoral penetration, safety, and efficacy of selinexor monotherapy for recurrent glioblastoma.
Patients and Methods:
Seventy-six adults with Karnofsky Performance Status≥60 were enrolled. Patients undergoing cytoreductive surgery received up to three selinexor doses (twice weekly) pre-operatively (Arm A; N=8 patients). Patients not undergoing surgery received 50mg/m2 (Arm B, N=24), or 60mg (Arm C, N=14) twice weekly, or 80mg once weekly (Arm D; N=30). Primary endpoint was six-month progression-free survival rate (PFS6).
Results:
Median selinexor concentrations in resected tumors from patients receiving pre-surgical selinexor was 105.4nM (range 39.7-291nM). In Arms B, C, and D, respectively, the PFS6 was 10% (95%CI, 2.79-35.9), 7.7% (95%CI, 1.17-50.6), and 17% (95%CI, 7.78-38.3). Measurable reduction in tumor size was observed in 19 (28%) and RANO-response rate overall was 8.8% (Arm B, 8.3% (95%CI, 1.0-27.0); C:7.7% (95%CI, 0.2-36.0); D:10% (95%CI, 2.1-26.5)), with one complete and two durable partial responses in Arm D. Serious adverse events (AEs) occurred in 26 (34%) patients; one (1.3%) was fatal. The most common treatment-related AEs were fatigue (61%), nausea (59%), decreased appetite (43%) and thrombocytopenia (43%), and were manageable by supportive care and dose modification. Molecular studies identified a signature predictive of response (AUC=0.88).
Conclusion:
At 80mg weekly, single-agent selinexor induced responses and clinically relevant PFS6 with manageable side effects requiring dose reductions. Ongoing trials are evaluating safety and efficacy of selinexor in combination with other therapies for newly diagnosed or recurrent glioblastoma.
Trial registration:
Keywords: selinexor, glioblastoma, clinical trial, exportin 1
INTRODUCTION
Glioblastoma (GBM) is the most common primary brain tumor in adults (1), with a poor prognosis (2). The karyopherin exportin-1 (XPO1) is a nuclear export protein that facilitates the transport of ~300 proteins harboring a leucine-rich nuclear export signal from the nucleus to the cytoplasm (3). It is overexpressed in many solid tumors, including gliomas, where its increased expression is associated with poor outcome (4-6). Selinexor is a novel, oral selective inhibitor of XPO1-mediated nuclear export (SINE) that crosses the blood-brain barrier and, since the current study for GBM was designed, has been approved by the US FDA for refractory multiple myeloma and relapsed/refractory diffuse large B–cell lymphoma (7). XPO1 inhibition directly causes nuclear retention and functional reactivation of tumor suppressor proteins (including TP53, RB1 and CDKN1B), reduces translation of oncogene mRNAs (including MYC, BCL2 and BCL6) by sequestering eIF4E-oncogene mRNA complexes in the nucleus and can indirectly modulate other pathways including PTEN and CDKN2A (8). In preclinical GBM models, selinexor reduced proliferation, sensitized cells to radiotherapy, and prolonged survival of mice with intracranial xenografts (9). Finally, a Phase I study of heavily pre-treated patients with progressive advanced stage or metastatic solid tumors demonstrated clinical benefit for some patients (10). Therefore, based on the anti-tumor activity observed in GBM models and a Phase 1 study with suitable tolerability (9,10), along with the potential importance of XPO1 in glioma biology, we conducted a phase 2 trial in recurrent GBM.
MATERIALS AND METHODS
Trial Design
The Efficacy and Safety of Selinexor (KPT-330) in Recurrent Glioblastoma [KING]) trial was an open-label, international, phase 2 study with four arms (Fig. 1). A surgical arm (Arm A) was designed to explore intra-tumoral pharmacokinetics and pharmacodynamics of selinexor treating patients with 1-3 doses of selinexor (50 mg/m2 twice weekly) beginning up to 12 days before cytoreductive surgery for recurrent tumor planned as part of routine care. Complete resection was not required, although eligibility did require that the size of tumor and extent of resection would be sufficient to provide tissue for the exploratory analyses in the clinical judgement of the investigator; the final pre-surgical dose was to be administered 2-24 hours pre-operatively. Intra-tumoral concentration ≥ 25 nM among the first 5 evaluable cases was required to continue enrollment. Medical arms (B, C, and D) explored different dosing schedules for patients not undergoing surgery. Initially, only Arm B (50 mg/m2 twice weekly) was part of the trial design; however, accrual was stopped on March 23, 2015, because of unacceptably frequent adverse events (AEs), particularly fatigue, anorexia, and thrombocytopenia. The study was amended with modified schedules, and patients were randomized 1:1 to either Arm C (60 mg flat dose twice weekly, n=14) or Arm D (80 mg flat dose once weekly, n=15). Randomization continued until July 22, 2016, when a prespecified interim analysis suggested better tolerability and efficacy for Arm D, which was expanded (n=30), whereas accrual to Arm C was terminated. There was no blinding to treatment which was intended to continue indefinitely or until disease progression or unacceptable toxicity.
Figure 1. CONSORT diagram.
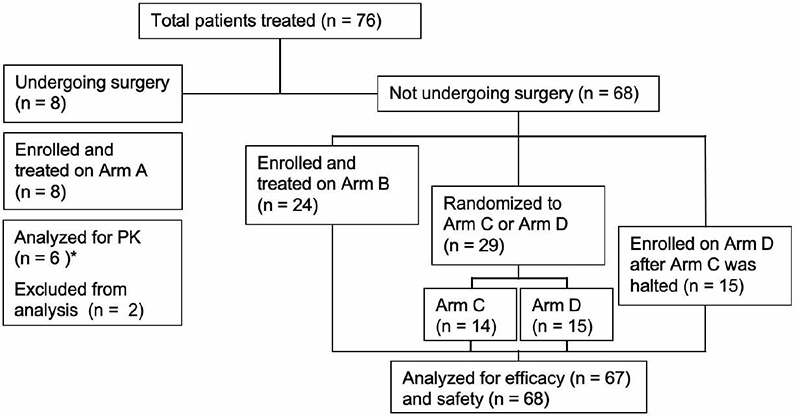
PK, pharmacokinetics. One patient from Arm C did not undergo efficacy evaluation.
*Patients did not receive a dose of selinexor on the same day of surgery
The study protocol was conducted following the Declaration of Helsinki and the International Conference on Harmonization–Good Clinical Practice. The study was approved by the institutional review board/equivalent at each participating center. All patients provided written informed consent.
Study participants
Adults male or female patients aged at least 18 years with a locally determined diagnosis of GBM (11) and recurrence/progression after at least radiotherapy (RT) and temozolomide were eligible. Karnofsky Performance Status(12) (KPS) ≥ 60 and adequate bone marrow, renal and hepatic function were required. Eligibility for Arms B-D also required recurrent radiographically measurable disease per the Response Assessment in Neuro-Oncology (RANO) criteria to allow evaluability for partial response (PR) or complete response (CR), and an interval of at least 12 weeks from completion of radiation therapy (unless histologically proven recurrence was detected on intervening resection) (13). Available pre-selinexor archived tissue for exploratory correlative studies was also required. Prior treatment with bevacizumab or other direct VEGF/ VEGFR inhibitors was exclusionary (Further detail is available in the Supplementary data).
Efficacy and Safety Assessments
Arm A was designed only to explore the intra-tumor pharmacokinetics and pharmacodynamics of selinexor. Arms B, C and D were designed to assess efficacy by the 6-month progression-free survival (PFS6) rate (with progression and assessments determined by the local investigator using the RANO criteria; no central reviews were performed). Secondary objectives included response rate (partial or complete, by RANO per the local investigator, with assessments performed approximately every 8 weeks), 6-cycle (defined as 28 days) PFS (6cPFS, with a window of ±14 days allowed around the 6-cycle visit) rate, median PFS, median overall survival (OS), and the evaluation of safety and tolerability. Molecular alterations associated with response to selinexor were explored by DNA and RNA sequencing and transcriptomal analyses on pre-treatment archival tumor samples (below).
All AEs occurring during the trial and up to 30 days after the last selinexor dose were documented, with toxicities graded according to NCI–CTCAE version 4.03. Study-related toxicities were managed using 5–hydroxytryptamine receptor 3 (5-HT3) antagonists and/or other anti-emetics, analgesics, short courses of low dose oral steroids, and anti-diarrheal agents, as recommended in the study protocol.
Pharmacokinetics (PK) were determined for patients enrolled in Arm A by measuring pre- and post-dose blood levels of selinexor compared against the selinexor concentration in resected tumor samples recovered at the time of surgery (see also Supplementary Methods).
Exploratory Molecular Correlative Studies
Details on immunostaining, exome sequencing, and RNA sequencing (RNAseq) performed on available resected tumors (Arm A) and paired pre-selinexor treatment archival specimens are available in the Supplementary Methods.
Sequencing was performed on archival tissue blocks from patients with adequate selinexor exposure (defined as at least 3 doses or treated for at least 21 days). RNAseq data were used to compare patients with benefit to those with resistance (n= 52; benefit defined as best overall response [BOR] of CR, PR, or durable [PFS>140 days] stable disease [SD]); resistance defined as BOR of progressive disease [PD] or non-durable [PFS <100 days] SD), as described in the Supplementary Methods. Exome sequencing data were used to determine associations between mutated genes with PFS and OS for all genes mutated in at least five patients using log-rank tests (additional details available in the Supplementary Methods).
Statistical Analysis
The primary endpoint was PFS6 among all patients undergoing efficacy assessment (modified Intent to Treat, mITT). Simon’s optimal two-stage design was used to calculate the sample size in each arm B-D. A true PFS6 rate above 30% was deemed relevant for further study, and a PFS6 below 9% was regarded as insufficient for additional investigations. Of 12 patients accrued during the first stage, if more than one was progression-free at 6 months, enrollment would proceed to the second stage to a total of 30 patients. With a one-sided type I error rate of 0.10 and a power of 90%, the null hypothesis would be rejected if five or more out of 30 patients were progression-free at 6 months. PFS was defined as the interval from treatment start to progression or death from any cause and OS to death from any cause. Median PFS and OS were calculated using the Kaplan-Meier method(14), and patients alive and/or without documented disease progression were right-censored for time to event analyses. Intra-arm efficacy comparisons were performed for overall response rate (ORR) using Fisher’s exact test and PFS using a log-rank test and Cox proportional hazards model.
Data lock occurred on May 4, 2020. This trial is registered with ClinicalTrials.gov, NCT01986348.
Data Availability
The data generated in this study are available within the article and its supplementary data files. Sequencing data are available on gene expression omnibus accession GSE186332.
RESULTS
Efficacy
There were 76 patients treated between March 10, 2014 and January 23, 2020 (arm A, 8; B, 24; C, 14; D, 30) (Fig 1). Patients had received a median of one prior therapy in addition to radiotherapy and temozolomide (range 1–8), and the median KPS was 90 (range 60–100). The median age at the time of enrollment was 56 years, and 71% of patients were men (Table 1).
Table 1.
Patient characteristics
| Arm A (N = 8) |
Arm B (N = 24) |
Arm C (N = 14) |
Arm D (N = 30) |
Total (N = 76) |
|
|---|---|---|---|---|---|
| Selinexor Dose | 50 mg/m2 BIW (n=7), 60 mg BIW (n=1)# |
50 mg/m2 BIW |
60 mg BIW | 80 mg QW | |
| Age (years)*, median (range) | 58.0 (43-65) | 50.5 (29-69) | 52.0 (27-65) | 56 (2-78) | 56 (20-78) |
| Sex, n (%) | |||||
| Male | 7 (87.5) | 19 (79.2) | 9 (64.3) | 19 (63.3) | 54 (71.1) |
| Female | 1 (12.5) | 5 (20.8) | 5 (35.7) | 11 (36.7) | 22 (28.9) |
| Prior lines of therapy†, median (range) | 1.5 (1-2) | 1 (1-2) | 1 (1-3) | 1 (1-8) | 1 (1-8) |
| Baseline Karnofsky Performance Status, n (%) | |||||
| ≤80% | 5 (62.5) | 9 (37.5) | 5 (35.7) | 14 (46.7) | 33(43.4) |
| >80% | 3 (37.5) | 15 (62.5) | 15 (62.5) | 16 (53.3) | 43 (56.6) |
At time of first dose.
Data missing for two patients in Arm C and one patient in Arm D.
Patient treated after protocol update in version 4.0 in which the dose was changed to 60mg flat. PFS6, 6-month progression-free survival; BIW, twice per week; QW, once per week. Arm A was primarily designed to determine intra-tumoral pharmacokinetics, and arms B-D tested efficacy of different dose schedules.
The modified intent to treat population (mITT, n=67) consisted of patients treated in the medical arms (B, C and D) evaluated for efficacy (excluding one patient from Arm C who did not undergo efficacy evaluation). The median time on treatment for these patients was 1.64 months (range= 1 day – 42.1 months, interquartile range [IQR] = 1.02–2.74 months). The most common cause of treatment discontinuation was disease progression (n=56, 83.6%).
The PFS6 was 10% (95% CI, 2.67 to 35.4), 7.7% (95% CI, 1.2 to 50.6), and 17.2% (95% CI, 7.78 to 38.3) for Arms B, C, and D, respectively (Table 2, Table S1, Fig. 2A). The median OS was 10.5 months (95% CI, 4.9 to 17.0) for patients in Arm B, 8.5 months (95% CI, 7.3 to not evaluable) for Arm C, and 10.2 months (95% CI, 7.0 to 15.4) for Arm D (Table 2, Tables S1 and S2, Fig. 2B). The overall response rate (partial or complete) was 8.3% (n=2, 95% CI, 1.0 to 27), 7.7% (n=1, 95% CI, 0.2 to 36.0), 10% (n=3, 95% CI, 2.1 to 26.5), respectively (Table 2). Notably, a measurable reduction in tumor size (regardless of formal RANO-based response that requires ≥50% reduction in cross-sectional area) was observed in 19 patients (28% overall), none of whom had increases in dexamethasone within 30 days before the greatest measured reduction in tumor size (Fig. 2C and D, and Supplementary Table S3, Supplemental Figs. S1 and S2).
Table 2.
Efficacy outcomes in mITT population
| Arm B (N = 24) |
Arm C (N = 13) |
Arm D (N = 30) |
|
|---|---|---|---|
| 6-month PFS* %, (95% CI) | 10.0 (2.7 - 35.4) | 7.7 (1.2 - 50.6) | 17.2 (7.8 - 38.3) |
| Progression Free at 6 Months, n (%) | 2 (8.3) | 1 (7.7) | 5 (16.7) |
| Median PFS, months (95% CI) | 1.6 (1.2- 3.2) | 1.9 (1.8- 14.9) | 1.9 (1.8- 3.0) |
| Median OS, months (95% CI) | 10.5 (4.9 - 17.0) | 8.5 (7.3 - NE) | 10.2 (7.0 - 15.4) |
| Best Overall Response, n (%) | 2 (8.3) | 1 (7.7) | 3 (10.0) |
| 95% CI | (1.0 - 27.0) | (0.2 - 36.0) | (2.1 - 26.5) |
| Complete Response, n (%) | 0 (0) | 0 (0) | 1 (3.3) |
| 95% CI | NE | NE | (0.1 - 17.2) |
| Partial Response, n (%) | 2 (8.3) | 1 (7.7) | 2 (6.7) |
| 95% CI | (1.0 - 27.0) | (0.2 - 36) | (0.8 - 22.1) |
| Stable Disease, n (%) | 6 (25.0) | 4 (30.8) | 7 (23.3) |
| 95% CI | (9.8 - 46.7) | (9.1 - 61.4) | (9.9 - 42.3) |
| Progressive Disease, n (%) | 15 (62.5) | 8 (61.5) | 17 (56.7) |
| 95% CI | (40.6 - 81.2) | (31.6 - 86.1) | (37.4 - 74.5) |
CI, confidence interval; NE, not evaluable; PFS, progression-free survival; OS, overall survival
survival rate point estimates are presented for 6-month PFS using the Kaplan-Meier method. One patient from Arm C who did not undergo efficacy evaluation is not included in the efficacy analyes.
Figure 2. Efficacy and survival of selinexor treatment in the modified intent to treat population.

(A) Overall survival and (B) disease-free survival in the mITT population, stratified by trial arm (excluding one patient from Arm C who did not undergo efficacy evaluation). (C) Waterfall plot shows the maximal reduction (or increase) for 63 patients treated in Arms B (n=23), C (n=13) and D (n=27), calculated as the change from baseline in sum of the products of the perpendicular diameters of the tumor, as determined by the local investigators using the response assessment for neuro-oncology criteria. (D) Swimmer plot of patients enrolled in Arm D. BIW, twice weekly; QW, once weekly.
Safety
The safety population consisted of all the treated patients in all 4 arms (76: arm A, 8; B, 24; C, 14; D, 30). Hematologic treatment related AEs (TRAEs) of any grade that occurred in ≥10% (Table 3) included, most commonly, thrombocytopenia (n=33, 43.4%), neutropenia (n=20, 26.3%), and anemia (n=13, 17.1%). Febrile neutropenia was not reported, and no bleeding events occurred in patients with Grade 3 or 4 thrombocytopenia. The most common non-hematological TEAEs were fatigue (n=46, 60.5%), nausea (n=45, 59.2%), decreased appetite (n=33, 43.4%), vomiting (n=23, 30.3%), dysgeusia (n=20, 26.3%), hyponatremia (n=15, 19.7%), decreased weight (n=13, 17.1%), constipation (n=11, 14.5%), blurred vision (n=8, 10.5%) and diarrhea (n=9, 11.8%) (Table 3). Nearly all of the AEs were reversible with dose modification and standard supportive care, as reported in other selinexor studies (15-17).
Table 3.
Treatment-related Adverse Events
| Arm A (N = 8) |
Arm B (N = 24) |
Arm C (N = 14) |
Arm D (N =30) |
Overall (N = 76) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade 1/2 | Grade 3 | Total | Grade 1/2 | Grade 3 | Total | Grade 1/2 | Grade 3 | Total | Grade 1/2 | Grade 3 | Total | Total | |
| Fatigue | 3 (37.5) | 1 (12.5) | 4 (50.0) | 10 (41.7) | 7 (29.2) | 17 (70.8) | 8 (57.1) | 2 (14.3) | 10 (71.4) | 14 (46.6) | 1 (3.3) | 15 (50.0) | 46 (60.5) |
| Nausea | 6 (75.0) | 0 | 6 (75.0) | 9 (37.5) | 1 (4.2) | 10 (41.7) | 9 (64.3) | 0 | 9 (64.3) | 20 (66.7) | 0 | 20 (66.7) | 45 (59.2) |
| Decreased appetite | 4 (50.0) | 0 | 4 (50.0) | 11 (45.8) | 0 | 11 (45.8) | 10 (71.5) | 0 | 10 (71.5) | 8 (26.7) | 0 | 8 (26.7) | 33 (43.4) |
| Thrombocytopenia | 5 (62.5) | 1 (12.5) | 6 (75.0) | 14 (58.3) | 2 (8.3) | 16 (66.7) | 4 (28.6) | 0 | 4 (28.6) | 6 (20.0) | 1 (3.3) | 7 (23.3) | 33 (43.4) |
| Vomiting | 1 (12.5) | 0 | 1 (12.5) | 7 (29.2) | 0 | 7 (29.2) | 5 (35.7) | 0 | 5 (35.7) | 10 (33.3) | 0 | 10 (33.3) | 23 (30.3) |
| Leukopenia | 0 | 0 | 0 | 5 (20.8) | 2 (8.3) | 7 (29.2) | 0 | 1 (7.1) | 1 (7.1) | 12 (40.0) | 1 (3.3) | 13 (43.3) | 21 (27.6) |
| Dysgeusia | 1 (12.5) | 0 | 1 (12.5) | 9 (37.5) | 0 | 9 (37.5) | 6 (42.9) | 0 | 6 (42.9) | 4 (13.3) | 0 | 4 (13.3) | 20 (26.3) |
| Neutropenia | 1 (12.5) | 0 | 1 (12.5) | 3 (12.5) | 4 (16.7) | 7 (29.2) | 0 | 2 (14.3) | 2 (14.3) | 8 (26.7) | 2 (6.7) | 10 (33.3) | 20 (26.3) |
| Hyponatremia | 2 (25.0) | 0 | 2 (25.0) | 9 (37.5) | 1 (4.2) | 10 (41.7) | 2 (14.3) | 0 | 2 (14.3) | 1 (3.3) | 0 | 1 (3.3) | 15 (19.7) |
| Anemia | 1 (12.5) | 0 | 1 (12.5) | 5 (20.8) | 0 | 5 (20.8) | 1 (7.1) | 0 | 1 (7.1) | 6 (20.0) | 0 | 6 (20.0) | 13 (17.1) |
| Weight decrease | 1 (12.5) | 0 | 1 (12.5) | 4 (16.7) | 0 | 4 (16.7) | 5 (35.7) | 1 (7.1) | 6 (42.9) | 2 (6.7) | 0 | 2 (6.7) | 13 (17.1) |
| Constipation | 0 | 0 | 0 | 2 (8.3) | 0 | 2 (8.3) | 4 (28.6) | 0 | 4 (28.6) | 5 (16.7) | 0 | 5 (16.7) | 11 (14.5) |
| Blurred Vision | 0 | 0 | 0 | 5 (20.8) | 0 | 5 (20.8) | 1 (7.1) | 0 | 1 (7.1) | 2 (6.7) | 0 | 2 (6.7) | 8 (10.5) |
| Diarrhea | 1 (12.5) | 1 (12.5) | 2 (25.0) | 3 (12.5) | 0 | 3 (12.5) | 0 | 0 | 0 | 4 (13.3) | 0 | 4 (13.3) | 9 (11.8) |
| Lymphopenia | 0 | 2 (25.0) | 2 (25.0) | 1 (4.2) | 1 (4.2) | 2 (8.3) | 0 | 0 | 0 | 0 | 3 (10.0) | 4 (13.3) | 8 (10.5) |
Serious AEs were experienced by 26 (34.2%) patients, most commonly, seizures in 6 (8%); syncope in 3 (4%); and fatigue, headache, pulmonary embolism, and urinary tract infection in 2 (3%) patients each. Eight of the 26 SAEs were considered related: decreased appetite (grade 2), diarrhea (grade 3), seizure (grade 2), pneumonia (grade 3), hyperlipasaemia (grade 3), hypophosphatemia (grade 4) and two events of fatigue (both grade 3). Additional grade 4 or 5 serious AEs were observed, but all were considered unrelated to selinexor and included one patient each with grade 4 hyperglycemia, grade 4 cerebral edema and grade 5 (fatal) pulmonary embolism.
Five (6.6%) patients discontinued treatments due to AEs: one each due to thrombocytopenia (without bleeding), pneumonia, anorexia, malaise, nausea/vomiting, weight loss, and low quality of life. Dose reductions were required in a total of 28.9% of patients due to AEs. The most common AEs resulting in dose reductions were fatigue in 10 (13.2%) patients, decreased appetite in 5 patients (6.6%), hyperlipasaemia in 2 patients (2.6%), hypophosphatemia in 2 patients (2.6%), leukopenia in 2 patients (2.6%), and thrombocytopenia in 2 patients (2.6%). There was no obvious correlation between on-target AEs and response.
Intra-tumoral Pharmacokinetics and Pharmacodynamics
Selinexor concentrations measured in the contrast-enhancing tumors ranged from 39.7 to 291 nM (median 105.4 nM, average 136 nM), whereas concentrations in the plasma 2 hours post-dosing ranged from 645nM to 1.62μM (median 835 nM) Tumor/plasma ratios ranged from 0.0616 to 0.190 (median 0.0914) (Supplementary Table S4 and Fig. S3). This is in the range of the IC50 (median 148 nM, average 166 nM) for patient-derived glioblastoma cell lines treated with selinexor(9).
To assess subcellular localization of tumor suppressor proteins exported by XPO1, immunohistochemistry (IHC) was performed on the post-treatment resected tumor and pre-treatment archival tumor tissue from a patient in arm A. There was a marked reduction in proliferation (Ki67+ cells, 29% ± 3.0% pre- vs 13% ± 0.8% post-treatment, p=0.012) and increase in apoptosis (cleaved caspase 3+ cells, 2% ± 0.7% pre- vs 28% ± 3.0% post-treatment, p=0.003). Furthermore, the post-treatment sections showed increased nuclear localization of the tumor suppressors PTEN, FOXO1, and TP53, along with increased expression of NGFR, a negative regulator of NF-κB induced by selinexor treatment (18,19) (Supplementary Fig. S4), consistent with the intended mechanism of action of selinexor.
RNAseq was used to compare global expression profiles of post-treatment resected tumors from three patients with archival tumor specimens from the same patients. All three post-treatment tumors showed marked increases in XPO1 RNA expression (average 2.34-fold increase; Padj = 1.54 × 10−5), which is a known pharmacodynamic marker indicating successful inhibition of XPO1 nuclear export activity (10) (Supplementary Fig. S5). Significant RNA-level increases of other genes known to be induced by selinexor treatment were also observed, including HSPA4L, SLC43A2 and the tumor suppressor ARRDC3 (20,21) (Supplementary Fig. S5).
Molecular Predictors of Response
In a post-hoc exploratory analysis (see also Supplementary Methods) to seek molecular markers of outcome, informative and quality exome sequencing and RNAseq were performed on resected tumor specimens at the time of diagnostic surgery before the recurrence from 52 study patients from all arms with adequate selinexor exposure and evidence of either clinical benefit or resistance defined above. Among the identified recurrently mutated genes, patients whose tumors harbored mutations in pancreatic and duodenal homeobox 1 (PDX1, n=5), E1A Binding Protein P400 (EP400, n=13) or Dedicator of Cytokinesis 8 (DOCK8, n=7) survived longer than patients with wild-type tumors (Supplementary Fig. S6). Mutations commonly observed in GBM were also observed but did not correlate with outcome, including IDH1 (as determined centrally, mutated in n=9 patients), TP53 (n=14), PTEN (n=14), EGFR (n=11), PIK3CA (n=5), RB1 (n=7), ATRX (n=6), and NF1 (n=8) (Supplementary Fig. S7).
RNAseq data were used to infer the activity for 6,203 master regulator proteins using the VIPER algorithm (22). The sequenced specimens were split into a discovery set of 7 clear responders (BOR of CR or PR) compared to 23 resistors (BOR of PD despite at least 30 days of treatment) and an internal validation set of the remaining patients with other, although less robust, suggestions of either selinexor resistance (BOR PD or or non-durable SD) or clinical benefit (BOR of durable SD). An ensemble of five different machine learning algorithms was used to generate an integrated predictive model for selinexor response in GBM. This model was based on the VIPER-inferred activity for three proteins that were activated in the responders compared to the non-responders in the discovery set, ZC3H12A (false discovery rate P-value [FDR]=6.45 x 10−11), RAB43 (FDR=3.81 x 10−10), and SOCS3 (FDR=3.16 x 10−9). The model achieved an integrated area under the receiver operating characteristic (ROC) curve of 0.88 (p<0.05, permutation test) for a Leave-one-out cross-validation analysis in the discovery set and correctly predicted 9 of 11 patients classified as experiencing clinical benefit and 7 of 11 patients classified as selinexor-resistant in the validation set (ROC-AUC = 0.67) (Supplementary Fig. S8).
DISCUSSION
We explored three different dosing schedules (arm B, 50mg/m2 twice weekly; C, 60 mg as a flat dose twice weekly; and D, 80 mg as a flat dose once weekly) in a multi-arm open-label trial of selinexor monotherapy for recurrent GBM. Although the PFS6 goal of 30% was not met, the null hypothesis was rejected for Arm D (PFS6 17.2%) which also employed the most tolerable dosing schedule of 80 mg once-weekly and was associated with a 10% RANO-defined response rate. Furthermore, tumor size was reduced in 28% of patients overall, and several remained on selinexor for more than 12 months, including one for 42 months at data lock. Taken together, we believe these results show that selinexor is an active drug in some patients with GBM and is worthy of further study.
The surgical substudy (Arm A) showed that intra-tumor selinexor concentration is in the range of the IC50 for GBM cells preclinically (Supplementary Fig. S3) (9). Importantly, selinexor is a covalent inhibitor, forming a reversible covalent bond (t½ ~24 hours) with Cys528 in XPO1, for a relatively long effective biological half-life of 48-72 hours, suggesting that dosing once weekly is reasonable (23,24).
Finally, pharmacodynamic studies of three sets of paired pre-and post-treatment tumors (Figs. S4 and S5) showed significant increases in XPO1 RNA levels, which indicates XPO1 protein activity was sufficiently inhibited, and feedback was induced to increase XPO1 transcription. This analysis also identified the significant induction of the tumor suppressor protein arrestin domain-containing 3 (25), induced by selinexor in triple-negative breast cancer cells to block tumor proliferation and migration (26). In addition to the above-described transcriptome analysis, immunohistochemistry on post-selinexor tissue samples demonstrated increased nuclear localization of the XPO1 cargo proteins TP53, FOXO1, and PTEN decreased proliferation markers, and increased levels of apoptosis, consistent with the reported mechanism of action of selinexor. Interestingly, selinexor also induced protein expression levels of nerve growth factor receptor (NGFR). This is similar to the induction observed in glioma models, where NGFR induction reduced free nuclear NF-κB levels, decreased stemness markers, and increased cell differentiation markers (27). Thus, the pharmacokinetic and pharmacodynamic results further support development of selinexor in the treatment of glioblastoma.
The interpretability of the drug penetration into the tumor is limited by the extent of the tissue resected. We did not systematically perform pharmacokinetic analyses on both enhancing tumor on brain imaging and non-enhancing tumor. Therefore, we cannot comment on the penetration into the surrounding, non-enhancing brain parenchyma that presumably contains microscopic disease. In addition, it is plausible that some of the pharmacodynamic effects described resulted not from selinexor but instead from molecular drift over time or intervening therapy between archival tumor sampling and initial of study treatment.
We also performed exome sequencing and transcriptome analysis to explore markers potentially associated with selinexor drug response in pre-dosed tumors. These studies identified PDX1, EP400, and DOCK8 mutations in the tumors of patients with longer survival (Supplementary Fig. S6). To our knowledge, the observed PDX1 mutations have not been previously identified in GBMs. Interestingly, the recurrent missense changes p.C18R and p.P33T mutations have been confirmed to impact PDX1-mediated transcription (28). Although PDX1 is a crucial regulator of pancreatic cell development and is well-characterized in pancreatic cancer, there are reports of its ectopic expression in other cancer types (29). Our data support further investigation of a role for PDX1 in GBM. Likewise, somatic DOCK8 mutations have been reported in various cancer types but are not characteristic of a particular malignancy or thought to be a recurrent feature of GBM. Notably, constitutional DOCK8 mutations underlie a rare combined immunodeficiency syndrome (DOCK8 syndrome) (30). Despite the association of IDH mutations with improved outcome in newly diagnosed glioma (31), we did not observe a correlation with survival (Supplementary Fig. S7). Moreover, none of the patients with durable disease control (PFS6) in Arm D had tumors harboring an IDH1 or IDH2 mutation by sequencing. As the study was not randomized, it is plausible that PDX1, EP400, and DOCK8 mutations were prognostic for longer survival in recurrent glioblastoma generally rather than predicting response to selinexor specifically.
Analysis of the transcriptome was used to infer protein activities in pre-dosed tumors and accurately classify patients likely to respond to selinexor treatment in both discovery and validation sets. Increased activity of three proteins that regulate different cellular pathways was observed in both sets. We speculate the combined activities of the proteins are associated with a GBM cell phenotypic state that is particularly responsive to XPO1 inhibition. If validated, this could be useful for the identification of patients most likely to benefit from selinexor. The three-protein signature consisted of the activities of the endoribonuclease ZC3H12A (also called regnase-1), the GTPase RAB43, and SOCS3, a direct inhibitor of JAK kinases. Notably, SOCS3 has previously been investigated in the context of GBM, where it was shown to be overexpressed in comparison to normal brain tissue and linked to radiotherapy sensitivity (32).
Moreover, SOCS3 promoter methylation has been explored as a biomarker of poor response in GBM (33). Since SOCS3 plays an integral role in controlling GBM cell survival, it was not surprising to identify an association between SOCS3 activity and selinexor response. The part of ZC3H12A is more complex, as it has also been shown to both promote and impede tumorigenesis, depending on the cancer type (34). Like XPO1, RAB43 regulates intracellular protein trafficking, as it controls anterograde endoplasmic reticulum-Golgi transport of nascent G-protein coupled receptors. Elucidating the links between outcomes on selinexor and high activity of RAB43, SOCS3 and ZC3H12A will require further mechanistic studies.
There were several limitations to our study. As all patients received study treatment, efficacy comparisons are against historic controls rather than internal randomization to a standard regimen (such as lomustine) for recurrent GBM. In addition, the nature, power, and quality of the molecular correlative analyses were limited by the number and quality of available biological material, as well as the strength of the clinical signal. For example, in the discovery analysis, the difference between selinexor-sensitive and resistant cases was more robust than in the validation set which was consequently more prone to error. Moreover, these were not statistically pre-specified analyses; rather, we endeavored to explore biomarkers in pre-treatment tumor tissue that might predict efficacy which could be confirmed in a future study with an independent set of tissue samples in a post-hoc, hypothesis generating, non pre-planned or statistically powered approach.
Nonetheless, overall, our results suggest that single-agent oral selinexor 80 mg once weekly warrants further study in GBM. As synergistic and additive activities in combination with DNA damaging agents and radiation therapy have been observed for selinexor (9,35-37), ongoing studies are investigating combination strategies in both newly diagnosed and recurrent GBM (NCT04216329 and NCT04421378) and will prospectively validate the potentially predictive biomarkers identified in the KING trial.
Supplementary Material
Translational Relevance.
Glioblastoma is an incurable primary brain cancer that demands new therapeutic approaches. Exportin-1 is a nuclear export protein overexpressed in many solid tumors, including gliomas, which correlates with prognosis. Selinexor is a first-in-class exportin-1 inhibitor with efficacy in various cancers. We conducted an international multi-arm clinical trial of selinexor for patients with recurrent glioblastoma and demonstrated adequate intra-tumoral drug penetration, and we observed clinically relevant disease control with manageable side effects requiring dose reductions. Molecular studies identified a signature predictive of response. Ongoing trials evaluate the safety and efficacy of selinexor in combination with other therapies for newly diagnosed and recurrent glioblastoma.
Acknowledgments
Karyopharm funded the study and provided selinexor supply. Tamar Aprahamian, PhD, of JetPub Scientific Communications, LLC, supported by funding from Karyopharm, provided drafts and editorial assistance to the authors during preparation of this manuscript. Authors were also supported in part by The William Rhodes and Louise Tilzer-Rhodes Center for Glioblastoma at New York-Presbyterian Hospital (A.B.L. and A.C.) and by National Institutes of Health (NIH)/National Cancer Institute (NCI) grants U01 CA217858 (A.C.), P30CA013696 (A.B.L. and A.C), UG1CA189960 (A.B.L.), S10 OD012351 (A.C.), and S10 OD021764 (A.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH/NCI.
This study was funded by Karyopharm Therapeutics.
Footnotes
Disclosures
A.B.L. received study-relevant honoraria, travel support, and research funding from Karyopharm and (in the last 12-months) also received research funding or other support from QED, Bayer, Orbus, Agios, Kadmon, VBI Vaccines, Beigene, Oncoceutics, Pfizer, Genentech/Roche, Millenium, Celldex, Novartis, BMS, AbbVie. Novocure, Northwest Biotherapeutics, Celgene, Aeterna Zentaris, Abbott Molecular; honoraria from Novocure, Orbus, Karyopharm, Sapience, and Bioclinica as a blinded independent reader of clinical and imaging data for a BMS-sponsored trial.
P.Y.W. reports research support from Agios, Astra Zeneca/Medimmune, Bayer, Beigene, Celgene, Eli Lily, Genentech/Roche, Kazia, MediciNova, Merck, Novartis, Nuvation Bio, Oncoceutics, Vascular Biogenics, VBI Vaccines, Vigeo and is on the Advisory Board at Agios, Astra Zeneca, Bayer, Boston Pharmaceuticals, CNS Pharmaceuticals, Immunomic Therapeutics, Imvax, Karyopharm, CNS Pharmaceuticals, Merck, Novartis, Oncoceutics, Tocagen, Vascular Biogenics, VBI Vaccines, Voyager, QED, Elevate Bio.
M.V.D.B. reports consulting fees from Abbvie, Celgene, Agios, Boehringer Ingelheim, Bayer, Carthera, Genenta, Nerviano, Boston pharmaceuticals and research funding from Abbvie.
S.R.P. is co-founder of NFlection Therapeutics and NF2 Therapeutics and consults for AstraZeneca and SonalaSense.
A.W. reports research funding and travel expenses from IPSEN.
K.L., C.J.W., H.C., S.T., and L.H., are employees of Karyopharm Therapeutics. Y.L., M.G.K., and S.S. are employees and shareholders of Karyopharm Therapeutics.
M.J.A. reports patents and royalties from DarwinHealth Inc.
A.C. is founder, equity holder, consultant, and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. M.J.A. is Chief Scientific Officer and equity holder at DarwinHealth, Inc. Columbia University is also an equity holder in DarwinHealth Inc. US patent numbers 10,777,299 and 10,790,040 have been awarded related to this work, assigned to Columbia University.
M.M-S. has served on advisory boards for Genmab, Roche, and Bayer and has received research grants from Karyopharm Therapeutics, PUMA Biotechnologies, and MSD.
A.L.G. and Y.S. have no disclosures to report.
References
- 1.Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro. Oncol Oxford University Press; 2019. page V1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. NLM (Medline); 2020;22:1073–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. Cell Press; 1997;90:1041–50. [DOI] [PubMed] [Google Scholar]
- 4.Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65:153–9. [DOI] [PubMed] [Google Scholar]
- 5.Liu X, Chong Y, Tu Y, Liu N, Yue C, Qi Z, et al. CRM1/XPO1 is associated with clinical outcome in glioma and represents a therapeutic target by perturbing multiple core pathways. J Hematol Oncol. 2016;9:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu S, Qiao Q, Li G. A Radiosensitivity Gene Signature and XPO1 Predict Clinical Outcomes for Glioma Patients. Front Oncol. Frontiers Media S.A.; 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.XPOVIO [package insert]. Karyopharm Therapeutics Inc. Newton, MA. 2019. [Internet]. [cited 2019 Dec 18]. Available from: https://www.karyopharm.com/wp-content/uploads/2019/07/NDA-212306-SN-0071-Prescribing-Information-01July2019.pdf [Google Scholar]
- 8.Azizian NG, Azizian NG, Li Y, Li Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol BioMed Central Ltd.; 2020. page 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green AL, Ramkissoon SH, McCauley D, Jones K, Perry JA, Hsu JH-R, et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015;17:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdul Razak AR, Mau-Soerensen M, Gabrail NY, Gerecitano JF, Shields AF, Unger TJ, et al. First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients With Advanced Solid Tumors. J Clin Oncol. American Society of Clinical Oncology; 2016;34:4142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. Springer; 2007;114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.KARNOFSKY DA. The bases for cancer chemotherapy. Stanford Med Bull. Stanford Med Bull; 1948;6:257–69. [PubMed] [Google Scholar]
- 13.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated Response Assessment Criteria for High-Grade Gliomas: Response Assessment in Neuro-Oncology Working Group. J Clin Oncol. 2010;28:1963–72. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 15.Chari A, Vogl DT, Gavriatopoulou M, Nooka AK, Yee AJ, Huff CA, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. Massachussetts Medical Society; 2019;381:727–38. [DOI] [PubMed] [Google Scholar]
- 16.N K, M M, F C, G F, A G, JSP V, et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): a single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol. Lancet Haematol; 2020;7. [DOI] [PubMed] [Google Scholar]
- 17.Gavriatopoulou M, Chari A, Chen C, Bahlis N, Vogl DT, Jakubowiak A, et al. Integrated safety profile of selinexor in multiple myeloma: experience from 437 patients enrolled in clinical trials. Leukemia. Nature Publishing Group; 2020;1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X, Hao Q, Liao P, Luo S, Zhang M, Hu G, et al. Nerve growth factor receptor negates the tumor suppressor p53 as a feedback regulator. Elife. eLife Sciences Publications Ltd; 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeSisto JA, Flannery P, Lemma R, Pathak A, Mestnik S, Philips N, et al. Exportin 1 inhibition induces nerve growth factor receptor expression to inhibit the NF-kB pathway in preclinical models of pediatric high-grade glioma. Mol Cancer Ther. American Association for Cancer Research Inc.; 2020;19:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crochiere M, Kashyap T, Kalid O, Shechter S, Klebanov B, Senapedis W, et al. Deciphering mechanisms of drug sensitivity and resistance to Selective Inhibitor of Nuclear Export (SINE) compounds. BMC Cancer. 2015;15:910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun H, Lin DC, Cao Q, Guo X, Marijon H, Zhao Z, et al. CRM1 inhibition promotes cytotoxicity in ewing sarcoma cells by repressing EWS-FLI1-dependent IGF-1 signaling. Cancer Res. American Association for Cancer Research Inc.; 2016;76:2687–97. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Hilda Ye B, et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. Nature Publishing Group; 2016;48:838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neggers JE, Vercruysse T, Jacquemyn M, Vanstreels E, Baloglu E, Shacham S, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol. Elsevier Ltd; 2015;22:107–16. [DOI] [PubMed] [Google Scholar]
- 24.Neggers JE, Vanstreels E, Baloglu E, Shacham S, Landesman Y, Daelemans D. Heterozygous mutation of cysteine528 in XPO1 is sufficient for resistance to selective inhibitors of nuclear export. Oncotarget. Impact Journals LLC; 2016;7:68842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soung YH, Pruitt K, Chung J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci Rep. Nature Publishing Group; 2014;4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soung YH, Kashyap T, Nguyen T, Yadav G, Chang H, Landesman Y, et al. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget. Impact Journals, LLC; 2017;8:52935–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeSisto JA, Flannery P, Lemma R, Pathak A, Mestnik S, Philips N, et al. Exportin 1 inhibition induces nerve growth factor receptor expression to inhibit the NF-kB pathway in preclinical models of pediatric high-grade glioma. Mol Cancer Ther. American Association for Cancer Research Inc.; 2020;19:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Sterr M, Ansarullah, Burtscher I, Böttcher A, Beckenbauer J, et al. Point mutations in the PDX1 transactivation domain impair human β-cell development and function. Mol Metab. Elsevier GmbH; 2019;24:80–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu J, Liu SH, Sanchez R, Nemunaitis J, Rozengurt E, Charles Brunicardi F. PDX1 associated therapy in translational medicine. Ann Transl Med. AME Publishing Company; 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined Immunodeficiency Associated with DOCK8 Mutations . N Engl J Med. New England Journal of Medicine (NEJM/MMS); 2009;361:2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 Mutations in Gliomas . N Engl J Med. New England Journal of Medicine (NEJM/MMS); 2009;360:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ventero MP, Fuentes-Baile M, Quereda C, Perez-Valeciano E, Alenda C, Garcia-Morales P, et al. Radiotherapy resistance acquisition in glioblastoma. Role of SOCS1 and SOCS3. PLoS One. Public Library of Science; 2019;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martini M, Pallini R, Luongo G, Cenci T, Lucantoni C, Larocca LM. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int J Cancer. Int J Cancer; 2008;123:2955–60. [DOI] [PubMed] [Google Scholar]
- 34.Mao R, Yang R, Chen X, Harhaj EW, Wang X, Fan Y. Regnase-1, a rapid response ribonuclease regulating inflammation and stress responses. Cell. Mol. Immunol Chinese Soc Immunology; 2017. page 412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kashyap T, Argueta C, Aboukameel A, Unger TJ, Klebanov B, Mohammad RM, et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget. Impact Journals LLC; 2016;7:78883–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ranganathan P, Kashyap T, Yu X, Meng X, Lai TH, McNeil B, et al. XPO1 inhibition using selinexor synergizes with chemotherapy in acute myeloid leukemia by targeting DNA repair and restoring topoisomerase IIα to the nucleus. Clin Cancer Res. American Association for Cancer Research Inc.; 2016;22:6142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner JG, Dawson JL, Grant S, Shain KH, Dalton WS, Dai Y, et al. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. J Hematol Oncol. BioMed Central Ltd.; 2016;9:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. Sequencing data are available on gene expression omnibus accession GSE186332.