

SUMMARY
Excess liver fat, called hepatic steatosis, is a leading risk factor for end-stage liver disease and cardiometabolic diseases but often remains undiagnosed in clinical practice because of the need for direct imaging assessments. We developed an abdominal MRI-based machine-learning algorithm to accurately estimate liver fat (correlation coefficients, 0.97–0.99) from a truth dataset of 4,511 middle-aged UK Biobank participants, enabling quantification in 32,192 additional individuals. 17% of participants had predicted liver fat levels indicative of steatosis, and liver fat could not have been reliably estimated based on clinical factors such as BMI. A genome-wide association study of common genetic variants and liver fat replicated three known associations and identified five newly associated variants in or near the MTARC1, ADH1B, TRIB1, GPAM, and MAST3 genes (p < 3 × 10−8). A polygenic score integrating these eight genetic variants was strongly associated with future risk of chronic liver disease (hazard ratio > 1.32 per SD score, p < 9 × 10−17). Rare inactivating variants in the APOB or MTTP genes were identified in 0.8% of individuals with steatosis and conferred more than 6-fold risk (p < 2 × 10−5), highlighting a molecular subtype of hepatic steatosis characterized by defective secretion of apolipoprotein B-containing lipoproteins. We demonstrate that our imaging-based machine-learning model accurately estimates liver fat and may be useful in epidemiological and genetic studies of hepatic steatosis.
Graphical Abstract

In brief
Haas et al. report a machine-learning algorithm used to precisely quantify liver fat, a leading driver of end-stage liver disease, from abdominal MRI imaging data of 36,703 UK Biobank participants. They identify common and rare genetic variants influencing liver fat and demonstrate utility for epidemiological studies.
INTRODUCTION
Hepatic steatosis, a condition defined by liver fat content of more than 5.5%, is a leading risk factor for chronic liver disease and is strongly associated with a range of cardiometabolic conditions.1–4 Recent studies have suggested a prevalence of up to 25% across global populations, with rates rapidly increasing in step with the global epidemics of obesity and diabetes.5,6 Although the condition is frequently undiagnosed in clinical practice, previous evidence indicates that avoidance of excessive alcohol intake, weight loss strategies including bariatric surgery, and emerging pharmacologic therapies can reduce liver fat and prevent progression to more advanced liver disease.7
Previous studies of hepatic steatosis suggest that systematic quantification in large cohorts may provide new biologic insights or improve clinical care but suffer from important limitations. First, the traditional approach dichotomizes individuals with hepatic steatosis into nonalcoholic fatty liver disease (NAFLD) or alcoholic fatty liver disease according to largely arbitrary thresholds.7,8 Second, studies of the clinical significance of hepatic steatosis have often been based on non-quantitative ultrasound assessments or physician diagnosis codes, which are known to introduce imprecision into downstream analyses.9–11 Third, genome-wide association studies (GWAS) of common variants for liver fat have been limited, hampered by time-consuming quantification of liver fat from abdominal computed tomography (CT) or MRI images and, thus, have analyzed only up to 16,492 individuals.12–15 By comparison, a recent GWAS of BMI, a quantitative trait easily measured in clinical practice, analyzed 681,275 individuals.16
Based on these prior results, three key areas of uncertainty remain. First, the extent to which a machine-learning algorithm can be trained to accurately quantify liver fat in a large group of individuals warrants additional study. Second, the association of clinical risk factors with hepatic steatosis, as well as the ability to predict liver fat content without direct imaging, have not been fully characterized in large studies of individuals not ascertained for any specific clinical indication. Third, whether an expanded set of individuals with precise liver fat quantification can enable new genetic discoveries using GWAS or a rare variant association study (RVAS) is largely unknown.
Here we address these areas of uncertainty by studying 36,703 middle-aged UK Biobank participants with extensive linked imaging, genetic, and clinical data (Figure 1). We develop a machine-learning algorithm that precisely quantifies liver fat using raw abdominal MRI images, achieving correlation coefficients of 0.97 and 0.99 in hold-out testing datasets. Using these data, we quantify significantly increased rates of hepatic steatosis among key subgroups, such as those with obesity or diabetes. Genetic analysis identified 8 common genetic variants associated at genome-wide levels of statistical significance, 5 of which are newly associated, and rare inactivating variants in the genes encoding apolipoprotein B (APOB) and microsomal triglyceride transfer protein (MTTP) that associate with significantly increased liver fat and steatosis.
Figure 1. Machine learning enables liver fat quantification and clinical and genetic analyses.
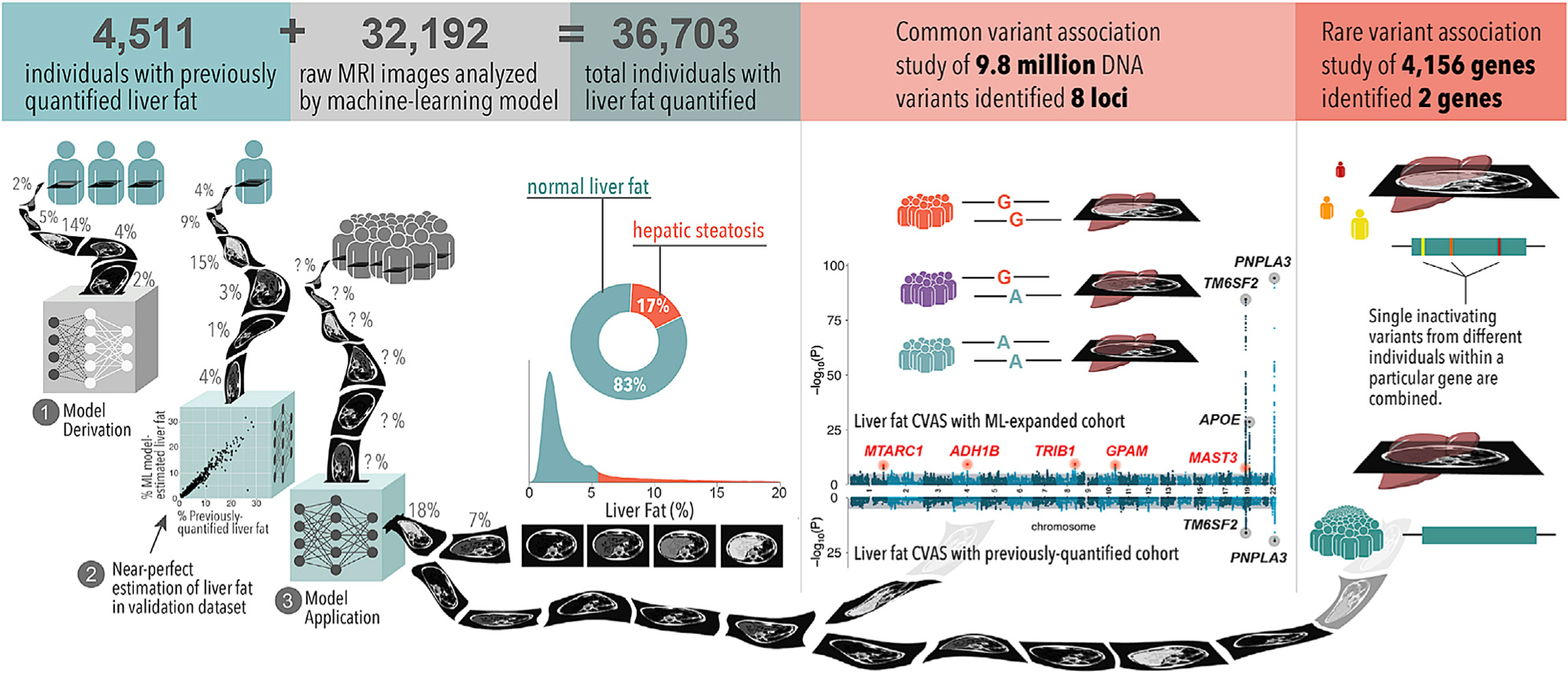
We developed a machine-learning model using a training set of 4,511 individuals with previously quantified liver fat from the UK Biobank. We applied this to estimate liver fat in an additional 32,192 individuals in the UK Biobank. Of the 36,703 total individuals with liver fat quantified, 17% met criteria for hepatic steatosis, defined as liver fat content greater than 5.5%. 1.6% of individuals had liver fat greater than 20% (not shown in the density plot). A common variant GWAS identified eight loci associated at genome-wide significance (p < 5.0 × 10−8), of which five are newly identified relative to previous studies (top Manhattan plot). None of these newly associated variants were identified in a common variant association study of those with liver fat quantified previously (bottom Manhattan plot). An RVAS identified inactivating variants in APOB and MTTP significantly (p < 1.2 × 10−5) associated with liver fat and steatosis.
RESULTS
A machine-learning model for quantification of hepatic fat
To study liver fat in 36,703 UK Biobank participants, we first developed a machine-learning algorithm that allowed precise quantification based on raw abdominal MRI data. We processed available images within a cloud-based computational environment, leveraging a subset of 4,511 participants with liver fat quantified previously by Perspectum Diagnostics.17 Using a two-stage method with deep convolutional neural networks (see STAR Methods for details), we trained an algorithm to quantify liver fat that achieved highly accurate quantification: in hold-out testing datasets, correlation coefficients were 0.97 and 0.99, and mean absolute errors were 0.50% and 0.41% in the two stages, with comparable performance in self-reported European and non-European study participants (Figure S1). As expected, the ability to quantify liver fat using direct imaging data was substantially higher than using clinical data alone. For example, within the hold-out testing dataset of 1,214 individuals, the correlation between BMI and liver fat was 0.42, improving to 0.58 in a model that incorporated 24 additional clinical factors and biomarker data, including liver-related biomarkers such as alanine aminotransferase (Figure S3). Having trained and validated the machine-learning algorithm, we next applied this model to quantify liver fat in the remaining 32,192 UK Biobank participants with raw MRI images available.
Liver fat is strongly associated with cardiometabolic diseases
Across all 36,703 participants studied, median liver fat was 2.2%, and 6,250 (17.0%) had liver fat greater than 5.5%, consistent with hepatic steatosis. Mean age at time of imaging was 64 years (range, 45–82), and 52% were female (Table S1). Liver fat was significantly (p value threshold = 0.05) increased in male versus female participants (median, 2.7 versus 2.0%; p = 5.6 × 10−220), those who reported alcohol consumption in excess of current United States clinical guidelines7 (median, 2.6 versus 2.2%; p = 3.1 × 10−14), and those with diagnosed diabetes (median, 4.9 versus 2.2%; p = 2.7 × 10−13). As expected, median liver fat was significantly higher among 93 individuals with a diagnosis of NAFLD in the electronic health record compared with the remainder of the population (median, 8.6 versus 2.2%, respectively; p = 8.4 × 10−14; Figure 2). 56 of 93 (60.2%) of those diagnosed with NAFLD met imaging-based criteria for hepatic steatosis versus 6,194 of 36,610 (16.9%) in the remainder of the population, corresponding to an adjusted odds ratio of 7.65 (95% confidence interval [CI], 5.02–11.67; p = 3.2 × 10−21).
Figure 2. Associations of clinical parameters with liver fat and hepatic steatosis in 36,703 individuals.

(A–C) The distribution of liver fat and prevalence of hepatic steatosis according to the presence of (A) electronic health record diagnosis of nonalcoholic fatty liver disease (NAFLD), (B) high waist-to-hip ratio, and (C) clinical categories of obesity. Hepatic steatosis was defined as liver fat greater than5.5%.17 High waist-to-hip ratio was defined at time of imaging as greater than 0.9 when male and greater than 0.85 when female.18 Weight categories were defined using BMI at time of imaging:19 underweight, BMI < 18.5 kg/m2; normal, 18.5 ≤ BMI < 25 kg/m2; overweight, 25 ≤ BMI < 30 kg/m2; obese, 30 ≤ BMI < 40 kg/m2; severely obese, BMI ≥ 40 kg/m2. For boxplots, boxes indicate interquartile range (IQR; 25th–75th percentiles), and whiskers indicate distances of 1.5 IQRs from box limits. For bar plots, error bars indicate upper bounds of 95% CI.
By stratifying individuals according to presence of hepatic steatosis, we observed significant (p value threshold = 0.05) enrichment of cardiometabolic risk factors in those with high liver fat (Table 1). For example, 13.8% of those with steatosis had been diagnosed with diabetes compared with 3.6% of those in the remainder (adjusted odds ratio, 4.21; 95% CI, 3.83–4.64; p = 1.1 × 10−189), and 45.1% of those with steatosis had been diagnosed with hypertension compared with 27.1% in the remainder (adjusted odds ratio, 2.24; 95% CI, 2.11–2.37; p = 6.1 × 10−161). We also examined the association of liver fat with circulating biomarkers collected at time of enrollment, noting that circulating triglycerides, liver-associated aminotransferases and glycemic indices were all significantly increased in those with steatosis.
Table 1.
Baseline characteristics of 36,703 UK Biobank participants with quantified liver fat, stratified by presence of hepatic steatosis
| Steatosis absent (n = 30,453) | Steatosis present (n = 6,250) | p value | |
|---|---|---|---|
| Female | 16,540 (54.3%) | 2,509 (40.1%) | 1.07 × 10−92 |
| Age at enrollment, years | 54.9 (7.51) | 54.7 (7.23) | 0.004 |
| Age at imaging, years | 64.3 (7.62) | 63.8 (7.23) | 9.0 × 10−7 |
| Self-reported ethnicity | |||
| White | 29,527 (97.0%) | 6,045 (96.7%) | 0.36 |
| Black | 185 (0.6%) | 29 (0.5%) | 0.18 |
| South Asian | 239 (0.8%) | 74 (1.2%) | 0.002 |
| Other Asian | 138 (0.5%) | 27 (0.4%) | 0.82 |
| Multiple, other or not provided | 364 (1.2%) | 75 (1.2%) | 0.98 |
| Coronary artery diseasea | 1,030 (3.4%) | 253 (4.0%) | 0.009 |
| Diabetesa | 1,094 (3.6%) | 862 (13.8%) | 1.6 × 10−234 |
| Hypertensiona | 8,264 (27.1%) | 2,821 (45.1%) | 2.5 × 10−175 |
| Obesity | 3,964 (13.0%) | 2,531 (40.5%) | <1 × 10−300 |
| Medications | |||
| Anti-hypertensive therapy | 3,555 (11.7%) | 1,385 (22.2%) | 1.8 × 10−108 |
| Lipid-lowering therapy | 4,287 (14.1%) | 1,265 (20.2%) | 3.1 × 10−35 |
| Anthropometric data | |||
| Weight, kg | 74.7 (13.8) | 86.6 (15.3) | <1 × 10−300 |
| Waist-to-hip ratio | 0.85 (0.08) | 0.91 (0.08) | <1 × 10−300 |
| BMI, kg/m2 | 25.9 (3.8) | 29.7 (4.4) | <1 × 10−300 |
| Body fat, % | 29.4 (8.1) | 32.6 (8.1) | 3.1 × 10−136 |
| Estimated untreated systolic blood pressure, mmHg | 136 (19.2) | 136 (19.2) | 143 (18.8) |
| Alcohol consumption | |||
| Weekly drinks, United States standard | 5.4 (6.1) | 5.9 (7.7) | 0.02 |
| Weekly drinks, United Kingdom standard | 9.4 (10.6) | 10.4 (13.6) | 0.02 |
| Excessive alcohol intake, United States | 1,559 (5.1%) | 456 (7.3%) | 5.9 × 10−12 |
| Excessive alcohol intake, United Kingdom | 7,417 (24.4%) | 1,649 (26.4%) | 7.1 × 10−4 |
| Liver-associated biomarker concentrations | |||
| Alanine aminotransferase, IU/L | 21.3 (12.0) | 31.4 (18.9) | <1 × 10−300 |
| Aspartate aminotransferase, IU/L | 25.2 (9.9) | 28.7 (12.9) | 8.7 × 10−174 |
| Gamma glutamyltransferase, IU/L | 31.3 (31.3) | 45.6 (42.6) | <1 × 10−300 |
| Estimated untreated lipid concentrations | |||
| Total cholesterol, mg/dL | 226 (40.3) | 230 (42.3) | 1.5 × 10−11 |
| LDL cholesterol, mg/dL | 143 (31.7) | 150 (32.8) | 9.2 × 10−65 |
| HDL cholesterol, mg/dL | 58.5 (14.6) | 49.5 (11.6) | <1 × 10−300 |
| Triglycerides, mg/dL | 117 [85–169] | 176 [129–249] | <1 × 10−300 |
| Glycemic biomarker concentrations | |||
| Glycated hemoglobin, % | 5.3 (0.4) | 5.5 (0.6) | 1.4 × 10−128 |
| Glucose, mg/dL | 89.2 (16.0) | 93.4 (23.1) | 6.1 × 10−45 |
| Liver fat, % | 2.0 [1.5–2.9] | 9.9 [7.1–14.2] | <1 × 10−300 |
Liver fat was quantified in 36,703 UK Biobank participants from machine learning of MRI data using previous commercial vendor measurements in a subset of 4,511 individuals. Columns show participants grouped according to whether they had evidence of hepatic steatosis, defined as liver fat greater then 5.5%.17 Rows show measurements at the initial UK Biobank assessment visit, with values corresponding to number (%), mean (SD), or median [IQR]. p values correspond to unadjusted comparisons between presence or absence of steatosis, assessed via chi-square test or Wilcoxon rank-sum test (for categorical and continuous variables, respectively). Obesity was defined as BMI of 30 kg/m or greater.2,19 Excessive alcohol intake, United States was defined as alcohol intake exceeding American Association for the Study of Liver Disease guidelines for NAFLD definition.7 Excessive alcohol intake, United Kingdom was defined as alcohol intake exceeding the UK Chief Medical Officers’ recommendations.20 Estimated untreated lipid measurements and blood pressure were according to adjustments described previously.21,22 See also Tables S1 and S2.
Disease status assessed at time of MRI imaging visit.
Despite the correlation of liver fat with cardiometabolic risk factors, clinicians would not be able to reliably estimate liver fat without direct imaging assessment. For example, a broad range of values was observed across BMI categorizations used in clinical practice (Figure 2). In those with severe obesity (BMI ≥ 40 kg/m2), median liver fat was 9.8%, and 254 of 361 (70.4%) met criteria for steatosis, but measured liver fat varied greatly from 0.5%–31.5%. Even among those with normal weight in whom median liver fat was 1.6%, 470 of 14,307 (3.3%) still had imaging evidence of hepatic steatosis. Similarly, only 4,854 of 17,730 (27.3%) with an elevated waist-to-hip ratio, a measure of central adiposity, had hepatic steatosis.
GWAS identifies 5 newly associated loci
We first confirmed prior studies noting a significant inherited component to liver fat,12,23,24 estimating that up to 30% of the observed variance is explained by measured genetic variants when considered in aggregate using the BOLT-REML method.25 To identify the specific variants most strongly contributing to this heritability, we performed a common variant GWAS, assessing the relationship of each of 9.8 million common (minor allele frequency > 1%) genetic variants and liver fat percentage using the BOLT-LMM algorithm.26 Given that 97% of individuals with liver fat quantified were self-reported European (Table S1) and the potential for small numbers of individuals of distinct ancestries to introduce confounding by population stratification, we restricted these analyses to 32,974 individuals of European ancestries selected by genetic principal-component analysis27 of self-reported ethnicity (STAR Methods). Minimal evidence of test statistic inflation was observed, with λ = 1.10 and LD (linkage disequilibrium) score regression intercept,28 a measure of inflation that accounts for polygenicity, of 1.02 (Figure S4).
Given the highly right-skewed distribution of measured liver fat, we applied an inverse-normal transformation to liver fat residuals to meet standard GWAS algorithm assumptions of normally distributed phenotype residuals. This resulted in a Gaussian distribution with mean of zero and SD of 1 (Figure S5). Beta coefficients derived from regression models of this transformed and standardized phenotype with SD units are reported. To improve clinical interpretability, we also provide effect estimates in units of absolute liver fat percentage points and odds ratios for hepatic steatosis.
The GWAS identified eight loci in which common genetic variants were significantly associated with increased liver fat at a p value threshold of less than 5.0 × 10−8, including five not identified previously at genome-wide levels of statistical significance (Figure 3; Table 2). The two most significantly associated variants confirm known associations13,29 of the p.I148M missense variant in the gene encoding patatin-like phospholipase domain-containing protein 3 (PNPLA3) and the p.E167K missense variant in the gene encoding transmembrane 6 super-family member 2 (TM6SF2). In our study, the effect size of the TM6SF2 variant (beta = 0.29 SD, p = 2.8 × 10−85) was somewhat larger than that of the PNPLA3 variant (beta = 0.19 SD, p = 5.6 × 10−95), consistent with a previous study of UK Biobank participants14 but distinct from other studies of liver fat measured via CT15 or NAFLD diagnosed via liver biopsy,30 where the PNPLA3 variant had a larger effect size than that of TM6SF2. Further genetic studies across diverse populations and NAFLD-related phenotypes are needed to quantify the relative effect of these two variants. More recently, two studies reported the p.R130C variant in the gene encoding apolipoprotein E (APOE) associated with increased liver fat,14,15 with this same variant subsequently linked to end-stage liver disease (cirrhosis) as well.31 This variant was similarly associated with increased liver fat in our analysis (beta = 0.12 SD, p = 1.5 × 10−29), corresponding to an increase in liver fat of 0.51 percentage points and an odds ratio for steatosis of 1.40. For each of these three variants, application of the recently described PolyFun fine-mapping algorithm32 provided support for the theory that the missense variants are likely to be causal, with a posterior inclusion probability (PIP) of 94% or greater for each (Table S3).
Figure 3. Common variant GWAS of liver fat in 32,974 individuals identifies eight loci.

Associations of 9.8 million common (alternate allele frequency > 1%) genetic variants with inverse normal transformed liver fat, quantified from MRI data using machine learning, in 32,974 individuals from the UK Biobank were assessed using linear mixed models. Results of each variant association are shown with chromosome and base pair position of the variant on the x axis and −log10(p value) of the association with liver fat on the y axis. The lead variants at each of 8 genome-wide significant loci are indicated by orange points. A gray line indicates the genome-wide significance threshold (p = 5 × 10−8). See also Figure S4.
Table 2.
Eight common genetic variants associated with increased liver fat indices
| Lead variant | Chr. | Position (hg19) | Nearest gene | Consequence | Effect allele | Other allele | Effect allele freq. | Effect on liver fat, beta (95% CI) p value | Effect on liver fat, % (95% CI) p value | Effect on hepatic steatosis, OR (95% CI) p value |
|---|---|---|---|---|---|---|---|---|---|---|
| Newly associated variants | ||||||||||
| rs2642438 | 1 | 220970028 | MTARC1 | missense (P.T165A) | G | A | 0.70 | 0.05 (0.04–0.07) p = 2 × 10−9 | 0.22 (0.14–0.29) p = 3 × 10−9 | 1.17 (1.11–1.22) p = 6 × 10−11 |
| rs1229984 | 4 | 100239319 | ADH1B | missense (p.H48R) | C | T | 0.98 | 0.16 (0.11–0.21) p = 7 × 10−10 | 0.51 (0.29–0.72) p = 3 × 10−6 | 1.37 (1.18–1.59) p = 3 × 10−5 |
| rs112875651 | 8 | 126506694 | TRIB1 | intergenic | G | A | 0.61 | 0.05 (0.03–0.07) p = 4 × 10−10 | 0.19 (0.13–0.26) p = 2 × 10−8 | 1.10 (1.06–1.15) p = 9 × 10−6 |
| rs2250802 | 10 | 113921354 | GPAM | intronic | G | A | 0.27 | 0.05 (0.04–0.07) p = 1 × 10−9 | 0.24 (0.17–0.31) p = 1 × 10−10 | 1.13 (1.08–1.18) p = 1 × 10−7 |
| rs56252442 | 19 | 18229208 | MAST3 | intronic | T | G | 0.25 | 0.05 (0.03–0.07) p = 3 × 10−8 | 0.18 (0.1–0.25) p = 3 × 10−6 | 1.09 (1.04–1.14) p = 3 × 10−4 |
| Previously associated variants | ||||||||||
| rs58542926 | 19 | 19379549 | TM6SF2 | missense (p.E167K) | T | C | 0.07 | 0.29 (0.26–0.32) p = 3 × 10−85 | 1.37 (1.25–1.49) p = 1 × 10−104 | 1.90 (1.78–2.04) p = 1 × 10−75 |
| rs429358 | 19 | 45411941 | APOE | missense (p.R130C) | T | C | 0.85 | 0.12 (0.10–0.14) p = 2 × 10−29 | 0.51 (0.42–0.60) p = 2 × 10−28 | 1.40 (1.32–1.49) p = 2 × 10−26 |
| rs738409a | 22 | 44324727 | PNPLA3 | missense (p.I148M) | G | C | 0.21 | 0.19 (0.18–0.21) p = 6 × 10−95 | 0.88 (0.81–0.96) p = 1 × 10−106 | 1.59 (1.52–1.66) p = 7 × 10−83 |
A common variant genome-wide association study (GWAS) was performed to measure associations of 9.8 million common (alternate allele frequency > 1%) genetic variants with liver fat, quantified from MRI data using machine learning, in 32,974 individuals from the UK Biobank. Rows show the variant with the smallest p value (lead variant) at each of 8 loci associated with liver fat below the genome-wide significance threshold p value of 5 × 10−8 assessed using inverse normal transformed liver fat. “Newly associated” indicates variants not reported previously to be associated with liver fat at genome-wide significance. “Previously associated” indicates previously reported variants.13,14,29 The first 8 columns show information on each lead variant, including position, frequency, and consequence. “Effect on liver fat, beta” shows the effect of each variant on inverse normal transformed liver fat in SD units, assessed using a linear mixed model. For clinical interpretability, “Effect on liver fat, %” shows the effect of each variant in units of absolute liver fat percentage points, and “Effect on hepatic steatosis, OR” shows the effect of each variant on the risk of hepatic steatosis (liver fat > 5.5%)17 in odds ratio units, assessed using linear and logistic regression, respectively, in the same 32,974 individuals.
Beyond replicating previous liver fat results, our GWAS identified five associated common variants not identified previously at genome-wide levels of statistical significance. Among these five newly associated genetic variants, the effect on liver fat percentage ranged from 0.18–0.51 percentage points, and the odds ratio for hepatic steatosis per allele ranged from 1.09–1.37 (Table 2). First, a variant in the gene encoding mitochondrial amidoxime reducing component 1 (MTARC1) was associated with an increase in liver fat (beta = 0.05 SD, p = 1.7 × 10−9, corresponding to 0.22 percentage points), with PolyFun fine-mapping supporting the p.T165A missense variant as causal (PIP = 0.91). We and others similarly identified this variant as associated with an increased risk of cirrhosis.33–37 Second, the p.H48R missense variant (PIP > 0.99) in the gene encoding alcohol dehydrogenase 1B (class I), beta polypeptide (ADH1B), was associated with a 0.51 percentage point increase in liver fat (beta = 0.16 SD, p = 7.0 × 10−10). ADH1B plays a key role in oxidation of ethanol to acetaldehyde, with this variant linked previously to decreased rates of alcohol aversion, increased alcohol consumption, and increased rates of liver fibrosis.38–41 Third, an intergenic variant near the gene encoding tribbles pseudokinase 1 (TRIB1) was associated with a 0.19 percentage point increase in liver fat (beta = 0.05 SD, p = 3.8 × 10−10), with fine-mapping nominating a set of three variants 30–60 kb downstream of the gene in the 95% credible set (Table S3). Variants near this gene have been associated previously with circulating triglyceride concentrations, with functional studies suggesting a role in regulating hepatic lipogenesis.42–45 Fourth, an intronic variant in the gene encoding glycerol-3-phosphate acyltransferase, mitochondrial (GPAM) was associated with a 0.24 percentage point increase in liver fat (beta = 0.05 SD, p = 1.4 × 10−9). Fine-mapping highlighted 17 variants in or near GPAM within the 95% credible set, including the p.V43I missense variant with the highest PIP (0.26). This gene was associated previously with liver triglyceride content in murine overexpression and knockout experiments.46,47 Fifth, an intronic variant in the gene encoding microtubule-associated serine/threonine kinase 3 (MAST3) was associated with a 0.18 percentage point increase in liver fat (beta = 0.05 SD, p = 2.7 × 10−8). Fine-mapping failed to resolve the causal variant at this locus, with nine variants included in the 95% credible set: seven intronic variants in MAST3, a gene linked to inflammatory bowel disease48 but with an unknown role in liver fat metabolism, and two missense variants (PIP 0.03 and 0.02) in MPV17 mitochondrial inner membrane protein-like 2 (MPV17L2) and IFI30 lysosomal thiol reductase (IFI30), respectively.
Our machine-learning imaging-based analyses expanded the number of individuals with liver fat quantification from 4,040 to 32,974, providing increased power to enable the GWAS discovery effort. Taking the most strongly associated variant, the p.I148M missense variant in PNPLA3, as an example, the p value for association decreased from 2.3 × 10−20 when performing a GWAS in only 4,040 individuals to 5.6 × 10−95 when using 32,974 participants (Table S4). Moreover, although each of the five newly identified variants had directionally consistent evidence of association in the GWAS limited to 4,040 individuals with previously quantified liver fat (p values ranging from 0.16–3.6 × 10−4), none met the standard threshold for genome-wide statistical significance of p < 5 × 10−8 (Table S4).
We next sought to replicate additional variants reported previously to affect liver fat or risk of NAFLD (Table S5). A missense variant in the gene encoding the glucokinase regulator (GCKR)12,14,15,23 showed a suggestive association with liver fat below the threshold for genome-wide statistical significance (p = 4.1 × 10−7), as did a variant near the gene encoding membrane bound O-acyltransferase domain-containing 7 (MBOAT7;49–51 p = 8.8 × 10−6). Consistent with prior reports suggesting that an inactivating variant in the gene encoding hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) relates more strongly to advanced forms of liver disease,52–54 we did not observe an association with liver fat in our study population (p = 0.40).
Given a known important role of alcohol intake on liver fat, we performed two sets of sensitivity analyses. First, we repeated the GWAS after exclusion of individuals who reported having stopped drinking alcohol or who reported alcohol consumption in excess of United States NAFLD or United Kingdom guidelines. Second, we repeated the GWAS, adjusting for self-reported number of alcoholic drinks consumed per week. In both cases, results for the 8 variants identified were largely similar, suggesting that these variants have a consistent effect on liver fat independent of alcohol consumption (Table S6). For the p.H48R missense variant in ADH1B, the effect size was somewhat reduced, but an association with increased liver fat remained in all sensitivity analyses (p = 5.3 × 10−5 to 3.4 × 10−9). This observation for the ADH1B variant is consistent with a recent study focused on nonalcoholic steatohepatitis (NASH), a more advanced form of fatty liver disease that also includes significant liver inflammation.41 Additional studies that probe the interplay of this variant, alcohol consumption, and liver disease are needed to understand the mechanistic basis of this relationship.
To test for replication of the GWAS associations in independent cohorts, we analyzed liver fat, as assessed by an alternate imaging modality (CT), in 3,284 participants of the Framingham Heart Study Offspring and Third Generation cohorts and 4,195 participants of the Multi-Ethnic Study of Atherosclerosis (MESA) study. In the Framingham Heart Study cohorts, the average age at time of imaging was 52, and 48% were female; in MESA, the average age was 61, and 51% were female. Although the CT measures of hepatic fat based on liver attenuation cannot be directly converted to units of liver fat percentage, 7 of 8 variants’ associations were directionally consistent, and 5 were nominally significant (p < 0.05; Table S7).
Beyond association with liver fat indices, we sought additional validation of the variants identified by GWAS using liver biomarkers assessed at time of study enrollment and clinical diagnosis in the medical record. In UK Biobank, we analyzed up to 362,910 UK Biobank participants, excluding those included in the abdominal MRI substudy. We first determined associations with the liver-associated biomarkers alanine aminotransferase (ALT) and aspartate aminotransferase (AST). All eight variants were robustly (p value threshold = 0.00625 = 0.05/8 variants) associated with increased ALT (p = 0.0002 to <1 × 10−300), and 7 of the 8 variants were associated with increased AST at nominal levels of statistical significance (p < 0.05; Table S8). We next examined association of the GWAS variants with a recorded clinical diagnosis of NAFLD or NASH in the UK Biobank and the Mass General Brigham Biobank, a hospital-based bio-repository.55 2,225 of 362,910 participants in the UK Biobank and 4,129 of 30,573 participants of the Mass General Brigham Biobank had been diagnosed with NAFLD or NASH. In a meta-analysis of these two studies, 7 of the 8 variants were strongly (p value threshold = 0.00625) associated with increased risk, with odds ratios ranging from 1.08–1.43 (p = 0.0003–3.1 × 10−23; Table S9). The remaining variant, rs56252442 near MAST3, was directionally consistent but did not achieve statistical significance (p = 0.32).
Polygenic score associated with chronic liver diseases
Recognizing that each of the 8 common variants individually are estimated to have a modest effect on liver fat percentage or risk of steatosis, we next combined information from each into a weighted polygenic score. Within the discovery study population of 32,974 UK Biobank individuals, this polygenic score explained 3.5% of the observed variance in liver fat percentage. To determine the relationship of the polygenic score to chronic liver diseases, we calculated it in 361,852 UK Biobank participants who were not included in the liver fat imaging substudy and had not been diagnosed with liver disease at time of enrollment. Over a median follow-up of 8.6 years, the polygenic score was strongly associated with a new diagnosis code of NAFLD entered into the medical record during follow-up, with a hazard ratio (HR) per SD score increment (HR/SD) of 1.33 (95% CI, 1.27–1.39, p = 5.6 × 10−36; Figure 4). Individuals who developed NAFLD had a median polygenic score in the 62nd percentile of the distribution compared with the 50th percentile for the remainder of the population. The polygenic score significantly improved discrimination when added to a baseline model comprised of age, age squared, sex, genotyping array, and the first 10 principal components of genetic variation, with C-statistic increasing from 0.55 to 0.60 (p = 4.4 × 10−34). Beyond NAFLD, the polygenic score was also associated with an increased risk of more advanced forms of liver disease: NASH (HR/SD, 1.67; p = 1.1 × 10−17), cirrhosis (HR/SD, 1.41; p = 1.6 × 10−32), and hepatocellular carcinoma (HR/SD, 1.72; p = 8.4 × 10−17), with each showing improvement in C-statistic over the baseline model (p < 2.6 × 10−15; Table S10). Based on prior observations of an association between liver disease risk-increasing alleles of variants in the PNPLA3 and TM6SF2 genes and decreased cholesterol,13,56 we determined the relationship of the polygenic score to estimated untreated low-density lipoprotein (LDL) cholesterol concentrations. Each SD increment in the score was associated with a 1.9 mg/dL (95% CI, 1.7–2.0; p = 6.4 × 10−245) decrease in LDL cholesterol concentrations, illustrating a tradeoff rooted in rates of hepatic lipid secretion with potentially important implications for drug development.
Figure 4. Polygenic score comprised of eight common genetic variants associated with risk of liver disease.

A single polygenic score for each individual was calculated by additively combining the 8 common lead genome-wide association study (GWAS) variants identified in Figure 3 via the number of liver-fat-increasing variants present in each individual, each weighted by their GWAS effect size estimate. (A) Associations between the polygenic score and incident disease occurrence after UK Biobank enrollment were assessed using a Cox proportional hazards model in 361,852 individuals who were not included in the discovery GWAS of imaging data and who did not have prevalent liver disease at time of enrollment, adjusting for age at enrollment, age at enrollment squared, sex, the first 10 principal components of genetic variation, and genotyping array. Hazard ratios (HRs) of incident disease per SD increase in the polygenic score are shown; error bars represent 95% confidence interval (CI).
(B) Rates of incident disease in each decile of the polygenic score are shown; error bars represent 95% CI.
See also Table S10.
Rare inactivating variants in APOB and MTTP
For the subset of 18,013 UK Biobank participants with liver fat quantified and exome sequencing available, we next investigated whether rare inactivating DNA variants might affect liver fat or risk of steatosis. Observed variants were included in this analysis based on a minor allele frequency of less than 0.1% and a prediction to cause premature truncation of a protein (nonsense), insertions or deletions that scramble protein translation (frameshift), or disruption of the messenger RNA splicing process (splice site), as annotated by the LOFTEE (loss-of-function transcript effect estimator) algorithm.57 Because such variants do not occur with adequate frequency to detect individual variant-phenotype relationships, we performed a “collapsing burden” RVAS. In this approach, the observed liver fat residuals for carriers of any inactivating variant for a given gene are compared with individuals without inactivating variants in this gene. This analysis was restricted to 4,156 genes with at least 10 carriers of inactivating variants observed, resulting in an exome-wide Bonferroni-corrected p value for statistical significance of 1.2 × 10−5 (0.05/4,156).
Inactivating variants in the genes encoding apolipoprotein B (APOB) or microsomal triglyceride transfer protein (MTTP), both known to play key roles in lipid homeostasis, were associated with significantly increased liver fat. Among 23 carriers of inactivating variants in APOB, liver fat was substantially increased compared with 17,990 individuals without such a variant (beta = 1.15 SD; p = 1.41 × 10−7). This corresponded to a median liver fat of 8.3% versus 2.2% for carriers and noncarriers, respectively, and an odds ratio for hepatic steatosis in carriers of 6.3 (95% CI, 2.7–14.5; p = 1.80 × 10−5; Figure 5). Consistent with RVAS providing complementary and often non-overlapping information to GWAS, no common variant in the APOB gene was associated with increased liver fat (p > 0.001 for all).
Figure 5. RVAS of liver fat in 18,013 individuals.

18,013 unrelated individuals with exome sequencing data and liver fat estimation available were grouped according to whether they carried any rare variant predicted to inactivate a given gene (carriers) or not (noncarriers). Rare inactivating variants were defined as predicted to cause premature truncation of a protein (nonsense), insertions or deletions that scramble protein translation (frameshift), or disruption of the messenger RNA splicing process (splice site) with an alternate allele frequency of less than 0.1%. Association of carrier status for each gene with inverse normal transformed liver fat, quantified from MRI data using machine learning, was assessed using linear regression. Genes with fewer than 10 inactivating variant carriers were excluded to increase the likelihood of having sufficient statistical power to detect an effect, resulting in 4,156 genes in the analysis and a significance threshold of p = 1.2 × 10−5 (0.05/4,156 genes tested).
(A) Quantile-quantile (QQ) plot with expected p values of each gene from a uniform distribution are shown on the x axis and corresponding observed p values of each gene on the y axis. A gray line indicates the significance threshold (observed p = 1.20 × 10−5).
(B) Liver fat distribution in carriers and noncarriers of inactivating variants in the APOB or MTTP genes. A gray line indicates the hepatic steatosis threshold (liver fat = 5.5%).
(C) Prevalence of hepatic steatosis in carriers and noncarriers of inactivating variants in APOB or MTTP. For boxplots, boxes indicate IQR (25th–75th percentiles), and whiskers indicate distances of 1.5 IQRs from box limits. For bar plots, error bars indicate upper bounds of 95% CI.
See also Tables S11 and S12.
Significant prior genetic and pharmacologic data implicate APOB in hepatic fat accumulation. Apolipoprotein B is an integral component of lipoprotein particles that export fat out of the liver.58 Individuals with two copies of inactivating variants in APOB (human knockouts) suffer from the Mendelian condition homozygous familial hypobetalipoproteinemia, characterized by near-absent levels of circulating apolipoprotein B and LDL cholesterol but significantly increased rates of hepatic steatosis.59,60 Similarly, prior studies of individuals with heterozygous familial hypobetalipoproteinemia, carrying one copy of an inactivating variant in APOB, suggest an increased risk of steatosis, albeit with variable penetrance.61–64 More recently, a candidate gene-based analysis suggested enrichment of pathogenic APOB variants in individuals with NAFLD-associated hepatocellular carcinoma.65 Pharmacologic knockdown of the APOB gene via the antisense oligonucleotide mipomersen is approved for treatment of severe hypercholesterolemia but is infrequently used in clinical practice because of high rates of hepatic steatosis in clinical trials.66
Inactivating variants in the MTTP gene were also significantly associated with increased liver fat (beta = 1.17 SD, p = 1.0 × 10−6) among 19 carriers of inactivating variants versus 17,994 individuals without such a variant. This corresponded to a median liver fat percentage of 6.9% versus 2.2% for carriers and noncarriers, respectively, and an odds ratio for hepatic steatosis of 11.0 (95% CI, 3.9–30.9; p = 4.8 × 10−6) for carriers (Figure 5). A post hoc analysis of GWAS results noted a common missense variant in MTTP, p.I128T (previously associated with LDL cholesterol56), associated with increased liver fat at subthreshold statistical significance (beta = 0.04 SD, p = 3.70 × 10−5). No relationship was observed between this common variant and the presence of an inactivating variant in MTTP (R2 < 0.01).
The association of inactivating variants in MTTP with liver fat is also highly consistent with known biology. MTTP plays a central role in secretion of apolipoprotein B-containing lipoproteins from the liver. Individuals with two inactivating MTTP variants suffer from the Mendelian disorder abetalipoproteinemia, characterized by absence of circulating apolipoprotein B and increased rates of hepatic steatosis.67,68 Similar to APOB inhibition, a pharmacologic inhibitor of MTTP activity is approved for treatment of severe hypercholesterolemia, but clinical use is limited by increased hepatic fat with its use.69
To further determine the phenotypic consequences of inactivating variants in APOB and MTTP, we analyzed an expanded set of 168,600 UK Biobank participants with exome sequencing data available (regardless of availability of abdominal MRI data). Of these 168,600 individuals, 130 (0.08%) had an inactivating variant in APOB. Liver-related biomarker concentrations were increased in these individuals: 35% higher ALT and 14% higher AST; p = 9.6 × 10−13 and 1.7 × 10−5, respectively (Table S11). In contrast to higher values of aminotransferases, carriers of inactivating APOB variants had markedly lower levels of circulating lipoproteins: 38% lower apolipoprotein B, 44% lower LDL cholesterol, and 45% lower triglycerides (p = 7.8 × 10−18 to 3.7 × 10−113). This was associated with a 74% reduction in risk of coronary artery disease (p = 0.04), consistent with our recent report in an expanded dataset.70 Similar to prior data suggesting that inactivating MTTP variants affect circulating biomarkers only when both copies are affected via recessive inheritance,59 no differences in liver-related biomarkers or lipid concentrations were noted when comparing 90 heterozygous carriers of MTTP variants with 168,510 noncarriers (Table S11).
These RVAS results highlight a discrete molecular subtype of hepatic steatosis driven by rare genetic variation. 13 of 3,273 (0.4%) of individuals with steatosis on imaging had an inactivating variant in APOB, characterized by 6-fold increased risk of steatosis but markedly lower circulating lipid concentrations and reduced risk of coronary artery disease. Similarly, 14 of 3,273 (0.4%) of individuals with steatosis had an inactivating variant in MTTP, characterized by 11-fold increased risk of steatosis in the context of defects in apolipoprotein B secretion. Of note, 0 of the 27 individuals with steatosis and an APOB or MTTP inactivating variant reported alcohol intake in excess of United States guidelines compared with 7% in the remainder of individuals with steatosis, and only 22% were obese compared with 40% in the remainder of individuals with steatosis (Table S12). Although these observations were not statistically significant in the context of small sample sizes and warrant investigation in larger studies, they are consistent with a disproportionate genetic rather than environmental predisposition among these individuals.
DISCUSSION
Our analysis describing quantification of liver fat in 36,703 middle-aged participants in the UK Biobank, the majority of whom were of European ancestry, using a machine-learning algorithm trained on a small subset with previously quantified values has several implications for biologic discovery and clinical medicine.
First, the highly accurate estimation of liver fat enabled by a high-throughput machine-learning algorithm extends prior efforts and is likely to be broadly generalizable across a diverse spectrum of important phenotypes. In hold-out testing datasets, our model-based liver fat assessment was highly correlated with liver fat quantified previously by a commercial vendor, with correlation coefficients of 0.97 and 0.99. Previous efforts have similarly shown feasibility of using a convolutional neural net framework to automate liver fat quantification using CT or MRI images in clinical practice.71 Such efforts may be of particular value for liver fat because, in routine clinical practice, liver fat noted from ultrasound or CT imaging is typically reported in qualitative rather than quantitative terms that lack precision and accuracy.11 Beyond the liver, we recently validated a machine-learning model to quantify the diameter of the aorta using cardiac MRI data, enabling discovery of 93 associated genetic variants.72 These and other studies73,74 suggest that machine-learning approaches to rapidly quantify phenotypes in rich imaging datasets are likely to yield important new scientific insights, particularly when extended to complex features derived from dynamic tissues, such as a beating heart, or latent phenotypes not currently measured in clinical practice.
Second, we demonstrate that, although correlated with many cardiometabolic traits, liver fat cannot be readily predicted using information available in clinical practice. Our large-scale study confirmed significantly increased liver fat in important clinical groups, such as those with diabetes or severe obesity. These observations suggest that future research might validate clinical prediction tools, potentially including a polygenic score, that identify subgroups of individuals in whom screening for hepatic steatosis is warranted or those with known steatosis who are most likely to progress to cirrhosis.75 Outside of focused screening, abdominal imaging is very common across a wide range of clinical indications. Application of a machine-learning algorithm to alert ordering clinicians of an incidental finding of hepatic steatosis may enable measures that prevent progression to more advanced liver disease, such as treatment of hepatitis C infection, alcohol avoidance, dietary interventions, or bariatric surgery in those with severe obesity.7 This approach has proven useful in identifying individuals with subclinical atherosclerosis on chest CT imaging, and reporting this atherosclerosis as an incidental finding is now recommended in clinical guidelines.76,77
Third, our common variant GWAS using our estimated liver fat dataset identified eight significantly associated genetic variants. None of the five newly associated variants were identified using the subset of 4,040 individuals with liver fat quantified without machine learning. Variants identified were largely overlapping with recent analyses of UK Biobank imaging data using a complementary automated segmentation U-net approach or quantification by a different commercial vendor published during review of this manuscript.78,79 We note compelling biology underlying most of the associated variants and provide proof of concept that a polygenic score composed of the eight variants was associated with risk of liver diseases. Additional studies are needed to develop absolute risk estimators for NAFLD and related diseases, ideally integrating clinical risk factors, a polygenic score, and rare predisposing variants.
Fourth, an RVAS, despite a relatively small sample size of 18,013 individuals with liver fat and exome sequencing data available, identified associations of inactivating variants in APOB and MTTP with liver fat. These observations recapitulate results observed in pharmacologic studies of APOB or MTTP inhibition as a treatment for hypercholesterolemia: those with inactivating variants in APOB had strikingly lower lipid concentrations, but this came at the expense of increased aminotransferase concentrations and a more than 6.3-fold increase in rates of hepatic steatosis. Given that elevated liver biomarkers or increased hepatic fat are commonly observed adverse reactions to novel drug candidates, in many cases leading to termination of drug development programs, our approach to using genetics to predict hepatotoxicity may prove valuable. Moreover, our results suggest that a subset of candidate treatments for hepatic steatosis may have adverse effects by increasing circulating lipids. Thus, prioritization of drug targets, such as MTARC1, where genetic studies suggest inhibition will protect against liver disease without increasing cholesterol concentrations or risk of cardiovascular disease,33,37 may be warranted.
Limitations of the study
Our results should be interpreted in the context of several potential limitations. First, participants of the UK Biobank imaging study tend to be healthier than the general population, and 97% were of self-reported European ancestry. Although our algorithm for liver fat estimation appeared to perform comparably well in non-European participants (Figure S1), additional research is needed to investigate generalizability and trans-ancestry portability. Second, diagnostic codes entered into the electronic health record were used to study the relationship between a clinical diagnosis of NAFLD and liver fat based on imaging. Because such codes are known to be imperfect, future studies involving biopsy-confirmed cases of NAFLD are warranted. Third, because imaging of UK Biobank participants occurred recently and not at time of enrollment, we were not able to directly compare the predictive power of liver fat versus other clinical or biomarker predictors with respect to future risk of cardiometabolic or liver diseases.
We applied a machine-learning algorithm to quantify liver fat in 36,703 participants in the UK Biobank, identifying 17% of the population with evidence of hepatic steatosis despite lack of a recorded clinical diagnosis of fatty liver disease, enabling new genetic discoveries with potential implications for new mechanistic pathways underlying risk for liver disease in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Amit Khera (avkhera@mgh.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Summary statistics for the liver fat GWAS have been deposited in the GWAS Catalog at https://www.ebi.ac.uk/gwas/ and are publicly available under accession number GCST90029073. The machine learning model architectures and weights have been deposited in the ML4H GitHub at https://github.com/broadinstitute/ml4h repository and are publicly available in the ML4H model zoo under the name liver_fat_from_mri_ukb. Liver fat quantification data has been returned to the UK Biobank and can be accessed via application to the UK Biobank at https://www.ukbiobank.ac.uk/. DOIs and accession numbers are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| UK Biobank, including previously quantified liver fat and liver fat quantified via machine learning | 17,80; this paper | https://www.ukbiobank.ac.uk/; df-22402 |
| Liver fat common variant genome-wide association study summary statistics | This paper | https://www.ebi.ac.uk/gwas/ Study: GCST90029073 |
| Liver fat previously quantified in Framingham Heart Study | 81–83 | https://www.ncbi.nlm.nih.gov/gap/; Dataset: pht005157.v3.p13 |
| Liver fat previously quantified in Multi-Ethnic Study of Atherosclerosis | 84,85 | https://www.ncbi.nlm.nih.gov/gap/; Dataset: pht002104.v2.p3 |
| Software and algorithms | ||
| BOLT-LMM version 2.3.4 | 26 | https://alkesgroup.broadinstitute.org/BOLT-LMM/BOLT-LMM_manual.html |
| BOLT-REML in BOLT-LMM version 2.3.4 | 25 | https://alkesgroup.broadinstitute.org/BOLT-LMM/BOLT-LMM_manual.html |
| R statistical software version 3.5 | 86 | http://www.R-project.org/ |
| Michigan Imputation Server version 1.1 | 87 | http://imputationserver.sph.umich.edu/index.html |
| Liver fat machine learning algorithm architecture and weights | This paper | https://github.com/broadinstitute/ml4h (liver_fat_from_mri_ukb in model zoo) |
| PolyFun version 1.0.0 | 32 | https://github.com/omerwe/polyfun |
| SuSiE version 0.9.53 | 88 | https://stephenslab.github.io/susie-paper/ |
| Ensembl Variant Effect Predictor (VEP) version 96 | 89 | https://uswest.ensembl.org/info/docs/tools/vep/index.html |
| LOFTEE | 57 | https://uswest.ensembl.org/info/docs/tools/vep/index.html |
| Python 3 | 90 | https://www.python.org/ |
| tensorflow version 2.1 | 91 | https://www.tensorflow.org/ |
| ML4H version 0.0.1 | 92 | https://github.com/broadinstitute/ml4h |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study cohorts
UK Biobank
The UK Biobank is a prospective cohort study that enrolled 502,617 individuals aged 40–69 years of age from across the United Kingdom between 2006 and 2010.80 As part of the study protocol, a subset of individuals underwent detailed imaging including abdominal MRI93 between 2014 and 2019, an average of 9.3 years after enrollment visit. Participants who underwent imaging tended to be healthier than those who did not, as reflected by lower rates of obesity, coronary artery disease, and diabetes (Table S1).
Framingham Heart Study
The Framingham Heart Study is a multigenerational prospective cohort study that enrolled individuals free of cardiovascular disease beginning in 1948. Here, we analyze 3,284 individuals in the Offspring and Third Generation cohorts (enrollment beginning in 1971 and 2002, respectively)81,82 with genotype data available who underwent multidetector abdominal CT for liver fat quantification as previously described.83
Multi-Ethnic Study of Atherosclerosis (MESA)
The Multi-Ethnic Study of Atherosclerosis (MESA) study is a prospective cohort that enrolled individuals free of cardiovascular disease between 2000 and 2002.84 4,195 individuals who underwent multidetector CT for liver fat quantification85 and had genetic data available and were used in analyses described below.
Mass General Brigham Biobank
Mass General Brigham Biobank is a hospital-based biorepository with genetic data linked to clinical records.55 Patients were defined as having NAFLD or NASH according to diagnosis codes in the electronic health care record (Table S2) and were compared to controls without such diagnoses as described below.
Informed Consent and Study Approval
The UK Biobank study was approved by the Research Ethics Committee (reference 16/NW/0274) and informed consent was obtained from all participants. Analysis of UK Biobank data was conducted under application 7089 and was approved by the Mass General Brigham institutional review board. Framingham HeartStudy and MESA genotype and phenotype data were retrieved for analysis from NCBI dbGAP under procedures approved by the Mass General Brigham institutional review board. Mass General Brigham Biobank participants each provided written informed consent and analysis was approved by the Mass General Brigham institutional review board.
METHOD DETAILS
Sample inclusion
Liver fat quantification in UK Biobank
We first quantified liver fat in UK Biobank participants with abdominal MRI imaging available. The UK Biobank abdominal imaging protocol was first performed with gradient echo imaging; a subset of participants had liver fat quantified by Perspectum Diagnostics as previously described.17 Beginning in 2018, imaging was switched to the “iterative decomposition of water and fat with echo asymmetry and least-squares estimation” (IDEAL) protocol. A subset of participants underwent both imaging protocols.
To determine liver fat percentage from abdominal MRI images, we used 2D Convolutional Neural Networks (CNNs) to estimate liver fat percentage from abdominal MRI in 38,706 individuals. The imaging protocol in UK Biobank was switched from gradient echo to IDEAL mid-study, and liver fat was previously quantified by Perspectum Diagnostics only in individuals imaged using the gradient echo protocol.17 To be able to infer liver fat from both protocols, we therefore used a two-model approach with “teacher-student” models. The “teacher” model was a 2D CNN trained on individuals who underwent the gradient echo imaging protocol. The gradient echo protocol consisted of acquiring 10 images;17 to avoid potential errors in estimation that could arise from using a different number of images, we restricted the participants used for model training to individuals who had 10 images, resulting in 3,210 used for model training and 1,215 held out for model testing. The truth data for this model were liver fat values previously quantified by Perspectum Diagnostics from gradient echo imaging protocols which were made available to UK Biobank researchers. Liver fat values for the remaining 5,496 participants with gradient echo imaging and 10 images were estimated using this model.
To estimate liver fat in participants imaged using the IDEAL protocol, we also trained a 2D CNN “student” model in the participants who had undergone both the gradient echo and IDEAL imaging protocols. The IDEAL protocol included 36 images with largest image pixel value < 1024; of the 1,441 individuals who had both imaging protocols and these 36 images, 1,057 were used for training and 384 were held out for testing. The truth data for this model was liver fat in the gradient echo protocol, which was inferred from the “teacher” model. Liver fat values for the remaining 28,595 participants with IDEAL imaging and 36 images were inferred using this model. In total, we estimated liver fat for 34,091 participants with these two models. For both models, model prediction of liver fat < 0.1% was set to missing. Two participants in the testing datasets – one in the teacher model and one in the student model – had missing predictions, resulting in final testing datasets of 1,214 and 383 participants respectively.
To combine the previously-quantified liver fat and results of the two models, we first used the previously-quantified liver fat estimates provided by the UK Biobank where available. When previously-quantified liver fat was unavailable, we preferentially used the liver fat estimates from the teacher model. When teacher model liver fat estimates were unavailable, we used the liver fat estimates from the student model. For subsequent analyses of liver fat, we filtered to 36,703 individuals in UK Biobank with genetic data and liver imaging available. Final sources of liver fat were: n = 4,511 previously-quantified, n = 4,971 estimated from gradient echo protocol, n = 27,221 estimated from IDEAL protocol.
Common variant association studies
We next performed a common variant genome-wide association study (GWAS) of liver fat on a subset of 32,974 UK Biobank participants. We excluded samples that had no imputed genetic data, a genotyping call rate < 0.98, a mismatch between submitted and inferred sex, sex chromosome aneuploidy, exclusion from kinship inference, excessive third-degree relatives, or that were outliers in heterozygosity or genotype missingness rates, all of which were previously defined centrally by the UK Biobank.27 Due to the small percentage of non-European samples (Table S1), to avoid artifacts from population stratification we restricted our GWAS to a subset of samples of European ancestries, selected by self-reported British, Irish, or ‘Any other white’ ethnic background with removal of individuals who were outliers based on principal components of genetic variation analysis (PCA), identified using the R package aberrant as previously described.94 We did not remove related individuals from this analysis, but rather used a linear mixed model able to account for cryptic relatedness in common variant association studies.26
To further validate the common variants associated with liver fat in the GWAS, we studied association of single variants as well as a composite 8-variant polygenic score with liver disease and/or blood biomarkers alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in individuals in the UK Biobank who did not undergo imaging and therefore were not part of the discovery cohort. Sample quality control was performed by excluding samples that had no imputed genetic data, a genotyping call rate < 0.95, a mismatch between submitted and inferred sex, sex chromosome aneuploidy, exclusion from kinship inference, excessive third-degree relatives, and outliers in heterozygosity or genotype missingness rates, and restricting to the PCA-selected European subset. We also removed one of each pair of related individuals (2nd degree or closer, KING coefficient > 0.0884), and those which were part of the liver fat GWAS to avoid sample overlap, resulting in up to 362,910 individuals available for analysis. For associations of polygenic score with liver diseases or LDL cholesterol, we additionally excluded individuals who had any of the four diseases investigated or hepatitis B or C infection documented by time of enrollment, resulting in 361,852 participants in the analysis.
Rare variant association study
To assess the relationship of rare inactivating variants with liver fat and related traits, we studied the subset of 168,600 UK Biobank participants with whole exome sequencing data available. Sample quality control was performed by excluding samples that had no imputed genetic data, a genotyping call rate < 0.95, a mismatch between submitted and inferred sex, sex chromosome aneuploidy, exclusion from kinship inference, excessive third-degree relatives, or that were outliers in heterozygosity or genotype missingness rates, and restricting to the PCA-selected European subset as well as removing one of each pair of related individuals (2nd degree or closer, KING coefficient > 0.0884). We first analyzed the relationship between rare inactivating variants and liver fat in 18,013 individuals with both whole exome sequencing and abdominal MRI imaging data available. Next, to understand the relationship between inactivating variants in two genes, APOB and MTTP, and related biomarkers and disease states, we analyzed the full set of up to (depending on biomarker availability) 168,600 participants with exome sequencing data available.
UK Biobank phenotypes
Baseline characteristics of the 36,703 UK Biobank participants are shown in Table S1. Owing to discrepant definitions of a standard drink between U.S. and UK guidelines, we report two sets of numbers. First, we report number of drinks according to the U.S. definition, where 1 drink = 14 g ethanol,7,95 according to the following conversions: red or white wine, 0.84 drinks/glass; beer, 1.29 drinks/pint; liquor, 0.68 drinks/measure; fortified wine, 0.7 drinks/glass; other alcohol, 1 drink/glass. For participants who reported consuming alcohol monthly rather than weekly, monthly alcohol consumption was converted to weekly by multiplying by 0.23. For U.S. guidelines, excessive alcohol intake was defined according to the U.S. American Association for the Study of Liver Diseases (AASLD) guidelines for NAFLD – greater than 14 weekly drinks if female or greater than 21 weekly drinks if male.7 Second, we report alcohol intake according to the UK definition, where 1 drink = 8 g of ethanol.95,96 For UK consumption, excessive alcohol intake was defined according to the UK Chief Medical Officers guideline – greater than 14 weekly drinks regardless of gender.20
Physician diagnosis of NAFLD and other diseases were defined using ICD codes, and self-report and procedure codes where applicable (Table S2). Hepatic steatosis was defined as liver fat > 5.5%, as determined previously for UK Biobank using the original previously-quantified liver fat values.17 High waist-to-hip ratio was defined as greater than 0.9 if male and greater than 0.85 if female.18 Weight categories were defined using BMI: underweight, BMI < 18.5 kg/m2; normal, 18.5 £ BMI < 25 kg/m2; overweight, 25 £ BMI < 30 kg/m2; obese, 30 £ BMI < 40 kg/m2; severely obese, BMI 3 40 kg/m2 as previously defined.19 Body fat percentage was estimated using bioelectrical impedance analysis. Untreated blood lipid measurements and blood pressure were estimated by adjusting for lipid-lowering medication use or anti-hypertensive medication use, respectively, as previously described.21,22 Variables with > 3% difference in proportion of missing data between compared groups are indicated.
QUANTIFICATION AND STATISTICAL ANALYSIS
Liver fat quantification in UK Biobank participants using a new machine learning algorithm
Input MRI images were prepared by stacking each time slice from the abdominal MRI according to their instance number into a 3D tensor. Images were normalized per individual to have a mean of 0 and a standard deviation of 1 for each MRI. The teacher model for the gradient echo modality had 10 channels corresponding to the 10 instances in the gradient echo protocol with height and width of 160 pixels, while the student model for the IDEAL protocol had input images of 36 channels, height of 256 and width of 232 pixels.
The 2D CNNs were optimized with backpropagation and Adaptive Moment stochastic gradient descent (ADAM). We used a batch size of 5 for the student model and 8 for the teacher model, a learning rate of 2e–4, and the ADAM variant of stochastic gradient descent in our analysis as outlined previously.97 The models were implemented in tensorflow version 2.191 using the ML4H modeling framework.92 The python package hyperopt90 was used for Bayesian hyperparameter optimization of the model architecture to select the width, depth, activation function, and the size of each residual block in the CNN. The final architecture consisted of two layers of convolution followed by three residual blocks of 2 convolutions in parallel whose outputs are concatenated and max-pooled reducing the size of the representation by a factor of 4 after each block. To explore the extent to which our trained models focused on the liver tissue, we assembled saliency maps on 100 test set MRIs. As expected, the model was highly attuned to liver parenchyma tissue and attention layering was thus deferred. The output of the final convolutional block is flattened and processed by two fully-connected layers and finally fed to the output regression neuron. All non-linear activations functions in the model are rectified linear units.
Performance on the held-out testing sets was assessed based on Pearson correlation coefficient and mean absolute error for each model (Figure S1). To determine whether our model was prone to overfitting, we generated learning curves that show model’s loss parameters according to epoch on the training set and on a held-out set of dataset of images distinct from the final test set. Each epoch was defined as a full pass over the training set MRIs. By the end of training, we noted consistent loss in the training and validation dataset, suggestive of no evidence of overfitting (Figure S2). As an additional sensitivity analysis, we performed 10-fold cross validation within subsets of the training datasets, noting nearly identical performance in the held-out testing dataset as for the model developed using the full training datasets. For the teacher model, we observed a mean Pearson correlation coefficient across each of 10-folds of 0.975 (values in each fold: 0.970, 0.976, 0.976, 0.976, 0.976, 0.977, 0.976, 0.976, 0.974, 0.976) and an average mean absolute error across each of 10-folds of 0.50% (values in each fold: 0.57%, 0.49%, 0.53%, 0.46%, 0.50%, 0.49%, 0.48%, 0.50%,0.52%, 0.51%). For the student model, we observed a mean Pearson correlation coefficient across each of 10-folds of 0.983 (values in each fold: 0.985, 0.985, 0.978, 0.974, 0.984, 0.982, 0.981, 0.986, 0.985, 0.987) and an average mean absolute error of 0.58% (values in each fold: 0.53%, 0.54%, 0.69%, 0.65%, 0.56%, 0.58%, 0.62%, 0.56%, 0.52%, 0.52%).
To compare the performance of our machine learning, image-based model for liver fat quantification to an approach using clinical and anthropometric factors, we developed and tested a multivariable regression model. A beta distribution was selected based on effective modeling of liver fat percentages as a series of proportions in the interval (0,1).98 We therefore constructed a beta regression model of liver fat using clinical and anthropometric factors in the same derivation and testing sets used to develop the machine learning model. We selected available anthropometrics, biomarkers associated with metabolic function and liver function or injury, as well as measurements of total body or abdominal fat available in UK Biobank. Only traits which were nominally (p value < 0.05) associated with liver fat in univariable analysis were included in the beta regression model. Variables which were not associated with liver fat and were therefore excluded from the beta regression model were: total bilirubin, direct bilirubin and indirect bilirubin. Final variables included in the model were: body-mass index, waist circumference, hip circumference, total body fat mass, total body fat percent, age at baseline, sex, height, weight, trunk fat mass, trunk fat percent, waist-to-hip ratio, LDL cholesterol, total cholesterol, HDL cholesterol, triglycerides, systolic blood pressure, alkaline phosphatase, alanine aminotransferase (ALT), aspartate aminotransferase (AST), ALT/AST, gamma glutamyltransferase, hemoglobin A1c, random glucose, and C-reactive protein. Lipid measures were adjusted for lipid-lowering medication use and blood pressure was adjusted for anti-hypertensive medication use, as previously described.21,22 Measurements at time of imaging assessment were available for BMI, height, weight, waist circumference, hip circumference, waist-to-hip ratio and systolic blood pressure and preferentially used in this regression analysis, while the remainder of predictors were measured at time of study enrollment. Missing values were imputed using the aregImpute function in the R package Hmisc. We constructed a variable dispersion beta regression model using 3,210 individuals with liver fat previously quantified by Perspectum Diagnostics.17 This model was constructed using the betareg package in R, optimizing the mean and precision link functions to cloglog and log, respectively, using AIC & BIC comparisons. Performance of the model was evaluated by the Pearson correlation between previously quantified liver fat and predicted liver fat in the held-out testing dataset of 1,214 individuals (Figure S3).
Association of liver fat with clinical characteristics
To determine the relationship between clinical/anthropometric characteristics (sex, excessive alcohol consumption, physician diagnosis of NAFLD, physician diagnosis of diabetes) on median liver fat, or the effects of hepatic steatosis on triglyceride concentrations, we performed median regression. Similarly, we used logistic regression to evaluate the effects of physician diagnosis of NAFLD on hepatic steatosis, and hepatic steatosis on diabetes or hypertension diagnosis. In both median and logistic regression, we included sex, birth year, age at imaging, age at imaging squared and MRI machine serial number as covariates.
Genetic analyses
UK Biobank genotyping and variant quality control
UK Biobank samples were genotyped on either the UK BiLEVE or UK Biobank Axiom arrays, then imputed into the Haplotype Reference Consortium and UK10K + 1000 Genomes panels. We excluded genotyped variants with call rate < 0.95, imputed variants with INFO score < 0.3, and imputed or genotyped variants with minor allele frequency < 1% in the UK Biobank population. Variant positions were denoted in GRCh37/hg19 coordinates. Principal components of genetic variation were calculated centrally by UK Biobank in all participants as previously described.27
Phenotype transformation
Because liver fat is not normally distributed and nor are its residuals with respect to clinical covariates, we transformed the input liver fat phenotype to a rank-based output for the GWAS and RVAS analyses. This approach has commonly used in previous GWAS of quantitative traits with skewed distributions, including body-mass index, a previous study of liver fat, and lipid concentrations.12,56,99,100 First, we took the residuals of liver fat in a linear model that included sex, year of birth, age at time of MRI, age at time of MRI squared, genotyping array, MRI device serial number, and the first ten principal components of genetic variation. Then, we performed the inverse normal transform on the residuals from this model, yielding a standardized output with mean 0 and standard deviation of 1. Results using this transformation of the liver fat phenotype are reported as ‘Betas’ and have a unit of standard deviations.
Common variant genome-wide association study
We performed a GWAS of the inverse normal transformed liver fat residuals in 32,974 individuals, applying linear mixed models with BOLT-LMM (version 2.3.4) to account for ancestry, cryptic population structure, and sample relatedness.26 The default European linkage disequilibrium panel provided with BOLT was used and the first ten principal components of genetic variation were included as covariates. We measured heritability in the same samples using BOLT-REML (BOLT-LMM version 2.3.4 with the –reml flag). Variants with BOLT-LMM p value < 5 × 10−8 were considered to be genome-wide significant. Loci were defined by 2 MB windows (1 MB distance from the most-significant variant in either direction). The most strongly associated variant at each locus is referred to as the lead variant. We determined the effects of each of the eight lead variants on liver fat % and presence of hepatic steatosis (liver fat >5.5%) using linear and logistic regression, respectively, in the same 32,974 individuals in the GWAS, adjusting for sex, year of birth, age at time of MRI, age at time of MRI squared, genotyping array, MRI device serial number, and the first ten principal components of genetic variation. We repeated the GWAS in the subset of 4,040 individuals with previously-quantified liver fat who passed the GWAS sample quality control.
We applied the PolyFun (functionally-informed fine-mapping) algorithm32 to fine map the 8 genome-wide significant loci discovered in the GWAS. We mapped a window surrounding the lead variant at each locus based on visual inspection of the region as sufficiently large enough to cover all possible associated signals while excluding overlapping associations from nearby independent regions. For these 8 regions, we independently applied the PolyFun algorithm for fine-mapping of causal variant(s) with functional enrichment priors estimated from the baseline-LF 2.2.UKB annotations (pre-computed from PolyFun). This is a broad set of coding, conserved, regulatory, and LD-related annotations and has ~19 million UK Biobank imputed SNPs with minor allele frequency > 0.1%.32 We used SuSiE as the underlying fine-mapping method,88 set the maximum number of causal variants of 10, and used in-sample linkage disequilibrium information from individuals in the GWAS.
GWAS replication.
We replicated the GWAS findings in the Framingham Heart Study and the Multi-Ethnic Study of Atherosclerosis (MESA). In the Framingham cohort (Offspring Cohort and Third Generation Cohort), we examined whether the 8 variants associate with hepatic steatosis on CT imaging. Genotyping was imputed to the HapRef consortium using the Michigan Imputation Server.87 After imputation, variants with allele frequency < 0.01% and those with an imputation score < 0.3 were excluded from analysis. Liver fat was assessed by computing the liver-to-phantom ratio of the average Hounsfield units of three liver measurements to average Hounsfield units of three phantom measurements (to correct for inter-individual differences in penetration), as previously described.83 This liver fat phenotype was inverse normal rank transformed prior to genetic analysis. We tested the association of all 8 variants with liver-to-phantom ratio adjusting for age, sex and ten principal components of genetic variationusing a linear mixed model (BOLT-LMM) to control for relatedness among individuals.
In the Multi-Ethnic Study of Atherosclerosis cohort (MESA), genotypes were imputed to the HapRef consortium using the Michigan Imputation Server.87 After imputation, variants with allele frequency < 0.01% and those with an INFO score < 0.3 were excluded from analysis. Liver fat was measured as the mean of three attenuation measurements, two in the right lobe of the liver and one in the left lobe,85 without use of phantom measurement normalization. Liver fat measurements were inverse normal rank transformed prior to analysis. We tested the association of the top GWAS variants with mean liver attenuation with adjustment for age, sex and five principal components of genetic variation.
Individuals with higher liver fat have lower liver-to-phantom ratios and liver attenuation measurements. To increase interpretability and for consistency with UK Biobank results, we therefore report beta estimates in the Framingham study and MESA where positive values correspond to increased liver fat. Effect estimates from the Framingham study and MESA were combined via fixed-effect meta-analysis; heterogeneity was assessed and random-effects models were used when evidence of heterogeneity was noted (phet < 0.05).
Association of GWAS variants with liver biomarkers and disease.
We examined the association of the top GWAS variants with blood biomarkers assessed at time of study enrollment alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in UK Biobank using linear regression of each biomarker (in U/L) adjusting for sex, year of birth, age at enrollment and age at enrollment squared, genotyping array and the first ten principal components of genetic variation.
We also examined the association of the top GWAS variants with physician diagnosis of NAFLD/NASH in UK Biobank and Mass General Brigham Biobank. Disease definitions are provided in Table S2. In UK Biobank, association of each top GWAS variant was assessed using logistic regression of disease status with sex, year of birth, age at enrollment and age at enrollment squared, genotyping array and the first ten principal components of genetic variationas covariates. In the Mass General Brigham Biobank, genotyping was performed using an Illumina MEGA array. Variants were imputed to the HapRef consortium using the Michigan Imputation Server. Variants with multinucleotide alleles and those with call rate less than 90% were excluded prior to imputation. After imputation, variants with allele frequency < 0.01% and those with an INFO score < 0.3 were excluded from analysis. Association of each top GWAS variant was assessed using logistic regression of disease status with age, sex and five principal components of genetic variationas covariates. Effect estimates from UK Biobank and Mass General Brigham Biobank were combined via fixed-effect meta-analysis; heterogeneity was assessed and random-effects models were used when nominal heterogeneity was noted (phet < 0.05).
Polygenic score analysis.
We constructed a single polygenic score for each individual by additively combining the 8 lead GWAS variants based on number of liver-fat increasing variants present in each individual, each weighted by their GWAS effect size estimate.
We tested for association between the score and incident disease occurrence after UK Biobank enrollment using a Cox model in the same set of individuals used to test associations between single GWAS variants and NAFLD/NASH. We excluded individuals who had any of the four diseases investigated or hepatitis B or C infection documented at time of enrollment, resulting in 361,852 participants in the analysis. We focused on the association of the score with liver diseases; given previously reported association of liver fat variants with circulating lipids,56 we also examined association of the score with circulating LDL cholesterol using linear regression. LDL cholesterol was adjusted for lipid-lowering medication to estimate untreated values as above;22 liver disease definitions are listed in Table S2. All polygenic score analyses were adjusted for age at enrollment, age at enrollment squared, sex, the first ten principal components of genetic variation, and genotyping array. We also quantified the proportion of individuals who developed each disease during study follow-up stratified by PRS decile. C statistics were calculated for a baseline model (age at enrollment, age at enrollment squared, sex, the first ten principal components of ancestry, and genotyping array) and a baseline plus polygenic score model; a likelihood ratio test was used to assess whether the two models were significantly different.
Rare variant association study
In the subset of individuals with whole exome sequencing available, we identified rare (minor allele frequency < 0.1%) inactivating variants in each gene. Sequencing data from the “Functionally Equivalent” gene sequencing dataset was annotated using the LOFTEE plugin for the Ensembl Variant Effect Predictor (VEP) software (version 96.0).89 LOFTEE applies a set of filters to identify high-confidence inactivating variants based on predicted impact on the resulting transcript.57 High-confidence inactivating variants include those predicted to cause premature truncation of a protein (nonsense), insertions or deletions (indels) of DNA that scramble protein translation beyond the variant site (frameshift) and point mutations at sites of pre-messenger ribonucleic acid splicing that alter the splicing process (splice-site).
We aggregated the inactivating variants identified within each gene into a rare variant burden analysis: individuals were considered as an inactivating variant carrier for a particular gene if they had one or more inactivating variants in the gene, and a non-carrier otherwise. We tested the association of inactivating variant carrier status for each gene with inverse normal transformed liver fat as described above (see Phenotype Transformation) using linear regression with the first ten principal components of genetic variation as covariates. We removed genes with fewer than 10 inactivating variant carriers to increase the likelihood of having sufficient statistical power to detect an effect, leaving 4,156 genes in the analysis. To determine the effects of APOB or MTTP inactivating variants on blood biomarkers or disease outcomes, we used linear or logistic regression, respectively, adjusting for sex, year of birth, age at enrollment and age at enrollment squared, genotyping array and the first ten principal components of genetic variation. LDL cholesterol and triglycerides were adjusted for lipid-lowering medication to estimate untreated values as previously described.22
Statistical analyses were conducted using R version 3.5 software.86
Supplementary Material
Highlights.
A machine-learning algorithm precisely quantified liver fat in 36,703 individuals
17% of imaged participants had excess liver fat but were largely undiagnosed
8 common genetic variants and polygenic score associated with liver fat and liver disease
Rare variants in APOB or MTTP genes highlight a molecular subtype of steatosis
ACKNOWLEDGMENTS
This research was conducted using the UK Biobank resource, application 7089. Funding support was provided by NIH grants 1K08HG010155 and 1U01HG011719 (to A.V.K.) from the National Human Genome Research Institute; 1R01HL092577, R01HL128914, K24HL105780 (to P.T.E), and R01HL071739 (to M.B.) from the National Heart, Lung, and Blood Institute; 5P42ES010337 (to R.L.) from the National Institute of Environmental Health Sciences; 5UL1TR001442 (to R.L.) from the National Center for Advancing Translational Sciences; R01DK106419, P30DK120515 (to R.L.), and K23 DK122104 (to T.G.S.) from the National Institute of Diabetes and Digestive and Kidney Diseases. This work was also supported by CA170674P2 (to R.L.) from the Department of Defense Peer Reviewed Cancer Research Program, a Hassenfeld Scholar Award from Massachusetts General Hospital (to A.V.K.), a Merkin Institute Fellowship from the Broad Institute of MIT and Harvard (to A.V.K.), a John S. LaDue Memorial Fellowship and Sarnoff Scholar Award (to J.P.P.), a sponsored research agreement from IBM Research (to A.P.), and American Association for the Study of Liver Diseases Foundation Clinical and Translational Research Awards (to V.A. and T.G.S.). MESA and the MESA SHARe projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020 D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N 92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420 and supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. Funding for SHARe genotyping was provided by NHLBI contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. The authors thank the other investigators, the staff, and the participants of the MESA study for valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
DECLARATION OF INTERESTS
M.E.H. is currently an employee and shareholder of Regeneron Pharmaceuticals. J.P.P. has served as a consultant for Maze Therapeutics. R.L. serves as a consultant or advisory board member for Arrowhead Pharmaceuticals; AstraZeneca; Boehringer-Ingelheim; Bristol Myers Squibb; Celgene; Cirius; CohBar; Galmed; Gemphire; Gilead; Glympse bio; Intercept; Ionis; Inipharma; Merck; Metacrine, Inc.; NGM Biopharmaceuticals; Novo Nordisk; Pfizer; and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly and Company, Galmed Pharmaceuticals, Genfit, Gilead, Intercept, Janssen, Madrigal Pharmaceuticals, NGM Biopharmaceuticals, Novartis, Pfizer, pH Pharma, and Siemens. He is also co-founder of Liponexus, Inc. A.Y.Z. is an employee of Color Health. J.R.H. was an employee of Color Health and is currently an employee of Maze Therapeutics. K.E.C. serves on the advisory boards of Novo Nordisk and BMS, has consulted for Gilead, and has received grant funding from BMS, Boehringer-Ingelheim, and Novartis. T.G.S. has served as a consultant for Aetion. A.P. is employed as a Venture Partner at GV, a venture capital group within Alphabet; he is also supported by a grant from Bayer AG to the Broad Institute, focused on machine learning for clinical trial design. S.N.F. and P.B. are supported by grants from Bayer AG and IBM applying machine learning in cardiovascular disease. P.B. has served as a consultant to Novartis. P.T.E. is supported by a grant from Bayer AG to the Broad Institute, focused on the genetics and therapeutics of cardiovascular diseases. P.T.E. has also served on advisory boards or consulted for Bayer AG, Quest Diagnostics, MyoKardia, and Novartis. A.V.K. has served as a scientific advisor to Sanofi, Amgen, Maze Therapeutics, Navitor Pharmaceuticals, Sarepta Therapeutics, Verve Therapeutics, Veritas International, Color Health, Third Rock Ventures, and Columbia University (NIH); received speaking fees from Illumina, MedGenome, Amgen, and the Novartis Institute for Biomedical Research; and received sponsored research agreements from the Novartis Institute for Biomedical Research and IBM Research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2021.100066.
REFERENCES
- 1.Allen AM, Therneau TM, Larson JJ, Coward A, Somers VK, and Kamath PS (2018). Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: A 20 year-community study. Hepatology 67, 1726–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caussy C, Reeder SB, Sirlin CB, and Loomba R (2018). Noninvasive, Quantitative Assessment of Liver Fat by MRI-PDFF as an Endpoint in NASH Trials. Hepatology 68, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loomba R, Friedman SL, and Shulman GI (2021). Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564. [DOI] [PubMed] [Google Scholar]
- 4.Speliotes EK, Massaro JM, Hoffmann U, Vasan RS, Meigs JB, Sahani DV, Hirschhorn JN, O’Donnell CJ, and Fox CS (2010). Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: the Framingham Heart Study. Hepatology 51, 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loomba R, and Sanyal AJ (2013). The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol 10, 686–690. [DOI] [PubMed] [Google Scholar]
- 6.Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, Wai-Sun Wong V, Yilmaz Y, George J, Fan J, and Vos MB (2019). Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 69, 2672–2682. [DOI] [PubMed] [Google Scholar]
- 7.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, and Sanyal AJ (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357. [DOI] [PubMed] [Google Scholar]
- 8.Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, Ratziu V, and McCullough A (2011). Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 54, 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexander M, Loomis AK, Fairburn-Beech J, van der Lei J, Duarte-Salles T, Prieto-Alhambra D, Ansell D, Pasqua A, Lapi F, Rijnbeek P, et al. (2018). Real-world data reveal a diagnostic gap in non-alcoholic fatty liver disease. BMC Med. 16, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanyal AJ (2018). Putting non-alcoholic fatty liver disease on the radar for primary care physicians: how well are we doing? BMC Med. 16, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YN, Fowler KJ, Hamilton G, Cui JY, Sy EZ, Balanay M, Hooker JC, Szeverenyi N, and Sirlin CB (2018). Liver fat imaging-a clinical overview of ultrasound, CT, and MR imaging. Br. J. Radiol 91, 20170959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, et al. ; NASH CRN; GIANT Consortium; MAGIC Investigators; GOLD Consortium (2011). Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 7, e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, Vogt TF, Hobbs HH, and Cohen JC (2014). Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 46, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parisinos CA, Wilman HR, Thomas EL, Kelly M, Nicholls RC, McGonigle J, Neubauer S, Hingorani AD, Patel RS, Hemingway H, et al. (2020). Genome-wide and Mendelian randomisation studies of liver MRI yield insights into the pathogenesis of steatohepatitis. J. Hepatol 73, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer ND, Kahali B, Kuppa A, Chen Y, Du X, Feitosa MF, Bielak LF, O’Connell JR, Musani SK, Guo X, et al. (2021). Allele-specific variation at APOE increases nonalcoholic fatty liver disease and obesity but decreases risk of Alzheimer’s disease and myocardial infarction. Hum. Mol. Genet 30, 1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, and Visscher PM; GIANT Consortium (2018). Meta-analysis of genome-wide association studies for height and body mass index in :700000 individuals of European ancestry. Hum. Mol. Genet 27, 3641–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilman HR, Kelly M, Garratt S, Matthews PM, Milanesi M, Herlihy A, Gyngell M, Neubauer S, Bell JD, Banerjee R, and Thomas EL (2017). Characterisation of liver fat in the UK Biobank cohort. PLoS ONE 12, e0172921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.World Health Organization (2011). Waist circumference and waist-hip ratio: report of a WHO expert consultation, Geneva, 8–11 December 2008 (World Health Organization; ). [Google Scholar]
- 19.National Institutes of Health (1998). Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults–The Evidence Report. Obes. Res 6 (Suppl 2), 51S–209S. [PubMed] [Google Scholar]
- 20.Department of Health (2016). UK Chief Medical Officers’ Low Risk Drinking Guidelines (Williams Lea; ). [Google Scholar]
- 21.Ehret GB, Ferreira T, Chasman DI, Jackson AU, Schmidt EM, Johnson T, Thorleifsson G, Luan J, Donnelly LA, Kanoni S, et al. ; CHARGE-EchoGen consortium; CHARGE-HF consortium; Well-come Trust Case Control Consortium (2016). The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat. Genet 48, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel AP, Wang M, Fahed AC, Mason-Suares H, Brockman D, Pelletier R, Amr S, Machini K, Hawley M, Witkowski L, et al. (2020). Association of Rare Pathogenic DNA Variants for Familial Hypercholesterolemia, Hereditary Breast and Ovarian Cancer Syndrome, and Lynch Syndrome With Disease Risk in Adults According to Family History. JAMA Netw. Open 3, e203959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, Kahali B, Carr JJ, Harris TB, Jhun MA, et al. (2013). Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 58, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loomba R, Schork N, Chen C-H, Bettencourt R, Bhatt A, Ang B, Nguyen P, Hernandez C, Richards L, Salotti J, et al. ; Genetics of NAFLD in Twins Consortium (2015). Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 149, 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loh P-R, Bhatia G, Gusev A, Finucane HK, Bulik-Sullivan BK, Pollack SJ, de Candia TR, Lee SH, Wray NR, Kendler KS, et al. ; Schizophrenia Working Group of Psychiatric Genomics Consortium (2015). Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet 47, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loh P-R, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, et al. (2015). Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet 47, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, Daly MJ, Price AL, and Neale BM; Schizophrenia Working Group of the Psychiatric Genomics Consortium (2015). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, and Hobbs HH (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 40, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, Burt AD, Bedossa P, Palmer J, Liu YL, et al. ; EPoS Consortium Investigators (2020). Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort☆. J. Hepatol 73, 505–515. [DOI] [PubMed] [Google Scholar]
- 31.Emdin CA, Haas M, Ajmera V, Simon TG, Homburger J, Neben C, Jiang L, Wei WQ, Feng Q, Zhou A, et al. (2021). Association of Genetic Variation With Cirrhosis: A Multi-Trait Genome-Wide Association and Gene-Environment Interaction Study. Gastroenterology 160, 1620–1633.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weissbrod O, Hormozdiari F, Benner C, Cui R, Ulirsch J, Gazal S, Schoech AP, van de Geijn B, Reshef Y, Márquez-Luna C, et al. (2020). Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet 52, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emdin CA, Haas ME, Khera AV, Aragam K, Chaffin M, Klarin D, Hindy G, Jiang L, Wei WQ, Feng Q, et al. ; Million Veteran Program (2020). A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 16, e1008629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Innes H, Buch S, Hutchinson S, Guha IN, Morling JR, Barnes E, Irving W, Forrest E, Pedergnana V, Goldberg D, et al. (2020). Genome-Wide Association Study for Alcohol-Related Cirrhosis Identifies Risk Loci in MARC1 and HNRNPUL1. Gastroenterology 159, 1276–1289.e7. [DOI] [PubMed] [Google Scholar]
- 35.Luukkonen PK, Juuti A, Sammalkorpi H, Penttilä AK, Orešič M, Hyötyläinen T, Arola J, Orho-Melander M, and Yki-Järvinen H (2020). MARC1 variant rs2642438 increases hepatic phosphatidylcho-lines and decreases severity of non-alcoholic fatty liver disease in humans. J. Hepatol 73, 725–726. [DOI] [PubMed] [Google Scholar]
- 36.Mann JP, Pietzner M, Wittemans LB, Rolfe EL, Kerrison ND, Imamura F, Forouhi NG, Fauman E, Allison ME, Griffin JL, et al. (2020). Insights into genetic variants associated with NASH-fibrosis from metabolite profiling. Hum. Mol. Genet 29, 3451–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider CV, Schneider KM, Conlon DM, Park J, Vujkovic M, Zandvakili I, Ko YA, Trautwein C, Center R, Carr RM, et al. (2021). A genome-first approach to mortality and metabolic phenotypes in MTARC1 p.Ala165Thr (rs2642438) heterozygotes and homozygotes. Med (N Y) 2, 851–863.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bosron WF, and Li TK (1986). Genetic polymorphism of human liver alcohol and aldehyde dehydrogenases, and their relationship to alcohol metabolism and alcoholism. Hepatology 6, 502–510. [DOI] [PubMed] [Google Scholar]
- 39.Edenberg HJ (2007). The genetics of alcohol metabolism: role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 30, 5–13. [PMC free article] [PubMed] [Google Scholar]
- 40.Li D, Zhao H, and Gelernter J (2011). Strong association of the alcohol dehydrogenase 1B gene (ADH1B) with alcohol dependence and alcohol-induced medical diseases. Biol. Psychiatry 70, 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vilar-Gomez E, Sookoian S, Pirola CJ, Liang T, Gawrieh S, Cummings O, Liu W, and Chalasani NP (2020). ADH1B*2 Is Associated With Reduced Severity of Nonalcoholic Fatty Liver Disease in Adults, Independent of Alcohol Consumption. Gastroenterology 159, 929–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, et al. (2008). Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet 40, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burkhardt R, Toh S-A, Lagor WR, Birkeland A, Levin M, Li X, Robblee M, Fedorov VD, Yamamoto M, Satoh T, et al. (2010). Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J. Clin. Invest 120, 4410–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishizuka Y, Nakayama K, Ogawa A, Makishima S, Boonvisut S, Hirao A, Iwasaki Y, Yada T, Yanagisawa Y, Miyashita H, et al. ; Jichi Medical University Promotion Team of Large-Scale Human Genome Bank for All over Japan (2014). TRIB1 downregulates hepatic lipogenesis and glycogenesis via multiple molecular interactions. J. Mol. Endocrinol 52, 145–158. [DOI] [PubMed] [Google Scholar]
- 45.Bauer RC, Sasaki M, Cohen DM, Cui J, Smith MA, Yenilmez BO, Steger DJ, and Rader DJ (2015). Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα. J. Clin. Invest 125, 3809–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hammond LE, Gallagher PA, Wang S, Hiller S, Kluckman KD, Posey-Marcos EL, Maeda N, and Coleman RA (2002). Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol. Cell. Biol 22, 8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindén D, William-Olsson L, Ahnmark A, Ekroos K, Hallberg C, Sjögren HP, Becker B, Svensson L, Clapham JC, Oscarsson J, and Schreyer S (2006). Liver-directed overexpression of mitochondrial glycerol-3-phosphate acyltransferase results in hepatic steatosis, increased triacylglycerol secretion and reduced fatty acid oxidation. FA-SEB J. 20, 434–443. [DOI] [PubMed] [Google Scholar]
- 48.Labbé C, Goyette P, Lefebvre C, Stevens C, Green T, Tello-Ruiz MK, Cao Z, Landry AL, Stempak J, Annese V, et al. (2008). MAST3: a novel IBD risk factor that modulates TLR4 signaling. Genes Immun. 9, 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD, Brosch M, Rosendahl J, Berg T, Ridinger M, et al. (2015). A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet 47, 1443–1448. [DOI] [PubMed] [Google Scholar]
- 50.Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, Borén J, Montalcini T, Pujia A, Wiklund O, et al. (2016). The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 150, 1219–1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teo K, Abeysekera KWM, Adams L, Aigner E, Anstee QM, Banales JM, Banerjee R, Basu P, Berg T, Bhatnagar P, et al. ; EU-PNAFLD Investigators; GOLD Consortium (2021). rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatol 74, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, Kozlitina J, Stender S, Wood GC, et al. (2018). A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med 378, 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, de Boer YS, Koh C, Chen Y, Du X, et al. ; for the Nonalcoholic Steatohepatitis Clinical Research Network (2019). 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 69, 1504–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gellert-Kristensen H, Nordestgaard BG, Tybjaerg-Hansen A, and Stender S (2020). High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase-Lowering Effect of a HSD17B13 Variant. Hepatology 71, 56–66. [DOI] [PubMed] [Google Scholar]
- 55.Karlson EW, Boutin NT, Hoffnagle AG, and Allen NL (2016). Building the Partners HealthCare Biobank at Partners Personalized Medicine: Informed Consent, Return of Research Results, Recruitment Lessons and Operational Considerations. J. Pers. Med 6, E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, et al. ; Charge Diabetes Working Group; EPIC-InterAct Consortium; EPIC-CVD Consortium; GOLD Consortium; VA Million Veteran Program (2017). Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet 49, 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. ; Genome Aggregation Database Consortium (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ipsen DH, Lykkesfeldt J, and Tveden-Nyborg P (2018). Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci 75, 3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee J, and Hegele RA (2014). Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J. Inherit. Metab. Dis 37, 333–339. [DOI] [PubMed] [Google Scholar]
- 60.Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, Peretti N, Dumortier J, Zoulim F, Fontanges T, et al. (2014). Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J. Hepatol 61, 891–902. [DOI] [PubMed] [Google Scholar]
- 61.Cefalù AB, Pirruccello JP, Noto D, Gabriel S, Valenti V, Gupta N, Spina R, Tarugi P, Kathiresan S, and Averna MR (2013). A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler. Thromb. Vasc. Biol 33, 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rimbert A, Pichelin M, Lecointe S, Marrec M, Le Scouarnec S, Barrak E, Croyal M, Krempf M, Le Marec H, Redon R, et al. (2016). Identification of novel APOB mutations by targeted next-generation sequencing for the molecular diagnosis of familial hypobetalipoproteinemia. Atherosclerosis 250, 52–56. [DOI] [PubMed] [Google Scholar]
- 63.Schonfeld G, Patterson BW, Yablonskiy DA, Tanoli TSK, Averna M, Elias N, Yue P, and Ackerman J (2003). Fatty liver in familial hypobetalipoproteinemia: triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J. Lipid Res 44, 470–478. [DOI] [PubMed] [Google Scholar]
- 64.Wishingrad M, Paaso B, and Garcia G (1994). Fatty liver due to heterozygous hypobetalipoproteinemia. Am. J. Gastroenterol 89, 1106–1107. [PubMed] [Google Scholar]
- 65.Pelusi S, Baselli G, Pietrelli A, Dongiovanni P, Donati B, McCain MV, Meroni M, Fracanzani AL, Romagnoli R, Petta S, et al. (2019). Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep 9, 3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, Chin W, Tribble DL, and McGowan M (2012). Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 126, 2283–2292. [DOI] [PubMed] [Google Scholar]
- 67.Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, and Wetterau JR (2000). The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu. Rev. Nutr 20, 663–697. [DOI] [PubMed] [Google Scholar]
- 68.Sharp D, Blinderman L, Combs KA, Kienzle B, Ricci B, Wager-Smith K, Gil CM, Turck CW, Bouma ME, Rader DJ, et al. (1993). Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature 365, 65–69. [DOI] [PubMed] [Google Scholar]
- 69.Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, and Rader DJ (2007). Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med 356, 148–156. [DOI] [PubMed] [Google Scholar]
- 70.Peloso GM, Nomura A, Khera AV, Chaffin M, Won H-H, Ardissino D, Danesh J, Schunkert H, Wilson JG, Samani N, et al. (2019). Rare Protein-Truncating Variants in APOB, Lower Low-Density Lipoprotein Cholesterol, and Protection Against Coronary Heart Disease. Circ Genom Precis Med 12, e002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang K, Mamidipalli A, Retson T, Bahrami N, Hasenstab K, Blansit K, Bass E, Delgado T, Cunha G, Middleton MS, et al. ; members of the NASH Clinical Research Network (2019). Automated CT and MRI Liver Segmentation and Biometry Using a Generalized Convolutional Neural Network. Radiol Artif Intell 1, 180022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pirruccello JP, Chaffin MD, Fleming SJ, Arduini A, Lin H, Khurshid S, Chou EL, Friedman SN, Bick AG, Weng LC, et al. (2020). Deep learning enables genetic analysis of the human thoracic aorta. bioRxiv. 10.1101/2020.05.12.091934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Agrawal S, Klarqvist MDR, Diamant N, Ellinor PT, Mehta NN, Philippakis A, Ng K, Batra P, and Khera AV (2021). Association of machine learning-derived measures of body fat distribution in > 40,000 individuals with cardiometabolic diseases. medRxiv. 10.1101/2021.05.07.21256854. [DOI] [Google Scholar]
- 74.Meyer HV, Dawes TJW, Serrani M, Bai W, Tokarczuk P, Cai J, de Marvao A, Henry A, Lumbers RT, Gierten J, et al. (2020). Genetic and functional insights into the fractal structure of the heart. Nature 584, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ajmera V, Park CC, Caussy C, Singh S, Hernandez C, Bettencourt R, Hooker J, Sy E, Behling C, Xu R, et al. (2018). Magnetic Resonance Imaging Proton Density Fat Fraction Associates With Progression of Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 155, 307–310.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hecht HS, Cronin P, Blaha MJ, Budoff MJ, Kazerooni EA, Narula J, Yankelevitz D, and Abbara S (2017). 2016 SCCT/STR guidelines for coronary artery calcium scoring of noncontrast noncardiac chest CT scans: A report of the Society of Cardiovascular Computed Tomography and Society of Thoracic Radiology. J. Thorac. Imaging 32, W54–W66. [DOI] [PubMed] [Google Scholar]
- 77.Pakdaman MN, Rozanski A, and Berman DS (2017). Incidental coronary calcifications on routine chest CT: Clinical implications. Trends Cardiovasc. Med 27, 475–480. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y, Basty N, Whitcher B, Bell JD, Sorokin EP, van Bruggen N, Thomas EL, and Cule M (2021). Genetic architecture of 11 organ traits derived from abdominal MRI using deep learning. eLife 10, e65554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jamialahmadi O, Mancina RM, Ciociola E, Tavaglione F, Luukkonen PK, Baselli G, Malvestiti F, Thuillier D, Raverdy V, Männistö V, et al. (2021). Exome-Wide Association Study on Alanine Aminotransferase Identifies Sequence Variants in the GPAM and APOE Associated With Fatty Liver Disease. Gastroenterology 160, 1634–1646.e7. [DOI] [PubMed] [Google Scholar]
- 80.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. (2015). UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, and Castelli WP (1979). An investigation of coronary heart disease in families. The Framingham offspring study. Am. J. Epidemiol 110, 281–290. [DOI] [PubMed] [Google Scholar]
- 82.Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, D’Agostino RB Sr., Fox CS, Larson MG, Murabito JM, et al. (2007). The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am. J. Epidemiol 165, 1328–1335. [DOI] [PubMed] [Google Scholar]
- 83.Speliotes EK, Massaro JM, Hoffmann U, Foster MC, Sahani DV, Hirschhorn JN, O’Donnell CJ, and Fox CS (2008). Liver fat is reproducibly measured using computed tomography in the Framingham Heart Study. J. Gastroenterol. Hepatol 23, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR Jr., Kronmal R, Liu K, et al. (2002). Multi-Ethnic Study of Atherosclerosis: objectives and design. Am. J. Epidemiol 156, 871–881. [DOI] [PubMed] [Google Scholar]
- 85.Zeb I, Li D, Nasir K, Katz R, Larijani VN, and Budoff MJ (2012). Computed tomography scans in the evaluation of fatty liver disease in a population based study: the multi-ethnic study of atherosclerosis. Acad. Radiol 19, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.R Core Team (2019). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- 87.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. (2016). Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang G, Sarkar A, Carbonetto P, and Stephens M (2020). A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. B Stat. Methodol 82, 1273–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Van Rossum G, and Drake FL (2009). Python 3 Reference Manual (CreateSpace; ). [Google Scholar]
- 91.Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al. (2016). TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. arXiv, ar-Xiv:1603.04467. https://arxiv.org/abs/1603.04467. [Google Scholar]
- 92.Sarma GP, and Reinertsen E; ML4CVD Group (2020). Physiology as a Lingua Franca for Clinical Machine Learning. Patterns (N Y) 1, 100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Littlejohns TJ, Holliday J, Gibson LM, Garratt S, Oesingmann N, Alfaro-Almagro F, Bell JD, Boultwood C, Collins R, Conroy MC, et al. (2020). The UK Biobank imaging enhancement of 100,000 participants: rationale, data collection, management and future directions. Nat. Commun 11, 2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haas ME, Aragam KG, Emdin CA, Bick AG, Hemani G, Davey Smith G, and Kathiresan S; International Consortium for Blood Pressure (2018). Genetic Association of Albuminuria with Cardiometabolic Disease and Blood Pressure. Am. J. Hum. Genet 103, 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.World Health Organization Global Health Observatory (2018). Standard drink defined by country. https://apps.who.int/gho/data/view.main.56470.
- 96.National Institute for Health and Care Excellence (2011). Alcohol Use Disorders: diagnosis, assessment and management of harmful drinking (high-risk drinking) and alcohol dependence. https://www.nice.org.uk/guidance/cg115.
- 97.Kingma DP, and Ba J (2017). Adam: A Method for Stochastic Optimization. arXiv,arXiv:1412.6980. https://arxiv.org/abs/1412.6980. [Google Scholar]
- 98.Ferrari S, and Cribari-Neto F (2004). Beta Regression for Modelling Rates and Proportions. J. Appl. Stat 31, 799–815. [Google Scholar]
- 99.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. ; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang J, Loos RJF, Powell JE, Medland SE, Speliotes EK, Chasman DI, Rose LM, Thorleifsson G, Steinthorsdottir V, Mägi R, et al. (2012). FTO genotype is associated with phenotypic variability of body mass index. Nature 490, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics for the liver fat GWAS have been deposited in the GWAS Catalog at https://www.ebi.ac.uk/gwas/ and are publicly available under accession number GCST90029073. The machine learning model architectures and weights have been deposited in the ML4H GitHub at https://github.com/broadinstitute/ml4h repository and are publicly available in the ML4H model zoo under the name liver_fat_from_mri_ukb. Liver fat quantification data has been returned to the UK Biobank and can be accessed via application to the UK Biobank at https://www.ukbiobank.ac.uk/. DOIs and accession numbers are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| UK Biobank, including previously quantified liver fat and liver fat quantified via machine learning | 17,80; this paper | https://www.ukbiobank.ac.uk/; df-22402 |
| Liver fat common variant genome-wide association study summary statistics | This paper | https://www.ebi.ac.uk/gwas/ Study: GCST90029073 |
| Liver fat previously quantified in Framingham Heart Study | 81–83 | https://www.ncbi.nlm.nih.gov/gap/; Dataset: pht005157.v3.p13 |
| Liver fat previously quantified in Multi-Ethnic Study of Atherosclerosis | 84,85 | https://www.ncbi.nlm.nih.gov/gap/; Dataset: pht002104.v2.p3 |
| Software and algorithms | ||
| BOLT-LMM version 2.3.4 | 26 | https://alkesgroup.broadinstitute.org/BOLT-LMM/BOLT-LMM_manual.html |
| BOLT-REML in BOLT-LMM version 2.3.4 | 25 | https://alkesgroup.broadinstitute.org/BOLT-LMM/BOLT-LMM_manual.html |
| R statistical software version 3.5 | 86 | http://www.R-project.org/ |
| Michigan Imputation Server version 1.1 | 87 | http://imputationserver.sph.umich.edu/index.html |
| Liver fat machine learning algorithm architecture and weights | This paper | https://github.com/broadinstitute/ml4h (liver_fat_from_mri_ukb in model zoo) |
| PolyFun version 1.0.0 | 32 | https://github.com/omerwe/polyfun |
| SuSiE version 0.9.53 | 88 | https://stephenslab.github.io/susie-paper/ |
| Ensembl Variant Effect Predictor (VEP) version 96 | 89 | https://uswest.ensembl.org/info/docs/tools/vep/index.html |
| LOFTEE | 57 | https://uswest.ensembl.org/info/docs/tools/vep/index.html |
| Python 3 | 90 | https://www.python.org/ |
| tensorflow version 2.1 | 91 | https://www.tensorflow.org/ |
| ML4H version 0.0.1 | 92 | https://github.com/broadinstitute/ml4h |