

Abstract
Corticosterone, the stress hormone, exacerbates alcohol-associated tissue injury, but the mechanism involved is unknown. We examined the role of the glucocorticoid receptor (GR) in corticosterone-mediated potentiation of alcohol-induced gut barrier dysfunction and systemic response. Hepatocyte-specific GR deficient (GRΔHC) and intestinal epithelial-specific GR deficient (GRΔIEC) mice were fed ethanol, combined with corticosterone treatment. Intestinal epithelial tight junction integrity, mucosal barrier function, microbiota dysbiosis, endotoxemia, systemic inflammation, liver damage, and neuroinflammation were assessed. Corticosterone potentiated ethanol-induced epithelial tight junction disruption, mucosal permeability, and inflammatory response in GRΔHC mouse colon; these effects of ethanol and corticosterone were absent in GRΔIEC mice. Gut microbiota compositions in ethanol-fed GRΔHC and GRΔIEC mice were similar to each other. However, corticosterone treatment in ethanol-fed mice shifted the microbiota composition to distinctly different directions in GRΔHC and GRΔIEC mice. Ethanol and corticosterone synergistically elevated the abundance of Enterobacteriaceae and E. coli and reduced the abundance of Lactobacillus in GRΔHC mice but not in GRΔIEC mice. In GRΔHC mice, corticosterone potentiated ethanol-induced endotoxemia and systemic inflammation, but these effects were absent in GRΔIEC mice. Interestingly, ethanol-induced liver damage and its potentiation by corticosterone were observed in GRΔHC mice but not in GRΔIEC mice. GRΔIEC mice were also resistant to ethanol and corticosterone-induced inflammatory response in the hypothalamus. These data indicate that the intestinal epithelial GR plays a central role in alcohol and corticosterone-induced gut barrier dysfunction, microbiota dysbiosis, endotoxemia, systemic inflammation, liver damage, and neuroinflammation. This study identifies a novel target for potential therapeutic for alcohol-associated tissue injury.
Keywords: Tight junction, inflammation, hepatitis, hypothalamus, endotoxemia
Introduction
Chronic alcohol use has a causal relationship with at least 60 diseases, including alcoholic liver disease (ALD) and neurodegenerative disorders. Almost all heavy drinkers develop fatty liver, but only up to 35% of them develop hepatitis, and 10–20% develop cirrhosis (1), which supports the “second-hit” or “multiple hits” models to describe ALD pathogenesis (2). Alcohol misuse is comorbid with chronic stress disorders (CSD) (3–5). Alcohol use and CSD feedback into each other and exacerbate clinical symptoms. At present, no targeted therapy exists for CSD, ALD, or ALD-CSD comorbidity (6). Glucocorticoid analogs are the empirical anti-inflammatory therapy for alcoholic hepatitis but are ineffective in many patients and unsuitable for intermediate or long-term treatments. The mortality rate in steroid-resistant patients is 45% or higher (7). Dysregulated hypothalamic-pituitary-adrenal (HPA) axis and elevated plasma cortisol levels have been reported in alcoholics and ALD patients (3, 8–10). Elevated plasma corticosterone was also demonstrated in the experimental models of ALD (11–14). However, it is unclear whether the elevated glucocorticoid is a defense mechanism against alcoholic tissue injury or involved in the alcohol-associated tissue injury mechanism. A recent study showed that chronic restraint stress (CRS) and corticosterone treatment exacerbate ethanol-induced gut permeability, endotoxemia, and liver damage (15), suggesting that elevated glucocorticoid is involved in the mechanism of alcohol-associated tissue injury. Therefore, chronic stress and glucocorticoids are the potential second hits in the pathogenesis of alcohol-associated diseases.
Evidence indicates that endotoxemia is the common denominator in the pathogenesis of stress (15–17) and alcohol-associated diseases (18–20). Gut barrier dysfunction, altered gut microbiota composition (dysbiosis), and consequent endotoxemia are crucial steps in the mechanism of alcohol-induced systemic inflammation and multi-organ injury (21). Increased production of intestinal microbial lipopolysaccharide (LPS) due to dysbiosis and disruption of barrier function leads to LPS absorption into the portal circulation, targeting Kupffer cells and hepatocytes in the liver. The LPS-activated liver cells release a variety of pro-inflammatory cytokines and chemokines (22–24). Once released into the systemic circulation, LPS targets multiple organs by inducing a systemic inflammatory response. Dysbiosis of gut microbiota is associated with stress (25–27) and alcohol misuse (28). Human studies have highlighted the increased abundance of Enterobacteriaceae and decreased abundance of Akkermansia in alcohol use disorder patients without organ disease (29), and higher abundance of Haemophilus, Lactobacillus, and Bifidobacterium, and a lower abundance of Bilophila and Oscillospira in alcohol use disorder with hepatitis (30, 31). Therefore, a distinct composition of gut microbiota is expected in ALD-CSD comorbidity.
Stress is well known to activate the hypothalamic-pituitary-adrenal (HPA) axis, and the sustained elevation of cortisol plays a crucial role in the stress-induced pathophysiology (32–36). Alcohol misuse is known to disturb the HPA axis and elevate plasma glucocorticoids (9, 36, 37). Restraint stress and alcohol feeding in rodents elevate plasma corticosterone (15, 38). Glucocorticoids play a role in neuro-immune functions (39, 40); however, their role in alcohol-induced peripheral tissue injury is unclear. Cortisol and corticosterone actions are mediated by the glucocorticoid receptor (GR) (41). Colon and ileum express high levels of GR (41). Therefore, it is essential to dissect the role of GR in stress-alcohol interactions.
The primary component of the intestinal mucosal barrier function is the epithelial tight junction (TJ), which prevents diffusion of LPS from the gut lumen into the mucosa and subsequent absorption into the systemic circulation. The TJ is a multiprotein complex consisting of various proteins, including occludin, claudins, ZO-1, and cingulin (42–44). The TJ protein complex is fastened to the actomyosin belt at the apical end of the epithelial cells. Ethanol and its metabolite, acetaldehyde, synergistically disrupt the intestinal epithelial TJ by involving multiple intracellular signaling mechanisms (20, 45, 46), a crucial step involved in alcohol-induced endotoxemia. The adherens junction (AJ) consists of E-cadherin, β-catenin, and other proteins (47). It is located beneath the TJ and indirectly regulates the integrity of TJ (48).
In the present study, we examined the role of the glucocorticoid receptor (GR) in the alcohol and corticosterone-induced gut barrier dysfunction and systemic response using tissue-specific GR-knockout mice.
Materials and Methods
Chemicals and antibodies
Maltose dextrin was purchased from Bioserv (Flemington, NJ). Regular Lieber DeCarli ethanol diet (Dyet no. 710260) was purchased from Dyets Inc. (Bethlehem, PA). Hoechst 33342 dye was purchased from Life Technologies (Grand Island, NY). Anti-ZO-1 and anti-occludin antibodies were purchased from Invitrogen (Carlsbad, CA). Anti-E-cadherin and anti-β-catenin antibodies were purchased from BD Biosciences (Billerica, MA). AlexaFlour-488-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG were purchased from Molecular Probes (Eugene, OR). All other chemicals were purchased from either Sigma-Aldrich (St Louis, MO) or Thermo Fisher Scientific (Tustin, CA).
Animals and diets
B6.Cg-Nr3c1tma1.1jda/J (strain #021021) or GRFlox/Flox, B6.Cg.Tg(Vil1-cre)997Gum/J (strain #004586) or VillinCre, and B6.FVB(129)-Tg(Alb1-cre)1Dlr/J (strain #016832) or AlbCre mice were purchased from Jackson Laboratories (Sacramento, CA). GRFlox/Flox mice are floxed mutant mice that possess loxP sites flanking exon 3 of the Nr3c1 gene. VillinCre mice express Cre recombinase in villus and crypt epithelial cells of the intestine. AlbCre mice express Cre recombinase in hepatocytes in the liver. GRFlox/Flox-AlbCre (GRΔHC) lack GR gene exclusively in the hepatocytes, whereas and GRFlox/Flox-VillinCre (GRΔIEC) mice lack GR gene exclusively in the intestinal epithelium, including all types of cells in the intestinal epithelium. All animal experiments were performed according to the protocols approved by the University of Tennessee Health Science Center Institutional Animal Care and Use Committee. Animals were housed in an institutional animal care facility with 12:2-h light-dark cycles. All mice had free access to regular laboratory chow and water until the start of experiments. During the experiments, they were fed a liquid diet as described below.
Ethanol feeding
Adult (8 to 10 weeks old; male and female) GRΔHC and GRΔIEC mice were fed Lieber–DeCarli liquid diet for 4 weeks. Animals were gradually adapted to an ethanol-containing diet (0% for 2 days, 1% for 2 days, and 2% ethanol for 2 days, followed by 4% ethanol for 1 week, 5% for 1 week and 6% for 1 week). Control groups were pair-fed diets containing maltodextrin isocaloric to ethanol. Some groups of PF and EF mice were injected with corticosterone (25 mg/kg/day, s.c.). Control groups were injected with the vehicle. In all experiments, animals were maintained in pairs to facilitate body temperature maintenance and prevent social isolation. Body weights were recorded twice a week. Only the corticosterone group showed body weight changes significantly higher compared to other groups (Fig. S1). At the end of the experiment, the gut permeability was measured as described below. Blood samples were collected by cardiac puncture (into heparinized tubes) and centrifuged at 3,000 × g for 10 minutes at 4°C to prepare plasma samples. Pieces of distal colon and liver were fixed in buffered formalin or cryofixed in OCT, and the remainder of the tissues was stored in RNAlater for RNA isolation. Brain tissues were collected, and the hypothalamus was separated. Liver weights, normalized to body weights, were significantly higher in corticosterone with ethanol feeding group of GRΔHC mice; in GRΔIEC mice, liver weights were higher in corticosterone groups with or ethanol feeding (Fig. S2).
Gut permeability in vivo
At the end of each set of experiments, mice were intravenously injected with FITC-inulin (50 mg/ml solution: 2 μl/g body weight) via tail vein using a restrainer. One hour after injection, blood samples were collected by cardiac puncture under isoflurane anesthesia to prepare plasma. Mice were euthanized by cervical dislocation under isoflurane anesthesia. Luminal contents of intestinal segments were flushed with 0.9% saline. Fluorescence in plasma and luminal flushing samples was measured using a fluorescence plate reader. Fluorescence values in the luminal flushing were normalized to fluorescence values in corresponding plasma samples and calculated as percent of inulin load. This method measures the serosal-to-lumen flux of inulin, which involves no physical manipulation of the intestinal segment, and has been consistently correlated with epithelial TJ disruption (15, 49).
Immunofluorescence microscopy
Cryosections (10 μm thickness) of the colon were fixed in acetone and methanol mixture (1:1) at −20°C for 2 minutes and rehydrated in phosphate-buffered saline (PBS; 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM disodium hydrogen phosphate, and 1.8 mM potassium dihydrogen phosphate, pH 7.4). Sections were permeabilized with 0.2% Triton X-100 in PBS for 10 minutes and blocked in 4% nonfat milk in Triton–Tris buffer (150 mM sodium chloride containing 10% Tween-20 and 20 mM Tris, pH 7.4). It was then incubated for 1 hour with the primary antibodies (mouse monoclonal anti-occludin and rabbit polyclonal anti-ZO-1 antibodies or mouse monoclonal E-cadherin and rabbit polyclonal anti-β-catenin antibodies), followed by incubation for 1 hour with secondary antibodies (AlexaFluor-488-conjugated anti-mouse IgG and cy3-conjugated anti-rabbit IgG antibodies) and 10 min incubation with Hoechst 33342. The fluorescence was examined using a Zeiss 710 confocal microscope (Carl Zeiss Microscopy, Jena, Germany), and images from x–y sections (1 μm) were collected using LSM 5 Pascal software (Carl Zeiss Microscopy). Images were stacked using the software Image J (NIH, Bethesda, MD) and processed by Adobe Photoshop (Adobe Systems Inc., San Jose, CA). All images for tissue samples from the different groups were collected and processed under the identical laser, gain, optical thickness, and contrast conditions.
Quantitative RT-PCR
Total RNA (1.5 μg) was used to generate cDNAs using the ThermoScript RT-PCR system to synthesize the first strand (Invitrogen). Quantitative PCR (qPCR) reactions were performed using cDNA mix (cDNA corresponding to 35 ng RNA) with 300 nmoles of primers in a final volume of 25 μl of 2× concentrated RT2 Real-Time SYBR Green/ROX master mix (Qiagen) in an Applied Biosystems QuatStudio 6-Flex Real-Time PCR instrument (Norwalk, CT, USA). The cycle parameters were: 50 °C for 2 min, one denaturation step at 95 °C for 10 min and 40 cycles of denaturation at 95 °C for 10s followed by annealing and elongation at 60 °C. The relative gene expression of each transcript was normalized to GAPDH using the ΔΔCt method. Sequences of primers used for qPCR are provided in Table S1.
Microbiome analyses
Microbiome analyses of colonic flushing were performed by 16S rRNA sequencing and metagenomic analyses, as described recently (15, 50).
DNA extraction and Illumina MiSeq sequencing:
Colonic flushing samples were resuspended in 500 μL of TNES buffer containing 200 units of lyticase and 100 μL of 0.1/0.5 (50/50 Vol.) zirconia beads. Incubation was performed for 20 min at 37°C. Following mechanical disruption using ultra-high-speed bead beating, 20 μg of proteinase K was added to all samples, and they were incubated overnight at 55°C with agitation. Total DNA was extracted using chloroform-isoamyl-alcohol mixture (25:24:1), and total DNA concentration per mg stool was determined by qRT-PCR. Purified DNA samples were sent to the Argonne National Laboratory (Lemont, IL) for amplicon sequencing using the NextGen Illumina MiSeq platform. Blank samples passed through the entire collection, extraction, and amplification process remained free of DNA amplification.
Bioinformatics:
Sequencing data were processed and analyzed using QIIME (Quantitative Insights into Microbial Ecology) 1.9.1 Sequences were first demultiplexed, then denoised, and clustered into sequence variants. For bacteria, we rarified to a depth of 10,000 sequences. Representative bacterial sequences were aligned via PyNAST, with taxonomy assigned using the RDP Classifier. Processed data were then imported into Calypso 8.84 for further analysis and data visualization (51). The Shannon index was used to quantify α-diversity (52, 53). Bray-Curtis analysis was used to quantify β-diversity, and the differences were compared using PERMANOVA with 999 permutations. For fungi, sequences were aligned, and taxonomy was assigned using the UNITE metabarcoding (dynamic setting) database (54). Fungal OTUs were rarified at a depth of 300 sequences for α-diversity using Chao1 and β-diversity based on Jaccard dissimilarities at the OTU level (55). As with bacteria, beta diversity was then assessed using permutational multivariate analysis of variance (PERMANOVA). To quantify the relative abundance of taxa between groups (56), we utilized ANOVA adjusted using the Bonferroni correction and FDR for multiple comparisons. We used linear discriminant analysis of effect size (LEfSe) to test for significance and perform high-dimensional biomarker identification (57).
Plasma endotoxin assay
Plasma endotoxin concentrations were measured, as described before (15), using Pierce LAL Chromogenic Endotoxin Quantitation Kit (Thermo Scientific, Cat# 88282).
Plasma cytokine assay
Plasma cytokine and chemokine were measured with a Duoset ELISA kit per manufacturer instructions (R&D system, Minneapolis, MN). Briefly, fifty microliters of plasma were incubated in the capture antibody-coated microplates overnight, followed by incubation with detection antibody for 2 hours. It was then incubated with horseradish peroxidase-conjugated streptavidin for 20 min and with substrate solution for 20 min. The reaction was stopped by stop-solution, and absorbance was measured at 450 nm with wavelength correction at 570 nm.
Liver Histopathology
Liver tissues were fixed in 10% buffered formalin, and 8 μm-thick paraffin-embedded sections were stained with hematoxylin and eosin (H&E), as described before (15). Stained sections were imaged in a Nikon 80Ti microscope (Nikon Instruments, Inc., Melville, NY) using a 10x objective lens and a color camera.
Plasma Transaminase Assay
Plasma alanine transaminase (ALT) and aspartate transaminase (AST) activities were measured by colorimetric assay using ALT (Cayman, Cat# 700260) and AST (Cayman, Cat# 7012640) assay kits, according to Vendor’s instructions (15).
Triglyceride Assay
Liver triglycerides were estimated, as described before (15), using the GPO method and an assay kit from Beckman Coulter (Brea, CA). Briefly, lipids were extracted by digesting the tissue with 3 M potassium hydroxide (in 65% EtOH) for 1 hour at 70°C followed by 24 hours at room temperature. Triglycerides were measured by enzymatic hydrolysis into glycerol and free fatty acids, followed by colorimetric measurement of glycerol (at 540 nm wavelength). Triglyceride values were expressed as mg/g liver tissue.
Statistical analyses
All data are expressed as Mean ± SEM. The differences among multiple groups were first analyzed by ANOVA (Prism 6.0). When a statistical significance was detected, Tukey’s t-test was used to determine the statistical significance between multiple testing groups and the corresponding control. Statistical significance was established at 95%.
Results
Deletion of intestinal epithelial GR attenuates alcohol and corticosterone-induced disruption of TJ and AJ
Stress is comorbid with alcohol-associated diseases and disorders (3–5). A recent study showed that stress exacerbates ethanol-induced intestinal epithelial barrier dysfunction, endotoxemia, and liver damage (15); this effect of stress could be reproduced by the administration of corticosterone, the primary stress hormone. To determine the role of tissue-specific GR in ethanol and corticosterone-induced gut barrier dysfunction and liver damage, we generated hepatocyte-specific (GRΔHC) and intestinal epithelial-specific (GRΔIEC) mice by crossing GRFlox/Flox mice with AlbCre or VillinCre mice. GRΔHC and GRΔIEC mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) with or without the combination of corticosterone (Cort) treatment.
Confocal immunofluorescence microscopy for occludin and ZO-1 (major TJ proteins) shows the qualitative changes in the TJ integrity. The interaction between occludin and ZO-1 is necessary to maintain TJ dynamics and epithelial barrier function (58). Therefore, co-localization of occludin and ZO-1 at the intercellular junctions is a well-established indicator of TJ integrity (44, 59). Disruption of TJ by various injurious factors results in loss or redistribution of occludin and ZO-1 from the intercellular junctions. Current data show that occludin and ZO-1 are co-localized at the intercellular junctions of the colonic epithelium of pair-fed GRΔHC and GRΔIEC mice (Fig. 1a), indicating the presence of intact TJ. In GRΔHC mice, ethanol feeding reduced the junctional distribution of occludin and ZO-1, which was confirmed by semi-quantitation of ZO-1 (Fig. 1b) and occludin (Fig. 1c) fluorescence at the intercellular junctions. These data indicate that ethanol disrupts colonic epithelial TJ in GRΔHC mice. Corticosterone administration did not appear to change the occludin and ZO-1 distribution, but fluorescence densities indicated a slight reduction of junctional occludin and ZO-1 by corticosterone. Fluorescence images indicate that corticosterone exacerbated the ethanol-induced loss of junctional occludin and ZO-1 (Fig. 1a). However, ZO-1 (Fig. 1b) and occludin (Fig. 1c) fluorescence densities indicated no difference in the levels of these proteins in the ethanol-fed mouse colon with or without corticosterone treatment. This finding suggests that qualitative rather than quantitative differences in colonic epithelial TJ exist in the vehicle and corticosterone-treated ethanol-fed GRΔHC mice. Strikingly, ethanol and corticosterone-induced redistribution of occludin and ZO-1 fluorescence were absent in GRΔIEC mouse colon; instead, ZO-1 density was significantly higher in ethanol-fed mouse colon, and ethanol slightly reduced occludin density.
Figure 1: Deletion of intestine epithelial GR attenuates alcohol and corticosterone-induced disruption of intestinal epithelial apical junctional complexes.
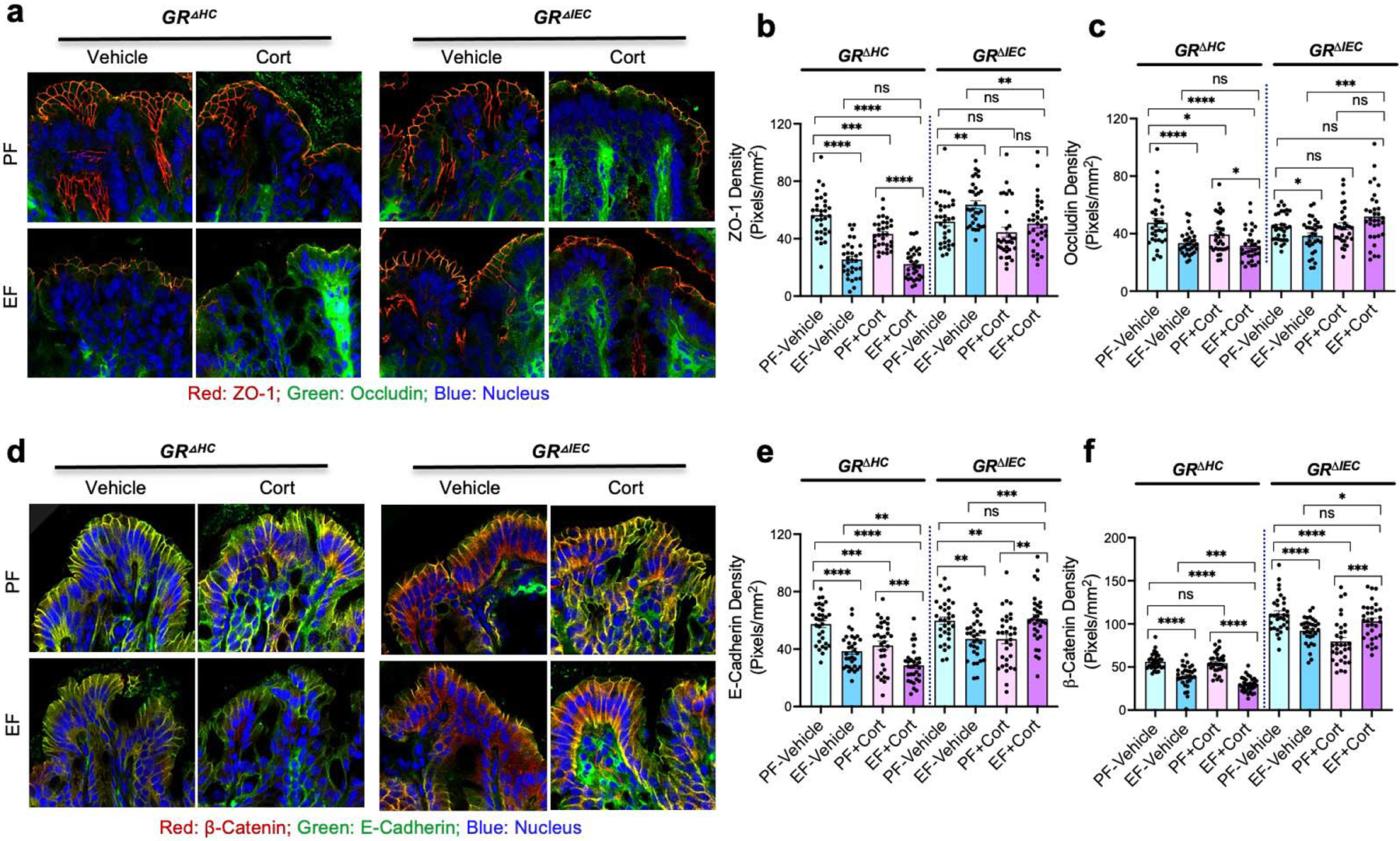
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. a-c: Tight junction integrity was assessed by immunofluorescence staining of colon cryosections for occludin and ZO-1 (a; green, occludin; red, ZO-1; blue, nucleus) and confocal microscopy. The ZO-1 (b) and occludin (c) fluorescence densities were measured at the epithelial junctions. d-f: Adherens junction integrity was assessed by staining colon sections for E-cadherin and β-catenin (b; green, E-cadherin; red, β-catenin; blue, nucleus) and measuring E-cadherin (e) and β-catenin (f) densities at the epithelial junctions. Values in panels b, c, e, and f are Mean ± SEM (n = 6); * = p<0.05, ** = p<0.01, *** = p<0.005, and **** = p<0.001 for significant difference between the indicated groups; “ns” = not significant.
E-cadherin and β-catenin are major AJ proteins, and the interaction (co-localization) between these proteins is essential to maintain AJ integrity. Reduced amounts, redistribution, or separation from each other indicate the loss of AJ integrity. The current data show that E-cadherin and β-catenin are co-localized in the colonic epithelial junctions in pair-fed GRΔHC and GRΔIEC mouse colons, indicating the presence of intact AJ in these mice (Fig. 1d). Ethanol reduced E-cadherin and β-catenin fluorescence in the GRΔHC mouse colonic epithelium, and corticosterone treatment potentiated these effects of ethanol. The effects of ethanol and corticosterone were confirmed by quantitation of E-cadherin (Fig. 1e) and β-catenin (Fig. 1f) fluorescence densities. The effects of ethanol and corticosterone on E-Cadherin and β-catenin distribution were much reduced in the GRΔIEC mouse colon. The β-catenin fluorescence in the GRΔIEC mouse colon was higher in all groups compared to the GRΔHC mouse colon.
These data indicate that ethanol and corticosterone-mediated TJ and AJ disruption in mouse colon requires intestinal epithelial GR, but not the hepatocyte GR. While changes in the interaction between occludin and ZO-1 are an absolute requirement to maintain TJ integrity, changes in the cellular levels of these proteins may change the interaction between them. Therefore, the localization of these proteins at the junctions (Fig. 1a) is a better indicator of the differences in the TJ integrity in different groups. The measurement of fluorescence densities at the junctions (Fig. 1b & 1c) supports the interpretation of images. Similarly, localization of E-cadherin and β-catenin at the junctions is an indicator of AJ integrity. Again, the fluorescence density measurements provide supporting information. Since the animal groups were pair-fed, the alcohol intake and calorie intake are similar in all groups; the differences in the results are mainly due to the ethanol, corticosterone, and strain differences.
Intestinal epithelial GR is required for alcohol and corticosterone-mediated gut permeability and mucosal inflammatory response
Ethanol feeding increased mucosal permeability to FITC-inulin (6 kDa) in GRΔHC mouse colon (Fig. 2a) and ileum (Fig. 2b). Corticosterone administration by itself slightly increased the colonic mucosal permeability to inulin, but it significantly potentiated ethanol-induced increase in mucosal permeability. These effects of ethanol and corticosterone were absent in GRΔIEC mice. Disruption of epithelial junctions and increase in mucosal permeability leads to diffusion of toxins from the intestinal lumen into the lamina propria, which induces an inflammatory response. We measured mRNA for cytokines and chemokine in the colonic mucosa. Data show that mRNA for TNFα (Fig. 3a), IL-1β (Fig. 3b), IL-6 (Fig. 3c), and MCP1 (Fig. 3d) was significantly elevated by ethanol feeding in GRΔHC mice. Corticosterone treatment also significantly increased mRNA for these cytokines and MCP1. Corticosterone potentiated the ethanol effects on cytokine expression; ethanol-induced increase in TNFα and IL-1β mRNA was synergistic. These data indicate that the effects of ethanol and corticosterone on the intestinal barrier dysfunction and mucosal inflammatory response require intestinal epithelial GR.
Figure 2: Intestinal epithelial GR is required for alcohol and corticosterone-induced gut permeability.
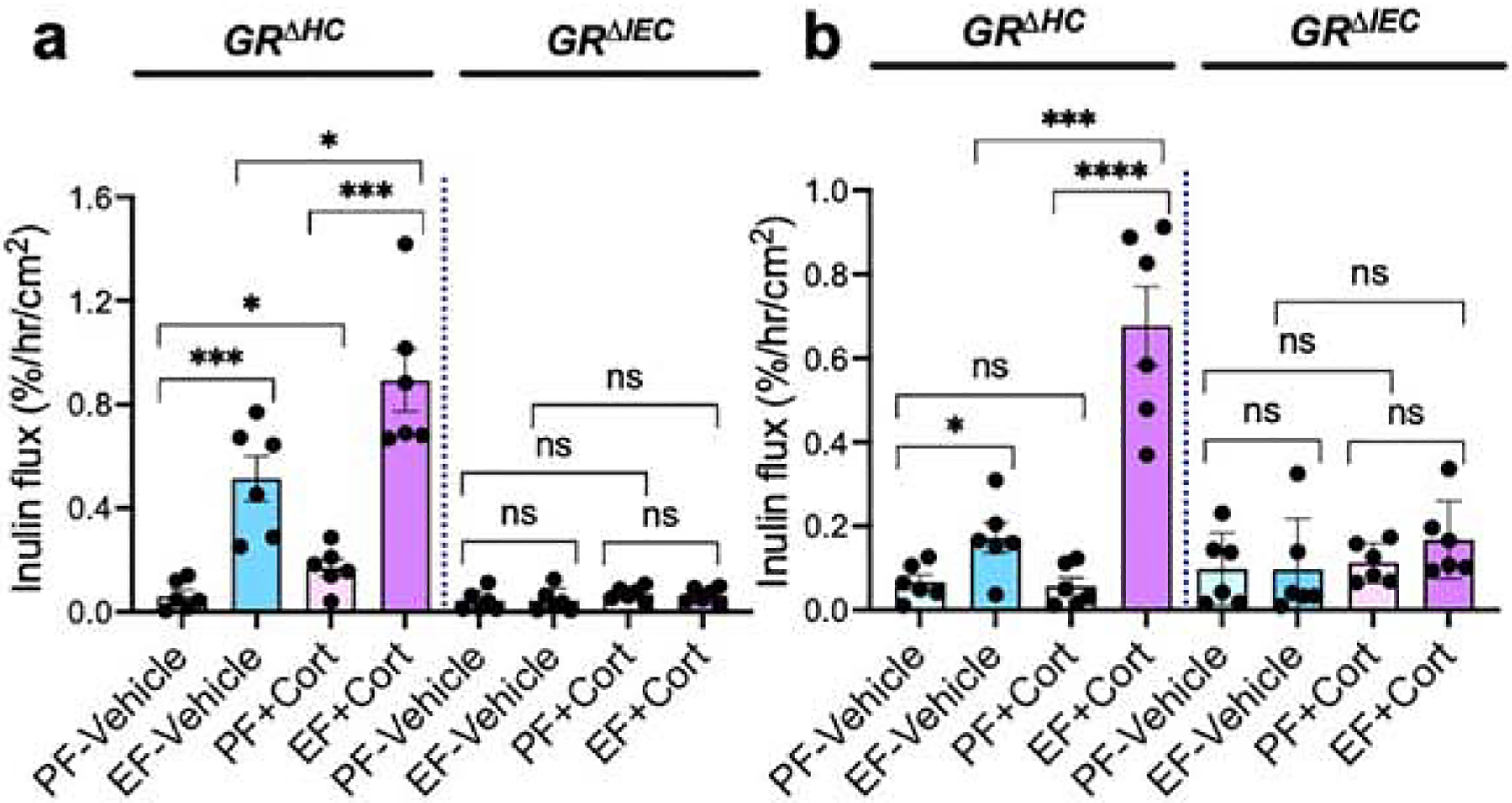
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. a & b: Mucosal permeability in the colon (a) and ileum (b) was evaluated by measuring the vascular-to-luminal flux of FITC-inulin in vivo. Values are Mean ± SEM (n = 6); * = p<0.05, ** = p<0.01, *** = p<0.005, and **** = p<0.001 for significant difference between the indicated groups; “ns” = not significant.
Figure 3: Intestinal epithelial GR is required for alcohol and corticosterone-induced mucosal inflammatory response in the colon.
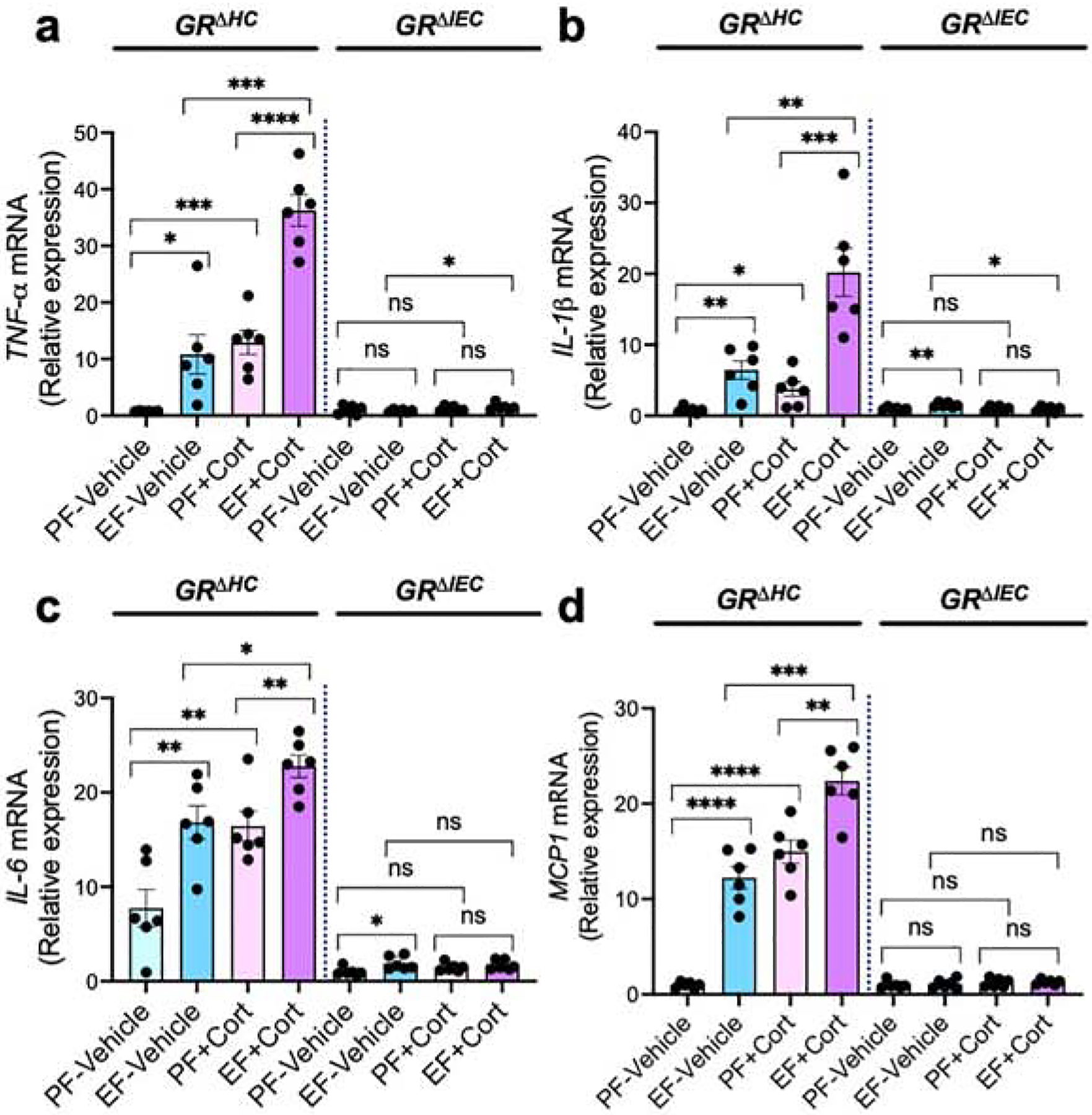
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. RNA isolated from colonic mucosa was analyzed for mRNA specific for TNFα (a), IL-1β (b), IL-6 (c), and MCP1 (d) by RT-qPCR. Values are Mean ± SEM (n = 6); * = p<0.05, ** = p<0.01, *** = p<0.005, and **** = p<0.001 for significant difference between the indicated groups; “ns” = not significant.
Deletion of GR from the intestinal epithelium modifies alcohol and corticosterone-induced dysbiosis of gut microbiota.
Both ethanol and corticosterone are known to alter the gut microbiota composition. Our recent study indicated that a combination of ethanol and corticosterone treatment further disturbed the gut microbiota diversity, composition, and abundance in the mouse intestine (15). Here we examined the potential role of GR in ethanol and corticosterone-induced dysbiosis of gut microbiota by 16S ribosomal RNA (rRNA) sequencing and taxonomic analyses. As shown before, alcohol and corticosterone themselves altered the taxonomic composition of colonic microbiota in both GRΔHC and GRΔIEC mice (Fig. 4a). There was a significant difference between GRΔHC and GRΔIEC mice when ethanol feeding was combined with corticosterone treatment; a relatively high abundance of Proteobacteria and Bacteroides was present in GRΔHC mice, whereas the abundance of Firmicutes was very high in GRΔIEC mice. Evaluation of α-diversity at the genus level by Shannon index analysis showed that ethanol (Fig. 4b) and corticosterone (Fig. 4c) reduced microbial diversity in both GRΔHC and GRΔIEC mice, although the effect of ethanol was not statistically significant. However, significant differences in microbial diversity were detected between GRΔHC and GRΔIEC mice treated with ethanol and corticosterone (Fig. 4d). Ethanol and corticosterone combined treatment caused a significant reduction in α-diversity in GRΔHC mice. On the contrary, in GRΔIEC mice, alcohol and corticosterone increased α-diversity. Analysis of β-diversity by Bray-Curtis plotting showed that the microbiota diversities in GRΔHC and GRΔIEC mice under basal conditions are different from each other (Fig. 4e), whereas ethanol feeding attenuated this difference. Similarly, corticosterone reduced the microbiota differences between GRΔHC and GRΔIEC mice (Fig. 4f). Whereas ethanol-fed GRΔHC and GRΔIEC mice exhibited similar microbial diversity, combining it with corticosterone treatment caused a remarkable shift in the microbiota diversity in distinct directions (Fig. 4g). Spearman’s correlation at the genus level displayed as a heatmap confirmed substantial differences between the ethanol and corticosterone-treated GRΔHC and GRΔIEC mice (Fig. 4h); the differences were minimal in ethanol-fed or corticosterone-treated GRΔHC and GRΔIEC mice (Fig. S3). Spearman’s correlation and linear discriminant analysis effect size (LEfSe; Fig. S4–S6) indicated that ethanol feeding along with corticosterone treatment selectively altered genera of bacteria that are distinctly different between GRΔHC and GRΔIEC mice. Some striking differences include a high abundance of Lactobacillus and Clostridiales in GRΔIEC mice and a high abundance of Bacteroides, Enterococcus, Pantoea, Pseudomonas, and Erwinia in GRΔHC mice. The differences between GRΔHC and GRΔIEC mice in Enterobacteriaceae, E. coli, and Lactobacillus changes were confirmed by RT-qPCR of 16S rDNA in the colonic flushing. Data show that, in GRDHC mice, both ethanol and corticosterone significantly increased the abundance of Enterobacteriaceae, which was several folds higher when ethanol feeding was combined with corticosterone treatment (Fig. 4i). Such effects of ethanol and corticosterone on Enterobacteriaceae were absent in GRΔIEC mice, except that corticosterone significantly reduced Enterobacteriaceae abundance. In GRΔHC mice, both ethanol and corticosterone significantly elevated (18–20 folds) the abundance of E. coli in the colon (Fig. 4j); a combination of ethanol feeding with corticosterone treatment induced a robust increase in E. coli abundance (6.5 folds over ethanol group and 118 folds over pair-fed group). On the other hand, both ethanol and corticosterone reduced the abundance of Lactobacillus in GRΔHC mice (Fig. 4k), whereas the abundance of Lactobacillus was maintained high among all groups of GRΔIEC mice. Results suggest that intestinal epithelial GR plays a significant role in maintaining gut microbiota composition.
Figure 4: Deletion of GR from the intestinal epithelium modifies alcohol and corticosterone-induced alteration of gut microbiota composition.
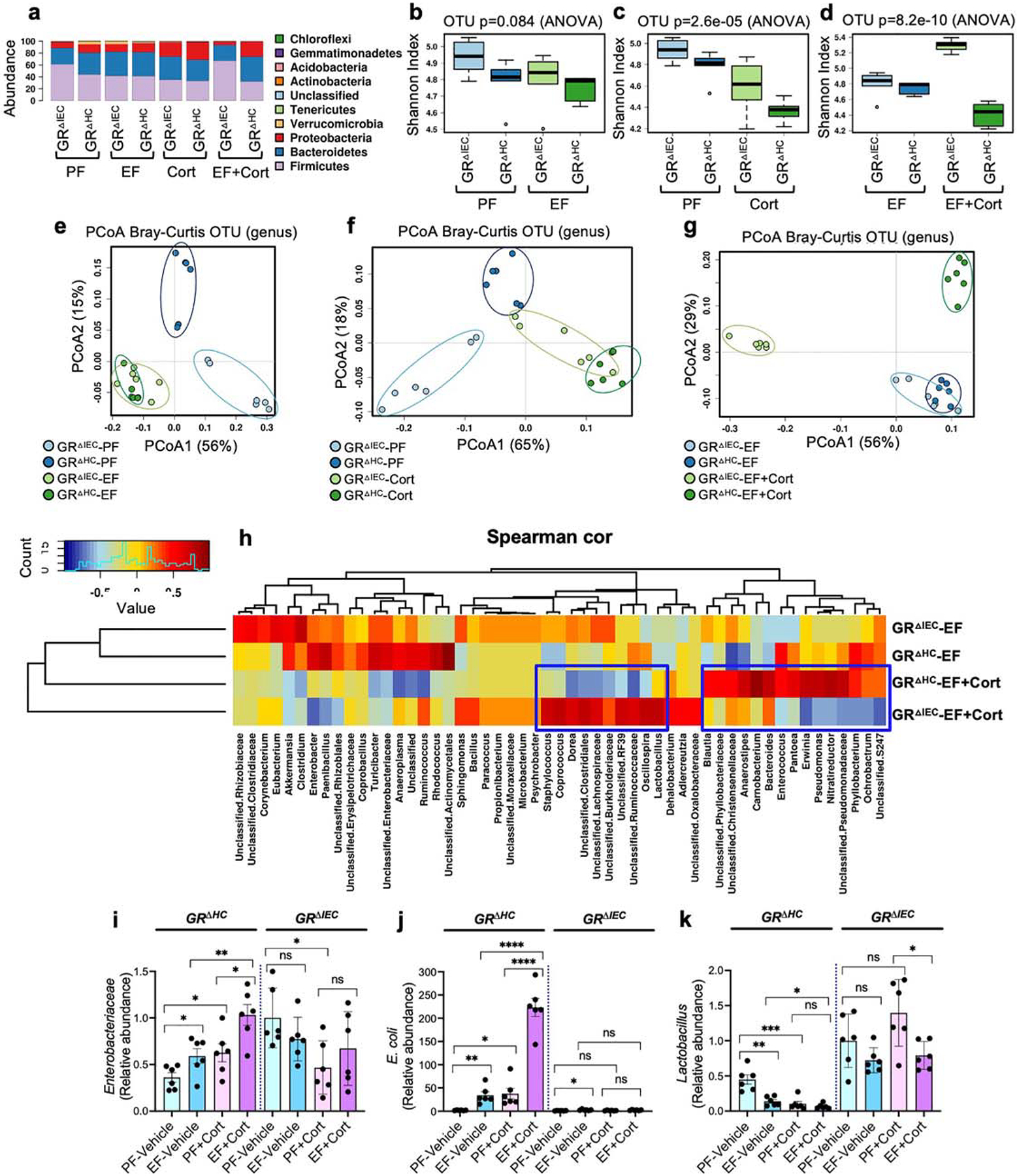
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. The composition of microbiota in colonic flushing was analyzed by 16S rRNA-sequencing and metagenomics. a: The relative abundance of different phyla of bacteria. b-d: The Shannon Index was used to quantify α-diversity. e-g: Principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity analysis was performed to determine β-diversity. h: Spearman’s correlation of microbiota at the genus level in different experimental groups. Blue boxes identify some of the genera of bacteria with significant differences between the GRΔHC and GRΔIEC EF+Cort groups. i-k: 16S ribosomal DNA from flushing samples were analyzed for the abundance of Enterobacteriaceae (i), E. coli (j), and Lactobacillus (k) by RT-qPCR. Values are mean ± SEM (n = 6); * = p<0.05, ** = p<0.01, and *** = p<0.005 for significant difference between the indicated groups; “ns” = not significant.
Role of intestinal epithelial GR in alcohol and corticosterone-induced endotoxemia and systemic inflammation
Dysbiosis of gut microbiota and epithelial TJ disruption lead to LPS absorption, causing endotoxemia. Analysis of endotoxemia by measuring plasma LPS showed that ethanol and corticosterone caused a significant elevation of plasma LPS in GRΔHC mice (Fig. 5a); this increase was much higher when ethanol feeding was combined with corticosterone administration. However, in GRΔIEC mice, ethanol and corticosterone failed to increase plasma LPS, indicating that ethanol and corticosterone-induced endotoxemia requires the intestinal epithelial GR. Endotoxemia targets all organ systems and leads to the systemic inflammatory response (60). We assessed systemic inflammation by measuring TNFα, IL-6, and MCP1 in the plasma by ELISA. Data show that ethanol significantly elevated plasma TNFα (Fig. 5b), IL-6 (Fig. 5c), and MCP1 (Fig. 5d) in GRΔHC mice. Corticosterone did not significantly alter plasma cytokines but induced a slight elevation of MCP1. Corticosterone, however, potentiated ethanol-induced increase in plasma TNFα, IL-6, and MCP1. In GRΔIEC mice, ethanol slightly elevated plasma TNFα and IL-6, but the combined effect of ethanol and corticosterone on plasma cytokines and chemokine was absent. These data indicate that intestinal epithelial GR plays a crucial role in ethanol and corticosterone-induced endotoxemia and systemic inflammation.
Figure 5: Intestinal glucocorticoid receptor determines alcohol and corticosterone-induced endotoxemia and systemic inflammation.
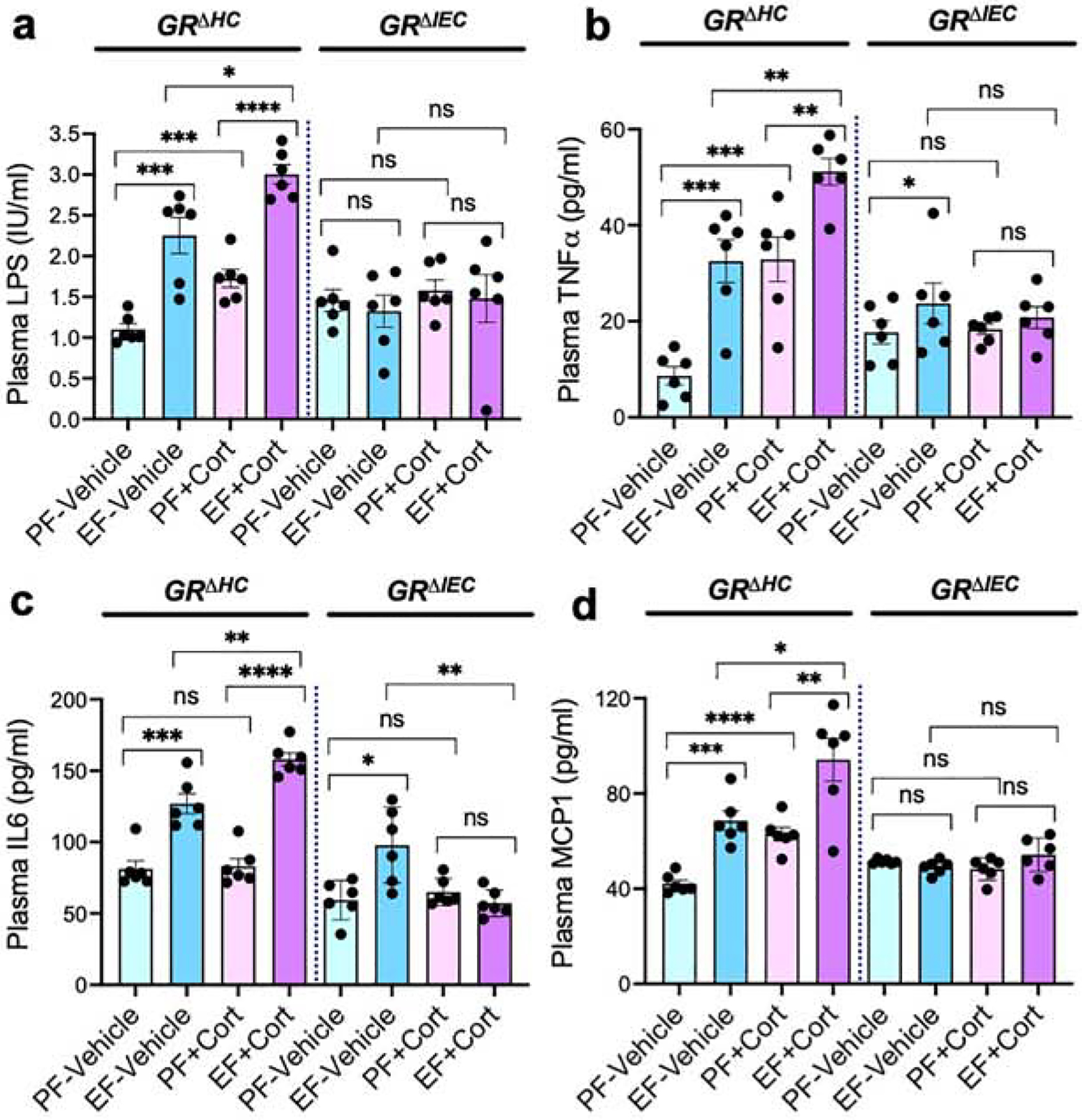
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. a: Endotoxemia was assessed by measuring plasma LPS levels. b-d: Levels of TNFα (b), IL-6 (c), and MCP1 (d) were measured in plasma by ELISA. Values in graphs are mean ± SEM (n = 4); * = p<0.05, ** = p<0.01, and *** = p<0.005 for significant difference between the indicated groups; “ns” = not significant.
Intestinal epithelial GR is an essential requirement in alcohol and corticosterone-induced liver damage.
A previous study demonstrated that chronic stress and corticosterone exacerbate ethanol-induced liver damage in mice (15). Here we confirm this corticosterone effect on liver damage in GRΔHC mice. Corticosterone exacerbates ethanol-induced increase in plasma ALT (Fig. 6a) and AST (Fig. 6b). Liver damage was confirmed by histopathology, which showed a larger number of vacuole-like spots presumably representing lipid droplets in the GRΔHC mice (Fig. 6c). Ethanol and corticosterone-induced steatosis were assessed by measuring liver triglyceride, which showed potentiation of ethanol-induced increase in liver triglyceride by corticosterone (Fig. 6d). The inflammatory response in the liver was assessed by measuring TNFα, IL-1β, and IL-6 mRNA. Data show that ethanol elevated TNFα (Fig. 6e), IL-1β (Fig. 6f), and IL-6 (Fig. 6g) mRNA in GRΔHC mouse liver and corticosterone significantly potentiated these effects of ethanol. The ethanol and corticosterone-induced liver damage in GRΔHC mice indicates that GR in the hepatocyte is not involved in alcohol and corticosterone-induced liver damage. Interestingly, corticosterone alone significantly elevated liver cytokine mRNA under our experimental conditions. On the other hand, ethanol and corticosterone-induced elevation of plasma AST/ALT, liver triglyceride, and liver cytokine mRNA were absent in GRΔIEC mice. These results indicate that GR in the intestinal epithelium, but not hepatocyte GR, is required for alcohol and corticosterone-induced liver damage.
Figure 6: Glucocorticoid receptor in the intestine, but not that in the liver, determines alcohol and corticosterone-induced liver damage.
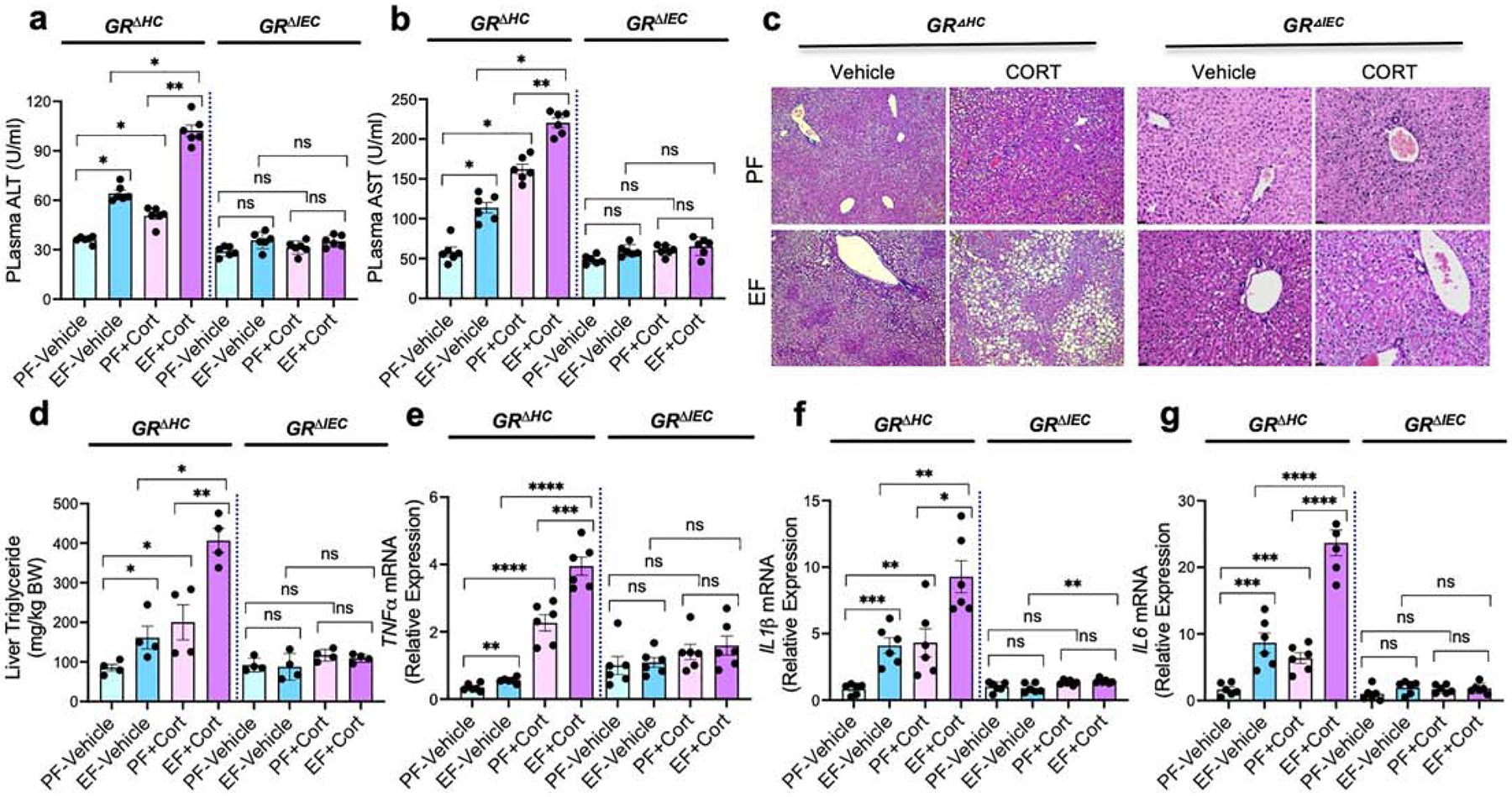
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. a & b: Plasma was analyzed for ALT (a) and AST (b) activities. c: Liver histopathology was performed by H & E staining and bright field microscopy. d: Steatosis was assessed by measuring liver triglyceride content. e-g: Inflammatory responses in the liver were determined by measuring specific mRNA for TNFα (e), IL-1β (f), and IL-6 (g). Values in graphs are mean ± SEM (n = 6); * = p<0.05, ** = p<0.01, **** = p<0.005, and **** = p<0.001 for significant difference between the indicated groups; “ns” = not significant.
Intestinal epithelial GR plays an essential role in alcohol and corticosterone-induced neuroinflammation.
Alcohol misuse is associated with neuroinflammation (61). Alteration of the HPA axis and sustained elevation of plasma glucocorticoid is a common finding in alcohol use disorders (3, 8–10). We assessed inflammatory response in the hypothalamus by measuring IL-6 and MCP1 mRNA, the representative cytokine and chemokine. In GRΔHC mice, both ethanol and corticosterone significantly elevated IL-6 (Fig. 7a) and MCP1 (Fig. 7b) mRNA in the hypothalamus. However, the increase in these cytokine and chemokine mRNA by combined ethanol and corticosterone treatment was significantly higher than the sum of the individual effects of ethanol and corticosterone. These data indicate that ethanol and corticosterone synergistically induce an inflammatory response in the hypothalamus. Interestingly, ethanol and corticosterone-induced inflammatory response in the hypothalamus was absent in GRΔIEC mice, indicating that GR receptor activation in the intestinal epithelium is a key step in ethanol and corticosterone-induced neuroinflammation.
Figure 7: Glucocorticoid receptor in the intestine, but not that in the liver, determines alcohol and corticosterone-induced neuroinflammation.
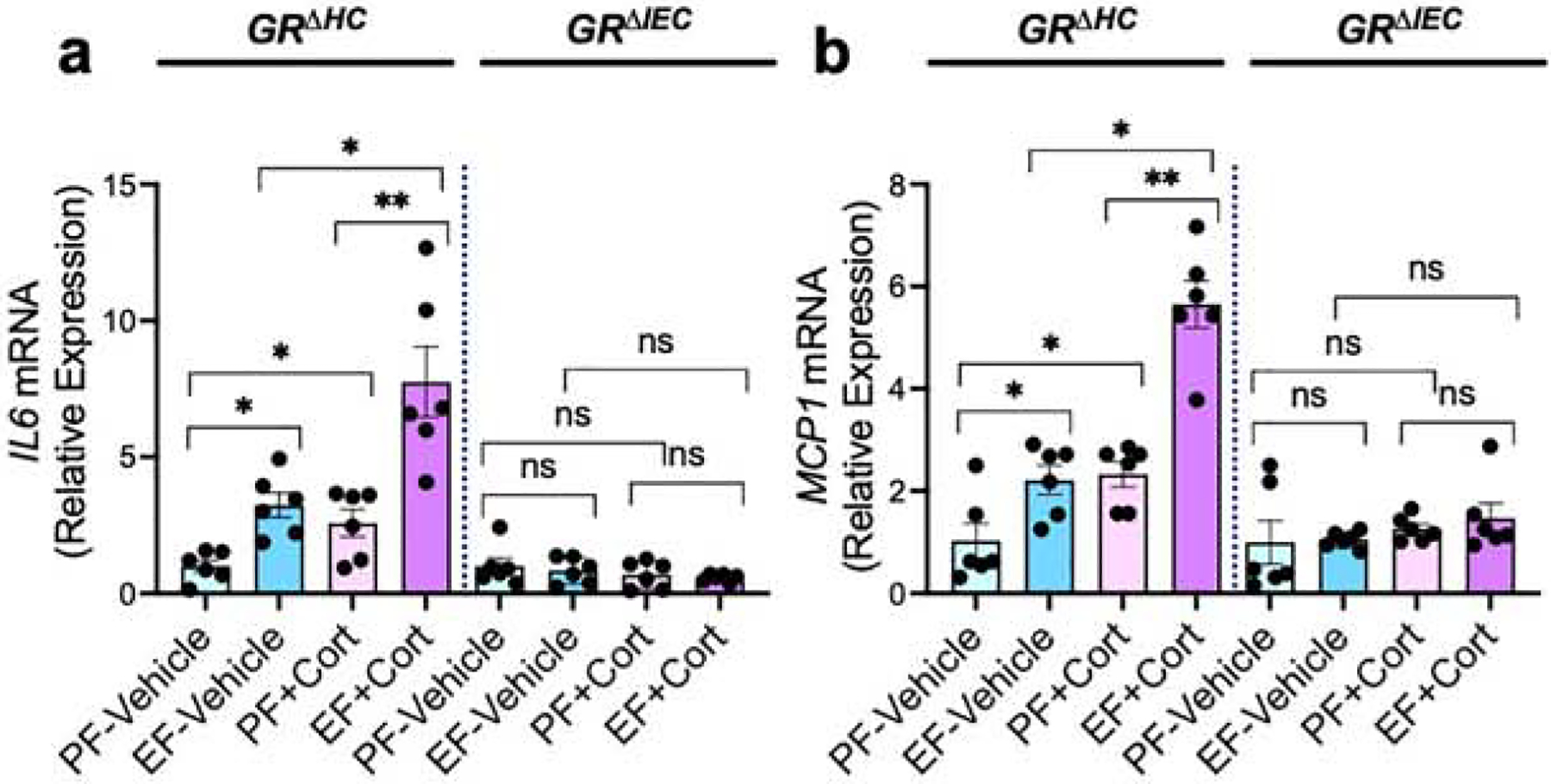
Adult GRΔHC and GRΔIEC mice were fed a liquid diet with (EF) or without (PF) ethanol for four weeks. In some groups, animals were injected with corticosterone (Cort) daily; the other groups received the vehicle. Inflammatory responses in the hypothalamus were assessed by measuring IL-6 (a) and MCP1 (b)-specific mRNA. Values in graphs are mean ± SEM (n = 6); * = p<0.05 and ** = p<0.01 for significant difference between the indicated groups; “ns” = not significant.
Discussion
Chronic stress and elevated plasma glucocorticoids are co-morbid with alcohol-associated diseases. Gut barrier dysfunction and dysbiosis of gut microbiota are crucial steps in the pathogenesis of alcohol-associated diseases. Therefore, investigating the alcohol and glucocorticoid targets in the intestinal epithelium is essential to identify therapeutic targets to treat ALD-CSD co-morbidity. The present study shows that intestinal epithelial GR plays a crucial role in alcohol and corticosterone-induced gut injury, liver damage, and neuroinflammation. Deletion of GR from the intestinal epithelial cells attenuates alcohol and corticosterone-induced: 1) epithelial TJ disruption, mucosal barrier dysfunction, and mucosal inflammatory response, 2) alteration of gut microbiota composition, 3) endotoxemia and systemic inflammation, 4) liver damage, and 5) neuroinflammation. Deletion of GR from the hepatocytes, however, failed to block alcohol and corticosterone effects. Overall, this study identifies intestinal epithelial GR as a potential initial target in the pathogenesis of alcohol-associated tissue injury.
Chronic alcohol consumption has previously been shown to alter the HPA axis and cause sustained elevation of plasma corticosterone (9, 62). Chronic ethanol elevates plasma corticosterone in mice, and corticosterone potentiates ethanol-induced disruption of intestinal epithelial TJ and AJ (15). However, the role of GR in these effects of ethanol or corticosterone is unknown. Here we show that deletion of GR from the intestinal epithelium attenuates alcohol and corticosterone-induced TJ and AJ disruption, indicating that GR-mediated gene expression changes in the intestinal epithelium may determine the alcohol-induced epithelial TJ disruption and mucosal barrier dysfunction. Epithelial barrier dysfunction, assessed by measuring the vascular-to-luminal flux of FITC-inulin in vivo, showed that GR deletion from the intestinal epithelium also blocks alcohol and corticosterone-induced barrier dysfunction. Epithelial barrier dysfunction is known to cause diffusion of luminal microbial toxins across the epithelium into the lamina propria of intestinal mucosa leading to initiation of inflammatory response in the mucosa. The synergistic elevation of cytokine and chemokine mRNA in GRΔHC mice but not in GRΔIEC mice indicated that the intestinal epithelial GR is required for alcohol and corticosterone-induced intestinal mucosal inflammatory response. Therefore, the intestinal epithelium is a crucial target for altered HPA axis and elevated plasma glucocorticoids caused by chronic alcohol consumption.
The second major factor involved in alcohol-associated endotoxemia is dysbiosis of gut microbiota. Numerous studies have shown that alcohol use alters the abundance and composition of gut microbiota (29–31). Our recent study demonstrated that corticosterone further modulates alcohol-induced alteration of gut microbiota in mice (15). However, the precise mechanism involved in alcohol or stress-induced dysbiosis of gut microbiota is poorly defined. Therefore, we asked the question of whether GR played a role in alcohol and corticosterone-induced microbiota dysbiosis. Our data show that the diversity of microbiota within the group was significantly high in pair-fed, ethanol-fed, corticosterone-treated, and ethanol + corticosterone-treated GRΔIEC mice, indicating the absence of intestinal epithelial GR increases microbiota diversity under all conditions. Analyses of β-diversity indicated that microbiota composition in GRΔHC is distinctly different from GRΔIEC mice; ethanol feeding and corticosterone treatment reduced the differences in microbiota diversity between the strains. However, the microbiota composition in GRΔHC and GRΔIEC mice was distinctly different from each other when ethanol feeding was combined with corticosterone treatment. Spearman’s correlation and LDA analysis confirmed the vast difference between microbiota composition in ethanol + corticosterone-treated GRΔHC and GRΔIEC mice. Some striking differences included a high abundance of genera under the Enterobacteriaceae family in GRΔHC mice and a high abundance of Lactobacillus in GRΔIEC mice. Furthermore, RT-qPCR confirmed that ethanol and corticosterone synergistically increased Enterobacteriaceae and E. coli in GRΔHC mice but not in GRΔIEC mice. On the other hand, ethanol and corticosterone drastically reduced the abundance of Lactobacillus in GRΔHC mice, whereas a high abundance of Lactobacillus was maintained in all groups of GRΔIEC mice. These data suggest that the absence of intestinal epithelial GR may prevent ethanol and corticosterone-induced increase in pathogenic bacteria such as E. coli and maintain a high abundance of beneficial bacteria such as Lactobacillus species.
Dysbiosis of gut microbiota and epithelial barrier dysfunction leads to endotoxemia. Here we show that corticosterone potentiates alcohol-induced elevation of plasma LPS in GRΔHC mice but not in GRΔIEC mice. Endotoxemia is known to cause systemic inflammation. The present study shows that ethanol and corticosterone-induced elevation of plasma cytokines and chemokine is absent in GRΔIEC mice, suggesting that intestinal epithelial GR is necessary for alcohol and corticosterone-induced endotoxemia and systemic inflammation. Thus, alteration of intestinal epithelial functions is a primary step in alcohol-induced endotoxemia and systemic inflammation, mediated by the glucocorticoids and intestinal epithelial GR. In the enterocytes, GR signaling may sensitize ethanol and acetaldehyde-induced TJ disruption and barrier dysfunction. Our previous study showed that corticosterone sensitizes Caco-2 cell monolayers for ethanol and acetaldehyde-induced TJ disruption and barrier dysfunction (15). The mechanism involved in the intestinal epithelial GR-mediated regulation of gut microbiota composition is unclear at this time. GR in specific cell types in the intestinal epithelium may indirectly regulate the microbiota composition. More detailed studies are required to answer this question and explore a new avenue of investigation.
The previous study showed that stress and corticosterone exacerbate alcohol-induced liver damage. Here we show that the absence of intestinal epithelial GR attenuates alcohol and corticosterone-induced liver damage. Interestingly, the deletion of GR from the hepatocytes did not block alcohol and corticosterone-induced steatosis, histopathologic lesions, or inflammatory responses in the liver. These results indicate that intestinal events such as microbiota dysbiosis and epithelial barrier dysfunction, and consequent endotoxemia are essential steps in the mechanism of alcohol and corticosterone-induced liver injury. Therefore, interruption of intestinal epithelial GR signaling may attenuate alcohol-induced liver damage.
Alcohol is well known to alter the HPA axis and elevate plasma corticosterone (3, 8–10); however, the mechanism involved in this effect of alcohol is unknown. The previous study showed a synergistic increase in the inflammatory response in the hypothalamus by ethanol and corticosterone (15). Here we show that ethanol and corticosterone-induced elevation of IL-6 and MCP1 mRNA in the hypothalamus is absent in GRΔIEC mice, indicating that intestinal epithelial GR is necessary for alcohol and corticosterone-induced neuroinflammation and alteration of the HPA axis. These findings suggest that two-way interactions at the gut-brain axis are involved in alcohol-associated diseases and disorders. Our study indicates that glucocorticoid, on the one hand, is required for gut barrier disruption and dysbiosis of gut microbiota. On the other hand, events mediated by intestinal epithelial GR are required for the alcohol-induced inflammatory response in the hypothalamus. In this study, the sex-dependent differences were not addressed in depth. Although female mice showed slightly higher sensitivity to alcohol, the effects of GR deficiency were the same in male and female mice.
Currently, the precise mechanism of GR-induced signaling in the intestinal epithelium is unclear. GR is a nuclear receptor, activation of which leads to altered gene expression. GR plays a role in the pathogenesis of the non-alcoholic fatty liver disease (63, 64); however, the mechanisms involved is unclear. Various intestinal epithelial cells express GR and regulate multiple cellular functions such as amino acid metabolism (65) and dendritic cell function (66). The amelioration of alcohol and corticosterone effects by the deletion of intestinal epithelial GR but not hepatocyte GR suggest the GR-mediated gene expression changes in the hepatocytes and enterocytes may vary. In the intestinal epithelium, GR signaling may regulate gene expression in multiple cells, including enterocytes, Paneth cells, and enteroendocrine cells leading to barrier dysfunction and dysbiosis of microbiota. Data also suggest that GR-mediated gene expression changes in the hepatocyte are not sufficient to cause liver damage. On the other hand, GR signaling in the intestinal epithelial cells is required for alcohol-induced fat deposition and inflammatory response in the liver.
In summary, the results of this study demonstrate that cellular changes caused by corticosterone and intestinal epithelial GR are initial steps in the mechanism of alcohol-induced gut barrier dysfunction, dysbiosis of gut microbiota, and systemic response. This study identifies intestinal epithelial GR as a therapeutic target for treating alcoholic tissue injury. The exacerbating effects of corticosterone on alcohol effects question the steroid-based therapy in alcoholic diseases. Prednisolone is a primary treatment for severe alcoholic hepatitis (67); however, it offers only a short-term solution. Prednisolone is not helpful for intermediate or long-term survival (68). The primary mechanism of prednisolone therapy is its anti-inflammatory activity. Data from our present study demonstrate that glucocorticoids elevated by alcohol use or administered exogenously caused gut barrier dysfunction, endotoxemia, systemic inflammation, and multiple organ damage. Therefore, it is not surprising that prednisolone is not an effective treatment strategy for the long-term survival of alcoholic hepatitis patients.
Supplementary Material
Acknowledgments
This study was supported primarily by a grant from the Veterans Administration I01-BX003014 and partly by NIH grants R01-AA12307 and R01-DK55532.
Abbreviations
- AJ
adherens junction
- ALD
alcoholic liver disease
- ALT
alanine transaminase
- AST
aspartate transaminase
- Cort
corticosterone
- EF
ethanol-fed
- GR
glucocorticoid receptor
- HC
hepatocyte
- IEC
intestinal epithelial cell
- IL-1 & 6
interleukin-1 & 6
- LPS
lipopolysaccharide
- MCP1
monocyte chemoattractant protein-1
- OTU
operational taxonomic unit
- PF
pair-fed
- PCoA
principal coordinates analysis
- TJ
tight junction
- TER
transepithelial electrical resistance
- TNF
tumor necrosis factor
- ZO
zonula occludens
Footnotes
Disclosures: Authors have nothing to disclose.
Data Availability
Data from this study were submitted to FigureShare: https://figshare.com/s/f59848310945fce97da1
References
- 1.Osna NA, Donohue TM Jr., and Kharbanda KK (2017) Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res 38, 147–161 [PMC free article] [PubMed] [Google Scholar]
- 2.Tsukamoto H, Machida K, Dynnyk A, and Mkrtchyan H (2009) “Second hit” models of alcoholic liver disease. Semin Liver Dis 29, 178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allan CA, and Cooke DJ (1985) Stressful life events and alcohol misuse in women: a critical review. J Stud Alcohol 46, 147–152 [DOI] [PubMed] [Google Scholar]
- 4.Debell F, Fear NT, Head M, Batt-Rawden S, Greenberg N, Wessely S, and Goodwin L (2014) A systematic review of the comorbidity between PTSD and alcohol misuse. Soc Psychiatry Psychiatr Epidemiol 49, 1401–1425 [DOI] [PubMed] [Google Scholar]
- 5.Young-Wolff KC, Kendler KS, and Prescott CA (2012) Interactive effects of childhood maltreatment and recent stressful life events on alcohol consumption in adulthood. J Stud Alcohol Drugs 73, 559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagy LE, Ding WX, Cresci G, Saikia P, and Shah VH (2016) Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology 150, 1756–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smart L, Gobejishvili L, Crittenden N, Barve S, and McClain CJ (2013) Alcoholic Hepatitis: Steroids vs. Pentoxifylline. Curr Hepat Rep 12, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar M, Gupta GK, Wanjari SJ, Tak V, Ameta M, and Nijhawan S (2019) Relative Adrenal Insufficiency in Patients with Alcoholic Hepatitis. J Clin Exp Hepatol 9, 215–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephens MA, and Wand G (2012) Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res 34, 468–483 [PMC free article] [PubMed] [Google Scholar]
- 10.Wand GS, and Dobs AS (1991) Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrinol Metab 72, 1290–1295 [DOI] [PubMed] [Google Scholar]
- 11.Gao Y, Zhou Z, Ren T, Kim SJ, He Y, Seo W, Guillot A, Ding Y, Wu R, Shao S, Wang X, Zhang H, Wang W, Feng D, Xu M, Han E, Zhong W, Zhou Z, Pacher P, Niu J, and Gao B (2019) Alcohol inhibits T-cell glucose metabolism and hepatitis in ALDH2-deficient mice and humans: roles of acetaldehyde and glucocorticoids. Gut 68, 1311–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumari S, Mittal A, and Dabur R (2016) Moderate alcohol consumption in chronic form enhances the synthesis of cholesterol and C-21 steroid hormones, while treatment with Tinospora cordifolia modulate these events in men. Steroids 114, 68–77 [DOI] [PubMed] [Google Scholar]
- 13.Van Thiel DH, Gavaler JS, and Lester R (1978) Alcohol-induced ovarian failure in the rat. J Clin Invest 61, 624–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkatesan S, and Simpson KJ (1989) Fatty liver and plasma corticosterone levels in chronically alcohol- and pair-fed rats. Biochem Soc Trans 17, 1114–1115 [DOI] [PubMed] [Google Scholar]
- 15.Shukla PK, Meena AS, Dalal K, Canelas C, Samak G, Pierre JF, and Rao R (2021) Chronic stress and corticosterone exacerbate alcohol-induced tissue injury in the gut-liver-brain axis. Sci Rep 11, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Covelli V, Passeri ME, Leogrande D, Jirillo E, and Amati L (2005) Drug targets in stress-related disorders. Curr Med Chem 12, 1801–1809 [DOI] [PubMed] [Google Scholar]
- 17.de Punder K, and Pruimboom L (2015) Stress induces endotoxemia and low-grade inflammation by increasing barrier permeability. Front Immunol 6, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, and Gillevet P (2009) Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res 33, 1836–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shukla PK, Meena AS, Rao V, Rao RG, Balazs L, and Rao R (2018) Human Defensin-5 Blocks Ethanol and Colitis-Induced Dysbiosis, Tight Junction Disruption and Inflammation in Mouse Intestine. Sci Rep 8, 16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao R (2009) Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50, 638–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris NL, Ippolito JA, Curtis BJ, Chen MM, Friedman SL, Hines IN, Haddad GE, Chang SL, Brown LA, Waldschmidt TJ, Mandrekar P, Kovacs EJ, and Choudhry MA (2015) Alcohol and inflammatory responses: summary of the 2013 Alcohol and Immunology Research Interest Group (AIRIG) meeting. Alcohol 49, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seabra V, Stachlewitz RF, and Thurman RG (1998) Taurine blunts LPS-induced increases in intracellular calcium and TNF-alpha production by Kupffer cells. J Leukoc Biol 64, 615–621 [DOI] [PubMed] [Google Scholar]
- 23.Thomas P, Lazure DA, Moussa R, Bajenova O, Burke PA, Ganguly A, and Forse RA (2006) Identification of two novel LPS-binding proteins in Kupffer cells: implications in TNF-alpha production. J Endotoxin Res 12, 352–357 [DOI] [PubMed] [Google Scholar]
- 24.Weinhold L, Schulze-Specking A, and Decker K (1991) Signal transduction in endotoxin-stimulated synthesis of TNF-alpha and prostaglandin E2 by rat Kupffer cells. Role of extracellular calcium ions and protein kinase C. Biol Chem Hoppe Seyler 372, 829–834 [DOI] [PubMed] [Google Scholar]
- 25.Chi H, Cao W, Zhang M, Su D, Yang H, Li Z, Li C, She X, Wang K, Gao X, Ma K, Zheng P, Li X, and Cui B (2021) Environmental noise stress disturbs commensal microbiota homeostasis and induces oxi-inflammmation and AD-like neuropathology through epithelial barrier disruption in the EOAD mouse model. J Neuroinflammation 18, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lv WJ, Wu XL, Chen WQ, Li YF, Zhang GF, Chao LM, Zhou JH, Guo A, Liu C, and Guo SN (2019) The Gut Microbiome Modulates the Changes in Liver Metabolism and in Inflammatory Processes in the Brain of Chronic Unpredictable Mild Stress Rats. Oxid Med Cell Longev 2019, 7902874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westfall S, Lomis N, Kahouli I, Dia SY, Singh SP, and Prakash S (2017) Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cell Mol Life Sci 74, 3769–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Litwinowicz K, Choroszy M, and Waszczuk E (2020) Changes in the composition of the human intestinal microbiome in alcohol use disorder: a systematic review. Am J Drug Alcohol Abuse 46, 4–12 [DOI] [PubMed] [Google Scholar]
- 29.Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B, Ward DV, Grabherr F, Gerner RR, Pfister A, Enrich B, Ciocan D, Macheiner S, Mayr L, Drach M, Moser P, Moschen AR, Perlemuter G, Szabo G, Cassard AM, and Tilg H (2018) Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 67, 891–901 [DOI] [PubMed] [Google Scholar]
- 30.Ciocan D, Rebours V, Voican CS, Wrzosek L, Puchois V, Cassard AM, and Perlemuter G (2018) Characterization of intestinal microbiota in alcoholic patients with and without alcoholic hepatitis or chronic alcoholic pancreatitis. Sci Rep 8, 4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciocan D, Voican CS, Wrzosek L, Hugot C, Rainteau D, Humbert L, Cassard AM, and Perlemuter G (2018) Bile acid homeostasis and intestinal dysbiosis in alcoholic hepatitis. Aliment Pharmacol Ther 48, 961–974 [DOI] [PubMed] [Google Scholar]
- 32.Henry JP (1993) Psychological and physiological responses to stress: the right hemisphere and the hypothalamo-pituitary-adrenal axis, an inquiry into problems of human bonding. Integr Physiol Behav Sci 28, 369–387; discussion 368 [DOI] [PubMed] [Google Scholar]
- 33.Kathol RG, Noyes R, and Lopez A (1988) Similarities in hypothalamic-pituitary-adrenal axis activity between patients with panic disorder and those experiencing external stress. Psychiatr Clin North Am 11, 335–348 [PubMed] [Google Scholar]
- 34.Pervanidou P, and Chrousos GP (2018) Early-Life Stress: From Neuroendocrine Mechanisms to Stress-Related Disorders. Horm Res Paediatr 89, 372–379 [DOI] [PubMed] [Google Scholar]
- 35.Russell G, and Lightman S (2019) The human stress response. Nat Rev Endocrinol 15, 525–534 [DOI] [PubMed] [Google Scholar]
- 36.Sillaber I, and Henniger MS (2004) Stress and alcohol drinking. Ann Med 36, 596–605 [DOI] [PubMed] [Google Scholar]
- 37.Blaine SK, Milivojevic V, Fox H, and Sinha R (2016) Alcohol Effects on Stress Pathways: Impact on Craving and Relapse Risk. Can J Psychiatry 61, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gameiro GH, Gameiro PH, Andrade Ada S, Pereira LF, Arthuri MT, Marcondes FK, and Veiga MC (2006) Nociception- and anxiety-like behavior in rats submitted to different periods of restraint stress. Physiol Behav 87, 643–649 [DOI] [PubMed] [Google Scholar]
- 39.Haddad JJ (2004) Alcoholism and neuro-immune-endocrine interactions: physiochemical aspects. Biochem Biophys Res Commun 323, 361–371 [DOI] [PubMed] [Google Scholar]
- 40.Choudhry MA, Li X, and Chaudry IH (2006) A role for corticosterone in impaired intestinal immunity and barrier function in a rodent model of acute alcohol intoxication and burn injury. J Neuroimmune Pharmacol 1, 428–434 [DOI] [PubMed] [Google Scholar]
- 41.Sheppard KE (2002) Nuclear receptors. II. Intestinal corticosteroid receptors. Am J Physiol Gastrointest Liver Physiol 282, G742–746 [DOI] [PubMed] [Google Scholar]
- 42.Anderson JM, and Van Itallie CM (2009) Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 1, a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Itallie CM, and Anderson JM (2014) Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol 36, 157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao R (2009) Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci 1165, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samak G, Chaudhry KK, Gangwar R, Narayanan D, Jaggar JH, and Rao R (2015) Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. Biochem J 465, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samak G, Gangwar R, Meena AS, Rao RG, Shukla PK, Manda B, Narayanan D, Jaggar JH, and Rao R (2016) Calcium Channels and Oxidative Stress Mediate a Synergistic Disruption of Tight Junctions by Ethanol and Acetaldehyde in Caco-2 Cell Monolayers. Sci Rep 6, 38899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coopman P, and Djiane A (2016) Adherens Junction and E-Cadherin complex regulation by epithelial polarity. Cell Mol Life Sci 73, 3535–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell HK, Maiers JL, and DeMali KA (2017) Interplay between tight junctions & adherens junctions. Exp Cell Res 358, 39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shukla PK, Meena AS, Manda B, Gomes-Solecki M, Dietrich P, Dragatsis I, and Rao R (2018) Lactobacillus plantarum prevents and mitigates alcohol-induced disruption of colonic epithelial tight junctions, endotoxemia, and liver damage by an EGF receptor-dependent mechanism. Faseb J, fj201800351R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pierre JF, Akbilgic O, Smallwood H, Cao X, Fitzpatrick EA, Pena S, Furmanek SP, Ramirez JA, and Jonsson CB (2020) Discovery and predictive modeling of urine microbiome, metabolite and cytokine biomarkers in hospitalized patients with community acquired pneumonia. Sci Rep 10, 13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion MJ, Berger B, and Krause L (2017) Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 33, 782–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, and Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes JB, Hellmann JJ, Ricketts TH, and Bohannan BJ (2001) Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 67, 4399–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bengtsson-Palme J, Richardson RT, Meola M, Wurzbacher C, Tremblay ED, Thorell K, Kanger K, Eriksson KM, Bilodeau GJ, Johnson RM, Hartmann M, and Nilsson RH (2018) Metaxa2 Database Builder: enabling taxonomic identification from metagenomic or metabarcoding data using any genetic marker. Bioinformatics 34, 4027–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, Rosenbaum M, and Gordon JI (2013) The long-term stability of the human gut microbiota. Science 341, 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lozupone C, Lladser ME, Knights D, Stombaugh J, and Knight R (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5, 169–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elias BC, Suzuki T, Seth A, Giorgianni F, Kale G, Shen L, Turner JR, Naren A, Desiderio DM, and Rao R (2009) Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. The Journal of biological chemistry 284, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson JM, and Van Itallie CM (2009) Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 1, a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, and Moller K (2008) Human endotoxemia as a model of systemic inflammation. Curr Med Chem 15, 1697–1705 [DOI] [PubMed] [Google Scholar]
- 61.Erickson EK, Grantham EK, Warden AS, and Harris RA (2019) Neuroimmune signaling in alcohol use disorder. Pharmacol Biochem Behav 177, 34–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rose AK, Shaw SG, Prendergast MA, and Little HJ (2010) The importance of glucocorticoids in alcohol dependence and neurotoxicity. Alcohol Clin Exp Res 34, 2011–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mueller KM, Themanns M, Friedbichler K, Kornfeld JW, Esterbauer H, Tuckermann JP, and Moriggl R (2012) Hepatic growth hormone and glucocorticoid receptor signaling in body growth, steatosis and metabolic liver cancer development. Mol Cell Endocrinol 361, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woods CP, Hazlehurst JM, and Tomlinson JW (2015) Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol 154, 94–103 [DOI] [PubMed] [Google Scholar]
- 65.Flynn NE, Bird JG, and Guthrie AS (2009) Glucocorticoid regulation of amino acid and polyamine metabolism in the small intestine. Amino Acids 37, 123–129 [DOI] [PubMed] [Google Scholar]
- 66.Yang X, Geng J, and Meng H (2020) Glucocorticoid receptor modulates dendritic cell function in ulcerative colitis. Histol Histopathol 35, 1379–1389 [DOI] [PubMed] [Google Scholar]
- 67.Patel R, and Gossman W (2020) Alcoholic Liver Disease. In StatPearls, Treasure Island (FL) [Google Scholar]
- 68.Im GY (2019) Acute Alcoholic Hepatitis. Clin Liver Dis 23, 81–98 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from this study were submitted to FigureShare: https://figshare.com/s/f59848310945fce97da1